

Abstract
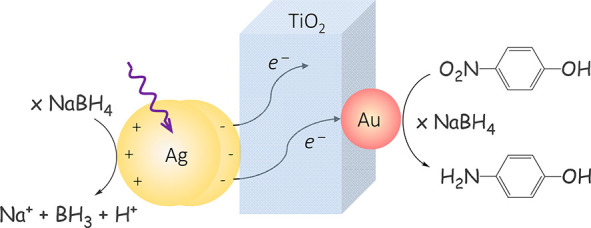
Plasmon hot carriers are interesting for photoredox chemical synthesis but their direct utilization is limited by their ultrafast thermalization. Therefore, they are often transferred to suitable accepting materials that expedite their lifetime. Solid-state photocatalysts are technologically more suitable than their molecular counterparts, but their photophysical processes are harder to follow due to the absence of clear optical fingerprints. Herein, the journey of hot electrons in a solid-state multimetallic photocatalyst is revealed by a combination of ultrafast visible and infrared spectroscopy. Dynamics showed that electrons formed upon silver plasmonic excitation reach the gold catalytic site within 700 fs and the electron flow could also be reversed. Gold is the preferred site until saturation of its 5d band occurs. Silver-plasmon hot electrons increased the rate of nitrophenol reduction 16-fold, confirming the preponderant role of hot electrons in the overall catalytic activity and the importance to follow hot carriers’ journeys in solid-state photosystems.
Keywords: Plasmon, hot-electron, photocatalysis, multimetallic system, ultrafast dynamics
Light is used in a multitude of processes, from energy and chemical production to therapy. The development of energy efficient lighting devices and sunlight abundance have made light a versatile and cost-effective reagent. Plasmonic materials have optical absorptions that exceed 10-fold their cross-section, making them uniquely suitable light converters into electric carriers. Optical excitation of the localized surface plasmon resonance (LSPR) creates hot carriers1,2 that can be used in chemical transformations3−8 and photovoltaics.9−11 Their relaxation liberates thermal energy that can be used in photothermal processes, such as catalysis,12,13 desalination,14,15 cancer therapy,16 and sensing.17
Plasmon hot carriers with energy up to the absorbed photon energy18 multiply rapidly, decreasing their average thermal energy.19 This ultrafast charge relaxation process limits their direct utilization.18,20 Their transference to a suitable acceptor, such as semiconductor, can extend their lifetime2,11,21,22 as well as proton-coupled electron transfer events.23 Semiconductors have characteristically low surface reactivity; therefore, the hot carriers are subsequently transferred to a catalytic site, e.g., metallic nanoparticles (NPs).4,5
Solid-state systems are stabler and better for application in photocatalysis than their molecular counterparts. Understanding energy and hot carrier flow in plasmonic solid-state nanostructures is essential to exploit their potential. Aslam et al.24,25 were able to control the energy flow on a multimetallic nanostructure composed of silver nanocubes and platinum. However, the hot carriers’ journeys in solid-state systems remain elusive because their photophysical processes are noticeably harder to follow, due to the absence of clear optical fingerprints. Charge transfer to the catalyst is often deduced by signal quenching.5
Herein, we report an ultrafast spectroscopic study of plasmon-created hot electron flow in a multimetallic nanostructure consisting of Ag-TiO2–Au, where silver, TiO2, and gold act as a photosensitizer, electron relay, and catalyst, respectively. Dynamics showed that plasmon-formed hot electrons on silver take 700 fs to reach the catalytic site through the electron relay. The catalyst is the favoured site until the saturation of the gold 5d band. The plasmon-mediated process increases 16-fold the rate of nitrophenol reduction, confirming the presence of accumulative charge separation26 necessary to fulfil the requirements on multicharge photoredox processes. Gold is considered the noblest of the metals,27 but Haruta et al.28 demonstrated that gold at sub-10 nm is active due to depopulation of the 5d band caused by hybridization with the 6sp band.29 Since then, gold has been found active in several reactions, e.g., nitrogen30 and alkane activation,31 carbonylations,32 acetylene hydrochlorination,33 and selective CO2 reduction catalyst in aqueous media.7,34
The complete multimetallic system consisted of gold NPs sandwiched between TiO2 layers. The sandwich structure was immersed in an aqueous colloidal solution to attach silver NPs. A schematic overview of the system, as well as the associated absorption spectra of the system, can be found in Figure 1c and 1d, respectively. Further preparation and characterization can be found in the Supporting Information (SI).
Figure 1.

Morphology and optical properties. TEM images of the silver NPs and the size distribution as an insert (a); TEM images of the gold NPs on TiO2 and the size distribution as an insert (b); schematic overview of the full multimetallic system (c); and UV–vis absorbance spectra of the full multimetallic system and the gold and silver absorbances as inserts (d).
