

Summary
Leucocyte recruitment is critical during many acute and chronic inflammatory diseases. Chemokines are key mediators of leucocyte recruitment during the inflammatory response, by signalling through specific chemokine G‐protein‐coupled receptors (GPCRs). In addition, chemokines interact with cell‐surface glycosaminoglycans (GAGs) to generate a chemotactic gradient. The chemokine interleukin‐8/CXCL8, a prototypical neutrophil chemoattractant, is characterized by a long, highly positively charged GAG‐binding C‐terminal region, absent in most other chemokines. To examine whether the CXCL8 C‐terminal peptide has a modulatory role in GAG binding during neutrophil recruitment, we synthesized the wild‐type CXCL8 C‐terminal [CXCL8 (54–72)] (Peptide 1), a peptide with a substitution of glutamic acid (E) 70 with lysine (K) (Peptide 2) to increase positive charge; and also, a scrambled sequence peptide (Peptide 3). Surface plasmon resonance showed that Peptide 1, corresponding to the core CXCL8 GAG‐binding region, binds to GAG but Peptide 2 binding was detected at lower concentrations. In the absence of cellular GAG, the peptides did not affect CXCL8‐induced calcium signalling or neutrophil chemotaxis along a diffusion gradient, suggesting no effect on GPCR binding. All peptides equally inhibited neutrophil adhesion to endothelial cells under physiological flow conditions. Peptide 2, with its greater positive charge and binding to polyanionic GAG, inhibited CXCL8‐induced neutrophil transendothelial migration. Our studies suggest that the E70K CXCL8 peptide, may serve as a lead molecule for further development of therapeutic inhibitors of neutrophil‐mediated inflammation based on modulation of chemokine–GAG binding.
Keywords: chemokine, CXCL8, glycosaminoglycan, inflammation, neutrophil migration, structure–function, synthetic chemistry
Martínez‐Burgo et al. describe the development of a C‐terminal CXCL8 peptide based on chemokine–glycosaminoglycan (GAG) interactions which reduces neutrophil adhesion and migration during inflammation. The model proposes the therapeutic potential of the peptide to modulate chemokine function by displacing chemokine from cell surface GAG potentially interfering with the formation of the chemokine gradient.

Abbreviations
- BSA
bovine serum albumin
- GAG
glycosaminoglycan
- GPCR
G‐protein‐coupled receptor
- HBSS
Hank's balanced salt solution
- HMECs
human microvascular endothelial cells
- HUVECs
human umbilical vein endothelial cells
- ICAM
intercellular adhesion molecule type 1
- MALDI‐TOF
matrix‐assisted laser desorption/ionization time‐of‐flight
- POSAT
Prolong Organ Survival After Transplantation (project acronym)
- PTM
post‐translational modification
- RP‐HPLC
reverse‐phase high‐performance liquid chromatography
- RU
resonance units/response units
- SA
streptavidin
- SPR
surface plasmon resonance
- TNF‐α
tumour necrosis factor‐α
Introduction
Leucocyte recruitment, a hallmark of the inflammatory response, is a crucial component of many acute and chronic inflammatory situations.1, 2, 3 Chemokines are small, soluble chemotactic proteins that co‐ordinate leucocyte recruitment.4 They can be expressed in response to pro‐inflammatory mediators such as the cytokines tumour necrosis factor (TNF), interferon‐γ or interleukin‐1β. Chemokines recruit leucocytes to a site of injury, by binding to the endothelium via glycosaminoglycans (GAGs), forming a chemokine gradient and activating integrins, which allow leucocyte adhesion. In addition, chemokines are involved in many other processes such as angiogenesis, proliferation, development and the control of leucocyte mobilization from primary or secondary lymphoid organs.5, 6, 7, 8, 9 Chemokine function depends, among many other factors, on their signalling via specific chemokine G‐protein‐coupled receptors (GPCRs). The interaction between a chemokine and its receptor is an attractive therapeutic target in many diseases, including rheumatoid arthritis,10, 11, 12 psoriasis,13 or in acute and chronic organ damage after ischaemia reperfusion injury following transplantation.14, 15
Studies that have focused on the chemokine interaction with GPCRs have led to the development of several neutralizing antibodies, modified chemokines and antagonists.16, 17, 18, 19, 20, 21 However, to date, only two chemokine receptor antagonists have been fully validated and marketed as therapeutics: Maraviroc (a CCR5 antagonist) and AMD3100 (a CXCR4 antagonist).22, 23, 24 These two antagonists are not used as anti‐inflammatory drugs, but rather as a human immunodeficiency virus entry inhibitor, and as a haematopoietic stem cell mobilizer during transplantation, respectively. The challenge of targeting chemokines in anti‐inflammatory therapy arises primarily from the apparent redundancy within the human chemokine system.25, 26
In addition to the well‐characterized, high‐affinity interaction of chemokines with their specific GPCRs, chemokine activity in vivo also depends on their interaction with GAGs, such as endothelial heparan sulphate.21, 27 GAGs are ubiquitously present on cell surfaces and in the extracellular matrix. They are thought to inhibit chemokine diffusion, recruiting chemokines at high concentration forming a gradient towards the site of injury.28, 29, 30 The highly sulphated and acidic GAGs bind to basic residues within chemokines largely through electrostatic forces, but also through Van der Waals interactions and hydrogen bonding. This usually involves residues such as arginine, lysine or histidine, which typically form the BBXB or (B)BXX(X/B)BXXB(B) peptide sequence signature, where B is a basic amino acid residue and X is a non‐conserved amino acid, which is present in virtually all chemokines.27 The importance of the chemokine–GAG interaction is highlighted by studies that have selectively targeted either GAG or GPCR binding domains. For example, chemokines with increased GAG binding but decreased GPCR binding, show anti‐inflammatory activity in in vivo models of CXCL8/neutrophil‐driven inflammation presumably by disrupting the natural chemokine gradient.31
Levels of CXCL8 significantly increase during the inflammatory response associated with ischaemia reperfusion injury,32, 33 which can lead to acute kidney injury34, 35 and transplant rejection.36, 37, 38 CXCL8 expressed at high concentrations on the endothelial GAG surface at the site of injury contributes to neutrophil firm arrest, by activation of integrins.39 Therefore, modulation of a CXCL8 haptotactic gradient might have potential in ameliorating the ischaemia reperfusion injury and therefore improve organ function.30, 32, 34 Therapeutic targeting of CXCL8 and its association with heparan sulphate has been investigated in numerous neutrophil‐driven inflammatory diseases such as chronic obstructive pulmonary disease, Crohn's disease and psoriasis.40 A CXCL8‐based decoy protein named PA401, with decreased GPCR binding and increased GAG binding, decreased CXCL8‐mediated neutrophil recruitment in in vivo studies, suggesting its translational potential for the treatment of respiratory diseases such as chronic obstructive pulmonary disease or cystic fibrosis.41
The C‐terminal α‐helical region of CXCL8 is known to be critical for GAG binding (Fig. 1), largely due to its positive electrostatic charge giving it micromolar affinity for negatively charged GAGs.29, 42, 43, 44 This binding is mediated by core residues H18, K20, R60, K64, K67 and R68, as shown in Fig. 1, where known CXCL8‐receptor binding residues are also highlighted.
Figure 1.
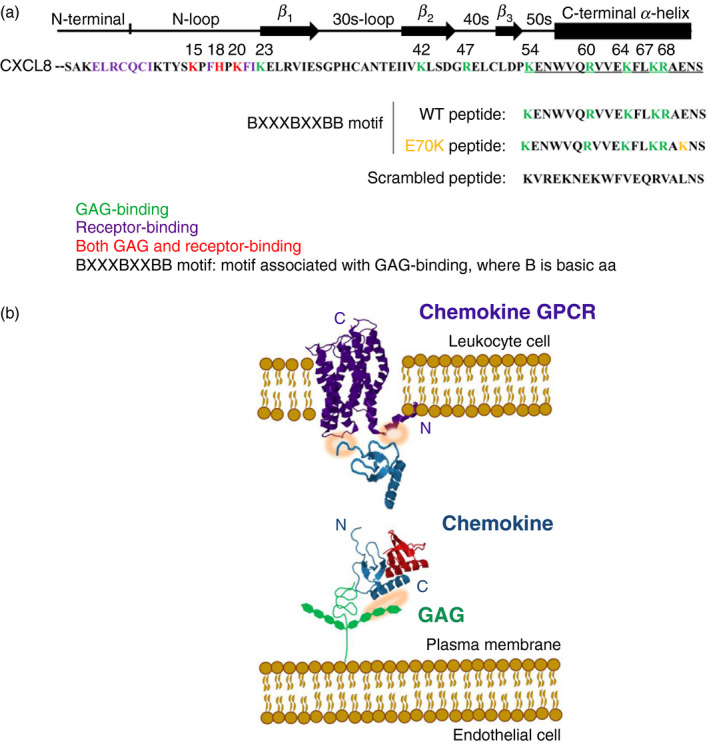
(a) CXCL8 active sequence. (b) Schematic representation of the chemokine binding to the endothelial glycosaminoglycan (GAG) and to the leucocyte chemokine G‐protein coupled receptor (GPCR). (a) Sequence of the most common active CXCL8 form (amino acids 28–99), with 72 amino acids. Green: GAG‐binding residues. Purple: GPCR‐binding residues. Red: residues involved in both GAG‐ and receptor‐binding. Underlined amino acids: C‐terminal α‐helix region selected for chemical synthesis. (b) Schematic representation of chemokine (Protein Data Bank ID 1IL‐8/CXCL8) interaction with endothelial surface through GAG (residues involved highlighted in orange), which enables subsequent high‐affinity chemokine binding to leucocyte CXCR1/2 GPCR receptor (Protein Data Bank ID 2LNL; also highlighted in orange). Chemokine monomer is shown in blue and the dimer is depicted with one molecule in blue and the other in red. Note that illustration shows one potential scenario of chemokine binding.
In this study, we aimed to assess whether the CXCL8 C‐terminal peptide (54–72) could modulate CXCL8 function. We synthesized the CXCL8 wild‐type C‐terminal region (54–72) (wild‐type peptide, Peptide 1), a peptide with substitution of glutamic acid (E) 70 with lysine (K), in order to increase the peptide positive charge, and hence its GAG‐binding potential (Peptide 2), and a scrambled peptide containing the wild‐type amino acids (Peptide 3; Fig. 1). The biophysical properties of the peptides and their potential biological functions, using in vitro cytokine‐mediated neutrophil flow‐based adhesion and transendothelial migration studies, were investigated.
Materials and methods
Human neutrophil isolation
Primary neutrophils were isolated from whole blood of healthy volunteers. Ethical approval to obtain blood from healthy volunteers was granted by the County Durham and Tees Valley Research Ethics Committee (12/NE/0121). Primary neutrophils were isolated by dextran sedimentation (Dextran T500; Pharmacosmos, Holbaek, Denmark) and centrifugation on Percoll (GE Healthcare, Buckinghamshire, UK) density gradients as previously described.45
Synthesis of chemokine peptides
The chemokine C‐terminal peptides (Peptides 1–3) were synthesized on Rink Amide resin using Fmoc solid‐phase peptide synthesis on a CEM Liberty 1 single‐channel microwave peptide synthesizer equipped with a Discover microwave unit, as described earlier.46 After synthesis, peptides were acetylated at the N‐terminal (20% acetic anhydride), having amide at the C‐terminal. They were then cleaved from the resin, and crude peptides were purified by semi‐prep reverse‐phase high‐performance liquid chromatography (RP‐HPLC). Then peptides were characterized by matrix‐assisted laser desorption/ionization time‐of‐flight (MALDI‐TOF) using an Autoflex II ToF/ToF mass spectrometer (Bruker Daltonik GmbH, Coventry, UK), and using the Pep‐Calc calculator to analyse the sequence47 and the obtained mass spectrometry spectra. Following this, analytical RP‐HPLC was used to examine the pure peptide. Chemokine peptides were initially synthesized at Durham University Chemistry Department (Durham, UK), and further synthesized by ISCA Biochemicals (Exeter, UK) (>95% purity).
