

Abstract
The rapid growth of serious infections caused by antibiotic resistant bacteria, especially the nosocomial ESKAPE pathogens, has been acknowledged by Governments and scientists and is one of the world’s major health problems. Various strategies have been and are currently investigated and developed to reduce and/or delay the bacterial resistance. One of these strategies regards the design and development of antimicrobial hybrids and conjugates. This unprecedented critical review, in which our continuing interest in the synthesis and evaluation of the bioactivity of imidazole derivatives is testified, aims to summarise and comment on the results obtained from the end of the 1900s until February 2020 in studies conducted by numerous international research groups on the synthesis and evaluation of the antibacterial properties of imidazole-based molecular hybrids and conjugates in which the pharmacophoric constituents of these compounds are directly covalently linked or connected through a linker or spacer. In this review, significant attention was paid to summarise the strategies used to overcome the antibiotic resistance of pathogens whose infections are difficult to treat with conventional antibiotics. However, it does not include literature data on the synthesis and evaluation of the bioactivity of hybrids and conjugates in which an imidazole moiety is fused with a carbo- or heterocyclic subunit.
Keywords: imidazoles, molecular hybrids, molecular conjugates, antibacterials, antibiotic resistance, synthesis, bioactivity, antibiotics
1. Introduction
Serious infections caused by antibiotic resistant bacteria, especially the six nosocomial ESKAPE pathogens Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp., are one of the world’s major healthcare problems in the 21st century and are a cause of morbidity and mortality [1]. Overconsumption and misuse of antibiotics and exposure to infections in hospitals has caused the emergence of multi-drug resistant bacteria many of which, according to the World Health Organization (WHO) list, are Gram-negative pathogens [2]. In fact, their structure consists of a protective extra outer membrane that antibiotics have great difficulty going through. Gram-positive bacteria lack this important layer.
In order to reduce and/or delay bacterial resistance, various strategies have been investigated and developed. One of these concerns the development of new antibacterial drugs [3,4] and the structural modification of existing antibiotics, but the studies in this area are very expensive and time consuming. Another effective strategy involves the use of the combination therapy, i.e., the use of two or more drugs to restore or to increase the efficacy of both drugs against the bacterial pathogens that are resistant to ordinary antibiotics. However, none of the expected benefits of following this strategy have thus far been observed in in vivo studies [5]. A third clinically employed strategy concerns the deactivation of the mechanism of resistance through the use of a combination of a β-lactam antibiotic with a β-lactamase inhibitor adjuvant. Finally, another strategy to which much attention has been paid in recent years, presenting a great opportunity for progress, concerns design and development of antimicrobial hybrids [6] and conjugates. According to Klahn’s definition [7], antimicrobial hybrids are molecules that contain two discrete functional elements, both having antibacterial activity, which can be linked through a spacer. Due to the dual targeting, resistance development can be significantly impaired, the pharmacokinetic properties can be better compared to combination therapies with the single antibacterial elements, and the antibacterial activity is often greater than the sum of the antibacterial activity of each element. On the other hand, still according to Klahn [7], in antibacterial conjugates, a single functional moiety controls the accumulation of the other part of the conjugate by mediating an active transport into the bacterial cell or blocking the efflux from it and stimulating the accumulation of the second moiety that acts as an antibiotic through antimicrobial peptides, cell penetrating peptides, lipopeptides or siderophore vectors. Unfortunately, in some recent papers, this classification of antibacterial agents in hybrids and conjugates has not been followed and the antibacterial substances in which a single pharmacophore is connected to a non-bacterial subunit through a spacer or linker have been named both as antibacterial hybrids and conjugates. Nevertheless, in this unprecedented critical review we have defined an antibiotic hybrid as a synthetic combination of two or more covalently linked pharmacophores belonging to an established agent known to elicit an antibacterial effect.
This review, with 261 references, in which we testified our continuing interest in the synthesis and bioactivity of imidazole derivatives [8,9,10,11,12,13,14,15], aims to summarise and comment on the main results obtained from the end of 1900s until the end of February 2020 in studies conducted by numerous international research groups on the synthesis and evaluation of the antibacterial properties of imidazole-based molecular hybrids and conjugates in which their discrete functional elements are directly covalently linked or are connected through a linker or spacer. The review, in which significant attention was paid to summarise the strategies used to overcome the antibiotic resistance of pathogens whose infections are difficult to treat with conventional antibiotics, was organised in the following main sections: (i) heterocyclic conjugates and hybrids bearing nitroimidazole moiety; (ii) coumarin/imidazole hybrids and conjugates; (iii) furanchalcone/imidazole hybrids; (iv) hybrids based on benzofuran, quinazolinone, and imidazolium moieties; (v) 1H-imidazoles containing azetidinone derivatives; (vi) pyrrole/imidazole and indole/imidazole hybrids; (vii) pyrazole/imidazole hybrids; (viii) 1,2,3-triazole/imidazole hybrids and conjugates; (ix) hybrids of imidazole derivatives and 5-membered heterocycles containing oxygen and nitrogen; (x) 1,8-naphthalimide/imidazole hybrids; (xi) bis-imidazoles; (xii) pyridine/imidazole hybrids; (xiii) quinoline/imidazole hybrids; (xiv) pyrimidine/imidazole hybrids; (xv) addendum.
However, this review did not include the synthesis and evaluation of the bioactivity of molecular hybrids and conjugates in which imidazole moieties are fused with carbo- or hetero-cyclic subunits or contain only (un)functionalised acyclic groups. Furthermore, no literature data were reported on the synthesis of imidazole containing hybrids and conjugates for which antimicrobial assessments were not carried out.
Occasionally, the original biological data and their standard errors have been rounded off to provide readers with comparable and more statistically consistent results. For the same reason, whenever possible, MIC and (sometimes) IC50 values have been reported on a molar basis, regardless of the data format in the original papers.
2. Heterocyclic Conjugates and Hybrids Bearing Nitroimidazole Moiety
Nitroimidazole derivatives are a class of antimicrobial drugs that are used as effective therapeutic agents for treatment of infections caused by Gram-negative and Gram-positive bacteria and protozoa such as Giardia, Lamblia, and Entamoeba histolytica [16]. Metronidazole [2-(2-methyl-5-nitro-1H-imidazol-1-yl)ethan-1-ol, Flagyl®, Pfizer] (1) (Figure 1) is an antibiotic used to treat a wide variety of infections caused by anaerobic Gram-negative bacteria, such as Helicobacter pylori, and for treatment of infections caused by Clostridium difficile [17], a Gram-positive anaerobic bacillus that causes life-threating severe diarrhoea, abdominal pain, and fever [18].
Figure 1.
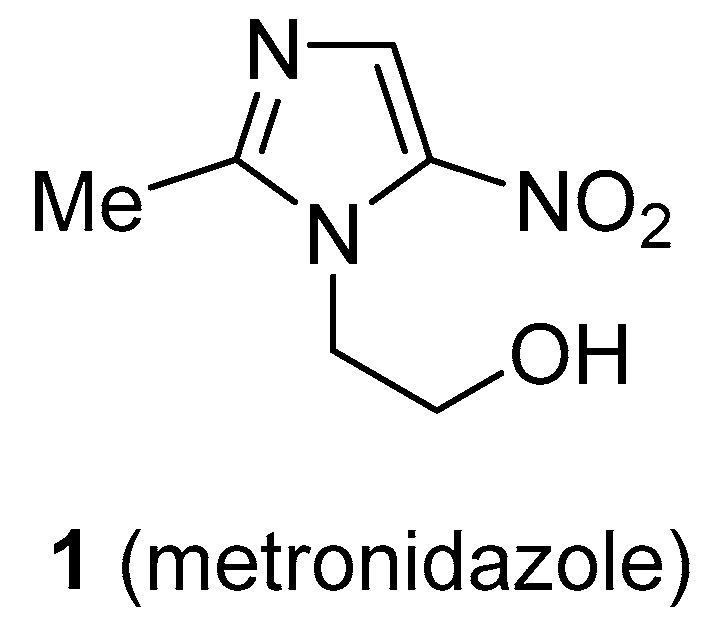
Structure of metronidazole.
The mechanism of action 1 and other nitroimidazoles involves the conversion of this prodrug, via an anaerobic 1-electron reduction of the nitro group, to a short-lived nitro radical anion, which is unstable and decomposes to give a nitrite anion and an imidazole radical. These toxic radical species can inhibit DNA synthesis and cause DNA strand breaks leading to cell death [16,19,20]. Unfortunately, metronidazole resistance has been observed in many pathogenic Gram-negative anaerobic bacteria, e.g., carbapenem resistant P. aeruginosa, K. pneumoniae, carbapenem-resistant A. baumannii, Escherichia coli, 3rd generation cephalosporin-resistant, and fluoroquinolone-resistant Neisseria gonorrhoeae, Chlamydia trachomatis, and Yersinia pestis [21,22]. In fact, Gram-negative bacteria are generally more resistant to multiple antibiotics than Gram-positive bacteria because their outer membrane comprises a complex lipopolysaccharide whose lipid moiety acts as an endotoxin [23]. Therefore, in order to overcome this serious problem, in the last two decades much attention has been paid to the design and synthesis of nitroimidazole hybrids and conjugates having antibacterial activity higher than that of compound 1 and possibly not involving bacterial resistance.
In 1999, Demirayak and coworkers [24] synthesised six 5-nitroimidazole/pyrrole hybrids of general formula 2 in yields ranging from 63 to 83% by reaction of 2-(2-methyl-5-nitro-1H-imidazol-1-yl)ethan-1-amine dihydrochloride (3) with equimolar amounts of the appropriate 1,4-diones, i.e., compounds 4, 5, and 6a–d, and 2 eq of AcONa in AcOH under reflux for 0.5 h, under the conventional Paal-Knorr pyrrole synthesis conditions [25] (Scheme 1).
Scheme 1.

Synthesis of 5-nitroimidazole/pyrrole hybrids 2a–f.
From the evaluation of the antibacterial activity of hybrids 2 against E. coli and multi-drug resistant S. aureus, it turned out that compounds 2a–d possessed MIC values of 20–40 µM against S. aureus, while all hybrids 2a–f had MIC values of 40–70 µM against E. coli. However, the activity of all hybrids 2 was significantly lower than that of ceftriaxone (4 and 0.1 µM against S. aureus and E. coli, respectively), a cephalosporin that was used as a reference compound [24].
In 2007, Shafiee and coworkers [26], considering that (Z)-2-[(5-nitrofuran-2-yl)methylene] benzofuran-3(2H)-ones 7 (Figure 2) exhibits very good in vitro antibacterial properties against Gram-positive (S. aureus ATCC 25923) and a Gram-negative bacterium (Caulobacter crescentus NA 1000) [27], synthesised (Z)-2-[(1-methyl-5-nitro-1H-imidazol-2-yl)methylene]benzofuran-3(2H)-ones 8 and (Z)-2-[(1-methyl-4-nitro-1H-imidazol-5-yl)methylene] benzofuran-3(2H)-ones 9 and tested the in vitro antibacterial properties of these nitroimidazole hybrids against the Gram-positive bacteria S. aureus, methicillin-resistant S. aureus (MRSA), Staphylococcus epidermidis, and Bacillus subtilis, and the Gram-negative bacterium K. pneumoniae.
Figure 2.
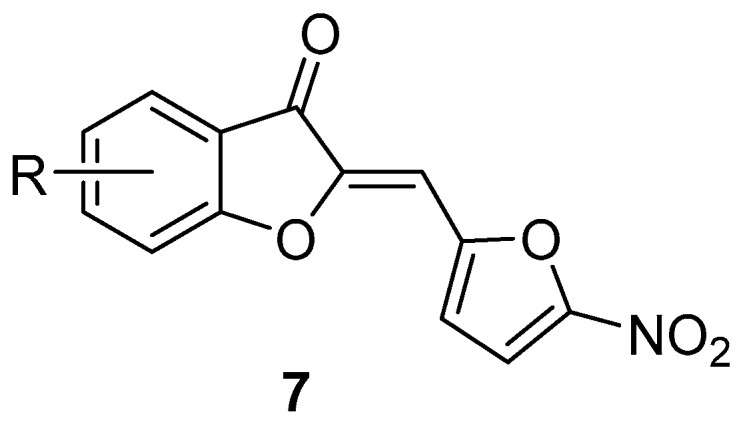
Structure of (Z)-2-[(5-nitrofuran-2-yl)methylene]benzofuran-3(2H)-ones 7.
Hybrids 8 and 9 were synthesised as outlined in Scheme 2. 1-Methyl-5-nitro-1H-imidazole-2-carbaldehyde (10) was condensed with 3(2H)-benzofuranones 11 in AcOH at 100 °C for 6 h in the presence of a catalytic amount of sulfuric acid or by treatment with Ac2O in the presence of AcONa at 100 °C for 1.5 h affording compounds 8 in yields ranging from 42 to 62%.
Scheme 2.

Synthesis of hybrids 8 and 9.
Instead, hybrids 9 were synthesised in yields ranging from 26 to 62% by the reaction of 1-methyl-4-nitro-1H-imidazole-5-carbaldehyde (12) with benzofuran-3(2H)-ones 11 in Ac2O in the presence of AcONa at 100 °C for 1.5 h [26].
Figure 3 shows the chemical structures and yields of some representative compounds 8 and 9, which were synthesised using the reactions depicted in Scheme 2.
Figure 3.

Structures and yields of some representative compounds 8 and 9 synthesised according to Scheme 2.
Compounds 11, which were key intermediates for the synthesis of hybrids 8 and 9, were in turn prepared starting from salicylic acids 13 via the route shown in Scheme 3 that involved the ring closure of intermediates 14 in refluxing Ac2O/AcOH followed by hydrolysis of the resulting compounds 15 in a mixture of HCl/H2O/MeOH (1:10:40) under reflux for 1 h [26].
Scheme 3.

Synthesis of 3(2H)-benzofuranones 11.
Interestingly, most hybrids 8 showed a remarkable antibacterial activity against Gram-positive bacteria, whereas hybrids 9 exhibited no effect against selected bacteria. Compound 8m (R = 5-NO2) revealed to be from 1.7 to 14 times less active than 8a (R = H) against Gram-positive bacteria and 8n (R = 6,7-(OMe)2) was found to be the most active hybrid against these bacteria [26].
In 2008, in the context of a study to find new molecules for treatment of infections caused by the microaerophilic Gram-negative bacterium clarithromycin resistant H. pylori, the root cause of gastric and duodenal ulcers, Foroumadi and coworkers discovered that 5-nitroimidazole/(1,3,4-thiadiazol-2-yl)-morpholine-1,1-dioxide hybrids 16 and related compounds were able to inhibit the growth of two clinical metronidazole sensitive and metronidazole resistant H. pylori strains [28]. For example, in a bacterial growth inhibition assay in which the disk diffusion method was used, 4-[5-(1-methyl-5-nitro-1H-imidazol-2-yl)-1,3,4-thiadiazol-2-yl] thiomorpholine-1,1-dioxide (16a) at a concentration of 8 μg/disc turned out to have inhibition zone diameters of 32 and 27 mm against these two bacterial strains, respectively.
The synthesis of hybrid 16a was achieved via the route depicted in Scheme 4, in which the first step was the conversion of aldehyde 10 to thiosemicarbazone 17. Oxidative cyclisation of 17 in the presence of NH4Fe(SO4)2·12H2O [29] provided compound 18, which by diazotation in HCl in the presence of Cu powder [30] gave 2-chloro-5-(1-methyl-5-nitro-1H-imidazol-2-yl)-1,3,4-thiadiazole 19. The latter compound was treated with thiomorpholine in dioxane and the resulting compound 20 was oxidised with excess of 30% H2O2 in AcOH at 55–60 °C to give hybrid 16a [28]. Unfortunately, the yields of this synthetic route were not reported.
Scheme 4.

Synthesis of 4-[5-(1-methyl-5-nitro-1H-imidazol-2-yl)-1,3,4-thiadiazol-2-yl] thiomorpholine-1,1-dioxide (16a).
However, biological data showed that 4-[5-(5-nitrofuran-2-yl)-1,3,4-thiadiazol-2-yl] thiomorpholine-1,1-dioxide (16b) (Figure 4), an analogue of 16a, was more potent than this hybrid [28].
Figure 4.

Structure of 4-[5-(5-nitrofuran-2-yl)-1,3,4-thiadiazol-2-yl]thiomorpholine-1,1-dioxide (16b).
In 2009, Atia reported the synthesis of numerous hybrids containing a 2-nitroimidazole moiety starting from metronidazole (1) [31]. Such hybrids included three 4-arylidene-2-[1-(2-chloroethyl)-5-nitro-1H-imidazole-2-yl]oxazol-5(4H)-ones 21, three 3′-amino-5′-arylidene-1-(2-chloroethyl)-5-nitro-3′,5′-dihydro-1H,4′H-(2,2′-biimidazol)-4′-ones 22, nine 5′-arylidene-3′-arylideneamino-1-(2-chloroethyl)-5-nitro-3′,5′-dihydro-1H,4′H-(2,2′-biimidazol)-4′-ones 23, three ethyl N-[4-arylidene-1′-(2-chloroethyl)-5′-nitro-5-oxo-4,5-dihydro-1H,1′H-(2,2′-biimidazol)-1-yl] glycinates 24, three 2-{[4-arylidene-1′-(2-chloroethyl)-5′-nitro-5-oxo-4,5-dihydro-1H,1′H-(2,2′-biimidazol)-1-yl]amino}acetohydrazides 25, three 2-{[4-arylidene-1′-(2-chloroethyl)-5′-nitro-5-oxo-4,5-dihydro-1H,1′H-(2,2′-biimidazol)-1-yl]glycyl}hydrazine-1-carbothioamides 26, three 5′-arylidene-1-(2-chloroethyl)-5-nitro-3′-{[(5-sulfanyl-1,3,4-oxadiazol-2-yl)methyl]amino}-3′,5′-dihydro-1H,4′H-(2,2′-biimidazol)-4′-ones 27, three 3′-{[(4-amino-5-sulfanyl-4H-1,2,4-triazol-3-yl)methyl] amino}-5′-arylidene-1-(2-chloroethyl)-5-nitro-3′,5′-dihydro-1H,4′H-(2,2′-biimidazol)-4′-ones 28, three 5′-arylidene-1-(2-chloroethyl)-5-nitro-3′-{[(5-sulfanyl-1,3,4-thiadiazol-2-yl)methyl]amino}-3′,5′-dihydro-1H,4′H-(2,2′-biimidazol)-4′-ones 29, and three 5′-arylidene-1-(2-chloroethyl)-5-nitro-3′-{[(5-sulfanyl-4H-1,2,4-triazol-3-yl)methyl]amino}-3′,5′-dihydro-1H,4′H-[2,2′-biimidazol]-4′-ones 30.
The synthesis of these compounds was achieved as shown in Scheme 5. In particular, the reaction of compound 1 with SOCl2 provided 1-(2-chloroethyl)-2-methyl-5-nitro-1H-imidazole 31, which was treated with 1 eq of KMnO4 and 1 eq of NaHCO3 in water under reflux giving rise to carboxylic acid 32 in 55% yield. The latter compound was treated with an equimolar amount of SOCl2 in benzene under reflux to give the 5-chloroformyl derivative 33 in 90% yield. The subsequent reaction of 33 with an equimolar amount of glycine (34) in the presence of a 10% aqueous solution of NaOH gave compound 35 in 80% yield. Compound 35 was then treated with an equimolar amount of aryl aldehydes 36 in a mixture of Ac2O and AcOH providing compounds 21 in yields ranging from 51 to 57%. The next step involved reaction with a large molar excess of 99% hydrazine hydrate in dry pyridine under reflux for 20 h gave compounds 22 in 35–47% yields, which by treatment with equimolar amounts of aryl aldehydes 36 in EtOH under reflux produced Schiff’s bases 23 in 68–86% yield. On the other hand, the reaction of compound 21 with an equimolar amount of sodium in absolute EtOH for 2 h followed by addition of an equimolar amount of ethyl bromoacetate (37) and heating the resulting mixture under reflux for 5 h afforded compounds 24 in 56–66% yield. Acetohydrazides 25, which were obtained in 64–75% yield by reaction of compounds 24 with an equimolar amount of 99% hydrazine hydrate in EtOH under reflux for 8 h, were converted to compounds 27 in 63–69% yield by treatment with equimolar amounts of CS2 and KOH in EtOH under reflux for 3 h. Instead, the reaction of compounds 25 with an equimolar amount of CS2, followed by treatment of the resulting crude products with 99% hydrazine hydrate afforded hybrids 28 in yields ranging from 48 to 55%. Compounds 24 were also converted in 35–42% yield to compounds 26 by treatment with thiosemicarbazide and the reaction of the latter derivatives with 4% aqueous NaOH for 4 h, followed by acidification with conc. HCl, provided hybrids 30 in yields ranging from 43 to 61%. Instead, the reaction of compounds 26 with cold conc. sulfuric acid at room temperature for 24 h afforded hybrids 29 in yields ranging from 62 to 78% (Scheme 5). Unfortunately, both the structures in the Schemes and IUPAC names in the original paper were incorrectly referred to as derivatives of a regioisomer of 1 [31].
Scheme 5.

Synthesis of hybrids 21–30 starting from metronidazole (1).
Hybrids 21–30 were tested for their antibacterial activity by the agar disc-diffusion method against S. aureus, E. coli, and Proteus mirabilis, a facultative anaerobic Gram-negative bacterium, at a concentration of 1 mM in DMSO and it was found that: (i) all tested compounds except 23b (Ar = 4-nitrophenyl, Ar1 = 3-nitrophenyl) displayed activity against P. mirabilis; (ii) all tested compounds except 22a (Ar = 4-nitrophenyl) and 23d (Ar = 4-bromophenyl, Ar1 = p-tolyl) were active towards E. coli; and (iii) several hybrids including 29a (Ar = 4-nitrophenyl) exhibited high inhibition potency against S. aureus [31].
Still in 2009, Rawat and coworkers [32] conducted a study concerning the development of compounds capable of overcoming the phenomenon of resistance to metronidazole (1) [33,34] and in this context they synthesised metronidazole/1,2,3-triazole conjugates 38 and tested the antibacterial activity of these compounds against Gram-negative E. coli and P. aeruginosa and Gram-positive S. aureus. Compounds 38 were synthesised via the route shown in Scheme 6, in which the key intermediate, 1-(2-azidoethyl)-2-methyl-5-nitro-1H-imidazole (39), was prepared in two steps from 1. The subsequent reaction of 39 with the appropriate 1-alkynes 40 in a 1:1 mixture of t-BuOH and water in the presence of sodium ascorbate and CuSO4 ·5H2O afforded hybrids 38 in yields ranging from 52 to 67%.
Scheme 6.

Synthesis of metronidazole/1,2,3-triazole conjugates 38.
{1-[2-(2-Methyl-5-nitro-1H-imidazol-1-yl)ethyl]-1H-1,2,3-triazol-4-yl}methanol (38a) (R = H) and compound 38o (R = p-ClC6H4) were found to exhibit potent activity against E. coli and P. aeruginosa. The IC50 values of 38a against these bacteria were of 360 and 710 nM, respectively, and the MIC values of 38o were 8 and 55 nM, respectively. Tetracycline, which was used as a reference drug, possessed IC50 values of 200 and 140 nM, respectively, against these two bacterial strains. Interestingly, compound 38a also exhibited antibacterial activity against S. aureus and S. epidermidis with IC50 values of 1.4 μM. Potent activity against E. coli and P. aeruginosa (IC50 = 200 nM) was also displayed by compound 38l (R = 4-(CHO)C6H4), but it was inactive against Gram-positive bacteria. Compounds 38b (R = Me), 38c (R = Ph), 38d (R = 2-MeC6H4), 38i (R = 2-(NO2)C6H4), 38j (R = 4-(NO2)C6H4), 38f (R = 2-MeC6H4), 38h (R = 3-MeC6H4), 38k (R = 2-(CHO)C6H4), 38m (R = 4-(MeCO)C6H4), and 38n (R = 2-ClC6H4) had no activity towards the tested Gram-positive and Gram-negative bacteria [32].
In 2010, Saadeh, Mubarak, and coworkers [35] investigated the antimicrobial activity of some 5-nitroimidazole/3-sulfanyl-1,2,4-triazole hybrids of general formula 41, which were synthesised via the route shown in Scheme 7 starting from metronidazole tosylate (42) [36] and 1,2,4-triazole-3-thiones 43 [37].
Scheme 7.

Synthesis of 5-nitroimidazole/3-sulfanyl-1,2,4-triazole hybrids 41a–e and 5-nitroimidazole/1,2,4-triazole-3-thione hybrids 44a–e.
Specifically, compound 42 was treated with compounds 43 in DMF at 75–80 °C in the presence of K2CO3 and KI affording a mixture of 5-nitroimidazole/3-sulfanyl-1,2,4-triazole hybrids 41 and 5-nitroimidazole/1,2,4-triazole-3-thione hybrids 44 in 26–32% and 10–14% yield, respectively. These compounds, which were separated by flash chromatography, were tested for their antibacterial activity against bacterial species including P. aeruginosa, E. coli, S. aureus, and Clostridium sporogenes, a Gram-positive anaerobic bacterium, and it was found that many of them had not detectable activity at concentration as high as 0.5 mg/cm3. Nevertheless, hybrids 41c and 41e turned out to exhibit antibacterial activity against C. sporogenes with MIC values of 18 ± 6 and 17 ± 6 µM, respectively, lower than that of metronidazole (1) (43 ± 16 µM), which was used as the reference compound [35].
Still in 2010, Sahu and coworkers reported that (S)-N-{[3-(3-fluoro-4-{4-[2-(2-methyl-5-nitro-1H-imidazol-1-yl)ethyl]piperazin-1-yl}phenyl)-2-oxooxazolidin-5-yl]methyl}acetamide (45), a 5-nitroimidazole/oxazolidinone hybrid, was a potent antibacterial compound against Bacillus cereus MTCC 430, a facultative anaerobic Gram-positive bacterium, with an MIC value of 200 nM [38]. Compound 45 was synthesised in 59% yield by reaction of the piperazine derivative 46 with 1-bromomethyl-2-methyl-5-nitro-1H-imidazole (47) in Et3N at 0 °C in the presence of hydroxybenzotriazole (HOBt) and (3-dimethylaminopropyl)-1-ethylcarbodiimide (EDC) (Scheme 8) [38].
Scheme 8.

Synthesis of (S)-N-{[3-(3-fluoro-4-{4-[2-(2-methyl-5-nitro-1H-imidazol-1-yl)ethyl]-piperazin-1-yl}phenyl)-2-oxooxazolidin-5-yl]methyl}acetamide (45).
In 2011, Foroumadi and coworkers [39] synthesised hybrid (S)-N-{[3-[3-fluoro-4-[4-[5-(1-methyl-5-nitro-1H-imidazol-2-yl)-1,3,4-thiadiazol-2-yl]-1-piperazinyl]phenyl-2-oxooxazolidin-5-yl]methyl}acetamide (48) and described the experimental details of the preparation of the key intermediate of this hybrid, the piperazine derivative (S)-46, starting from 3,4-difluoronitrobenzene (49). Scheme 9 shows the synthetic procedure used for the synthesis of (S)-46 from 49. The latter compound was converted in three steps and 44% yield to benzyl 4-(4-{[(benzyloxy) carbonyl]amino}-2-fluorophenyl)piperazine-1-carboxylate (50). The subsequent reaction of 46 with 1.6 M n-BuLi–hexane in THF at −78 °C for 1.5 h followed by addition of commercially available (R)-glycidyl butyrate (51), stirring of the resulting mixture at room temperature for 3.5 h, addition of saturated aqueous NH4Cl, and extraction with AcOEt provided compound 52 in 82% yield.
Scheme 9.

Synthesis of (S)-N-({3-[3-fluoro-4-(piperazin-1-yl)phenyl]-2-oxooxazolidin-5-yl} methyl)acetamide hydrochloride (S)-(46).
Such intermediate was then treated with methanesulfonyl chloride in CH2Cl2 in the presence of Et3N at 0 °C for 1.5 h and at room temperature for 3 h and then with potassium phthalimide (53) in MeCN under reflux for 48 h affording phthalimide 54 in 66% yield. Deprotection of the latter compound by treatment with 40% MeNH2 in water and EtOH under reflux for 6.5 h followed by reaction with Ac2O and pyridine provided benzyl (S)-4-{4-[5-(acetamidomethyl)-2-oxooxazolidin-3-yl]-2-fluorophenyl}piperazine-1-carboxylate (55) in 33% yield. The next step involved the reaction of 55 with a catalytic amount of 10% Pd on carbon in MeOH and CH2Cl2 under hydrogen atmosphere for 48 h, which gave rise to compound (S)-46 in 91% yield (Scheme 9) [39]. Finally, compound (S)-46 was converted to target (S)-48 in 80% yield by treatment with chlorothiadiazole 19 in EtOH in the presence of Et3N (Scheme 10) [39].
Scheme 10.

Synthesis of (S)-N-{[3-[3-fluoro-4-[4-[5-(1-methyl-5-nitro-1H-imidazol-2-yl)-1,3,4-thiadiazol-2-yl]-1-piperazinyl]phenyl-2-oxooxazolidin-5-yl]methyl}acetamide (S)-(48) from (S)-46 and chlorothiadiazole 19.
Foroumadi and coworkers also tested the antibacterial activity of hybrid (S)-48 and discovered that this compound displayed potent activity at non-cytotoxic concentrations that was higher than that of the reference drugs against the tested Gram-positive bacteria, i.e., B. subtilis PTCC 1023, Corynebacterium glutamicum ATCC 13032, Micrococcus luteus ATCC 9341, MRSA 3, MRSA 5, MRSA 17, Staphylococcus aureus ATCC 6538p, S. lentus ATCC 29070, S. saprophyticus ATCC 15305, S. warneri ATCC 27836, and S. xylosus ATCC 29971 [39]. For example, (S)-48 had MIC values of 180 nM against B. subtilis PTCC 1023 and against S. aureus ATCC 6538p, of 44 nM against MRSA 17 and S. saprophyticus ATCC 15305, and of 11 nM against S. warneri ATCC 27836 [39].
Still in 2011, Foroumadi and coworkers reported the synthesis and in vitro antibacterial activity of three new 2-[(1-methyl-4-nitro-1H-imidazol-5-yl)sulfonyl]-1,3,4-thiadiazoles 56 [40]. These hybrids were prepared via the route outlined in Scheme 11, in which 4(5)-bromo-5(4)-nitro-1H-imidazole (57) [41] was treated with diazomethane in Et2O affording a mixture of 4-bromo-1-methyl-5-nitro-1H-imidazole (58) and 5-bromo-1-methyl-4-nitro-1H-imidazole (59), in which the latter compound was the major component. Treatment of 59 with the appropriate 1,3,4-thiadiazole-2-thiols 60 in EtOH under reflux in the presence of KOH afforded compounds 61. Finally, a mixture of 61 and 3 eq of m-chloroperbenzoic acid and 3 eq of NaHCO3 in CH2Cl2 was stirred at room temperature for three days providing the required hybrids 56a–c in 82–85% yield.
Scheme 11.

Synthesis of 2-[(1-methyl-4-nitro-1H-imidazol-5-yl)sulfonyl]-1,3,4-thiadiazoles 56a–c.
It was then found that 2-[(1-methyl-4-nitro-1H-imidazol-5-yl)sulfonyl]-5-(5-nitrofuran-2-yl)-1,3,4-thiadiazole (56b) and 2-[(1-methyl-4-nitro-1H-imidazol-5-yl)sulfonyl]-5-(1-methyl-5-nitro-1H-imidazol-2-yl)-1,3,4-thiadiazole (56c) possessed antibacterial activity against K. pneumoniae with MIC values of 41 and 80 μM, respectively, and E. coli with an MIC value of 41 and 40 μM, respectively. However, compounds 56b and 56c turned out to be less active than the reference drug norfloxacin (780 and 410 nM against K. pneumoniae and E. coli, respectively). Furthermore, N-{5-[(1-methyl-4-nitro-1H-imidazol-5-yl)sulfonyl]-1,3,4-thiadiazol-2-yl}acetamide (56a) showed no activity against Gram-positive and Gram-negative bacteria [40].
In 2012, two novel series of 5-nitroimidazole/1,3,4-oxadiazole hybrids of general formula 62 and 63 were synthesised from Gong, Zhu, and coworkers starting from commercially available 2-methyl-5-nitro-1H-imidazole 64 using the protocol depicted in Scheme 12 [42]. Compound 65, which was obtained by reaction of 64 with ethyl chloroacetate (66) in acetone in the presence of K2CO3, was converted to 2-(2-methyl-5-nitro-1H-imidazol-1-yl)acetohydrazide (67) by hydrazinolysis in MeOH. Subsequent treatment of 67 with an equimolar amount of benzoic acids 68a–i in phosphoryl chloride under reflux for 10–16 h provided hybrids 62a–i. Instead, the reaction of 67 with phenylacetic acids 69 in refluxing POCl3 gave hybrids 63. Unfortunately, the yields of hybrids 62 and 63 were not reported [42].
Scheme 12.