Silver NPs were prepared via a modified version of the polyol process35 that yields spherical NPs with a mean diameter of 22 nm (TEM analysis, Figure 1a). DLS measurements (Figure S3) showed larger particle size because the hydrodynamic radius is sensitive to the PVP capping layer, undetectable by TEM. The PVP layer avoids Fermi level pinning upon charge injection to passivate the TiO2 surface.36 The silver NPs exhibited a single LSPR peak at 404 nm (Figure S4). Gold NPs were formed by evaporation and subsequent annealing of a 1 nm gold layer. The procedure yielded uniform gold NPs with an average diameter of 5 nm (TEM analysis, Figure 1b), thus within gold active range.28 Upon assembling, the Ag NPs LSPR red-shifted by 16 nm (Figure 1d) due to the change in the dielectric environment.37 The ratio between silver and gold was determined by SEM EDS (Table S1), using 10 randomly selected sample spots. The average silver/gold ratio was found to be 8.4 ± 1.1, indicating a uniform distribution of silver and gold on the prepared films and excess of photosensitizer silver NPs.
Transient absorption spectroscopy was used to gain an insight into the charge carrier dynamics. An excitation wavelength of 435 nm was chosen as the best comprise between exciting silver near its LSPR and having a low response resulting from gold interband transitions,38 which occur in the same spectral region (Figure S12). The studies were performed using the same photon density (200 μW and fixed aperture), enabling a direct comparison of the samples. The selected wavelength also avoids any signal related to the excitation of the TiO2 bandgap or trap states (see Figures S14 and S15).
The plasmonic response of silver NPs in the visible regime was studied in the complete system (Figure 2c) and without gold catalyst (Ag-TiO2; Figure 2b). An additional sample comprised of silver NPs on ZrO2 was used as a noninjectable reference11 (Figure 2a). For both silver and gold NPs, light excitation resulted in a change in the internal electronic structure of the NPs that broadens the plasmon resonance, resulting in the characteristic transient signal comprised of a bleach around the NPs LSPR surrounded by two positive winglets. The signal decay is known to be dominated by electron–phonon scattering (e-ph),39 the dominant cooling pathway for hot electrons following ultrafast electron–electron scattering.39Figure 2e shows the kinetics at 470 nm (where the silver transient bleach occurs) for Ag-ZrO2, Ag-TiO2, and the full multimetallic system. Signal fitting was performed using the instrument response function (IRF) convoluted with a monoexponential decay and an infinite lifetime when TiO2 is present, as signals did not decay to zero. The rising-edge region at 470 nm is assigned to electron–electron scattering events,11,23 but in the present case the determined time constant was below the instrumental resolution (ca. 150 fs); thus, one can only state that those events happen in less than 150 fs, as expected.1 Kinetic analysis of the decay part revealed a faster decay component for Ag-ZrO2, corresponding to e-ph scattering, of 1.1 ps, in comparison to Ag-TiO2 and Ag-TiO2–Au, of 1.7 and 1.5 ps, respectively. The reduced e-ph scattering time observed on ZrO2 is attributed to the blue-shift of the silver LSPR on ZrO2 caused by the difference in dielectric constant between ZrO2 and TiO2,40 resulting in excitation taking place further away from the LSPR and consequently giving rise to lower electron temperature and faster cooling. Furthermore, the kinetic traces not decaying to zero for Ag-TiO2 and Ag-TiO2–Au within the experimental delay line (ca. 5 ns) indicates the presence of a residual amount of unrecombined holes in silver, suggesting electron injection taking place from silver into TiO2. This explanation is further supported by the appearance of a broad signal from 620 to 740 nm for the Ag-TiO2 and Ag-TiO2–Au samples (Figure 2b and 2c), attributed to electrons in the TiO2 conduction band,21 as confirmed by measuring the transient response of bare TiO2 when pumped above the bandgap at 330 nm (Figure S13).
Figure 2.

Transient absorption spectra of Ag-ZrO2 (a), Ag-TiO2 (b), and Ag-TiO2–Au (c). Resulting transient absorption spectra after subtracting the data of panel b from c (d). Kinetic traces extracted at 470 nm from panels a, b, and c (e), and traces of the gold signal at 550 nm (bleach) and 680 nm (winglet) (f).