Circular dichroism spectroscopy
Far‐UV circular dichroism spectroscopy was conducted using a Jasco J‐810 spectropolarimeter (Jasco GmbH, Gross‐Umstadt, Germany) in the range of 240–197 nm wavelength, with a 1‐mm path length and a 500‐μl quartz cuvette. Peptide samples (Peptide 1, Peptide 2 or Peptide 3) were diluted 5–100 μm in phosphate‐buffered saline. For the measurements, 300 μl peptide solution was transferred to a cuvette. All data collection was taken at room temperature, and the mean spectrum derived from five to ten scans was corrected by subtraction of the buffer blank, as previously reported.48 For samples of peptide combined with heparin (Sigma‐Aldrich, St Louis, MO), the spectrum was also corrected by subtraction of a heparin blank. Scans were conducted at 50 nm/min, 1 nm data pitch, 5 mdeg sensitivity and with a 2‐second response.49
Surface plasmon resonance
Surface plasmon resonance (SPR) was performed using a BIAcore X100 as previously described.50 The running buffer used was HBS‐P (10 mm HEPES pH 7·4, 150 mm NaCl, 0·005% Tween‐20). Unless otherwise stated all reagents were from GE Healthcare (Uppsala, Sweden). To allow immobilization onto the streptavidin (SA)‐coated chip, biotinylated GAG heparin was obtained as previously described50, 51, 52 (generously provided by Prof. Hughes Lortat‐Jacob's Laboratory, Institute of Structural Biology, Grenoble, France). Mono‐biotinylation at the reducing end of the GAG is important for correct presentation when immobilized. Between 5 and 20 μg/ml biotinylated heparin in 300 mm NaCl was injected at 10 μl/min for 30 seconds followed by a 2 m NaCl wash to remove unbound heparin. Injections were repeated until a total resonance units of 200 was achieved. Following preparation of the chip surface, SPR assays assessed the GAG‐binding properties of CXCL8; and synthesized peptides (Peptide 1, Peptide 2 and Peptide 3). A range of CXCL8 concentrations (50–1000 nm; CN‐09; Almac, Edinburgh, UK) were flowed across the chip at 5 μl/min for 5 min followed by a 500‐second dissociation phase and their resonance units were measured. The same conditions were applied to the peptides analysed at concentrations from 2500 nm to 10 000 μm. After every chemokine or chemokine peptide measurement, regeneration buffer was used to remove sample from the chip surface (10 mm HEPES, 2 m NaCl, 50 mm EDTA, 0·005% Tween‐20). Binding was calculated by subtraction of the resonance units of the SA flow cell from the resonance units of the GAG‐SA flow cell. Data analysis was performed using BIA evaluation 4.1.
Solute diffusion gradient chemotaxis and transendothelial chemotaxis of neutrophils
Chemotaxis experiments were performed using a Transwell system (Falcon, BD Biosciences, Oxford, UK), as previously reported.53 First, 24‐well companion plates (Falcon, BD Biosciences) were blocked with 1 ml 1% bovine serum albumin (BSA) (Sigma‐Aldrich)/ RPMI (Lonza, Wokingham, UK) per well for 1 hr before the assay to prevent chemokine binding and consequent decreased chemokine concentration. Then, 800 μl of 10 nm chemokine, after optimization (data not shown) and as earlier described,54, 55, 56 or chemokine peptide in a range of 0·1–10 000 nm in 1% BSA/RPMI was added to each well. Cell culture inserts (3‐μm pore size; Falcon, BD Biosciences) formed the transwell upper chamber where 500 μl of 3 × 105 primary neutrophils in 1% BSA/RPMI were added. Wells containing 1% BSA/RPMI only on the transwell bottom chamber were used as a negative control. The plate was then incubated at 37° for 90 min. After incubation, cells that had fully migrated to the transwell lower chamber were counted by flow cytometry as a ratio to the known number of counting beads. For transendothelial chemotaxis, 3 days before the assay human microvascular endothelial cells (HMECs; ATCC CRL‐3243)57, 58 were seeded onto the transwell upper chamber using 500 μl of 2 × 105 HMECs per insert in MCDB‐131 media (10372019) (Thermo Fisher, Waltham, MA) with 10% fetal bovine serum as earlier described.59, 60 MCDB‐131 medium was then carefully aspirated before the assay. Anti‐intercellular adhesion molecule type 1 (ICAM‐1) blocking monoclonal antibody (HA58) (eBioscience; Thermo Fisher, Waltham, MA) and IgG1 κ isotype control (MOPC‐21) (BD Biosciences, San Jose, CA) were used to treat the HMEC layer at 20 μg/ml in 0·5% BSA/phosphate‐buffered saline for 30 min at room temperature.
Calcium signalling
Intracellular calcium was measured loading cells with Indo‐1, AM (Thermo Fisher Waltham, MA). For each tube, 3 million neutrophils were used. Freshly isolated neutrophils were first left to rest in an incubator for about 15 min, and then used for the experiment. Cells were washed in Hank's balanced salt solution (HBSS; Sigma‐Aldrich) and resuspended at 10 million cells/ml. Then, cells were washed in HBSS supplemented with 1 mm CaCl2, 1 mm MgCl2, 1% fetal bovine serum (volume/volume). Once cells were washed, they were loaded with 3 μm indo‐1, AM, and incubated for 30 min at 37° covered in foil. After the 30 min of indo‐1, AM incubation, cells were washed with supplemented HBSS at 400 g for 5 min, then resuspended at 3 million cells per 1·5 ml in their corresponding FACS tube and left to rest for 30 min at 37° before analysis. Calcium flux was measured by FACS‐Fortessa flow cytometry, using UV filter 530/30. Once settings were adjusted with unstained cells at low flow rate, the stained cells were run. As baseline, stained untreated cells (HBSS only) were first run for 1 min at medium flow. Then 1 μl HBSS or chemokine was added for 4 min, and then 8 μl ionomycin (I0634) (Sigma‐Aldrich) was added for 2 min. Cells were studied for the effect of CXCL8 on calcium flux and compared with the effect of CXCL8 combined with Peptide 1, Peptide 2 or Peptide 3. Calculation of intracellular calcium concentrations, measured in terms of the light emission as a ratio of fluorescence intensities at 340 and 380 nm, was carried out using the equation [calcium (nmol/l)] = K d × (R − R min)/(R max − R), where K d (844 nmol/l) is the dissociation constant of calcium bound to the fluorochrome61 and R is the peak intracellular calcium flux in response to the additive (chemokine or chemokine peptide). The basal concentration (HBSS, negative control) was subtracted to calculate the values.
Flow‐based neutrophil adhesion
In order to evaluate the neutrophil adhesion in response to chemokine or chemokine peptide under physiological in vitro conditions, the Venaflux platform (Cellix Ltd., Dublin, Ireland) was used, in a similar way to previous studies.62, 63, 64 To accommodate an endothelial layer on the biochip platform for neutrophil perfusion, the Vena8 Endothelial+ chip was initially coated with 10 μl 100 μg/ml fibronectin (Sigma‐Aldrich). Coated biochip was stored in a closed humidified chamber overnight at 4°. On the first day, Human Umbilical Vein Endothelial Cells (HUVECs; C‐12203; PromoCell, Heidelberg, Germany) were treated in a 75‐cm2 flask with 1 ng/ml TNF (210‐TA‐010; R&D Systems, Minneapolis, MN) overnight at 37°.65 Next day, the fibronectin‐coated Vena8 Endo+ biochip was seeded with 10 μl of HUVECs (at 1·5 million per 100 μl), used as negative control, or with TNF‐stimulated HUVECs, as positive control. A HUVEC layer was generated within 1–1·5 hr of seeding. For this, the addition of 40 μl of extra culture medium to each channel reservoir was required 10–15 min after HUVEC seeding to humidify the channel and generate the endothelial layer. Afterwards, chemokine treatment was carried out. The seeded biochip channel was treated with chemokine (20 nm), chemokine peptide (50 nm; Peptide 1, Peptide 2 or Peptide 3); or low‐molecular‐weight heparin, tinzaparin (50 nm; Leo Pharmaceuticals, Ballerup, Denmark), to analyse their potential role in displacing the chemokine from GAG.66 In parallel, different CXCR1/2 antagonists [repertaxin (Cayman Chemical, Cambridge, UK) and SB225002 (SML0716; Sigma‐Aldrich)], and CXCR2 antagonist SB265610 (SML0421; Sigma‐Aldrich) were used at 50 nm – to analyse their role in displacing the chemokine from GPCR67 – treating neutrophils before the assay. A 10‐μl treatment was inserted into each channel, followed by careful addition of 40 μl of the treatment on to each channel reservoir. The effect of each treatment on the neutrophil flow‐based adhesion was evaluated using the Venaflux platform; 3 × 105 primary neutrophils were flowed per ml through each biochip channel and analysed. Cell adhesion analysis was performed using imagej Analysis Software. Cell adhesion count for each treatment was calculated from the average of five standard fields of view of adhered neutrophils.
Data analysis
Data were analysed using prism7c software (GraphPad Software Inc., La Jolla, CA). Each graph column denotes mean and each bar indicates standard error of the mean (SEM). P values were calculated using one‐way statistical analysis of variance followed by Bonferroni's post hoc test, with significant differences when P < 0·05, highly significant when P < 0·01, and extremely significant when P < 0·001 or P < 0·0001.
Results
Design, synthesis and biophysical characterization of CXCL8 C‐terminal peptide
The wild‐type C‐terminal region of CXCL8 [CXCL8 (54–72)] (Peptide 1), the E70K peptide (Peptide 2), and a scrambled peptide with the same amino acids as the wild‐type peptide in a random order, (Peptide 3), were synthesized using Fmoc solid‐phase peptide synthesis on Rink Amide resin (see Supplementary material, Fig. S1). The purified peptides were characterized by MALDI‐TOF and analytical RP‐HPLC. A summary of yields and purity for the three peptides is shown in Table 1. Circular dichroism was used to determine the structure of synthesized peptides alone and in comparison with peptides combined with heparin. All peptides showed an extended, non‐helical or random coil structure, different to the α‐helix structure of this region within full‐length CXCL8. However, Peptide 1 and Peptide 2 in solution with heparin showed a minor change in structure, not seen with Peptide 3, indicating a potential interaction between CXCL8‐derived peptide and heparin (see Supplementary material, Fig. S2).
Table 1.
