

Abstract
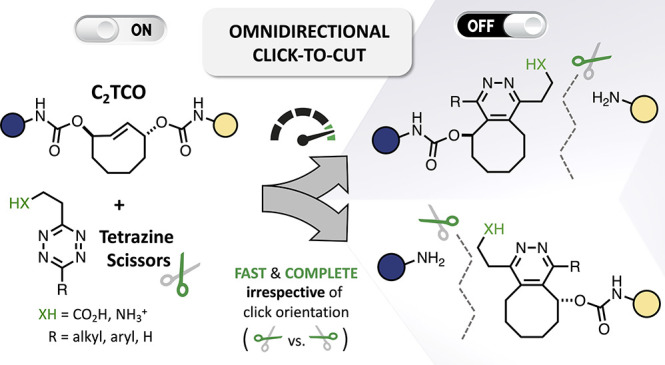
Bioorthogonal chemistry is bridging the divide between static chemical connectivity and the dynamic physiologic regulation of molecular state, enabling in situ transformations that drive multiple technologies. In spite of maturing mechanistic understanding and new bioorthogonal bond-cleavage reactions, the broader goal of molecular ON/OFF control has been limited by the inability of existing systems to achieve both fast (i.e., seconds to minutes, not hours) and complete (i.e., >99%) cleavage. To attain the stringent performance characteristics needed for high fidelity molecular inactivation, we have designed and synthesized a new C2-symmetric trans-cyclooctene linker (C2TCO) that exhibits excellent biological stability and can be rapidly and completely cleaved with functionalized alkyl-, aryl-, and H-tetrazines, irrespective of click orientation. By incorporation of C2TCO into fluorescent molecular probes, we demonstrate highly efficient extracellular and intracellular bioorthogonal disassembly via omnidirectional tetrazine-triggered cleavage.
Introduction
Biologically compatible click chemistries have become powerful tools for the design, synthesis, and tracking of labeled (bio)molecules.1−6 The toolbox of these bioorthogonal reactions has expanded substantially in the past decade, providing chemists and chemical biologists with highly selective methods to achieve efficient ligation in complex biological environments.7 Nevertheless, the application of bioorthogonal chemistries to solve important clinical problems has remained at an early stage, still focused on exploratory models. While this is in large part a function of the tremendous biological complexity that confronts any new chemical tools, it is also the case that a range of important reaction capabilities have not yet been invented (vide infra).
Among these chemistries, the inverse electron demand Diels–Alder (IEDDA)-initiated reaction of tetrazines (Tz) and trans-cyclooctenes (TCO) is increasingly used, chiefly due to its exceptionally high reaction rates but also as a consequence of significant molecular versatility.8,9 The highly tunable Tz/TCO click reaction has thus become a method of choice for time-critical applications and fast bioorthogonal ligation even at low concentrations of both reactants. Applications range from the synthesis of molecular probes and rapid radiolabeling of biomolecules10−15 to bioorthogonal ligations carried out in live cells16−19 and even in vivo, enabling pretargeting strategies for molecular imaging and therapy.20−24 The development of fluorogenic turn-on tetrazines (Figure 1a, upper panel) has enabled no-wash cellular imaging,25−28 even at super-resolution as shown very recently.29
Figure 1.

(a) Bioorthogonal ligation of tetrazines (Tz) and trans-cyclooctenes (TCO) for the fluorogenic labeling of biomolecules, targeting compounds or ligands (top) and Tz-triggered cleavage from trans-cyclooctene modified in allylic position (release-TCO, rTCO) for the bioorthogonal activation of prodrugs or rTCO-caged biomolecules (bottom). (b) Concept of bioorthogonal cleavage to disassemble molecular probes (top) or inactivate fused biomolecules. (c) Limitations of the IEDDA pyridazine elimination with emphasis on Tz/TCO click rates, release yield, and release kinetics.
In parallel, the concept of bioorthogonal bond-cleavage has further expanded the repertoire of chemical methods to control the function of (bio)molecules. In recent years, several reactions of this type have been developed, including metal-induced as well as click-triggered bond-cleavage.30,31 Robillard and co-workers were first to show the release of amino-functionalized compounds from trans-cyclooctene modified in allylic position (release-TCO, rTCO) upon IEDDA reaction with tetrazines and coined the term click-to-release.32 In this IEDDA-initiated pyridazine elimination, the Tz reacts with rTCO to form a dihydropyridazine intermediate that can spontaneously lead to release of the leaving group in allylic position via a 1,4-elimination. Application of this reaction has emerged in recent years mainly in the field of targeted drug delivery. On the basis of their initial report, Robillard et al. designed antibody-drug conjugates comprising a cleavable TCO linker and achieved targeted drug release in a xenograft mouse model upon pretargeting of the antibody conjugate followed by systemic administration of the Tz.33,34 An inverse strategy was developed by Mejia Oneto et al. using Tz-modified hydrogels that can locally activate systemically dosed rTCO-modified prodrugs.35,36 In addition, rTCO-lysine was successfully incorporated into proteins for the design of caged biomolecules that can be activated upon reaction with Tz (Figure 1a, lower panel).37,38
In contrast to these and other turn-ON applications, bioorthogonal cleavage/release has not yet been implemented for controlled inactivation of probes or (bio)molecules in biological environments. In this light, we envision bond-cleavage reactions to be powerful tools to develop new turn-OFF methods and deactivation strategies for chemical biology and biomedical research. Bioorthogonal cleavage of fluorescent probes (Figure 1b) might enable superior methods for applications that require the removal or deactivation of the dye after analysis, such as multiplexed imaging.39 Compared to other methods using either (i) harsh destaining/quenching conditions,40−42 (ii) complex DNA-based cycling and imaging techniques,43−47 or (iii) the combination of clickable fluorophores and quenchers,48 bioorthogonal cleavage would allow rapid, gentle, biocompatible removal of the dye by using small molecules as chemical scissors. More broadly, the incorporation of cleavable linkers into (bio)molecules would enable molecular inactivation for spatiotemporal control of biological function (Figure 1b).
Envisaging the IEDDA pyridazine elimination for bioorthogonal disassembly of molecular probes, we noted the current limitations of this chemistry, which remains unable to achieve both fast (i.e., seconds to minutes) and complete (>99%) cleavage. Unlike target activation approaches, in which a relatively broad range of yields may generate discernible signal and/or relevant downstream activity, the combination of fast AND complete cleavage/release is essential for turn-off strategies to reach minimal residual signals and/or function within a useful time frame.
We have recently uncovered the critical role of tautomerization and isomerization events after ligation—the postclick reaction network—and identified intramolecular cyclization as the main reason for incomplete release when using first-generation bis-alkyl-tetrazines, such as 3,6-dimethyltetrazine (DMT, 1) with rTCO. The blocking of this undesired pathway by N-methyl substitution at the leaving carbamate moiety enabled us to achieve complete release.49 Nonetheless, the relatively low click reactivity of bis-alkyl-tetrazines (when compared to other Tz) and the slow release of the resulting dihydropyridazine intermediates limit further applications (Figure 1c). Improved click kinetics have been achieved by TCO-triggered cleavage of Tz-carbamates, as reported very recently by Robillard et al. Applying this inverse approach for bioorthogonal turn-off, however, is limited due to only slow (>24 h) and incomplete release (67–93%).50 Likewise, second-generation 2-pyrimidyl-substituted tetrazines 2(51) and 3(52) (Figure 1c) achieve higher click rates with rTCO and accelerated release, especially when using the aminoethyl-substituted Tz 3 (PymK).52 These improvements are offset by the penalty of incomplete release (<80%)—consistent with previous findings showing limited rTCO cleavage with aryl-tetrazines32,33,37—even though no intramolecular cyclization was observed.52 We thus set out to elucidate the postclick reaction network for aryl-tetrazines and develop new chemical tools that facilitate fast and complete cleavage for bioorthogonal ON/OFF control.