The injection is apparent from the responses of the multimetallic system in the visible regime but is considerably overlapped with the other signals. Therefore, we opted to perform similar experiments but probing in the mid-infrared regime, where free charge carriers in the conduction band of TiO2 yield a characteristic, strong, featureless absorption,41 enabling us to study electron injection into TiO2 without overlaps (Figure 3a). Kinetic traces extracted at 5200 nm can be found in Figure 3b. A time of approximately 220 fs associated with charge injection from silver into the TiO2 conduction band was found by the fitting of the rising edge with a monoexponential growth function. This time was virtually unaffected by the addition of gold NPs (ca. 240 fs), signifying that electrons are indeed relayed via TiO2. The lower signal magnitude for the full system suggests rapid transference of some electrons from TiO2 to gold NPs. The measured decay of the sample with and without gold was fitted using two-exponential decay functions and an infinite lifetime component. Addition of gold resulted in a decrease of both time components from 4.1 to 3.0 ps and from 600 to 550 ps, respectively, and most importantly a significant decrease in the ΔA values. In the absence of gold, the measured decay is attributed to recombination processes across the TiO2–Ag interface. The difference in decay kinetics and signal amplitude upon gold addition indicates the appearance of a new channel that competes for the hot electrons in the TiO2 conduction band, which was intuitively assigned to electrons being transferred from TiO2 to gold. This is somewhat probable considering the energetics of the complete system, with the Fermi level of gold located at energies below the silver Fermi level and TiO2 conduction band edge position.
Figure 3.
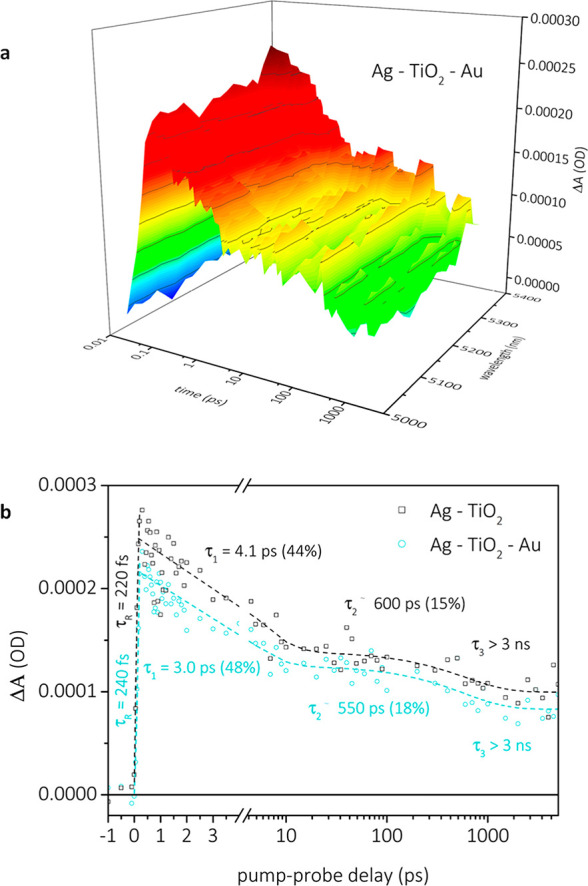
Transient infrared absorption spectra of Ag-TiO2–Au (a). Kinetic traces at 5200 nm extracted from TIRAS spectra (b).
Note that the observed TIRAS signal for electrons in the TiO2 conduction band does not establish their origin, which can come from several processes, namely, (i) enhancement by the near-field of the silver NPs; (ii) enhancement by the near-field of the gold NPs; (iii) plasmon-induced resonant energy transfer (PIRET), and (iv) hot carriers generated directly in the silver NPs. We observed experimentally (SI; section on Reverse electron flow, pages 21–25) that the electron flow could be reversed by exciting the system at 550 nm (gold LSPR resonance). The signal trends were similar to what was observed when silver was excited, suggesting that electrons come from plasmon excitation. Additionally, and as aforementioned, the residual signal associated with unrecombined holes in the plasmonic structure that was excited is only consistent with a plasmon-mediated electron transfer mechanism.
While decreases in lifetimes of electrons on a semiconductor relay (TiO2 here) have been reported before upon addition of a catalytic site that electrons can preferentially move to,5,6 the process has remained elusive and without direct proof. The use of gold here, however, allows for direct observations of changes in gold’s electronic structure reflected by its transient absorption response following silver excitation. Figure 2d shows the difference spectrum between the full multimetallic system and Ag-TiO2 pumped at identical fluences, and the associated kinetics extracted at the bleach and high-wavelength winglet of this difference spectrum is presented in Figure 2f. The corresponding transient absorption spectrum of Au-TiO2 pumped at 435 nm and kinetic analyses can be found in Figures S16 and S17, respectively, for comparison. Three important differences between these spectra are to be noted. First, the signal amplitude in the difference spectrum extracted from Ag-TiO2–Au is significantly higher than the one observed when pumping Au-TiO2 pumped at 435 nm, indicating a larger hot carrier population. Second, the monoexponential fitting of the signal decay that provides information about the e-ph scattering time39 revealed a significantly increased e-ph cooling time, from 1.4 to 1.6 ps on Au-TiO2 pumped at 435 nm (Figure S17) to 2.2–2.4 ps (Figure 2f) for the full system. The changes were rationalized as that higher electron density on gold in the full system will result in slower cooling, similar to the electron–phonon bottleneck reported for other systems.42,43 Third, and possibly the most important of all, there is a clear time lag (ca. 450–500 fs) in the rising edge component of the complete system, which is not present on the signal of Au-TiO2 when pumped at 435 nm, that shows the expected edge rising at time zero with a time constant outside our instrument time resolution. The time lag in the ΔA signal was assigned to the travel time of hot electrons originated from the silver-plasmon to reach the TiO2–Au interface. Once at the TiO2–Au interface, the electrons take roughly 190 fs to move from TiO2 to gold, indicating that most hot electrons initially created at the plasmonic silver site reach the gold catalytic site in 650–700 fs. It is recognized here that this time is extremely fast, and assuming a 1 μm TiO2 thickness, which roughly corresponds to the minimal thickness for screen-printed TiO2 films, an electron travelling velocity in the order of 106 m s–1 is required. Thermal velocities in the order of 105 m s–1 of electrons at room temperature in bulk semiconductors have been reported.44 The electrons having larger energies than just thermal energy could explain the higher velocity. We speculate, however, that the presence of silver NPs in the mesoporous TiO2 channels is more likely to cause this rapid electron transfer, as this could significantly decrease the Ag–Au distance, which would subsequently also result in lower required electron velocities.
