

SUMMARY
Neuroblastoma (NB), derived from the neural crest (NC), is the most common pediatric extracranial solid tumor. Here, we establish a platform that allows the study of human NBs in mouse-human NC chimeras. Chimeric mice were produced by injecting human NC cells carrying NB relevant oncogenes in utero into gastrulating mouse embryos. The mice developed tumors composed of a heterogenous cell population that resembled that seen in primary NBs of patients but were significantly different from homogeneous tumors formed in xenotransplantation models. The human tumors emerged in immunocompetent hosts and were extensively infiltrated by mouse cytotoxic T cells, reflecting a vigorous host anti-tumor immune response. However, the tumors blunted the immune response by inducing infiltration of regulatory T cells and expression of immune-suppressive molecules similar to escape mechanisms seen in human cancer patients. Thus, this experimental platform allows the study of human tumor initiation, progression, manifestation, and tumor-immune-system interactions in an animal model system.
In Brief
Cohen et al. present a novel system to model human cancer in immunocompetent mice. This is accomplished by injecting human pluripotent stem cell-derived neural crest cells (hNCC) into early mouse embryos, generating mouse-human neural crest chimeras. Oncogenes are then induced, allowing transformation of the hNCCs to form neuroblastoma.
Graphical Abstract
INTRODUCTION
Based on the discovery of immune checkpoints and the success of checkpoint blockade agents, the new generation of cancer immunotherapies have resulted in remarkable advances in cancer treatment. However, the fraction of patients who respond to immune therapy is generally around 20% for the most common solid tumors (Ribas and Wolchok, 2018), calling for model systems that would facilitate the study of the parameters that allow tumors to escape immune inhibition. Mouse transgenic and syngeneic models were extensively used to discover the basic core principles of immuno-oncology, and the use of these animal models has significantly advanced cancer research. However, species-specific differences between humans and mice may make it problematic to relate such results obtained in transgenic mouse models to human cancer. Furthermore, transgenic tumors often present very weak immune responses. Alternative experimental approaches for studying human cancers use xenotransplantation models that involve the implantation of human cancer cells or primary tumors into immunocompromised mice. Although these models have yielded a wealth of information on the biology of human cancer and therapeutic strategies, they pose several limitations. Because only end-stage tumor cells, often adapted to cell growth in culture or harvested from patients, are transplanted into host animals that are immune deficient, these xenotransplantation models do not allow investigation of tumor initiation and the anti-tumor immune reactions or tumor immune evasion. Thus, so far, it has been impossible to study human cancer formation in immunocompetent animals.
Interspecies chimeras represent a promising experimental system for studying human development and disease and may provide the most physiologically relevant environment to study human disease in an in vivo context by overcoming some of the limitations of conventional xenotransplantation animal models (Wu et al., 2016; Suchy and Nakauchi 2017; Soldner and Jaenisch, 2018). Both pluripotent and committed stem cells have been used as donor cells to generate interspecies chimeras. Injection of pluripotent rat stem cells (PSCs) into mouse blastocysts resulted in chimeric mice with rat donor cells contributing to all tissues (Kobayashi et al., 2010). By contrast, human PSCs introduced into mouse blastocysts resulted in very low if any functional incorporation of the human donor cells into the host embryo (Gafni et al., 2013; Theunissen et al., 2016; Wu et al., 2017; Yang et al., 2017), with no postnatal chimeras having been generated.
Interspecies postnatal chimeras have been produced by introducing multipotent, lineage-restricted stem cells into post-implantation mouse embryos or neonates. For example, human glial progenitors injected into the neonatal mouse integrated into the host brain and showed enhanced synaptic plasticity and learning in the chimeric mice (Han et al., 2013; Windrem et al., 2017). Similarly, hPSC-derived neurons transplanted into an Alzheimer’s disease (AD) mouse model displayed signs of neurodegeneration manifesting as cell death and pathological features typically observed in AD patients (Espuny-Camacho et al., 2017). Also, hPSC-differentiated β-like precursor cells transplanted into the neonatal mouse pancreas led to functional in vivo engraftment of human β-like cells (Ma et al., 2018). We have, based on previous in utero manipulations of mouse embryos (Jaenisch, 1985; Huszar et al., 1991), generated mouse-human neural crest chimeras (Cohen et al., 2016). Neural crest cells (NCCs) are multipotent, emerge from the neural tube at gastrulation, and generate a wide variety of lineages, including peripheral neurons, enteric neurons, Schwann cells, melanocytes, and cells of the adrenal medulla (Bronner and LeDouarin, 2012). Neural crest (NC)-related deficiencies are the cause of multiple human diseases, and the term “neurocristopathies” has been proposed to denote syndromes or tumors involving NCCs. NC-derived malignancies include cancers, such as melanoma, neuroblastoma, and neurofibromatosis (Vega-Lopez et al., 2018), and the use of hPSC-derived NCCs for modeling human NC diseases is an attractive in vitro experimental approach (Fattahi et al., 2016; Huang et al., 2016). We have previously demonstrated that hPSC-derived NCCs, when injected into the gastrulating mouse embryo, migrate along the dorso-lateral migration route contributing to the pigment system of the mouse, suggesting that this platform may be used to model neurocristopathies in vivo (Cohen et al., 2016).