Results and Discussion
The “Aryl-Tz Problem”
Upon the IEDDA reaction of Tz and rTCO-conjugates, the initial click product A, a 4,5-dihydropyridazine (4,5-DHP) is known to be able to tautomerize to either the 1,4-dihydropyridazine B1 or its 2,5-isomer B2; release proceeds via 1,4-elimination from the B1 tautomer, while formation of B2 leads to a nonreleasing and thus undesired intermediate (Figure 2a).49,53 In theory, aryl substituents could alter these equilibria to interfere with release via either preferential formation of the nonreleasing B2 tautomer or inefficient elimination from B1. In prior work, we discovered that modified Tz capable of intramolecular proton donation (Tz-acids) can significantly accelerate tautomerization and subsequent elimination, a capability shared by pH-independent ammonium-functionalized Tz.52 Directed tautomerization to B1 is, however, only possible if the directing group (XH = NH3+, COOH) is in ‘head-to-head’ position to the leaving group on the TCO scaffold (Figure 2a), while the reciprocal ‘head-to-tail’ click product leads to preferred formation of B2. We have previously shown that this results in (i) biphasic release when using Tz-monoacids (fast via B1, and slow via B2 ⇆ A ⇆ B1) and (ii) slow release (hours) when using a Tz-diacid at physiologic pH.49
Figure 2.
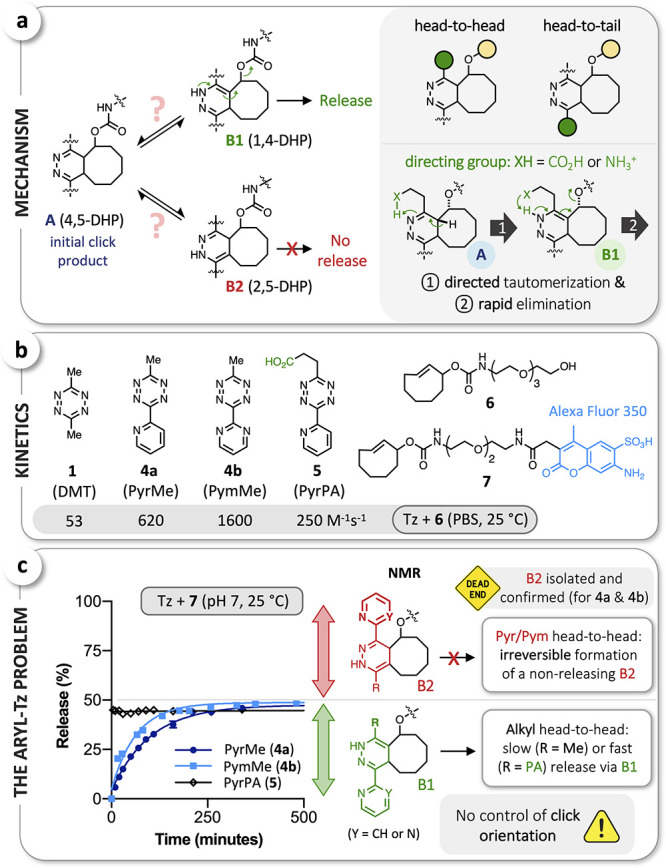
(a) The critical role of postclick tautomerization (left; DHP = dihydropyridazine); directed and accelerated formation of B1 as well as enhanced 1,4-elimination with directing groups (CO2H, NH3+) in head-to-head position (right). (b) Stopped-flow measurements verified the significantly higher click reactivity of aryl-Tz 4a, 4b, and 5 with the water-soluble rTCO-derivative 6. (c) Investigation of the reactions of PyrMe (4a), PymMe (4b), and PyrPA (5) with rTCO-Alexa Fluor 350 (7) by LCMS revealed the irreversible formation of B2 tautomers with Pyr/Pym in head-to-head position, as confirmed by NMR for the reactions of 4a and 4b with rTCO-glycine49 (see Supporting Information).
In both cases, the slow release step takes up to several hours, which not only increases the risk of oxidation of the DHP-intermediates, thus preventing further release,49 but also makes the Tz-acid/rTCO combination unsuitable for turn-off applications that require complete and rapid cleavage (i.e., minutes, not hours).
Alkyl-aryl-Tz are known to be significantly more reactive in the initial click step than bis-alkyl-Tz,32,54 which we verified in this study by measuring the second order rate constants for the reactions of DMT (1), PyrMe (4a), PymMe (4b), and PyrPA (5, PA = propionic acid) with the PEGylated rTCO 6, observing a rate enhancement of up to 30-fold (4b vs 1, Figure 2b). Similar to our previous study,49 we used an rTCO-AlexaFluor 350 conjugate (7, Figure 2b) to further investigate the release step. Reactions were carried out in buffered aqueous solution at pH 7 and 25 °C, selecting concentrations of PyrMe (4a), PymMe (4b), or PyrPA (5) (200 μM) and 7 (125 μM) such that the click reaction was done in less than 4 min, allowing temporal monitoring of postclick reactions by LCMS. With PyrMe (4a) and PymMe (4b), we observed slow monophasic release that reached a yield of approximately 50% within ∼8 h. For PyrPA (5), an initial fast phase of release (too rapid to resolve by HPLC) reached a very similar release yield (Figure 2c), whereupon no further release was observed (>24h). We inferred that these different kinetic profiles result from the methyl group (slow release) and the propionic acid (PA) moiety (fast release) being in head-to-head position, as we had demonstrated for PA-functionalized alkyl Tz,49 implying in turn that the nonreleasing fraction is due to formation of the respective head-to-tail intermediates with 2-pyridyl (Pyr) or 2-pyrimidyl (Pym) in head-to-head position. To verify these hypotheses, we reacted PyrMe (4a) and PymMe (4b) with rTCO-glycine49 (see Supporting Information), allowed the release reactions to proceed to completion, and isolated the nonreleasing isomers. NMR analysis confirmed the aryl-substituent in the head-to-head position, as hypothesized, and revealed these undesired products to be respective B2 tautomers (Figure 2c). We were thus able to show that tautomerization is no longer an equilibrium (cf. Figure 2a) and that incomplete release for aryl-Tz is due to the preferential and irreversible formation of a stable B2 tautomer for one of the two possible click orientations (Pyr/Pym in head-to-head). This key finding is in line with the observations of van Kasteren et al., who achieved approximately 80% release with PymK (3) and invoked the preferred formation of the releasing adduct with the ethylammonium moiety in head-to-head position (due to preclick interaction of NH3+ with the carbamate functionality of the leaving group) to explain this increased conversion.52
Design and Synthesis of C2TCO
In principle, a strategy to bias click orientation even more strongly in favor of the head-to-head geometry could further improve release performance, though at the risk of kinetic penalties imposed by added steric bulk. Recognizing the limits of unstructured molecular interactions in aqueous solution and the strict priority of complete fast release, we instead envisioned a C2-symmetric trans-cyclooctene (C2TCO) with leaving groups at each of the two allylic positions of TCO. Upon click reaction, the directing group (CO2H, NH3+) on the Tz inevitably aligns head-to-head with one of the two release positions, irrespective of the click orientation, leading to fast and complete omnidirectional cleavage (Figure 3a). As axially configured rTCO is known to be significantly more reactive than its equatorial isomer,32 we aimed for the axial configuration of both leaving groups. Starting from 1,3-cyclooctadiene (8), we were able to prepare the desired C2TCO-diol (bis-axial 11) in three steps (Figure 3b). Tetraphenylporphyrin (TPP)-sensitized photooxygenation of 8 followed by DBU-catalyzed in situ Kornblum-DeLaMare rearrangement55 afforded lactol 9 (63%), which was then reduced by reaction with sodium triacetoxyborohydride56 to obtain cyclooct-2-ene-1,4-diol (10) with an anti/syn-ratio of 10/1 (45%). Flow-photoisomerization of 10 and continuous active removal of trans-cyclooctene products by using Ag(I) immobilized on tosic silica gel (TAg silica)57 finally gave bis-axial-11 (47%) and bis-equatorial-11 (30%). 13C NMR analysis of the separated products showed only four signals for each compound, strongly indicating the C2-symmetry of both obtained TCOs (Figure 3b, overlay of 13C NMR spectra), which results from the rigid “crown” (or twist) conformation58−60 of the trans-cyclooctene core structure.