The dependence of the ΔA signal in the mid-infrared regime, attributed to free charge carriers on TiO2,41,45 on the pump fluence is presented in Figure 4a. Since electrons reach gold on ultrafast time scales (<1 ps), these measurements were performed only up until a 10 ps pump–probe delay. Twenty measurements at each power were performed, and the obtained average decay was fitted with a monoexponential function on the time scale of the measurement. The ΔA signal at 0.6 ps was extracted as the transient absorption measurements had shown the maximum amplitude of the gold plasmonic response to occur at this time (Figure 2d and 2f). As shown in Figure 4b, a clear linear increase in signal intensity for Ag-TiO2 and Au-TiO2 is observed upon increasing the pump fluence. As expected, the increase observed for Ag-TiO2 is significantly steeper than for Au-TiO2, i.e., at 1.12 μW–1 versus 0.22 μW–1, since the LSPR of silver is excited at 435 nm, resulting in a larger formation of hot, injectable electrons than in case of gold, where direct interband transitions are primarily excited at 435 nm.38 Interestingly, extrapolating the fitted line for Au-TiO2 to a theoretical zero power value yields a nonzero ΔA value. We speculate that this is a result of plasmon-induced interfacial charge transfer46 or direct interfacial charge transfer.47 Both processes happen within 10s of fs and are beyond the time resolution we can achieve. The fact that this is observed only when gold is present is likely due to the direct contact between gold and TiO2 and the absence of capping ligands, which, as observed for the Ag-TiO2 sample, seem to inhibit these processes.
Figure 4.

Kinetic traces at three different pump-powers, extracted at 5200 nm for the Ag-TiO2–Au system (a), power dependence measurements, kinetics extracted at 5200 nm (b), and proposed model to account for the observed results in the power dependence measurements (c). The gap between silver and TiO2 in (c) represents the PVP layer that prevents Fermi level pinning upon charge injection.
An evaluation of the response obtained for the full multimetallic system shows the presence of two distinguishable regimes, i.e., a low power regime, in which an increase of the pump fluence results in an increase of the ΔA signal of 0.35 μW–1, and a high-power regime, where a 1.16 μW–1 increase in the ΔA signal is observed. We attribute this sudden change in slope around 125 μW to the saturation of the partly unfilled 5d states of gold.29 Gold is considered the noblest of the metals due to its largely filled 5d band,27 which reduces its ability to adsorb molecules and accept an electrical charge.48−50 The detected saturation point does not necessarily equate to the point at which the gold 5d band is filled; rather, it suggests that after this point it is hard to transfer more charge to gold nanoparticles. Further substantiation for this assignment is that the saturation point is sensitive to the gold concentration, shifting to higher pulse energies with an increase of gold concentration. Once at gold, hot electrons should remain there for a significant time since the electronic barrier between the TiO2 conduction band and gold is ca. 0.8 eV and thus not thermally accessible to enable fast back electron transfer.51,52 After the saturation point, transference to gold is largely suppressed, and thus, the injected electrons originated from silver-plasmons remain in TiO2. The small increase in slope at pump powers below the saturation point indicates that while it is energetically favourable for electrons to move to gold (ca. 0.8 eV), not all injected electrons reach the gold NPs. The nearly identical slopes obtained after the saturation point illustrate that further injected electrons remain on TiO2 until recombination occurs.
The overall process is summarized in Figure 4c. Upon light absorption by silver NPs, plasmons were created and relaxed into hot carriers, omitted from the scheme due to space constraints. The thermalized hot electrons were injected into TiO2 in 220–240 fs through the PVP layer,36 used to prevent Fermi level pinning. The PVP layer is expected to affect electron transfer,53 but assessing its influence is outside the scope of this work. The injected hot electrons were subsequently transferred to the catalytic gold site within ca. 450–500 fs after their injection into TiO2. The process was continued until saturation of the gold electronic structure occurred.