Summary of yield and purity obtained for each synthesized peptide
| Peptide | Chemokine region | Yielda | Purityb |
|---|---|---|---|
| WT (Peptide 1) | WT C‐terminal | 60·4% | Approx. 95% |
| E70K (Peptide 2) | E70K C‐terminal | 10·4% | Approx. 95% |
| Scrambled (Peptide 3) | Scrambled from C‐terminal | 12·7% | Approx. 95% |
Yield is calculated comparing the dry mass of pure peptide with the mass of crude peptide [theoretical mass at 100% yield based on the 0·1 mmol resin (0·1 mmol peptide) = 100% peptide = mass of peptide (mg)].
Purity is obtained from analytical high performance liquid chromatography.
Binding of CXCL8 C‐terminal peptides to GAG‐heparin
To assess the GAG‐binding ability of synthesized C‐terminal peptides, SPR binding studies were performed. We first evaluated the binding of CXCL8 to a heparin‐coated chip following established protocols.68 Then, binding of each synthesized peptide was studied, to evaluate affinity for heparin. Heparin‐CXCL8 SPR confirmed binding68, 69 as shown in Fig. 2. Peptide binding was only detectable at much higher concentrations of Peptides 1 and 3 (10 mm), >104‐fold higher than with full‐length CXCL8 (the sensorgram with magnified y‐axis of binding of Peptides 1 and 3 is shown in the Supplementary material, Fig. S3). The E70K peptide (Peptide 2; charge +4), showed significant binding at lower concentrations (5 mm) than the other peptides (charge +2), but this was still a much higher concentration than full‐length CXCL8 (Fig. 2).
Figure 2.
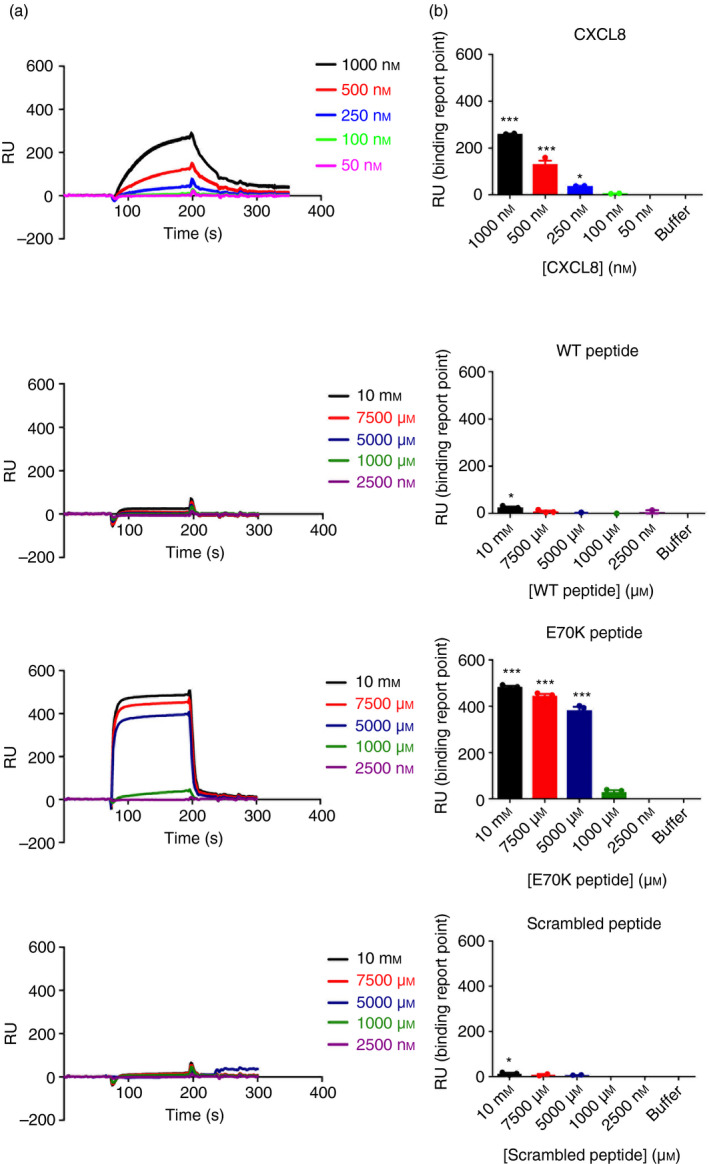
Surface plasmon resonance of CXCL8 peptide–heparin binding. (a) Surface plasmon resonance sensorgram shows heparin–CXCL8 binding in the range of 50–1000 nm CXCL8, and heparin–CXCL8 peptide binding in the range of 2·5–10 000 μm peptide. Chemokine or peptide were flowed at 5 μl/min over the chip. (b) Binding shown for each chemokine or peptide concentration. Sensorgram with magnified y‐axis of binding of wild‐type (WT) peptide, and scrambled peptide is shown in Supplementary material (Fig. S3). Data were analysed by one‐way analysis of variance (P < 0·0001) followed by Bonferroni post‐hoc test. *P < 0·05, ***P < 0·001. Data are representative of three independent experiments over a single heparin‐coated SA chip.
CXCL8 C‐terminal peptides do not interfere with GPCR‐mediated signalling
The peptides were predicted to bind endothelial GAGs. To determine whether the peptides also had a role in GPCR‐binding, all three peptides were evaluated by CXCL8‐diffusion gradient chemotaxis and CXCL8‐mediated calcium signalling. The peptides had no significant effect on CXCL8‐diffusion gradient chemotaxis (Fig. 3). Data on CXCL8‐mediated neutrophil calcium signalling was consistent with the diffusion gradient chemotaxis. Neutrophil calcium increased in response to CXCL8 stimulation, but no change was seen with the peptides alone. The combination of CXCL8 with each of the synthesized peptides did not affect calcium flux compared with CXCL8 alone (Fig. 4). Hence, data suggested that the peptides do not interfere with chemokine–GPCR binding.
Figure 3.
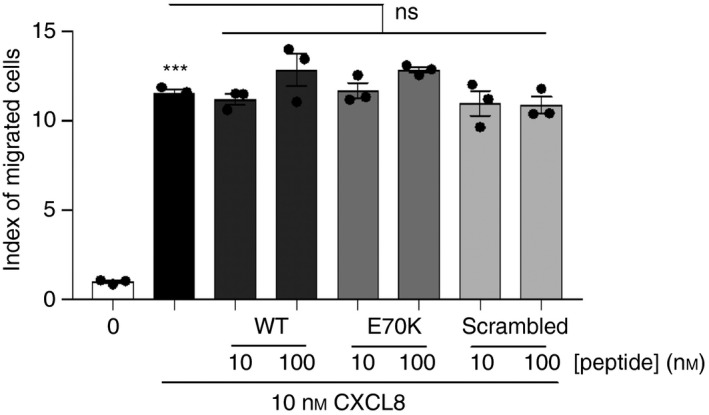
Diffusion gradient migration in response to CXCL8 combined with each peptide. For positive control, 10 nm CXCL8 was used. Synthesized CXCL8 C‐terminal peptides (10 or 100 nm) showed no interference with neutrophil migration in the absence of endothelial glycosaminoglycan (GAG) surface, which suggests no binding to CXCR1/2 receptors. Wild‐type (WT)/Peptide 1 (KENWVQRVVEKFLKRAENS); E70K/Peptide 2 (KENWVQRVVEKFLKRAKNS); or scrambled/Peptide 3 (KVREKNEKWFVEQRVALNS) were studied. Index of migrated cells or chemotaxis index (CI) is the ratio between the total number of migrated neutrophils and the number of neutrophils that migrated non‐specifically, and was calculated for each treatment. Data were analysed by one‐way analysis of variance (P < 0·0001) followed by Bonferroni post‐hoc test. ***P < 0·001 shows significant migration in response to CXCL8 compared with negative control; ns, not significant. Representative data of three independent experiments (n = 3), each performed in triplicate.
Figure 4.
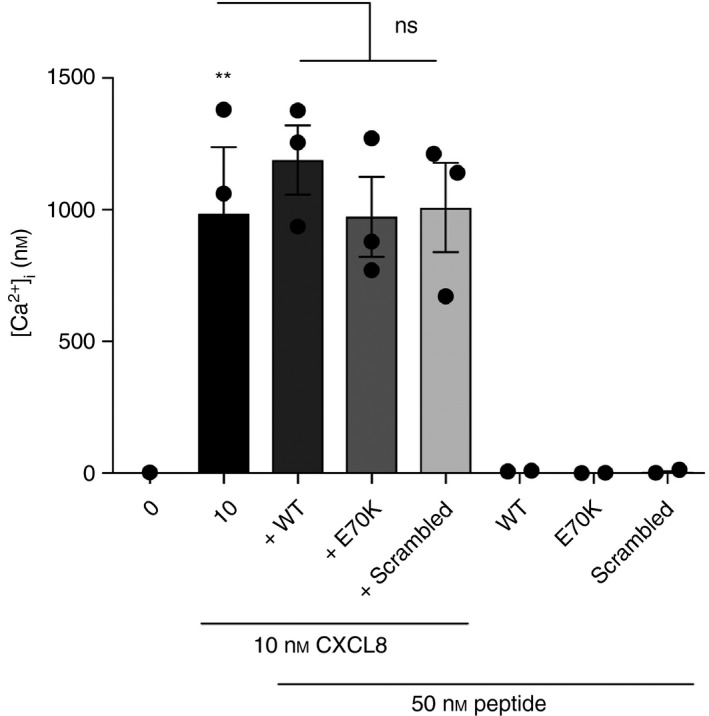
Calcium flux in response to CXCL8 combined with each peptide. Intracellular calcium ([Ca2+]i) was measured in response to CXCL8, or CXCL8 combined with each peptide [wild‐type (WT)/Peptide 1: KENWVQRVVEKFLKRAENS; E70K/Peptide 2: KENWVQRVVEKFLKRAKNS; or scrambled/Peptide 3: KVREKNEKWFVEQRVALNS]. Primary blood neutrophils were labelled with Indo‐1, AM. Then, cells were analysed in response to Hank's balanced salt solution (HBSS) only (negative control), 10 nm CXCL8 (positive control) or CXCL8 combined with each peptide at 50 nm, within the range of 10–100 nm. Data were analysed by one‐way analysis of variance (P < 0·0001) followed by Bonferroni post‐hoc test. **P < 0·01 shows significant calcium flux in response to CXCL8 compared with the negative control. ns, no significant. Data are representative of three independent experiments (n = 3).
C‐terminal peptides inhibit neutrophil flow‐based adhesion to endothelial cells
A schematic representation of the endothelial biochip seeding, and subsequent leucocyte flow‐based adhesion is shown in Fig. 5. Primary neutrophil adhesion in response to TNF‐stimulated, CXCL8‐treated HUVECs was used as positive control. Cytokine‐mediated neutrophil flow‐based adhesion was reduced in the presence of 50 nm of all three peptides (wild‐type peptide and scrambled peptide P < 0·01; E70K peptide P < 0·001). Similarities between the peptides suggest that short positively charged peptides, all containing Lys and Arg residues, interfere non‐specifically or with functional redundancy with chemokine‐activated neutrophil adhesion to the endothelium under physiological flow conditions (Fig. 5).
Figure 5.
