

Abstract
Conclusions from previously reported articles have revealed that many commonly used pharmaceutical excipients, known to be pharmacologically inert, show effects on drug transporters and/or metabolic enzymes. Thus, the pharmacokinetics (absorption, distribution, metabolism and elimination) of active pharmaceutical ingredients are possibly altered because of their transport and metabolism modulation from the incorporated excipients. The aim of this review is to present studies on the interaction of various commonly-used excipients on pre-systemic metabolism by CYP450 enzymes. Excipients such as surfactants, polymers, fatty acids and solvents are discussed. Based on all the reported outcomes, the most potent inhibitors were found to be surfactants and the least effective were organic solvents. However, there are many factors that can influence the inhibition of CYP450, for instance type of excipient, concentration of excipient, type of CYP450 isoenzyme, incubation condition, etc. Such evidence will be very useful in dosage form design, so that the right formulation can be designed to maximize drug bioavailability, especially for poorly bioavailable drugs.
Keywords: Pharmaceutical excipients, metabolism, cytochrome P450
1. Introduction
The most popular route for drug delivery is oral administration because of pain avoidance, ease of ingestion, patient compliance and versatility of drug candidates. Moreover, the manufacturing for oral drug delivery systems is less expensive as the production process is simple and there are no requirements for sterile conditions [1]. The growth rate of the oral drug delivery market between 2010 and 2017 was 10.3% [2]. Despite all the benefits of oral delivery, poor bioavailability of oral formulations is a limiting factor that can alter the efficacy and therapeutic effect [3]. Various factors are contributing to low oral bioavailability including physiological factor, high gastric emptying time, the effect of food, intestinal barrier and enzymatic degradation of drugs (Table 1). First-pass metabolism is one of the key factors responsible for poor bioavailability. The extensive metabolism of drugs prior to reaching the systemic circulation is known as the first-pass metabolism. After oral administration, the drug is absorbed by the gastrointestinal tract (GIT) and transported to the liver through the portal veins. Then, the drug is metabolized in the liver before reaching systemic circulation, resulting in a low available concentration at the intended target site (Figure 1). Due to insufficient plasma concentrations, the bioavailability of the drug is significantly reduced and therefore a high dose of the drug is required [4].
Table 1.
Examples of drugs with very poor bioavailability with reported reasons.
| Drugs | Pharmacological Class | Bioavailability (%) | Reasons | References |
|---|---|---|---|---|
| Alendronate | Bisphosphonates | 0.59–0.78 | Poor solubility and absorption | [5,6,7] |
| Atorvastatin | Statins | 14 | P-gp and CYP450 activities | [8,9,10] |
| Bromocriptine | Dopamine receptor agonists | 5–10 | Extensive first-pass effect | [11,12,13] |
| Clodronate | Bisphosphonates | 1 | Poor solubility and absorption | [5,6,7] |
| Cytarabine | Antimetabolites | 20 | Intestinal and hepatic first-pass | [14] |
| Domperidone | D2 receptor antagonists | 15 | Gut and liver first-pass | [15] |
| Doxorubicin | Anthracycline antibiotics | 5 | Hepatic and intestinal metabolism | [16] |
| Budesonide | Corticosteroids | 11 | Hepatic first-pass effect | [17] |
| Etidronate | Bisphosphonates | 5 | Poor solubility and absorption | [6,7,13] |
| Felodipine | Calcium channel blockers | 15 | P-gp and CYP450 activities | [17] |
| Isradipine | Calcium channel blockers | 15 | P-gp and CYP450 activities | [18] |
| Fluvastatin | Statins | 20 | P-gp and CYP450 activities | [8,9,10] |
| Nimodipine | Calcium Channel blockers | 13 | P-gp and CYP450 activities | [19] |
| Hyoscine | Antispasmodics | 20 | Hepatic metabolism | [20] |
| Ketamine | Dissociative anesthetics | 20 | Hepatic and intestinal metabolism | [21] |
| Lovastatin | Statins | <5 | P-gp and CYP450 activities | [8,9,10] |
| Morphine | Opioids | 20–33 | Gut and liver first-pass | [22] |
| Pyridostigmine | Acetylcholinesterase inhibitors | 14 | Poor absorption | [23] |
| Naloxone | Opioid antagonists | 2–10 | Extensive first-pass but 90% absorption | [24] |
| Naltrexone | Opiate antagonists | 5–40 | First-pass, enterohepatic recycling | [10] |
| Pamidronate | Bisphosphonates | 1 | Poor solubility and absorption | [5,6,7] |
| Pravastatin | Statins | 17–34 | P-gp and CYP450 activities | [8,9,10] |
| Prochlorperazine | Phenothiazines | 20 | Intestinal and hepatic first-pass | [25] |
| Risedronate | Bisphosphonates | <1 | Poor solubility and absorption | [5,6,7] |
| Selegiline | Monoamine oxidase type B inhibitors | 20 | Extensive first-pass | [26] |
| Simvastatin | Statins | 5–48 | P-gp and CYP450 activities | [8,9,10] |
| Sumatriptan | Serotonin receptor agonists | 20 | Hepatic first-pass | [27] |
| Tacrine | Cholinesterase inhibitors | 10–30 | Hepatic first-pass | [28] |
| Terbutaline | Adrenergic receptor agonists | 9–21 | Extensive first-pass and poor absorption | [29] |
| Lidocaine | Local anesthetics | 3 | Hepatic first-pass effect | [30] |
| Tiludronate | Bisphosphonates | 6 | Poor solubility and absorption | [5,6,7] |
Figure 1.

Schematic diagram depicting the route of poor bioavailability after oral administration of the drugs.
The principle enzymes responsible for first-pass metabolism or biotransformation of drug molecules are cytochrome P450 (CYP450). CYP450 includes a superfamily of hemeproteins which is categorized into families and subfamilies based on amino acid sequence homology [31]. They are allotted a family number (for instance, CYP1 or CYP2) followed by a subfamily letter (e.g., CYP1A or CYP2C) and are distinguished by a number for the individual enzyme or isoforms (e.g., CYP1A2 or CYP2C19) [31]. According to Coller et al., the most commonly prescribed drugs in the United States (USA) are reported to be metabolized by CYP1, CYP2 and CYP3 families. The most common enzymes responsible for oxidation of approximately 79% of these drugs are CYP2C9, CYP2D6, CYP2C19 and CYP3A4/5 [32].
2. Traditional Strategies to Overcome Pre-Systemic Metabolism
Several novel, as well as traditional, methods are being investigated to circumvent drug metabolism. Prodrugs, enzyme inhibitors, polymeric excipients, self-emulsifying drug delivery systems (SEDDS) and liposomes have been investigated over the years as inhibitors to metabolic enzymes.
2.1. Prodrug Approaches
Prodrug is one of the common approaches investigated, according to the literature. Approximately 10% of the drugs in the pharmaceutical market are categorized as prodrugs and one third of small molecular weight (Mw) drugs are classified as prodrugs [33,34,35,36,37,38]. Prodrugs are pharmacologically inert substances which undergo conversion, once inside the body, to release the active ingredient for its therapeutic effect [39,40]. Prodrugs successfully overcome the drug physicochemical and biopharmaceutical obstacles, thus enhancing their pharmacokinetic properties such as oral absorption and metabolism [41,42].
2.2. Enzyme Inhibitors
Another promising strategy to reduce the pre-systemic metabolism is the co-administration of CYP450 inhibitors with orally administered drugs [43]. Probably, the best-known enzyme inhibitor is grapefruit juice, which significantly improves the oral bioavailability of many drugs [44,45]. Food–drug interaction is a common occurrence and can be significant when the drug’s pharmacokinetics are altered. The classic example is the interaction between grapefruit juice and felodipine. Felodipine is known to undergo high pre-systemic metabolism resulting in very low absolute bioavailability with an average of 15%. A study showed the inhibitory effect of grapefruit juice flavonoids such as quercetin, naringenin and naringin on CYP3A4, using felodipine as a substrate [45]. The concentration-dependent inhibition was examined when the flavonoids were co-incubated with felodipine [45]. Moreover, many studies reported furanocoumarins, present in grapefruit juice, as an inhibitor for CYP450 [46,47,48,49,50,51,52,53,54]. Bergamottin inhibits CYP1A2, CYP1B1, CYP 2A6, CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4 and CYP3A5. The dihydroxybergamottin (DHB) and paradisins inhibit CYP1A2, CYP1B1, CYP2C9, CYP2C19, 2D6 and CYP3A4 [55].
Besides grapefruit juice, many drugs in the market such as ketoconazole, Troleandomycin, saquinavir, diltiazem and fluconazole exhibit an enzyme inhibitory effect resulting in improved bioavailability of many drugs [43,56]. For instance, the oral bioavailability of alfentanil was increased 19-fold with troleandomycin [57]. However, co-administration of enzyme inhibitors can lead to adverse drug effects which also includes fatal events. For example, when clarithromycin (inhibitor) is co-administered with astemizole, terfenadine or cisapride (substrates), a severe ventricular arrhythmia may occur [58].
3. Other Approaches
These include polymeric excipients, self-emulsifying drug delivery systems (SEDDS) and liposomes. Normally, polymers are used as enteric coatings for different types of drugs to overcome hydrolytic instability [59]. Some polymers, such as polycarbophil and carbomer, demonstrate inhibitory effects on trypsin, which decrease pre-systemic metabolism in the intestine. That means drugs incorporated in such polymers can be protected from pre-systemic degradation by trypsin [60]. Similarly, most of the studies on SEDDS and liposomes focus on increased solubility by these systems and their protective property towards pre-systemic metabolism is hardly examined [43].
3.1. Novel Approach to Overcome Pre-Systemic Metabolism
The addition of commonly-used pharmaceutical excipients can be a potential solution to these problems. Pharmaceutical excipients are ingredients other than the active pharmaceutical ingredient (API) present in a finished pharmaceutical drug formulation. These are frequently used as lubricants, diluent, binders, flavorings, coating and coloring agents for the formulation. These substances are often therapeutically inert [61].
The functional roles of pharmaceutical excipients include modulating bioavailability and solubility of APIs, increasing the stability of APIs in the dosage form, maintaining the osmolarity and/or pH of the liquid formulations, preventing dissociation and aggregation, etc. Recently, addition of pharmaceutical excipients in drug formulations have gained attention which can alter the pharmacokinetics of drugs, resulting in improved bioavailability [61]. The aim of this review article is to survey the current literature and evaluate the effect of different pharmaceutical excipients on metabolic enzymes. This review looks at the effect on surfactant, polymers, etc. excipients on the expression of cytochrome P450 enzymes.