The multimetallic system photocatalytic activity was evaluated using the 6e– reduction reaction between 4-nitrophenol (4-NP) and NaBH4 (Figure 5a).54,55 Since silver and gold NPs have both been reported to catalyze this reaction in the absence of light, the reaction is ideal to assess any changes in catalytic activity upon excitation of the silver LSPR. Figure 5b shows the setup used in these experiments.56 Tests were performed in dark and with light and using 100-fold excess of NaBH4, to extract the apparent reaction rate from the pseudo-first-order rate kinetics (SI, page 5).54 As is evident, in the case of Au-TiO2, the apparent rate is increased by a factor of 1.35, whereas in the case of Ag-TiO2, an increase of 4.3 times was observed upon laser excitation, which is in line with the laser excitation being performed closer to the silver LSPR (Figure 5c, top panel, and Figures S32–S34). We attribute these rate enhancements to an increase in electron density on the NPs due to the holes on the metal NPs being regenerated by the hydride source, while injected electrons also return to the NP, resulting in an overall larger number of electrons. The slower increase observed for gold is in good agreement with the results presented in Figure 4, showing a significantly lower electron injection for Au-TiO2 than for Ag-TiO2, as expected based on the relative LSPR peaks of gold and silver.
Figure 5.

Proposed reaction scheme for the reduction of 4-nitrophenol under light irradiation of the prepared multimetallic system (a) and the setup used to carry out the catalytic measurements (b). Observed apparent catalytic rates, determined via UV–vis spectroscopy (c), with the top panel depicting monometallic systems with light and the bottom panel depicting the full multimetallic system.
Upon testing the full system (Figure 5c bottom panel), a 16-fold rateincrease was observed when illumination at 445 nm is applied, and an apparent rate constant of 1.85 × 10–3 s–1 is found, surpassing by one order of magnitude the combined reactivity of the two individual systems. The results obtained here are in good quantitative agreement with those obtained by Hajfathalian et al.57 on AuCu bimetallic structures. This study, however, adds an active support , TiO2, which acts as an electron relay, to the complete system, thereby resembling more closely the design of nature’s photosynthetic system.58 The role of gold as a catalytic site and as a means to increase the electron lifetime can be inferred from these and the ultrafast transient absorption results. Note that in all cases the only product detected was 4-aminophenol,54,55 as confirmed by the appearance of the peak at around 300 nm and an isosbestic point at ca. 320 nm (Figures S35–S42). It should be mentioned that due to experimental limitations concerning CW laser power output, it was not possible to reach electronic saturation of gold and observe reduced kinetics. However, even if that were possible, we speculate that reduced kinetics would be observed before gold saturation due to mass transfer limitations.
In summary, the journey of a hot electron in a solid-state photocatalyst was followed in real-time. Ultrafast studies revealed that plasmon-generated hot electron transfer from the photosensitizer (silver NPs) to the catalytic site (gold NPs) via a semiconductor relay (TiO2) takes roughly 700 fs. The presence of gold was shown to enhance charge separation, contributing to a 16-fold increase in photocatalytic activity in the studied model reaction. The studies also revealed gold NP electronic saturation, confirming effective electron transport to the catalytic site and limited capacity of the gold 5d band. The observations revealed that multimetallic photoredox systems containing plasmonic materials are highly effective in generating and utilizing hot carriers, and thus, we forecast their widespread utilization in photocatalysis and photovoltaics.
Acknowledgments
The authors would like to thank Uppsala University for financial support. J.S. acknowledges the support by the Swedish Research Council (2019-03597). K.Z. acknowledges the support by the Danish Council for Independent Research (7026-0037B) and the Swedish Research Council (2017-05337).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.nanolett.0c03344.