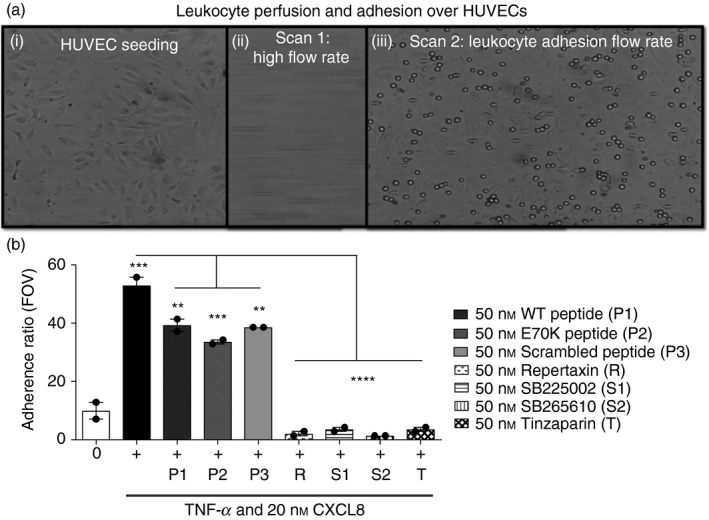
Schematic representation of leucocyte perfusion and adhesion over primary human umbilical vein endothelial cells (HUVECs). (a) (i) First, HUVECs were seeded over the fibronectin‐coated biochip. (ii) Next, leucocytes were loaded onto the endothelial layer and initially perfused at a high flow rate, −10 dynes/cm2 for 10 seconds, to allow leucocyte circulation over the chip (negative flow, towards pump). (iii) Leucocyte adhesion was then analysed at a more physiological flow rate, −0·5 dynes/cm2 for 3 min. Leucocytes were fluorescently labelled using 1 μm (DIOC 6)3. (b) Flow‐based adhesion of primary neutrophils in the presence of different modulators. Negative control is untreated HUVECs (fibronectin only). Positive control is tumour necrosis factor (TNF) ‐stimulated HUVECs with 20 nm CXCL8 (100 μg/ml fibronectin, 1 ng/ml TNF/TNF‐α). CXCL8 (20 nm) and CXCL8 peptide (50 nm) were added over TNF‐stimulated HUVECs and neutrophil adhesion was analysed after 1 hr of treatment. HUVECs were treated with low‐molecular‐weight heparin (LMWH) tinzaparin at 50 nm for 1 hr before performing the assay. Neutrophils were treated with each CXCR1&2 antagonist [Repertaxin (R); or SB225002 (S1)] or CXCR2 antagonist (SB265610) (S2) at 50 nm for 1 hr before the assay. Adherence ratio, obtained from the average of five fields of view per channel of chip, is the ratio between the total number of adhered neutrophils and the number of neutrophils that adhered non‐specifically. Wild‐type (WT)/Peptide 1 (P1) is KENWVQRVVEKFLKRAENS; E70K/Peptide 2 (P2) is KENWVQRVVEKFLKRAKNS; scrambled/Peptide 3 (P3) is KVREKNEKWFVEQRVALNS. Data were analysed by one‐way analysis of variance (P < 0·0001) followed by Bonferroni post‐hoc test. **P < 0·01, ***P < 0·001, ****P < 0·0001. Representative data of three independent experiments (n = 3).
Further studies performed with the low‐molecular‐weight heparin tinzaparin showed significant chemokine displacement and inhibition of flow‐based chemokine‐mediated neutrophil adhesion (P < 0·0001).
In addition, studies using the CXCR1/2 chemokine receptor antagonists repertaxin, SB225002 or SB265610 led to significant inhibition of GPCR‐chemokine binding as shown by significantly reduced neutrophil flow‐based adhesion (P < 0·0001).
E70K peptide inhibits neutrophil transendothelial migration
To further investigate CXCL8 C‐terminal peptide binding to endothelial GAG, their potential to block CXCL8‐mediated transendothelial neutrophil migration was evaluated. There was no significant effect of Peptide 1 or Peptide 3 on neutrophil transendothelial chemotaxis. Peptide 2, E70K, reduced CXCL8‐mediated neutrophil transendothelial migration (P < 0·001; Fig. 6; see Supplementary material, Fig. S4). Primary neutrophils express several cell‐surface proteins involved in endothelial adhesion, in addition to high levels of the CXCL8 receptors, CXCR1 and CXCR2 (see Supplementary material, Fig. S5). This may partly explain why CXCL8‐displacing peptides do not fully inhibit neutrophil migration. To determine whether blocking the function of other proteins involved in transendothelial migration would further interfere in the process, we combined the E70K peptide with an ICAM‐1 blocking monoclonal antibody. As previously described, blocking ICAM‐1 alone did not affect neutrophil transendothelial migration.70 When ICAM‐1 blockade was combined with E70K there was a significant reduction in neutrophil endothelial transmigration; however, this was not greater that E70K alone, suggesting no synergistic interaction (Fig. 7). This proposes the therapeutic potential of E70K peptide to modulate chemokine function by interfering with chemokine–GAG binding, potentially interfering with the formation of the chemokine gradient.
Figure 6.
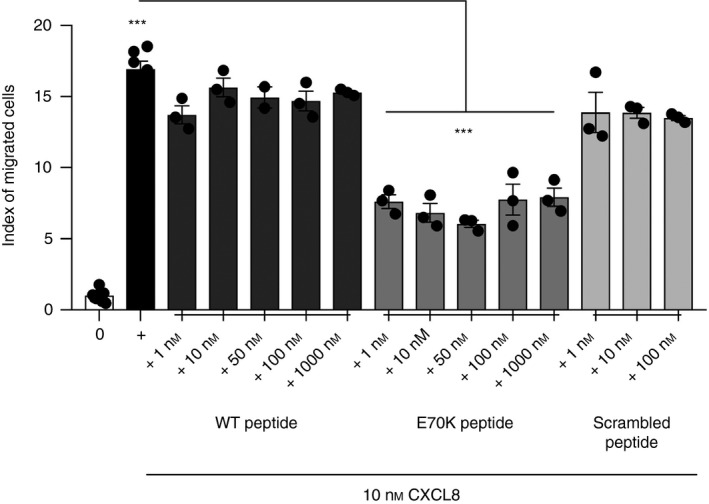
Neutrophil transendothelial migration directed by CXCL8 combined with peptide. Neutrophil response to CXCL8 (10 nm), or to CXCL8 combined with each peptide, at 1–1000 nm [wild‐type (WT)/Peptide 1: KENWVQRVVEKFLKRAENS; E70K/Peptide 2: KENWVQRVVEKFLKRAKNS; or scrambled/Peptide 3: KVREKNEKWFVEQRVALNS] was measured. Cell counts were performed using counting beads by flow cytometry. Index of migrated cells or chemotaxis index (CI) is the ratio between the total number of migrated neutrophils and the number of neutrophils that migrated non‐specifically. Further titration of peptides is shown in the Supplementary material (Fig. S4). Data were analysed by one‐way analysis of variance (P < 0·0001) followed by Bonferroni post‐hoc test. ***P < 0·001 on black column indicates significant migration in response to CXCL8 compared with negative control. Data are representative of two independent experiments (n = 2) from different primary neutrophil preparations, each performed in triplicate.
Figure 7.
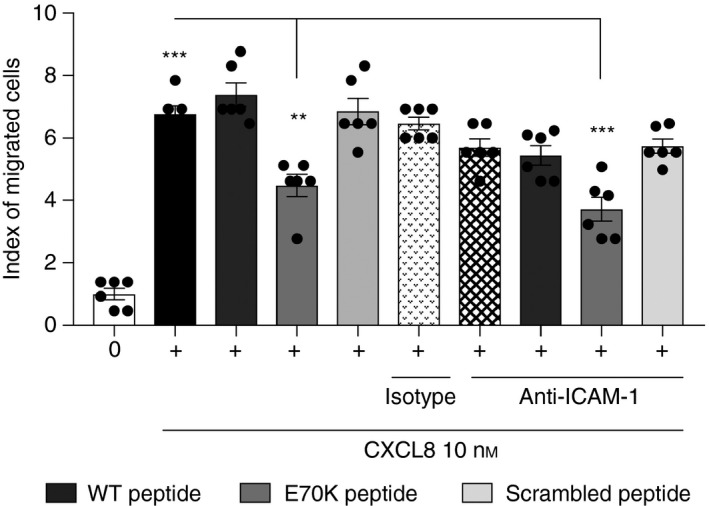
Neutrophil transendothelial migration directed by CXCL8 can be inhibited by the E70K peptide. A similar effect was shown when the peptide was combined with intercellular adhesion molecule type 1 (ICAM‐1) blocking antibody. Neutrophil response to CXCL8 (10 nm), or to CXCL8 combined with each peptide, at 50 nm [wild‐type (WT)/Peptide 1: KENWVQRVVEKFLKRAENS; E70K/Peptide 2: KENWVQRVVEKFLKRAKNS; or scrambled/Peptide 3: KVREKNEKWFVEQRVALNS] was measured. Human microvascular endothelial cells (HMECs) were treated with ICAM‐1 blocking antibody. Cell counting was performed using a counting chamber. Index of migrated cells or chemotaxis index (CI) is the ratio between the total number of migrated neutrophils and the number of neutrophils that migrated non‐specifically. Data were analysed by one‐way analysis of variance (P < 0·0001) followed by Bonferroni post‐hoc test. **P < 0·01. ***P < 0·001. *** in black column indicates significant migration in response to CXCL8 compared with negative control. Data are representative of three independent experiments (n = 3) from different primary neutrophil preparations, each performed in duplicate.
Discussion
Targeting chemokine–GPCR binding has been clinically approved for two indications. However, there are numerous examples in pre‐clinical studies that suggest they have great potential to modify inflammatory responses during disease.22, 23, 24, 71 The regulation of chemokine function by GAG binding using chemokine peptides in vivo has previously been investigated,9, 41, 72 but its translational potential has not been fully explored. Here, to better understand the regulation of chemokine function by GAG binding, chemokine‐derived peptides were synthesized. All peptides showed low‐affinity, but significant, GAG binding in a charge‐dependent manner, presumably through electrostatic interactions. Chemotaxis and calcium signalling studies confirmed that peptides lacked GPCR antagonist function. The C‐terminal peptides showed a significant reduction in flow‐based neutrophil adhesion; however, no difference was observed between the peptides. This suggests that integrin‐mediated neutrophil–endothelium adhesion, which is stimulated by cytokines, can be modulated by all the positively charged peptides tested under physiological flow rate. GAG binding of these peptides may not require a defined three‐dimensional structure. Neutrophil transendothelial chemotaxis assays showed that only Peptide 2, with its higher positive charge, significantly reduced neutrophil migration. Peptide 2 has a charge of +4, which is higher than the wild‐type peptide (Peptide 1) or scrambled peptide (Peptide 3; charge +2). We propose that the higher charge increases the affinity for GAG binding, and this contributes to chemokine displacement from cell surface GAGs disrupting the chemokine gradient (Fig. 8).
Figure 8.