Synthesis of 5-nitroimidazole/1,3,4-oxadiazole hybrids 62a–i and 63a–i.
All these hybrids were then screened for their antibacterial activities against two Gram-negative bacterial strains, E. coli and P. aeruginosa, and two Gram-positive bacterial strains, B. subtilis and S. aureus, by the MTT assay [43]. Hybrids 62e, 62h, and 62i were found to possess significant antibacterial activities with MIC values of 4.9–17 µM against E. coli ATCC 35128, but hybrids 63e, 63h, and 63i displayed less potent activity with MIC values of 42–160 µM. Compounds 62h and 62i turned out to be the most potent among the tested hybrids [42].
In 2013, Geng, Zhou, and coworkers [44] synthesised, characterised, and evaluated the antimicrobial activity of a series of hybrids of nitroimidazoles and berberine (70) (Figure 5), the major isoquinoline alkaloid isolated from a number of medicinal plants including Coptis chinensis Franch. and Berberis spp. [45] that has been widely used as a drug for treatment of many diseases and exhibits antibacterial activity against MRSA [46], E. coli [47], S. epidermidis [48], Vibrio vulnificus [49], Streptococcus agalactiae [47], Acromonas hydrophila [47], and Edwardsiella ictaluri [47].
Figure 5.

Structures of berberine (70) and hybrids 71.
As shown in Scheme 13, the target berberine/nitroimidazole hybrids 71a–k (Figure 5) were prepared from commercially available halobenzyl chlorides 72a–e, diethanolamine (73), and berberrubine (74), one of the major metabolites of the naturally occurring alkaloid berberine with appreciable anti-ulcerative colitis effect [47].
Scheme 13.

Synthesis of nitroimidazole/berberine hybrids 71a–k.
N-Benzylation of diethanolamine with compounds 72 produced intermediates 75, which were treated with PBr3 in CHCl3 to afford dibromides 76. The subsequent reaction between 1 eq of compounds 76 and 1 eq of 4-nitro-1H-imidazole or 2-methyl-5-nitro-1H-imidazole in MeCN at 60 °C in the presence of 2 eq of K2CO3 provided compounds 77a–k in modest to good yields. Finally, target hybrids 71a–k were obtained in 23–39% yields by treatment of compounds 77 with 0.98 eq of berberrubine (74) at 110 °C in DMF for 20 h. Compound 74 was in turn obtained in 88% yield by demethylation of berberine (70) at 190 °C under reduced pressure for 15 min [44].
Notably, most of the target hybrids turned out to exhibit effective antibacterial activity against Gram-positive and Gram-negative bacterial strains and their potency against MRSA was superior or comparable to that of reference drugs norfloxacin, chloramphenicol [50], and berberine. In particular, hybrid 71g had low inhibitory concentration towards the Gram-negative bacteria Shigella dysenteriae and Proteus vulgaris ATCC 6896 with MIC values of 6 µM for both bacteria. Instead, the MIC values for the reference drug chloramphenicol were 100 µM towards both bacterium species, while the MIC values for norfloxacin towards S. dysenteriae and P. vulgaris were 13 and 25 µM, respectively. It is also worth noting that hybrids bearing 4-nitroimidazole moiety, such as 71b and 71f displayed antimicrobial activities lower than those of hybrids bearing 5-nitroimidazole moiety such as 71g [44].
Still in 2013, Zhu and coworkers reported that the Schiff’s base derivative 78 bearing a 5-nitroimidazole moiety exhibited effective inhibitory activity towards E. coli ATCC 35218 (MIC = 7.1 μM) that was similar to that of both standards used, kanamycin B and penicillin G (6.5 and 9.4 μM, respectively) [51]. (E)-N’-[1-(4-Bromophenyl)-2-(2-methyl-5-nitro-1H-imidazol-1-yl)ethylidene] isonicotinohydrazide (78) was synthesised via a two-step protocol in which the first step involved the preparation of the key intermediate, 1-(4-bromophenyl)-2-(2-methyl-5-nitro-1H-imidazol-1-yl)ethan-1-one (79) by treatment of 2-methyl-5-nitro-1H-imidazole (64) with 2-bromo-1-(4-bromophenyl)ethan-1-one (80) in MeCN in the presence of K2CO3 and phase transfer catalyst tetra-n-butylammonium bromide (TBAB), and the second step involved the Ni(NO3)2·6H2O-catalysed reaction of 79 with hydrazide 81 in EtOH at room temperature (Scheme 14) [51].
Scheme 14.

Synthesis of hybrid 78.
In 2014, Gu and coworkers synthesised methyl (1R,4aS)-7-isopropyl-1,4a-dimethyl-9-[2-(2-methyl-5-nitro-1H-imidazol-1-yl)ethyl]-2,3,4,4a,9,13c-hexahydro-1H-dibenzo[a,c]carbazole-1-carboxylate (82), a novel hybrid of 2-methyl-5-nitro-1H-imidazole (64) and a 1H-dibenzo [a,c]carbazole derivative, from dehydroabietic acid (83) (Scheme 15) and evaluated the antimicrobial activity of this hybrid against Gram-positive bacteria B. subtilis CGMCC 1.1162 and S. aureus CGMCC 1.1361 and Gram-negative bacteria E. coli CGMCC 1.1571 and Pseudomonas fluorescens CGMCC 1.182086 [52]. The developed synthetic route involved the conversion of naturally occurring 83, a compound of which many derivatives have been shown to possess interesting biological activities [53,54], to intermediate 84 as described in the literature [55]. N-Alkylation of the latter compound by treatment with a large molar excess of 1,2-dibromoethane (85) in the presence of NaOH and TBAB afforded the N-bromoethyl carbazole derivative 86 in 56% yield. Finally, the target hybrid 82 was obtained in 58% yield by reaction of 86 with 2 eq of nitroimidazole 64 in MeCN under reflux in the presence of 5 eq of K2CO3 and 1 eq of KI.
Scheme 15.

Synthesis of methyl (1R,4aS)-7-isopropyl-1,4a-dimethyl-9-[2-(2-methyl-5-nitro-1H-imidazol-1-yl)ethyl]-2,3,4,4a,9,13c-hexahydro-1H-dibenzo[a,c]carbazole-1-carboxylate (82).
Gu and coworkers then found that hybrid 82 displayed potent antibacterial activity against B. subtilis CGMCC 1.1162 with an MIC value 1.6 μM that was comparable to that of the reference antibiotic amikacin (1.5 μM). Hybrid 82 also turned out to possess significant antibacterial activity against S. aureus CGMCC 1.1361, but low activity against E. coli CGMCC 1.1571 and P. fluorescens CGMCC 1.182086 [52].
In 2015, Sangani, Zhu, and coworkers [56] synthesised Schiff’s base derivatives 87a–j bearing 5-nitroimidazole and pyrazole moieties by Ni(NO3)2·6H2O-catalysed reaction of 5-aryloxypyrazole-4-carbaldehydes 88a–j [57] with 2-(2-methyl-5-nitro-1H-imidazol-1-yl) acetohydrazide (67) [58] in EtOH at room temperature. As shown in Scheme 16, this reaction provided hybrids 87a–j in 75–88% yield.
Scheme 16.

Synthesis of hybrids 87a–j.
These compounds were tested for their antibacterial properties against E. coli ATCC 25922, P. aeruginosa ATCC 27853, B. subtilis ATCC 530, and S. aureus ATCC 25923 and for inhibition of E. coli FabH activity, and many of these hybrids proved to be effective against the applied bacterial strains. Compounds 87h (MIC = 6.2 μM) and 87i (MIC = 6.4 μM) showed higher activity against E. coli than other hybrids and their activities were comparable to that of antibiotic kanamycin B (MIC = 6.5 μM). Furthermore, 87i turned out to show the most effective inhibition (IC50 = 4.6 ± 0.2 μM) by binding into the active site of E. coli FabH receptor with minimum binding energy [56].
Still in 2015, Cooper and coworkers [59], taking into account that resistance to metronidazole (1) had been observed in Gram-positive and Gram-negative anaerobic bacteria [21,56], reasoned that suitable metronidazole derivatives consisting of metronidazole/triazole conjugates (Mtz-triazoles) might exhibit potent activity against anaerobic bacteria for which compound 1 is used as a treatment. Thus, they synthesised metronidazole/triazole conjugates 89 via the route shown in Scheme 17.
Scheme 17.

Synthesis of metronidazole/triazole conjugates 89 and 1-(prop-2-yn-1-yl)-1H-pyrazole (90g).
Specifically, azide 39, which was prepared in high yield from mesylate prepared from 1, was submitted to a CuSO4·5H2O/sodium ascorbate-catalysed 1,3-dipolar cycloaddition to a library of 1-alkynes 90 in MeOH at 45 °C, affording Mtz-triazoles 89 in yields ranging from 9 to 97%. All 1-alkynes were commercially available with the exception of 90g, which was prepared in 16% yield by reaction of pyrazole (91) with propargyl bromide (92) and K2CO3 in toluene in the presence of TBAB (Scheme 17) [59].
Compounds 89 were then tested for their activity under aerobic conditions against C. difficile, microaerophilic H. pylori, and other microorganisms including ESKAPE pathogens (MRSA, E. coli, K. pneumoniae, P. aeruginosa, and A. baumannii). For example, the MIC values of Mtz-triazoles 89a (R = Ph), 89b (R = 4-MeOC6H4), 89j (R = 2-pyridyl), 89n (R = 3-thienyl), and 89t (R = CH2NH2) were higher than 100 μM against the representative ESKAPE pathogens, but 89a and 89q (R = CH2OH) had MIC values higher than 210 μM against a panel of eight additional drug resistant S. aureus strains, vancomycin resistant E. faecium, and multidrug-resistant, penicillin non-susceptible Gram-positive Streptococcus pneumoniae. It is worth noting that this lack of activity contrasted with what was reported in 2009 by Rawat and coworkers [32], but it was consistent with the reported inactivity of 378 Mtz-triazoles against E. coli that was observed in the study of Eckmann and coworkers [48]. It must also be pointed out that: (i) several metronidazole/triazole conjugates 89 displayed potent activity against multiple strains of C. difficile, including two major pathogenic strains of NAP1/027 and a VPI 10463 strain associated with epidemics; and (ii) cross-resistance to metronidazole was observed against stable metronidazole resistant C. difficile strains [59].
In a study carried out in 2015, Geng, Zhou, and coworkers synthesised and characterised quinolone-based metronidazole derivatives 93a–l and tested these hybrids for their antibacterial activities towards the Gram-negative bacteria E. coli, Proteus hauseri [60], P. aeruginosa, and Salmonella enterica and the Gram-positive bacteria MRSA, S. aureus, B. subtilis, and M. luteus [61].
As shown in Scheme 18, compounds 93 were synthesised by N-alkylation of commercially available 4-quinolone-2-carboxylic acids 94a–c with 2-(chloromethyl)oxirane (95) in MeCN at room temperature, followed by treatment of the resulting compounds 96 with formic acid to adjust the pH value to 5.5–6.5 and by addition of an equimolar amount of 2-methyl-5-nitro-1H-imidazoles 97 in MeCN at 80 °C for 20 h in the presence of K2CO3. Hybrids 93a–l were so obtained in yields ranging from 20 to 32% [61].
Scheme 18.

Synthesis of quinolone/imidazole hybrids 93a–l.
It is noteworthy that most of them exhibited good antibacterial activity towards the above-mentioned Gram-positive and Gram-negative bacteria, and that 8-chloro-1-cyclopropyl-6-fluoro-7-(3-{[2-hydroxy-3-(2-methyl-5-nitro-1H-imidazol-1-yl)propyl]amino}pyrrolidin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (93i) (Figure 6) possessed antibacterial activity against Gram-negative P. aeruginosa with an MIC value of 460 nM, which was lower than that of the reference drugs chloramphenicol [50] (50 μM), norfloxacin (13 μM), ciprofloxacin (3 μM), and clinafloxacin (3 μM) [61].
Figure 6.

Structure of hybrid 93i.
The interaction of hybrid 93i with P. aeruginosa DNA and Cu2+ ion was also investigated, and it was established that 93i could intercalate into P. aeruginosa DNA through Cu2+ ion bridge to form a steady 93i-Cu2+-DNA ternary complex, which might further block DNA replication to exert the powerful bioactivities [61].
In a study carried out in 2015 by Hurdle and coworkers, the synthesis of twenty-six N-substituted metronidazole/tetramic acid hybrids 98 was reported, and clear evidence was provided that the localisation of metronidazole by a lactobacilli-inspired tetramic acid motif improves treatment outcomes in the hamster model of C. difficile infection [62]. Four leads, i.e., 98a (R = isobutyl), 98b (R = 4-phenylbenzyl), 98c (R = 1-naphthylmethyl), and 98d (R = 1-methyl-3-(1H)-indolylmethyl), were found to be more effective than metronidazole (1) in C. difficile-infected animals retaining the mode of action of metronidazole and demonstrated lack of propensity for de novo resistance. This study also suggested a role for the tetramic acid motif for colon-specific drug delivery. Furthermore, the structure-activity relationship of metronidazole/tetramic acid hybrids showed that substitution at the 5-position of the tetramic core was an important factor for activity, as the hybrid lacking a 5-substituent, i.e., 98e (R = H), was, on a molar basis, more than 80 times less active than 98a [62].
Compounds 98 were synthesised via the route shown in Scheme 19.
Scheme 19.

Synthesis of N-substituted metronidazole/tetramic acid hybrids 98.
In particular, α-aminoacid methyl ester hydrochlorides 99 were treated with 2-nitrobenzenesulfonyl chloride (nosyl chloride) (100) in CH2Cl2 in the presence of Et3N, and the resulting compounds 101 treated with metronidazole (1), PPh3, and diethyl azodicarboxylate (DEAD) in THF under N2 atmosphere to afford compounds 102. These crude intermediates were treated with a large molar excess of 4-methoxybenzenethiol (103) in CH2Cl2 in the presence of K2CO3 and the resulting crude compounds 104 treated with 2,2,6-trimethyl-4H-1,3-dioxin-4-one (105) in dry toluene under reflux for 2 h to give compounds 106. Finally, treatment of 106 with 2 eq of Amberlyst A-26 (OH) resin in dry MeOH overnight at room temperature under N2 atmosphere provided the required hybrids 98 in yields ranging from 19 to 67% (Scheme 19) [62].
Still in 2015, Crossley and coworkers developed a successful methodology for the synthesis of amino-acid linked porphyrin-nitroimidazole antibiotics targeting Porphyromonas gingivalis [63], the Gram-negative anaerobic periodontal pathogen that requires porphyrin supplementation for growth. The study carried out by these authors involved the synthesis of L-amino acid-linked deuterioporphyrin-nitroimidazole DPIX-Lys(Boc)-Nim adducts 107a and 107b via the reaction sequence shown in Scheme 20. In particular, the reaction of 1.03 eq of aminoimidazole 3 [64] with a solution of 1 eq of Fmoc-Lys(Boc)-OH 108 (1 eq) in DMF and CH2Cl2 in the presence of 3.1 eq of N,N’-diisopropylethylamine (DIPEA) and 1.03 eq of O-(benzotriazol-1-yl)-N,N,N’,N’-tetramethyl uronium hexafluorophosphate (HBTU) at room temperature for 3 h gave Fmoc-Lys(Boc)-Nim 109 in 80% yield. At the same time, DPIX monomethyl esters 110a and 110b were prepared in 49% yield by controlled hydrolysis of DPIX dimethyl ester with 4 M HCl in refluxing methanol. Subsequently, compound 109 was Fmoc deprotected by treatment with 1.2 eq of DBU and the resulting amino derivative was treated with the carboxylic group of monomethyl esters 110 affording adducts 111a and 111b in 90% yield. Finally, treatment of the latter regioisomeric compounds with a molar excess of LiOH in a mixture of MeOH, THF, and water overnight at room temperature provided free carboxylic acids 107a and 107b in quantitative yield (Scheme 20) [63].
Scheme 20.

Synthesis of L-aminoacid-linked porphyrin-nitroimidazole adducts DPIX-Lys(Boc)-Nim adducts 107a and 107b.
Remarkably, DPIX-Lys(Boc)-Pro-Nim adducts 107a and 107b with a proline-lysine linker bridging metronidazole to deuterioporphyrin IX (DPIX 2) retarded the growth but did not kill P. gingivalis at 20 μM and, unlike metronidazole, they did not kill a range of other anaerobic bacteria isolated from the human gastro-intestinal tract [63].
In 2017, Li, Zhou, and coworkers synthesised novel naphthalimide-derived metronidazoles 112a–g and tested their in vitro antibacterial activities towards Gram-positive and Gram-negative bacteria [65]. The synthesis of hybrids 112 was carried out via the reaction sequence shown in Scheme 21, in which commercially available 4-bromo-1,8-naphthalic anhydride (namely, 6-bromo-1H-benzo[de]isoquinoline-1,3(2H)-dione) (113) was the starting material.
Scheme 21.

Synthesis of naphthalimide-derived metronidazoles 112a–g.
Compound 113 was treated with aqueous ammonia producing intermediate 114 in 92% yield. The subsequent reaction of 114 with chloroacetone (115) in DMF in the presence of K2CO3 afforded compound 116 in 62% yield. The latter was treated with bromine in AcOH at 60 °C for 3 h providing compound 117 in 53% yield. The coupling of 117 with 2-methyl-5-nitro-1H-imidazole (64) in DMF in the presence of K2CO3 gave in 64% yield the nitroimidazole derivative 118, which was reduced with NaBH4 in EtOH at 60 °C for 6 h affording compound 119 in a moderate yield. Finally, target hybrids 112a–g were obtained in 40–60% yield by reaction of 119 with amines 120a–g in DMSO at 90 °C for 4 h under N2 atmosphere using K2CO3 as base and Cu2O as catalyst [65].
Among these hybrids, compound 112b (R1 = R2 = H; n = 2) proved capable of not only exhibiting effective inhibition towards the growth of both P. vulgaris (MIC = 2 μM), a Gram-negative bacterium that inhabits the intestinal tract of humans and animals, and S. dysenteriae (MIC = 10 μM), a Gram-negative fluoroquinolone-resistant bacterium that can cause shigellosis (bacillary dysentery), but also of rapidly killing the tested strains and to prevent development of bacterial resistance. Interestingly, hybrid 112b also proved capable of intercalating into calf thymus DNA to form a steady supramolecular complex, which might block DNA replication to display the antibacterial activity and being effectively transported by human serum albumin [65].
In 2018, Yang, Luo, and coworkers [66], considering that indolin-2-ones are known to possess antibacterial activity [67,68], synthesised (E)-3-[(1-methyl-5-nitro-1H-imidazol-2-yl)methylene] indolin-2-one (121), a hybrid of indolin-2-one and 1-methyl-5-nitro-1H-imidazole. Such a compound was obtained in 69% yield by reaction of indolin-2-one (122) with 1.2 eq of 1-methyl-5-nitro-1H-imidazole-2-carbaldehyde (10) and 1.5 eq of piperidine in MeOH under reflux (Scheme 22).
Scheme 22.

Synthesis of (E)-3-[(1-methyl-5-nitro-1H-imidazol-2-yl)methylene]indolin-2-one (121) and (E)-3-[(1-methyl-5-nitro-1H-imidazol-2-yl)methylene]-5-nitroindolin-2-one (123).
Hybrid 121 was found to exhibit antibacterial activity against S. aureus strains with MIC values of 7 μM and 4 μM on methicillin-sensitive S. aureus ATCC 25923 and MRSA ATCC 33591, respectively [66].
The protocol employed for the synthesis of 121 was then used for the synthesis of (E)-3-[(1-methyl-5-nitro-1H-imidazol-2-yl)methylene]-5-nitroindolin-2-one (123) in 75% yield from 10 and 5-nitroindolin-2-one (124) (Scheme 22). Hybrid 123 was found to possess remarkable antibacterial activity against E. coli ATCC 25922, P. aeruginosa ATCC 27853, and vancomycin-resistant Enterococcus strain B148, and its efficacy against ATCC 25923 was, on a molar basis, 18 times higher than that of 121 [66].
In 2018, Cai, Zhou, and coworkers synthesised berberine/nitroimidazole hybrids 125 via the reaction sequence outlined in Scheme 23, in which berberine (70) was the starting material [69]. Berberrubine (74), which was obtained in 90% yield by selective demethylation of 70, was reduced with NaBH4 in MeOH generating compound 126 in 57% yield. Formylation of 126 by treatment with HMTA in trifluoroacetic acid produced aldehyde 127, which was condensed with 2-(2-methyl-4-nitro-1H-imidazol-1-yl)acetonitrile 128 in the presence of piperidine affording compound 129. Finally, O-alkylation of the latter compound with alkyl/aryl halides 130 provided hybrids 125 in yields ranging from 13 to 50%.
Scheme 23.

Synthesis of berberine/nitroimidazole hybrids 125.
Cai, Zhou, and coworkers next evaluated the antibacterial activities of hybrids 125 against MRSA, E. faecalis, S. aureus, S. aureus ATCC 25923, B. subtilis ATCC 6633, M. luteus ATCC 4698, K. pneumoniae, E. coli, E. coli ATCC 25922, P. aeruginosa, P. aeruginosa ATCC 27853, and A. baumannii and found that, among these compounds, tetrahydroberberine-9-(2-fluorobenzyl)-12-[2-(2-methyl-5-nitroimidazolyl)acrylonitrile (125p) (R1 = 2-FC6H4, n = 1) a berberine/nitroimidazole hybrid with an improved aqueous solubility (4.54 ± 0.08 μg/mL), not only exhibited strong antibacterial activity against drug resistant E. coli with an MIC value of 3 µM and was 33 times more potent than norfloxacin, but also displayed low toxicity towards RAW 264.7 mouse cancer cells. Cai, Zhou, and coworkers also investigated the antibacterial mechanism and found that compound 125p might target E. coli DNA polymerase II through the formation of hydrogen bonds, effectively permeability-resistant E. coli cell membrane, and intercalate into DNA isolated from resistant E. coli to form 125p–DNA complex, thus blocking DNA replication [69]. They also reported that resistance in E. coli, Klebsiella spp., and Enterobacter spp. is mainly related to the production of extended-spectrum β-lactamase, but other resistance mechanisms are also possible [69].
Still in 2018, studies on the synthesis and evaluation of the antimicrobial activity of molecular hybrids and conjugates containing nitroimidazole moiety were also carried out by other research teams. Kumar and coworkers [70], in continuation of their studies on the synthesis of new anti-tubercular scaffolds [71,72], synthesised nitroimidazole/7-chloroquinoline conjugates 131a–d (Scheme 24) and assessed both their activities towards Mycobacterium tuberculosis and their cytotoxicity towards the J774 murine macrophage cell line.
Scheme 24.

Synthesis of nitroimidazole/7-chloroquinoline conjugates 131a–d.
The synthesis of compounds 131 began with the preparation of 4-piperazinyl-7-chloroquinoline (132) by the site-selective reaction of 4,7-dichloroquinoline (133) with a molar excess of piperazine (134) in Et3N at 120 °C for 10–12 h [73]. Subsequently, a solution of 1 eq of 132 was treated at 0 °C with a suspension of 2 eq of NaH in DMF, followed by the addition of 1.3 eq of 1-(bromoalkyl)-2-methyl-5-nitro-1H-imidazoles 135. The resulting mixture was stirred at room temperature for 2 h affording conjugates 131a–d in 62–68% yield. Compounds 135 were in turn prepared by reaction of 1 eq of 2-methyl-5-nitro-1H-imidazole (64) with 1.2 eq of the required 1,ω-dibromoalkanes 136 in DMF at room temperature in the presence of 2 eq of K2CO3 [70].
The anti-mycobacterial properties of conjugates 131 were subsequently evaluated, and it was found that the activity of these compounds was not higher than that of the standard drug isoniazid. Nevertheless, these compounds had appreciable activity with minimal cytotoxicity. 7-Chloro-4-{4-[4-(2-methyl-5-nitro-1H-imidazol-1-yl)butyl]piperazin-1-yl}quinoline (131b) proved to be the most potent among these conjugates with a IC50 value of 5.1 µM [70].
In the same year, Zhou and coworkers [74] investigated the antibacterial activities of naphthalimide/nitroimidazole hybrids 137a–d and 138a–j towards resistant A. baumannii, a clinically important nosocomial aerobic Gram-negative pathogen that can cause various infective diseases [75]. The synthesis of hybrids 137 and 138 was designed taking into account that: (i) naphthalimides can effectively inhibit the bacterial growth and that these compounds, in combination with nitrogen heterocycles, exhibit potent antimicrobial activities also towards bacterial resistant strains [76,77,78,79]; and (ii) nitroimidazoles are a class of antimicrobial drugs that have a remarkable spectrum of activity against anaerobic Gram-positive and Gram-negative bacteria [16].
Naphthalimide/nitroimidazole hybrids 137a–d and 138a–j were synthesised in yields ranging from 30 to 60% by the route outlined in Scheme 25. In particular, compound 119, which was prepared from commercially available 113 according to the protocol illustrated in Scheme 21, was treated with alicyclic amines 139a–d in 2-methoxyethanol under reflux for 4 h to afford hybrids 137a–d. Instead, hybrids 138a–j were prepared by reaction between 119 and N-alkylpiperazines 140 in 2-methoxyethanol under reflux and N2 atmosphere [74].
Scheme 25.

Synthesis of naphthalimide/nitroimidazole hybrids 137a–d and 138a–j.
Hybrids 137 and 138 were then tested for their in vitro antibacterial activities against five Gram-positive bacterial strains (MRSA, S. aureus, S. aureus ATCC 25923, S. aureus ATCC 29213, and E. faecalis) and six Gram-negative bacterial strains (E. coli, E. coli ATCC 25922, K. pneumoniae, P. aeruginosa, P. aeruginosa ATCC 27853, and A. baumannii). The obtained results showed that these hybrids were able to inhibit the growth of the tested strains and that their effectiveness, except that of compound 137a, was better than that of compound 119. It was also found that hybrid 138e showed high antibacterial activity (MIC = 13 µM) against resistant A. baumannii with rapid killing effect and no obvious resistance development [74]. Furthermore, it was discovered that when 138e was used in combination with chloramphenicol [50], norfloxacin, or clinafloxacin, the antibacterial potency of this compound against resistant A. baumannii was improved [74].
In 2019, Zang, Zhang, Zhou, and coworkers turned their attention to the development of novel compounds possessing the ability to combat the resistance of Gram-positive pathogen MRSA [80]. The resistance is due to the acquisition of mecA gene, which, unlike of any PBP (penicillin binding protein) normally produced by S. aureus, encodes the protein PBP2a that has a low affinity for β-lactam antibiotics such as penicillin and methicillin [80]. In particular, the above-mentioned researchers directed their efforts towards the development of novel structural candidates of enone-bridged indole/nitroimidazole scaffolds. In fact, satisfactory antimicrobial activity of this type of conjugates had already been highlighted [81]. Furthermore, it was kept in mind that α,β-unsaturated carbonyl derivatives are linkers commonly used as functional structures for drug design [82].
The target enone bridged indole/nitroimidazole conjugates 141a–c, 142a–i, and 143a–f were synthesised as outlined in Scheme 26. In particular, 2-methyl-5-nitro-1H-imidazole (64) was treated with chloroacetone (115) in the presence of K2CO3 to give compound 144 in 77% yield. The reaction of 144 with 1H-indoles-3-carbaldehydes 145a–c in toluene using piperidine and AcOH as catalysts afforded compounds 146a–c, which were used as key intermediates in the synthesis of conjugates 141, 142, and 143. Compounds 145a–c were in turn obtained by the Vilsmeier-Haack reaction of indoles 147a–c. Treatment of intermediates 146 with the chloroacetyl derivatives 148 and 149 in MeCN in the presence of K2CO3 afforded conjugates 142 and 143, respectively. Compounds 143a–f were obtained in 36–48% yield and compounds 142a–i were prepared in yields ranging from 36 to 72%. Indole/nitroimidazole conjugates 141a–c were instead prepared in satisfactory yields by reaction of 146 with the appropriate alkyl halides 130 in MeCN in the presence of K2CO3 [80].
Scheme 26.

Synthesis of enone bridged indole/nitroimidazole conjugates 141a–c, 142a–i, and 143a–f.
The results of the evaluation of the antimicrobial activity of conjugates 141a–c, 142a–i, and 143a–f showed that most of these conjugates displayed inhibitory efficiency towards Gram-positive bacteria. It was also discovered that (E)-3-(2-methyl-5-nitro-1H-imidazol-1-yl)-4-{1-[2-oxo-2-(piperidin-1-yl)ethyl]-1H-indol-3-yl}but-3-en-2-one (142b) possessed a satisfactory inhibitory activity on MRSA (MIC = 2 μM) and could effectively prevent the development of bacterial resistance, could intercalate into DNA, and also permeate MRSA membrane. It is worth noting that this hybrid also exhibited low cytotoxicity towards normal lung epithelial cell line BEAS-2B [80].
Concluding this section of the review it finally deserves to be mentioned that, in 2016, Kedar and coworkers designed and synthesised a set of new double nitroimidazoles and that one of these compounds, bis[1-(2-methyl-5-nitro-1H-imidazol-1-yl)propan-2-yl]-2,2′-(piperazine-1,4-diyl) diacetate (150) exhibited significant antibacterial activity against E. coli and S. aureus higher than those evaluated for other tested compounds [83].
Compound 150 was synthesised by reaction of chloroacetyl chloride (151) with secnidazole [1-(2-methyl-5-nitro-1H-imidazol-1-yl)propan-2-ol] (152) in pyridine at 0 °C for 5–10 min, followed by treatment of the resulting compound 153 with piperazine (134) in MeOH (Scheme 27). Compound 152 is an antibiotic approved in the USA by the FDA for the treatment of bacterial vaginosis in adult women.
Scheme 27.

Synthesis of double nitroimidazole 150.
The zones of inhibition of 150 against E. coli at the concentrations of 50, 100, 300, and 500 μg/mL were 2, 7, 10, and 14 mm, respectively, while those of ofloxacin, the standard drug, were 13, 16, 24, and 31 mm, respectively. On the other hand, the zones of inhibition of 150 against S. aureus at the concentrations of 100, 300, and 500 μg/mL were 5, 12, and 14 mm, respectively, while those of ofloxacin were 16, 20, and 28 mm, respectively [83].
3. Coumarin/Imidazole Hybrids and Conjugates
Synthetic and naturally occurring coumarins have been the subject of various studies concerning their antibacterial properties [84,85,86,87,88,89,90] and it has been observed that although these compounds do not display relevant activity (MIC ≥ 128 μg/mL), they are capable of modulating the antibiotic resistance through a mechanism that most likely involves inhibition of bacterial pump efflux [91]. Furthermore, in recent years, in order to enhance the antibacterial activities of these substances, coumarin/imidazole hybrids and conjugates have been designed, synthesised, and tested in vitro.
In 2016, Zhou and coworkers [92] synthesised and characterised 7-[2-hydroxy-3-(1H-imidazol-1-yl)propoxy]-4-methyl-2H-chromen-2-ones 154a–e via the route shown in Scheme 28, in which compound 155, which was prepared in two steps form resorcinol (156), was the key intermediate. Treatment of 155 with the appropriate imidazoles 157 in EtOH using K2CO3 as base afforded hybrids 154 in yields ranging from 24 to 61%.
Scheme 28.

Synthesis of hybrids 154a–e.
These compounds were next tested for their antibacterial activities against MRSA, B. subtilis, M. luteus ATCC 4698, E. coli JM 109, P. aeruginosa ATCC 27853, and S. dysenteriae and it was found that 154a–c and 154e did not display significant antibacterial properties. Instead, hybrid 154d exhibited activity against the tested Gram-positive bacteria with an MIC value of 93 μM against MRSA, B. subtilis, and M. luteus. Noticeably, the MIC value of 154d against B. subtilis turned out to be comparable to that of the reference drug chloramphenicol (99 μM) but higher than that of norfloxacin (MIC = 13 μM) [92].
Zhou and coworkers also prepared compounds 158a–e bearing bis-imidazolyl ethanol groups starting from phloroglucinol (159) (Scheme 29) via a three-step route, but found that these substances did not show appreciable antibacterial activity against the above-mentioned bacterial pathogens [92].
Scheme 29.

Synthesis of hybrids 158a–e.
In 2018, Shastri and coworkers [93] synthesised 4-(4,5-diphenyl-1H-imidazol-2-yl)-2H-chromen-2-ones 160a–g in good to excellent yields by cyclocondensation of benzil (161) with 4-formylcoumarins 162a–g [94] and AcONH4 in AcOH using both conventional heating conditions and microwave irradiation (Scheme 30). As expected, the yields of the microwave promoted cyclocondensation reactions were higher than those of the reactions carried out using conventional heating conditions. Hybrids 160a–f were then converted to 4-(4,5-diphenyl-1-tosyl-1H-imidazol-2-yl)-2H-chromen-2-ones 163a–f by treatment with 1.5 eq of TsCl in CH2Cl2 at 0–5 °C in the presence of Et3N (Scheme 30).
Scheme 30.