3.2. Effect of Excipients
Substances that can enhance or inhibit cytochrome P450 activity can alter the rate of drug metabolism, leading to an increase or decrease in drug bioavailability. There are many studies on new materials as drug delivery vehicles, including vesicles, block copolymer micelles, degradable polymer particles, dendrimers, polymer prodrugs and lipid nanoparticles; however, the effect of excipients used is often ignored. For instance, liquid acetaminophen, which contains propylene glycol, is less toxic than solid preparations with no propylene glycol. The main reason for acute hepatic failure in Europe and the United States is acetaminophen [62,63]. Its toxicity is due to the reductive metabolism via CYP2E1 [64]. Liquid formulation of acetaminophen contains a solubilizing agent called propylene glycol, which is used to dissolve the drug in aqueous solution [65]. Since children ingest liquid formulation and are less vulnerable to its toxicity, a single-blinded cross-over study was conducted to compare metabolism of solid and liquid acetaminophen 15 mg/kg dose by CYP2E1, using 15 healthy adults as volunteers. As a result, the measured AUCs for the metabolites were 16% lower than solid formulation. This is because propylene glycol is a competitive antagonist to CYP2E1; hence, it shows the protective effect in liquid formulation [65].
One method by which excipients may alter the drug metabolism is by inhibiting CYP450 enzymes present in cellular microsomes [61].
3.3. Surfactants
Surfactants, also known as surface-active agents, possess hydrophilic (polar) and hydrophobic (non-polar) characteristics. The hydrophobic (non-polar) part is referred to as the tail group and the hydrophilic (polar) part as the head group. Surfactants are normally used to increase the solubility of the drugs and to decrease the interfacial tension between the drug and the medium [66]. Surfactants can be categorized into four different groups: anionic, cationic, zwitterionic and non-ionic surfactants (Table 2). Anionic surfactants carry a negative charge, whereas cationic surfactants carry a positive charge on their hydrophilic head. Zwitterionic surfactants have the potential to carry both positive and negative charges [66].
Table 2.
Summary of the effect of surfactants on CYP activities.
| Surfactants | Substrates | Mechanism of Action | Structures | Type | References |
|---|---|---|---|---|---|
| Brij 35 | 7-ethoxycoumarin | Increased CYP3A4 inhibition with increased surfactant concentration |
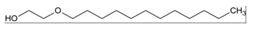
|
Non-ionic | [74] |
| Brij 58 | Rabeprazole | Significant inhibition of drug degradation by CYP enzymes |
|
Non-ionic | [81] |
| CTAB | 7-ethoxycoumarin | Increased CYP3A4 inhibition with increased surfactant concentration |
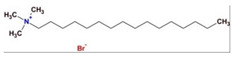
|
Cationic | [74] |
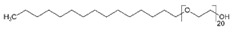
| Kollidon 12 PF | 7-ethoxycoumarin | Increased CYP3A4 inhibition with increased surfactant concentration |
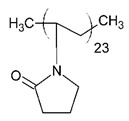
|
Non-ionic | [74] |
| Lutrol F68 NF | 7-ethoxycoumarin | Increased CYP3A4 inhibition with increased surfactant concentration |
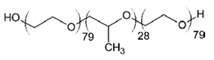
|
Non-ionic | [74] |
| Octyl-B-D-glucopyranoside | 7-ethoxycoumarin | Increased CYP3A4 inhibition with increased surfactant concentration |
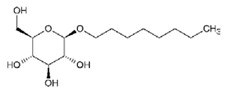
|
Non-ionic | [74] |
| SDS | 7-ethoxycoumarin | Increased CYP3A4 inhibition with increased surfactant concentration |
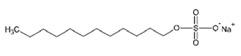
|
Anionic | [74] |
| Solutol HS 15 | 7-ethoxycoumarin | Increased CYP3A4 inhibition with increased surfactant concentration |
|
Non-ionic | [74] |
| Triton X-100 reduced | 7-ethoxycoumarin | Increased CYP3A4 inhibition with increased surfactant concentration |
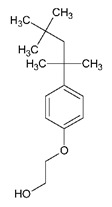
|
Non-ionic | [74] |
| Polysorbate 80 | Testosterone Diclofenac |
Increased CYP3A4 and CYP2C9 inhibition in concentration-dependent manner |
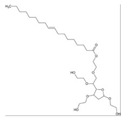
|
Non-ionic | [68] |
| TPGS | Testosterone Diclofenac |
Increased CYP3A4 and CYP2C9 inhibition in concentration-dependent manner |
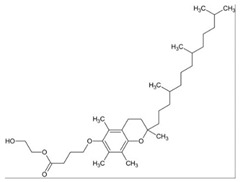
|
Non-ionic | [68] |
| Sucrose laurate | Testosterone Diclofenac |
Increased CYP3A4 and CYP2C9 inhibition in concentration-dependent manner |
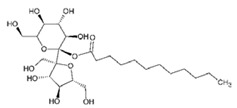
|
Non-ionic | [68] |
| Gelucire 44/14 | Rabeprazole | Significant inhibition of drug degradation by CYP enzymes | Lauroyl polyoxyl-32 glycerides (C9H14N2) | Non-ionic | [81] |
| Polyoxyl 40 Stearate | Midazolam | Strong inhibition of rCYP3A4 |
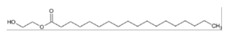
|
Non-ionic | [61] |
| Pluronic F68 | Midazolam | Strong inhibition of rCYP3A4 |
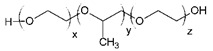
|
Non-ionic | [61] |
Cremophor EL is a heterogeneous non-ionic surfactant made up of castor oil and ethylene oxide with a molar ratio of 1:35 [67]. Cremophor EL helps in solubilizing many hydrophobic drugs which include photosensitizers, immunosuppressive agents, sedatives, anesthetics and anticancer drugs (experimental) [67]. Different in vitro studies have been reported for the impact of Cremophor EL on the metabolism of drugs [68,69,70]. In 2010, a study focused on the effect of Cremophor EL on CYP3A4 and CYP2C9 mediated metabolism of testosterone and diclofenac, respectively [68]. Cremophor EL was tested at different concentrations (0.01–100 mM) using human liver microsomes in vitro. As a result, the inhibitory effect of Cremophor EL was found to be concentration dependent. The half-maximal inhibitory concentration (IC50) of the CYP3A4- and CYP2C9-mediated metabolism were determined as 0.60 and 0.03 mM, respectively.
Mudra et al. further showed that solubilizing agents inhibited verapamil-N-demethylase activity in vitro and in situ. The rate of verapamil-N-demethylation was reduced in the presence of Cremophor EL, suggesting moderate inhibition of CYP3A4: 20.7% and 21.8%, in situ and in vitro at 47.5 μg/mL, respectively [69]. The inhibition of CYP450 by polyethoxylated solubilizing agents (e.g., Cremophor EL, Tween 80) can be attributed to the collective evidence that supports the following hypothesis: The drug absorption is altered in the presence of polyethoxylated solubilizing agents due to agent-produced membrane fluidization, causing in local environment perturbation required for protein to function [70,71,72,73]. Similarly, the outcomes in this article are reliable with agent-induced fluidization of microsomal membrane resulting in perturbation of the enzyme micro-environment, thus decreasing CYP3A4 function.
To support this hypothesis, another study on Cremophor EL presented results consistent with the previous studies. In vitro metabolism of 7-ethoxycoumarin was studied at three different concentration of the surfactant (0.03%, 0.06% and 0.1% w/v). Various enzymes are responsible for metabolism of ethoxycoumarin, but the major enzymes are CYP450 1A2, CYP450 1A1 and CYP450 2B. Increasing the concentration of Cremophor EL decreased the metabolic activity of these enzymes [68,74]. The CYP450 activities were reduced from 97% to 93% when surfactant concentration was increased from 0.03% to 0.10%. This is because the surfactant concentration above its critical micellar concentration (CMC) value disrupts the CYP450 enzymes membrane, causing an inhibitory effect. In contrast, below its CMC value, there is less disruption [74].
In 2004, Gonzalez et al. investigated the effect surfactants have on the metabolism of the CYP3A2 substrate by Midazolam (MDZ) by determining the intrinsic clearance (Clint) of MDZ in rat hepatocyte and rat microsome systems [75]. In the rat hepatocytic system, the Clint of MDZ decreased significantly above its critical micellar concentration (CMC) at 0.03% (Clint/0.03% = 70.3% ± 2.1%) and 0.3% (Clint/0.3% = 54.9% ± 2.2%), whereas the Clint of MDZ was not altered significantly below its CMC (at 0.0003% and 0.003%). Similar outcomes were observed with rat liver microsomes; significant decrease in Clint of MDZ at 0.03% (Clint/0.03% = 89.5 ±3.7 µL/min/mg protein) and 0.3% (Clint/0.3% = 35.0 ±0.8 µL/min/mg protein) of the Cremophor EL. Thus, the addition of Cremophor EL reduced the intrinsic clearance of MDZ by inhibiting CYP450 in both systems [75].
Taxol is an active ingredient used for refractory ovarian cancer, lung cancer and breast cancer. It is readily metabolized by the CYP450 system in the liver. A study conducted by Carlos et al. (1994) illustrated that Cremophor EL had the most significant effect on taxol metabolism, by the CYP450 system, compared to other co-administered drugs (diphenhydramine, cimetidine and dexamethasone). In human and rat liver microsomes, the formation of 6α-hydroxytaxol was completely prevented by Cremophor EL at 20 µL/mL. In human liver slices, Cremophor EL reduced the formation of 6α-hydroxytaxol as well as the ratio of metabolite to parent drug at 20 µL/mL [76]. However, at 2 µL/mL, Cremophor EL showed very little effect. These results suggest that Cremophor EL indirectly reduces the taxol uptake by the liver. To conclude from all the studies, the inhibition of CYP450 enzymes by Cremophor EL seems to be dependent on the concentration of surfactant, type of isoenzyme and type of microsomal assay.
Cremophor RH-40 is found in many oral drug formulations, which is mainly used to improve solubilization of the drugs, such as Tegretol, Anafranil and Sandimmun Neoral. Cremophor RH-40 has a different molar content of ethylene oxide (45 mol) than Cremophor EL [77]. The inhibition of CYP450 enzymes by Cremophor RH-40 has been reported in many studies [63]. Christiansen et al. examined the effect of non-ionic surfactants on CYP3A4 and CYP2C9 enzymes [68]. The inhibitory action of Cremophor RH40 (IC50 = 0.80 mM) was found to be less than Cremophor EL (IC50 = 0.60 mM) for CYP3A4, whereas stronger inhibition was reported with a similar IC50 value (0.03 mM) for CYP2C9. To understand the action of Cremophor RH40 in biotransformation, it was examined for its inhibitory effect on CYP3A4 in vitro as well as in vivo [61]. According to the in vitro study conducted by Ren et al. (2008), the inhibition rate of CYP3A4 by Cremophor RH40 was 99.40%. Furthermore, single and multiple doses of Cremophor RH40 were examined in male Sprague-Dawley rats for its impact on MDZ metabolism. In comparison to control saline, Cremophor RH40 increased the area under curve (AUC0-∞) of MDZ up to 1.1-fold and decreased the production of 1′-Hydroxymidazolam (1′-OH-MDZ) up to 0.44-fold in a single dose. The MDZ AUC0-∞ was increased by 1.69-fold and 1’-OH-MDZ AUC0-∞ was decreased by 0.9-fold in a multiple-dose regimen [61].