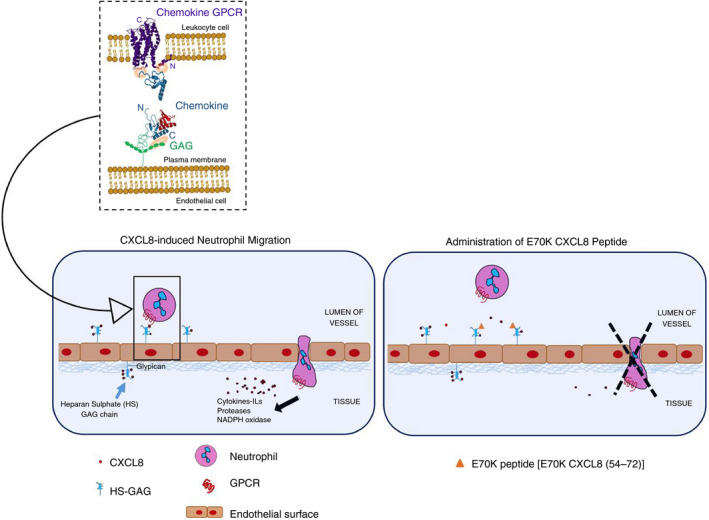
The proposed modulatory activity of E70K CXCL8 peptide in in vitro models of neutrophil flow‐based adhesion and migration during inflammation. This model proposes the therapeutic potential of E70K peptide to modulate chemokine function by displacing chemokine from cell surface glycosaminoglycan, potentially interfering with the formation of the chemokine gradient.
Alternative approaches to enhance the peptide–GAG binding to increase its ability to displace chemokine could include further substitution of positively charged residues in the CXCL8 GAG‐binding region; study of potential folding of unfolded states of the truncated chemokine region; or the development of cyclic peptides;73, 74 or stapled peptides to stabilize an α‐helical structure.75 Furthermore, the inclusion of non‐standard amino acids is another strategy to increase the peptide stability against proteolytic cleavage.76 Also, it might be of interest to study potential peptide oligomerization, as it could further increase GAG binding.29, 42, 43, 77, 78 These strategies might facilitate the impairment of the chemokine‐mediated neutrophil recruitment to ameliorate the injury associated with neutrophil‐mediated inflammation, such as in ischaemia reperfusion injury during transplantation, or in rheumatoid arthritis.79
Mice express only CXCL8 homologues, KC/CXCL1 and MIP‐2/CXCL2. The human CXCL8 C‐terminal peptide used (54–72 amino acids) shares 32% identity and 21% identity with murine homologues (within KC/CXCL1 and MIP‐2/CXCL2), respectively.80 This makes targeting C‐terminal domain function in mouse models more difficult. In order to study the potential role of E70K peptide in vivo, a murine air pouch model of inflammation was used as optimized previously by our group.81, 82 However, no significant effect was observed (data not shown), which may reflect the degree of sequence difference described above; or it might have an inhibitory effect only in a specific environment. Alternative animal models, such as a humanized mouse model,83 or additional physiological studies could further probe the translational role of peptides.
Moreover, analysis of the effect of CXCL8‐derived peptides on other factors such as N‐formyl‐l‐methionyl‐l‐leucyl‐phenylalanine, leukotriene B4, C5a;84 immunochemically related chemokines, e.g. neutrophil chemoattractant CXCL1, or CXCL9; and on other GAGs, may unravel further functionality of synthetic peptides. It is also worth noting that chemokine peptides are usually associated with favourable properties such as low toxicity and low immunogenicity, which contributes to their increasing recognition as potential candidates for novel drugs.85, 86
Taken together, this approach shows the ability of CXCL8 (54–72) to bind GAG, and to significantly reduce chemokine‐mediated neutrophil adhesion. In addition, the E70K CXCL8 peptide also showed a significant reduction in neutrophil transendothelial migration. This might be due to E70K's higher positive charge and higher binding affinity for polyanionic GAG. The ability of chemokine peptides to bind GAG and regulate chemokine function requires further work to determine if they have the potential to ameliorate acute or chronic neutrophil‐driven organ damage.
Disclosure
The authors declare no conflicts of interest.
Authors contributions
BM‐B performed research, analysed the data and wrote the manuscript. SA, NSS, JAK, TP, DK, EP and SLC provided intellectual input in the design of study. SA, NSS, EP and SLC helped with the writing of the article.
Supporting information
Figure S1. Schematic representation of chemistry for wild‐type CXCL8 C‐terminal peptide.
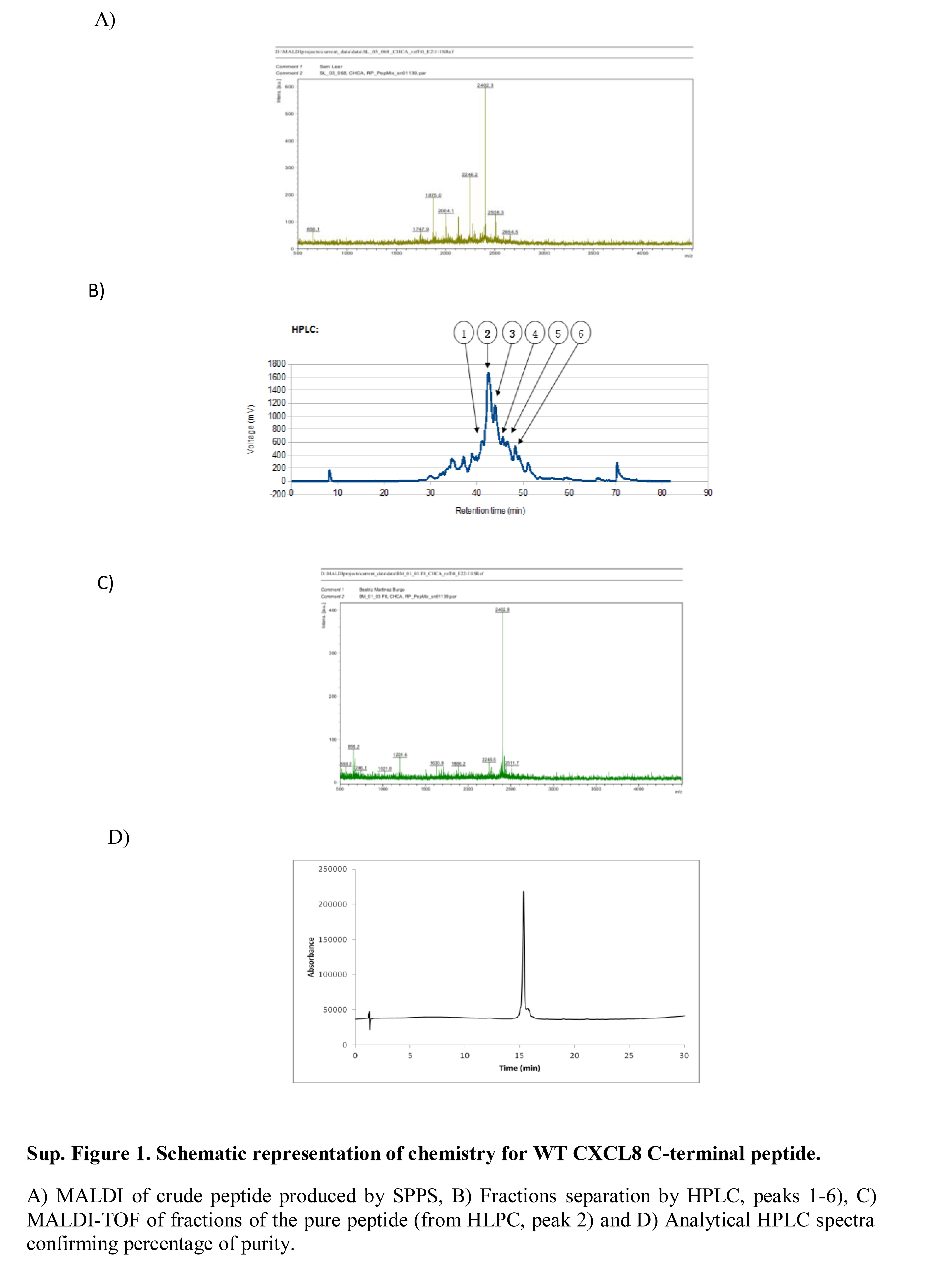{kind=link}
Figure S2. Circular dichroism of each peptide alone or combined with heparin.
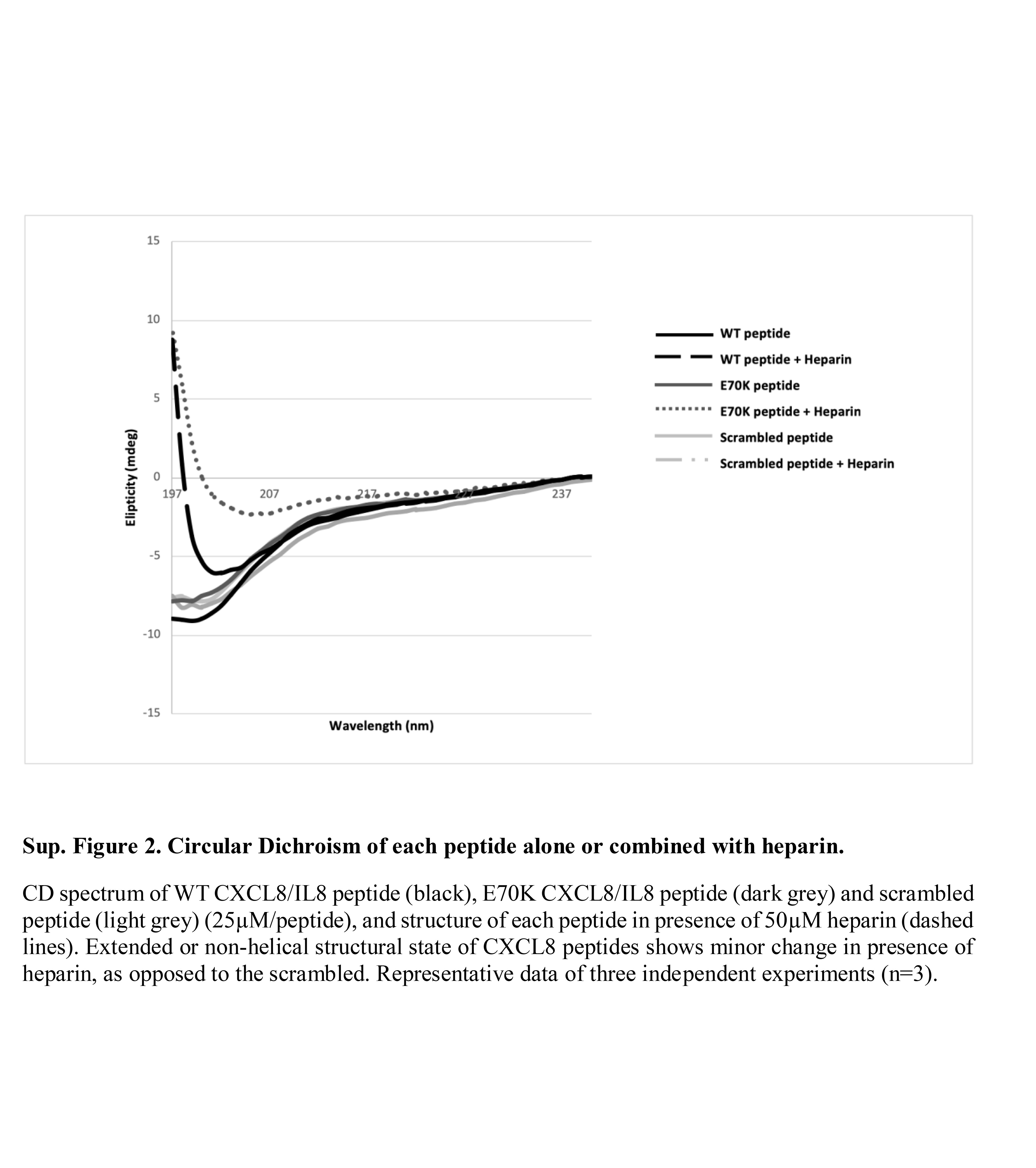{kind=link}
Figure S3. Surface plasmon resonance of heparin‐CXCL8 peptide at 5 μl/min.