Detailed description of the sample preparation, structural characterization, transient absorption experiments, and catalytic experiments (PDF)
Author Contributions
J.S. conceived and planned the project in liaison with L.v.T. The experimental work was performed by L.v.T. and J.M. and was supported by Y.T. (transient experiments). L.v.T. analyzed and treated the data and also prepared the figures in liaison with J.S.. The manuscript was drafted by J.S. and L.v.T. with input from all authors. The final version was read and agreed upon by all the authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Brongersma M. L.; Halas N. J.; Nordlander P. Plasmon-induced hot carrier science and technology. Nat. Nanotechnol. 2015, 10, 25–34. 10.1038/nnano.2014.311. [DOI] [PubMed] [Google Scholar]
- Sá J.; et al. Direct observation of charge separation on Au localized surface plasmons. Energy Environ. Sci. 2013, 6, 3584–3588. 10.1039/c3ee42731e. [DOI] [Google Scholar]
- Linic S.; Christopher P.; Ingram D. B. Plasmonic-metal nanostructures for efficient conversion of solar to chemical energy. Nat. Mater. 2011, 10, 911–921. 10.1038/nmat3151. [DOI] [PubMed] [Google Scholar]
- Mubeen S.; et al. An autonomous photosynthetic device in which all charge carriers derive from surface plasmons. Nat. Nanotechnol. 2013, 8, 247–252. 10.1038/nnano.2013.18. [DOI] [PubMed] [Google Scholar]
- Pavliuk M. V.; et al. Nano-hybrid plasmonic photocatalyst for hydrogen production at 20% efficiency. Sci. Rep. 2017, 7, 8670. 10.1038/s41598-017-09261-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavliuk M. V.; et al. Hydrated electron generation by excitation of copper localized surface plasmon resonance. J. Phys. Chem. Lett. 2019, 10, 1743–1749. 10.1021/acs.jpclett.9b00792. [DOI] [PubMed] [Google Scholar]
- Welch A. J.; et al. Nanoporous gold as a highly selective and active carbon dioxide reduction catalyst. ACS Appl. Energy Mater. 2019, 2, 164–170. 10.1021/acsaem.8b01570. [DOI] [Google Scholar]
- DuChene J. S.; et al. Optical excitation of a nanoparticle Cu/p-NiO photocathode improves reaction selectivity for CO2 reduction in aqueous electrolytes. Nano Lett. 2020, 20, 2348–2358. 10.1021/acs.nanolett.9b04895. [DOI] [PubMed] [Google Scholar]
- Garcia de Arquer F. P.; et al. Photoelectric energy conversion of plasmon-generated hot carriers in metal-insulator-semiconductor structures. ACS Nano 2013, 7, 3581–3588. 10.1021/nn400517w. [DOI] [PubMed] [Google Scholar]
- Nakamura K.; et al. Properties of plasmons-induced photoelectric conversion on a TiO2/NiO p–n junction with Au nanoparticles. J. Phys. Chem. Lett. 2016, 7, 1004–1009. 10.1021/acs.jpclett.6b00291. [DOI] [PubMed] [Google Scholar]
- Hattori Y.; et al. Simultaneous hot electron and hole injection upon excitation of gold surface plasmon. J. Phys. Chem. Lett. 2019, 10, 3140–3146. 10.1021/acs.jpclett.9b01085. [DOI] [PubMed] [Google Scholar]
- Swearer D. F.; et al. Light-driven chemical looping for ammonia synthesis. ACS Energy Lett. 2019, 4, 1505–1512. 10.1021/acsenergylett.9b00860. [DOI] [Google Scholar]
- Zhou L.; et al. Light-driven methane dry reforming with single atomic site antenna-reactor plasmonic photocatalysts. Nat. Energy 2020, 5, 61–70. 10.1038/s41560-019-0517-9. [DOI] [Google Scholar]
- Dongare P. D.; et al. Solar thermal desalination as a nonlinear optical process. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 13182–13187. 10.1073/pnas.1905311116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alabastri A.; et al. Resonant energy transfer enhances solar thermal desalination. Energy Environ. Sci. 2020, 13, 968–976. 10.1039/C9EE03256H. [DOI] [Google Scholar]
- Rastinehad A. R.; et al. Gold nanoshell-localized photothermal ablation of prostate tumors in a clinical pilot device study. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 18590–18596. 10.1073/pnas.1906929116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu G. Dual-functional plasmonic photothermal biosensor for highly accurate severe acute respiratory syndrome coronavirus 2 detection. ACS Nano 2020, 14, 5268. 10.1021/acsnano.0c02439. [DOI] [PubMed] [Google Scholar]
- Lehmann J.; Merschdorf M.; Pfeiffer W.; Thon A.; Voll S.; Gerber G. Surface plasmon dynamics in silver nanoparticles studied by femtosecond time-resolved photoemission. Phys. Rev. Lett. 2000, 85, 2921–2924. 10.1103/PhysRevLett.85.2921. [DOI] [PubMed] [Google Scholar]