These results are consistent with the recent study on a Cremophor RH40-based self-micro emulsifying drug delivery system (SMEDDS) [78]. A significant decrease in 1′-OH-MDZ production was observed (p < 0.05), when the concentration of Cremophor RH40 was increased from 0.05% to 3% w/v. Studies have revealed that chemotherapeutic substances can downregulate the CYP3A gene expression [79,80]. Therefore, Western blot analysis was performed to examine the effect of SMEDDS on the CYP3A activity. As a result, Cremophor RH40-based SMEDDS reduced the CYP3A protein expression levels at dilutions ranging from 1:50 to 1:100. Another study reported a similar significant inhibition of CYP3A4 and CYP2C19 enzymes in vitro by Cremophor RH40 when rabeprazole was used as a probe drug (p < 0.05) [81]. It was concluded that these inhibitory effects involved direct action or micelle formation to disrupt CYP activities.
Recently, Tween 80 has been reported to inhibit different isoenzymes of CYP450: 3A4, 2C9, 1A1, 1A2 and 2B. Tween 80 (polysorbate 80) is the most commonly-used hydrophilic non-ionic surfactant with the ability to improve the solubility of compounds [69]. Tween 80 is derived from oleic acid and polyethoxylated sorbitol and has been used in preclinical and clinical drug formulations [69]. Moderate inhibition of CYP3A4 and CYP2C9 by Tween 80 was determined. The IC50 value for CYP3A4 and CYP2C9 was found to be 0.40 and 0.04 mM, respectively. Christiansen et al. and Rao et al. illustrated the inhibition of CYP450 to be a concentration-dependent manner. For example, when the concentration of Tween 80 in SMEDDS was increased from 0.05% to 3% (w/v), the 1′-OH-MDZ production was reduced from ~80% to 30%. The in vitro metabolism of 7-ethoxycoumarin was reduced from 85% to 65% when the concentration of Tween 80 was increased from 0.03% to 0.10% [68].
The incubation of Tween 80 with rat hepatocytes reduced Clint of MDZ significantly, above its CMC (Clint/0.03% = 75.2% ±1.6%; Clint/0.3% 79.2% ±1.5%, p < 0.05), whereas the inhibitory effect of Tween 80 was reported below its CMC with rat liver microsomes (Clint/0.003% = 70% ± 5.7%, Clint/0.03% = 66.9% ± 1.0%, Clint/0.3% = 8.24% ± 0.28%, p < 0.05) [78]. A similar study reported the inhibition of CYP3A4 by approximately 20% with a Tween 80 concentration of 50 mM [61]. Mudra et al. measured the impact of Tween 80 on intestinal verapamil-N-demethylation activity in-situ and in vitro. The inhibition rate was found to be 56.3% (in-situ) and 13.5% (in vitro) at 25 μg/mL, reflecting the reduction of CYP3A activity [69].
Tween 20 (Polysorbate 20) and Tween 80 are composed of the same hydrophilic group but different hydrophobic groups. Tween 20 has a mixture of palmitic, stearic, lauric and myristic acids, whereas Tween 80 contains oleic, linoleic and stearic acids [61]. According to Ren et al. and Randall et al., Tween 20 is a more potent inhibitor than Tween 80. The production of 1′-OH-MDZ was inhibited by around 80% at 50 mM Tween 20 concentration. It also decreased the metabolism of 7-ethoxycoumarin from 66% to 56% at a concentration ranging from 0.03% to 0.10% v/v [61,74].
Triton X-100 (TX-100) is an octylphenol polyethoxylated non-ionic surfactant. Its structure consist of poly(ethylene glycol) with a 4-(1,1,3,3-tetramethylbutyl)phenyl group [82]. TX-100 has been reported as a strong inhibitor for CYP450 enzymes. It reduced the metabolism of CYP3A4 substrates: 7-ethoxycoumarin by 54% and midazolam by 99.8% [74,81]. The interaction of TX-100 with CYP450 in Prochilodus scrofa was studied using antioxidant and mono-oxygenase system. The CYP content at 0.05 mM of TX-100 was found to be 0%, explaining the stronger inhibition of CYP450. Similarly, competitive inhibition of CYP1A1 by TX-100 was reported far below its CMC value (250 μM) [83].
Furthermore, multiple studies pointed out the ability of other surfactants to interfere with CYP450, which can lead to improved bioavailability of drugs. The surfactants that were reported to show inhibitory actions are listed in Table 2.
3.4. Polymers
Polymers are macromolecular compounds and constitute a large and diverse group of substances, including synthetic polymers, semi-synthetic polymers, natural polymers and fermentation products (Table 3). These polymers are commonly used as excipients in pharmaceutical dosage forms; parenterally, orally, nasally, rectally, intravaginally, inhalationally and topically, on the oral mucosa and in ophthalmic preparations [84].
Table 3.
Different types of polymers with some examples.
| Polymers | Examples |
|---|---|
| Natural | Sodium alginate Gelatin Chitosan |
| Semi-synthetic | Cellulose derivatives |
| Synthetic | Polyethylene glycols Poloxamers Polyactides Polyamides Acrylic acid polymers |
| Fermentation products | Xanthan gum |
Lihui Qiu et al. evaluated the in vitro inhibition of six CYP isoforms by different mPEGx-PCLx (methoxy poly(ethylene glycol)-poly(ε-caprolactone)) amphiphilic copolymer micelles: mPEG2k-PCL2k, mPEG2k-PCL3.5k, mPEG2k-PCL5k and mPEG2k-PCL10k. The inhibitory effect was found to be concentration-dependent in manner [85]. All CYP450 enzymes were significantly inhibited by mPEG2k-PCLx when the concentration was increased to 1000 μg/mL. For example, the CYP activities were reduced to 68.7% for CYP2D2, 64.6% for CYP2C6, 40.4% and 33.5% for CYP3A2/1, 38.1% for 2B1, 26.2% for 2C11 and 9.4% for 1A2 by mPEG2k-PCL2k at 1000 μg/mL. The extent of inhibition, ranked downwards, was as follows: mPEG2k-PCL2k > mPEG2k-PCL3.5k > mPEG2k-PCL5k > and mPEG2k-PCL10k [85].
Similarly, Martin and co-workers investigated the effect of 10 commonly-used polymers on seven CYP isoforms (2E1, 3A4, 3A5, 2C9, 2C19, 1A2 and 2D6). As shown in Table 4, nine out of ten polymers inhibited CYP activities. Cytochromes 2E1, 3A5, 2C9, 2C19 and 2D6 were inhibited by polyethylene glycol (PEG), while 2E1, 3A4, 3A5 and 2C9 were downregulated by pluronic F68. Pluronic F127, polyvinyl acetate (PVA) and sodium carboxymethyl cellulose (NaCMC) inhibited 2E1, 2E1 and 1A2, respectively. Hydroxypropyl methylcellulose (HPMC) inhibited 2E1 and 3A5, whereas polyvinyl pyrrolidone (PVP) downregulated 3A4 and 1A2. Other polymers, such as Kollicoat, in this study also inhibited specific enzymes, apart from hydroxypropyl cellulose (HPC), which exhibited no effect on any of the enzymes [86].
Table 4.
Effect of different polymers on activities of CYP 2E1, 3A5, 2C9, 2C19, 1A2 and 2D6.
| Polymer | IC50 Values (μM) | ||||||
|---|---|---|---|---|---|---|---|
| CYP2E1 | CYP3A4 | CYP3A5 | CYP2C9 | CYP2C19 | CYP1A2 | CYP2D6 | |
| PEG | 75.3 ± 2.1 | - | 78.0 ± 17.8 | 365.6 ± 32.8 | 139.0 ± 22.4 | - | 409.6 ± 34.5 |
| F68 | 203.7 ± 48.3 | 59.1 ± 13.6 | 209.9 ± 29.7 | 244.8 ± 13.2 | - | - | - |
| F127 | 218.9 ± 13.3 | - | - | - | - | - | - |
| NaCMC | - | - | - | - | - | 224.7 ± 14.8 | |
| HPC | - | - | - | - | - | - | - |
| HPMC | 253.5 ± 17.9 | - | 19.4 ± 0.6 | - | - | - | - |
| PVA | 548.9 ± 30.4 | - | - | - | - | - | - |
| Kollicoat | 598.1 ± 26.1 | - | - | - | - | 10.0 ± 3.9 | 89.9 ± 2.9 |
| HG | 141.2 ± 14.1 | - | - | - | - | 40.9 ± 8.4 | - |
| PVP | - | 107.3 ± 11.2 | - | - | - | 78.3 ± 4.2 | - |
Huang et al. used the Hill equation and Lineweaver–Burk plots to determine the inhibitory effect of pluronic F68 (F68) on CYP3A4. The dose-dependent inhibition was reported with the Ki and IC50 values for F68 averaged 0.16 and 0.11 mg/mL, respectively [87]. The mechanism of action was based on the previously reported inhibitory effect study, which includes the direct interaction with CYP450 enzymes, cell membrane disruption and alteration of cell membrane [75].
Another study tested 22 commonly used excipients, which included sodium alginate, PEG4000, PEG1000, PEG6000 and PEG 2000. All the listed polymers inhibited 1′-OH-MDZ formation in the following order (from strong to weak): Sodium alginate > PEG4000 > PEG1000 > PEG6000 > PEG2000 [61]. The physical and chemical nature of each excipient clearly play a major role in their inhibitory capacity. Thus, sodium alginate was the most effective inhibitor compared to the other polymers due to its ability to disrupt CYP3A4 activity strongly. On the other hand, the outcomes of this study were limited to CYP3A4. As previously mentioned, there are many families and subfamilies of CYP450. There is a possibility that the least effective polymer (PEG2000) could be the most effective on other CYP450 isoenzymes.
Pluronic P85 (P85) is a block copolymer consisting of two equal polyoxyethylene chains joined by a polyoxypropylene chain. A study revealed that P85 strongly inhibited norverapamil formation by CYP3A in a concentration-dependent manner [88]. The study included enterocyte-based metabolism, where the P85 concentrations used were 0.01% and 0.1% v/v. Compared to other polymers in the study, P85 reduced the formation of norverapamil by around 25% and 50% at 0.01% and 0.1% v/v, respectively [88].