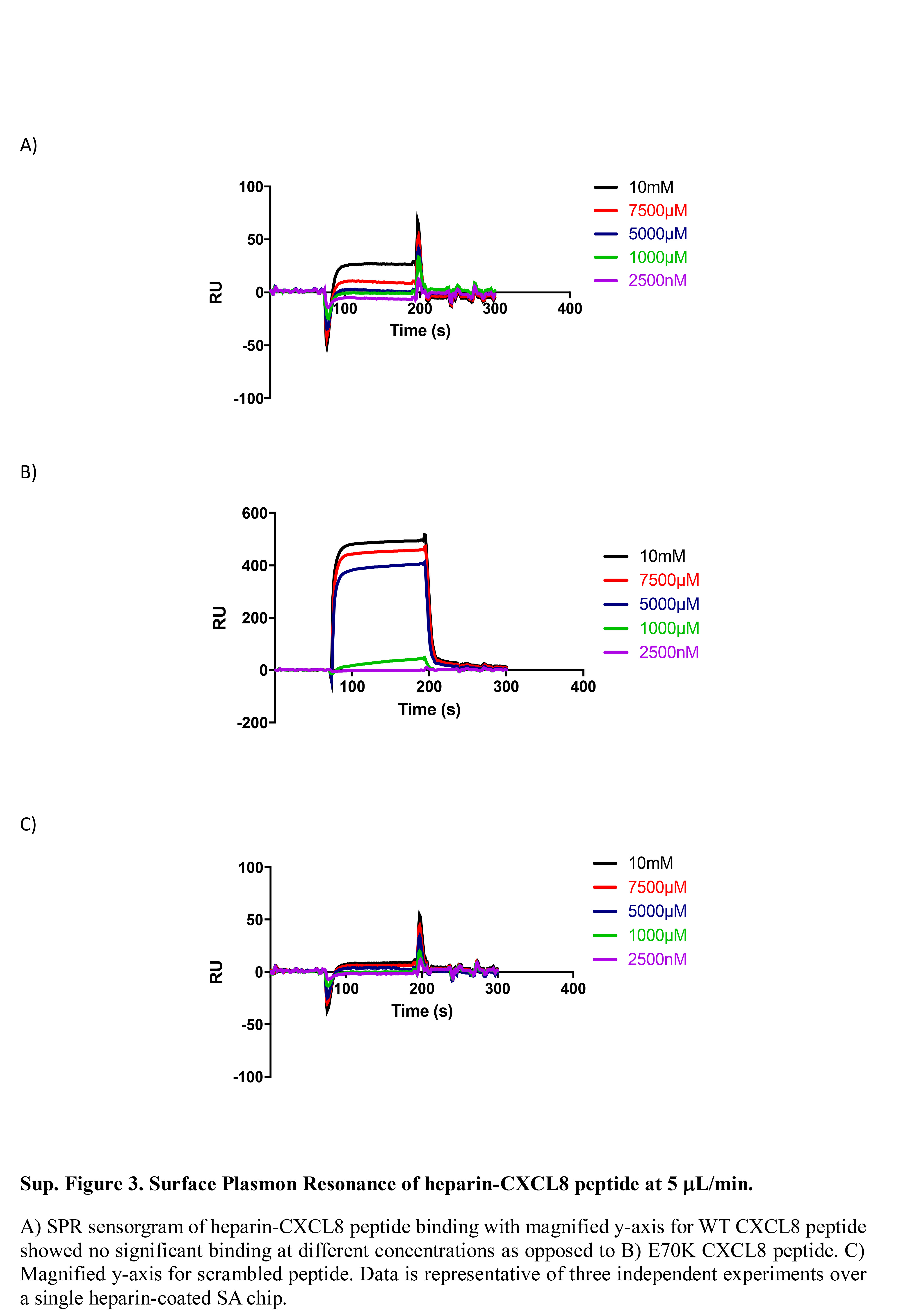{kind=link}
Figure S4. Neutrophil transendothelial migration directed by CXCL8 combined with peptide (extended).
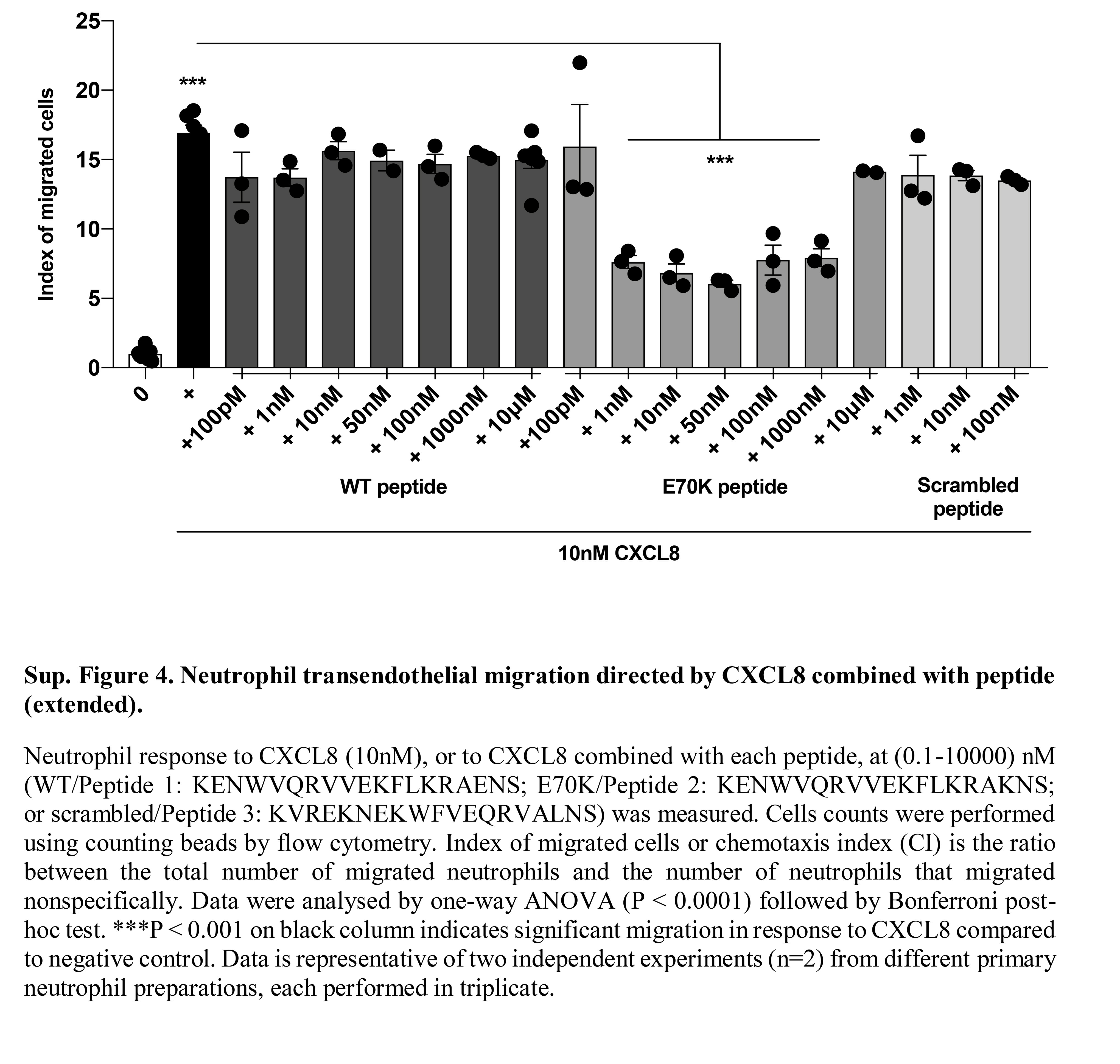{kind=link}
Figure S5. Cell‐surface expression of neutrophil antigens.
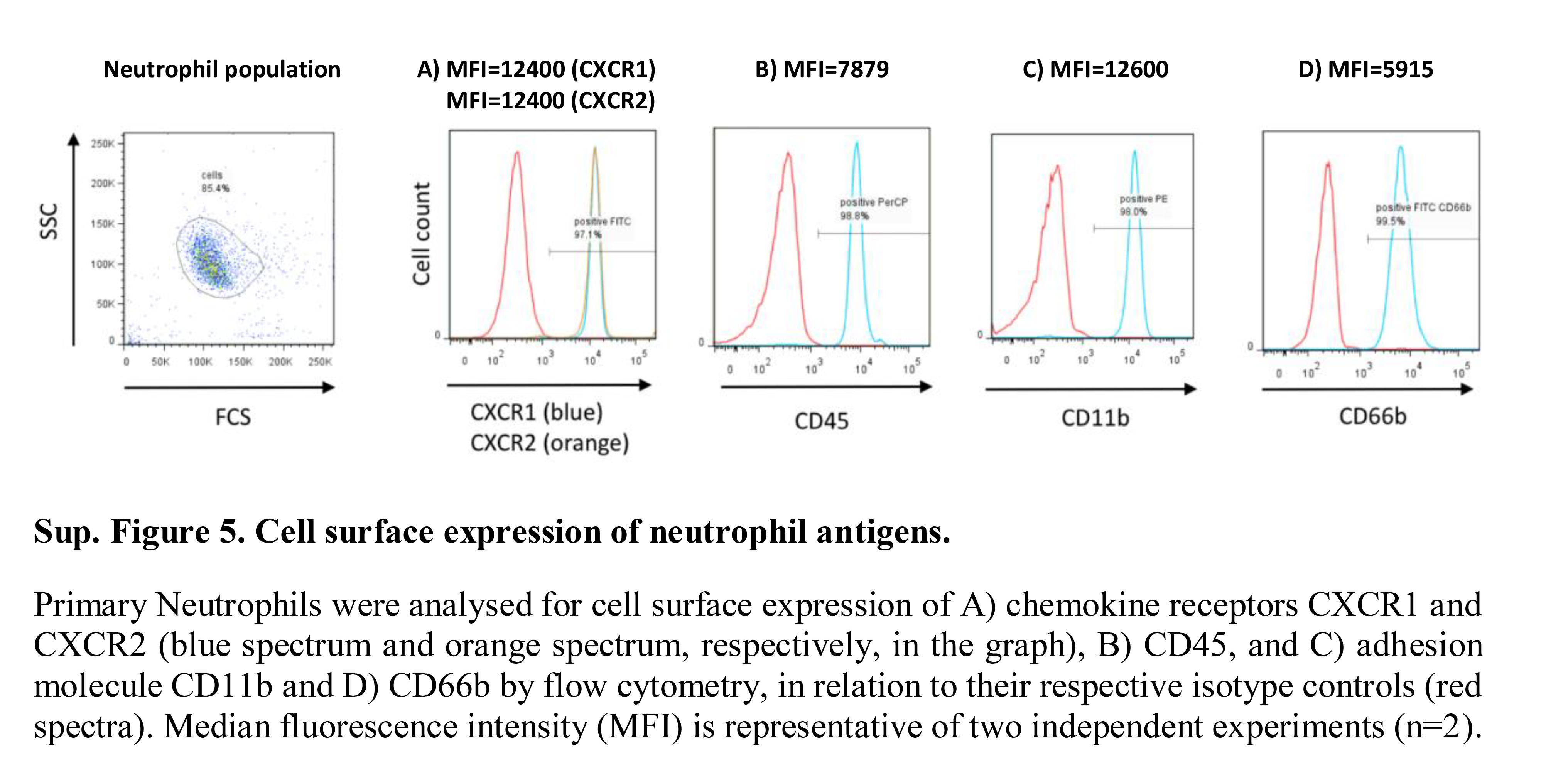{kind=link}
Acknowledgements
This work has been supported by a Marie Skłodowska‐Curie Grant from the European Commission (POSAT 606979, FP7‐PEOPLE‐2013‐ITN). Biophysical work received generous support from the EPSRC Bridging the Gaps Fund at Durham University Biophysical Sciences Institute. EP and SLC are grateful to the Biophysical Sciences Institute for seed corn funding. Durham University mass spectrometry was facilitated by service manager Dr Jackie Mosely. The authors thank Dr Elizabeth H.C. Bromley (Durham University, UK), and Dr Helen Waller and Prof Jeremy H. Lakey (ICaMB, Newcastle University, UK) for helpful discussions regarding the circular dichroism analysis. The authors would also like to thank Dr Helen Waller and Prof Jeremy H. Lakey, and Dr Tim Fagge (Biacore, Edinburgh, UK) for their instrumental SPR guidance. The authors acknowledge Mr Jonathan Scott (ICM, Newcastle University, UK) for his help with neutrophil isolation. This work was also supported by the NIHR Blood and Transplant Research Unit in Organ Transplantation at the University of Cambridge in partnership with Newcastle University and the Newcastle NIHR Biomedical Research Centre in Ageing and Long‐Term Conditions.