- Tagliabue G.; et al. Quantifying the role of surface plasmon excitation and hot carrier transport in plasmonic devices. Nat. Commun. 2018, 9, 3394. 10.1038/s41467-018-05968-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett-Tapley G. L.; et al. Plasmon-mediated catalytic oxidation of sec-phenethyl and benzyl alcohols. J. Phys. Chem. C 2011, 115, 10784–10790. 10.1021/jp202769a. [DOI] [Google Scholar]
- Furube A.; et al. Ultrafast plasmon-induced electron transfer from gold nanodots into TiO2 nanoparticles. J. Am. Chem. Soc. 2007, 129, 14852–14853. 10.1021/ja076134v. [DOI] [PubMed] [Google Scholar]
- Tagliabue G. Ultrafast of hot-hole injection modifies hot-electron dynamics in Au/p-GaN heterostructures. Nat. Mater. 2020, 1. 10.1038/s41563-020-0737-1. [DOI] [PubMed] [Google Scholar]
- Hattori Y.; et al. Light-induced ultrafast proton-couple electron transfer responsible for H2 evolution on silver plasmonics. Mater. Today 2018, 21, 590–593. 10.1016/j.mattod.2018.05.002. [DOI] [Google Scholar]
- Aslam U.; Chavez S.; Linic S. Controlling energy flow in multimetallic nanostructures for plasmonic catalysis. Nat. Nanotechnol. 2017, 12, 1000–1006. 10.1038/nnano.2017.131. [DOI] [PubMed] [Google Scholar]
- Sykes M. E.; et al. Enhanced generation and anisotropic Coloumb scattering of hot electrons in an ultra-broadband plasmonic nanopatch metasurface. Nat. Commun. 2017, 8, 986. 10.1038/s41467-017-01069-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarström L. Accumulative charge separation for solar fuels production: coupling light-induced single electron transfer to multielectron catalysis. Acc. Chem. Res. 2015, 48, 840–850. 10.1021/ar500386x. [DOI] [PubMed] [Google Scholar]
- Hammer B.; Nørskov J. K. Why gold is the noblest of all the metals. Nature 1995, 376, 238–240. 10.1038/376238a0. [DOI] [Google Scholar]
- Haruta M.; et al. Gold catalysts prepared by coprecipitation for low-temperature oxidation of hydrogen and carbon monoxide. J. Catal. 1989, 115, 301–309. 10.1016/0021-9517(89)90034-1. [DOI] [Google Scholar]
- Zhang P.; Sham T. K. X-ray studies of the structure and electronic behaviour of alkanethiolate-capped gold nanoparticles: the interplay of size and surface effects. Phys. Rev. Lett. 2003, 90, 245502. 10.1103/PhysRevLett.90.245502. [DOI] [PubMed] [Google Scholar]
- Shi M.-M.; et al. Au sub-nanoclusters on TiO2 towards highly efficient and selective electrocatalyst for N2 conversion to NH3 at ambient conditions. Adv. Mater. 2017, 29, 1606550. 10.1002/adma.201606550. [DOI] [PubMed] [Google Scholar]
- Sá J.; et al. Activation of alkanes by gold-modified lanthanum oxide. ChemCatChem 2011, 3, 394–398. 10.1002/cctc.201000285. [DOI] [Google Scholar]
- Goguet A.; et al. Increased dispersion of supported gold during methanol carbonylation conditions. J. Am. Chem. Soc. 2009, 131, 6973–6975. 10.1021/ja9021705. [DOI] [PubMed] [Google Scholar]
- Kaiser S. K.; et al. Nanostructuring unlocks high performance of platinum single-atom catalysts for stable vinyl chloride production. Nat. Catal. 2020, 3, 376–385. 10.1038/s41929-020-0431-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuChene J. S.; et al. Hot hole collection and photoelectrochemical CO2 reduction with plasmonic Au/p-GaN photocathodes. Nano Lett. 2018, 18, 2545–2550. 10.1021/acs.nanolett.8b00241. [DOI] [PubMed] [Google Scholar]
- Silvert P.-Y.; et al. Preparation of colloidal silver dispersions by the polyol process. J. Mater. Chem. 1996, 6, 573–577. 10.1039/JM9960600573. [DOI] [Google Scholar]
- García de Arquer F. P.; et al. Photoelectric energy conversion of plasmon-generated hot carriers in metal-insulator-semiconductor structures. ACS Nano 2013, 7, 3581–3588. 10.1021/nn400517w. [DOI] [PubMed] [Google Scholar]
- Kelly K. L.; et al. The Optical Properties of Metal Nanoparticles: The Influence of Size, Shape, and Dielectric Environment. J. Phys. Chem. B 2003, 107, 668–677. 10.1021/jp026731y. [DOI] [Google Scholar]
- Bernardi M.; et al. Theory and computation of hot carriers generated by surface plasmon polaritons in noble metals. Nat. Commun. 2015, 6, 1–9. 10.1038/ncomms8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voisin C.; et al. Ultrafast electron dynamics and optical nonlinearities in metal nanoparticles. J. Phys. Chem. B 2001, 105, 2264–2280. 10.1021/jp0038153. [DOI] [Google Scholar]