3.5. Fatty Acids
Fatty acids (FA) are the main components of phospholipids, triacylglycerols and many complex lipids. FAs are present in the majority of dietary fat in humans. Different foods provide a variety of fatty acids. In addition, the human body can synthesize them, either from other fatty acids or nonlipid precursors, e.g., glucose. In recent years, studies have revealed that various FAs have a wide range of microbicidal activity against several Gram-positive and Gram-negative bacteria, as well as enveloped viruses, including S. aureus and N. gonorrhoeae [89,90]. For instance, lauric acid (LA) was found to be the most potent inhibitor for the growth of Gram-positive bacteria. These direct or indirect inhibitory effects are due to the destabilization of the bacterial cell membrane caused by fatty acids [91,92,93,94,95].
Besides their potent microbicidal activity, FAs can also reduce pre-systemic metabolism of drugs by inhibiting CYP450 enzymes. The effect of saturated and unsaturated fatty acids on CYP3A4, 2E1, 2D6, 2C19, 2C9, 2C8, 2B6, 2A6 and 1A2 were studied using midazolam 1-hydroxylation, chlorzoxazone 6-hydroxylation, dextromethorphan O-demethylation, mephenytoin 4-hydroxylation, diclofenac 4-hydroxylation, amodiaquine N-deethylation, bupropion hydroxylation, coumarin 7-hydroxylation and phenacetin O-deethylation, respectively. As a result, out of all the saturated fatty acids, lauric acid showed potential inhibition of 2B6. Myristic acid inhibited 1A2 (IC50 = 15.8 μM), 2B6 (IC50 = 10.7 μM), 2C8 (IC50 = 13.3 μM) and 2C9 (IC50 = 36.1 μM). One of the mono-unsaturated fatty acids, oleic acid, inhibited all the CYP isoforms except for CYP2E1, CYP2C19 and CYP2A6. Similarly, arachidonic acid inhibited activities of CYP3A4, CYP2C19, CYP2C9, CYP2C8, CYP2B6 and CYP1A2. Other FAs also showed a distinct inhibitory effect on different isoforms: gondoic acid inhibited all except 2C8; linoleic acid inhibited CYP2B6, CYP2C8 and CYP2C9; linolenic acid inhibited CYP1A2, CYP2B6, CYP2C8 and 2C9; and timnodonic acid inhibited 2C8 (Table 5) [96]. Amongst all the isoenzymes, the majority of FAs inhibited CYP2C8 activity. This can be attributed to the large active site on CYP2C8, which allows different sized substrates to accommodate. It also has a peripheral FA binding site that can alter the dynamics of the main active site, affecting the reaction catalyzed by this enzyme. Moreover, it is responsible for the transformation of polyunsaturated FAs to epoxide products (signaling agents). Hence, the unsaturated fatty acids are potent inhibitors for CYP enzymes than saturated fatty acids [97,98].
Table 5.
Effect of fatty acids on nine CYPs: 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2E1 and 3A4.
| Fatty Acid | Absolute IC50 (μM) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1A2 | 2A6 | 2B6 | 2C8 | 2C9 | 2C19 | 2D6 | 2E1 | 3A4 | |
| Arachidonic acid | 9.7 | 21.4 | 4.6 | 1.3 | 3.3 | 23.2 | 18.3 | 61.7 | 11.7 |
| Behenic acid | >30 | >30 | >30 | >30 | >30 | >30 | >30 | >30 | >30 |
| Cervonic acid | 6.3 | 11.7 | 6.7 | 1.2 | 2.6 | 15.8 | 5.6 | 44.4 | 7.5 |
| Gondoic acid | 16.2 | 81.9 | 16.6 | 6.0 | 17 | >100 | >100 | >100 | >100 |
| Lauric acid | >100 | >100 | 21.5 | 42.9 | >100 | >100 | >100 | >100 | >100 |
| Linoleic acid | 13.3 | 28.9 | 7.1 | 1.0 | 7.4 | 55.8 | 17.5 | 58.9 | 18.5 |
| α-Linolenic acid | 8.8 | 13.5 | 9.7 | 4.4 | 10.6 | 53.3 | 34.3 | 67.2 | 36.9 |
| Myristic acid | 15.8 | >100 | 10.7 | 13.3 | 36.1 | >100 | >100 | >100 | >100 |
| Nervonic acid | >11.1 | >11.1 | >11.1 | >11.1 | >11.1 | >11.1 | >11.1 | >11.1 | >11.1 |
| Oleic acid | 11.2 | 25 | 8.2 | 4.4 | 5.7 | 98.9 | 18.1 | 83.8 | 11.4 |
| Palmitic acid | >100 | >100 | 90.5 | >100 | >100 | >100 | >100 | >100 | >100 |
| Palmitoleic acid | 7.8 | 36.2 | 8 | 9.7 | 11.9 | 58.1 | 30.3 | 72.1 | 26.5 |
| Stearic acid | >33.3 | >33.3 | >33.3 | >33.3 | >33.3 | >33.3 | >33.3 | >33.3 | >33.3 |
| Timnodonic acid | 8.2 | 17.4 | 5.9 | 1.5 | 3.8 | 13.8 | 5.7 | 77.4 | 16 |
The results are consistent with a published study on inhibitory effect of saturated and polyunsaturated fatty acids by Yao and co-workers. Two saturated (palmitic acid and stearic acid) and five polyunsaturated fatty acids (linoleic acid, linolenic acid, arachidonic acid, eicosapentaenoic acid and docosahexaenoic acid) were examined for their effect on six isoforms of CYPs (1A2, 3A4, 2C9, 2C19, 2E1 and 2D6). As shown in Table 6, saturated fatty acids showed no inhibitory effect, while all polyunsaturated fatty acids inhibited all six isoforms [99]. These polyunsaturated fatty acids had little inhibitory effect at low concentrations, whereas there was a complete inhibition of all isoforms at 200 μM. One potential explanation based on the results is that, at high concentration, polyunsaturated fatty acids disrupt the microsomal membrane, which prevents the binding of the drug to the active site of the CYP450 enzyme [99]. However, other studies have reported that the CYP enzymes can also catalyze the metabolism of polyunsaturated fatty acids. Thus, fatty acids can act as a common substrate for the active site and compete with drugs to bind with CYP enzymes [100,101]. Therefore, the mechanism of inhibition remains unknown.
Table 6.
Effect of saturated and unsaturated fatty acids where, saturated fatty acids showed NI (no inhibition) and unsaturated inhibited all CYP isoforms.
| Fatty Acids | IC50 | |||||
|---|---|---|---|---|---|---|
| CYP1A2 | CYP2C9 | CYP2C19 | CYP2D6 | CYP2E1 | CYP3A4 | |
| Palmitic acid | NI | NI | NI | NI | NI | NI |
| Stearic acid | NI | NI | NI | NI | NI | NI |
| Linoleic acid | 74 | 4.1 | 15 | 192 | 113 | 49 |
| Linolenic acid | 52 | 8.1 | 9.3 | 151 | 82 | 61 |
| Arachidonic acid | 37 | 3.5 | 4.8 | 113 | 67 | 48 |
| Eicosapentaenoic acid | 41 | 4.4 | 4.4 | 127 | 53 | 54 |
| Docosahexaenoic acid | 41 | 2.9 | 6.7 | 122 | 65 | 34 |
4. Co-Solvents/Solvents
In the last few decades, the effect of organic solvents (such as acetonitrile) on CYP-mediated metabolism has been reported by many researchers [102,103,104,105,106,107,108,109]. Most of the in vitro studies include organic solvents to dissolve the compounds and prepare the samples for analysis [110,111,112,113,114]. However, at higher concentrations, these organic solvents inhibit different isoforms of CYPs. Li et al. evaluated the inhibitory effect of five organic solvents (dimethyl sulfoxide (DMSO), acetonitrile, methanol, ethanol and acetone) on five CYP isoforms [115]. All organic solvents at 10% v/v inhibited CYPs activities, whereas >10% inhibition of CYP2D and >40% inhibition of CYP2E (except acetonitrile) was reported at 1% v/v. Overall, the organic solvents showed concentration-dependent inhibition. The inhibitory effect of organic solvents is summarized in Figure 2. DMSO and ethanol showed highest inhibitory effect amongst all organic solvents.
Figure 2.

Illustrates the inhibition of CYP2C, CYP2D, CYP2E, CYP3A and CYP1A by acetonitrile, methanol, ethanol, acetone and DMSO at 1% and 10% concentrations.
Another study demonstrated similar results with DMSO. When diazepam was dissolved in DMSO, the rCYP3A4 activity was inhibited in a mixed-type or competitive manner [116]. The inhibition of rCYP3A4 was concluded to be concentration- and time-dependent. The DMSO inhibitory constant (Ki) value for the formation of temazepam and nordiazepam was found to be 24 and 6 mM, respectively.
In 2009, the effect of co-solvent on diclofenac and S-warfarin metabolism by CYP2C9 was studied using ethanol [117]. The inhibitory effect was determined to be substrate dependent. The inhibition of S-warfarin metabolism required less ethanol concentration compared to diclofenac metabolism. Ethanol could inhibit S-warfarin metabolism at a low concentration of 17 mM (0.1% v/v), whereas it required 510 mM (3.0% v/v) of ethanol to competitively inhibit CYP2C9. Ethanol also presented concentration-dependent inhibition for both substrates: S-warfarin and diclofenac.
Eight organic solvents have been studied for their effects on CYP3A4 activity using pooled human microsomes [102]. The hydrophilic organic solvents used for this study were DMSO, acetonitrile, methanol, 1-propanol, ethanol, polyethylene glycol, propylene glycol and N,N-dimethylformamide. All organic solvents, except methanol, had significantly decreased testosterone 6β-hydroxylation activity, which reflected the inhibition of CYP3A4. In contrast, only DMSO and polyethylene glycol showed potential inhibitory effect on midazolam 1′-hydroxylation activity. Similar to this study, DMSO showed strongest inhibition for both metabolic activities. Moreover, the inhibition of testosterone 6β-hydroxylation activity by DMSO was in a concentration-dependent manner: from 10% to 50% inhibition at from 0.1% to 1% v/v concentration. The summary of all the reported organic solvents is shown in Figure 3.
Figure 3.

Roadmap of various reported organic solvents based on their inhibitory effect on CYP450 system. The size of each circle represents the potency of various organic solvents and the lines depict the order of inhibition: from DMSO being the most potent to N,N-dimethylformamide being the least.
5. Effect of Excipients on CYP450 Expression
As discussed above, the alteration of CYP450 activities by excipients were mainly through direct inhibition. However, studies also revealed that some excipients can alter the metabolic mechanism via mRNA/protein expression regulation [61,118], thus modulating absorption and metabolism of Class 3 drugs (high aqueous solubility and low intestinal permeability). Gene expression of the main metabolizing enzymes can be modulated by many environmental toxins or drugs, which can affect the toxicity and efficacy of co-medicated drugs and cause drug–drug interactions [119,120,121,122]. Recently, Tompkins et al. investigated the effect of nineteen excipients on regulation of CYP3A4 expression in human liver and colon cells [118]. CYP3A4 is a highly inducible isoenzyme and is mainly regulated by a xenobiotic receptor named Pregnane X receptor (PXR), at the transcription level [123,124]. This study also included a PXR activation assay to predict the effect of excipients on CYP3A4 expression. As a result, all the excipients failed to activate the CYP2B6 promoter used for PXR activation assay. Instead, few excipients (HPMC, pregelatinized starch and polysorbate-80) showed a decrease in the multi-drug resistance gene (MDR1) and CYP3A4 expression (Table 7). These results suggest that excipients stimulate their effect via an alternate route.