Contributor Information
Neil S. Sheerin, Email: Neil.Sheerin@newcastle.ac.uk.
Simi Ali, Email: simi.ali@ncl.ac.uk.
References
- 1. Vestweber D. How leukocytes cross the vascular endothelium. Nat Rev Immunol 2015; 15:692–704. [DOI] [PubMed] [Google Scholar]
- 2. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 2013; 13:159–75. [DOI] [PubMed] [Google Scholar]
- 3. Mocsai A. Diverse novel functions of neutrophils in immunity, inflammation, and beyond. J Exp Med 2013; 210:1283–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Adams DH, Lloyd AR. Chemokines: leucocyte recruitment and activation cytokines. Lancet 1997; 349:490–5. [DOI] [PubMed] [Google Scholar]
- 5. Lo DJ, Weaver TA, Kleiner DE, Mannon RB, Jacobson LM, Becker BN et al Chemokines and their receptors in human renal allotransplantation. Transplantation 2011; 91:70–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blanchet X, Langer M, Weber C, Koenen RR, von Hundelshausen P. Touch of chemokines. Front Immunol 2012; 3:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Weber M, Hauschild R, Schwarz J, Moussion C, de Vries I, Legler DF et al Interstitial dendritic cell guidance by haptotactic chemokine gradients. Science 2013; 339:328–32. [DOI] [PubMed] [Google Scholar]
- 8. Kufareva I, Salanga CL, Handel TM. Chemokine and chemokine receptor structure and interactions: implications for therapeutic strategies. Immunol Cell Biol 2015; 93:372–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vanheule V, Boff D, Mortier A, Janssens R, Petri B, Kolaczkowska E et al CXCL9‐derived peptides differentially inhibit neutrophil migration in vivo through interference with glycosaminoglycan interactions. Front Immunol 2017; 8:530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Proost P, Struyf S, Loos T, Gouwy M, Schutyser E, Conings R et al Coexpression and interaction of CXCL10 and CD26 in mesenchymal cells by synergising inflammatory cytokines: CXCL8 and CXCL10 are discriminative markers for autoimmune arthropathies. Arthritis Res Ther 2006; 8:R107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grainger R, McLaughlin RJ, Harrison AA, Harper JL. Hyperuricaemia elevates circulating CCL2 levels and primes monocyte trafficking in subjects with inter‐critical gout. Rheumatology (Oxford) 2013; 52:1018–21. [DOI] [PubMed] [Google Scholar]
- 12. Brennan FM, Zachariae CO, Chantry D, Larsen CG, Turner M, Maini RN et al Detection of interleukin 8 biological activity in synovial fluids from patients with rheumatoid arthritis and production of interleukin 8 mRNA by isolated synovial cells. Eur J Immunol 1990; 20:2141–4. [DOI] [PubMed] [Google Scholar]
- 13. Bonifati C, Ameglio F. Cytokines in psoriasis. Int J Dermatol 1999; 38:241–51. [DOI] [PubMed] [Google Scholar]
- 14. Barker CE, Ali S, O'Boyle G, Kirby JA. Transplantation and inflammation: implications for the modification of chemokine function. Immunology 2014; 143:138–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stroo I, Stokman G, Teske GJ, Raven A, Butter LM, Florquin S et al Chemokine expression in renal ischemia/reperfusion injury is most profound during the reparative phase. Int Immunol 2010; 22:433–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jo M, Jung ST. Engineering therapeutic antibodies targeting G‐protein‐coupled receptors. Exp Mol Med 2016; 48:e207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol 2014; 11:255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dobaczewski M, Gonzalez‐Quesada C, Frangogiannis NG. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J Mol Cell Cardiol 2010; 48:504–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moschovakis GL, Bubke A, Friedrichsen M, Ristenpart J, Back JW, Falk CS et al The chemokine receptor CCR7 is a promising target for rheumatoid arthritis therapy. Cell Mol Immunol 2018. 10.1038/s41423-018-0056-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jacobson O, Weiss ID. CXCR4 chemokine receptor overview: biology, pathology and applications in imaging and therapy. Theranostics 2013; 3:1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thompson S, Martinez‐Burgo B, Sepuru KM, Rajarathnam K, Kirby JA, Sheerin NS et al Regulation of chemokine function: the roles of GAG‐binding and post‐translational nitration. Int J Mol Sci 2017; 18:E1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lieberman‐Blum SS, Fung HB, Bandres JC. Maraviroc: a CCR5‐receptor antagonist for the treatment of HIV‐1 infection. Clin Ther 2008; 30:1228–50. [DOI] [PubMed] [Google Scholar]
- 23. Van Der Ryst E. Maraviroc – a CCR5 Antagonist for the Treatment of HIV‐1 Infection. Front Immunol 2015; 6:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cashen AF, Nervi B, DiPersio J. AMD3100: CXCR4 antagonist and rapid stem cell‐mobilizing agent. Future Oncol 2007; 3:19–27. [DOI] [PubMed] [Google Scholar]
- 25. Yoshie O. Chemokine receptors as therapeutic targets. Nihon Rinsho Meneki Gakkai Kaishi 2013; 36:189–96. [DOI] [PubMed] [Google Scholar]
- 26. Dyer DP, Salanga CL, Johns SC, Valdambrini E, Fuster MM, Milner CM et al The anti‐inflammatory protein TSG‐6 regulates chemokine function by inhibiting chemokine/glycosaminoglycan interactions. J Biol Chem 2016; 291:12627–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Johnson Z, Proudfoot AE, Handel TM. Interaction of chemokines and glycosaminoglycans: a new twist in the regulation of chemokine function with opportunities for therapeutic intervention. Cytokine Growth Factor Rev 2005; 16:625–36. [DOI] [PubMed] [Google Scholar]
- 28. Murphy PM. Neutrophil receptors for interleukin‐8 and related CXC chemokines. Semin Hematol 1997; 34:311–8. [PubMed] [Google Scholar]
- 29. Gangavarapu P, Rajagopalan L, Kolli D, Guerrero‐Plata A, Garofalo RP, Rajarathnam K. The monomer‐dimer equilibrium and glycosaminoglycan interactions of chemokine CXCL8 regulate tissue‐specific neutrophil recruitment. J Leukoc Biol 2012; 91:259–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bedke J, Nelson PJ, Kiss E, Muenchmeier N, Rek A, Behnes CL et al A novel CXCL8 protein‐based antagonist in acute experimental renal allograft damage. Mol Immunol 2010; 47:1047–57. [DOI] [PubMed] [Google Scholar]
- 31. Gschwandtner M, Strutzmann E, Teixeira MM, Anders HJ, Diedrichs‐Mohring M, Gerlza T et al Glycosaminoglycans are important mediators of neutrophilic inflammation in vivo . Cytokine 2017; 91:65–73. [DOI] [PubMed] [Google Scholar]
- 32. Bertini R, Allegretti M, Bizzarri C, Moriconi A, Locati M, Zampella G et al Noncompetitive allosteric inhibitors of the inflammatory chemokine receptors CXCR1 and CXCR2: prevention of reperfusion injury. Proc Natl Acad Sci USA 2004; 101:11791–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Frangogiannis NG. The role of the chemokines in myocardial ischemia and reperfusion. Curr Vasc Pharmacol 2004; 2:163–74. [DOI] [PubMed] [Google Scholar]
- 34. Cugini D, Azzollini N, Gagliardini E, Cassis P, Bertini R, Colotta F et al Inhibition of the chemokine receptor CXCR2 prevents kidney graft function deterioration due to ischemia/reperfusion. Kidney Int 2005; 67:1753–61. [DOI] [PubMed] [Google Scholar]
- 35. Elmoselhi H, Mansell H, Soliman M, Shoker A. Circulating chemokine ligand levels before and after successful kidney transplantation. J Inflamm (Lond) 2016; 13:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bedke J, Kiss E, Schaefer L, Behnes CL, Bonrouhi M, Gretz N et al Beneficial effects of CCR1 blockade on the progression of chronic renal allograft damage. Am J Transplant 2007; 7:527–37. [DOI] [PubMed] [Google Scholar]
- 37. Bonavia A, Singbartl K. A review of the role of immune cells in acute kidney injury. Pediatr Nephrol 2018; 33:1629–39. [DOI] [PubMed] [Google Scholar]
- 38. Jang HR, Rabb H. Immune cells in experimental acute kidney injury. Nat Rev Nephrol 2015; 11:88–101. [DOI] [PubMed] [Google Scholar]
- 39. Gerszten RE, Garcia‐Zepeda EA, Lim YC, Yoshida M, Ding HA, Gimbrone MA Jr et al MCP‐1 and IL‐8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature 1999; 398:718–23. [DOI] [PubMed] [Google Scholar]
- 40. Yang XD, Corvalan JR, Wang P, Roy CM, Davis CG. Fully human anti‐interleukin‐8 monoclonal antibodies: potential therapeutics for the treatment of inflammatory disease states. J Leukoc Biol 1999; 66:401–10. [DOI] [PubMed] [Google Scholar]
- 41. Adage T, del Bene F, Fiorentini F, Doornbos RP, Zankl C, Bartley MR et al PA401, a novel CXCL8‐based biologic therapeutic with increased glycosaminoglycan binding, reduces bronchoalveolar lavage neutrophils and systemic inflammatory markers in a murine model of LPS‐induced lung inflammation. Cytokine 2015; 76:433–41. [DOI] [PubMed] [Google Scholar]
- 42. Falsone A, Wabitsch V, Geretti E, Potzinger H, Gerlza T, Robinson J et al Designing CXCL8‐based decoy proteins with strong anti‐inflammatory activity in vivo . Biosci Rep 2013; 33:e00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kuschert GS, Coulin F, Power CA, Proudfoot AE, Hubbard RE, Hoogewerf AJ et al Glycosaminoglycans interact selectively with chemokines and modulate receptor binding and cellular responses. Biochemistry 1999; 38:12959–68. [DOI] [PubMed] [Google Scholar]
- 44. Joseph PR, Mosier PD, Desai UR, Rajarathnam K. Solution NMR characterization of chemokine CXCL8/IL‐8 monomer and dimer binding to glycosaminoglycans: structural plasticity mediates differential binding interactions. Biochem J 2015; 472:121–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dransfield I, Buckle AM, Savill JS, McDowall A, Haslett C, Hogg N. Neutrophil apoptosis is associated with a reduction in CD16 (Fcγ RIII) expression. J Immunol 1994; 153:1254–63. [PubMed] [Google Scholar]