- Robertson J. High dielectric constant oxides. Eur. Phys. J.: Appl. Phys. 2004, 28, 265–291. 10.1051/epjap:2004206. [DOI] [Google Scholar]
- Antila L. J.; et al. Hunting for the elusive shallow traps in TiO2 anatase. Chem. Commun. 2015, 51, 10914–10916. 10.1039/C5CC02876K. [DOI] [PubMed] [Google Scholar]
- Nozik A. J. Quantum dot solar cells. Phys. E 2002, 14, 115–120. 10.1016/S1386-9477(02)00374-0. [DOI] [Google Scholar]
- Fu J.; et al. Hot carrier cooling mechanisms in halide perovskites. Nat. Commun. 2017, 8, 1–9. 10.1038/s41467-017-01360-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anwar F.; Mahbub R.; Satter S. S.; Ullah S. M. Effect of Different HTM Layers and Electrical Parameters on ZnO Nanorod-Based Lead-Free Perovskite Solar Cell for High-Efficiency Performance. Int. J. Photoenergy 2017, 2017, 9846310–9846318. 10.1155/2017/9846310. [DOI] [Google Scholar]
- Panayotov D. A.; Burrows S. P.; Morris J. R. Infrared Spectroscopic Studies of Conduction Band and Trapped Electrons in UV-Photoexcited, H-Atom n-Doped, and Thermally Reduced TiO2. J. Phys. Chem. C 2012, 116, 4535–4544. 10.1021/jp2053103. [DOI] [Google Scholar]
- Wu K.; et al. Efficient hot-electron transfer by a plasmon-induced interfacial charge-transfer transition. Science 2015, 349, 632–635. 10.1126/science.aac5443. [DOI] [PubMed] [Google Scholar]
- Kolb D. M.; Przasnyski M.; Gerischer H. Optical interfacial electron transfer between metal adatoms and a semiconductor electrode. Z. Phys. Chem. 1974, 93, 1–14. 10.1524/zpch.1974.93.1-6.001. [DOI] [Google Scholar]
- Soares J. M. C.; Morrall P.; Crossley A.; Harris P.; Bowker M. Catalytic and noncatalytic CO oxidation on Au/TiO2 catalysts. J. Catal. 2003, 219, 17–24. 10.1016/S0021-9517(03)00194-5. [DOI] [Google Scholar]
- Liu H.; Mun B. S.; Thornton G.; Isaacs S. R.; Shon Y.-S.; Ogletree D. F.; Salmeron M. Electronic structure of ensembles of gold nanoparticles: size and proximity effects. Phys. Rev. B: Condens. Matter Mater. Phys. 2005, 72, 155430. 10.1103/PhysRevB.72.155430. [DOI] [Google Scholar]
- Reinecke B. N.; Kuhl K. P.; Ogasawara H.; Li L.; Voss J.; Abild-Pedersen F.; Nilsson A.; Jaramillo T. F. Elucidating the electronic structure of supported gold nanoparticles its relevance to catalysis by means of hard X-ray photoelctron spectroscopy. Surf. Sci. 2016, 650, 24–33. 10.1016/j.susc.2015.12.025. [DOI] [Google Scholar]
- Lahiri D.; Subramanian V.; Shibata T.; Wolf E. E.; Bunker B. A.; Kamat P. V. Photoinduced transformations at semicondutor/metal interfaces: X-ray absorption studies of titania7gold films. J. Appl. Phys. 2003, 93, 2575. 10.1063/1.1544068. [DOI] [Google Scholar]
- Takai A.; Kamat P. V. Capture, store, and discharge shittling photogenerated electrons across TiO2-silver interface. ACS Nano 2011, 5, 7369–7376. 10.1021/nn202294b. [DOI] [PubMed] [Google Scholar]
- Park J. Y.; Lee H.; Renzas J. R.; Zhang Y.; Somorjai G. A. Probing hot electron flow generated on Pt nanoparticles with Au/TiO2 Schottky diodes during catalytic CO oxidation. Nano Lett. 2008, 8, 2388–2392. 10.1021/nl8012456. [DOI] [PubMed] [Google Scholar]
- Li M.; Chen G. Revisiting catalytic model reaction p-nitrophenol/NaBH4 using metallic nanoparticles coated on polymeric spheres. Nanoscale 2013, 5, 11919–11927. 10.1039/c3nr03521b. [DOI] [PubMed] [Google Scholar]
- Aditya T.; Pal A.; Pal T. Nitroarene reduction: a trusted model reaction to test nanoparticle catalysts. Chem. Commun. 2015, 51, 9410–9431. 10.1039/C5CC01131K. [DOI] [PubMed] [Google Scholar]
- Hariprasad E.; Radhakrishnan T. P. A Highly Efficient and Extensively Reusable “Dip Catalyst” Based on a Silver-Nanoparticle-Embedded Polymer Thin Film. Chem. - Eur. J. 2010, 16, 14378–14384. 10.1002/chem.201001679. [DOI] [PubMed] [Google Scholar]
- Hajfathalian M.; et al. Photocatalytic enhancements to the reduction of 4-nitrophenol by resonantly excited triangular gold-copper nanostructures. J. Phys. Chem. C 2015, 119, 17308–17315. 10.1021/acs.jpcc.5b04618. [DOI] [Google Scholar]
- Barber J.; Andersson B. Revealing the blueprint of photosynthesis. Nature 1994, 370, 31–34. 10.1038/370031a0. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.