Table 7.
Effect of excipients on PXR activation, CYP3A4 and MDR1. change.
| Excipients | Fa2N4 | HPH | LS174T | |||
|---|---|---|---|---|---|---|
| mRNA | Protein | mRNA | Protein | CYP3A4 | MDR1 | |
| HPMC | ↑ | ↓ | = | x | ↓ | ↓a |
| Pregelatinized starch | = | = | ↓ | x | ↓ | ↓ |
| Croscarmellose sodium | ↑ | = | ↑ | x | ↓a | ↓a |
| Crospovidone | ↑a | ↓ | = | x | ↓ | ↓a |
| Polysorbate-80 | ↓ | ↓ | ↓ | ↓ | = | = |
All the excipients failed to activate PXR but reduced expression of CYP3A4 and MDR1. ↑: increase in expression, ↓: decrease in expression, =: no change, x: not measured, a: no significant.
Based on a previous study, Takeshita et al. evaluated the effect of plasticizers on PXR-mediated transcription by the luciferase receptor, PXR-coactivator interaction, PXR knockdown, CYP3A4 activity assay and PCR analysis of CYP3A4 enzyme expression [125]. Tompkins et al. (2010) used only two plasticizers which showed no effect on PXR or CYP3A4 induction. However, this study was focused on eight pharmaceutical plasticizers: acetyl tributyl citrate (ATBC), acetyl triethyl citrate, tributyl citrate, triethyl citrate, diethyl phthalate, dibutyl phthalate, triacetin and dibutyl sebacate. As a result, ATBC, dibutyl phthalate and acetyl triethyl citrate activated PXR. ATBC being the most potent transcription inducer showed dose-dependent activation. However, ATBC induced only intestinal CYP3A4 expression and not hepatic expression. This could be due to the PXR splice variants that vary in their gene targets, expression patterns, ligands, biological functions, subcellular localization and protein interactions [125]
6. Conclusions
Pharmaceutical excipients play an important role in pharmaceutical products and are often presumed to be pharmacologically inert. However, there is growing evidence that they can change the pharmacokinetics of APIs through various mechanisms, such as P-gp inhibition and CYP450 inhibition. In this review, we present recent research concerning the effects of common pharmaceutical excipients on pre-systemic metabolism by phase I metabolic enzymes (CYP450). According to our review, more than 40 commonly-used excipients were revealed to interfere with different isoforms of CYP450 in vitro, although very few have been assessed in humans. Based on the evidence, the mechanism of action was mainly found to be direct inhibition of the enzymes. Out of all the various excipients, surfactants were the most potent inhibitors due to their ability to cause perturbation of the enzyme’s microenvironment. Despite many similarities in the results from different articles, there appears to be a need for a robust approach to integrate the in vitro data that can predict pharmacokinetic changes in humans. Further research investigations are warranted to shed light on this issue.
Acknowledgments
In this section you can acknowledge any support given which is not covered by the author contribution or funding sections. This may include administrative and technical support, or donations in kind (e.g., materials used for experiments).
Abbreviations
| API | Active Pharmaceutical Ingredient |
| AUC | Area Under the Curve |
| CMC | Critical Micellar Concentration |
| CTAB | Cetyltrimethylammonium Bromide |
| CYP450 | Cytochrome P450 |
| DHB | Dihydroxybergamottin |
| DMSO | Dimethyl Sulfoxide |
| FA | Fatty Acids |
| GIT | Gastrointestinal Tract |
| HPC | Hydroxypropyl Cellulose |
| HPMC | Hydroxypropyl Methylcellulose |
| IC50 | Inhibitory Concentration |
| Ki | Inhibitory Constant |
| Clint | Intrinsic Clearance |
| mRNA | Messenger RNA |
| MDZ | Midazolam |
| Mw | Molecular weight |
| MDR1 | Multi-drug Resistance Gene |
| mPEGx-PCLx | Methoxy Poly(ethylene glycol)-poly(ε-caprolactone) (mPEGx-PCLx) |
| ATBC | Acetyl Tributyl Citrate |
| F68 | Pluronic F68 |
| PEG | Polyethylene Glycol |
| PVA | Polyvinyl Acetate |
| PVP | Polyvinyl Pyrrolidone |
| PXR | Pregnane X receptor |
| SDS | Sodium Dodecyl Sulfate |
| SEDSS | Self-emulsifying Drug Delivery System |
| SMEDDS | Self-micro Emulsifying Drug Delivery System |
| NaCMC | Sodium Carboxymethyl Cellulose |
| NI | No Inhibition |
| TPGS | D -α-Tocopherol Polyethylene Glycol 1000 Succinate |
| TX-100 | Triton X-100 |
Author Contributions
Conceptualization, A.E.; J.B. and R.P.; methodology, A.E. and R.P.; writing—original draft preparation, R.P.; writing—review and editing, A.E. and J.B.; visualization, R.P.; supervision, A.E. and J.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Sastry S.V., Nyshadham J.R., Fix J.A. Recent technological advances in oral drug delivery—A review. Pharm. Sci. Technol. Today. 2000;3:138–145. doi: 10.1016/S1461-5347(00)00247-9. [DOI] [PubMed] [Google Scholar]
- 2.Major Advances in Oral Drug Delivery over the Past 15 Years. [(accessed on 24 July 2020)]; Available online: https://www.americanpharmaceuticalreview.com/Featured-Articles/148747-Major-Advances-in-Oral-Drug-Delivery-over-the-Past-15-Years/
- 3.Zhang W., Li Y., Zou P., Wu M., Zhang Z., Zhang T. The Effects of Pharmaceutical Excipients on Gastrointestinal Tract Metabolic Enzymes and Transporters—An Update. AAPS PharmSciTech. 2016;18:830–843. doi: 10.1208/s12248-016-9928-8. [DOI] [PubMed] [Google Scholar]
- 4.Pathak K., Raghuvanshi S. Oral Bioavailability: Issues and Solutions via Nanoformulations. Clin. Pharmacokinet. 2015;54:325–357. doi: 10.1007/s40262-015-0242-x. [DOI] [PubMed] [Google Scholar]
- 5.Ezra A., Golomb G. Administration routes and delivery systems of bisphosphonates for the treatment of bone resorption. Adv. Drug Deliv. Rev. 2000;42:175–195. doi: 10.1016/S0169-409X(00)00061-2. [DOI] [PubMed] [Google Scholar]
- 6.Cremers S., van Hogezand R., Bänffer D., den Hartigh J., Vermeij P., Papapoulos S.E., Hamdey N.A.T. Absorption of the oral bisphosphonate alendronate in osteoporotic patients with Crohn’s disease. Osteoporos. Int. 2005;16:1727–1730. doi: 10.1007/s00198-005-1911-7. [DOI] [PubMed] [Google Scholar]
- 7.Lambrinoudaki I., Christodoulakos G., Botsis D. Women’s health and disease: Gynaecologic, endocrine, and reproductive issues. Ann. N. Y. Acad. Sci. 2006;1092:397–402. doi: 10.1196/annals.1365.036. [DOI] [PubMed] [Google Scholar]
- 8.Reinoso R.F., Sanchez Navarro A., Garcia M.J., Prous J.R. Preclinical pharmacokinetics of statins. Clin. Exp. Pharmacol, Physiol. 2002;24:593–613. [PubMed] [Google Scholar]
- 9.Garcia M., Reinoso R., Sanchez Navarro A., Prous J. Clinical pharmacokinetics of statins. Clin. Exp. Pharmacol. Physiol. 2003;24:457–481. doi: 10.1358/mf.2003.25.6.769652. [DOI] [PubMed] [Google Scholar]
- 10.Buse J. Statin Treatment in Diabetes Mellitus. Clin. Diabetes. 2003;21:168–172. doi: 10.2337/diaclin.21.4.168. [DOI] [Google Scholar]
- 11.Degim I., Acartürk F., Erdogan D., Demirez Lortlar N. Transdermal Administration of Bromocriptine. Biol. Pharm. Bull. 2003;26:501–505. doi: 10.1248/bpb.26.501. [DOI] [PubMed] [Google Scholar]
- 12.Turner J., Glass B., Agatonovic-Kustrin S. Prediction of drug bioavailability based on molecular structure. Anal. Chim. Acta. 2003;485:89–102. doi: 10.1016/S0003-2670(03)00406-9. [DOI] [Google Scholar]
- 13.Niemi R., Turhanen P., Vepsäläinen J., Taipale H., Järvinen T. Bisphosphonate prodrugs: Synthesis and in vitro evaluation of alkyl and acyloxymethyl esters of etidronic acid as bioreversible prodrugs of etidronate. Eur. J. Pharm. Sci. 2000;11:173–180. doi: 10.1016/S0928-0987(00)00099-3. [DOI] [PubMed] [Google Scholar]
- 14.Joseph N.M., Sharma P.K. Cross-linked nanoparticles of cytarabine: Encapsulation, storage and in vivo release. Afr. J. Pharm. Pharmacol. 2007;1:10–13. [Google Scholar]
- 15.Ahmad N., Keithferris J., Gooden E., Abell T. Making a case for domperidone in the treatment of gastrointestinal motility disorders. Curr. Opin. Pharmcol. 2006;6:571–576. doi: 10.1016/j.coph.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 16.Sturgill M., August D., Brenner D. Hepatic Enzyme Induction with Phenobarbital and Doxorubicin Metabolism and Myelotoxicity in the Rabbit. Cancer Investig. 2000;18:197–205. doi: 10.3109/07357900009031824. [DOI] [PubMed] [Google Scholar]
- 17.Dorne J., Walton K., Renwick A. Human variability in CYP3A4 metabolism and CYP3A4-related uncertainty factors for risk assessment. Food. Chem. Toxicol. 2003;41:201–224. doi: 10.1016/S0278-6915(02)00209-0. [DOI] [PubMed] [Google Scholar]
- 18.Tse F., Jaffe J. Pharmacokinetics of PN 200-110 (isradipine), a new calcium antagonist, after oral administration in man. Eur. J. Clin. Pharmacol. 1987;32:361–365. doi: 10.1007/BF00543970. [DOI] [PubMed] [Google Scholar]
- 19.Choi J., Burm J. Enhanced nimodipine bioavailability after oral administration of nimodipine with morin, a flavonoid, in rabbits. Arch. Pharm. Res. 2006;29:333–338. doi: 10.1007/BF02968580. [DOI] [PubMed] [Google Scholar]