Synthesis of conjugates 160a–g and 163a–f.
Conjugates 160 and 163 were then evaluated for their antimicrobial activities against Gram-positive Bacillus flexus and Gram-negative Pseudomonas spp. bacterial strains using ciprofloxacin (MIC = 2.1 μM) as the reference drug. Hybrids 163 were found to have MIC values lower than those of the corresponding conjugates 160 and compounds 163d and 163f had an MIC value 300–400 nM against both B. flexus and Pseudomonas spp. Instead, the MIC values the corresponding conjugates 160d and 160f against B. flexus were 0.9 and 1.4 μM, respectively, and against Pseudomonas spp., they were 0.9 and 1.2 μM, respectively [93].
It was finally observed that sodium (Z)-4-(2-hydroxyphenyl)-4-(4,5-diphenyl-1H-imidazol-2-yl)but-3-enoates 164a–f (Figure 7), which were obtained by treatment of conjugates 160a–f with 25% NaOH in EtOH at 80 °C, were, on a molar basis, highly potent and effective as hybrids 163 against both bacterial strains [93].
Figure 7.

Structure of sodium (Z)-4-(2-hydroxyphenyl)-4-(4,5-diphenyl-1H-imidazol-2-yl)but-3-enoates 164a–f.
Still in 2018, Wang and coworkers synthesised numerous coumarin derivatives containing imidazole skeleton and tested twenty-two of these hybrids of general formula 165 for their antibacterial activities against two Gram-negative bacteria, E. coli and Flavobacterium columnare, and two Gram-positive bacteria, S. aureus and S. agalactiae, using the fluoroquinolone antibiotics enrofloxacin and norfloxacin as reference drugs [95]. 7-Hydroxycoumarin (166), which was used as starting material in the synthesis of compounds 165 was prepared in 72% yield by reaction of ethyl chloroacetate (66) with PPh3 in EtOH, followed by treatment with 2,4-dihydroxybenzaldehyde (167) and KOH at 80 °C (Scheme 31).
Scheme 31.

Synthesis of coumarin derivatives 165 containing imidazole skeleton.
The subsequent reaction of 166 with K2CO3 in acetone under reflux in the presence of Et3N, followed by the addition of 2 eq of α,ω-dibromoalkanes 136 provided intermediate coumarin bromides 168. Finally, the reaction of compounds 168 with 2 eq of 1H-imidazoles 169 and K2CO3 in MeCN at room temperature provided hybrids 165 (Scheme 31) [95].
Investigations into the antibacterial activities of these hybrids were then undertaken and the obtained results showed that compounds 165c (n = 8; R = H), 165g (n = 8, R = 2-Me), and 165k (n = 8, R = 4-Me) exhibited remarkable activities against F. columnare and S. agalactiae. Hybrid 165g (n = 8, R = 2-Me) with MIC value of 2 and 4 μM, respectively, turned out to be the most potent among the tested hybrids and its potency was comparable to or higher than that of enrofloxacin. However, this compound did not exhibit significant antibacterial activity against E. coli. By contrast, potent activity against E. coli was exhibited by hybrid 165f (n = 6, R = 2-Me), which had an MIC value of 8 μM [95]. It was finally demonstrated that the potent antibacterial activities of the above-mentioned hybrids were probably correlated to their FabI and FabK inhibitory activities. FabI and Fabk are bacterial enoyl–acyl carrier protein (ACP) reductase that catalyse the rate limiting step of bacterial fatty acid biosynthesis [96].
Finally, in 2020, Shastri and coworkers [97] continued their studies undertaken two years earlier on the synthesis and evaluation of the antimicrobial properties of coumarin/imidazoles conjugates and in this context they synthesised three new coumarin/imidazole conjugates of general formula 170, 171, and 172 using AcOH-catalysed four component cyclocondensation reactions and evaluated the antimicrobial activity of these compounds against Gram-positive B. flexus and Gram-negative Pseudomonas spp. bacterial strains. The optimised synthesis of conjugates 170 was carried out in good yields (> 65%) by treatment of 4-formylcoumarins 162 with benzil (161), p-substituted anilines 173 and AcONH4 in the presence of AcOH using conventional heating at 100 °C as well under microwave irradiation (Scheme 32).
Scheme 32.

Synthesis of coumarin/imidazole conjugates 170.
Coumarin/imidazole conjugates 172 were similarly synthesised in good to excellent yields by reaction of aldehydes 162 with benzil (161), p-aminobenzoic acid (173), and AcONH4 in AcOH at 100 °C (Scheme 33). Conjugates 171 were then converted to the corresponding methyl esters 172 by reaction with MeOH in the presence of conc. sulfuric acid (Scheme 33) [97].
Scheme 33.

Synthesis of coumarin/imidazole conjugates 171 and 172.
The results of antibacterial assays showed that conjugates 170 bearing an unsubstituted 1-phenylimidazole moiety exhibited excellent antibacterial activities against both bacterial strains with MIC values ranging from 200 to 860 nM. On the other hand, coumarin/imidazole conjugates 171 bearing a 1-(4-carboxy-substituted phenyl) moiety exhibited a decrease in the bacterial activity, but when they were converted to the corresponding methyl esters 172 they revealed improved activity compared to the corresponding compounds 170 [97].
4. Furanchalcone/Imidazole Hybrids
In 2019, Araque, Cardona-G, and coworkers reported the synthesis and evaluation of the antimicrobial activity in silico of six methylimidazolium/furanchalcone hybrids 175 that are molecules with two structural domains having different biological functions [98]. Furans, which are constituents of the furanchalcone domain have been shown to exhibit significant antibacterial activity [99,100,101]. Furthermore, chalcone and its derivatives have proven to be potent antibacterial agents [102].
As shown in Scheme 34, hybrids 175 were prepared by ultrasonic irradiation-assisted Claisen-Schmidt condensation reaction of 4-hydroxyacetophenone (176) with furfural (177), which provided (E)-3-furan-2-yl-1-(4-hydroxyphenyl)prop-2-en-1-one (178) in 88% yield. The subsequent microwave-promoted Williamson etherification of the latter compound with the appropriate α,ω-dibromoalkanes 136 afforded compounds 179 in 51–80% yields. Finally, microwave-assisted N-alkylation of 1-methyl-1H-imidazole (180) with bromides 179 provided the required hybrids 175 in 60–98% yield [98].
Scheme 34.

Synthesis of methylimidazolium/furanchalcone hybrids 175.
These compounds were then tested for their antimicrobial activities against the Gram-negative bacteria E. coli and P. aeruginosa, and the Gram-positive bacteria S. aureus, S. agalactiae, Streptococcus mutans, Bacillus cereus, and B. subtilis subsp. spizizenii and it was found among these hybrids compound 175d (n = 6) exhibited high activity against S. aureus ATCC 25923 (IC50 = 14.6 ± 0.5 mM) and S. mutans (clinical isolate) (IC50 = 18.7 ± 0.7 mM). However, the antibacterial activities of hybrids 175 against all the tested bacterial strains were lower in comparison to that of the standard drug oxytetracycline (IC50 = 14.0 ± 0.7 µM), but they were better than those of the lead compounds, i.e., furanchalcone, 1-methylimidazolium, or their mixture [98].
5. Hybrids Based on Benzofuran, Quinazolinone, and Imidazolium Moieties
Over the past two decades, several benzofuran derivatives have been prepared and introduced as antibacterial agents [103]. A lot of research has also been conducted on the synthesis of quinazolinone derivatives with potent antimicrobial activity especially against Gram-positive strains [104,105].
In 2017, Khodarahmi and coworkers hypothesised that the potency of these pharmacophores could be enhanced by incorporating them into hybrids containing imidazolium moieties [106]. Thus, they synthesised and characterised 3-{1-(benzofuran-2-yl)-2-[(2-methyl-4-oxoquinazolin-3(4H)-yl) amino]ethyl}-1-methyl-1H-imidazol-3-ium chlorides 181a–i and tested their in vitro activities against three Gram-positive bacteria (S. aureus PTCC 1023, B. subtilis PTCC 1023, and Listeria monocitogenes PTCC 1165) and three Gram-negative bacteria (E. coli PTCC 1338, P. aeruginosa PTCC 1074, and Salmonella Enteritidis (namely, S. enterica subsp. enterica sv. Enteritidis) PTCC 1091) using fluoroquinolone antibiotic ciprofloxacin as the reference compound. Hybrids 181a–i were synthesised via the route outlined in Scheme 35.
Scheme 35.

Synthesis of 3-{1-(benzofuran-2-yl)-2-[(2-methyl-4-oxoquinazolin-3(4H)-yl)amino] ethyl}-1-methyl-1H-imidazol-3-ium chlorides 181a–i.
In particular, a solution of 1-(benzofuran-2-yl)-2-bromoethan-1-one derivatives 182 in THF was treated with a mixture of quinazolin-4(3H)-ones 183, Et3N, and THF under reflux for 6–10 h and the resulting 3-{[2-(benzofuran-2-yl)-2-oxoethyl]amino}-2-methylquinazolin-4(3H)-ones 184 were reduced with NaBH4 in MeOH at room temperature for 12 h affording intermediates 185 in yields ranging from 59 to 67%. Treatment of the latter compounds with SOCl2 in CHCl3 under reflux for 4–8 h provided 3-{[2-(benzofuran-2-yl)-2-chloroethyl]amino}-2-methylquinazolin-4(3H)-ones 186, which finally were converted to hybrids 181 in 40–60% yield based on compounds 185 by reaction with 1-methyl-1H-imidazole (180) in MeCN under reflux for 24–36 h. Compounds 183 were in turn prepared by reaction of anthranilic acids 187 with Ac2O under reflux for 3–8 h, followed by treatment of the resulting 2-methyl-4H-benzo[d][1,3]oxazin-4-ones 188 with a solution of hydrazine hydrate in EtOH under reflux for 3–6 h (Scheme 36). On the other hand, the preparation of compounds 182 was carried out by treatment of salicylaldehydes 189 with chloroacetyl chloride (115) in acetone under reflux in the presence of K2CO3, followed by reaction of the resulting (benzofuran-2-yl)ethanones 190 with a solution of bromine in AcOH (Scheme 36) [106].
Scheme 36.

Synthesis of compounds 183a–c and 182a–c.
Hybrids 181 were then tested for their antimicrobial activities against the above-mentioned strains of microorganisms, and hybrid 181e turned out to be the most active against S. aureus and B. subtilis with MIC values of 29 and 58 μM, respectively, which were, however, higher than those of ciprofloxacin. The MIC values of this antibiotic against these two bacterial species were 12 and 24 μM, respectively. All other hybrids 181 had MIC values against the tested bacterial strains that ranged from 230 to 1200 μM [106].
6. 1H-Imidazoles Containing Azetidin-2-one (β-lactam) Derivatives
Azetidin-2-one (β-lactam) antibiotics have been widely used for treatment of a wide variety of infections mainly caused by aerobic Gram-negative bacteria [107,108,109]. These drugs, which act by binding to and inactivating the enzymes required for bacterial cell wall synthesis, have been the subject of several reviews concerning their structures, synthesis, and mode of action [110,111,112,113]. However, for some time a rapid appearance of a great number of bacteria presenting resistance to these agents has been observed and numerous investigations have been conducted in order to elucidate and counter these phenomena of resistance [114,115,116,117].
β-Lactamases, which are serine-dependent enzymes produced by the bacteria in defence against all classes of β-lactam antibiotics, particularly in Gram-negative bacteria are a major determinant of resistance [118]. Production of class-A, class-B, and class-C enzymes by the bacteria causes inefficiency in some cases of β-lactam antibiotics. In order to overcome this resistance, several β-lactamase inhibitors have been developed and used in clinics in combination with β-lactam antibiotics. In this context, in 2004, Venkatesan and coworkers synthesised 6-methylidene-penem carboxylic acid sodium salts 191a–e (Scheme 37) as broad-spectrum β-lactamase inhibitors and tested these compounds against various β-lactamase producing isolates [119].
Scheme 37.

Synthesis of imidazole substituted 6-methylidene-penems 194a–e and 191a–e.
Sodium salts 191a–e were synthesised by a process (Scheme 37), in which 4-nitrobenzyl (5R,6S)-6-bromo-7-oxo-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylate (192), which was used as the starting material, was prepared from commercially available 6-aminopenicillanic acid (193) by a multistep procedure reported in the literature [120].
The anhydrous MgBr2-mediated aldol condensation reaction between 192 and the appropriately substituted aldehydes 36a–c in MeCN at −20 °C in the presence of Et3N under N2 atmosphere followed by treatment with Ac2O in one portion, warming to 0 °C, and stirring for 27 h provided acetoxy bromohydrins 194. Finally, the latter compounds were treated with activated Zn powder activated in an amount that was four times the weight of the substrate bromohydrins. The reaction, which was carried out at room temperature in a mixture of MeCN, THF (1:2), and 0.5 M phosphate buffer (pH 6.5) led to the (Z)-stereoselective formation of compounds 191. Interestingly, this elimination reaction with a rather surprising stereochemical result also involved the deprotection of the carboxyl functionality of compounds 194 [119].
Methylidene-penem derivatives 191a–e were next tested in vitro against TEM-1 β-lactamase, Imi-1 (class A), CerA (class B), and AmpC (class C) enzymes for their inhibitory ability and, except for compound 191e, which was found to be less active against Imi-1, these compounds turned out to be potent inhibitors of both class-A and class-C enzymes. Interestingly, they had a spectrum of activity that was broader than that of the inhibitors then available in the market. It was also found that compound 191b in vivo enhanced the activity of piperacillin (a broad-spectrum β-lactam antibiotic of the ureidopenicillin class) against E. coli LSU 80-8, a TEM-1 producing organism. The in vivo ED50 value of piperacillin and 191b (4:1 ratio) in a murine acute lethal infection model with E. coli LSU 80-8 was 43 ± 5 mg/kg, while the ED50 value of piperacillin alone was >128 mg/kg [119].
Some years after this study, Pagadala and coworkers described a practical and efficient synthesis of imidazole containing bisazetidinones 195a–j and 196a–j (Figure 8) and evaluated the in vitro antibacterial activity of these compounds against the Gram-positive bacteria B. subtilis, P. vulgaris, and S. aureus and the Gram-negative bacteria E. coli and K. pneumoniae using the penicillin antibiotic ampicillin as the standard drug [121].
Figure 8.

Structures of imidazole containing bisazetidinones 195 and 196.
Compounds 195a–j were synthesised in excellent yields through the reactions illustrated in Scheme 38.
Scheme 38.

Synthesis of compounds 195a–j.
In accordance with a previously reported procedure [122], p-nitrobenzaldehyde (197) was treated with 1,2-diaminoethane (198) in the presence of zeolite under microwave irradiation in the absence of solvent to furnish compound 199 in a good yield. The nitro groups of this compound were then selectively reduced according to a literature procedure [123], which involved the reaction of this substrate with a suspension of zinc powder in MeOH at room temperature in the presence of hydrazine glyoxylate. The latter compound was prepared by neutralising slowly equal molar amounts of hydrazine hydrate and glyoxylic acid monohydrate (200). Condensation of the resulting diamine 201 with the appropriately substituted aryl aldehydes 36 in refluxing EtOH afforded bis-imines 202a–j. Finally, the [2+2] cycloaddition reaction (wrongly named by the authors as a Staudinger reaction) that involved bis-imines 202a–j and ketene generated in situ from chloroacetyl chloride (151) and Et3N provided bisazetidinones 196a–j (Scheme 38) [121].
On the other hand, a similar reaction sequence involving the [2+2] cycloaddition reaction of bis-imines 196a–j and ketene generated in situ from chloroacetyl chloride (151) and Et3N, was employed for the synthesis of imidazole containing bisazetidinones 196a–j in excellent yields (Scheme 39). Bis-imines 203a–j were, in turn, prepared by reaction of 198 with 3-nitrobenzaldehyde (204) in the presence of zeolite under microwave irradiation and selective reduction of the nitro groups of the resulting compound 205 followed by condensation of the appropriate aldehydes 36 with diamine 206 resulting from the reduction reaction (Scheme 39) [121].
Scheme 39.

Synthesis of compounds 196a–j.
The antibacterial data of the synthesised imidazole containing bisazetidinones showed that at 100 µg/mL, compounds 195a, 195b, and 196b were highly active against B. subtilis. Compound 196a was highly active against P. vulgaris and S. aureus, 195b and 196c proved to be highly active against E. coli and 195a and 196b displayed the high activity against K. pneumoniae, which turned out to be almost equivalent to that of ampicillin [121]. Instead, hybrids 195d, 196a, 196c, and 196d, exhibited moderate activity against B. subtilis [121].
In 2018, Noori and coworkers synthesised imidazole derivatives 207a–e bearing β-lactam moiety and evaluated their antibacterial activity against S. aureus, Enterococcus faecalis, E. coli, Streptococcus pyogenes, and K. pneumoniae [124]. The synthesis of these hybrids, which in disagreement with Klahn’s definition [7] were named by Noori as conjugates, was carried out in a modest overall yield from lophine (2,4,5-triphenyl-1H-imidazole) (208) via the reaction sequence shown in Scheme 40.
Scheme 40.

Synthesis of compounds 207a–e.
Specifically, N-acylation of 208 (lophine) with chloroacetyl chloride (151) in acetone in the presence of anhydrous Na2CO3 afforded compound 209 in 35% yield. Subsequent treatment of 209 with 2 eq of hydrazine hydrate in acetone in the presence of anhydrous Na2CO3 provided compound 210 in 32% yield, which was converted into the Schiff base derivatives 211a–e by reaction with the appropriate aldehydes 36 in EtOH under reflux in the presence of a catalytic amount of TsOH.
Compounds 211a–e, which were obtained in 35–60% yield, were then treated with 130 in dioxane in the presence of Et3N providing the required hybrids 207a–e in yields ranging from 18 to 33% [124].
Antibacterial activity tests showed that compound 207d at a concentration of 900 µM was active against E. coli and E. faecalis with an inhibition zone of 15 mm and 20 mm, respectively, while the inhibition zone of the standard drug ceftazidime, a semisynthetic cephalosporin antibiotic, against these two bacterial species was 10 and 15 mm, respectively. However, 207d did not exhibit antibacterial activity against S. aureus and K. pneumoniae. Good activity against all the bacterial species examined was instead displayed by hybrid 207c (Ar = 4-MeOC6H4), which at a concentration of 900 µM had inhibition zones with diameters of 23, 20, 30, 42, and 37 mm against S. aureus, K. pneumoniae, E. coli, E. faecalis, and S. pyogenes, respectively [124].
7. Pyrrole/Imidazole and Indole/Imidazole Hybrids
Several pyrrole/imidazole hybrids have been isolated from marine sponges [125,126] and some of these alkaloids have been found to exhibit significant antimicrobial properties. Over the past two decades, several reviews summarising the isolation, synthesis, and biological properties of some of these substances have been published [126,127,128,129,130,131,132]. For example, nagelamide J (212) (Figure 9), was isolated in 2007 from an Okinawan marine sponge Agelas spp. and was found to exhibit antimicrobial activity against the yeast Cryptococcus neoformans with an MIC value of 21 μM [133]. However, to our knowledge, the total synthesis of this hybrid has not been reported so far.
Figure 9.

Structures of nagelamide J (212) and sceptrin (213).
Instead, many data on sceptrin (213) (Figure 9), which is the first naturally occurring dimeric pyrrole/imidazole hybrid, are available in literature. This compound, which was isolated in 1981 from the sponge Agelas sceptrum [134] and in 1993 from the South Pacific sponge Agelas mauritiana [135], was found to be an antimicrobial agent with a bacteriostatic rather than bactericidal effect on exponentially growing E. coli cells at the MIC [135]. Since 2004, numerous total syntheses of 213 have been reported in the literature [136,137,138,139,140]
In 2014, Ma, Wang, and coworkers synthesised ent-213 [140] while targeting the originally reported absolute stereochemistry of this alkaloid [134]. The multi-step synthesis of ent-213 (Scheme 41), in which L-glutamic acid (214) was used as the source of chirality, involved the conversion of alcohol 215 to dihydrofuran 216 via a three-step reaction sequence. The subsequent electrophilic selenation and treatment with azidoimidazole 217 provided selenide 218, which by oxidative elimination of the phenylselenyl group and treatment with PPh3 was converted to 219. The unconventional [2+2] cycloaddition reaction of the latter compound according to the photoredox method developed by Yoon [141] provided cyclobutane 220. In particular, the reaction involved treatment of a degassed DMF solution of 219 with tris[2-phenylpyridinate-C2,N]iridium(III) [Ir(ppy)3] (3 mol%) followed by irradiation with light generated from an 11 W CFL bulb at 23 °C. The resulting crude compound 220 was treated with propane-1,3-dithiol (221) and TiCl4 in CH2Cl2 at −78 °C affording dithiane 222 in 43% yield. A subsequent nine-step reaction sequence provided bis-azide 223, which was oxidised using Dess-Martin periodinane. Treatment of the resulting unstable mesyl aldehyde with Boc-guanidine⋅TFA (224) provided aminoimidazole 225. The azido groups of the latter compound were next reduced to amino groups, which were amidated by treatment with 4-bromo-1H-pyrrole-2-carboxylic acid (226) in DMF in the presence of EDC and HOBt. Finally, removal of the protecting groups of the resulting compound 227 provided ent-sceptrin in 0.4% yield based on L-glutamic acid (213) [140].
Scheme 41.

Total synthesis of ent-sceptrin (ent-213).
In 2014, Ma, Wang, and coworkers synthesised ent-213 [140] while targeting the originally reported absolute stereochemistry of this alkaloid [134]. The multi-step synthesis of ent-213 (Scheme 41), in which L-glutamic acid (214) was used as the source of chirality, involved the conversion of alcohol 215 to dihydrofuran 216 via a three-step reaction sequence. The subsequent electrophilic selenation and treatment with azidoimidazole 217 provided selenide 218, which by oxidative elimination of the phenylselenyl group and treatment with PPh3 was converted to 219. The unconventional [2+2] cycloaddition reaction of the latter compound according to the photoredox method developed by Yoon [141] provided cyclobutane 220. In particular, the reaction involved treatment of a degassed DMF solution of 219 with tris[2-phenylpyridinate-C2,N]iridium(III) [Ir(ppy)3] (3 mol%) followed by irradiation with light generated from an 11 W CFL bulb at 23 °C. The resulting crude compound 220 was treated with propane-1,3-dithiol (221) and TiCl4 in CH2Cl2 at −78 °C affording dithiane 222 in 43% yield. A subsequent nine-step reaction sequence provided bis-azide 223, which was oxidised using Dess-Martin periodinane. Treatment of the resulting unstable mesyl aldehyde with Boc-guanidine⋅TFA (224) provided aminoimidazole 225. The azido groups of the latter compound were next reduced to amino groups, which were amidated by treatment with 4-bromo-1H-pyrrole-2-carboxylic acid (226) in DMF in the presence of EDC and HOBt. Finally, removal of the protecting groups of the resulting compound 227 provided ent-sceptrin in 0.4% yield based on L-glutamic acid (213) [140].
In 2010, Bourguet-Kondracki and coworkers isolated 2-aminoimidazole alkaloid clathridimine (228) (Figure 10) from the Mediterranean calcareous sponge Clathrina clathrus and found that this metabolite exhibited selective anti-E. coli activity and that its zinc complex showed anti-S. aureus activity [142]. Unfortunately, this alkaloid has not yet been synthesised.
Figure 10.

Structure of clathridimine (228).
Recently, unnatural pyrrole/imidazole hybrids have also been the subject of synthetic and biological studies. In 2018, Chawla and Kapoor [143] synthesised 1-[1-(1-aryl-4,5-diphenyl-1H-imidazol-2-yl)-2-methyl-5-phenyl-1H-pyrrol-3-yl]ethan-1-ones 229a–l in moderate to excellent yields by microwave-promoted Bi(NO3)3·5H2O-catalysed reaction of the appropriate 1-aryl-4,5-diphenyl-1H-imidazole-2-amines 230 [144] with phenacyl bromide (231) and 2,4-pentandione (232) (Scheme 42).
Scheme 42.

Synthesis of 1-[1-(1-aryl-4,5-diphenyl-1H-imidazol-2-yl)-2-methyl-5-phenyl-1H-pyrrol-3-yl]ethan-1-ones 229a–l.
Hybrids 229 were next evaluated for their antibacterial activities against S. aureus MTCC 96, B. subtilis MTCC 121, E. coli MTCC 614, and P. aeruginosa MTCC 2453 using ciprofloxacin as standard drug, and it was found that compounds 229c, 229h, 229k, and 229l revealed significant activity that was better against the Gram-positive bacteria rather than the Gram-negative bacteria [143]. The zones of inhibition of 229c, 229h, and 229l at 100 μg/mL towards S. aureus MTCC 96 were 24, 25, and 24 mm, respectively, while the zones of inhibition of these same compounds towards P. aeruginosa MTCC 2453 were 17, 19, and 20 mm, respectively [143].
In 2004, a study on the synthesis and antimicrobial activities of some imidazole/indole hybrids was carried out by Benkli and coworkers [145]. These hybrids included 1-substituted 3-(4,5-diaryl-1H-imidazol-2-yl)-2-(1H-imidazol-1-yl)-1H-indoles 233a–l, which were prepared in 48–72% yield by treatment of equimolar amounts of 1-substituted 2-(1H-imidazol-1-yl)-1H-indole-3-carbaldehydes 234 with the appropriate benzil derivatives 235 and AcONH4 in AcOH under reflux for 2 h (Scheme 43).
Scheme 43.

Synthesis of 1-substituted 3-(4,5-diaryl-1H-imidazol-2-yl)-2-(1H-imidazol-1-yl)-1H-indoles 233a–l.
Among the twelve synthesised hybrids, the most significant antibacterial activity was exhibited by compound 233j, which was prepared in 72% yield and had an MIC value (63 μM) against E. coli that was comparable to that of the control antibiotic chloramphenicol succinate (72 μM) [145].
More recently, Channe Gowda and coworkers synthesised imidazole/indole hybrids 236 and 237 via the multi-step route outlined in Scheme 44 [146].
Scheme 44.

Synthesis of imidazole/indole hybrids 236 and 237.
Imidazole 238 was treated with a molar excess of hydrazine hydrate to give compound 239, which was treated with N-protected tryptophan 240, EDC, and HOBt using 4-methylmorpholine (NMM) as a base to provide compound 241. This intermediate was treated with TFA and the resulting compound 242 was treated with the required aryl isocyanates 243 or aryl isothiocyanates 244 providing ureas 236 and thioureas 237, respectively. These imidazole/tryptophan hybrids were tested for their antibacterial properties against E. coli and S. aureus using streptomycin as the reference drug. The obtained results showed that hybrids 236a (R = 4-FC6H4), 236b (R = 4-(NO2)C6H4), 237a (R = 4-FC6H4), and 237b (R = 4-(NO2)C6H4) exhibited inhibitory activity higher than that of streptomycin and that urea derivatives 236 were more active than thiourea derivatives 237. It was also observed that the electronic properties of hybrids 236 and 237 had a close relationship with the biological properties and that hybrids 236 and 237 bearing phenyl groups 4-substituted by electron-withdrawing groups (NO2 > F > Cl > OMe > Me) had antimicrobial activity higher than that of the reference drug [146].
Over the past two decades, much attention was also paid to the synthesis and evaluation of the antimicrobial properties of oroidin [(E)-N-[3-(2-amino-1H-imidazol-4-yl)allyl]-4,5-dibromo-1H-pyrrole-2-carboxamide] (245a) [147,148,149,150,151,152], a naturally occurring pyrrole–2-aminoimidazole alkaloid, which was originally isolated from sponges of the genus Agelas [153,154,155,156]. A recent synthesis of 245a involved the reaction of (E)-4-(3-aminoprop-1-en-1-yl)imidazole-2-amine (246) with 4,5-dibromo-1H-pyrrole-2-carboxylic acid (247a), the coupling reagent N,N,N′,N′-tetramethyl-O-(benzotriazol-1-yl)uronium tetrafluoroborate (TBTU) in NMM, and DMF at room temperature for 6 h (Scheme 45) [152]. This reaction produced naturally occurring hybrid 245a in 25% yield [152].
Scheme 45.

Synthesis of oroidin (245a) from (E)-4-(3-aminoprop-1-en-1-yl)imidazole)-2-amine (246) and 4,5-dibromo-1H-pyrrole-2-carboxylic acid (247a).
Interestingly, naturally occurring hybrid 245a at a concentration of 50 μM exhibited noticeably high activity against the Gram-positive bacteria S. aureus (>90% inhibition of growth) (IC50 = 31 µM) and E. faecalis (approximately 50% inhibition of growth), but was inactive against the Gram-negative bacterium E. coli [152].
Tammela, Mašič, and coworkers also synthesised naturally occurring (E)-N-[3-(2-amino-1H-imidazol-4-yl)allyl]-1H-pyrrole-2-carboxamide (clathrodin) (245b) (Figure 11) in 18% yield by TBTU-mediated reaction of 246 with 1H-pyrrole-2-carboxylic acid (247b) in NMM and DMF at room temperature [152]. They also found that 245b exhibited activity below the hit threshold (>80% inhibition of growth at a concentration of 50 μM) against all the microbial strains tested [152].
Figure 11.

Structures of clathrodin (245b) and 1H-pyrrole-2-carboxylic acid (247b).
8. Pyrazole/Imidazole Hybrids
Over the past two decades, research has also been conducted on pyrazole-based compounds possessing significant anti-microbial activity [157,158,159,160]. Some of these studies focused in particular on the synthesis and evaluation of the in vitro activity of imidazole/pyrazole hybrids.
In 2004, Menozzi, La Colla, and coworkers synthesised 1,5-disubstituted 4-[(1H-imidazol-1-yl)(phenyl)methyl]-1H-pyrazoles 248e–o and 248p–u via the routes depicted in Scheme 46 and Scheme 47, respectively [161]. In particular, ethyl aroylacetates 249a–d were treated with N,N-dimethylformamide dimethyl acetal (250) and the resulting compounds 251a–d were treated with the appropriate arylhydrazines 252 affording pyrazoles 253e–o. The alcohols, which were obtained by reduction of these compounds with diisobutylaluminum hydride (DIBAL-H) in toluene or with LiAlH4 in Et2O, were oxidised with pyridinium chlorochromate (PCC) in CH2Cl2 and the resulting aldehydes 254e–o were treated with phenylmagnesium bromide yielding alcohols 255e–o. Finally, treatment of these intermediates with N,N’-carbonyldiimidazole (CDI) in dry toluene under reflux for 4 h provided hybrids 248e–o in moderate to good yields (Scheme 46).
Scheme 46.

Synthesis of 1,5-disubstituted 4-[(1H-imidazol-1-yl)(phenyl)methyl]-1H-pyrazoles 248e–o.
Scheme 47.

Synthesis of 1,5-disubstituted 4-[(1H-imidazol-1-yl)(phenyl)methyl]-1H-pyrazoles 248p–u.
Hybrids 248p–u (Scheme 47) were instead prepared by reaction of 2-[(dimethylamino)methylene]-1,3-diphenylpropane-1,3-dione (256) [161] with the appropriate arylhydrazine hydrochlorides 252 in refluxing EtOH. Reduction of the resulting ketones 257 with LiAlH4 in Et2O led to alcohols 255, which by treatment with CDI provided hybrids 248 [161].
Hybrids 248p–u were proven to be weakly efficacious against S. aureus whereas other tested hybrids showed no activity. Furthermore, all hybrids 248 turned out to be inactive towards Gram-negative Salmonella spp. In addition, hybrids 248p–u were proven to be the most potent derivatives against M. tuberculosis strains resistant to drugs commonly employed in the antitubercular therapy, and their activity was superior to that of clotrimazole and econazole, which were used as reference drugs [161].
In 2008, Mamolo and coworkers described the synthesis of 4-(1H-imidazol-1-yl)-3,5-diaryl-4,5-dihydro-1H-pyrazole derivatives 258a–t and tested the antitubercular activity of these hybrids against M. tuberculosis H37Rv [162]. The three-step synthesis of compounds 258 (Scheme 48) was carried out by treating substituted 2-bromoacetophenones 259 with imidazole (260) in THF providing 1-aryl-2-(1H-imidazol-1-yl)ethan-1-ones 261. The subsequent condensation of these compounds with a series of substituted benzaldehydes 36 in refluxing toluene in the presence of piperidine, followed by treatment of the resulting α,β-unsaturated ketones 262 with EtOH solutions of hydrazines 263 (hydrazine hydrate, methylhydrazine sulfate, phenylhydrazine, and 4-fluorophenylhydrazine), produced hybrids 258a–t in yields ranging from 15 to 89%.
Scheme 48.