The goal of this work was to determine the feasibility of modeling a NC-derived human cancer in chimeric mice, using hPSC-derived NCCs. We chose to model human neuroblastoma (NB), an embryonal cancer derived from the developing NC, with tumors arising in the sympathetic ganglia and adrenal medulla (Cheung and Dyer, 2013). We generated hPSC-derived hNCCs expressing the doxycycline (Dox)-inducible NB-relevant oncogenes MYCN (Brodeur et al., 1984; Seeger et al., 1985) and ALKF1174L (Chen et al., 2008; George et al., 2008; Janoueix-Lerosey et al., 2008; Mossé et al., 2008). Introduction of these hNCCs into mouse embryos led to the formation of tumors resembling primary human NBs. Most significantly, the tumors developed in immunocompetent host mice, enabling the analysis of human tumor-immune system interactions and immune escape mechanisms in the experimental animal model system. We observed that the host mounts an anti-tumor immune response that is blunted by tumor-induced immune inhibitory mechanisms. Our mouse-human chimeric system facilitates the study of critical interactions between tumor cells and the immune system, in contrast to conventional xenotransplantation assays that rely on immunocompromised host animals and thus preclude meaningful study of the immune response during cancer development.
RESULTS
Generation of Human Pluripotent Cells Expressing Conditional Oncogenes
Previous data have shown that human NC cells injected into E8.5 embryos migrate along the dorso-lateral route and can contribute to the host pigment system in postnatal mice (Cohen et al., 2016). In these earlier studies, we did not investigate whether the human donor cells could also migrate along the medial NC route and contribute to internal structures (Trainor, 2005). To this end, GFP-labeled hPSC-derived hNCCs, which demonstrate characteristics of the NC, were microinjected into E8.5 mouse embryos as previously described (Figures S1A–S1C; Jaenisch, 1985; Cohen et al., 2016), and chimeric embryos were analyzed for human cell contribution (Figures S1D–S1F). 24 h post injection, hNCCs were found within the embryos together with the host NCCs and expressing the NC markers p75 and HNK1 (Figure S1D). At 5 to 6 days post injection, human eGFP-positive cells were found to differentiate and migrate into NC-derivative tissues of the developing peripheral nervous system (PNS), such as dorsal root ganglia (DRG) and the trigeminal ganglion (Figures S1E and S1F). Immunostaining for peripherin, a PNS marker, confirmed the proper integration of human cells within the host DRGs (Figure S1E, right). To assess the hNCC contribution to postnatal mice, we dissected DRGs of adult chimeric mice (confirmed by coat-color contribution) and individually tested the DRGs by genomic qPCR (Cohen et al., 2016). Human donor cell contribution was found in 12.4% ± 2.4% of tested DRGs (n = 35–39 per animal; Figure S1G), indicating that hNCCs can contribute, in addition to the pigment lineage, to PNS lineages of adult chimeric mice. However, we failed to detect donor hNCCs in structures of the autonomic nervous system, such as in the intestine or the adrenal gland, likely due to low donor cell contribution to these structures, as their colonization requires a long migration period through the developing embryo. Nevertheless, the contribution of donor cells to the autonomic nervous system was enhanced by providing the donor NCCs with a proliferative advantage through the expression of tumor-relevant oncogenes, as shown below.