- 20.Bennett M., Lucas V., Brennan M., Hughes A., O’Donnell V., Wee B. Using anti-muscarinic drugs in the management of death rattle: Evidence-based guidelines for palliative care. Palliat. Med. 2002;16:369–374. doi: 10.1191/0269216302pm584oa. [DOI] [PubMed] [Google Scholar]
- 21.Kharasch E., Labroo R. Metabolism of Ketamine Stereoisomers by Human Liver Microsomes. Anesthesiology. 1992;77:1201–1207. doi: 10.1097/00000542-199212000-00022. [DOI] [PubMed] [Google Scholar]
- 22.Manoir B., Bourget P., Langlois M., Szekely B., Fischler M., Chauvin M. Evaluation of the pharmacokinetic profile and analgesic efficacy of oral morphine after total hip arthroplasty. Eur. J. Anaesthesiol. 2006;23:748–754. doi: 10.1017/S0265021506000731. [DOI] [PubMed] [Google Scholar]
- 23.Aquilonius S., Eckernas S., Hartvig P., Lindstrom B., Osterman P. Pharmacokinetics and oral bioavailability of pyridostigmine in man. Eur. J. Clin. Pharmacol. 1980;18:423–428. doi: 10.1007/BF00636797. [DOI] [PubMed] [Google Scholar]
- 24.Kleiman-Wexler R.L., Adair C.G., Ephgrave K.S. Pharmacokinetics of naloxone: An insight into the locus of effect on stress-ulceration. J. Pharmacol. Exp. Ther. 1989;251:435–438. [PubMed] [Google Scholar]
- 25.Finn A., Collins J., Voyksner R., Lindley C. Bioavailability and Metabolism of Prochlorperazine Administered via the Buccal and Oral Delivery Route. J. Clin. Pharmacol. 2005;45:1383–1390. doi: 10.1177/0091270005281044. [DOI] [PubMed] [Google Scholar]
- 26.Clarke A., Jankovic J. Selegiline orally disintegrating tablet in the treatment of Parkinson’s disease. Therapy. 2006;3:349–356. doi: 10.2217/14750708.3.3.349. [DOI] [Google Scholar]
- 27.Cossons V.F., Fuseau E. Mixed effect modelling of sumatriptan pharmacokinetics during drug development: II. From healthy subjects to phase 2 dose ranging in patients. J. Pharmacokinet. Pharmacodyn. 1999;27:149–171. doi: 10.1023/A:1020601906027. [DOI] [PubMed] [Google Scholar]
- 28.Jann M., Shirley K., Small G. Clinical Pharmacokinetics and Pharmacodynamics of Cholinesterase Inhibitors. Clin. Pharmacokinet. 2002;41:719–739. doi: 10.2165/00003088-200241100-00003. [DOI] [PubMed] [Google Scholar]
- 29.Nakhat P., Kondawar A., Babla I., Rathi L., Yeole P. Studies on buccoadhesive tablets of terbutaline sulphate. Indian J. Pharm. Sci. 2007;69:505. doi: 10.4103/0250-474X.36934. [DOI] [Google Scholar]
- 30.Isohanni M.H., Neuroven P.J., Olkkola K.T. Effect of fluvoxamine and erythromycin on the pharmaco- kinetics of oral lidocaine. Basic Clin. Pharmacol. Toxicol. 2006;99:168–172. doi: 10.1111/j.1742-7843.2006.pto_482.x. [DOI] [PubMed] [Google Scholar]
- 31.Nelson D.R. The cytochrome P450 homepage. Hum. Genomics. 2009;4:59–65. doi: 10.1186/1479-7364-4-1-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zanger U., Turpeinen M., Klein K., Schwab M. Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation. Anal. Bioanal. Chem. 2008;392:1093–1108. doi: 10.1007/s00216-008-2291-6. [DOI] [PubMed] [Google Scholar]
- 33.Markovic M., Ben-Shabat S., Keinan S., Aponick A., Zimmermann E., Dahan A. Lipidic prodrug approach for improved oral drug delivery and therapy. Med. Res. Rev. 2018;39:579–607. doi: 10.1002/med.21533. [DOI] [PubMed] [Google Scholar]
- 34.Huttunen K., Raunio H., Rautio J. Prodrugs—from Serendipity to Rational Design. Pharmacol. Rev. 2011;63:750–771. doi: 10.1124/pr.110.003459. [DOI] [PubMed] [Google Scholar]
- 35.Dahan A., Wolk O., Agbaria R. Provisional in-silico biopharmaceutics classification (BCS) to guide oral drug product development. Drug Des. Devel. Ther. 2014;8:1563. doi: 10.2147/DDDT.S68909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rautio J., Kumpulainen H., Heimbach T., Oliyai R., Oh D., Järvinen T. Prodrugs: Design and clinical applications. Nat. Rev. Drug Discov. 2008;7:255–270. doi: 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- 37.Clas S., Sanchez R., Nofsinger R. Chemistry-enabled drug delivery (prodrugs): Recent progress and challenges. Drug Discov. Today. 2014;19:79–87. doi: 10.1016/j.drudis.2013.08.014. [DOI] [PubMed] [Google Scholar]
- 38.Zawilska J., Wojcieszak J., Olejniczak A. Prodrugs: A challenge for the drug development. Curr. Pharmacol. Rep. 2013;65:1–14. doi: 10.1016/S1734-1140(13)70959-9. [DOI] [PubMed] [Google Scholar]
- 39.Dahan A., Zimmermann E., Ben-Shabat S. Modern Prodrug Design for Targeted Oral Drug Delivery. Molecules. 2014;19:16489–16505. doi: 10.3390/molecules191016489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han H., Amidon G. Targeted prodrug design to optimize drug delivery. AAPS PharmSciTech. 2000;2:48–58. doi: 10.1208/ps020106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amidon G., Leesman G., Elliott R. Improving intestinal absorption of water-insoluble compounds: A membrane metabolism strategy. J. Pharm. Sci. 1980;69:1363–1368. doi: 10.1002/jps.2600691203. [DOI] [PubMed] [Google Scholar]
- 42.Stella V. Prodrugs: Some Thoughts and Current Issues. J. Pharm. Sci. 2010;99:4755–4765. doi: 10.1002/jps.22205. [DOI] [PubMed] [Google Scholar]
- 43.Pereira de Sousa I., Bernkop-Schnürch A. Pre-systemic metabolism of orally administered drugs and strategies to overcome it. J. Control. Release. 2014;192:301–309. doi: 10.1016/j.jconrel.2014.08.004. [DOI] [PubMed] [Google Scholar]
- 44.Bailey D., Malcolm J., Arnold O., David Spence J. Grapefruit juice–drug interactions. Br. J. Clin. Pharmacol. 1998;46:101–110. doi: 10.1046/j.1365-2125.1998.00764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fasinu P., Choonara Y., Khan R., Du Toit L., Kumar P.K., Ndesendo V. Flavonoids and Polymer Derivatives as CYP3A4 Inhibitors for Improved Oral Drug Bioavailability. J. Pharm. Sci. 2013;102:541–555. doi: 10.1002/jps.23382. [DOI] [PubMed] [Google Scholar]
- 46.Lin H., Kent U., Hollenberg P. The Grapefruit Juice Effect Is Not Limited to Cytochrome P450 (P450) 3A4: Evidence for Bergamottin-Dependent Inactivation, Heme Destruction, and Covalent Binding to Protein in P450s 2B6 and 3A5. J. Pharmacol. Exp. Ther. 2004;313:154–164. doi: 10.1124/jpet.104.079608. [DOI] [PubMed] [Google Scholar]
- 47.Tassaneeyakul W., Guo L., Fukuda K., Ohta T., Yamazoe Y. Inhibition Selectivity of Grapefruit Juice Components on Human Cytochromes P450. Arch. Biochem. Biophys. 2000;378:356–363. doi: 10.1006/abbi.2000.1835. [DOI] [PubMed] [Google Scholar]
- 48.Girennavar B., Poulose S., Jayaprakasha G., Bhat N., Patil B. Furocoumarins from grapefruit juice and their effect on human CYP 3A4 and CYP 1B1 isoenzymes. Bioorg. Med. Chem. 2006;14:2606–2612. doi: 10.1016/j.bmc.2005.11.039. [DOI] [PubMed] [Google Scholar]
- 49.Paine M., Criss A., Watkins P. Two Major Grapefruit Juice Components Differ in Intestinal CYP3A4 Inhibition Kinetic and Binding Properties. Drug Metab. Dispos. 2004;32:1146–1153. doi: 10.1124/dmd.104.000547. [DOI] [PubMed] [Google Scholar]
- 50.Paine M., Criss A., Watkins P. Two Major Grapefruit Juice Components Differ in Time to Onset of Intestinal CYP3A4 Inhibition. J. Pharmacol. Exp. Ther. 2004;312:1151–1160. doi: 10.1124/jpet.104.076836. [DOI] [PubMed] [Google Scholar]
- 51.Greenblatt D. Time course of recovery of cytochrome p450 3A function after single doses of grapefruit juice. Clin. Pharmacol. Ther. 2003;74:121–129. doi: 10.1016/S0009-9236(03)00118-8. [DOI] [PubMed] [Google Scholar]
- 52.Ohnishi A., Matsuo H., Yamada S., Takanaga H., Morimoto S., Shoyama Y. Effect of furanocoumarin derivatives in grapefruit juice on the uptake of vinblastine by Caco-2 cells and on the activity of cytochrome P450 3A4. Br. J. Pharmacol. 2000;130:1369–1377. doi: 10.1038/sj.bjp.0703433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eagling V.A., Profit L., Back D.J. Inhibition of the CYP3A4-mediated metabolism and P-glycoprotein-mediated transport of the HIV-1 protease inhibitor saquinavir by grapefruit juice components. Br. J. Clin. Pharmacol. 2001;48:543–552. doi: 10.1046/j.1365-2125.1999.00052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fukuda K., Ohta T., Yamazoe Y. Grapefruit Component Interacting with Rat and Human P450 CYP3A: Possible Involvement of Non-Flavonoid Components in Drug Interaction. Biol. Pharm. Bull. 1997;20:560–564. doi: 10.1248/bpb.20.560. [DOI] [PubMed] [Google Scholar]
- 55.Hanley M., Cancalon P., Widmer W., Greenblatt D. The effect of grapefruit juice on drug disposition. Expert Opin. Drug Metab. Toxicol. 2011;7:267–286. doi: 10.1517/17425255.2011.553189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Galetin A., Gertz M., Brian Houston J. Contribution of Intestinal Cytochrome P450-Mediated Metabolism to Drug-Drug Inhibition and Induction Interactions. Drug Metab. Pharmacokinet. 2010;25:28–47. doi: 10.2133/dmpk.25.28. [DOI] [PubMed] [Google Scholar]