Synthesis of 4-(1H-imidazol-1-yl)-3,5-diaryl-4,5-dihydro-1H-pyrazole derivatives 258a–t.
All compounds 258 were evaluated for antitubercular activity against M. tuberculosis H37Rv using a micro-dilution Resazurin assay [163], and it was found that they showed good antimycobacterial activity, which in the case of hybrids 258m, 258n, 258o, 258q, 258r, and 258t reached MIC values of 7–10 μM [162].
In 2011, Isloor and coworkers synthesised and characterised 3-aryl-4-(4,5-diaryl-1H-imidazol-2-yl)-1H-pyrazoles 264a–j and methyl (Z)-2-(3-{[(E)-(3-aryl-1H-pyrazol-4-yl)] methylene}amino)-5-oxo-2-sulfanylideneimidazolidin-4-ylidene]acetates 265a–d [164]. Hybrids 264 were synthesised in good yields by refluxing 3-aryl-1H-pyrazole-4-carbaldehydes 266 (1 eq) with 1,2-diketones 235 (1 eq) and AcONH4 (5 eq) in AcOH for 6–7 h (Scheme 49).
Scheme 49.

Synthesis of 3-aryl-4-(4,5-diaryl-1H-imidazol-2-yl)-1H-pyrazoles 264a–j.
Hybrids 265 were instead prepared in good yields by reaction of aldehydes 266 with thiosemicarbazide (267) followed by treatment of the resulting thiosemicarbazones 268 with equimolar amounts of dimethyl acetylenedicarboxylate (269) in MeOH under reflux for 1 h (Scheme 50) [164].
Scheme 50.

Synthesis of hybrids 265a–d.
The results of screening tests of the antibacterial properties of hybrids 264 and 265 against E. coli, S. aureus, B. subtilis, Salmonella Typhimurium (namely, S. enterica subsp. enterica sv. Typhimurium), Clostridium perfringens, and P. aeruginosa showed that methyl (Z)-2-{3-[({(E)-[3-(4-methylsulfanyl)phenyl]-1H-pyrazol-4-yl}]methylene)amino]-5-oxo-2-sulfanylideneimidazolidin-4-ylidene}acetate (265c) exhibited excellent activity against P. aeruginosa and C. perfringens at concentrations of 2.5 and 1.2 µM, respectively, compared to the standard drug streptomycin [164]. Instead, the remaining hybrids were found to possess moderately good activity against all six bacterial strains tested [164].
Still in 2011, Padmavathi and coworkers prepared N-(4-aryl-1H-imidazol-2-yl)-4-phenyl-1H-pyrazole-3-carboxamides 270a–c via the three-step route shown in Scheme 51, which involved the use of 4-aryl-1H-imidazol-2-amines 271 as the starting material [165].
Scheme 51.

Synthesis of N-(4-aryl-1H-imidazol-2-yl)-4-phenyl-1H-pyrazole-3-carboxamides 270a–c.
Treatment of amines 271a–c with cinnamoyl chloride (272) in toluene under reflux gave (E)-N-(4-aryl-1H-imidazol-2-yl)cinnamamides 273a–c in 76–81% yield. Subsequently, CH2Cl2 solutions of these compounds were treated with an Et2O solution of diazomethane at −15 to −20 °C in the presence of Et3N for 42–48 h affording compounds 274a–c in 73–77% yield. Finally, these compounds were converted to the required hybrids 270a–c in 63–68% yield by treatment with 1.2 eq of chloranil in xylene under reflux for 24–25 h [165].
Padmavathi and coworkers then found that, when compared to the standard drug chloramphenicol, hybrid 270c at a concentration of 270 μM displayed significant antibacterial activity and its inhibition zones against S. aureus, B. subtilis, P. aeruginosa, and K. pneumoniae were 34, 35, 32, and 39 mm, respectively. Instead, the inhibition zones of chloramphenicol at 310 μM against these bacterial species were 35, 38, 30, and 42 mm, respectively [165].
More recently, Narayana and coworkers synthesised ten 4-(2-aryl-4,5-diphenyl-1H-imidazol-1-yl)-1,5-dimethyl-2-phenyl-1,2-dihydro-3H-pyrazol-3-ones 275 via atom-economic, one-pot reaction of equimolar amounts of benzil (161), aryl aldehydes 36, commercially available 4-aminoantipyrine (276), and AcONH4 in glacial acetic acid, in the presence of a catalytic amount of ZnO nanoparticles (ZnO Nps) (Scheme 52) [166]. The preparation of this catalyst, which could be reused even after four catalytic cycles, was performed according to a documented procedure [166]. The cyclocondensation reaction, when carried out at 60 °C, provided hybrids 275 in 76–82% yield, but higher yields (86–90%) were obtained by carrying out the cyclocondensation reaction under microwave irradiation using a domestic microwave oven with a continuous irradiation power.
Scheme 52.

Synthesis of 4-(2-aryl-4,5-diphenyl-1H-imidazol-1-yl)-1,5-dimethyl-2-phenyl-1,2-dihydro-3H-pyrazol-3-ones 275.
All hybrids 275 were evaluated for their antibacterial activities against S. aureus and E. coli by resazurin reduction assay and demonstrated significant activity. The IC50 values of compounds 275c (Ar1 = 4-FC6H4) and 275g (Ar1 = 4-MeOC6H4), which were found to be the most active among the tested hybrids, were 46 and 53 μM, respectively, for S. aureus, and 88 and 23 μM, respectively, for E. coli [166].
9. 1,2,3- and 1,2,4-Triazole/Imidazole Hybrids and Conjugates
Over the last few decades, numerous 1,2,3-triazole-containing hybrids and conjugates possessing antibacterial activity have been identified [167,168,169,170,171,172,173,174] and some of them have been cited and commented on in an interesting review published in 2019 by Aisa and coworkers [174]. Unfortunately, this review did not include the synthesis and antibacterial properties of 1,2,3-triazole/imidazole hybrids and conjugates. Nevertheless, still in 2019, Gao, Wang, Xiao, and Huang summarised recent advances on the promising antibacterial activities of 1,2,4-triazole hybrids against both drug-sensitive and drug-resistant pathogens [175].
In this section of the review, we have summarised and commented the data on antibacterial 1,2,3- and 1,2,4-triazole/imidazole hybrids and conjugates, which were described in the literature from 2015 until the end of February 2020, noting that few of them [32,59] have already been discussed in Section 1 of the present review.
In 2015, Nikalje and coworkers synthesised 3-(2-aryl-4,5-diphenyl-1H-imidazol-1-yl)-1H-1,2,4-triazole-5-carboxylic acid derivatives 277 and evaluated the in vitro antibacterial activities against E. coli, S. aureus, S. pneumoniae, and S. pyogenes using ampicillin as a reference drug [176]. As shown in Scheme 53, compounds 277 were synthesised in 66–93% yield by CAN catalysed the four-component cyclocondensation reaction of benzil (161), aryl aldehydes 36, commercially available 3-amino-1H-1,2,4-triazole-5-carboxylic acid (278), and AcONH4 in EtOH under reflux for 3–4 h.
Scheme 53.

Synthesis of 3-(2-aryl-4,5-diphenyl-1H-imidazol-1-yl)-1H-1,2,4-triazole-5-carboxylic acid derivatives 277.
The results of the in vitro antibacterial data of compounds 277 showed that all these substances possessed moderate activity (MIC range = 130–1200 μM) when compared with ampicillin (MIC range = 290–720 μM). It was specifically found that 277e (Ar1 = 4-ClC6H4) showed higher activity (MIC = 140 μM) against E. coli, 277b (Ar1 = Ph) (MIC = 150 μM) against S. pneumoniae, and 277n (Ar1 = 2,4-(MeO)2C6H3) (MIC = 130 μM) against S. pyogenes than ampicillin (290 μM). Furthermore, against S. aureus, 277n was the most active among the tested compounds (MIC = 210 μM) compared to ampicillin (720 μM) [176].
In 2019, Subhashini and coworkers synthesised eight imidazole-linked mono-1,2,3-triazoles 279 and eight imidazole-linked bis-1,2,3-triazoles 280 and tested the antibacterial properties of these hybrids against S. aureus, B. cereus, E. coli, P. vulgaris, and the fungal species Aspergillus fumigatus using ampicillin as a reference drug [177]. Hybrids 279 were synthesised via the three-step procedure shown in Scheme 54 that involved the reaction of 4-hydroxybenzaldehyde (281) with propargyl bromide (92) in DMF in the presence of K2CO3.
Scheme 54.

Synthesis of imidazole/1,2,3-triazole hybrids 279.
The Cu(I)-catalysed click reaction between the resulting 1-alkyne 282 and aryl azides 283 in DMF in the presence of sodium ascorbate as a reduction reagent gave rise to 4-[(1-aryl-1H-1,2,3-triazol-4-yl)methoxy]benzaldehydes 284. Finally, the microwave-mediated reaction of these derivatives (1 eq) with benzil (161) (1 eq) and AcONH4 (4 eq) in EtOH in the presence of a catalytic amount of iodine provided hybrids 279 in yields ranging from 90 to 95% (Scheme 54). It was also observed that when this multicomponent reaction was carried out using conventional heat sources at 80 °C for 4–5 h hybrids 279 were obtained in yields ranging from 60 to 64% [177].
Imidazole-linked bis-1,2,3-triazoles derivatives 280 were instead prepared via the route depicted in Scheme 55, in which the order of the sequence of reactions that were used for the synthesis of hybrids 284 was reversed.
Scheme 55.

Synthesis of imidazole/1,2,3-triazole hybrids 280.
In particular, 4-hydroxybenzaldehyde (281) was treated with benzil (161) and AcONH4 in AcOH in the presence of a catalytic amount of iodine under microwave irradiation at 180 W for 3–5 min, affording trisubstituted imidazole 285. Bis-propargylation of 285 with propargyl bromide (92) in DMF in the presence of K2CO3 gave compound 286, which underwent microwave-promoted Cu(I)-catalysed click reaction with azides 283 in DMF, providing hybrids 280 in good to excellent yields [177].
Hybrids 279 and 280 were next characterised and tested for their antibacterial activity and it was found that compounds 279c (R1 = R = H; R3 = Me), 279h (R1 = R2 = H; R3 = NO2), 280d (R1 = OMe; R2 = R3 = H), and 280e (R1 = OMe; R2 = H; R3 = NO2) were capable to inhibit the bacterial growth more effectively than other hybrids with MIC values ranging from 11 to 69 μM. For example, the MIC values of 280e against E. coli and B. cereus were 11 and 12 μM, respectively, whereas the MIC values of ampicillin against these two bacterial strains were 31 and 24 μM, respectively. Furthermore, the MIC values of 279h against S. aureus, B. cereus, E. coli, and P. vulgaris were 30, 26, 19, and 46 μM, respectively [177].
Still in 2019, Verma and coworkers synthesised in moderate to excellent yields 4-{[1-(4,5-diaryl-1H-imidazol-2-yl)naphthalen-2-yl)oxy]methyl}-1-substituted-1H-1,2,3-triazoles 287 by condensation reaction of α-diketones 235 with an equimolar amount of 1,2,3-triazole -substituted naphthaldehydes 288 and a large molar excess of AcONH4 in glacial acetic acid under reflux for 8–12 h (Scheme 56) [178].
Scheme 56.

Synthesis of 2,4,5-trisubstituted 1H-imidazole/1,2,3-triazole hybrids 287.
The evaluation of hybrids 287 for their in vitro antibacterial activities against B. subtilis, S. epidermidis, E. coli, and P. aeruginosa using ciprofloxacin as a positive control showed that compound 287h (R1 = H; Ar = 4-BrC6H4) was the most active against S. epidermidis (MIC = 5.2 μM) and that compound 287e (R1 = H; Ar = 2-NO2C6H4) was highly active against E. coli (MIC = 5.5 μM). The MIC value of ciprofloxacin against all four bacterial strains tested was 4.7 μM. It was also found that hybrid 287e was anchored in the binding site of DNA gyrase topoisomerase II of E. coli [178].
Nine 1,2,3-triazole-substituted naphthaldehydes 288 were in turn prepared in 87–92% yield by treating 2-(prop-2-yn-1-yloxy)-1-naphthaldehyde (289) with the appropriate aryl azides 290, 10 mol% CuSO4·5H2O, and 20 mol% sodium ascorbate in a 4:1 mixture of DMF and water at 45 °C for 5–8 h (Scheme 57) [178].
Scheme 57.

Synthesis of 1,2,3-triazole substituted naphthaldehydes 288.
10. Hybrids of Imidazole Derivatives and 5-Membered Heterocycles Containing Oxygen and Nitrogen
For several years, studies have been carried out to identify isoxazolidine derivatives with significant antibacterial properties [179,180,181,182]. In 2004, Rangappa and coworkers synthesised with high regioselectivity isoxazolidine/imidazole hybrids 291a–j and tested their antibacterial activities against S. aureus, E. coli, and B. subtilis by using streptomycin as a positive control [183]. The synthesis of these hybrids (Scheme 58) was accomplished by treatment of 2-butyl-4(5)-chloro-1H-imidazole-5(4)-carbaldehyde (292) [184,185], a compound currently commercially available, with nitro compounds 293a and 293b and zinc dust, using histidine as catalyst, according to a previously reported procedure [186]. The resulting nitrones (Z)-294a and (Z)-294b (that were incorrectly reported as (E)-stereoisomers), which were contaminated by the corresponding (E)-stereoisomers, (E)-294a and (E)-294b, respectively, were subjected to 1,3-dipolar cycloaddition reaction with a molar excess of 1-alkenes 295 in a toluene or xylene solution giving rise to hybrids 291a–j in yields ranging from 59 to 72% together with minor amounts of the corresponding regioisomers 296a–j.
Scheme 58.

Synthesis of isoxazolidine/imidazole hybrids 291a–j.
The results of the evaluation of the antibacterial activity of hybrids 291a–j showed that compounds 291c, 291d, 291h, and 291i exhibited about 50–60% inhibition when compared with streptomycin and that the remaining hybrids were not effective against S. aureus, E. coli, and B. subtilis. For example, the zone of inhibition of S. aureus for compound 291c at a concentration of 25 mM was 21 mm, while that for streptomycin was 35 mm [183].
In 2005, Frank and Kalluraya synthesised 1,3,4-oxadiazole/imidazole hybrids 297a–l in good yields by reaction of 2-(2-methyl-5-nitro-1H-imidazol-1-yl)acetohydrazide (67) with the appropriate ketones 298a–l in glacial AcOH under reflux for 1 h, followed by treatment of the resulting compounds 299a–l with a large molar excess of Ac2O under reflux for 4 h (Scheme 59) [187]. Compound 67 was in turn prepared in 67% yield by a two-step reaction sequence, in which 2-methyl-5-nitro-1H-imidazole (64) was the starting material (Scheme 59, cf. Scheme 12).
Scheme 59.

Synthesis of 1,3,4-oxadiazole/imidazole hybrids 297a–l.
Hybrids 297a–l when screened for their antibacterial activities against S. aureus, P. aeruginosa, E. coli, and B. subtilis using nitrofurazone as the standard drug turned out to display significant activity. For example, hybrid 297l had an MIC value of 640 nM for each of these bacterial strains, while nitrofurazone had an MIC value of 2.5 μM for S. aureus, P. aeruginosa, and B. subtilis and of 1.3 μM for E. coli [187].
In 2007, Kalluraya and coworkers synthesised 2-aryl-5-[(2-methyl-4-nitro-1H-imidazol-1-yl)methyl]-1,3,4-oxadiazoles 300a–l in moderate yields by treatment of hydrazide 67 with the appropriate arenecarboxylic acids 301a–l in the presence of POCl3 under microwave irradiation or using the conventional heating method in an oil bath (Scheme 60) [188]. Higher yields of hybrids 300 (54–75%) were obtained when the reaction was carried out using microwave irradiation.
Scheme 60.

Synthesis of 2-aryl-5-[(2-methyl-4-nitro-1H-imidazol-1-yl)methyl]-1,3,4-oxadiazoles 300a–l.
The antibacterial activities of hybrids 300a–l was then evaluated at 10 mg/mL concentration against S. aureus, K. pneumoniae, E. coli, and P. aeruginosa using ciprofloxacin as a positive standard. The obtained results indicated that compounds 300e and 300l exhibited good activity against K. pneumoniae, E. coli, and P. aeruginosa, compound 300h showed good activity against E. coli and P. aeruginosa, compound 300i showed good activity against P. aeruginosa and K. pneumoniae, and compound 300j showed good activity against K. pneumoniae [188]. For example, a diameter of zone inhibition of 28 mm was found for hybrids 300e and 300i against P. aeruginosa, while that of ciprofloxacin was 22 mm [188].
11. 1,8-Naphthalimide/Imidazole Hybrids
Several 1,8-naphthalimide derivatives are known as antibacterial agents [76,189,190,191]. In recent years, special attention has been paid to antibacterial 1,8-naphthalimide/imidazole hybrids [79,192,193]. In 2011, Zhang and Zhou synthesised 1,8-naphthalimide-derived imidazoles 302a–d and 303a–d via the route shown in Scheme 61 [76].
Scheme 61.

Synthesis of 1,8-naphthalimide-derived imidazoles 302a–d and 303a–d.
Specifically, compounds 304, which were prepared in 41–78% yield by treatment of 4-bromo-1,8-naphthalimide (114) with a molar excess of α,ω-dibromoalkanes 136 in DMF in the presence of K2CO3, were treated with a mixture of imidazole (260) and NaH in THF providing hybrids 302a–d in yields that ranged from 58 to 69%. Finally, the reaction of these compounds with 1-(bromomethyl)-2,4-difluorobenzene (305) in MeCN under reflux provided hybrids 303a–d in good yields [76].
It deserves to be mentioned that in 2013 the synthesis of hybrids 302 and 303 via the route shown in Scheme 61 was repeated in detail by Lv, Zhou, and coworkers [77], and that in 2011 and in 2013, hybrids 302a–d and 303a–d were evaluated for their in vitro antimicrobial activities against Gram-positive bacteria S. aureus ATCC 25923, MRSA N315, B. subtilis ATCC 6633, M. luteus ATCC 4698, and Gram-negative bacteria P. hauseri, [60] E. coli JM 109, P. aeruginosa, and Salmonella Typhi (namely, S. enterica subsp. enterica sv. Typhi) [76,77,194]. Naphthalimide-based imidazoles 302a and 302b were found able to inhibit the growth of all bacterial strains tested with MIC values of 20–83 μM and hybrids 303a and 303b turned out to display better potency than other their analogues against the tested strains [76,77]. The MIC values of these two hybrids were 7 and 3 μM, respectively, against MRSA and this result suggested that they could be used as potential anti-MRSA agents in comparison with clinical drugs chloramphenicol [50] MIC = 12 μM) and orbifloxacin (MIC = 3 μM) [76]. Notably, compound 302b with (CH2)4 linker exhibited efficacy against E. coli and P. aeruginosa with MIC values of 20 and 40 μM, respectively, which are comparable to that of the standard drug chloramphenicol (25 and 50 μM) [77].
In 2016, Cai, Zhou, and coworkers carried out the synthesis and biological evaluation of Schiff base-linked imidazolyl naphthalimides 306a–c, 307, 308a–e, and 309a–k and tested the in vitro antibacterial activities of these compounds against the Gram-positive bacteria MRSA, S. aureus, B. subtilis, and M. luteus and the Gram-negative bacteria E. coli DH52, E. coli JM109, S. dysenteriae, P. aeruginosa, P. hauseri [60], and S. Typhi [194,195].
The target hybrids were synthesised as outlined in Scheme 62. Compound 310, which was obtained in 87% yield by reaction of anhydride 113 with hydrazine hydrate in EtOH at room temperature, was converted to naphthalimides 311a–c in 79–84% yield by treatment with the appropriate alicyclic amines 312 in ethylene glycol monomethyl ether under reflux and N2 atmosphere. The subsequent condensation reaction of 311a–c with 2-butyl-5-chloro-1H-imidazole-4-carbaldehyde (292) in toluene under reflux in the presence of 1 mol% P4O10 under N2 atmosphere provided hybrids 307a–c in 34–8% yields. Compound 306 was instead obtained by the condensation reaction of 310 with 292 in an unspecified yield. Finally, target compounds 308 were synthesised in moderate yields by reaction of compound 311c with the corresponding 1-alkylimidazolecarbaldehydes 313 and imidazolyl naphthalimides 309 were obtained by treatment of 311c with the appropriate substituted 1-benzylimidazolecarbaldehydes 314 in toluene at 100 °C using P4O10 as dehydrating agent. Unfortunately, even in this case, the yields of the reactions were not reported [195].
Scheme 62.

Synthesis of Schiff base-linked imidazolyl naphthalimides 306, 307a–c, 308a–e, and 309a–k.
All the above-mentioned target Schiff base-linked imidazolyl naphthalimides were tested for their in vitro antimicrobial activity against the Gram-positive bacteria MRSA, S. aureus, B. subtilis, and M. luteus and against the Gram-negative bacteria E. coli DH152, E. coli JM109, S. dysenteriae, P. aeruginosa, P. hauseri [60], and S. Typhi [194] using chloramphenicol [50] and norfloxacin as reference drugs. The results of preliminary screening tests showed that naphthalimides 307a–c possessed antibacterial activity higher than that of compound 306, and that 307c exhibited the highest inhibition activity among these three compounds. It was also observed that the activity of hybrids 308a–c was not much different from that of 307c, whereas the antibacterial efficacies of hybrids 308d and 308e were better than that of compound 307c against some tested strains. Interestingly, hybrid 308e, which had an MIC value of 10 µM against E. coli JM109, was 10 times more potent than clinical drug chloramphenicol. It is also worth mentioning that: (i) hybrid 309b proved capable of inhibiting the growth of B. subtilis at the concentration of 7 µM, was 14 times more potent than chloramphenicol and with activity comparable to that of norfloxacin; and (ii) hybrid 309i had an MIC value of 3 µM against MRSA and was 17 and 7 times more potent than chloramphenicol and norfloxacin, respectively [195].
12. Bis-Imidazoles
Over the past few decades, considerable efforts have been directed toward the synthesis and evaluation of the biological properties of bis-imidazole derivatives, a class of heterocyclic compounds displaying significant levels of antifungal, antibacterial, and antimycobacterial activities [196,197,198,199,200,201,202,203].
In 2007, Mamolo and coworkers synthesised 1-aryl-3-(1H-imidazol-1-yl)-2-[(1H-imidazol-1-yl)methyl]propan-1-ones 315a–h, 1-aryl-3-(1H-imidazol-1-yl)-2-[(1H-imidazol-1-yl)methyl] propan-1-ols 316b,f, and 1-(5-substituted-thiophen-2-yl)-3-(1H-imidazol-1-yl)-2-[(1H-imidazol-1-yl)methyl]propan-1-ones 317a–c via the reaction sequences shown in Scheme 63 [198]. Compounds 315 were prepared in 41–65% yield by the Mannich reaction of the appropriately 4-substituted acetophenones 318 with paraformaldehyde (319) and dimethylamine hydrochloride in AcOH. Subsequent irradiation of a water/EtOH solution of the resulting compounds 320a–h with 4 eq of imidazole (260) at 250 W for 6 min in a domestic microwave oven provided bis-imidazoles 315a–h in yields ranging from 32 to 71%. This reaction could also be carried out using conventional heating under reflux for 12–16 h, but the yields of the required bis-imidazoles were quite lower. Bis-imidazoles 316b and 316f were instead prepared in 30 and 36% yield, respectively, by treatment of 315b and 315f with NaBH4 in MeOH at room temperature for 3 h. Finally, bis-imidazole derivatives 317a, 317b, and 317c were synthesised in 69, 73, and 48% yield, respectively, from the appropriate thienyl methyl ketones 321 using compounds 322a–c as key intermediates [198].
Scheme 63.

Synthesis of 1-aryl-3-(1H-imidazol-1-yl)-2-[(1H-imidazol-1-yl)methyl]propan-1-ones 315a–h, 1-aryl-3-(1H-imidazol-1-yl)-2-[(1H-imidazol-1-yl)methyl]propan-1-ols 316b,f, and 1-(5-substituted-thiophen-2-yl)-3-(1H-imidazol-1-yl)-2-[(1H-imidazol-1-yl)methyl]propan-1-ones 317a–c.
Compounds 315a–h and 316b,f were then tested for their in vitro killing activities against M. tuberculosis H37Rv in comparison with antibiotic rifampicin and were found to exhibit a moderate activity with MIC values in the range 22–200 μM. It was particularly noted that the antimycobacterial activity of 1-[(1,1′-biphenyl)-4-yl]-3-(1H-imidazol-1-yl)-2-[(1H-imidazol-1-yl) methyl]propan-1-one (315h) was comparable to its antifungal activity towards clinical isolates of Candida albicans 3038 and C. glabrata 123, suggesting the presence of similar enzymatic interactions. Bis-imidazoles 317a–c exhibited instead moderate antifungal activity against the strain of C. albicans 3038 [198].
In 2010, as part of a study concerning the evaluation of the bioactivity of structurally modified derivatives of fluconazole, Zhou and coworkers synthesised amine-derived bis-imidazoles 323a–i and hydrochloride salts 324a–d and evaluated the in vitro activities of all these compounds against S. aureus ATCC 29213, S. aureus N 315, B. subtilis ATCC 21216, E. coli ATCC 25922, P. aeruginosa ATCC 27853, and P. hauseri ATCC 13315 [60,199]. The synthesis of these hybrids was carried out as shown in Scheme 64.
Scheme 64.

Synthesis of amine-derived bis-imidazoles 323a–i and hydrochlorides 324a–d.
A mixture of 1 eq of halobenzyl halides 325 and 1.2 eq of diethylethanolamine (73) in MeCN was stirred at 50 °C providing compounds 75 in excellent yields. The subsequent reaction of these diols with PBr3 in CHCl3 at room temperature for 2 h and then at 60 °C provided compounds 76 in 73–96% yield. Finally, treatment of a THF solution of the latter compounds with the sodium salts prepared from NaH and the appropriate 1H-imidazoles 157 gave amine-derived bis-imidazoles 323a–i in yields ranging from 58 to 87%. Compounds 323a–d were then converted to the corresponding hydrochlorides 324a–d in good yields by treatment with HCl in a 4:1 mixture of Et2O and CHCl3 [199].
Notably, compounds 323a–c exhibited remarkable antibacterial activities against all the tested bacterial strains and, among these hybrids, compound 323b with an MIC value of 11 μM against P. aeruginosa proved to be, on a molar basis, 18 times more potent than the reference drug chloramphenicol. Remarkably, a significant improvement in the bacterial activity was in general observed when some of the above-mentioned azole compounds were converted to the corresponding water-soluble hydrochlorides 324. The MIC values of these salts ranged from 3 to 150 μM [199].
In 2013, Al-Mohammed, Abdullah, and coworkers synthesised bis-imidazole sulfonamides 326a–c by reaction of diethanolamine (73) with the appropriate 4-substituted benzenesulfonyl chlorides 327, followed by treatment of the resulting compounds 328 with imidazole (260) under basic conditions (Scheme 65) [200].
Scheme 65.

Synthesis of bis-imidazole sulfonamides 326a–c.
The in vitro antibacterial activities of compounds 326a–c against E. coli, S. aureus, S. Typhimurium, B. subtilis, P. aeruginosa, Rhodococcus ruber, Acinetobacter calcoaceticus, E. faecalis, S. pyogenes, and S. epidermidis were evaluated using the microbroth dilution assay using amoxicillin and kanamycin B as reference drugs, and it was found that the majority of the compounds investigated possessed significant antibacterial activity towards most of these microorganisms. It was specifically found that N,N-bis[2-(1H-imidazol-1-yl)ethyl]-4-methoxybenzenesulfonamide (326b) had an MIC value of 800 µM towards P. aeruginosa and an MIC value of 270 µM towards B. subtilis, while the MIC value of kanamycin B for both of these bacterial species was < 100 µM. Furthermore, bis-imidazoles 326a and 326b demonstrated inhibitory effects ranging between 830 and 930 µM against the Gram-positive bacterium E. faecalis, while the antibiotic kanamycin B was inactive against this bacterium at the concentration range of this study (<1 mM) [200].
In 2015, Al-Mohammed, Alias, and Abdullah synthesised imidazolium geminal dicationic ionic liquids 329a–f in good to excellent yields by reaction of N,N-bis[2-(1H-imidazol-1-yl)ethyl]-4-methylbenzenesulfonamide (326a) with a molar excess of the appropriate alkyl bromides or chlorides (130) in MeCN at 20–55 °C under an N2 atmosphere (Scheme 66) [201]. Compounds 329d–f were then converted to the corresponding bis(trifluoromethylsulfonyl)amides 330d–f in good to excellent yields by treatment with lithium bis(trifluoromethanesulfonyl)imide [LiNTf2] in deionised water at room temperature (Scheme 66) [201]. Interestingly, all the ionic liquids thus prepared turned out to be miscible with water, acetone, AcOEt, THF, and CHCl3, but immiscible with hexane.
Scheme 66.

Synthesis of imidazolium geminal dicationic ionic liquids 329a–f and 330d–f.
Notably, most of these compounds were found to possess significant antibacterial activity against six Gram-negative bacteria, namely E. coli, S. Typhimurium, P. aeruginosa, A. calcoaceticus, S. pyogenes, and S. aureus, and against four Gram-positive bacteria, namely B. subtilis, R. ruber, E. faecalis, and S. epidermidis. High antibacterial toxicity against all the tested bacteria was exhibited by the ionic liquid 329c with acetonitrile substituent. Moreover, ionic liquids 329d and 329e demonstrated bacterial inhibition of E. faecalis with an MIC value of 410 and 360 µM, respectively. It was also found that all ionic liquids, except 1,1′-[(tosylazanediyl)bis(ethane-2,1-diyl)]bis(3-propargyl-1H-imidazol-3-ium) bromide (329b), possessed significant antibacterial activities against β-lactam resistant P. aeruginosa with MIC values ranging from 200 to 500 µM. Finally, it was observed that 329c exhibited considerable antibacterial activity (MIC = 100 µM) against B. subtilis, a bacterial species that required a high dose (680 µM) of amoxicillin [201].
In 2020, Mangalagiu, Mangalagiu, and coworkers designed, synthesised, and evaluated the antimycobacterial activity of bis(imidazole)pyridines 331a–g [202]. The efficient and straightforward synthesis of these new quaternary salts was carried out by an approach in which a solution of 3 eq of 1H-imidazole (260) or 4-nitro-1H-imidazole (332) in a mixture of THF and DMF was treated with a suspension of 3.6 eq of NaH in THF and 1 eq of 2,6-bis(chloromethyl)pyridine (333) at room temperature. The resulting compounds 334a–g were then treated with a solution of 2.4–2.8 eq of α-halocarbonyl derivatives 335a–g giving rise to the required quaternary salts 331a–g in yields ranging from 50 to 92% (Scheme 67) [202].
Scheme 67.

Synthesis of bis(imidazole)-pyridines 331a–g.
These derivatives, which were soluble at a high concentration (200 μM) in microbiological medium, were evaluated for their in vitro antimycobacterial activity against M. tuberculosis H37Rv grown under aerobic conditions. Among the tested quaternary ammonium salts, 1,1′-[pyridine-2,6-diylbis(methylene)]bis{3-[2-(4-chlorophenyl)-2-oxoethyl]-1H-imidazol-3-ium} bromide (331e) and 1,1′-[pyridine-2,6-diylbis(methylene)]bis{3-[2-(4-nitrophenyl)-2-oxoethyl]-1H-imidazol-3-ium} bromide (331f) turned out to have an excellent anti-TB activity against M. tuberculosis H37Rv under aerobic condition with MIC values of 51 and 58 μM, respectively, and IC50 values of 34 and 38 μM, respectively. Furthermore, they proved to be not cytotoxic and exhibited a very good intracellular activity [202].
13. Pyridine/Imidazole Hybrids
In 2013, in the context of a study on the identification and development of new antibacterial substances effective against resistant bacterial strains, Renganathan and coworkers synthesised four imidazole-pyridine fluorophores, i.e., 2,6-bis[4-(4,5-diphenyl-1H-imidazol-2-yl)phenyl]pyridine (2PBI) (336), 3,5-bis[4-(4,5-diphenyl-1H-imidazol-2-yl)phenyl]pyridine (3PBI) (337), 2-[4-(tert-butyl) phenyl]-6-[4-(4,5-diphenyl-1H-imidazol-2-yl)phenyl]pyridine (TPBI) (338), and 2-[4-(4,5-diphenyl-1H-imidazol-2-yl)phenyl]-6-(4-methoxyphenyl)pyridine (MPBI) (339) (Figure 12), by using Pd(PPh3)4-catalysed Suzuki reactions followed by AcOH-mediated three component cyclisation reactions [203].
Figure 12.

Structures of imidazole-pyridine fluorophores 2PBI (336), 3PBI (337), TPBI (338), and MPBI (339).
As shown in Scheme 68, compounds 336 and 337 were synthesised by Pd(PPh3)-catalysed cross-coupling reaction of 2.0 eq of 4-formylboronic acid (340) with 1.0 eq of 2,6-dibromopyridine (341) and 1.0 eq of 3,5-dibromopyridine (342), respectively, in a mixture of THF and water at 70 °C for 12 h using K2CO3 as base followed by treatment of the resulting compounds 343 and 344, respectively, with 2 eq of benzil (161) and 8 eq of AcONH4 in AcOH under reflux for 4 h. In this way, compounds 336 and 337 were obtained in 40 and 41% overall yield, respectively [203].
Scheme 68.