- 46. Lear S, Munshi T, Hudson AS, Hatton C, Clardy J, Mosely JA et al Total chemical synthesis of lassomycin and lassomycin‐amide. Org Biomol Chem 2016; 14:4534–41. [DOI] [PubMed] [Google Scholar]
- 47. Lear S, Cobb SL. Pep‐Calc.com: a set of web utilities for the calculation of peptide and peptoid properties and automatic mass spectral peak assignment. J Comput Aided Mol Des 2016; 30:271–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chaffey BT, Mitchell E, Birch MA, Lakey JH. A generic expression system to produce proteins that co‐assemble with alkane thiol SAM. Int J Nanomedicine 2008; 3:287–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Greenfield NJ. Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc 2006; 1:2876–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sarrazin S, Bonnaffe D, Lubineau A, Lortat‐Jacob H. Heparan sulfate mimicry: a synthetic glycoconjugate that recognizes the heparin binding domain of interferon‐gamma inhibits the cytokine activity. J Biol Chem 2005; 280:37558–64. [DOI] [PubMed] [Google Scholar]
- 51. Sadir R, Baleux F, Grosdidier A, Imberty A, Lortat‐Jacob H. Characterization of the stromal cell‐derived factor‐1α‐heparin complex. J Biol Chem 2001; 276:8288–96. [DOI] [PubMed] [Google Scholar]
- 52. Saesen E, Sarrazin S, Laguri C, Sadir R, Maurin D, Thomas A et al Insights into the mechanism by which interferon‐γ basic amino acid clusters mediate protein binding to heparan sulfate. J Am Chem Soc 2013; 135:9384–90. [DOI] [PubMed] [Google Scholar]
- 53. Ali S, Fritchley SJ, Chaffey BT, Kirby JA. Contribution of the putative heparan sulfate‐binding motif BBXB of RANTES to transendothelial migration. Glycobiology 2002; 12:535–43. [DOI] [PubMed] [Google Scholar]
- 54. Mortier A, Berghmans N, Ronsse I, Grauwen K, Stegen S, Van Damme J et al Biological activity of CXCL8 forms generated by alternative cleavage of the signal peptide or by aminopeptidase‐mediated truncation. PLoS One 2011; 6:e23913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dyer DP, Thomson JM, Hermant A, Jowitt TA, Handel TM, Proudfoot AE et al TSG‐6 inhibits neutrophil migration via direct interaction with the chemokine CXCL8. J Immunol 2014; 192:2177–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vacchini A, Mortier A, Proost P, Locati M, Metzemaekers M, Borroni EM. Differential effects of posttranslational modifications of CXCL8/interleukin‐8 on CXCR1 and CXCR2 internalization and signaling properties. Int J Mol Sci 2018; 19:E3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Salcedo R, Resau JH, Halverson D, Hudson EA, Dambach M, Powell D et al Differential expression and responsiveness of chemokine receptors (CXCR1‐3) by human microvascular endothelial cells and umbilical vein endothelial cells. FASEB J 2000; 14:2055–64. [DOI] [PubMed] [Google Scholar]
- 58. Schraufstatter IU, Trieu K, Zhao M, Rose DM, Terkeltaub RA, Burger M. IL‐8‐mediated cell migration in endothelial cells depends on cathepsin B activity and transactivation of the epidermal growth factor receptor. J Immunol 2003; 171:6714–22. [DOI] [PubMed] [Google Scholar]
- 59. Carter NM, Ali S, Kirby JA. Endothelial inflammation: the role of differential expression of N‐deacetylase/N‐sulphotransferase enzymes in alteration of the immunological properties of heparan sulphate. J Cell Sci 2003; 116:3591–600. [DOI] [PubMed] [Google Scholar]
- 60. Naemi FM, Carter V, Kirby JA, Ali S. Anti‐donor HLA class I antibodies: pathways to endothelial cell activation and cell‐mediated allograft rejection. Transplantation 2013; 96:258–66. [DOI] [PubMed] [Google Scholar]
- 61. Bassani JW, Yuan W, Bers DM. Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes. Am J Physiol 1995; 268:C1313–9. [DOI] [PubMed] [Google Scholar]
- 62. Zhou Y, Kucik DF, Szalai AJ, Edberg JC. Human neutrophil flow chamber adhesion assay. J Vis Exp 2014; 51410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shetty S, Weston CJ, Adams DH, Lalor PF. A flow adhesion assay to study leucocyte recruitment to human hepatic sinusoidal endothelium under conditions of shear stress. J Vis Exp 2014; 51330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lamberti G, Prabhakarpandian B, Garson C, Smith A, Pant K, Wang B et al Bioinspired microfluidic assay for in vitro modeling of leukocyte–endothelium interactions. Anal Chem 2014; 86:8344–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lidington EA, Moyes DL, McCormack AM, Rose ML. A comparison of primary endothelial cells and endothelial cell lines for studies of immune interactions. Transpl Immunol 1999; 7:239–46. [DOI] [PubMed] [Google Scholar]
- 66. Harvey JR, Mellor P, Eldaly H, Lennard TW, Kirby JA, Ali S. Inhibition of CXCR4‐mediated breast cancer metastasis: a potential role for heparinoids? Clin Cancer Res 2007; 13:1562–70. [DOI] [PubMed] [Google Scholar]
- 67. Ha H, Debnath B, Neamati N. Role of the CXCL8‐CXCR1/2 axis in cancer and inflammatory diseases. Theranostics 2017; 7:1543–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gerlza T, Hecher B, Jeremic D, Fuchs T, Gschwandtner M, Falsone A et al A combinatorial approach to biophysically characterise chemokine‐glycan binding affinities for drug development. Molecules 2014; 19:10618–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Adage T, Konya V, Weber C, Strutzmann E, Fuchs T, Zankl C et al Targeting glycosaminoglycans in the lung by an engineered CXCL8 as a novel therapeutic approach to lung inflammation. Eur J Pharmacol 2015; 748:83–92. [DOI] [PubMed] [Google Scholar]
- 70. Issekutz AC, Rowter D, Springer TA. Role of ICAM‐1 and ICAM‐2 and alternate CD11/CD18 ligands in neutrophil transendothelial migration. J Leukoc Biol 1999; 65:117–26. [DOI] [PubMed] [Google Scholar]
- 71. Szekanecz Z, Koch AE. Successes and failures of chemokine‐pathway targeting in rheumatoid arthritis. Nat Rev Rheumatol 2016; 12:5–13. [DOI] [PubMed] [Google Scholar]
- 72. Vanheule V, Janssens R, Boff D, Kitic N, Berghmans N, Ronsse I et al The positively charged COOH‐terminal glycosaminoglycan‐binding CXCL9(74–103) peptide inhibits CXCL8‐induced neutrophil extravasation and monosodium urate crystal‐induced gout in mice. J Biol Chem 2015; 290:21292–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gang D, Kim DW, Park HS. Cyclic peptides: promising scaffolds for biopharmaceuticals. Genes (Basel) 2018; 9:E557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Pohl E, Heine A, Sheldrick GM, Dauter Z, Schneider TR, Wilson KS et al Comparison of different X‐ray data‐collection systems using the crystal structure of octreotide. Acta Crystallogr D Biol Crystallogr 1995; 51:60–8. [DOI] [PubMed] [Google Scholar]
- 75. Tan YS, Lane DP, Verma CS. Stapled peptide design: principles and roles of computation. Drug Discov Today 2016; 21:1642–53. [DOI] [PubMed] [Google Scholar]
- 76. Liu M, Li X, Xie Z, Xie C, Zhan C, Hu X et al D‐peptides as recognition molecules and therapeutic agents. Chem Rec 2016; 16:1772–86. [DOI] [PubMed] [Google Scholar]
- 77. Webb LMC, Clark‐Lewis I, Alcami A. The γ herpesvirus chemokine binding protein binds to the N terminus of CXCL8. J Virol 2003; 77:8588–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Andreoni F, Ogawa T, Ogawa M, Madon J, Uchiyama S, Schuepbach RA et al The IL‐8 protease SpyCEP is detrimental for Group A Streptococcus host‐cells interaction and biofilm formation. Front Microbiol 2014; 5:339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. McNaughton EF, Eustace AD, King S, Sessions RB, Kay A, Farris M et al Novel anti‐inflammatory peptides based on chemokine‐glycosaminoglycan interactions reduce leukocyte migration and disease severity in a model of rheumatoid arthritis. J Immunol 2018; 200:3201–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tanino Y, Coombe DR, Gill SE, Kett WC, Kajikawa O, Proudfoot AE et al Kinetics of chemokine‐glycosaminoglycan interactions control neutrophil migration into the airspaces of the lungs. J Immunol 2010; 184:2677–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ali S, Robertson H, Wain JH, Isaacs JD, Malik G, Kirby JA. A non‐glycosaminoglycan‐binding variant of CC chemokine ligand 7 (monocyte chemoattractant protein‐3) antagonizes chemokine‐mediated inflammation. J Immunol 2005; 175:1257–66. [DOI] [PubMed] [Google Scholar]
- 82. O'Boyle G, Mellor P, Kirby JA, Ali S. Anti‐inflammatory therapy by intravenous delivery of non‐heparan sulfate‐binding CXCL12. FASEB J 2009; 23:3906–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. O'Boyle G, Fox CR, Walden HR, Willet JD, Mavin ER, Hine DW et al Chemokine receptor CXCR3 agonist prevents human T‐cell migration in a humanized model of arthritic inflammation. Proc Natl Acad Sci USA 2012; 109:4598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Boneschansker L, Yan J, Wong E, Briscoe DM, Irimia D. Microfluidic platform for the quantitative analysis of leukocyte migration signatures. Nat Commun 2014; 5:4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ezerzer C, Dolgin M, Skovorodnikova J, Harris N. Chemokine receptor‐derived peptides as multi‐target drug leads for the treatment of inflammatory diseases. Peptides 2009; 30:1296–305. [DOI] [PubMed] [Google Scholar]
- 86. Pamies D, Hartung T. 21st century cell culture for 21st century toxicology. Chem Res Toxicol 2017; 30:43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Schematic representation of chemistry for wild‐type CXCL8 C‐terminal peptide.
Figure S2. Circular dichroism of each peptide alone or combined with heparin.
Figure S3. Surface plasmon resonance of heparin‐CXCL8 peptide at 5 μl/min.
Figure S4. Neutrophil transendothelial migration directed by CXCL8 combined with peptide (extended).
Figure S5. Cell‐surface expression of neutrophil antigens.