Figure 3.

(a) Design of a C2-symmetric trans-cyclooctene enabling directed tautomerization and enhanced elimination for fast and complete cleavage irrespective of the click orientation. (b) Synthesis of bis-axial and bis-equatorial C2TCO-diol (11) and overlay of schematic 13C NMR spectra of the separated and purified compounds, which indicate C2-symmetry of both isomers resulting from the rigid “crown” (or twist) conformation of the TCO core structure.
Tz-Triggered Cleavage of C2TCO
In preparing to investigate the click and release reactions of C2TCO, we took into account that its unbiased click with Tz poses a characterization challenge. While the randomness of omnidirectional release enhances bioorthogonal turn-off applications, reacting a nonsymmetric C2TCO derivative with nonsymmetric Tz yields problematically complex mixtures. To facilitate chromatographic analysis, we therefore sought to modify both allylic OH-groups to obtain symmetrical probes.
Activation of both OH-functionalities as p-nitrophenyl (PNP) carbonates afforded an inseparable mixture of C2TCO-bis(PNP)-carbonate and p-nitrophenol (see Supporting Information), but we were able to obtain the bis-imidazolium-carbamate 12 as an activated intermediate by reacting bis-axial-11 with 1,1′-carbonyldiimidazole (CDI), similar to the activation of bioorthogonal cyclopropene tags,61 followed by N-methylation using methyl iodide (Figure 4a). In situ conversion of 12 with sarcosine methyl ester followed by saponification gave bis(sarcosinyl)-C2TCO (13). We incorporated sarcosine into this cleavable C2TCO-linker to prevent any potential formation of tricyclic dead-end isomers upon reaction with tetrazines.49 TSTU-mediated coupling of 13 with mono-Fmoc-1,3-diaminopropane and subsequent deprotection afforded bis-amine 14. This compound was reacted with AlexaFluor 594 (AF594)-NHS to obtain AF594-C2TCO-AF594 (15) as a fluorescently labeled C2TCO probe (Figure 4a) for subsequent investigations.
Figure 4.

(a) Modification of C2TCO (bis-axial-11) via bis-imidazolium-carbamate 12 to obtain bis(sarcosinyl)-C2TCO (13) and fluorescent AF594-C2TCO-AF594 (15) for further investigation of the Tz-triggered cleavage of C2TCO. (b) Reacting 15 with selected tetrazines at pH 7.4 and end point analysis by HPLC at 24 h revealed high cleavage efficiency for DMT and quantitative cleavage with tetrazines modified with a directing group (MPA (17), PyrPA (5), PymPA (18), PymK (3)). (c) Second order rate constants as determined by stopped-flow spectrophotometry showed decreased reactivity of C2TCO compared to rTCO (cf. Figure 2b), and significantly higher click kinetics for ammonium-functionalized PymK (3) compared to the respective Tz-acid PymPA (18). (d) Steric hindrance of the aryl substituent as well as interactions of directing groups affect the click kinetics with C2TCO.
As an initial assessment of cleavage efficiency, we reacted 15 with an excess of selected tetrazines in buffered solution at pH 7.4 and analyzed the reaction mixtures at an end point of 24 h by HPLC with fluorescence detection. We observed nearly complete cleavage with DMT (1) and quantitative cleavage (>99%) with the Tz-acids MPA (17), PyrPA (5), and PymPA (18) as well as with PymK (3). In contrast, reaction with bis(2-pyridyl)tetrazine (Pyr2Tz, 16) resulted in only 23% cleavage (Figure 4b), in line with previous observations for rTCO.32,33,37 We hypothesize that this is due to not only slow tautomerization but also limited elimination of the respective B1/B2 intermediate. The inability to achieve complete release with Pyr2Tz (16) highlights the advantages of a cleavage-directing group and an ongoing need for further molecular innovations.
We next sought to analyze release kinetics by serial HPLC measurement, which requires rapid click conversion to kinetically segregate click and release and enable monitoring of the release process exclusively. We thus first determined the second order rate constants for the click reaction of C2TCO with a range of tetrazines. Stopped-flow spectrophotometry in PBS revealed rate constants ranging from 3.3 M–1 s–1, for the reaction of MPA (17) with 13, to 240 M–1 s–1 for PymK (3) + 13 (Figure 4c). We hypothesize that the PA-carboxylate anion (pKa of PA-Tz: ∼4.549) leads to repulsion with the carbamate functionality of the leaving group, in contrast to the previously observed click enhancement caused by interaction of ammonium substituents.52 These effects (Figure 4d) might thus account for the 5-fold lower reactivity of MPA (17) (carboxylate-repulsion) compared to DMT and the 10-fold higher rate constant for PymK (3) (NH3+-interaction) than for PymPA (18) (Figure 4c). Notably, comparing the click reactions of PyrPA (5) with bis(sarcosinyl)-C2TCO (13) and the rTCO derivative 6 (Figure 2b) revealed a 23-fold lower reactivity of C2TCO in contrast to rTCO. We attributed this significant drop in reactivity to the increased steric hindrance of an additional allylic modification (Figure 4d), which is consistent with previous reports showing approximately 20-fold lower reactivity upon modification of one allylic position.32
H-Tetrazines for Fast and Complete Cleavage
To reduce the adverse effect of steric hindrance in reactions with C2TCO, we aimed to develop H-tetrazines (H-Tz) modified with CO2H and NH3+ directing groups. Similar reaction rates have been reported for aryl-Tz and H-Tz,54 but in the case of C2TCO, we anticipated that the significantly reduced steric demand of H-Tz might lead to even higher click reactivities (Figure 5a). H-Tz have been shown to lead to incomplete or even negligible release with rTCO;37,48,52 the mechanism of this effect is as yet unresolved but sufficiently robust for aryl-H-Tz that its pH dependence can be exploited to enforce nonrelease of rTCO species.48 In this case, however, given that reaction with C2TCO will lead to a directing group being in head-to-head position irrespective of the click orientation (analogous to aryl-Tz, cf. Figure 3a), we speculated that the rapid release-directing effect might supersede whatever pathways impede H-Tz-mediated release for rTCO. Thus, we prepared HPA (19) and HK (20)52 and proceeded to assess their global click/release performance. As for their pyridyl and pyrimidyl-substituted counterparts, HPA (19) and HK (20) achieved quantitative cleavage (>99%) and, moreover, significantly increased reaction rates (Figure 5a). As expected, HK (20) is more reactive, but HPA (19) showed even higher stability when using PBS solutions for serial experiments over prolonged timeframes (Supporting Information). At higher concentrations (up to 200 μM) of the fastest tetrazines (HK (20) and PymK (3)), we observed complete cleavage when analyzing the mixture after a reaction time of just 2 min, leaving us unable to disentangle the release rate from the initial click rate. The reaction of AF594-C2TCO-AF594 (15) with selected tetrazines at lower concentrations (10 μM C2TCO + 20 μM Tz) confirmed our impression that release/cleavage is no longer the rate-determining step, as observed release rates track closely with click kinetics. Even with less reactive tetrazines, such as MPA (17) or PymPA (18), we could not detect any postclick intermediates (e.g., A, B1, B2) but only the as-yet-unreacted starting material 15 and the cleaved product (see Supporting Information). On this time scale, directing groups appear to trigger near-instantaneous release, such that cleavage of C2TCO is controlled only by click kinetics and Tz concentration. For the fastest tetrazines tested, this enables fast and complete cleavage (>99%) with HK (20) and PymK (3) at just 20 μM concentration (Figure 5b). More generally, C2TCO-Tz pairs enable direct control of the time required for complete cleavage, as varying the Tz concentration can tune overall reaction rates to the demands of a particular application. While cleavage of the known cTCO-linker33,49 by reaction with DMT takes >1 h (using previously developed AF350-cTCO-Sar;49 see Supporting Information), complete bioorthogonal cut of C2TCO can be achieved within minutes (Figure 5c).