Synthesis of hybrids 2PBI (336) and 3PBI (337).
A different protocol was instead used to prepare compounds 338 and 339. As shown in Scheme 69, 341 was treated with 1.09 eq of 340 in a mixture of THF and water at 70 °C for 12 h in the presence of 5 mol% Pd(PPh3)4 and 3 eq of K2CO3 providing 4-(6-bromopyridin-2-yl)benzaldehyde (345) in 72% yield. The Pd(PPh3)4-catalysed Suzuki coupling of 345 with an equimolar amount of 4-(tert-butyl)phenylboronic acid (346) gave 4-{6-[4-(tert-butyl)phenyl]pyridin-2-yl}benzaldehyde (347) in 78% yield. The subsequent cyclisation reaction of this pyridine derivative with 1.09 eq of benzil (161) and 7.3 eq of AcONH4 in AcOH at 120 °C for 4 h then gave imidazole-pyridine fluorophore 338 in 53% yield. On the other hand, the Pd(PPh3)4-catalysed Suzuki coupling of 345 with 4-methoxyphenylboronic acid (348) provided compound 349 in 81% yield. This 2,6-diarylpyridine was then converted to 339 in 58% yield by cyclisation with benzil (161) and AcONH4 in AcOH at 120 °C for 4 h (Scheme 69) [203].
Scheme 69.

Synthesis of imidazole-pyridine fluorophores TPBI (338) and MPBI (339).
UV-Vis and fluorescence studies demonstrated that hybrids 336, 337, 338, and 339 had excellent fluorescence properties with high quantum efficiencies, they were sensitive to the polarity of the microenvironment, and that the effect of solvent polarity on their absorption behaviour was minimal. In addition, studies on the in vitro antibacterial activity of these fluorophores revealed that 339 had relatively good inhibitory power against Gram-negative strains (E. coli MTCC 2939, P. aeruginosa MTCC 1934, Aeromonas hydrophila MTCC 1739), while 338 exhibited antibacterial activity against Gram-positive Rhodococcus rhodochrous MTCC 265 [203].
In 2015, Gomha and coworkers [204] reported the synthesis of pyridines 350a–e and 351a–e containing imidazole moiety and bipyridine derivatives 352, 353, and 354 in good yields via one-pot multicomponent condensation reactions [9] and evaluated the in vitro antibacterial activities of these compounds against Gram-positive and Gram-negative bacteria.
Compounds 350 were prepared in 66–72% yield by one-pot condensation of 1-(4-methyl-1-phenyl-2-sulfanyl-1H-imidazol-5-yl)ethan-1-one (355) [205], the appropriate aryl aldehydes 36, malononitrile (356), and AcONH4 in AcOH under reflux (Scheme 70) [204].
Scheme 70.

Synthesis of compounds 350a–e.
Compounds 351 were prepared in similar manner in 66–76% yield by one-pot condensation of 355 with aldehydes 36, ethyl cyanoacetate (357), and a molar excess of AcONH4 in refluxing AcOH (Scheme 71) [204].
Scheme 71.

Synthesis of compounds 351a–e.
The protocol involving the one-pot cyclocondensation reaction of Scheme 71 was then extended to the synthesis of bispyridine derivatives 352, 353, and 354. In particular, 2 eq of 355 were treated with 1 eq of terephthaldehyde (358), 2 eq of malononitrile (356), and 16 eq of AcONH4 in AcOH under reflux for 6–8 h, providing 4,4′-(1,4-phenylene)bis[2-amino-6-(4-methyl-1-phenyl-2-sulfanyl-1H-imidazol-5-yl)nicotinonitrile] (352) in 68% yield (Scheme 72). When ethyl cyanoacetate (357) and diethyl malonate (359) replaced malononitrile, the cyclocondensation reaction gave rise to bispyridine derivatives 353 and 354, respectively, in 66–67% yield (Scheme 72) [204].
Scheme 72.

Synthesis of bipyridine derivatives 352, 353, and 354.
The in vitro antibacterial activity of all the hybrids so synthesised was evaluated against the Gram-positive bacteria S. pneumoniae and B. subtilis, and the Gram-negative bacteria P. aeruginosa and E. coli. Ampicillin and gentamicin were used as the reference drugs for the Gram-positive bacteria and the Gram-negative bacteria, respectively. Compounds 350c and 350d were found to exhibit high inhibitory activity against S. pneumoniae while compounds 350a, 350b, 350e, and 351a showed moderate inhibitory effects. Imidazole derivatives 350c, 350d, 351a, 351e, 352, 353, and 354 exhibited high inhibitory effects against B. subtilis. For example, the zone of inhibition of 350c was 20.8 ± 0.6 mm and that of ampicillin was 32.4 ± 0.3 mm. Furthermore, compounds 350c, 350d, 351a, 351e, and 354 exhibited high inhibitory effects against E. coli. For example, the zone of inhibition of 350d was 19.9 ± 0.4 mm and that of the reference compound, gentamicin, was 19.9 ± 0.3 mm [204].
Finally, in 2019, Narayana and coworkers synthesised 2-(2-aryl-4,5-diphenyl-(1H-imidazol-1-yl)-6-methylpyridines 360a–d by one-pot cyclocondensation of benzil (161) with aryl aldehydes 36a–j, 6-methylpyridin-2-amine (361), and AcONH4 in glacial acetic acid at 60 °C using conventional heating for 3 h or microwave irradiation for 20–25 min in the presence of ZnO nanoparticles (10 mol%) as effective catalyst (Scheme 73) [166]. The yields ranged from 60 to 72% using conventional heating and were higher (72–80%) when the reactions were carried out under MW irradiation. It is also worth noting that in the paper by Narayana and coworkers the structure of 361 was wrongly reported. Compounds 360a–d were then evaluated for their in vitro antibacterial activity using the resazurin reduction assay and it was found that the IC50 values of 360a towards S. aureus and E. coli were 96 and 47 µM, respectively [166].
Scheme 73.

Synthesis of 2-(2-aryl-4,5-diphenyl-1H-imidazol-1-yl)-6-methylpyridines 360a–d.
14. Quinoline/Imidazole Hybrids
Quinoline derivatives including 4-quinolones have attracted considerable attention because of their presence in the skeleton of several antibacterial drugs [206,207,208,209,210,211]. In recent years, efforts have also been directed to the design, synthesis, and evaluation of the antimicrobial properties of quinoline-imidazole derivatives.
In 2012, Parab and Dixit synthesised and characterised 2-chloro-3-formyl-7-methylquinoline/imidazole hybrids 362a–h based on 2-chloro-7-methylquinoline-3-carbaldehyde (363) and tested their antibacterial activities against the Gram-negative bacteria E. coli and P. aeruginosa and the Gram-positive bacteria Bacillus subtilis and B. megaterium [212]. As shown in Scheme 74, one of these hybrid, (Z)-3-(3-aminophenyl)-5-[(2-chloro-7-methyl-3-quinolinyl) methylene]-2-phenyl-3,5-dihydro-4H-imidazol-4-one (362a) was synthesised in 82% yield by refluxing 1 eq of 2-chloro-7-methylquinoline-3-carbaldehyde (363) and 1 eq of hippuric acid (364) in Ac2O with freshly prepared AcONa for 2 h, followed by addition of the resulting compound 365 to a solution of diamine 366a in EtOH containing few drop of glacial AcOH and subsequent heating. Compound 363 was in turn synthesised by a Vilsmeier–Haack reaction according to a procedure reported in the literature [213,214]. The other 2-chloro-7-methylquinoline-3-carbaldehyde-based imidazole derivatives, 362b–h, were prepared in a similar manner from 363 by using diamines 366b–h, but, unfortunately, the yields of many reactions illustrated in Scheme 74 were not reported.
Scheme 74.

Synthesis of 2-chloro-7-methylquinoline-3-carbaldehyde-based imidazole derivatives, 362a–h.
Nevertheless, compounds 362a–h were evaluated for their in vitro antibacterial activities and the results of these tests showed that hybrids 362a–c exhibited moderate inhibition against the above-mentioned Gram-negative bacterial species and especially against E. coli, while all hybrids displayed good activity against the Gram-positive organisms. In particular the inhibition zones of 362b and 362c against B. megaterium were 28 and 30 mm, respectively, while the inhibition zone of streptomycin, the reference drug, was 36 mm [212].
In 2014, Desai and coworkers described the synthesis and characterisation of (Z)-N-{4-[(2-chloroquinolin-3-yl)methylene]-5-oxo-2-phenyl-4,5-dihydro-1H-imidazol-1-yl} arenamides 367a–l (Scheme 75) and screened the in vitro antimicrobial activity of these quinoline based imidazole derivatives against E. coli MTCC 443, P. aeruginosa MTCC 1688, S. aureus MTCC 96, and S. pyogenes MTCC 442 [215]. (Z)-4-[(2-Chloroquinolin-3-yl)methylene]-2-phenyloxazol-5(4H)-one (368), which was the key synthetic intermediate, was prepared in 68% yield by microwave-promoted reaction of 2-chloroquinoline-3-carbaldehyde (369) [215] with hippuric acid (364). Compounds 367 were then prepared in 63–73% yield by heating 368 with equimolar amounts of the appropriate arene carbohydrazides 370 in pyridine in an oil bath at 150–155 °C for 6–8 h. Nevertheless, higher yields (69–78%) of compounds 367a–l were obtained by reaction between 368 and 370 under irradiation in a microwave oven at 400 W for 3–6 min.
Scheme 75.

Synthesis of (Z)-N-{4-[(2-chloroquinolin-3-yl)methylene]-5-oxo-2-phenyl-4,5-dihydro-1H-imidazol-1-yl}arenamides 367a–l.
The results of the evaluation of the antibacterial activity of hybrids 367a–l showed that: (i) compounds 367d and 367h exhibited activity (MIC = 50 μM) against E. coli higher than that of ampicillin, the reference drug (MIC = 290 μM); (ii) compounds 367h and 367j were highly potent against P. aeruginosa with MIC values of 100 and 110 μM, respectively; (iii) compounds 367f and 367i with an MIC value of 210 μM for each of them had remarkable antibacterial activity against S. aureus; and (iv) compound 367d showed significant potency (MIC = 100 μM) against S. pyogenes [215], an aerotolerant Gram-positive bacterium that is the cause of strep throat.
Still in 2014, Zhou and coworkers [216] designed and synthesised two novel series of 4-quinolone/imidazole hybrids 371 and 372. These compounds were prepared according to the route outlined in Scheme 76. In particular, arenamines 366 were treated with commercially available ethoxymethylene malonic acid ester 373 in EtOH under reflux providing compounds 374 in almost quantitative yields. Treatment of these intermediates with diphenyl ether under reflux for 2 h afforded quinolones 375 in yields ranging from 49 to 60%. The subsequent reaction of these compounds with a large molar excess of 2-(chloromethyl)oxirane (95) under reflux for 2–3 h in the presence of K2CO3 gave the ethyl 1-(oxiran-2-ylmethyl)-4-oxo-1,4-dihydroquinoline-3-carboxylate derivatives 376 in 81–95% yield. These compounds were then treated with 4-nitro-1H-imidazole (332) in EtOH under reflux for 2 h in the presence of K2CO3 providing compounds 377 in 80–99% yield. Finally, saponification of these derivatives with 3% NaOH at 100 °C for 2 h, followed by acidification with formic acid to pH 7 afforded hybrids 371 in yields ranging from 78 to 93%. Similarly, the reaction of compounds 376 with 2-methyl-5-nitro-1H-imidazole (64) in refluxing EtOH in the presence of K2CO3 provided compounds 378 in 81–89% yield. Finally, saponification of these derivatives followed by acidification produced hybrids 372 in yields ranging from 81 to 90% [216].
Scheme 76.

Synthesis of 4-quinolone/imidazole hybrids 371 and 372.
Subsequently, hybrids 371 and 372 and their direct precursors, compounds 377 and 378, respectively, were evaluated for their antibacterial activities against the Gram-positive bacteria S. aureus ATCC 25923, S. pneumoniae, S. aureus N315, M. luteus ATCC 4698, MRSA, B. subtilis ATCC 6633, and against the Gram-negative bacteria P. aeruginosa, E. coli DH52, S. dysenteriae, and S. Typhi [217] and it was found that many of the tested compounds, at low concentrations, exhibited good or even higher antimicrobial activities in comparison with the reference drugs chloramphenicol [50] and norfloxacin. It was also discovered that 1-[2-hydroxy-3-(2-methyl-5-nitro-1H-imidazol-1-yl)propyl]-4-oxo-7-trifluoromethyl-1,4-dihydroquinoline-3-carboxylic acid (372d) had in vitro an MIC value of 2 μM against MRSA and was more active than its ethyl ester precursor, 378d. Hybrid 372d also had an MIC value of 9 μM against S. pneumoniae and was more potent than chloramphenicol, which had an MIC value of 50 μM. The results of the study by Zhou and coworkers finally showed that hybrids bearing a 2-methyl-5-nitroimidazolyl moiety exhibited antibacterial activities higher than those of hybrids bearing a 4-nitroimidazolyl moiety [216]. Zhou and coworkers also investigated the mechanism of action of 372d by studying the interactions of this hybrid with calf thymus DNA and found that non-covalent interaction between 372d and TOPO IV DNA complex, especially hydrogen bonds between 372d and Ser79, was involved [216].
In 2015, Zhou and coworkers, as part of their studies on the synthesis and biological evaluation of quinolone/azole hybrids [61], synthesised several 4-quinolone/imidazole hybrids 379a–j and 380a–e as new types of antimicrobial agents [218]. Hybrids 379 were synthesised using the route shown in Scheme 77. In particular, norfloxacin (381a, R4 = Et) was treated with an equimolar amount of 2-(chloromethyl)oxirane (95) in MeCN at 50 °C for 10 h in the presence of NaHCO3 and then treated with formic acid to adjust the pH value to 5.5–6.5 providing 1-ethyl-6-fluoro-7-[4-(oxiran-2-ylmethyl)piperazin-1-yl]-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (382a) in 67% yield. Using similar experimental conditions compound 381b (R4 = cyclopropyl) was converted to carboxylic acid 382b in 64% yield. Subsequently, a mixture of 382a and 2-methyl-5-nitro-1H-imidazole (64) in MeCN was stirred at 75 °C in the presence of K2CO3, then cooled to room temperature and treated with formic acid to adjust the pH value to 5.5–6.5 providing compound 379a in 27% yield. A similar procedure was then used to prepare hybrids 379b–j from 382b and the appropriately substituted free-NH imidazoles [218].
Scheme 77.

Synthesis of hybrids 379a–j.
On the other hand, hybrids 380a–e were synthesised in two steps from the fluoroquinolone antibiotic clinafloxacin (381c) via the route shown in Scheme 78 that involved the use of a protocol similar to that developed for the synthesis of hybrids 379a–j [218].
Scheme 78.

Synthesis of hybrids 380a–e.
Hybrids 379a–j and 380a–e were then tested in vitro for their antimicrobial activities against the Gram-positive bacteria M. luteus ATCC 4698, MRSA N315, S. aureus ATCC 25923, and B. subtilis ATCC 2126 and against the Gram-negative bacteria P. aeruginosa, E. coli JM109, P. hauseri [60], and S. Typhi [217]. Chloramphenicol, norfloxacin, ciprofloxacin, and clinafloxacin were used as reference drugs. Most hybrids exhibited good bioactivities and compounds 379a, 379e, 379g, 379j, 380b, and 380c exhibited remarkable activity against MRSA with MIC values ranging from 0.39 to 6.6 µM, which were lower than that of chloramphenicol (MIC = 36 µM). With regard to units of measurement that were used to express MIC values, we wish to point out that in our opinion the values of these measurements were expressed by the authors using incorrect units instead of reporting them using µM values.
Notably, hybrid 380b was found to inhibit the growth of all bacterial strains tested with MIC values ranging from 97 nM to 1.4 µM and demonstrated excellent activity against E. coli with an MIC value of 97 nM. Moreover, the tested quinolone/imidazoles were found to inhibit induced bacterial resistance more slowly than clinical drug controls also exhibiting low cytotoxicity to human cells. The interactions of hybrid 380b, Cu2+, and MRSA DNA were also investigated, and it was discovered that 380b could intercalate into DNA through Cu2+ bridge to form a steady 380b, Cu2+, and MRSA DNA ternary complex, which might further block DNA replication. It was finally found that 380b could be effectively stored and carried by human serum albumin [218].
In 2016, Al-Qawasmeh, Al-Tel, and coworkers synthesised quinoline/imidazole hybrids 383a–h, 384a–h, 385a–h, and 386a–h and tested the activity of these 4-(4,5-diaryl-1H-imidazol-2-yl)-2-arylquinoline derivatives against a range of bacterial strains [219]. These hybrids were synthesised through the route shown in Scheme 79, in which the first step involved the condensation reaction of 1.0 eq of isatin (387) with 1.05 eq of acetophenones 318 in the presence of 3.2 eq of KOH and 20% aqueous EtOH under irradiation by MW with variable power to keep a constant temperature at 130 °C for 12 min, followed by acidification with AcOH.
Scheme 79.

Synthesis of quinoline/imidazole hybrids 383a–h, 384a–h, 385a–h, and 386a–h.
Esterification of the resulting carboxylic acids 388 with EtOH under microwave irradiation in the presence of conc. sulfuric acid produced esters 389, which were reduced using NaBH4 in EtOH, providing alcohols 390. Subsequent oxidation of these intermediates by treatment with SeO2 in dioxane under reflux gave aldehydes 391 in high yields. Finally, the reaction of these compounds with α-diketones 392 and AcONH4 in AcOH under microwave irradiation led to the required hybrids 383, 384, 385, and 386 [219].
These compounds were then tested against the Gram-negative bacteria E. coli ATCC 25922, ATCC strains of K. pneumoniae, P. vulgaris clinical isolate, P. aeruginosa clinical isolate, as well as against Gram-positive pathogens including two strains of S. aureus (methicillin-sensitive and methicillin-resistant) and S. epidermidis and turned out to be generally more active against Gram-positive bacterial species than Gram-negative bacteria, although most of these hybrids did inhibit the growth of E. coli and P. aeruginosa in a low MIC range. Only hybrids 384b, 384c, 384h, 386c, and 386h inhibited E. coli with MIC values less than 24–34 µM and were more potent than ceftriaxone, a third-generation cephalosporin, which was used as a positive control (56 µM). It was also observed that the most potent hybrids possessed hydrophobic and electron-withdrawing functionalities (384b, 384c, 386b, and 386c), such as halogens, on their aryl arms [219].
In 2018, Patel and coworkers synthesised imidazole derivatives 393a–f bearing quinoline nucleus and screened these compounds for their antimicrobial and antitubercular activities [220]. As outlined in Scheme 80, their optimised synthesis began with the reaction of 2-chloroquinoline-3-carbaldehydes 394 with phenols 395a and 395b in DMF in the presence of K2CO3. The subsequent reaction of the resulting 2-aryloxy-6-quinoline-3-carbaldehydes 396 with an equimolar amount of benzoin (397) or benzil (161) and 5 eq of AcONH4 in AcOH under reflux, in the presence of a catalytic amount of CAN, provided target hybrids 393 in yields ranging from 75 to 91%.
Scheme 80.

Synthesis of hybrids 393a–f.
These compounds were screened against three Gram-positive bacteria, i.e., B. subtilis MTCC 441, Clostridium tetani MTCC 449, and S. pneumoniae MTCC 1936, and three Gram-negative bacteria, i.e., E. coli MTCC 443, S. Typhi MTCC 98, and Vibrio cholerae MTCC 390, using ampicillin, ciprofloxacin, and chloramphenicol as standard antibacterial drugs. Among the tested compounds, hybrid 393c displayed high activity against B. subtilis and E. coli with MIC values of 120 μM comparable to that of chloramphenicol (MIC = 150 μM). Hybrids 393e and 393f displayed high activity against S. Typhi with MIC values of 220 and 210 μM, respectively, and hybrid 393c displayed activity against V. cholerae that, on a molar basis, was lower than that of ampicillin (MIC = 500 and 720 μM, respectively). It was also observed that the in vitro antitubercular activity of several hybrids 393 against the culture of M. tuberculosis H37Rv strain was generally significantly lower than that of the standard drug isoniazid, for which the percentage of inhibition at a concentration of 1.8 mM was 99%. Only hybrid 393c displayed 84% inhibition [220]. Molecular docking studies were also performed to highlight the interaction of hybrids 393 with GlcN-6-P synthase enzyme and it was found that the antimicrobial compounds 393a, 393c, and 393d exhibited the least binding energy among the tested hybrids [220].
In 2019, Insuasty, Abonia, and coworkers synthesised quinoline-based hydroxyimidazolium hybrids 398a–h and evaluated in vitro their antimicrobial activity against two Gram-negative microorganisms, E. coli and K. pneumoniae, Gram-positive S. aureus, and two acid fast slow-growing mycobacteria, Mycobacterium tuberculosis H37Rv and M. bovis BCG [221]. As shown in Scheme 81, the synthesis of hybrids 398 involved the conversion of 2-chloroquinoline-3-carbaldehydes 399 to 2-oxo-1,2-dihydroquinoline-3-carbaldehydes 400, followed by alkylation of the latter derivatives with n-BuBr or BnBr in DMF in the presence of K2CO3. Aldehydes 401 were then subjected to reaction with 3-butyl-1-methylimidazolium chloride (402), in MeCN at 80 °C in the presence of AcONa under ultrasonic irradiation to afford the required hybrids 398 in 60–91% yield [221,222]. Compounds 400 were in turn prepared by a Meth–Cohn reaction [223].
Scheme 81.

Synthesis of quinoline-based hydroxyimidazolium hybrids 398a–h.
Evaluation tests of antibacterial activity showed that almost all hybrids 398 exhibited low activity against the Gram-negative organisms, but hybrid 398b demonstrated a potent anti-staphylococcal activity with an MIC value of 5 μM. It was also found that hybrids 398a and 398b possessed significant inhibitory activity against M. tuberculosis H37Rv with MIC values of 46 and 24 μM, respectively. The cell wall of this bacterium, which is the causative agent of tuberculosis, has characteristics of both Gram-positive and Gram-negative bacteria. On the other hand, hybrid 398b at concentrations ranging from 30 to 500 μM demonstrated 100% inhibition of S. aureus and at 300 μM demonstrated 100% inhibition of K. pneumoniae, and hybrids 398d, 398e, and 398h displayed moderate growth inhibition against the virulent H37Rv strain with MIC values of 110 μM [221].
15. Pyrimidine/Imidazole Hybrids
In recent years, a large number of studies has been done on the synthesis and evaluation of the antibacterial properties of pyrimidine derivatives [224,225,226,227,228,229,230,231] and in this context significant attention has been paid to the identification, synthesis, and evaluation of the antibacterial properties of pyrimidine/imidazole hybrids with potentially significant activity [232,233,234,235].
In 2011, Rathod synthesised substituted ethyl 3-(4,5-diphenyl-1H-imidazol-2-yl)-6-methyl-2-oxo/sulfanylidene-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carboxylates 403a–g by condensation of benzil (161) with substituted ethyl 3-formyl-6-methyl-2-oxo/sulfanylidene-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carboxylates 404, and AcONH4 in the presence of acidic alumina and glacial AcOH under reflux for 12 h using conventional heating (Method A) or under microwave irradiation for 8 min (Method B) (Scheme 82) [232]. The yields of the required imidazolyl pyrimidines, which were higher using this second method, ranged from 67 to 79%.
Scheme 82.

Synthesis of imidazolyl pyrimidines 403a–g.
The antibacterial activity of compounds 403a–g was assayed against S. Typhi, P. aeruginosa, K. pneumoniae, and S. aureus using norfloxacin as the standard drug. Among the tested compounds at 100 µg/mL, 403d with a zone of inhibition of 12 mm was the most active against S. aureus. It was also observed that S. Typhi was highly sensitive to 403b (zone of inhibition = 12 mm), and that 403f exhibited maximum zone of inhibition (18 mm) against K. pneumoniae and minimum zone of inhibition against all other bacterial strains tested [232].
In 2014, Desai and coworkers described the synthesis of hybrid molecules 405 containing pyrimidine-based imidazole scaffolds and tested the antibacterial activity of these N-(4-arylidene-5-oxo-2-sulfanyl-4,5-dihydro-1H-imidazol-1-yl)-6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carboxamides against E. coli and S. aureus [233].
The synthesis of hybrids 405 was achieved as outlined in Scheme 83 starting from ethyl 6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carboxylate (406), which was prepared by reaction of benzaldehyde with urea and ethyl acetoacetate in dioxane under reflux in the presence of conc. HCl. The reaction of 406 with hydrazine hydrate in dioxane in the presence of a catalytic amount of conc. H2SO4 at 100 °C gave compound 407, which was treated with KSCN in acidic medium, affording carbothioamide 408. The subsequent reaction of 408 with chloroacetic acid (409) and AcONa in AcOH under reflux provided compound 410, which was eventually converted into the required hybrids 405 by a Knoevenagel-type condensation reaction with the appropriately substituted aryl aldehydes 36 in EtOH in the presence of EtONa. The reaction, which was carried out using a Dean-Stark apparatus, provided hybrids 405 with yields ranging from 58 to 80%. All these hybrids were presumed to mainly possess a Z-configuration [233].
Scheme 83.

Synthesis of hybrids 405.
The results of an antibacterial screening of compounds 405, in which ciprofloxacin was used as the standard drug, showed that (i) both 405b (Ar1 = 2-FC6H4) and 405d (R = 2-HOC6H4) exhibited good activity against E. coli and that the antimicrobial activity of hybrids 405f (Ar1 = 4-ClC6H4) and 405j (Ar1 = 2,6-Cl2C6H3) (MIC = 53 and 50 μM, respectively) against E. coli was lower than that of ciprofloxacin (75 μM); (ii) hybrid 405j had an MIC value of 25 μM against S. aureus that was, on a molar basis, six times lower than that of ciprofloxacin (150 μM) [233].
Finally, in 2016, Pathan and Rahatgaonkar [234] repeated the synthesis of imidazole/pyrimidine hybrids 403a–g, the preparation of which had been reported for the first time in 2011 by Rathod (cf. Scheme 82) [232], and studied the docking of these compounds into the active site of cytochrome P450 (14DM) 14α-sterol demethylase CPY51 enzyme. Hybrids 403a, 403b, 403c, 403d, which was the most active hybrid against S. aureus, and 403f were found to show the highest binding with P450 (14DM) in comparison to selective drug fluconazole [234].
16. Addendum
In recent years, some antibacterial molecular hybrids and conjugates bearing imidazole moiety, which have not been mentioned in the previous sections of this review, have been identified, synthesised, and evaluated for their bioactivity. The literature data of some of these compounds have been summarised below.
In 2011, Abdel-Wahab and coworkers [235] used 1-(5-methyl-2-phenyl-1H-imidazol-4-yl) ethan-1-one (411) [236] as a precursor for the synthesis of (Z)-2-{[(E)-1-(5-methyl-2-phenyl-1H-imidazol-4-yl)ethylidene]hydrazinylidene}thiazolidin-4-one (412) and (Z)-2-{[(E)-1-(5-methyl-2-phenyl-1H-imidazol-4-yl)ethyilidene]hydrazinylidene}-4-phenyl-2,5-dihydrothiazole (413). The reactions used for the synthesis of compounds 412 and 413 are shown in Scheme 84.
Scheme 84.

Synthesis of imidazole-based heterocycles 412 and 413.
These imidazole-based heterocycles proved to exhibit strong antibacterial activities against B. subtilis ATCC 6633 and S. aureus ATCC 29213, and hybrid 412 turned out to exhibit strong activity (MIC = 70 µM) also against K. pneumoniae ATCC 13883 [235].
In 2012, Kantevari and coworkers [237], taking into account that onychine (414) (Figure 13), a naturally occurring 4-azafluorenone (5H-indeno[1,2-b]pyridin-5-one) alkaloid isolated from the root of the plant Polyalthia debilis (Pierre) Finet & Gagnep. [238], exhibits antimicrobial activity against S. aureus, B. subtilis, and E. coli (MIC ≥ 500 μM) [239], synthesised 1-benzyl-2-butyl-4-chloroimidazole–embodied 4-azafluorenone hybrids 415.
Figure 13.
Structure of onychine (414).
These compounds were prepared in good yields by one pot condensation of 1-benzyl-2-butyl-4-chloro-1H-imidazole-5-carbaldeyde (416) with equimolar amounts of 1,3-indanedione (417) and methyl aryl ketones 418, and 2.5 eq of AcONH4 in DMF under reflux for 3–4 h (Scheme 85) [237].
Scheme 85.

Synthesis of 2-aryl-4-(1-benzyl-2-butyl-4-chloro-1H-imidazol-5-yl)-5H-indeno [1,2-b]pyridin-5-ones 415.
2-Aryl-4-(1-benzyl-2-butyl-4-chloro-1H-imidazol-5-yl)-5H-indeno[1,2-b]pyridin-5-ones 415 were then screened for their antibacterial activity against the Gram-positive bacteria B. subtilis and S. aureus and the Gram-negative bacteria E. coli, P. aeruginosa, and K. pneumoniae, and it was found that compounds 415a (Ar1 = Ph), 415c (Ar1 = 4-BrC6H4), 415f (Ar1 = 3,4,5-(MeO)3C6H2), 415g (Ar1 = 1-naphthyl), 415h (Ar1 = 2-naphthyl), 415j (Ar1 = 5-Br-2-thienyl), 415k (Ar1 = 2-furyl), 415l (Ar1 = 5-Br-2-pyridyl), and 415n (Ar1 = 2-dibenzothienyl) exhibited good activity towards S. aureus. Furthermore, compound 415k proved to display the highest zone of inhibition against S. aureus (15 mm), P. aeruginosa (14 mm), and K. pneumoniae (15 mm), while the standard drug gentamicin had inhibition zones of 15, 15, and 19 mm, respectively, against these bacterial strains [237].
In 2013, Kim and coworkers synthesised 3α-amino-5α-cholestane derivatives 419a–c containing imidazole rings and 3α,7α-diamino-5α-cholestane derivatives 420a–c containing imidazole rings starting from 5α-cholestane-3,7-dione (421) and evaluated the antimicrobial activity of these imidazole appended cholestane based-conjugates against a range of Gram-positive and Gram-negative bacterial strains [240]. The synthesis of conjugates 419 and 420 was carried out as illustrated in Scheme 86.
Scheme 86.

Synthesis of imidazole appended cholestane based-conjugates 419a–c and 420a–c.
Conjugates 419 were prepared by the one-step reductive amination of 421 with the appropriate primary amines hydrochlorides 422, Et3N, and sodium tris(2-ethylhexyloxy)borohydride [NaBH(OEh)3] in THF at room temperature. On the other hand, conjugates 420 were then obtained by treatment of compounds 419 with 3 eq of the appropriate amine hydrochlorides 422 and 3 eq of NaBH3CN in a 1:1 mixture of THF and MeOH at room temperature [240].
All the conjugates thus synthesised were evaluated for their antibacterial activities and it was found that compounds 419a–c showed activity against most of the Gram-positive bacteria and that 3-propylimidazole conjugate 419b was four times potent against S. aureus 77 and B. subtilis ATCC 6633 than compound 419a, whereas the latter compound exhibited two and four times higher potency against S. aureus 241 and E. faecalis ATCC 29212 strains, respectively, than compound 419b. Notably, 3,7-di(imidazole) conjugates 420a–c exhibited antimicrobial potency higher than imidazole conjugates 419a–c. In fact, compound 420b with MIC values from 4 to 8 μM showed 2–8 times higher activity than compound 419b against all Gram-positive bacteria (S. aureus ATCC 6538P, S. aureus giorgio (a methicillin-sensitive strain), S. aureus 77, S. aureus 241, S. epidermidis 887E, E. faecalis ATCC 29212, M. luteus ATCC 9341, Bacillus subtilis ATCC 6633, and B. licheniformis EMR). However, conjugates 419 and 420 did not exhibit significant activity against Gram-negative bacteria, which were resistant to these compounds [240].
In 2016, Zhou and coworkers [241], considering that berberine (70), an isoquinoline alkaloid isolated from C. chinensis and Berberis spp., is a versatile drug in treatment of common metabolic diseases [242] and is a broad spectrum antimicrobial agent [45,243,244,245], synthesised and characterised berberine-derived imidazoles 423a–j and evaluated their in vitro antibacterial activity against the Gram-negative bacteria E. coli, P. hauseri [60], P. aeruginosa, and S. Typhi [217] and the Gram-positive bacteria MRSA, S. aureus, B. subtilis, and M. luteus. As shown in Scheme 87 the synthesis of hybrids 423 was carried out by the rarely reported structural modification of the C-12 position in berberine backbone [246,247]. In particular, selective demethylation of berberine chloride (70) by heating at 190 °C under vacuum for 0.5 h provided berberrubine chloride (74) in 88% yield. The subsequent reaction of 5 eq of imidazoles 424a–j with 5 eq of formaldehyde aqueous solution and 1 eq of 74 in anhydrous n-butanol at 110 °C for 5 h in the presence of a catalytic amount of HCl afforded the required hybrids 423a–j in yields ranging from 42 to 55%.
Scheme 87.