- 57.Bylund J., Bueters T. Presystemic Metabolism of AZ’0908, A Novel mPGES-1 Inhibitor: An In Vitro and In Vivo Cross-Species Comparison. J. Pharm. Sci. 2013;102:1106–1115. doi: 10.1002/jps.23443. [DOI] [PubMed] [Google Scholar]
- 58.Zhou S., Yung Chan S., Cher Goh B., Chan E., Duan W., Huang M. Mechanism-Based Inhibition of Cytochrome P450 3A4 by Therapeutic Drugs. Clin. Pharmacokinet. 2005;44:279–304. doi: 10.2165/00003088-200544030-00005. [DOI] [PubMed] [Google Scholar]
- 59.Horn J., Howden C. Review article: Similarities and differences among delayed-release proton-pump inhibitor formulations. Aliment. Pharm. Ther. 2005;22:20–24. doi: 10.1111/j.1365-2036.2005.02714.x. [DOI] [PubMed] [Google Scholar]
- 60.Lueßen H., Rentel C., Kotzé A., Lehr C., de Boer A., Verhoef J. Mucoadhesive polymers in peroral peptide drug delivery. IV. Polycarbophil and chitosan are potent enhancers of peptide transport across intestinal mucosae in vitro. J. Control. Release. 1997;45:15–23. doi: 10.1016/S0168-3659(96)01536-2. [DOI] [Google Scholar]
- 61.Ren X., Mao X., Si L., Cao L., Xiong H., Qiu J. Pharmaceutical excipients inhibit cytochrome P450 activity in cell free systems and after systemic administration. Eur. J. Pharm. Biopharm. 2008;70:279–288. doi: 10.1016/j.ejpb.2008.03.019. [DOI] [PubMed] [Google Scholar]
- 62.Larson A., Polson J., Fontana R., Davern T., Lalani E., Hynan L. Acetaminophen-induced acute liver failure: Results of a United States multicenter, prospective study. Hepatology. 2005;42:1364–1372. doi: 10.1002/hep.20948. [DOI] [PubMed] [Google Scholar]
- 63.Marx J. TOXICOLOGY: Protecting Liver from Painkiller’s Lethal Dose. Science. 2002;298:341a–342a. doi: 10.1126/science.298.5592.341a. [DOI] [PubMed] [Google Scholar]
- 64.Zaher H., Buters J., Ward J., Bruno M., Lucas A., Stern S. Protection against Acetaminophen Toxicity in CYP1A2 and CYP2E1 Double-Null Mice. Toxicol. Appl. Pharm. 1998;152:193–199. doi: 10.1006/taap.1998.8501. [DOI] [PubMed] [Google Scholar]
- 65.USP A. Acetaminophen Oral Solution USP—PAI. [(accessed on 24 July 2020)]; Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4383763/
- 66.Sekhon B. Surfactants: Pharmaceutical and Medicinal Aspects. JPTRM. 2013;1:43–68. doi: 10.15415/jptrm.2013.11004. [DOI] [Google Scholar]
- 67.Gelderblom H., Verweij J., Nooter K., Sparreboom A. Cremophor EL. Eur. J. Cancer. 2001;37:1590–1598. doi: 10.1016/S0959-8049(01)00171-X. [DOI] [PubMed] [Google Scholar]
- 68.Christiansen A., Backensfeld T., Denner K., Weitschies W. Effects of non-ionic surfactants on cytochrome P450-mediated metabolism in vitro. Eur. J. Pharm. Biopharm. 2011;78:166–172. doi: 10.1016/j.ejpb.2010.12.033. [DOI] [PubMed] [Google Scholar]
- 69.Mudra D., Borchardt R. Absorption Barriers in the Rat Intestinal Mucosa. 3: Effects of Polyethoxylated Solubilizing Agents on Drug Permeation and Metabolism. J. Pharm. Sci. 2010;99:1016–1027. doi: 10.1002/jps.21836. [DOI] [PubMed] [Google Scholar]
- 70.Hugger E., Novak B., Burton P., Audus K., Borchardt R. A comparison of commonly used polyethoxylated pharmaceutical excipients on their ability to inhibit P-glycoprotein activity in vitro. J. Pharm. Sci. 2002;91:1991–2002. doi: 10.1002/jps.10176. [DOI] [PubMed] [Google Scholar]
- 71.Hugger E., Audus K., Borchardt R. Effects of Poly(ethylene glycol) on Efflux Transporter Activity in Caco-2 Cell Monolayers. J. Pharm. Sci. 2002;91:1980–1990. doi: 10.1002/jps.10175. [DOI] [PubMed] [Google Scholar]
- 72.Regev R., Assaraf Y.G., Eytan G.D. Membrane fluidization by ether, other anesthetics, and certain agents abolishes P-glycoprotein ATPase activity and modulates efflux from multidrug-resistant cells. Eur. J. Biochem. 1999;259:18–24. doi: 10.1046/j.1432-1327.1999.00037.x. [DOI] [PubMed] [Google Scholar]
- 73.Rege B.D., Kao J.P.Y., Polli J.E. Effects of nonionic surfactants on membrane transporters in Caco-2 cell monolayers. Eur. J. Pharm. Sci. 2002;16:237–246. doi: 10.1016/S0928-0987(02)00055-6. [DOI] [PubMed] [Google Scholar]
- 74.Randall K.P., Cheng S.W., Kotchevar A.T. Evaluation of surfactants as solubilizing agents in microsomal metabolism reactions with lipophilic substrates. In Vitro Cell. Dev. Biol. Anim. 2011;47:631–639. doi: 10.1007/s11626-011-9449-9. [DOI] [PubMed] [Google Scholar]
- 75.Bravo González R.C., Huwyler J., Boess F., Walter I., Bittner B. In vitro investigation on the impact of the surface-active excipients Cremophor EL, Tween 80 and Solutol HS 15 on the metabolism of midazolam. Biopharm. Drug. Dispos. 2004;25:37–49. doi: 10.1002/bdd.383. [DOI] [PubMed] [Google Scholar]
- 76.Jamis-Dow C., Klecker R., Katki A., Collins J. Metabolism of taxol by human and rat liver in vitro: A screen for drug interactions and interspecies differences. Cancer Chemother. Pharmacol. 1995;36:107–114. doi: 10.1007/BF00689193. [DOI] [PubMed] [Google Scholar]
- 77.Tayrouz Y. Pharmacokinetic and pharmaceutic interaction between digoxin and Cremophor RH40. Clin. Pharmacol. Ther. 2003;73:397–405. doi: 10.1016/S0009-9236(03)00059-6. [DOI] [PubMed] [Google Scholar]
- 78.Rao Z., Si L., Guan Y., Pan H., Qiu J., Li G. Inhibitive effect of cremophor RH40 or tween 80-based self-microemulsifying drug delivery system on cytochrome P450 3A enzymes in murine hepatocytes. J. Huazhong Univ. Sci. Med. 2010;30:562–568. doi: 10.1007/s11596-010-0543-0. [DOI] [PubMed] [Google Scholar]
- 79.Lim Y.P., Kuo S.C., Lai M.L. Inhibition of CYP3A4 expression by ketoconazole is mediated by the disruption of pregnane X receptor, steroid receptor coactivator-1, and hepatocyte nuclear factor 4alpha interaction. Pharmacogenet. Genom. 2009;19:11–24. doi: 10.1097/FPC.0b013e32831665ea. [DOI] [PubMed] [Google Scholar]
- 80.Fujita T., Yasuda S., Kamata Y. Contribution of down-regulation of intestinal and hepatic cytochrome P450 3A to increased absorption of cyclosporine A in a rat nephrosis model. J. Pharmacol. Exp. Ther. 2008;327:592–599. doi: 10.1124/jpet.108.142091. [DOI] [PubMed] [Google Scholar]
- 81.Ren S., Park M., Kim A., Lee B. In vitro metabolic stability of moisture-sensitive rabeprazole in human liver microsomes and its modulation by pharmaceutical excipients. Arch. Pharm. Res. 2008;31:406–413. doi: 10.1007/s12272-001-1171-z. [DOI] [PubMed] [Google Scholar]
- 82.Farsang E., Gaál V., Horváth O., Bárdos E., Horváth K. Analysis of Non-Ionic Surfactant Triton X-100 Using Hydrophilic Interaction Liquid Chromatography and Mass Spectrometry. Molecules. 2019;24:1223. doi: 10.3390/molecules24071223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Da Silva M., Meirelles N. Interaction of non-ionic surfactants with hepatic CYP in Prochilodus scrofa. Toxicol. In Vitro. 2004;18:859–867. doi: 10.1016/j.tiv.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 84.Karolewicz B. A review of polymers as multifunctional excipients in drug dosage form technology. Saudi Pharm. J. 2016;24:525–536. doi: 10.1016/j.jsps.2015.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Qiu L., Li Q., Huang J., Wu Q., Tu K., Wu Y. In vitro effect of mPEG2k-PCLx micelles on rat liver cytochrome P450 enzymes. Int. J. Pharm. 2018;552:99–110. doi: 10.1016/j.ijpharm.2018.09.052. [DOI] [PubMed] [Google Scholar]
- 86.Martin P., Giardiello M., McDonald T., Rannard S., Owen A. Mediation of in Vitro Cytochrome P450 Activity by Common Pharmaceutical Excipients. Mol. Pharm. 2013;10:2739–2748. doi: 10.1021/mp400175n. [DOI] [PubMed] [Google Scholar]
- 87.Huang J., Si L., Jiang L., Fan Z., Qiu J., Li G. Effect of pluronic F68 block copolymer on P-glycoprotein transport and CYP3A4 metabolism. Int. J. Pharm. 2008;356:351–353. doi: 10.1016/j.ijpharm.2007.12.028. [DOI] [PubMed] [Google Scholar]
- 88.Johnson B., Charman W., Porter C. An in vitro examination of the impact of polyethylene glycol 400, pluronic P85, and vitamin E d-a-tocopheryl polyethylene glycol 1000 succinate on P-glycoprotein efflux and enterocyte-based metabolism in excised rat intestine. AAPS PharmSciTech. 2002;4:193–205. doi: 10.1208/ps040440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Butt U., ElShaer A., Snyder L., Al-Kinani A., Le Gresley A., Alany R. Fatty Acid Based Microemulsions to Combat Ophthalmia Neonatorum Caused by Neisseria gonorrhoeae and Staphylococcus aureus. Nanomaterials. 2018;8:51. doi: 10.3390/nano8010051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bergsson G., Arnfinnsson J., Steingrímsson O., Thormar H. In Vitro Killing of Candida albicans by Fatty Acids and Monoglycerides. Antimicrob. Agents Chemother. 2001;45:3209–3212. doi: 10.1128/AAC.45.11.3209-3212.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yoon B., Jackman J., Valle-González E., Cho N. Antibacterial Free Fatty Acids and Monoglycerides: Biological Activities, Experimental Testing, and Therapeutic Applications. Int. J. Mol. Sci. 2018;19:1114. doi: 10.3390/ijms19041114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kabara J., Swieczkowski D., Conley A., Truant J. Fatty Acids and Derivatives as Antimicrobial Agents. Antimicrob. Agents Chemother. 1972;2:23–28. doi: 10.1128/AAC.2.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kabara J., Vrable R. Antimicrobial lipids: Natural and synthetic fatty acids and monoglycerides. Lipids. 1977;12:753–759. doi: 10.1007/BF02570908. [DOI] [PubMed] [Google Scholar]