Figure 5.
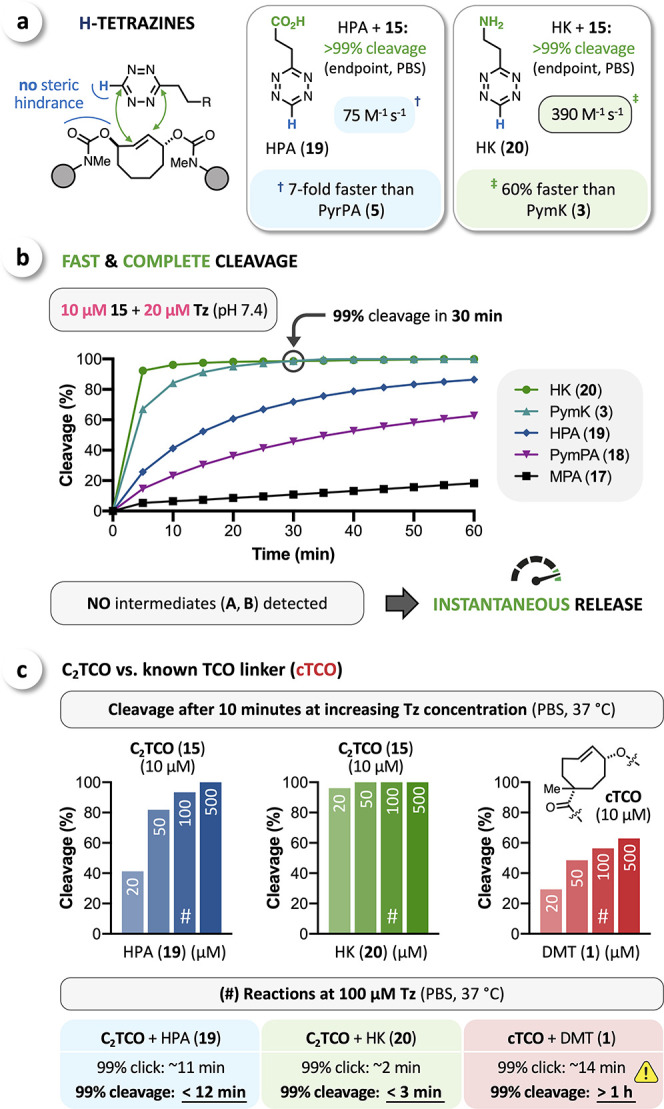
(a) H-tetrazines modified with a directing group enable complete cleavage of C2TCO with significantly improved click rates due to decreased steric hindrance. (b) Cleavage experiments at low concentrations (10 μM C2TCO + 20 μM Tz) revealed instantaneous release upon click (no intermediates detected) and complete overall cleavage (>99%) with HK (20) and PymK (3) after 30 min. (c) Cleavage of C2TCO triggered by HPA (19) or HK (20) is readily accelerated by increasing the Tz concentration enabling complete bioorthogonal cut in minutes (click = rate-determining). In contrast, the cleavage of a known TCO-linker (cTCO) triggered by DMT (1) requires >1 h to reach completeness (release = rate-determining).
Extracellular Cleavage of Antibody-C2TCO Conjugates
Having developed C2TCO as a new chemical tool to achieve fast and complete cleavage triggered by acid- or ammonium-functionalized Tz, we aimed to show its applicability in bioorthogonal turn-off strategies, such as the rapid disassembly of fluorescently labeled antibodies. Therefore, we modified bis(sarcosinyl)-C2TCO (13) to obtain DBCO-C2TCO-AF594 (21) as a stable multifunctional click- and cleavable fluorescent tag (Figure 6a; for synthetic procedures and details, see Supporting Information). The anti-EGFR-antibody cetuximab was labeled with 6-azidohexanoic acid sulfo-NHS ester, and the resulting cetuximab-N3 was then click-conjugated to 21 via strain-promoted azide–alkyne cycloaddition (Figure 6a).
Figure 6.
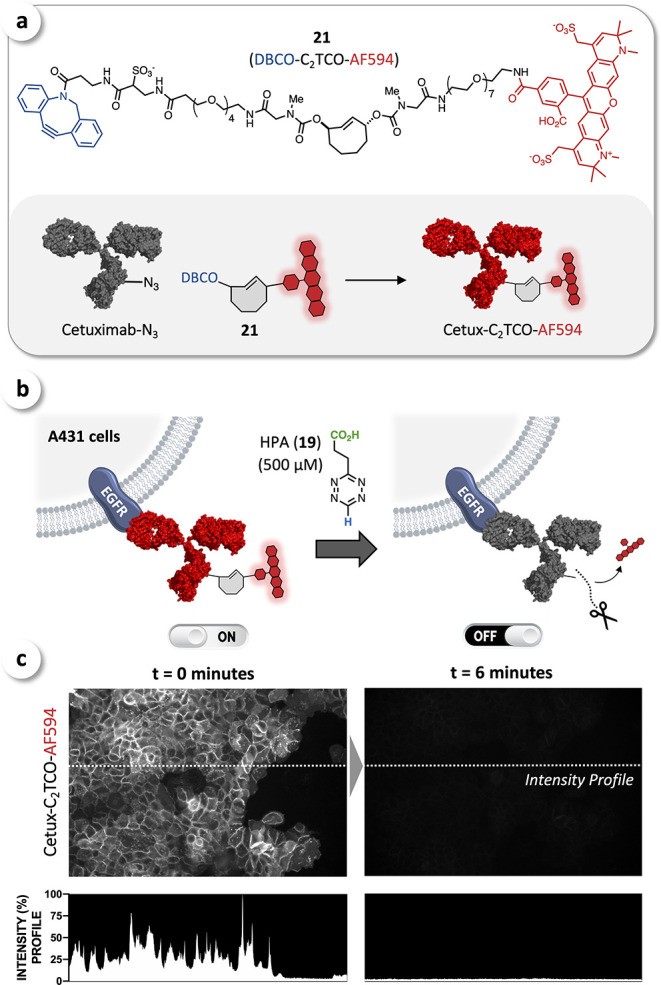
(a) DBCO-C2TCO-AF594 (21) as a click- and cleavable fluorescent tag for conjugation with azide-labeled antibodies. (b) Staining of EGFR-positive A431 cells with Cetux-C2TCO-AF594 (ON-state) and subsequent cleavage with HPA (19) (OFF-state). (c) Cell imaging confirmed near-quantitative bioorthogonal disassembly of Cetux-C2TCO-AF594 within 6 min.