Synthesis of C-12-substituted berberine/imidazole hybrids 423a–j.
Most of these hybrids turned out to exhibit good bioactivity against the tested bacteria and especially compound 423a with an MIC value of 2 μM against S. Typhi [217] was found to be much more active than the reference drugs berberine (1.4 mM), chloramphenicol [50] (100 μM), and norfloxacin (13 μM). In this study it was also revealed that hybrid 423a could effectively intercalate into calf thymus DNA to form 423a–DNA complex, which might further block DNA replication to exert the powerful antimicrobial activities [241].
Still in 2016, Idrees and coworkers synthesised and evaluated the antibacterial activity of N-(4-aryilidene-5-oxo-2-phenyl-4,5-dihydro-1H-imidazol-1-yl)-5-(benzofuran-2-yl)-1-phenyl-1H-pyrazole-3-carboxamides 425a–g [248]. These compounds were synthesised in 76–90% yield by treatment of 4-arylidene-2-phenyloxazol-5(4H)-ones 426 with an equimolar amount of 5-(benzofuran-2-yl)-1-phenyl-1H-pyrazole-3-carbohydrazides 427 in AcOH under reflux for 9 h (Scheme 88).
Scheme 88.

Synthesis of N-(4-aryilidene-5-oxo-2-phenyl-4,5-dihydro-1H-imidazol-1-yl)-5-(benzofuran-2-yl)-1-phenyl-1H-pyrazole-3-carboxamides 425a–g.
Compounds 425 were tested for their antimicrobial activity in vitro at different concentrations against Gram-positive bacteria Bacillus thuringiensis and S. aureus and Gram-negative bacteria E. coli and Enterobacter aerogenes. Chloramphenicol was used as a standard drug. It was found that compound 425a at a concentration of 230 μM showed excellent activity against B. thuringiensis (zone of inhibition = 18 mm) and moderate activity against S. aureus (zone of inhibition = 12 mm) and against E. coli (zone of inhibition = 15 mm). However, this compound was inactive against E. aerogenes at concentrations lower than 450 µM [248].
In 2017, Ishar and coworkers synthesised and characterised novel 3-(4,5-diphenyl-1H-imidazol-2-yl)-4H-chromen-4-ones 428 by one pot condensation of substituted 4-oxo-4H-chromene-3-carbaldehydes 429, benzil (161), and AcONH4 in refluxing AcOH under N2 atmosphere (Scheme 89) [249]. The subsequent reaction of compounds 428, which were obtained in yields ranging from 78 to 84%, with allyl bromide (430) in DMF at room temperature for 24 h in the presence of fused K2CO3 furnished 3-(1-allyl-4,5-diphenyl-1H-imidazol-2-yl)-4H-chromen-4-ones 431 in yields ranging from 50 to 62%.
Scheme 89.