- 94.Kabara J. Antimicrobial agents derived from fatty acids. J. Am. Oil Chem. Soc. 1984;61:397–403. doi: 10.1007/BF02678802. [DOI] [Google Scholar]
- 95.Kabara J., Conley A., Truant J. Relationship of Chemical Structure and Antimicrobial Activity of Alkyl Amides and Amines. Antimicrob. Agents Chemother. 1972;2:492–498. doi: 10.1128/AAC.2.6.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Palacharla R., Uthukam V., Manoharan A., Ponnamaneni R., Padala N., Boggavarapu R. Inhibition of cytochrome P450 enzymes by saturated and unsaturated fatty acids in human liver microsomes, characterization of enzyme kinetics in the presence of bovine serum albumin (0.1 and 1.0% w/v) and in vitro—in vivo extrapolation of hepatic clearance. Eur. J. Pharm. Sci. 2017;101:80–89. doi: 10.1016/j.ejps.2017.01.027. [DOI] [PubMed] [Google Scholar]
- 97.Schoch G., Yano J., Wester M., Griffin K., Stout C., Johnson E. Structure of Human Microsomal Cytochrome P450 2C8. J. Biol. Chem. 2003;279:9497–9503. doi: 10.1074/jbc.M312516200. [DOI] [PubMed] [Google Scholar]
- 98.Backman J., Filppula A., Niemi M., Neuvonen P. Role of Cytochrome P450 2C8 in Drug Metabolism and Interactions. Pharmacol. Rev. 2015;68:168–241. doi: 10.1124/pr.115.011411. [DOI] [PubMed] [Google Scholar]
- 99.Yao H., Chang Y., Lan S., Chen C., Hsu J., Yeh T. The inhibitory effect of polyunsaturated fatty acids on human CYP enzymes. Life Sci. 2006;79:2432–2440. doi: 10.1016/j.lfs.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 100.Rifkind A., Lee C., Chang T., Waxman D. Arachidonic acid metabolism by human cytochrome P450s 2C8, 2C9, 2E1, and 1A2: Regioselective oxygenation and evidence for a role for CYP2C enzymes in arachidonic acid epoxygenation in human liver microsomes. Arch. Biochem. Biophys. 1995;320:380–389. doi: 10.1016/0003-9861(95)90023-3. [DOI] [PubMed] [Google Scholar]
- 101.Yamazaki H. Effects of arachidonic acid, prostaglandins, retinol, retinoic acid and cholecalciferol on xenobiotic oxidations catalysed by human cytochrome P450 enzymes. Xenobiotica. 1999;29:231–241. doi: 10.1080/004982599238632. [DOI] [PubMed] [Google Scholar]
- 102.Iwase M., Kurata N., Ehana R., Nishimura Y., Masamoto T., Yasuhara H. Evaluation of the effects of hydrophilic organic solvents on CYP3A-mediated drug-drug interaction in vitro. Hum. Exp. Toxicol. 2006;25:715–721. doi: 10.1177/0960327106071979. [DOI] [PubMed] [Google Scholar]
- 103.Cotreau-Bibbo M.M., Von Moltke L.L., Greenblatt D.J. Influence of polyethylene glycol and acetone on the in vitro biotransformation of tamoxifen and alprazolam by human liver microsomes. J. Pharm. Sci. 1996;85:1180–1185. doi: 10.1021/js9601849. [DOI] [PubMed] [Google Scholar]
- 104.Draper A.J., Madan A., Parkinson A. Inhibition of coumarin 7-hydroxylase activity in human liver microsomes. Arch. Biochem. Biophys. 1997;341:47–61. doi: 10.1006/abbi.1997.9964. [DOI] [PubMed] [Google Scholar]
- 105.Chauret N., Gauthier A., Nicoll-Griffith D.A. Effect of common organic solvents on in vitro cytochrome P450-mediated metabolic activities in human liver microsomes. Drug Metab. Dispos. 1998;26:1–4. [PubMed] [Google Scholar]
- 106.Hickman D., Wang J.P., Wang Y., Unadkat J.D. Evaluation of the selectivity of in vitro probes and suitability of organic solvents for the measurement of human cytochrome P450 monooxygenase activities. Drug Metab. Dispos. 1998;26:207–215. [PubMed] [Google Scholar]
- 107.Busby W.F., Jr., Ackermann J.M., Crespi C.L. Effect of methanol, ethanol, dimethyl sulfoxide, and acetonitrile on in vitro activities of cDNA-expressed human cytochromes P-450. Drug Metab. Dispos. 1999;27:246–249. [PubMed] [Google Scholar]
- 108.Coller J.K., Somogyi A.A., Bochner F. Flunitrazepam oxidative metabolism in human liver microsomes: Involvement of CYP2C19 and CYP3A4. Xenobiotica. 1999;29:973–986. doi: 10.1080/004982599238056. [DOI] [PubMed] [Google Scholar]
- 109.Palamanda J., Feng W.W., Lin C.C., Nomeir A.A. Stimulation of tolbutamide hydroxylation by acetone and acetonitrile in human liver microsomes and in a cytochrome P-450 2C9-reconstituted system. Drug Metab. Dispos. 2000;28:38–43. [PubMed] [Google Scholar]
- 110.Tolonen A., Petsalo A., Turpeinen M., Uusitalo J., Pelkonen O. In vitro interaction cocktail assay for nine major cytochrome P450 enzymes with 13 probe reactions and a single LC/MSMS run: Analytical validation and testing with monoclonal anti-CYP antibodies. J. Mass Spectrom. 2007;42:960–966. doi: 10.1002/jms.1239. [DOI] [PubMed] [Google Scholar]
- 111.Youdim K., Lyons R., Payne L., Jones B., Saunders K. An automated, high-throughput, 384 well Cytochrome P450 cocktail IC50 assay using a rapid resolution LC–MS/MS end-point. J. Pharmaceut. Biomed. 2008;48:92–99. doi: 10.1016/j.jpba.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 112.Otten J., Hingorani G.P., Hartley D., Kragerud S., B. Franklin R. An In Vitro, High Throughput, Seven CYP Cocktail Inhibition Assay for the Evaluation of New Chemical Entities Using LC-MS/MS. Drug Metab. Lett. 2011;5:17–24. doi: 10.2174/187231211794455235. [DOI] [PubMed] [Google Scholar]
- 113.Kozakai K. Reliable high-throughput method for inhibition assay of 8 cytochrome P450 isoforms using cocktail of probe substrates and stable isotope-labeled internal standard. Drug Metab. Pharmacokinet. 2012;27:520–529. doi: 10.2133/dmpk.DMPK-12-RG-014. [DOI] [PubMed] [Google Scholar]
- 114.Chen Z., Zhang S., Long N., Lin L., Chen T., Zhang F. An improved substrate cocktail for assessing direct inhibition and time-dependent inhibition of multiple cytochrome P450s. Acta Pharmacol. Sin. 2016;37:708–718. doi: 10.1038/aps.2016.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li D., Han Y., Meng X., Sun X., Yu Q., Li Y. Effect of Regular Organic Solvents on Cytochrome P450-Mediated Metabolic Activities in Rat Liver Microsomes: Fig. 1. Drug Metab. Dispos. 2010;38:1922–1925. doi: 10.1124/dmd.110.033894. [DOI] [PubMed] [Google Scholar]
- 116.Nishiya Y., Nakamura K., Okudaira N., Abe K., Kobayashi N., Okazaki O. Effects of organic solvents on the time-dependent inhibition of CYP3A4 by diazepam. Xenobiotica. 2009;40:1–8. doi: 10.3109/00498250903337392. [DOI] [PubMed] [Google Scholar]
- 117.Tatsumi A., Ikegami Y., Morii R., Sugiyama M., Kadobayashi M., Iwakawa S. Effect of Ethanol on S-Warfarin and Diclofenac Metabolism by Recombinant Human CYP2C9.1. Biol. Pharm. Bull. 2009;32:517–519. doi: 10.1248/bpb.32.517. [DOI] [PubMed] [Google Scholar]
- 118.Tompkins L., Lynch C., Haidar S., Polli J., Wang H. Effects of Commonly Used Excipients on the Expression of CYP3A4 in Colon and Liver Cells. Pharm. Res. 2010;27:1703–1712. doi: 10.1007/s11095-010-0170-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Klaassen C., Slitt A. Regulation of Hepatic Transporters by Xenobiotic Receptors. Curr. Drug Metab. 2005;6:309–328. doi: 10.2174/1389200054633826. [DOI] [PubMed] [Google Scholar]
- 120.Meyer U. Overview of enzymes of drug metabolism. J. Pharmacokinet. Phar. 1996;24:449–459. doi: 10.1007/BF02353473. [DOI] [PubMed] [Google Scholar]
- 121.Lin J. Sense and Nonsense in the Prediction of Drug-Drug Interactions. Curr. Drug Metab. 2000;1:305–331. doi: 10.2174/1389200003338947. [DOI] [PubMed] [Google Scholar]
- 122.Wang H., LeCluyse E. Role of Orphan Nuclear Receptors in the Regulation of Drug-Metabolising Enzymes. Clin. Pharmacokinet. 2003;42:1331–1357. doi: 10.2165/00003088-200342150-00003. [DOI] [PubMed] [Google Scholar]
- 123.Kliewer S., Moore J., Wade L., Staudinger J., Watson M., Jones S. An Orphan Nuclear Receptor Activated by Pregnanes Defines a Novel Steroid Signaling Pathway. Cell. 1998;92:73–82. doi: 10.1016/S0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- 124.Lehmann J., McKee D., Watson M., Willson T., Moore J., Kliewer S. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J. Clin. Investig. 1998;102:1016–1023. doi: 10.1172/JCI3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Takeshita A., Igarashi-Migitaka J., Nishiyama K., Takahashi H., Takeuchi Y., Koibuchi N. Acetyl Tributyl Citrate, the Most Widely Used Phthalate Substitute Plasticizer, Induces Cytochrome P450 3A through Steroid and Xenobiotic Receptor. Toxicol. Sci. 2011;123:460–470. doi: 10.1093/toxsci/kfr178. [DOI] [PubMed] [Google Scholar]