We tested azido-antibodies at different degrees of labeling (DOL) and analyzed the C2TCO cleavage of the clicked conjugates after reaction with Tz. Unexpectedly, when clicked to the antibody, we noted a consistent trend toward lower C2TCO reactivity as the number of azides per antibody increased. Conversely, cleavage yields improved as a function of the concentration of 21 in the antibody labeling reaction (higher concentrations = faster intermolecular N3-DBCO click), strongly suggestive of an intramolecular reaction of antibody-bound N3-groups with C2TCO (see Supporting Information). We therefore recommend bioconjugation methods other than azide–alkyne chemistry when designing C2TCO probes with a higher DOL.
Nevertheless, at an average DOL of 0.9, Cetux-C2TCO-AF594 achieved >97% cleavage (for labeling procedures and details, see Supporting Information) and the respective probe was therefore carried forward for further investigation. EGFR-positive A431 cells were stained with Cetux-C2TCO-AF594, washed, and imaged to establish target-specific staining and baseline brightness (ON-state). Cells were then treated with HPA (19)62 at a concentration of 500 μM in PBS to ensure complete click in less than 3 min (Figure 6b). Serial imaging of the same field of view demonstrated rapid cleavage upon addition of HPA (19), with a dramatic reduction in brightness by the first measurement at 2 min (see Supporting Information) and near-complete elimination of the signal within 6 min of Tz addition (OFF-state). A quantitative line-intensity profile demonstrates the excellent cleavage efficiency, exhibiting no discernible difference between the regions with and without cells (Figure 6c). Hence, bioorthogonal disassembly of antibody-C2TCO probes will enable multiplexed imaging without any potential rebound in brightness as the dye is cleaved and thus removed upon reaction with Tz.
Intracellular Cleavage of Fluorescent C2TCO-Probes
Having shown the performance of C2TCO for extracellular cleavage of antibody conjugates, we next aimed to test whether C2TCO can be incorporated into probes that bind to intracellular targets. Therefore, we first investigated the stability of C2TCO under physiological conditions. Incubation of bis(sarcosinyl)-C2TCO (13) in full cell growth media (10% FBS) at 37 °C followed by titration with DMT revealed superb stability (>97%) of C2TCO for up to 48 h (see Supporting Information).
We have previously shown that the covalent BTK (Bruton’s tyrosine kinase) inhibitor ibrutinib (Ib) can be modified with several fluorescent dyes (e.g., silicon rhodamine, SiR) while maintaining selective binding to BTK.63,64 We thus prepared Ib-C2TCO-SiR (22, Figure 7a) and tested its cell uptake and binding to BTK in live HT1080 cells stably transfected with BTK-mCherry, followed by intracellular cleavage of C2TCO with HK (20) (Figure 7b). Live cells were incubated with 22 (2 μM), washed, fixed, and permeabilized (to facilitate efficient removal of released SiR-compounds, which would otherwise interfere with monitoring intracellular cleavage kinetics). Cell imaging by fluorescence microscopy confirmed uptake and selective binding of 22 to BTK, as shown by excellent colocalization of mCherry and SiR with a Pearson coefficient of 0.89 (Figure 7c, ON-state, top row). The cells were then treated with HK (20) (20 μM) for 30 min, rinsed with PBS, and imaged, showing efficient bioorthogonal turn-off (Figure 7c, OFF-state, bottom row). In control experiments with native (BTK-negative) HT1080 cells, we observed only minor nonspecific binding of 22 (see Supporting Information). Overall, we have been able to show that (i) C2TCO can be incorporated into molecular probes binding to intracellular targets and (ii) these compounds can be disassembled on-target via fast bioorthogonal cleavage, (iii) even after extended incubation (up to 5 h) in live cells demonstrating excellent intracellular stability.
Figure 7.
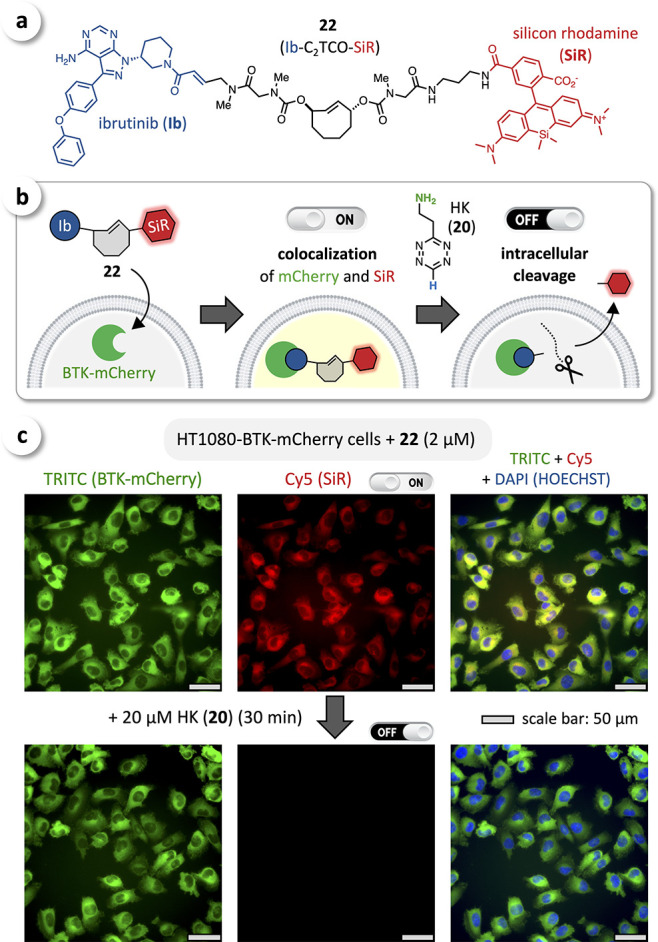
(a) Chemical structure of Ib-C2TCO-SiR (22) as a cleavable fluorescent ibrutinib conjugate. (b) Cellular uptake and binding to BTK of 22 upon treating HT1080-BTK-mCherry cells, and subsequent intracellular cleavage with HK (20). (c) Fluorescence microscopy imaging showed excellent colocalization of mCherry (green) and SiR (red), confirming selective binding of Ib-C2TCO-SiR (22) to BTK (ON-state, top row); efficient intracellular cleavage was observed upon treatment with 20 μM HK (20) for 30 min (OFF-state, bottom row).