Synthesis of 3-(4,5-diphenyl-1H-imidazol-2-yl)-4H-chromen-4-ones 428 and 3-(1-allyl-4,5-diphenyl-1H-imidazol-2-yl)-4H-chromen-4-ones 431.
Compounds 428 and 431 were then evaluated for their antibacterial activity against Gram-positive S. aureus MTCC 96 and B. subtilis MTCC 2451, and Gram-negative E. coli MTCC 82 and P. aeruginosa MTCC 2642. It was thus found that compound 431a (R1 = R2 = R3 = H) and compound 428c (R1 = R2 = H, R3 = Cl) exhibited excellent inhibitory activity against B. subtilis with MIC values of 2.4 and 3.2 μM, respectively. Compound 431a also displayed significant inhibitory activity against E. coli with an MIC value of 2.8 μM [249].
17. Conclusions
Over the past few decades, the growing demand for new antimicrobials for human use that can act against emerging pathogens and multidrug resistant bacterial strains non-treatable by the current antibiotics and responsible for the current increase in morbidity, mortality, and longer hospitalisation, has stimulated scientists to develop a variety of protocols for the synthesis of molecular hybrids and conjugates bearing imidazole moiety and to evaluate the antibacterial activity of these compounds.
In this unprecedented review with 261 references, we have illustrated and commented on the results obtained from the end of 1990s until the end of February 2020 in studies concerning the synthesis, characterisation, and evaluation of the antibacterial activities over 760 non-condensed molecular hybrids and conjugates bearing imidazole moiety, which include natural products such as oroidin (245a) [153,154,155,156], clathrodin (245b) [152], clathridimine (228) [142], and the enantiomer of the first naturally occurring dimeric pyrrole/imidazole hybrid of naturally occurring sceptrin (213) [134,135].
As regards the synthetic methodologies used in these studies, it is worth noting that multicomponent condensation reactions, a class of benign processes that are characterised by experimental simplicity and low cost, have frequently been used for the synthesis of a variety of hybrids and conjugates [9,93,97,145,164,166,176,177,178,219,220,232,237], and that such processes [250,251,252,253] as well as several other condensation reactions have often been preferentially carried out using microwave (MW)-promoted reactions instead of conventional heating sources [254,255,256,257,258]. In fact, as often highlighted by the published data, the advantages of microwave-promoted reactions [93,144,166,177,219,232] include significant shorter reaction times, higher yields, and product purity when compared to conventional thermal methods.
Even copper(I)-catalysed alkyne-azide cycloadditions [258,259] have been frequently employed for the efficient introduction of a 1,2,3-triazole moiety into the desired imidazole hybrids and conjugates [32,59,177,178]. On the other hand, classical Pd-catalysed Suzuki-type reactions have only been used for the preparation of imidazole-pyridine fluorophores 2PBI (336), 3PBI (337), TBPI (338), and MPBI (339) [203]. Instead, atom economic strategies involving Pd-catalysed regioselective direct (hetero)arylation reactions of heteroaromatics with (hetero)aryl halides or pseudohalides [9,10,11,12,13,14,15], which have been and are still frequently used in the chemistry of imidazole derivatives, as well as the van Leusen reaction based of tosylmethylisocyanides, which is one of the most appropriate strategies for the preparation of imidazole-based medicinal molecules [260], to the best of our knowledge, have never been used for the synthesis of hybrids and conjugates belonging to the classes of substances mentioned in this review.
Many results achieved in the study of the antibacterial properties of the hybrids and conjugates mentioned in this review are also worthy of mention. In fact, numerous compounds, some of which are mentioned below, were proven to exhibit better antibacterial properties than the reference compounds used as standards. In particular, 5-nitroimidazole/3-sulfanyl-1,2,4-triazole hybrids 41c and 41e proved to possess MIC values against C. sporogenes lower than that of metronidazole (1), the reference compound [35]. Nitroimidazole/berberine hybrid 71g had antibacterial potency against Gram-negative bacteria S. dysenteriae and P. vulgaris ATCC 6896 superior to that of the reference drugs norfloxacin and chloramphenicol, [44,50]. (E)-N’-(1-(4-Bromophenyl)-2-(2-methyl-5-nitro-1H-imidazol-1-yl)ethylidene)isonicotinohydrazide (78) was found to exhibit inhibitory activity against E. coli ATCC 35218 that was similar to that of both standards kanamycin B and penicillin G [51]. Quinolone-imidazole hybrid 93i possessed antibacterial activity against P. aeruginosa, one of the ESKAPE pathogens [261], with an MIC value that was significantly lower than that of the reference drugs chloramphenicol, norfloxacin, ciprofloxacin, and clinafloxacin [61]. Naphthalimide-derived metronidazole 112b proved capable to effectively inhibit the growth of both P. vulgaris and S. dysenteriae, to kill these bacteria, and to prevent bacterial resistance [65]. Naphthalimide-derived metronidazole 138e showed high activity against A. baumannii (an aerobic Gram-negative ESKAPE pathogen that causes serious infections in the lungs, blood, and brain) with rapid killing effect and no obvious resistance development [74,261]. Imidazole containing bisazetidinones 195a and 196b were shown to possess activity against the Gram-negative ESKAPE pathogen K. pneumoniae [262] that was almost equivalent to that of ampicillin [121], an antibiotic used to prevent and treat a number of bacterial infections. 1,8-Naphthalimide-derived imidazole 302b proved to exhibit activity against E. coli and P. aeruginosa that was comparable to that of the standard drug chloramphenicol [77]. 1,1′-[(Tosylazanediyl)bis(ethane-2,1-diyl)]bis(3-cyanomethyl-1H-imidazol-3-ium) chloride (329c) was found to be considerably more potent than antibiotic amoxicillin against Gram-positive B. subtilis [201]. Quinoline-based hydroxyimidazolium hybrid 398b demonstrated potent anti-staphylococcal activity and significant activity against M. tuberculosis H37Rv [223]. C-12 Substituted berberine-imidazole hybrid 423a was proven to exhibit activity against the Gram-negative bacterium S. Typhi [217] higher than that of the reference drugs berberine, chloramphenicol, and norfloxacin [241].
It should, however, be pointed out that, although some brilliant results have been achieved concerning the identification of molecular hybrids and conjugates bearing imidazole moiety that are capable of exercising in vitro effective and selective antibacterial properties also towards multidrug resistant bacteria, to the best of our knowledge none of the compounds mentioned in this review are currently subjected to clinical trials and are included in the list of antibiotics in the clinical pipeline in October 2019 [263]. However, this list contains morinidazole [(R)-1-(2-methyl-5-nitro-1H-imidazol-1-yl)-3-(morpholin-4-yl)propan-2-ol] (432) (Figure 14), a 5-nitroimidazole antimicrobial drug developed by Jiangsu Hansoh Pharmaceutical and approved in China for the treatment of anaerobic bacterial infections, including appendicitis and pelvic inflammatory disease caused by anaerobic bacteria [264].
Figure 14.
Structure of morinidazole (432).
Nevertheless, it is our belief that in the near future the current intense research activity in the field of this review [265] can allow to identify and develop novel imidazole-based antibacterial hybrids running into clinical trials and eventually for patient treatment. We hope that the data of this review will pave the way for innovative achievement in this field.
Acknowledgments
M.C. is grateful to Domenico Schillaci (Dipartimento di Scienze e Tecnologie Biologiche, Chimiche e Farmaceutiche, University of Palermo), for a useful discussion on microbiological subject.
Abbreviations
| Ac | acetyl |
| Ar | aryl |
| ATCC | American Type Culture Collection |
| Bn | benzyl |
| Boc | tert-butoxycarbonyl |
| BOM | benzyloxymethyl |
| Bu | butyl |
| CAN | cerium(IV) ammonium nitrate |
| Cbz | N-carboxybenzyl |
| CDI | 1,1′-carbonyldiimidazole |
| CFL | compact fluorescent lamp |
| CGMCC | China General Microbiological Culture Collection Center |
| m-CPBA | m-chloroperoxybenzoic acid |
| DBU | 1,5-diazabicyclo[5.4.0]undec-7-ene |
| DEAD | diethyl azodicarboxylate |
| DIBAL-H | diisobutylaluminum hydride |
| DIPEA | N,N-diisopropylethylamine |
| DMSO | dimethyl sulfoxide |
| DPIX | deuterioporphyrin |
| DMF | dimethylformamide |
| ED50 | half effective dose |
| EDC | 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| Eh | 2-ethylhexyl |
| eq | (stoichiometric) equivalent |
| Et | ethyl |
| FDA | Food and Drug Administration |
| Fmoc | fluorenylmethyloxycarbonyl |
| HOBt | hydroxybenzotriazole |
| HBTU | O-(benzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate |
| HMTA | hexamethylenetetramine |
| IC50 | half inhibitory concentration (bacterial growth or biochemical) |
| Ir(ppy)3 | tris(2-phenylpyridine)iridium |
| Me | methyl |
| MIC | minimum inhibitory concentration (bacterial growth) |
| MRSA | methicillin-resistant Staphylococcus aureus |
| Ms | methanesulfonyl |
| MTCC | Microbial Type Culture Collection (India) |
| Mtz | metronidazole |
| MW | microwave |
| Nim | 5-nitro-1H-imidazol-1-yl |
| NMM | 4-methylmorpholine |
| Nps | nanoparticles |
| n.r. | not reported |
| Ns | nosyl (p-nitrobenzesulfonyl) |
| PBP | penicillin binding protein |
| PCC | pyridinium chlorochromate |
| Ph | phenyl |
| PNB | p-nitrobenzyl |
| PNZ | p-nitrobenzyloxycarbonyl |
| Pr | propyl |
| Pro | proline |
| PTCC | Persian Type Culture Collection |
| Py | pyridyl |
| rt | room temperature |
| TBAB | tetrabutylammonium bromide |
| TBS | tert-butyldimethylsilyl |
| TBTU | N,N,N′,N′-tetramethyl-O-(benzotriazol-1-yl)uronium tetrafluoroborate |
| Tf | triflyl (trifluoromethanesulfonyl) |
| TFA | trifluoroacetic acid |
| THF | tetrahydrofuran |
| TIPS | trisopropylsilyl |
| Ts | tosyl (p-toluenesulfonyl) |
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References and Notes
- 1.Boucher H.W., Talbot G.H., Bradley J.S., Edwards J.E., Gilbert D., Rice L.B., Scheld M., Spellberg B., Bartlett J. Bad Bugs, No Drugs: No ESKAPE! An Update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 2.Breijyeh Z., Jubeh B., Karaman R. Resistance of Gram-Negative Bacteria to Current Antibacterial Agents and Approaches to Resolve It. Molecules. 2020;25:1340. doi: 10.3390/molecules25061340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rai J., Randhawa G.K., Kaur M. Recent advances in antibacterial drugs. Int. J. Appl. Basic Med. Res. 2013;3:3–10. doi: 10.4103/2229-516X.112229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jackson N., Czaplewski L., Piddock L.J.V. Discovery and development of new antibacterial drugs: Learning from experience? J. Antimicrob. Chemother. 2018;73:1452–1459. doi: 10.1093/jac/dky019. [DOI] [PubMed] [Google Scholar]
- 5.Gupta V., Datta P. Next-generation strategy for treating drug resistant bacteria: Antibiotic hybrids. Indian J. Med. Res. 2019;149:97–106. doi: 10.4103/ijmr.IJMR_755_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Decker M. Design of Hybrid Molecules for Drug Development. Elsevier; Oxford, UK: 2017. [Google Scholar]
- 7.Klahn P., Brönstrup M. Bifunctional antimicrobial conjugates and hybrid antimicrobials. Nat. Prod. Rep. 2017;34:832–885. doi: 10.1039/C7NP00006E. [DOI] [PubMed] [Google Scholar]
- 8.Rossi R., Ciofalo M. Current Advances in the Synthesis and Biological Evaluation of Pharmacologically Relevant 1,2,4,5-Tetrasubstituted-1H-Imidazole Derivatives. Curr. Org. Chem. 2019;23:2016–2101. doi: 10.2174/1385272823666191014154129. [DOI] [Google Scholar]
- 9.Rossi R., Angelici G., Casotti G., Manzini C., Lessi M. Catalytic Synthesis of 1,2,4,5-Tetrasubstituted 1H-Imidazole Derivatives: State of the Art. Adv. Synth. Catal. 2019;361:2737–2803. doi: 10.1002/adsc.201801381. [DOI] [Google Scholar]
- 10.Rossi R., Lessi M., Manzini C., Bellina F. Synthesis and Biological Profiles of 4,5-, 1,5-, and 1,2-Diaryl-1H-imidazoles. In: Yadav M.R., Murumkar P.R., Ghuge R.B., editors. Vicinal Diaryl Substituted Heterocycles. Elsevier; Amsterdam, The Netherlands: 2018. pp. 83–160. [Google Scholar]
- 11.Rossi R., Lessi M., Manzini C., Marianetti G., Bellina F. Direct (Hetero)arylation Reactions of (Hetero)arenes as Tools for the Step- and Atom-Economical Synthesis of Biologically Active Unnatural Compounds Including Pharmaceutical Targets. Synthesis. 2016;48:3821–3862. doi: 10.1055/s-0036-1588303. [DOI] [Google Scholar]
- 12.Bellina F., Rossi R. Regioselective Functionalization of the Imidazole Ring via Transition Metal-Catalyzed C-N and C-C Bond Forming Reactions. Adv. Synth. Catal. 2010;352:1223–1276. doi: 10.1002/adsc.201000144. [DOI] [Google Scholar]
- 13.Bonezzi K., Taraboletti G., Borsotti P., Bellina F., Rossi R., Giavazzi R. Vascular Disrupting Activity of Tubulin-Binding 1,5-Diaryl-1H-imidazoles. J. Med. Chem. 2009;52:7906–7910. doi: 10.1021/jm900968s. [DOI] [PubMed] [Google Scholar]
- 14.Bellina F., Rossi R. Recent advances in the synthesis of (hetero)aryl-substituted heteroarenes via transition metal-catalysed direct (hetero)arylation of heteroarene C-H bonds with aryl halides or pseudohalides, diaryliodonium salts, and potassium aryltrifluoroborates. Tetrahedron. 2009;65:10269–10310. doi: 10.1016/j.tet.2009.10.015. [DOI] [Google Scholar]
- 15.Bellina F., Cauteruccio S., Di Fiore A., Rossi R. Regioselective Synthesis of 4,5-Diaryl-1-methyl-1H-imidazoles Including Highly Cytotoxic Derivatives by Pd-Catalyzed Direct C-5 Arylation of 1-Methyl-1H-imidazole with Aryl Bromides. Eur. J. Org. Chem. 2008:5436–5445. doi: 10.1002/ejoc.200800738. [DOI] [Google Scholar]
- 16.Ang C.W., Jarrad A.M., Cooper M.A., Blaskovich M.A.T. Nitroimidazoles: Molecular Fireworks That Combat a Broad Spectrum of Infectious Diseases. J. Med. Chem. 2017;60:7636–7657. doi: 10.1021/acs.jmedchem.7b00143. [DOI] [PubMed] [Google Scholar]
- 17.Venugopal A.A., Johnson S. Current State of Clostridium difficile Treatment Options. Clin. Infect. Dis. 2012;55:S71–S76. doi: 10.1093/cid/cis355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kazanowski M., Smolarek S., Kinnarney F., Grzebieniak Z. Clostridium difficile: Epidemiology, diagnostic and therapeutic possibilities-a systematic review. Tech. Coloproctol. 2014;18:223–232. doi: 10.1007/s10151-013-1081-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson R.J., Groundwater P.W., Todd A., Worsley A.J. Antibacterial Agents. Chemistry, Mode of Action, Mechanisms of Resistance and Clinical Applications. Wiley; Chichester, UK: 2012. pp. 85–101. [Google Scholar]
- 20.Leiros H.-K.S., Kozielski-Stuhrmann S., Kapp U., Terradot L., Leonard G.A., McSweeney S.M. Structural basis of 5-nitroimidazole antibiotic resistance: The crystal structure of NimA from Deinococcus radiodurans. J. Biol. Chem. 2004;279:55840–55849. doi: 10.1074/jbc.M408044200. [DOI] [PubMed] [Google Scholar]
- 21.Kaakoush N.O., Asencio C., Mégraud F., Mendz G.L. A Redox Basis for Metronidazole Resistance in Helicobacter Pylori. Antimicrob. Agents Chemother. 2009;53:1884–1891. doi: 10.1128/AAC.01449-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peláez T., Cercenado E., Alcalá L., Marín M., Martín-López A., Martínez-Alarcón J., Catalán P., Sánchez-Somolinos M., Bouza E. Metronidazole resistance in Clostridium difficile is heterogeneous. J. Clin. Microbiol. 2008;46:3028–3032. doi: 10.1128/JCM.00524-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Exner M., Bhattacharya S., Christiansen B., Gebel J., Goroncy-Bermes P., Hartemann P., Heeg P., Ilschner C., Kramer A., Larson E., et al. Antibiotic resistance: What is so special about multidrug-resistant Gram-negative bacteria? GMS Hyg. Infect. Control. 2017;12:5. doi: 10.3205/dgkh000290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Demirayak S., Karaburun A.Ç., Kiraz N. Synthesis and antibacterial activities of some 1-[2-(substituted pyrrol-1-yl)ethyl]-2-methyl-5-nitroimidazole derivatives. Eur. J. Med. Chem. 1999;34:275–278. doi: 10.1016/S0223-5234(99)80062-2. [DOI] [Google Scholar]
- 25.Balakrishna A., Aguiar A., Sobral P.J.M., Wani M.Y., Almeida e Silva J., Sobral A.J.F.N. Paal–Knorr synthesis of pyrroles: From conventional to green synthesis. Cat. Rev.-Sci. Eng. 2018;61:84–110. doi: 10.1080/01614940.2018.1529932. [DOI] [Google Scholar]
- 26.Hadj-esfandiari N., Navidpour L., Shadnia H., Amini M., Samadi N., Faramarzi M.A., Shafiee A. Synthesis, antibacterial activity, and quantitative structure–activity relationships of new (Z)-2-(nitroimidazolylmethylene)-3(2H)-benzofuranone derivatives. Bioorg. Med. Chem. Lett. 2007;17:6354–6363. doi: 10.1016/j.bmcl.2007.09.062. [DOI] [PubMed] [Google Scholar]
- 27.Pires J.R., Saito C., Gomes S.L., Giesbrecht A.M., Amaral A.T.-d. Investigation of 5-Nitrofuran Derivatives: Synthesis, Antibacterial Activity, and Quantitative Structure-Activity Relationships. J. Med. Chem. 2001;44:3673–3681. doi: 10.1021/jm0101693. [DOI] [PubMed] [Google Scholar]
- 28.Mirzaei J., Siavoshi F., Emami S., Safari F., Khoshayand M.R., Shafiee A., Foroumadi A. Synthesis and in vitro anti-Helicobacter pylori activity of N-[5-(5-nitro-2-heteroaryl)-1,3,4-thiadiazol-2-yl]thiomorpholines and related compounds. Eur. J. Med. Chem. 2008;43:1575–1580. doi: 10.1016/j.ejmech.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 29.Foroumadi A., Mansouri S., Kiani Z., Rahmani A. Synthesis and in vitro antibacterial evaluation of N-[5-(5-nitro-2-thienyl)-1,3,4-thiadiazole-2-yl] piperazinyl quinolones. Eur. J. Med. Chem. 2003;38:851–854. doi: 10.1016/S0223-5234(03)00148-X. [DOI] [PubMed] [Google Scholar]
- 30.Pfeil E. Theorie und Praxis der Sandmeyerschen Reaktion. Angew. Chem. 1953;65:155–158. doi: 10.1002/ange.19530650603. [DOI] [Google Scholar]
- 31.Atia A.J.K. Synthesis and Antibacterial Activities of New Metronidazole and Imidazole Derivatives. Molecules. 2009;14:2431–2446. doi: 10.3390/molecules14072431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.[Negi] B., Kumar N., Rohilla R.K., Roy N., Rawat D.S. Synthesis and antibacterial activity evaluation of metronidazole-triazole conjugates. Bioorg. Med. Chem. Lett. 2009;19:1396–1398. doi: 10.1016/j.bmcl.2009.01.037. [DOI] [PubMed] [Google Scholar]
- 33.Meingassner J.G., Mieth H., Czok R., Lindmark D.G., Müller M. Assay Conditions and the Demonstration of Nitroimidazole Resistance in Tritrichomonas foetus. Antimicrob. Agents Chemother. 1978;13:1–3. doi: 10.1128/AAC.13.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Britz M.L., Wilkinson R.G. Isolation and Properties of Metronidazole-Resistant Mutants of Bacteroides fragilis. Antimicrob. Agents Chemother. 1979;16:19–27. doi: 10.1128/AAC.16.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saadeh H.A., Mosleh I.M., Al-Bakri A.G., Mubarak M.S. Synthesis and antimicrobial activity of new 1,2,4-triazole-3-thiol metronidazole derivatives. Monatsh. Chem. 2010;141:471–478. doi: 10.1007/s00706-010-0281-9. [DOI] [Google Scholar]
- 36.Clayton R., Ramsden C. N-Vinyl-Nitroimidazole Cycloadditions: Potential Routes to Nucleoside Analogues. Synthesis. 2005:2695–2700. doi: 10.1055/s-2005-872083. [DOI] [Google Scholar]
- 37.Zamani K., Faghihi K., Tofighi T., Shariatzadeh M.R. Synthesis and Antimicrobial Activity of Some Pyridyl and Naphthyl Substituted 1,2,4-Triazole and 1,3,4-Thiadiazole Derivatives. Turk. J. Chem. 2004;28:95–100. [Google Scholar]
- 38.Varshney V., Mishra N.N., Shukla P.K., Sahu D.P. Synthesis of nitroimidazole derived oxazolidinones as antibacterial agents. Eur. J. Med. Chem. 2010;45:661–666. doi: 10.1016/j.ejmech.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 39.Khalaj A., Nakhjiri M., Negahbani A.S., Samadizadeh M., Firoozpour L., Rajabalian S., Samadi N., Faramarzi M.A., Adibpour N., Shafiee A., et al. Discovery of a novel nitroimidazolyl-oxazolidinone hybrid with potent anti Gram-positive activity: Synthesis and antibacterial evaluation. Eur. J. Med. Chem. 2011;46:65–70. doi: 10.1016/j.ejmech.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 40.Letafat B., Mohammadhosseini N., Asadipour A., Foroumadi A. Synthesis and In vitro Antibacterial Activity of New 2-(1-Methyl-4-nitro-1H-imidazol-5-ylsulfonyl)-1,3,4-thiadiazoles. J. Chem. 2011;8:1120–1123. doi: 10.1155/2011/642071. [DOI] [Google Scholar]
- 41.Letafat B., Emami S., Aliabadi A., Mohammadhosseini N., Moshafi M.H., Asadipour A., Shafiee A., Foroumadi A. Synthesis and In-Vitro Antibacterial Activity of 5-Substituted 1-Methyl-4-nitro-1H-imidazoles. Arch. Pharm. 2008;341:497–501. doi: 10.1002/ardp.200800022. [DOI] [PubMed] [Google Scholar]
- 42.Li Y., Luo Y., Hu Y., Zhu D.-D., Zhang S., Liu Z.-J., Gong H.-B., Zhu H.-L. Design, synthesis and antimicrobial activities of nitroimidazole derivatives containing 1,3,4-oxadiazole scaffold as FabH inhibitors. Bioorg. Med. Chem. 2012;20:4316–4322. doi: 10.1016/j.bmc.2012.05.050. [DOI] [PubMed] [Google Scholar]
- 43.Riss T.L., Moravec R.A., Niles A.L., Duellman S., Benink H.A., Worzella T.J., Minor L. Cell Viability Assay. In: Sittampalam G.S., Grossman A., Brimacombe K., Arkin M., Auld D., Austin C.P., Baell J., Bejcek B., Caaveiro J.M.M., Chung T.D.Y., et al., editors. Assay Guidance Manual (Last Updated: June 1, 2020) Eli Lilly; Bethesda, MD, USA: 2004. pp. 1–25. [Google Scholar]
- 44.Zhang L., Chang J.-J., Zhang S.-L., Damu G.L.V., Geng R.-X., Zhou C.-H. Synthesis and bioactive evaluation of novel hybrids of metronidazole and berberine as new type of antimicrobial agents and their transportation behavior by human serum albumin. Bioorg. Med. Chem. 2013;21:4158–4169. doi: 10.1016/j.bmc.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 45.Gan R.-Y. Bioactivities of Berberine: An Update. Int. J. Modern Biol. Med. 2012;1:48–81. [Google Scholar]
- 46.Zhang D., Li A., Xie J., Ji C. In vitro antibacterial effect of berberine hydrochloride and enrofloxacin to fish pathogenic bacteria. Aquac. Res. 2010;41:1095–1100. doi: 10.1111/j.1365-2109.2009.02399.x. [DOI] [Google Scholar]
- 47.Yu X.-T., Xu Y.-F., Huang Y.-F., Qu C., Xu L.-Q., Su Z.-R., Zeng H.-F., Zheng L., Yi T.-G., Li H.-L., et al. Berberrubine attenuates mucosal lesions and inflammation in dextran sodium sulfate-induced colitis in mice. PLoS ONE. 2018;13:e0194069. doi: 10.1371/journal.pone.0194069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miyamoto Y., Kalisiak J., Korthals K., Lauwaet T., Cheung D.Y., Lozano R., Cobo E.R., Upcroft P., Upcroft J.A., Berg D.E., et al. Expanded therapeutic potential in activity space of next-generation 5-nitroimidazole antimicrobials with broad structural diversity. Proc. Natl. Acad. Sci. USA. 2013;110:17564–17569. doi: 10.1073/pnas.1302664110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brook I. Antimicrobial treatment of anaerobic infections. Exp. Opin. Pharmacother. 2011;12:1691–1707. doi: 10.1517/14656566.2011.576672. [DOI] [PubMed] [Google Scholar]
- 50.chloramphenicol is referred to as “chloromycin” in the original article.
- 51.Makawana J.A., Sun J., Zhu H.-L. Schiff’s base derivatives bearing nitroimidazole moiety: New class of antibacterial, anticancer agents and potential EGFR tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett. 2013;23:6264–6268. doi: 10.1016/j.bmcl.2013.09.086. [DOI] [PubMed] [Google Scholar]
- 52.Gu W., Qiao C., Wang S.-F., Hao Y., Miao T.-T. Synthesis and biological evaluation of novel N-substituted 1H-dibenzo[a,c]carbazole derivatives of dehydroabietic acid as potential antimicrobial agents. Bioorg. Med. Chem. Lett. 2014;24:328–331. doi: 10.1016/j.bmcl.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 53.González M.A., Pérez-Guaita D., Correa-Royero J., Zapata B., Agudelo L., Mesa-Arango A., Betancur-Galvis L. Synthesis and biological evaluation of dehydroabietic acid derivatives. Eur. J. Med. Chem. 2010;45:811–816. doi: 10.1016/j.ejmech.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 54.Manner S., Vahermo M., Skogman M.E., Krogerus S., Vuorela P.M., Yli-Kauhaluoma J., Fallarero A., Moreira V.M. New derivatives of dehydroabietic acid target planktonic and biofilm bacteria in Staphylococcus aureus and effectively disrupt bacterial membrane integrity. Eur. J. Med. Chem. 2015;102:68–79. doi: 10.1016/j.ejmech.2015.07.038. [DOI] [PubMed] [Google Scholar]
- 55.Gu W., Wang S. Synthesis and antimicrobial activities of novel 1H-dibenzo[a,c]carbazoles from dehydroabietic acid. Eur. J. Med. Chem. 2010;45:4692–4696. doi: 10.1016/j.ejmech.2010.07.038. [DOI] [PubMed] [Google Scholar]
- 56.Sangani C.B., Mak[a]wana J.A., Duan Y.-T., Tarpada U.P., Patel Y.S., Patel K.B., Dave V.N., Zhu H.-L. Design, synthesis, and antibacterial evaluation of new Schiff’s base derivatives bearing nitroimidazole and pyrazole nuclei as potent E. coli FabH inhibitors. Res. Chem. Intermed. 2015;41:10137–10149. doi: 10.1007/s11164-015-2018-1. [DOI] [Google Scholar]
- 57.Sangani C.B., Mungra D.C., Patel M.P., Patel R.G. Synthesis and in vitro antimicrobial screening of new pyrano[4,3-b]pyrane derivatives of 1H-pyrazole. Chin. Chem. Lett. 2012;23:57–60. doi: 10.1016/j.cclet.2011.09.012. [DOI] [Google Scholar]
- 58.Zhang L.-X., Liu Y., Cia L.-H., Hu Y.-J., Yin J., Hu P.-Z. Inhibitory study of some novel Schiff base derivatives on Staphylococcus aureus by microcalorimetry. Thermochim. Acta. 2006;440:51–56. doi: 10.1016/j.tca.2005.10.012. [DOI] [Google Scholar]
- 59.Jarrad A.M., Karoli T., Debnath A., Tay C.Y., Huang J.X., Kaeslin G., Elliott A.G., Miyamoto Y., Ramu S., Kavanagh A.M., et al. Metronidazole-triazole conjugates: Activity against Clostridium difficile and parasites. Eur. J. Med. Chem. 2015;101:96–102. doi: 10.1016/j.ejmech.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.reported as Bacillus proteus ATCC 13315 in the original paper.
- 61.Zhang L., Kannekanti V.K., Geng R.-X., Zhou C.-H. Design and biological evaluation of novel quinolone-based metronidazole derivatives as potent Cu2+ mediated DNA-targeting antibacterial agents. Bioorg. Med. Chem. Lett. 2015;25:3699–3705. doi: 10.1016/j.bmcl.2015.06.041. [DOI] [PubMed] [Google Scholar]
- 62.Cherian P.T., Wu X., Yang L., Scarborough J.S., Singh A.P., Alam Z.A., Lee R.E., Hurdle J.G. Gastrointestinal localization of metronidazole by a lactobacilli-inspired tetramic acid motif improves treatment outcomes in the hamster model of Clostridium difficile infection. J. Antimicrob. Chemother. 2015;70:3061–3069. doi: 10.1093/jac/dkv231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dingsdag S.A., Yap B.C.M., Hunter N., Crossley M.J. Amino acid-linked porphyrin-nitroimidazole antibiotics targeting Porphyromonas gingivalis. Org. Biomol. Chem. 2015;13:98–109. doi: 10.1039/C4OB01841A. [DOI] [PubMed] [Google Scholar]
- 64.Crossley M., Thordarson P., Hunter N., Yap B., Collyer C.A. Porphyrin Linked Metronidazole Against Gum Disease: Porphyromonas Gingivalis. WO 2006005137A1. World patent. 2006 Jan 19
- 65.Kang J., Tangadanchu V.K.R., Gopala L., Gao W.-W., Cheng Y., Liu H.-B., Geng R.-X., Li S., Zhou C.-H. Novel potentially antibacterial naphthalimide-derived metronidazoles: Design, synthesis, biological evaluation and supramolecular interactions with DNA, human serum albumin and topoisomerase II. Chin. Chem. Lett. 2017;28:1369–1374. doi: 10.1016/j.cclet.2017.04.002. [DOI] [Google Scholar]
- 66.Zhou Y., Ju Y., Yang Y., Sang Z., Wang Z., He G., Yang T., Luo Y. Discovery of hybrids of indolin-2-one and nitroimidazole as potent inhibitors against drug-resistant bacteria. J. Antibiot. (Tokyo) 2018;71:887–897. doi: 10.1038/s41429-018-0076-5. [DOI] [PubMed] [Google Scholar]
- 67.Gholamzadeh P., Mohammadi Ziarani G., Badiei A., Abolhassani Soorki A., Lashgari N. Efficient green synthesis of isoindigo derivatives using sulfonic-acid-functionalized nanoporous silica (SBA-Pr-SO3H) catalyst and study of their antimicrobial properties. Res. Chem. Intermed. 2013;39:3925–3936. doi: 10.1007/s11164-012-0909-y. [DOI] [Google Scholar]
- 68.Hosseinzadeh N., Hasani M., Foroumadi A., Nadri H., Emami S., Samadi N., Faramarzi M.A., Saniee P., Siavoshi F., Abadian N., et al. 5-Nitro-heteroarylidene analogs of 2-thiazolylimino-4-thiazolidinones as a novel series of antibacterial agents. Med. Chem. Res. 2013;22:2293–2302. doi: 10.1007/s00044-012-0224-6. [DOI] [Google Scholar]
- 69.Zhang G.-B., Maddili S.K., Tangadanchu V.K.R., Gopala L., Gao W.-W., Cai G.-X., Zhou C.-H. Discovery of natural berberine-derived nitroimidazoles as potentially multi-targeting agents against drug-resistant Escherichia coli. Sci. China Chem. 2018;61:557–568. doi: 10.1007/s11426-017-9169-4. [DOI] [Google Scholar]
- 70.Bhanot S., Viljoen A., Kremer L., Kumar V. Alkylated/aminated nitroimidazoles and nitroimidazole-7-chloroquinoline conjugates: Synthesis and anti-mycobacterial evaluation. Bioorg. Med. Chem. Lett. 2018;28:1309–1312. doi: 10.1016/j.bmcl.2018.03.021. [DOI] [PubMed] [Google Scholar]
- 71.Raj R., Biot C., Carrère-Kremer S., Kremer L., Guérardel Y., Gut J., Rosenthal P.J., Kumar V. 4-Aminoquinoline-β-Lactam Conjugates: Synthesis, Antimalarial, and Antitubercular Evaluation. Chem. Biol. Drug Des. 2014;83:191–197. doi: 10.1111/cbdd.12225. [DOI] [PubMed] [Google Scholar]
- 72.Singh A., Biot C., Viljoen A., Dupont C., Kremer L., Kumar K., Kumar V. 1H-1,2,3-triazole-tethered uracil-ferrocene and uracil-ferrocenylchalcone conjugates: Synthesis and antitubercular evaluation. Chem. Biol. Drug Des. 2017;89:856–861. doi: 10.1111/cbdd.12908. [DOI] [PubMed] [Google Scholar]
- 73.Bhovi M.G., Tanwar P., Yadav G.C. Synthesis and characterization of impurities present in an antimalarial drug piperaquine phosphate. Indian J. Heterocycl. Chem. 2010;19:215–220. [Google Scholar]
- 74.Kang J., Gopala L., Tangadanchu V.K.R., Gao W.-W., Zhou C.-H. Novel naphthalimide nitroimidazoles as multitargeting antibacterial agents against resistant Acinetobacter baumannii. Future Med. Chem. 2018;10:711–724. doi: 10.4155/fmc-2017-0160. [DOI] [PubMed] [Google Scholar]
- 75.Morris F.C., Dexter C., Kostoulias X., Uddin M.I., Peleg A.Y. The Mechanisms of Disease Caused by Acinetobacter baumannii. Front. Microbiol. 2019;10:1601. doi: 10.3389/fmicb.2019.01601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang Y.-Y., Zhou C.-H. Synthesis and activities of naphthalimide azoles as a new type of antibacterial and antifungal agents. Bioorg. Med. Chem. Lett. 2011;21:4349–4352. doi: 10.1016/j.bmcl.2011.05.042. [DOI] [PubMed] [Google Scholar]
- 77.Damu G.L.V., Wang Q., Zhang H., Zhang Y., Lv J., Zhou C. A series of naphthalimide azoles: Design, synthesis and bioactive evaluation as potential antimicrobial agents. Sci. China Chem. 2013;56:952–969. doi: 10.1007/s11426-013-4873-1. [DOI] [Google Scholar]
- 78.Lv J.-S., Peng X.-M., Kishore B., Zhou C.-H. 1,2,3-Triazole-derived naphthalimides as a novel type of potential antimicrobial agents: Synthesis, antimicrobial activity, interaction with calf thymus DNA and human serum albumin. Bioorg. Med. Chem. Lett. 2014;24:308–313. doi: 10.1016/j.bmcl.2013.11.013. [DOI] [PubMed] [Google Scholar]
- 79.Chen Y.-Y., Gopala L., Bheemanaboina R.R.Y., Liu H.-B., Cheng Y., Geng R.-X., Zhou C.-H. Novel Naphthalimide Aminothiazoles as Potential Multitargeting Antimicrobial Agents. ACS Med. Chem. Lett. 2017;8:1331–1335. doi: 10.1021/acsmedchemlett.7b00452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li Z.-Z., Tangadanchu V.K.R., Battini N., Bheemanaboina R.R.Y., Zang Z.-L., Zhang S.-L., Zhou C.-H. Indole-nitroimidazole conjugates as efficient manipulators to decrease the genes expression of methicillin-resistant Staphylococcus aureus. Eur. J. Med. Chem. 2019;179:723–735. doi: 10.1016/j.ejmech.2019.06.093. [DOI] [PubMed] [Google Scholar]
- 81.Roch M., Lelong E., Panasenko O.O., Sierra R., Renzoni A., Kelley W.L. Thermosensitive PBP2a requires extracellular folding factors PrsA and HtrA1 for Staphylococcus aureus MRSA β-lactam resistance. Commun. Biol. 2019;2:417. doi: 10.1038/s42003-019-0667-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li Z.-Z., Gopala L., Tangadanchu V.K.R., Gao W.-W., Zhou C.-H. Discovery of novel nitroimidazole enols as Pseudomonas aeruginosa DNA cleavage agents. Bioorg. Med. Chem. 2017;25:6511–6522. doi: 10.1016/j.bmc.2017.10.028. [DOI] [PubMed] [Google Scholar]
- 83.Girhepunje N.S., Kedar P.S., Ittadwar A.M., Dumore N.G. Design, Synthesis and Characterization of Some 5-Nitroimidazole Derivatives. Int. J. Pharm. Pharm. Res. 2016;6:456–480. [Google Scholar]
- 84.Kayser O., Kolodziej H. Antibacterial Activity of Simple Coumarins: Structural Requirements for Biological Activity. Z. Naturforsch. C Biosci. 1999;54:169–174. doi: 10.1515/znc-1999-3-405. [DOI] [PubMed] [Google Scholar]
- 85.De Souza S.M., Delle Monache F., Smânia A., Jr. Antibacterial Activity of Coumarins. Z. Naturforsch. C Biosci. 2005;60:693–700. doi: 10.1515/znc-2005-9-1006. [DOI] [PubMed] [Google Scholar]
- 86.Smyth T., Ramachandran V.N., Smyth W.F. A study of the antimicrobial activity of selected naturally occurring and synthetic coumarins. Int. J. Antimicrob. Agents. 2009;33:421–426. doi: 10.1016/j.ijantimicag.2008.10.022. [DOI] [PubMed] [Google Scholar]
- 87.Ostrov D.A., Hernández Prada J.A., Corsino P.E., Finton K.A., Le N., Rowe T.C. Discovery of Novel DNA Gyrase Inhibitors by High-Throughput Virtual Screening. Antimicrob. Agents Chemother. 2007;51:3688–3698. doi: 10.1128/AAC.00392-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Behrami A., Krasniqi I. Antibacterial activity of coumarine derivatives synthesized from 8-amino-4,7-dihydroxy-chromen-2-one and comparison with standard drug. J. Chem. Pharm. Res. 2012;4:2495–2500. [Google Scholar]
- 89.RamaGanesh C.K., Bodke Y.D., Venkatesh K.B. Synthesis and biological evaluation of some innovative coumarin derivatives containing thiazolidin-4-one ring. Indian J. Chem. 2010;49B:1151–1154. doi: 10.1002/chin.201051132. [DOI] [Google Scholar]
- 90.Tan N., Yazıcı-Tütüniş S., Bilgin M., Tan E., Miski M. Antibacterial Activities of Pyrenylated Coumarins from the Roots of Prangos hulusii. Molecules. 2017;22:1098. doi: 10.3390/molecules22071098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.De Araújo R.S.A., Barbosa-Filho J.M., Scotti M.T., Scotti L., da Cruz R.M.D., Falcão-Silva V.D.S., de Siqueira-Júnior J.P., Mendonça-Junior F.J.B. Modulation of Drug Resistance in Staphylococcus aureus with Coumarin Derivatives. Scientifica. 2016;2016:6894758. doi: 10.1155/2016/6894758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Peng X.-M., Kannekanti V.K., Damu G.L.V., Zhou C.-H. Coumarin-derived azolyl ethanols: Synthesis, antimicrobial evaluation and preliminary action mechanism. Sci. China Chem. 2016;59:878–894. doi: 10.1007/s11426-015-0351-0. [DOI] [Google Scholar]
- 93.Holiyachi M., Samundeeswari S., Chougala B.M., Naik N.S., Madar J., Shastri L.A., Joshi S.D., Dixit S.R., Dodamani S., Jalalpure S., et al. Design and synthesis of coumarin–imidazole hybrid and phenyl-imidazoloacrylates as potent antimicrobial and antiinflammatory agents. Monatsh. Chem. 2018;149:595–609. doi: 10.1007/s00706-017-2079-5. [DOI] [Google Scholar]
- 94.Holiyachi M., Shastri S.L., Chougala B.M., Shastri L.A. Effects of Base for the Efficient Synthesis of 4-Formylcoumarins and 4-Formylcarbostyrils. Synth. Commun. 2015;45:1002–1008. doi: 10.1080/00397911.2014.981754. [DOI] [Google Scholar]
- 95.Hu Y., Shen Y., Wu X., Tu X., Wang G.-X. Synthesis and biological evaluation of coumarin derivatives containing imidazole skeleton as potential antibacterial agents. Eur. J. Med. Chem. 2018;143:958–969. doi: 10.1016/j.ejmech.2017.11.100. [DOI] [PubMed] [Google Scholar]
- 96.Kitagawa H., Ozawa T., Takahata S., Iida M., Saito J., Yamada M. Phenylimidazole Derivatives of 4-Pyridone as Dual Inhibitors of Bacterial Enoyl-Acyl Carrier Protein Reductases FabI and FabK. J. Med. Chem. 2007;50:4710–4720. doi: 10.1021/jm0705354. [DOI] [PubMed] [Google Scholar]
- 97.Holiyachi M., Samundeeswari S., Chougala B.M., Naik N.S., Madar J.M., Shaikh F., Shastri L.A., Joshi S.D., Dixit S.R., Sunagar V.A., et al. Synthesis and molecular docking studies of coumarin-imidazole conjugates as potential antimicrobial agents. Indian J. Chem. 2020;59B:110–125. [Google Scholar]
- 98.Araque P., Herrera A., Montaño D., Yepes A., García E., Sepúlveda J., Torijano S., Cardona-G W. Antimicrobial Activity and In Silico Study of Methylimidazolium-Furanchalcone Hybrids and 1-Alkyl-3-methylimidazolium Salts. J. Chil. Chem. Soc. 2019;64:4547–4552. doi: 10.4067/S0717-97072019000304547. [DOI] [Google Scholar]
- 99.El-Obeid H.A., Elnima E.I., Al-Badr A.A. Synthesis and Antimicrobial Activity of New Furan Derivatives. Pharm. Res. 1985;2:42–43. doi: 10.1023/A:1016318106782. [DOI] [PubMed] [Google Scholar]
- 100.Kharb R., Birla S., Sharma A.K. Recent Updates on Antimicrobial Potential of Novel Furan Derivatives. Int. J. Pharm. Phytopharmacol. Res. 2014;3:451–459. [Google Scholar]
- 101.Vollaro A., Catania M.R., Iesce M.R., Sferruzza R., D’Abrosca B., Donnarumma G., De Filippis A., Cermola F., DellaGreca M., Buommino E. Antimicrobial and anti-biofilm properties of novel synthetic lignan-like compounds. New Microbiol. 2019;42:21–28. [PubMed] [Google Scholar]
- 102.Xu M., Wu P., Shen F., Ji J., Rakesh K.P. Chalcone derivatives and their antibacterial activities: Current development. Bioorg. Chem. 2019;91:103133. doi: 10.1016/j.bioorg.2019.103133. [DOI] [PubMed] [Google Scholar]
- 103.Khodarahmi G., Asadi P., Hassanzadeh F., Khodarahmi E. Benzofuran as a promising scaffold for the synthesis of antimicrobial and antibreast cancer agents: A review. J. Res. Med. Sci. The Off. J. Isfahan Univ. Med. Sci. 2015;20:1094–1104. doi: 10.4103/1735-1995.172835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jafari E., Khajouei M.R., Hassanzadeh F., Hakimelahi G.H., Khodarahmi G.A. Quinazolinone and quinazoline derivatives: Recent structures with potent antimicrobial and cytotoxic activities. Res. Pharm. Sci. 2016;11:1–14. [PMC free article] [PubMed] [Google Scholar]
- 105.Mohamed M.A., Ghanem H.M., Abd El-Ghaffar N.F., Mohamed S.S. Biological Evaluation and Molecular Docking of Substituted Quinazolinones as Antimicrobial Agents. Aust. J. Basic Appl. Sci. 2013;7:263–274. [Google Scholar]
- 106.Asadi P., Khodarahmi G., Jahanian-Najafabadi A., Saghaie L., Hassanzadeh F. Synthesis, characterization, molecular docking studies and biological evaluation of some novel hybrids based on quinazolinone, benzofuran and imidazolium moieties as potential cytotoxic and antimicrobial agents. Iran. J. Basic Med. Sci. 2017;20:975–989. doi: 10.22038/ijbms.2017.9260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pitout J.D.D., Sanders C.C., Sanders W.E., Jr. Antimicrobial Resistance with Focus on β-Lactam Resistance in Gram-Negative Bacilli. Am. J. Med. 1997;103:51–59. doi: 10.1016/S0002-9343(97)00044-2. [DOI] [PubMed] [Google Scholar]
- 108.Rokade Y., Dongre N., Engla G., Behera C., Sayyed R. Azetidinone (ß-Lactam) Derivatives: An Emerging Antimicrobials. Asian J. Microbiol. Biotechnol. Environ. Sci. 2009;11:109–114. [Google Scholar]
- 109.Konaklieva M.I. Molecular Targets of β-Lactam-Based Antimicrobials: Beyond the Usual Suspects. Antibiotics (Basel) 2014;3:128–142. doi: 10.3390/antibiotics3020128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Salunkhe D.S., Piste P.B. A Brief Review on Recent Synthesis of 2-Azetidinone Derivatives. Int. J. Pharm. Sci. Res. 2014;5:666–689. doi: 10.13040/IJPSR.0975-8232.5(3).666-89. [DOI] [Google Scholar]
- 111.Sarkar S., Chauhan R., Dwivedi J. Synthesis and Antibacterial Activity of Some Azetidinone Derivatives Containing 2-Amino 6,7 Substituted Benzothiazole. Turk. J. Pharm. Sci. 2015;12:39–44. [Google Scholar]