Conclusion
Although configured for click-triggered elimination, the outcome of the rTCO-Tz reaction nevertheless hinges on a complex array of factors, including environmental conditions, click orientation, and tautomerization events driven by the Tz side-chain(s). Informed by our finding that aryl-Tz preferentially generate a nonreleasing tautomer, we developed C2TCO as a new chemical tool to achieve fast and complete bioorthogonal cleavage. We show that its C2 symmetry is key to achieving omnidirectional cleavage with acid- and ammonium-functionalized Tz, irrespective of the orientation of the initial IEDDA click reaction. Increased steric hindrance due to the additional leaving group in the allylic position of the TCO scaffold can be circumvented by using less sterically demanding H-tetrazines modified with directing groups (CO2H, NH3+) that accelerate postclick tautomerization and enhance 1,4-elimination. We observed second order rate constants for the initial click reaction of up to ∼400 M–1 s–1 and instantaneous release that is no longer the rate-determining step. Complete cleavage (>99%) of C2TCO conjugates could thus be achieved within minutes even at low μM concentrations of both reactants and accelerated accordingly at higher Tz concentrations.
Due to the unbiased click with Tz, one cannot control which one of the two groups is released. While this is clearly not an obstacle for bioorthogonal turn-off applications, it would lead to randomization of the pyridazine moiety to either side of the cleaved linkage, highlighting the need for further developments that can recapitulate the chemical performance of C2TCO in the controlled delivery of molecular cargo.
Incorporating C2TCO into an antibody-dye conjugate, we successfully developed a strategy to achieve fast extracellular bioorthogonal cleavage within minutes. In a similar approach, using the covalent inhibitor ibrutinib as a targeting moiety, we were able to demonstrate fast intracellular cleavage of an ibrutinib-C2TCO-dye conjugate bound to its target (BTK).
Fast and complete disassembly is a prerequisite for bioorthogonal turn-off and efficient molecular inactivation. The development of C2TCO probes might thus enable not only new methods for advanced multiplexed imaging but also Tz-triggered cleavage in vivo, for instance, the removal of fluorescent tags or radionuclides from long-circulating compounds and thus the reduction of background signal or prolonged radiation of healthy tissue. Moreover, the design of C2TCO-fused (bio)molecules (e.g., bispecific active compounds or C2TCO-embedded molecular scaffolds/backbones) might allow temporal regulation of molecular function inside living cells by Tz-triggered cleavage, significantly expanding the scope of bioorthogonal ON/OFF control.
Acknowledgments
We thank Christian Hametner, Mario Rothbauer, and Emanuel Sporer for assistance with NMR measurements, cell imaging, and synthesis, respectively. We gratefully acknowledge financial support by the Austrian Science Fund (FWF): ZK 29-B21 (Young Independent Researcher Group, grant holder: H.M.).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c07922.
Synthetic procedures, compound characterization data, NMR spectra, click kinetics, release experiments, antibody labeling, cell culture, and fluorescence microscopy (PDF)
Author Contributions
$ The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. M.W. and M.H. contributed equally.
The authors declare the following competing financial interest(s): The authors declare the filing of a patent that covers parts of this research.
Supplementary Material
References
- Boyce M.; Bertozzi C. R. Bringing chemistry to life. Nat. Methods 2011, 8, 638–642. 10.1038/nmeth.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett J. C.; Bertozzi C. R. Cu-free click cycloaddition reactions in chemical biology. Chem. Soc. Rev. 2010, 39, 1272–1279. 10.1039/b901970g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj N. K. The future of bioorthogonal chemistry. ACS Cent. Sci. 2018, 4, 952–959. 10.1021/acscentsci.8b00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenry; Liu B. Bio-orthogonal click chemistry for in vivo bioimaging. Trends Chem. 2019, 1, 763–778. 10.1016/j.trechm.2019.08.003. [DOI] [Google Scholar]
- Carell T.; Vrabel M. Bioorthogonal chemistry—introduction and overview. Top. Curr. Chem. 2016, 374, 9. 10.1007/s41061-016-0010-x. [DOI] [PubMed] [Google Scholar]
- Fan X.; Li J.; Chen P. R. Bioorthogonal chemistry in living animals. Natl. Sci. Rev. 2017, 4, 300–302. 10.1093/nsr/nwx010. [DOI] [Google Scholar]
- Row R. D.; Prescher J. A. Constructing new bioorthogonal reagents and reactions. Acc. Chem. Res. 2018, 51, 1073–1081. 10.1021/acs.accounts.7b00606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer S.; Lang K. Tetrazines in inverse-electron-demand Diels–Alder cycloadditions and their use in biology. Synthesis 2017, 49, 830–848. 10.1055/s-0036-1588682. [DOI] [Google Scholar]
- Oliveira B. L.; Guo Z.; Bernardes G. J. L. Inverse electron demand Diels-Alder reactions in chemical biology. Chem. Soc. Rev. 2017, 46, 4895–4950. 10.1039/C7CS00184C. [DOI] [PubMed] [Google Scholar]
- Li Z.; Cai H.; Hassink M.; Blackman M. L.; Brown R. C. D.; Conti P. S.; Fox J. M. Tetrazine-trans-cyclooctene ligation for the rapid construction of 18F labeled probes. Chem. Commun. 2010, 46, 8043–8045. 10.1039/c0cc03078c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner T.; Keliher E. J.; Earley S.; Marinelli B.; Weissleder R. Synthesis and in vivo imaging of a 18F-labeled PARP1 inhibitor using a chemically orthogonal scavenger-assisted high-performance method. Angew. Chem., Int. Ed. 2011, 50, 1922–5. 10.1002/anie.201006579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner T.; Zeglis B. M. The inverse electron demand Diels-Alder click reaction in radiochemistry. J. Labelled Compd. Radiopharm. 2014, 57, 285–90. 10.1002/jlcr.3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.; Svatunek D.; Rohlfing K.; Liu Y.; Wang H.; Giglio B.; Yuan H.; Wu Z.; Li Z.; Fox J. Conformationally strained trans-cyclooctene (sTCO) enables the rapid construction of (18)F-PET probes via tetrazine ligation. Theranostics 2016, 6, 887–95. 10.7150/thno.14742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poty S.; Membreno R.; Glaser J. M.; Ragupathi A.; Scholz W. W.; Zeglis B. M.; Lewis J. S. The inverse electron-demand Diels-Alder reaction as a new methodology for the synthesis of 225Ac-labelled radioimmunoconjugates. Chem. Commun. 2018, 54, 2599–2602. 10.1039/C7CC09129J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Onzen A. H. A. M.; Rossin R.; Schenning A. P. H. J.; Nicolay K.; Milroy L.-G.; Robillard M. S.; Brunsveld L. Tetrazine—trans-cyclooctene chemistry applied to fabricate self-assembled fluorescent and radioactive nanoparticles for in vivo dual mode imaging. Bioconjugate Chem. 2019, 30, 547–551. 10.1021/acs.bioconjchem.9b00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj N. K.; Weissleder R. Biomedical applications of tetrazine cycloadditions. Acc. Chem. Res. 2011, 44, 816–827. 10.1021/ar200037t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K. S.; Budin G.; Reiner T.; Vinegoni C.; Weissleder R. Bioorthogonal imaging of Aurora kinase A in live cells. Angew. Chem., Int. Ed. 2012, 51, 6598–6603. 10.1002/anie.201200994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng T.; Hang H. C. Site-specific bioorthogonal labeling for fluorescence imaging of intracellular proteins in living cells. J. Am. Chem. Soc. 2016, 138, 14423–14433. 10.1021/jacs.6b08733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang H. S.; Jana S.; Blizzard R. J.; Meeuwsen J. C.; Mehl R. A. Access to faster eukaryotic cell labeling with encoded tetrazine amino acids. J. Am. Chem. Soc. 2020, 142, 7245–7249. 10.1021/jacs.9b11520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossin R.; Renart Verkerk P.; van den Bosch S. M.; Vulders R. C. M.; Verel I.; Lub J.; Robillard M. S. In vivo chemistry for pretargeted tumor imaging in live mice. Angew. Chem., Int. Ed. 2010, 49, 3375–3378. 10.1002/anie.200906294. [DOI] [PubMed] [Google Scholar]
- Zeglis B. M.; Sevak K. K.; Reiner T.; Mohindra P.; Carlin S. D.; Zanzonico P.; Weissleder R.; Lewis J. S. A pretargeted PET imaging strategy based on bioorthogonal Diels-Alder click chemistry. J. Nucl. Med. 2013, 54, 1389–96. 10.2967/jnumed.112.115840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossin R.; Läppchen T.; van den Bosch S. M.; Laforest R.; Robillard M. S. Diels–Alder Reaction for tumor pretargeting: In vivo chemistry can boost tumor radiation dose compared with directly labeled antibody. J. Nucl. Med. 2013, 54, 1989–1995. 10.2967/jnumed.113.123745. [DOI] [PubMed] [Google Scholar]
- Membreno R.; Houghton J.; Cook B.; Carnazza K.; Lewis J.; Zeglis B. The synthesis, radiolabeling, and characterization of 177Lu-labeled tetrazine radioligands for pretargeted radioimmunotherapy. J. Nucl. Med. 2016, 57, 1201–1201.26985060 [Google Scholar]
- Altai M.; Membreno R.; Cook B.; Tolmachev V.; Zeglis B. M. Pretargeted Imaging and Therapy. J. Nucl. Med. 2017, 58, 1553–1559. 10.2967/jnumed.117.189944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson J. C. T.; Meimetis L. G.; Hilderbrand S. A.; Weissleder R. BODIPY–Tetrazine Derivatives as Superbright Bioorthogonal Turn-on Probes. Angew. Chem., Int. Ed. 2013, 52, 6917–6920. 10.1002/anie.201301100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meimetis L. G.; Carlson J. C.; Giedt R. J.; Kohler R. H.; Weissleder R. Ultrafluorogenic coumarin-tetrazine probes for real-time biological imaging. Angew. Chem., Int. Ed. 2014, 53, 7531–7534. 10.1002/anie.201403890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieczorek A.; Werther P.; Euchner J.; Wombacher R. Green- to far-red-emitting fluorogenic tetrazine probes – synthetic access and no-wash protein imaging inside living cells. Chem. Sci. 2017, 8, 1506–1510. 10.1039/C6SC03879D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knorr G.; Kozma E.; Schaart J. M.; Németh K.; Török G.; Kele P. Bioorthogonally applicable fluorogenic cyanine-tetrazines for no-wash super-resolution imaging. Bioconjugate Chem. 2018, 29, 1312–1318. 10.1021/acs.bioconjchem.8b00061. [DOI] [PubMed] [Google Scholar]
- Werther P.; Yserentant K.; Braun F.; Kaltwasser N.; Popp C.; Baalmann M.; Herten D.-P.; Wombacher R. Live-cell localization microscopy with a fluorogenic and self-blinking tetrazine probe. Angew. Chem., Int. Ed. 2020, 59, 804–810. 10.1002/anie.201906806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Chen P. R. Development and application of bond cleavage reactions in bioorthogonal chemistry. Nat. Chem. Biol. 2016, 12, 129–137. 10.1038/nchembio.2024. [DOI] [PubMed] [Google Scholar]
- Tu J.; Xu M.; Franzini R. M. Dissociative bioorthogonal reactions. ChemBioChem 2019, 20, 1615–1627. 10.1002/cbic.201800810. [DOI] [PubMed] [Google Scholar]
- Versteegen R. M.; Rossin R.; ten Hoeve W.; Janssen H. M.; Robillard M. S. Click to release: Instantaneous doxorubicin elimination upon tetrazine ligation. Angew. Chem., Int. Ed. 2013, 52, 14112–14116. 10.1002/anie.201305969. [DOI] [PubMed] [Google Scholar]
- Rossin R.; van Duijnhoven S. M. J.; ten Hoeve W.; Janssen H. M.; Kleijn L. H. J.; Hoeben F. J. M.; Versteegen R. M.; Robillard M. S. Triggered drug release from an antibody–drug conjugate using fast “click-to-release” chemistry in mice. Bioconjugate Chem. 2016, 27, 1697–1706. 10.1021/acs.bioconjchem.6b00231. [DOI] [PubMed] [Google Scholar]
- Rossin R.; Versteegen R. M.; Wu J.; Khasanov A.; Wessels H. J.; Steenbergen E. J.; ten Hoeve W.; Janssen H. M.; van Onzen A. H. A. M.; Hudson P. J.; Robillard M. S. Chemically triggered drug release from an antibody-drug conjugate leads to potent antitumour activity in mice. Nat. Commun. 2018, 9, 1484. 10.1038/s41467-018-03880-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejia Oneto J. M.; Khan I.; Seebald L.; Royzen M. In vivo bioorthogonal chemistry enables local hydrogel and systemic pro-drug to treat soft tissue sarcoma. ACS Cent. Sci. 2016, 2, 476–482. 10.1021/acscentsci.6b00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czuban M.; Srinivasan S.; Yee N. A.; Agustin E.; Koliszak A.; Miller E.; Khan I.; Quinones I.; Noory H.; Motola C.; Volkmer R.; Di Luca M.; Trampuz A.; Royzen M.; Mejia Oneto J. M. Bio-orthogonal chemistry and reloadable biomaterial enable local activation of antibiotic prodrugs and enhance treatments against Staphylococcus aureus infections. ACS Cent. Sci. 2018, 4, 1624–1632. 10.1021/acscentsci.8b00344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Jia S.; Chen P. R. Diels-Alder reaction–triggered bioorthogonal protein decaging in living cells. Nat. Chem. Biol. 2014, 10, 1003–1005. 10.1038/nchembio.1656. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Li J.; Xie R.; Fan X.; Liu Y.; Zheng S.; Ge Y.; Chen P. R. Bioorthogonal chemical activation of kinases in living Systems. ACS Cent. Sci. 2016, 2, 325–331. 10.1021/acscentsci.6b00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzmann K.; Carter L. M.; Lewis J. S.; Aboagye E. O. Multiplexed imaging for diagnosis and therapy. Nat. Biomed. Eng. 2017, 1, 697–713. 10.1038/s41551-017-0131-8. [DOI] [PubMed] [Google Scholar]
- Schubert W.; Bonnekoh B.; Pommer A. J.; Philipsen L.; Böckelmann R.; Malykh Y.; Gollnick H.; Friedenberger M.; Bode M.; Dress A. W. M. Analyzing proteome topology and function by automated multidimensional fluorescence microscopy. Nat. Biotechnol. 2006, 24, 1270–1278. 10.1038/nbt1250. [DOI] [PubMed] [Google Scholar]