- 112.Askar F.W., Ali R.A., Al-Mouamin T.M. Synthesis of New Some Imidazole Derivatives Containing β-Lactam Ring. Baghdad Sci. J. 2016;13:307–316. doi: 10.21123/bsj.2016.13.2.2NCC.0307. [DOI] [Google Scholar]
- 113.Sankar P.S., Divya K., Padmaja A., Padmavathi V. Synthesis and Antimicrobial Activity of Azetidinone and Thiazolidinone Derivatives from Azolylindolyl Schiff’s Bases. Med. Chem. (Los Angeles) 2017;7:340–347. doi: 10.4172/2161-0444.1000478. [DOI] [Google Scholar]
- 114.Niccolai D., Tarsi L., Thomas R.J. The renewed challenge of antibacterial chemotherapy. Chem. Commun. 1997:2333–2342. doi: 10.1039/a704497f. [DOI] [Google Scholar]
- 115.Poole K. Resistance to β-lactam antibiotics. Cell. Mol. Life Sci. 2004;61:2200–2223. doi: 10.1007/s00018-004-4060-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fernandes R., Amador P., Prudêncio C. β-Lactams: Chemical structure, mode of action and mechanisms of resistance. Rev. Med. Microbiol. 2013;24:7–17. doi: 10.1097/MRM.0b013e3283587727. [DOI] [Google Scholar]
- 117.Thakuria B., Lahon K. The Beta Lactam Antibiotics as an Empirical Therapy in a Developing Country: An Update on Their Current Status and Recommendations to Counter the Resistance against Them. J. Clin. Diagn. Res. 2013;7:1207–1214. doi: 10.7860/JCDR/2013/5239.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shaikh S., Fatima J., Shakil S., Rizvi S.M.D., Kamal M.A. Antibiotic resistance and extended spectrum beta-lactamases: Types, epidemiology and treatment. Saudi J. Biol. Sci. 2015;22:90–101. doi: 10.1016/j.sjbs.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Venkatesan A.M., Agarwal A., Abe T., Ushirogochi H., Yamamura I., Kumagai T., Petersen P.J., Weiss W.J., Lenoy E., Yang Y., et al. Novel imidazole substituted 6-methylidene-penems as broad-spectrum β-lactamase inhibitors. Bioorg. Med. Chem. 2004;12:5807–5817. doi: 10.1016/j.bmc.2004.08.039. [DOI] [PubMed] [Google Scholar]
- 120.Osborne N.F., Atkins R.J., Broom N.J.P., Coulton S., Harbridge J.B., Harris M.A., Stirling-François I., Walker G. Synthesis of (5R)-(Z)-6-(1-Methyl-1,2,3-triazol-4-ylmethylene)penem-3-carboxylic acid, a Potent Broad Spectrum β-Lactamase Inhibitor, from 6-Aminopenicillanic Acid. J. Chem. Soc., Perkin Trans. 1. 1994:179–188. doi: 10.1039/P19940000179. [DOI] [Google Scholar]
- 121.Pagadala R., Uppalaiah K., Kamatala C.R., Meshram J.S. Synthesis and Antimicrobial Studies of Novel Imidazole Containing Bisazetidinones and Bisthiazolidinone Derivatives. J. Heterocycl. Chem. 2015;52:403–410. doi: 10.1002/jhet.1979. [DOI] [Google Scholar]
- 122.Pagadala R., Meshram J.S., Chopde H.N., Panyala N.R. A Zeolite Promoted Expeditious One-Pot Synthesis of 1-Arylmethyl-4, 5-dihydro-2-aryl-1H-imidazole. J. Heterocycl. Chem. 2010;47:350–353. doi: 10.1002/jhet.314. [DOI] [Google Scholar]
- 123.Raju B., Ragul R., Sivasankar B.N. A new reagent for selective reduction of nitro group. Indian J. Chem. 2009;48B:1315–1318. doi: 10.1002/chin.201002040. [DOI] [Google Scholar]
- 124.Noori S.D., Mousa M.N., Al-Jadaan S.A.N. Synthesis of a New Conjugates of Imidazole with Beta Lactam Moiety and Evaluation of its Expanded Antibacterial Activity. Orient. J. Chem. 2018;34:2495–2501. doi: 10.13005/ojc/340535. [DOI] [Google Scholar]
- 125.Forte B., Malgesini B., Piutti C., Quartieri F., Scolaro A., Papeo G. A Submarine Journey: The Pyrrole-Imidazole Alkaloids. Mar. Drugs. 2009;7:705–753. doi: 10.3390/md7040705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lindel T. Chemistry and Biology of the Pyrrole-Imidazole Alkaloids. In: Knölker H.-J., editor. The Alkaloids: Chemistry and Biology. Volume 77. Academic Press; Oxford, UK: 2017. pp. 117–219. [DOI] [PubMed] [Google Scholar]
- 127.Hoffmann H., Lindel T. Synthesis of the Pyrrole-Imidazole Alkaloids. Synthesis. 2003:1753–1783. doi: 10.1002/chin.200347248. [DOI] [Google Scholar]
- 128.Weinreb S.M. Some recent advances in the synthesis of polycyclic imidazole-containing marine natural products. Nat. Prod. Rep. 2007;24:931–948. doi: 10.1039/b700206h. [DOI] [PubMed] [Google Scholar]
- 129.Al-Mourabit A., Zancanella M.A., Tilvi S., Romo D. Biosynthesis, asymmetric synthesis, and pharmacology, including cellular targets, of the pyrrole-2-aminoimidazole marine alkaloids. Nat. Prod. Rep. 2011;28:1229–1260. doi: 10.1039/c0np00013b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wang X., Ma Z., Wang X., De S., Ma Y., Chen C. Dimeric pyrrole-imidazole alkaloids: Synthetic approaches and biosynthetic hypotheses. Chem. Commun. 2014;50:8628–8639. doi: 10.1039/C4CC02290D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Das J., Bhandari M., Lovely C.J. Isolation, Bioactivity, and Synthesis of Nagelamides. In: Atta-ur-Rahman, editor. Studies in Natural Products Chemistry. Volume 50. Elsevier; Amsterdam, The Netherlands: 2016. pp. 341–371. [Google Scholar]
- 132.Gjorgjieva M., Masic L.P., Kikelj D. Antibacterial and Antibiofilm Potentials of Marine Pyrrole-2-Aminoimidazole Alkaloids and their Synthetic Analogs. Mini-Rev. Med. Chem. 2018;18:1640–1658. doi: 10.2174/1389557516666160505120157. [DOI] [PubMed] [Google Scholar]
- 133.Araki A., Tsuda M., Kubota T., Mikami Y., Fromont J., Kobayashi J.i. Nagelamide J, a Novel Dimeric Bromopyrrole Alkaloid from a Sponge Agelas Species. Org. Lett. 2007;9:2369–2371. doi: 10.1021/ol0707867. [DOI] [PubMed] [Google Scholar]
- 134.Walker R.P., Faulkner D.J., Van Engen D., Clardy J. Sceptrin, an Antimicrobial Agent from the Sponge Agelas sceptrum. J. Am. Chem. Soc. 1981;103:6772–6775. doi: 10.1021/ja00412a052. [DOI] [Google Scholar]
- 135.Bernan V.S., Roll D.M., Ireland C.M., Greenstein M., Maiese W.M., Steinberg D.A. A study on the mechanism of action of sceptrin, an antimicrobial agent isolated from the South Pacific sponge Agelas mauritiana. J. Antimicrob. Chemother. 1993;32:539–550. doi: 10.1093/jac/32.4.539. [DOI] [PubMed] [Google Scholar]
- 136.Baran P.S., Zografos A.L., O’Malley D.P. Short Total Synthesis of (±)-Sceptrin. J. Am. Chem. Soc. 2004;126:3726–3727. doi: 10.1021/ja049648s. [DOI] [PubMed] [Google Scholar]
- 137.Birman V.B., Jiang X.-T. Synthesis of Sceptrin Alkaloids. Org. Lett. 2004;6:2369–2371. doi: 10.1021/ol049283g. [DOI] [PubMed] [Google Scholar]
- 138.Baran P.S., Li K., O’Malley D.P., Mitsos C. Short, Enantioselective Total Synthesis of Sceptrin and Ageliferin by Programmed Oxaquadricyclane Fragmentation. Angew. Chem. Int. Ed. 2006;45:249–252. doi: 10.1002/anie.200503374. [DOI] [PubMed] [Google Scholar]
- 139.O’Malley D.P., Li K., Maue M., Zografos A.L., Baran P.S. Total Synthesis of Dimeric Pyrrole-Imidazole Alkaloids: Sceptrin, Ageliferin, Nagelamide E, Oxysceptrin, Nakamuric Acid, and the Axinellamine Carbon Skeleton. J. Am. Chem. Soc. 2007;129:4762–4775. doi: 10.1021/ja069035a. [DOI] [PubMed] [Google Scholar]
- 140.Ma Z., Wang X., Wang X., Rodriguez R.A., Moore C.E., Gao S., Tan X., Ma Y., Rheingold A.L., Baran P.S., et al. Asymmetric syntheses of sceptrin and massadine and evidence for biosynthetic enantiodivergence. Science (New York) 2014;346:219–224. doi: 10.1126/science.1255677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yoon T.P. Visible Light Photocatalysis: The Development of Photocatalytic Radical Ion Cycloadditions. ACS Catal. 2013;3:895–902. doi: 10.1021/cs400088e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Roueé M.l., Domart-Coulon I., Ereskovsky A., Djediat C., Perez T., Bourguet-Kondracki M.-L. Cellular Localization of Clathridimine, an Antimicrobial 2-Aminoimidazole Alkaloid Produced by the Mediterranean Calcareous Sponge Clathrina clathrus. J. Nat. Prod. 2010;73:1277–1282. doi: 10.1021/np100175x. [DOI] [PubMed] [Google Scholar]
- 143.Chawla A., Kapoor V.K. Microwave Assisted One Pot Synthesis and Antimicrobial Activity of 2-(3′-Acetyl-2′-methyl-5′-phenyl)-pyrrol-1-yl-1,4,5-triphenyl-1H-imidazole Derivatives. Der Pharma Chem. 2018;10:27–31. [Google Scholar]
- 144.Chawla A., Kapoor V.K. Microwave Assisted Synthesis of 2-Amino-3,4,5-Trisubstituted Imidazolines Using Radiszewski Method, their Characterisation and Evaluation for Antioxidant Activity. Int. Res. J. Pharm. 2016;7:23–30. doi: 10.7897/2230-8407.0711123. [DOI] [Google Scholar]
- 145.Benkli K., Demirayak S., Gundogdu-Karaburun N., Kiraz N., Iscan G., Ucucu U. Synthesis and antimicrobial activities of some imidazole substituted indoles. Indian J. Chem. 2004;43B:174–179. doi: 10.1002/chin.200417111. [DOI] [Google Scholar]
- 146.Kumara H.K., Suhas R., Suyoga Vardhan D.M., Shobha M., Channe Gowda D. Imidazolo and tryptophan-imidazolo hybrid derived ureas/thioureas as potent bioactive agents-SAR and molecular modelling studies. Bioorg. Chem. 2019;86:34–38. doi: 10.1016/j.bioorg.2019.01.027. [DOI] [PubMed] [Google Scholar]
- 147.Olofson A., Yakushijin K., Horne D.A. Synthesis of Marine Sponge Alkaloids Oroidin, Clathrodin, and Dispacamides. Preparation and Transformation of 2-Amino-4,5-dialkoxy-4,5-dihydroimidazolines from 2-Aminoimidazoles. J. Org. Chem. 1998;63:1248–1253. doi: 10.1021/jo9718298. [DOI] [Google Scholar]
- 148.Lindel T., Hochgürtel M. Synthesis of the Marine Natural Product Oroidin and Its Z-Isomer. J. Org. Chem. 2000;65:2806–2809. doi: 10.1021/jo991395b. [DOI] [PubMed] [Google Scholar]
- 149.Berrée F., Girard-Le Bleis P., Carboni B. Synthesis of the marine sponge alkaloid oroidin and its analogues via Suzuki cross-coupling reactions. Tetrahedron Lett. 2002;43:4935–4938. doi: 10.1016/S0040-4039(02)00947-4. [DOI] [Google Scholar]
- 150.Richards J.J., Ballard T.E., Melander C. Inhibition and dispersion of Pseudomonas aeruginosa biofilms with reverse amide 2-aminoimidazole oroidin analogues. Org. Biomol. Chem. 2008;6:1356–1363. doi: 10.1039/b719082d. [DOI] [PubMed] [Google Scholar]
- 151.Rasapalli S., Kumbam V., Dhawane A.N., Golen J.A., Lovely C.J., Rheingold A.L. Total syntheses of oroidin, hymenidin and clathrodin. Org. Biomol. Chem. 2013;11:4133–4137. doi: 10.1039/c3ob40668g. [DOI] [PubMed] [Google Scholar]
- 152.Zidar N., Montalvão S., Hodnik Ž., Nawrot D.A., Žula A., Ilaš J., Kikelj D., Tammela P., Mašič L.P. Antimicrobial Activity of the Marine Alkaloids, Clathrodin and Oroidin, and Their Synthetic Analogues. Mar. Drugs. 2014;12:940–963. doi: 10.3390/md12020940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Forenza S., Minale L., Riccio R., Fattorusso E. New Bromo-pyrrole Derivatives from the Sponge Agelas oroides. J. Chem. Soc. D. 1971:1129–1130. doi: 10.1039/c29710001129. [DOI] [Google Scholar]
- 154.Hao E., Fromont J., Jardine D., Karuso P. Natural Products From Sponges of the Genus Agelas—On the Trail of a [2+2]-Photoaddition Enzyme. Molecules. 2001;6:130–141. doi: 10.3390/60100130. [DOI] [Google Scholar]
- 155.Richelle-Maurer E., De Kluijver M.J., Feio S., Gaudêncio S., Gaspar H., Gomez R., Tavares R., Van de Vyver G., Van Soest R.W.M. Localization and ecological significance of oroidin and sceptrin in the Caribbean sponge Agelas conifera. Biochem. Syst. Ecol. 2003;31:1073–1091. doi: 10.1016/S0305-1978(03)00072-3. [DOI] [Google Scholar]
- 156.Regalado E.L., Laguna A., Mendiola J., Thomas O.P., Nogueiras C. Bromopyrrole Alkaloids from the Caribbean Sponge Agelas cerebrum. Quim. Nova. 2011;34:289–291. doi: 10.1590/S0100-40422011000200022. [DOI] [Google Scholar]
- 157.Satheesha Rai N., Kalluraya B. A Novel synthesis of nitrofuran containing 1,3,4,5-tetrasubstituted pyrazoles via 1,3-dipolar addition reaction. Indian J. Chem. 2007;46B:375–378. [Google Scholar]
- 158.Bekhit A.A., Fahmy H.T.Y., Rostom S.A.F., Bekhit A.E.-D.A. Synthesis and biological evaluation of some thiazolylpyrazole derivatives as dual anti-inflammatory antimicrobial agents. Eur. J. Med. Chem. 2010;45:6027–6038. doi: 10.1016/j.ejmech.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 159.Mor S., Mohil R., Kumar D., Ahuja M. Synthesis and antimicrobial activities of some isoxazolyl thiazolyl pyrazoles. Med. Chem. Res. 2012;21:3541–3548. doi: 10.1007/s00044-011-9859-y. [DOI] [Google Scholar]
- 160.Pervaram S., Ashok D., Rao B.A., Sarasija M., Reddy C.V.R. Design and Synthesis of New 1,2,3-Triazole-pyrazole Hybrids as Antimicrobial Agents. Russ. J. Gen. Chem. 2017;87:2454–2461. doi: 10.1134/S1070363217100280. [DOI] [Google Scholar]
- 161.Menozzi G., Merello L., Fossa P., Schenone S., Ranise A., Mosti L., Bondavalli F., Loddo R., Murgioni C., Mascia V., et al. Synthesis, antimicrobial activity and molecular modeling studies of halogenated 4-[1H-imidazol-1-yl(phenyl)methyl]-1,5-diphenyl-1H-pyrazoles. Bioorg. Med. Chem. 2004;12:5465–5483. doi: 10.1016/j.bmc.2004.07.029. [DOI] [PubMed] [Google Scholar]
- 162.Zampieri D., Mamolo M.G., Laurini E., Scialino G., Banfi E., Vio L. Antifungal and antimycobacterial activity of 1-(3,5-diaryl-4,5-dihydro-1H-pyrazol-4-yl)-1H-imidazole derivatives. Bioorg. Med. Chem. 2008;16:4516–4522. doi: 10.1016/j.bmc.2008.02.055. [DOI] [PubMed] [Google Scholar]
- 163.Palomino J.-C., Martin A., Camacho M., Guerra H., Swings J., Portaels F. Resazurin Microtiter Assay Plate: Simple and Inexpensive Method for Detection of Drug Resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2002;46:2720–2722. doi: 10.1128/AAC.46.8.2720-2722.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Vijesh A.M., Isloor A.M., Telkar S., Peethambar S.K., Rai S., Isloor N. Synthesis, characterization and antimicrobial studies of some new pyrazole incorporated imidazole derivatives. Eur. J. Med. Chem. 2011;46:3531–3536. doi: 10.1016/j.ejmech.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 165.Padmavathi V., Prema kumari C., Venkatesh B.C., Padmaja A. Synthesis and antimicrobial activity of amido linked pyrrolyl and pyrazolyl-oxazoles, thiazoles and imidazoles. Eur. J. Med. Chem. 2011;46:5317–5326. doi: 10.1016/j.ejmech.2011.08.032. [DOI] [PubMed] [Google Scholar]
- 166.Jayashree A., Narayana B., Uppine G.B., Ghate V.M., Lewis S.A., Prakash B., Kunhanna S.B., Kumar M.S. ZnO Nanocatalyst Mediated Convergent Synthesis of Highly Substituted Imidazole and Imidazole-derived Bi-heterocyclic Scaffolds as Potential Antibacterial Agents. J. Heterocycl. Chem. 2019;56:2398–2410. doi: 10.1002/jhet.3627. [DOI] [Google Scholar]
- 167.Turan-Zitouni G., Kaplancıklı Z.A., Yıldız M.T., Chevallet P., Kaya D. Synthesis and antimicrobial activity of 4-phenyl/cyclohexyl-5-(1-phenoxyethyl)-3-[N-(2-thiazolyl)acetamido]thio-4H-1,2,4-triazole derivatives. Eur. J. Med. Chem. 2005;40:607–613. doi: 10.1016/j.ejmech.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 168.Sumangala V., Poojary B., Chidananda N., Fernandes J., Kumari N.S. Synthesis and Antimicrobial Activity of 1,2,3-Triazoles Containing Quinoline Moiety. Arch. Pharm. Res. 2010;33:1911–1918. doi: 10.1007/s12272-010-1204-3. [DOI] [PubMed] [Google Scholar]
- 169.Kumar D., [Negi] B., Khare G., Kidwai S., Tyagi A.K., Singh R., Rawat D.S. Synthesis of novel 1,2,3-triazole derivatives of isoniazid and their in vitro and in vivo antimycobacterial activity evaluation. Eur. J. Med. Chem. 2014;81:301–313. doi: 10.1016/j.ejmech.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 170.Kant R., Singh V., Nath G., Awasthi S.K., Agarwal A. Design, synthesis and biological evaluation of ciprofloxacin tethered bis-1,2,3-triazole conjugates as potent antibacterial agents. Eur. J. Med. Chem. 2016;124:218–228. doi: 10.1016/j.ejmech.2016.08.031. [DOI] [PubMed] [Google Scholar]
- 171.Aouad M.R., Mayaba M.M., Naqvi A., Bardaweel S.K., Al-blewi F.F., Messali M., Rezki N. Design, synthesis, in silico and in vitro antimicrobial screenings of novel 1,2,4-triazoles carrying 1,2,3-triazole scaffold with lipophilic side chain tether. Chem. Cent. J. 2017;11:117. doi: 10.1186/s13065-017-0347-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Maddili S.K., Katla R., Kannekanti V.K., Bejjanki N.K., Tuniki B., Zhou C.-H., Gandham H. Molecular interaction of novel benzothiazolyl triazolium analogues with calf thymus DNA and HSA-their biological investigation as potent antimicrobial agents. Eur. J. Med. Chem. 2018;150:228–247. doi: 10.1016/j.ejmech.2018.02.056. [DOI] [PubMed] [Google Scholar]
- 173.Bi F., Ji S., Venter H., Liu J., Semple S.J., Ma S. Substitution of terminal amide with 1H-1,2,3-triazole: Identification of unexpected class of potent antibacterial agents. Bioorg. Med. Chem. Lett. 2018;28:884–891. doi: 10.1016/j.bmcl.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 174.Bozorov K., Zhao J., Aisa H.A. 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg. Med. Chem. 2019;27:3511–3531. doi: 10.1016/j.bmc.2019.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Gao F., Wang T., Xiao J., Huang G. Antibacterial activity study of 1,2,4-triazole derivatives. Eur. J. Med. Chem. 2019;173:274–281. doi: 10.1016/j.ejmech.2019.04.043. [DOI] [PubMed] [Google Scholar]
- 176.Nikalje A.P.G., Ghodke M.S., Kalam Khan F.A., Sangshetti J.N. CAN catalyzed one-pot synthesis and docking study of some novel substituted imidazole coupled 1,2,4-triazole-5-carboxylic acids as antifungal agents. Chin. Chem. Lett. 2015;26:108–112. doi: 10.1016/j.cclet.2014.10.020. [DOI] [Google Scholar]
- 177.Subhashini N.J.P., Praveen Kumar E., Gurrapu N., Yerragunta V. Design and synthesis of imidazolo-1, 2,3-triazoles hybrid compounds by microwave-assisted method: Evaluation as an antioxidant and antimicrobial agents and molecular docking studies. J. Mol. Struct. 2019;1180:618–628. doi: 10.1016/j.molstruc.2018.11.029. [DOI] [Google Scholar]
- 178.Chauhan S., Verma V., Kumar D., Kumar A. Facile Synthesis, Antimicrobial Activity and Molecular Docking of Novel 2,4,5-Trisubstituted-1H-Imidazole-Triazole Hybrid Compounds. J. Heterocycl. Chem. 2019;56:2571–2579. doi: 10.1002/jhet.3655. [DOI] [Google Scholar]
- 179.Ravi Kumar K.R., Mallesha H., Rangappa K.S. Synthesis of Novel Isoxazolidine Derivatives and Their Antifungal and Antibacterial Properties. Arch. Pharm. 2003;336:159–164. doi: 10.1002/ardp.200390015. [DOI] [PubMed] [Google Scholar]
- 180.Ravi Kumar K.R., Mallesha H., Rangappa K.S. Synthesis and Characterization of 5-Substituted Novel Isoxazolidines Derived from 1,3-Dipolar Cycloaddition of Nitrones with Olefins: Studies of Antibacterial and Antifungal Activities. Synth. Commun. 2003;33:1545–1555. doi: 10.1081/SCC-120018773. [DOI] [Google Scholar]
- 181.Damodiran M., Sivakumar P.M., SenthilKumar R., Muralidharan D., Phani Kumar B.V.N., Doble M., Perumal P.T. Antibacterial Activity, Quantitative Structure-Activity Relationship and Diastereoselective Synthesis of Isoxazolidine Derivatives Via 1,3-Dipolar Cycloaddition of D-Glucose Derived Nitrone with Olefin. Chem. Biol. Drug Des. 2009;74:494–506. doi: 10.1111/j.1747-0285.2009.00887.x. [DOI] [PubMed] [Google Scholar]
- 182.Chiacchio M.A., Giofrè S.V., Romeo R., Romeo G., Chiacchio U. Isoxazolidines as Biologically Active Compounds. Curr. Org. Synth. 2016;13:726–749. doi: 10.2174/1570179412666150914195807. [DOI] [Google Scholar]
- 183.Sadashiva M.P., Mallesha H., Hitesh N.A., Rangappa K.S. Synthesis and microbial inhibition study of novel 5-imidazolyl substituted isoxazolidines. Bioorg. Med. Chem. 2004;12:6389–6395. doi: 10.1016/j.bmc.2004.09.031. [DOI] [PubMed] [Google Scholar]
- 184.Watson S.P. A Convenient Synthesis of 2-Butyl-4(5)-chloro-1H-imidazole-5(4)-carboxaldehyde. Synth. Commun. 1992;22:2971–2977. doi: 10.1080/00397919208021123. [DOI] [Google Scholar]
- 185.Griffiths G.J., Hauck M.B., Imwinkelried R., Kohr J., Roten C.A., Stucky G.C., Gosteli J. Novel Syntheses of 2-Butyl-5-chloro-3H-imidazole-4-carbaldehyde: A Key Intermediate for the Synthesis of the Angiotensin II Antagonist Losartan. J. Org. Chem. 1999;64:8084–8089. doi: 10.1021/jo9824910. [DOI] [PubMed] [Google Scholar]
- 186.Mallesha H., Ravi Kumar K.R., Vishu Kumar B.K., Mantelingu K., Rangappa K.S. Histidine as a catalyst in organic synthesis: A facile in situ synthesis of α,N-diarylnitrones. J. Chem. Sci. 2001;113:291–296. doi: 10.1007/BF02708648. [DOI] [Google Scholar]
- 187.Frank P.V., Kalluraya B. Synthesis of 1,3,4-oxadiazoles carrying imidazole moiety. Indian J. Chem. 2005;44B:1456–1459. doi: 10.1002/chin.200546120. [DOI] [Google Scholar]
- 188.Frank P.V., Girish K.S., Kalluraya B. Solvent-free microwave-assisted synthesis of oxadiazoles containing imidazole moiety. J. Chem. Sci. 2007;119:41–46. doi: 10.1007/s12039-007-0007-7. [DOI] [Google Scholar]
- 189.Luo Y.-L., Baathulaa K., Kannekanti V.K., Zhou C.-H., Cai G.-X. Novel benzimidazole derived naphthalimide triazoles: Synthesis, antimicrobial activity and interactions with calf thymus DNA. Sci. China Chem. 2015;58:483–494. doi: 10.1007/s11426-014-5296-3. [DOI] [Google Scholar]
- 190.Shaki H., Khosravi A., Gharanjig K., Mahboubi A. Investigation of synthesis, characterization, photophysical and biological properties of novel antimicrobial fluorescent naphthalimide derivatives. Mater. Technol. 2016;31:322–331. doi: 10.1179/1753555715Y.0000000058. [DOI] [Google Scholar]
- 191.Marinov M., Kostova I., Naydenova E., Stoyanov N. Synthesis and Antimicrobial Activity of 1,8-Naphthalimide Derivatives of Nalidixic Acid. J. Chem. Technol. Metall. 2019;54:1146–1156. [Google Scholar]
- 192.Al-Azzawi A.M., Hamd A.S. Synthesis and Antimicrobial Screening of New Naphthalimides Linked to Oxadiazole, Thiadiazole and Triazole Cycles. Int. J. Res. Pharm. Chem. 2014;4:283–290. [Google Scholar]
- 193.Al-Azzawi A.M., Sulaiman A. Synthesis, Characterization and Evaluation of Antibacterial Activity of Several New 1,8-Naphthalimides Containing Benzothiazole Moiety. Karbala J. Pharm. Sci. 2011:111–123. [Google Scholar]
- 194.reported as Bacillus typhi in the original paper.
- 195.Gong H.-H., Baathulaa K., Lv J.-S., Cai G.-X., Zhou C.-H. Synthesis and biological evaluation of Schiff base-linked imidazolyl naphthalimides as novel potential anti-MRSA agents. MedChemComm. 2016;7:924–931. doi: 10.1039/C5MD00574D. [DOI] [Google Scholar]
- 196.Elmorsy A., Hebishy A.M.S., Elwahy A., Abdelfattah M.S. Synthesis and Chemistry of Bis-imidazole Derivatives: A Review. GJSFR B Chem. 2018;18:25–31. [Google Scholar]
- 197.Zhao G., Yin F.-J., Ge H.-Y., Li S.-A. Synthesis, Structure and Antibacterial Properties of Bis-Imidazole-bis(naphthalene-1-yl-acetato)copper(II) Asian J. Chem. 2014;26:2550–2552. doi: 10.14233/ajchem.2014.15734. [DOI] [Google Scholar]
- 198.Zampieri D., Mamolo M.G., Vio L., Banfi E., Scialino G., Fermeglia M., Ferrone M., Pricl S. Synthesis, antifungal and antimycobacterial activities of new bis-imidazole derivatives, and prediction of their binding to P45014DM by molecular docking and MM/PBSA method. Bioorg. Med. Chem. 2007;15:7444–7458. doi: 10.1016/j.bmc.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 199.Fang B., Zhou C.-H., Rao X.-C. Synthesis and biological activities of novel amine-derived bis-azoles as potential antibacterial and antifungal agents. Eur. J. Med. Chem. 2010;45:4388–4398. doi: 10.1016/j.ejmech.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 200.Al-Mohammed N.N., Alias Y., Abdullah Z., Shakir R.M., Taha E.M., Hamid A.A. Synthesis and Antibacterial Evaluation of Some Novel Imidazole and Benzimidazole Sulfonamides. Molecules. 2013;18:11978–11995. doi: 10.3390/molecules181011978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Al-Mohammed N.N., Alias Y., Abdullah Z. Bis-imidazolium and benzimidazolium based gemini-type ionic liquids structure: Synthesis and antibacterial evaluation. RSC Adv. 2015;5:92602–92617. doi: 10.1039/C5RA13629F. [DOI] [Google Scholar]
- 202.Antoci V., Cucu D., Zbancioc G., Moldoveanu C., Mangalagiu V., Amariucai-Mantu D., Aricu A., Mangalagiu I.I. Bis-(imidazole/benzimidazole)-pyridine derivatives: Synthesis, structure and antimycobacterial activity. Future Med. Chem. 2020;12:207–222. doi: 10.4155/fmc-2019-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Nagarajan N., Vanitha G., Ananth D.A., Rameshkumar A., Sivasudha T., Renganathan R. Bioimaging, antibacterial and antifungal properties of imidazole-pyridine fluorophores: Synthesis, characterization and solvatochromism. J. Photochem. Photobiol. B Biol. 2013;127:212–222. doi: 10.1016/j.jphotobiol.2013.08.016. [DOI] [PubMed] [Google Scholar]
- 204.Abbas I., Gomha S., Elaasser M., Bauomi M. Synthesis and biological evaluation of new pyridines containing imidazole moiety as antimicrobial and anticancer agents. Turk. J. Chem. 2015;39:334–346. doi: 10.3906/kim-1410-25. [DOI] [Google Scholar]
- 205.Dhawas A.K., Thakare S.S., Thakare N.R. Synthesis and characterization of some new 1,4,5-trisubstituted imidazole-2-thiols derivatives. J. Chem. Pharm. Res. 2012;4:866–871. [Google Scholar]
- 206.Fu H.-G., Li Z.-W., Hu X.-X., Si S.-Y., You X.-F., Tang S., Wang Y.-X., Song D.-Q. Synthesis and Biological Evaluation of Quinoline Derivatives as a Novel Class of Broad-Spectrum Antibacterial Agents. Molecules. 2019;24:548. doi: 10.3390/molecules24030548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Pham T.D.M., Ziora Z.M., Blaskovich M.A.T. Quinolone antibiotics. MedChemComm. 2019;10:1719–1739. doi: 10.1039/C9MD00120D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Von Rosenstiel N., Adam D. Quinolone Antibacterials. An Update of their Pharmacology and Therapeutic Use. Drugs. 1994;47:872–901. doi: 10.2165/00003495-199447060-00003. [DOI] [PubMed] [Google Scholar]
- 209.Gómez C.M.M., Kouznetsov V.V. Recent Developments on Antimicrobial Quinoline Chemistry. In: Méndez-Vilas A., editor. Microbial Pathogens and Strategies for Combating Them: Science, Technology and Education. Formatex Research Center; Badajoz, Spain: 2013. pp. 666–677. [Google Scholar]
- 210.Kharb R., Kaur H. Therapeutic Significance of Quinoline Derivatives as Antimicrobial Agents. Int. Res. J. Pharm. 2013;4:63–69. doi: 10.7897/2230-8407.04311. [DOI] [Google Scholar]
- 211.Teng P., Li C., Peng Z., Anne Marie V., Nimmagadda A., Su M., Li Y., Sun X., Cai J. Facilely accessible quinoline derivatives as potent antibacterial agents. Bioorg. Med. Chem. 2018;26:3573–3579. doi: 10.1016/j.bmc.2018.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Parab R.H., Dixit B.C. Synthesis, Characterization and Antimicrobial Activity of Imidazole Derivatives Based on 2-chloro-7-methyl-3-formylquinoline. E-J. Chem. 2012;9:1188–1195. doi: 10.1155/2012/164235. [DOI] [Google Scholar]
- 213.Pawar R.A., Kohak A.L., Gogte V.G. Synthesis of Benzoxazolo-1,8- & 1,6-naphthapyridines, Benzoxazolylbenzo[h]quinoline & 1,9-Bisbenzoxazolophenanthroline. Indian J. Chem. 1976;14B:375–376. [Google Scholar]
- 214.Rajput A.P. Studies on Vilsmeier-Haack Reaction: Preparation and Synthetic Applications of Synthones 4-Chloro-2-arylaminothiazole-5-carboxaldehydes. Asian J. Chem. 2004;16:1374–1380. [Google Scholar]
- 215.Desai N.C., Maheta A.S., Rajpara K.M., Joshi V.V., Vaghani H.V., Satodiya H.M. Green synthesis of novel quinoline based imidazole derivatives and evaluation of their antimicrobial activity. J. Saudi Chem. Soc. 2014;18:963–971. doi: 10.1016/j.jscs.2011.11.021. [DOI] [Google Scholar]
- 216.Cui S.-F., Peng L.-P., Zhang H.-Z., Rasheed S., Kannekanti V.K., Zhou C.-H. Novel hybrids of metronidazole and quinolones: Synthesis, bioactive evaluation, cytotoxicity, preliminary antimicrobial mechanism and effect of metal ions on their transportation by human serum albumin. Eur. J. Med. Chem. 2014;86:318–334. doi: 10.1016/j.ejmech.2014.08.063. [DOI] [PubMed] [Google Scholar]
- 217.reported as Eberthella typhosa in the original paper.
- 218.Zhang L., Kannekanti V.K., Rasheed S., Geng R.-X., Zhou C.-H. Design, Synthesis, and Antimicrobial Evaluation of Novel Quinolone Imidazoles and Interactions with MRSA DNA. Chem. Biol. Drug Des. 2015;86:648–655. doi: 10.1111/cbdd.12532. [DOI] [PubMed] [Google Scholar]
- 219.Al-Qawasmeh R.A., Huthail B.B., Sinnokrot M.O., Semreen M.H., Odeh R.A., Abu-Zarga M.H., Tarazi H., Yousef I.A., Al-Tel T.H. Design, Synthesis and Qualitative Structure Activity Relationship Evaluations of Quinoline-Based Bisarylimidazoles as Antibacterial Motifs. Med. Chem. (Shariqah) 2016;12:563–573. doi: 10.2174/1573406412666160518142441. [DOI] [PubMed] [Google Scholar]
- 220.Shobhashana P.G., Prasad P., Kalola A.G., Patel M.P. Synthesis of Imidazole Derivatives Bearing Quinoline Nucleus Catalysed by CAN and their Antimicrobial, Antitubercular and Molecular Docking Studies. Res. J. Life Sci. Bioinform. Pharm. Chem. Sci. 2018;4:175–186. doi: 10.26479/2018.0403.15. [DOI] [Google Scholar]
- 221.Insuasty D., Vidal O., Bernal A., Marquez E., Guzman J., Insuasty B., Quiroga J., Svetaz L., Zacchino S., Puerto G., et al. Antimicrobial Activity of Quinoline-Based Hydroxyimidazolium Hybrids. Antibiotics. 2019;8:239. doi: 10.3390/antibiotics8040239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Laali K.K., Insuasty D., Abonia R., Insuasty B., Bunge S.D. Novel quinoline–imidazolium adducts via the reaction of 2-oxoquinoline-3-carbaldehyde and quinoline-3-carbaldehydes with 1-butyl-3-methylimidazolium chloride [BMIM][Cl] Tetrahedron Lett. 2014;55:4395–4399. doi: 10.1016/j.tetlet.2014.05.094. [DOI] [Google Scholar]
- 223.Meth-Cohn O. The Synthesis of Pyridines, Quinolines and Other Related Systems by the Vilsmeier and the Reverse Vilsmeier Method. Heterocycles (Sendai) 1993;35:539–557. doi: 10.3987/REV-92-SR4. [DOI] [Google Scholar]
- 224.Cieplik J., Stolarczyk M., Pluta J., Gubrynowicz O., Bryndal I., Lis T., Mikulewicz M. Synthesis and Antibacterial Properties of Pyrimidine Derivatives. Acta Pol. Pharm. 2015;72:53–64. [PubMed] [Google Scholar]
- 225.Sharma V., Chitranshi N., Agarwal A.K. Significance and Biological Importance of Pyrimidine in the Microbial World. Int. J. Med. Chem. 2014;2014:202784. doi: 10.1155/2014/202784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Dansena H., Dhongade H.J., Chandrakar K. Pharmacological Potentials of Pyrimidine Derivative: A Review. Asian J. Pharm. Clin. Res. 2015;8:171–177. [Google Scholar]
- 227.Ibraheem F., Saddique F.A., Aslam S., Mansha A., Farooq T., Ahmad M. Recent synthetic methodologies for pyrimidine and its derivatives. Turk. J. Chem. 2018;42:1421–1458. doi: 10.3906/kim-1806-8. [DOI] [Google Scholar]
- 228.Botta M., Artico M., Massa S., Gambacorta A., Marongiu M.E., Pani A., La Colla P. Synthesis, antimicrobial and antiviral activities of isotrimethoprim and some related derivatives. Eur. J. Med. Chem. 1992;27:251–257. doi: 10.1016/0223-5234(92)90009-P. [DOI] [Google Scholar]
- 229.Agarwal N., Srivastava P., Raghuwanshi S.K., Upadhyay D.N., Sinha S., Shukla P.K., Ram V.J. Chloropyrimidines as a New Class of Antimicrobial Agents. Bioorg. Med. Chem. 2002;10:869–874. doi: 10.1016/S0968-0896(01)00374-1. [DOI] [PubMed] [Google Scholar]
- 230.Prakash O., Bhardwaj V., Kumar R., Tyagi P., Aneja K.R. Organoiodine (III) mediated synthesis of 3-aryl/hetryl-5,7-dimethyl-1,2,4-triazolo[4,3-a]pyrimidines as antibacterial agents. Eur. J. Med. Chem. 2004;39:1073–1077. doi: 10.1016/j.ejmech.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 231.Desai N.C., Kotadiya G.M., Trivedi A.R. Studies on molecular properties prediction, antitubercular and antimicrobial activities of novel quinoline based pyrimidine motifs. Bioorg. Med. Chem. Lett. 2014;24:3126–3130. doi: 10.1016/j.bmcl.2014.05.002. [DOI] [PubMed] [Google Scholar]
- 232.Rathod A.K. Antifungal and Antibacterial activities of Imidazolylpyrimidines derivatives and their QSAR Studies under Conventional and Microwave-assisted. Int. J. PharmTech Res. 2011;3:1942–1951. [Google Scholar]
- 233.Desai N.C., Vaghani H.V., Rajpara K.M., Joshi V.V., Satodiya H.M. Novel approach for synthesis of potent antimicrobial hybrid molecules containing pyrimidine-based imidazole scaffolds. Med. Chem. Res. 2014;23:4395–4403. doi: 10.1007/s00044-014-1005-1. [DOI] [Google Scholar]
- 234.Pathan N.B., Rahatgaonkar A.M. Solid supported microwave induced synthesis of imidazole–pyrimidine hybrids: Antimicrobial evaluation and docking study as 14DM-CPY51 inhibitors. Arab. J. Chem. 2016;9:S100–S108. doi: 10.1016/j.arabjc.2011.02.013. [DOI] [Google Scholar]
- 235.Abdel-Wahab B.F., Awad G.E.A., Badria F.A. Synthesis, antimicrobial, antioxidant, anti-hemolytic and cytotoxic evaluation of new imidazole-based heterocycles. Eur. J. Med. Chem. 2011;46:1505–1511. doi: 10.1016/j.ejmech.2011.01.062. [DOI] [PubMed] [Google Scholar]
- 236.Veronese A.C., Cavicchioni G., Servadio G., Vecchiati G. Syntheses of 2-Arylimidazole Derivatives through Annelations Employing Benzylamines. J. Heterocycl. Chem. 1980;17:1723–1725. doi: 10.1002/jhet.5570170816. [DOI] [Google Scholar]
- 237.Addla D., [Bhukya] B., Sridhar B., Devi A., Kantevari S. Design, synthesis and antimicrobial evaluation of novel 1-benzyl 2-butyl-4-chloroimidazole embodied 4-azafluorenones via molecular hybridization approach. Bioorg. Med. Chem. Lett. 2012;22:7475–7480. doi: 10.1016/j.bmcl.2012.10.042. [DOI] [PubMed] [Google Scholar]
- 238.Prachayasittikul S., Manam P., Chinworrungsee M., Isarankura-Na-Ayudhya C., Ruchirawat S., Prachayasittikul V. Bioactive Azafluorenone Alkaloids from Polyalthia debilis (Pierre) Finet & Gagnep. Molecules. 2009;14:4414–4424. doi: 10.3390/molecules14114414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Koyama J., Morita I., Kobayashi N., Osakai T., Usuki Y., Taniguchi M. Structure–activity relations of azafluorenone and azaanthraquinone as antimicrobial compounds. Bioorg. Med. Chem. Lett. 2005;15:1079–1082. doi: 10.1016/j.bmcl.2004.12.059. [DOI] [PubMed] [Google Scholar]
- 240.Kim H.-S., Jadhav J.R., Jung S.-J., Kwak J.-H. Synthesis and antimicrobial activity of imidazole and pyridine appended cholestane-based conjugates. Bioorg. Med. Chem. Lett. 2013;23:4315–4318. doi: 10.1016/j.bmcl.2013.05.098. [DOI] [PubMed] [Google Scholar]
- 241.Wen S.-Q., Jeyakkumar P., Srinivasa Rao A., Zhang L., Zhou C.-H. Discovery of novel berberine imidazoles as safe antimicrobial agents by down regulating ROS generation. Bioorg. Med. Chem. Lett. 2016;26:2768–2773. doi: 10.1016/j.bmcl.2016.04.070. [DOI] [PubMed] [Google Scholar]
- 242.Wang H., Zhu C., Ying Y., Luo L., Huang D., Luo Z. Metformin and berberine, two versatile drugs in treatment of common metabolic diseases. Oncotarget. 2018;9:10135–10146. doi: 10.18632/oncotarget.20807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Yi Z.-B., Yan Y., Liang Y.-Z., Bao Z. Evaluation of the antimicrobial mode of berberine by LC/ESI-MS combined with principal component analysis. J. Pharm. Biomed. Anal. 2007;44:301–304. doi: 10.1016/j.jpba.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 244.Yan D., Jin C., Xiao X.-H., Dong X.-P. Antimicrobial properties of berberines alkaloids in Coptis chinensis Franch by microcalorimetry. J. Biochem. Bioph. Methods. 2008;70:845–849. doi: 10.1016/j.jbbm.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 245.Peng L., Kang S., Yin Z., Jia R., Song X., Li L., Li Z., Zou Y., Liang X., Li L., et al. Antibacterial activity and mechanism of berberine against Streptococcus agalactiae. Int. J. Clin. Exp. Pathol. 2015;8:5217–5223. [PMC free article] [PubMed] [Google Scholar]
- 246.Iwasa K., Lee D.-U., Kang S.-I., Wiegrebe W. Antimicrobial Activity of 8-Alkyl- and 8-Phenyl-Substituted Berberines and Their 12-Bromo Derivatives. J. Nat. Prod. 1998;61:1150–1153. doi: 10.1021/np980044+. [DOI] [PubMed] [Google Scholar]
- 247.Li R., Wu J., He Y., Hai L., Wu Y. Synthesis and in vitro evaluation of 12-(substituted aminomethyl) berberrubine derivatives as anti-diabetics. Bioorg. Med. Chem. Lett. 2014;24:1762–1765. doi: 10.1016/j.bmcl.2014.02.032. [DOI] [PubMed] [Google Scholar]
- 248.Idrees M., Nasare R.D., Siddiqui N.J. Synthesis and Antibacterial Screening of 4-Arylidene-5-oxo-imidazoles having Carboxamide Linkage with 5-(benzofuran-2-yl)-1-phenylpyrazole Moiety. Chem. Sci. Trans. 2016;5:1090–1095. doi: 10.7598/cst2016.1323. [DOI] [Google Scholar]
- 249.Sharma S., Sharma V., Singh G., Kaur H., Srivastava S., Ishar M.P.S. 2-(chromon-3-yl)imidazole derivatives as potential antimicrobial agents: Synthesis, biological evaluation and molecular docking studies. J. Chem. Biol. 2017;10:35–44. doi: 10.1007/s12154-016-0162-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Biggs-Houck J.E., Younai A., Shaw J.T. Recent advances in multicomponent reactions for diversity-oriented synthesis. Curr. Opin. Chem. Biol. 2010;14:371–382. doi: 10.1016/j.cbpa.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 251.Dömling A., Wang W., Wang K. Chemistry and Biology of Multicomponent Reactions. Chem. Rev. 2012;112:3083–3135. doi: 10.1021/cr100233r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Malinakova H.C. Recent advances in the discovery and design of multicomponent reactions for the generation of small-molecule libraries. Rep. Org. Chem. 2015:75–90. doi: 10.2147/ROC.S65115. [DOI] [Google Scholar]
- 253.Graebin C.S., Ribeiro F.V., Rogério K.R., Kümmerle A.E. Multicomponent Reactions for the Synthesis of Bioactive Compounds: A Review. Curr. Org. Synth. 2019;16:855–899. doi: 10.2174/1570179416666190718153703. [DOI] [PubMed] [Google Scholar]
- 254.Kappe C.O. Controlled Microwave Heating in Modern Organic Synthesis. Angew. Chem. Int. Ed. 2004;43:6250–6284. doi: 10.1002/anie.200400655. [DOI] [PubMed] [Google Scholar]
- 255.Bowman M.D., Holcomb J.L., Kormos C.M., Leadbeater N.E., Williams V.A. Approaches for Scale-Up of Microwave-Promoted Reactions. Org. Process. Res. Dev. 2008;12:41–57. doi: 10.1021/op700187w. [DOI] [Google Scholar]
- 256.Loftin A., Armstrong D. Review: Microwave-Promoted Organic Synthesis. Am. J. Undergrad. Res. 2009;8:5–7. doi: 10.33697/ajur.2009.012. [DOI] [Google Scholar]
- 257.Hügel H.M. Microwave Multicomponent Synthesis. Molecules. 2009;14:4936–4972. doi: 10.3390/molecules14124936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Kokel A., Schäfer C., Török B. Microwave-Assisted Reactions in Green Chemistry. In: Han B., Wu T., editors. Green Chemistry and Chemical Engineering. A Volume in the Encyclopedia of Sustainability Science and Technology. 2nd ed. Springer; New York, NY, USA: 2019. pp. 573–612. [Google Scholar]
- 259.Liang L., Astruc D. The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) “click” reaction and its applications. An overview. Coord. Chem. Rev. 2011;255:2933–2945. doi: 10.1016/j.ccr.2011.06.028. [DOI] [Google Scholar]
- 260.Zheng X., Ma Z., Zhang D. Synthesis of Imidazole-Based Medicinal Molecules Utilizing the van Leusen Imidazole Synthesis. Pharmaceuticals. 2020;13:37. doi: 10.3390/ph13030037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Santajit S., Indrawattana N. Mechanisms of Antimicrobial Resistance in ESKAPE Pathogens. Biomed. Res. Int. 2016;2016:2475067. doi: 10.1155/2016/2475067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Himo F., Lovell T., Hilgraf R., Rostovtsev V.V., Noodleman L., Sharpless K.B., Fokin V.V. Copper(I)-Catalyzed Synthesis of Azoles. DFT Study Predicts Unprecedented Reactivity and Intermediates. J. Am. Chem. Soc. 2005;127:210–216. doi: 10.1021/ja0471525. [DOI] [PubMed] [Google Scholar]
- 263.Butler M.S., Paterson D.L. Antibiotics in the clinical pipeline in October 2019. J. Antibiot. (Tokyo) 2020;73:329–364. doi: 10.1038/s41429-020-0291-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Cao C., Luo A., Wu P., Weng D., Zheng H., Wang S. Efficacy and safety of morinidazole in pelvic inflammatory disease: Results of a multicenter, double-blind, randomized trial. Eur. J. Clin. Microbiol. Infect. Dis. 2017;36:1225–1230. doi: 10.1007/s10096-017-2913-z. [DOI] [PubMed] [Google Scholar]
- 265.Ocakoglu K., Tasli H., Hosgor-Limoncu M., Lambrecht F.Y. Synthesis and antimicrobial activity of imidazolium salts. Trends Cancer Res. Chemother. 2018;1 doi: 10.15761/TCRC.1000103. [DOI] [Google Scholar]