- Lin J.-R.; Fallahi-Sichani M.; Sorger P. K. Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. Nat. Commun. 2015, 6, 8390. 10.1038/ncomms9390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J.-R.; Izar B.; Wang S.; Yapp C.; Mei S.; Shah P. M.; Santagata S.; Sorger P. K. Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. eLife 2018, 7, e31657. 10.7554/eLife.31657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saka S. K.; Wang Y.; Kishi J. Y.; Zhu A.; Zeng Y.; Xie W.; Kirli K.; Yapp C.; Cicconet M.; Beliveau B. J.; Lapan S. W.; Yin S.; Lin M.; Boyden E. S.; Kaeser P. S.; Pihan G.; Church G. M.; Yin P. Immuno-SABER enables highly multiplexed and amplified protein imaging in tissues. Nat. Biotechnol. 2019, 37, 1080–1090. 10.1038/s41587-019-0207-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullal A. V.; Peterson V.; Agasti S. S.; Tuang S.; Juric D.; Castro C. M.; Weissleder R. Cancer Cell Profiling by Barcoding Allows Multiplexed Protein Analysis in Fine-Needle Aspirates. Sci. Transl. Med. 2014, 6, 219ra9. 10.1126/scitranslmed.3007361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agasti S. S.; Liong M.; Peterson V. M.; Lee H.; Weissleder R. Photocleavable DNA Barcode–Antibody Conjugates Allow Sensitive and Multiplexed Protein Analysis in Single Cells. J. Am. Chem. Soc. 2012, 134, 18499–18502. 10.1021/ja307689w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S.-M.; Veneziano R.; Gordonov S.; Li L.; Danielson E.; Perez de Arce K.; Park D.; Kulesa A. B.; Wamhoff E.-C.; Blainey P. C.; Boyden E. S.; Cottrell J. R.; Bathe M. Multiplexed and high-throughput neuronal fluorescence imaging with diffusible probes. Nat. Commun. 2019, 10, 4377. 10.1038/s41467-019-12372-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giedt R. J.; Pathania D.; Carlson J. C. T.; McFarland P. J.; del Castillo A. F.; Juric D.; Weissleder R. Single-cell barcode analysis provides a rapid readout of cellular signaling pathways in clinical specimens. Nat. Commun. 2018, 9, 4550. 10.1038/s41467-018-07002-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J.; Oh J.; Ahmed M. S.; Carlson J. C. T.; Weissleder R. Ultra-fast cycling for multiplexed cellular fluorescence imaging. Angew. Chem., Int. Ed. 2020, 59, 6839–6846. 10.1002/anie.201915153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson J. C.; Mikula H.; Weissleder R. Unraveling tetrazine-triggered bioorthogonal elimination enables chemical tools for ultrafast release and universal cleavage. J. Am. Chem. Soc. 2018, 140, 3603–3612. 10.1021/jacs.7b11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Onzen A. H. A. M.; Versteegen R. M.; Hoeben F. J. M.; Filot I. A. W.; Rossin R.; Zhu T.; Wu J.; Hudson P. J.; Janssen H. M.; ten Hoeve W.; Robillard M. S. Bioorthogonal tetrazine carbamate cleavage by highly reactive trans-cyclooctene. J. Am. Chem. Soc. 2020, 142, 10955–10963. 10.1021/jacs.0c00531. [DOI] [PubMed] [Google Scholar]
- Fan X.; Ge Y.; Lin F.; Yang Y.; Zhang G.; Ngai W. S. C.; Lin Z.; Zheng S.; Wang J.; Zhao J.; Li J.; Chen P. R. Optimized tetrazine derivatives for rapid bioorthogonal decaging in living cells. Angew. Chem., Int. Ed. 2016, 55, 14046–14050. 10.1002/anie.201608009. [DOI] [PubMed] [Google Scholar]
- Sarris A. J. C.; Hansen T.; de Geus M. A. R.; Maurits E.; Doelman W.; Overkleeft H. S.; Codee J. D. C.; Filippov D. V.; van Kasteren S. I. Fast and pH-independent elimination of trans-cyclooctene by using aminoethyl-functionalized tetrazines. Chem. - Eur. J. 2018, 24, 18075–18081. 10.1002/chem.201803839. [DOI] [PubMed] [Google Scholar]
- Versteegen R. M.; ten Hoeve W.; Rossin R.; de Geus M. A. R.; Janssen H. M.; Robillard M. S. Click-to-release from trans-cyclooctenes: Mechanistic insights and expansion of scope from established carbamate to remarkable ether cleavage. Angew. Chem., Int. Ed. 2018, 57, 10494–10499. 10.1002/anie.201800402. [DOI] [PubMed] [Google Scholar]
- Karver M. R.; Weissleder R.; Hilderbrand S. A. Synthesis and evaluation of a series of 1,2,4,5-tetrazines for bioorthogonal conjugation. Bioconjugate Chem. 2011, 22, 2263–2270. 10.1021/bc200295y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza J. M.; Brocksom T. J.; McQuade D. T.; de Oliveira K. T. Continuous endoperoxidation of conjugated dienes and subsequent rearrangements leading to C–H oxidized synthons. J. Org. Chem. 2018, 83, 7574–7585. 10.1021/acs.joc.8b01307. [DOI] [PubMed] [Google Scholar]
- Evans D. A.; Chapman K. T.; Carreira E. M. Directed reduction of beta-hydroxy ketones employing tetramethylammonium triacetoxyborohydride. J. Am. Chem. Soc. 1988, 110, 3560–3578. 10.1021/ja00219a035. [DOI] [Google Scholar]
- Darko A.; Boyd S. J.; Fox J. M. Large-scale flow photochemical synthesis of functionalized trans-cyclooctenes using sulfonated silica gel. Synthesis 2018, 50, 4875–4882. 10.1055/s-0037-1610240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allinger N. L.; Sprague J. T. Conformational analysis. LXXXIV. Study of the structures and energies of some alkenes and cycloalkenes by the force field method. J. Am. Chem. Soc. 1972, 94, 5734–5747. 10.1021/ja00771a034. [DOI] [Google Scholar]
- Leong M. K.; Mastryukov V. S.; Boggs J. E. Structure and conformation of cyclopentene, cycloheptene and trans-cyclooctene. J. Mol. Struct. 1998, 445, 149–160. 10.1016/S0022-2860(98)00421-9. [DOI] [Google Scholar]
- Taylor M. T.; Blackman M. L.; Dmitrenko O.; Fox J. M. Design and synthesis of highly reactive dienophiles for the tetrazine–trans-cyclooctene ligation. J. Am. Chem. Soc. 2011, 133, 9646–9649. 10.1021/ja201844c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Šečkutė J.; Cole C. M.; Devaraj N. K. Live-cell imaging of cyclopropene tags with fluorogenic tetrazine cycloadditions. Angew. Chem., Int. Ed. 2012, 51, 7476–7479. 10.1002/anie.201202122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetrazine selection for this experiment took into account: i) the general goal of demonstrating complementary utility for both acid/ammonium-functionalized Tz in biological applications; ii) Tz stability, as the probes for antibody labeling and cleavage were shipped from Vienna to Boston for bioconjugation and cellular imaging.
- Turetsky A.; Kim E.; Kohler R. H.; Miller M. A.; Weissleder R. Single cell imaging of Bruton’s Tyrosine Kinase using an irreversible inhibitor. Sci. Rep. 2015, 4, 4782. 10.1038/srep04782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E.; Yang K. S.; Kohler R. H.; Dubach J. M.; Mikula H.; Weissleder R. Optimized near-IR fluorescent agents for in vivo imaging of Btk expression. Bioconjugate Chem. 2015, 26, 1513–1518. 10.1021/acs.bioconjchem.5b00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.