To determine whether hNCCs can also be induced to form tumors in addition to contributing to host developmental lineage, we generated hPSCs that conditionally overexpress MYCN and ALKF1174L, two oncogenic mutations that occur in primary patient neuroblastoma tumors and lead to tumor formation in genetically engineered mouse models (GEMMs) of the same disease. The MYCN oncogene is frequently amplified in high-risk NB and correlates with an aggressive phenotype and a poor prognosis (Brodeur et al., 1984; Seeger et al., 1985). Overexpression of MYCN leads to the formation of murine NB (Weiss et al., 1997). Several mutations in the anaplastic lymphoma kinase (ALK) gene are involved in the development of sporadic and familial neuroblastoma (Chen et al., 2008; George et al., 2008; Janoueix-Lerosey et al., 2008; Mossé et al., 2008). One of the common ALK mutations results in a cytosine-to-adenine change in exon 23, causing a phenylalanine-to-leucine substitution (F1174L) within the kinase domain, triggering abnormal proliferation of neuroblasts. Indeed, the combination of MYCN and ALKF1174L (Th-MYCN+ALKF1174L) overexpression induces NB tumors with high penetrance in a genetically engineered mouse model (Berry et al., 2012). To conditionally overexpress MYCN and ALKF1174L in hNCCs, we transduced the Dox-inducible lentiviral constructs FUW-TetO-ALKF1174L-t2A-tdTomato and FUW-TetO-MYCN-3×2A-NeoR (Figure S2E) into two hPSC lines (WIBR#3 hESCs and AA#1 hiPSCs; Cohen et al., 2016). To facilitate tracing of the cells after injection, we used hPSCs that harbored an eGFP reporter inserted into the AAVS1 locus, as well as Luciferase and LacZ reporter constructs in the AAVS1 locus of hESCs (Figures S2A-S2D; Hockemeyer et al., 2009). Using established protocols, the hPSCs were differentiated into hNCCs (Cohen et al., 2016) and tested for Dox-dependent oncogene activation and their effect on cell growth (Figure 1A). The addition of Dox to the hNCC cultures activated oncogene expression as indicated by immunofluorescence (IF), by the expression of the tdTomato fluorescence-reporter, and by RNA expression (Figures 1B and 1C). MYCN and ALKF1174L expression and constitutive activation of mutated ALK by phosphorylation were confirmed by western blot analyses (Figure 1D). Oncogene activation led to significant increases in hNCC proliferation and colony formation in soft agar, characteristics typical of transformed cells (Figures 1E and 1F). Finally, to test whether oncogene activation could promote tumor-like behavior in vivo, the hNCCs were injected into the flanks of immunocompromised mice. Figure 1G shows that Dox-dependent expression of the two oncogenes was sufficient to generate hNCC-derived xenograft tumors in immunocompromised mice. Our results suggest that the combined expression of NB-relevant oncogenes into hPSCs is sufficient to induce the malignant transformation of hNCC-derived cells.
Figure 1. hPSCs-Derived hNCCs Prone to Generate NB.

(A) A schematic representation of the experiment. Lentiviral transgenes of FUW-TetO-ALKF1174L-t2A-tdTomato and FUW-TetO-MYCN-3×2A-NeoR were introduced to hPSCs for the controlled overexpression of ALKF1174L and MYCN. hPSCs were differentiated into hNCCs, and oncogenes were activated by Dox.
(B) Addition of Dox to the cultured hNCCs induces strong expression of MYCN, ALKF1174L, and lentiviral-dependent tdTomato (bottom, nuclear staining; middle, peripheral staining; and top, whole-cell fluorescence, respectively). Scale bars indicate 10 μm.
(C) Oncogene activation was monitored for gene overexpression using qRT-PCR.
(D) Oncogenes overexpression was confirmed by western blot. The Dox treatment also induced self-activation of ALK (pALK Tyr1604; Kelly NB cell line serve as positive control).
(E and F) hNCCs with activated oncogenes formed significantly more colonies in soft agar (E, n = 28), and (F) exhibited significantly high rates of cell proliferation.
(G) Tumor formation of Dox treated hNCCs following injection into flanks of immune-compromised mice (tumor growth visible by IVIS, right, and after postmortem dissection, right).
Data presented as means; error bars represent SD; *** p value < 0.0001; (A) and (E): t test; (F): two-way ANOVA.
Tumor Formation in Mouse-Human Neural Crest Chimeras
To investigate whether hNCCs carrying the inducible MYCN and ALKF1174L oncogenes could generate tumors in mouse-human NC chimeras, the cells were microinjected in utero into developing mouse embryos at E8.5. To activate the oncogenes, Dox was added to the drinking water of pregnant females beginning at day E9.5 (Figure 2A). To trace the human cells during mouse development, we inspected the embryos at 6 days after injection and observed eGFP+ donor cell clumps, suggesting proliferation of the oncogene-activated cells (Figure S3). Females pregnant with injected embryos and postnatal pups were treated continuously with Dox to assure oncogene activation, and all animals were monitored for tumor formation. We first detected Luciferase activity by non-invasive bioluminescent imaging in 3- to 6-month-old mice derived from the injected embryos, consistent with proliferation of the injected hNCC-derived cells (Figures 2B and 2C). A total of 14% of the injected mice (n = 198) developed locally invasive abdominal masses, mostly in the retroperitoneum (n = 28) and the adrenal gland (n = 1), typical locations for human NB formation (see Figures 2C and S4A; Table S1). Similar to the Th-MYCN+ALKF1174L GEMM NB model, where tumors form in thoracic and abdominal paraspinal ganglia and the adrenal glands, but do not form distant metastasis (Berry et al., 2012), no macroscopic metastatic tumors were detected in the mouse-human chimeras. Tumors arising in chimeric mice (referred to as chimeric-derived human neuroblastoma [CHNB]) were collected between 3 and 15 months of age (10.4 ± 3.9 months; see Figure 4A; Table S1) and found to express eGFP, confirming human donor cell origin. Moreover, the tumors were positive for MYCN and ALK expression and expressed tdTomato, verifying oncogene expression in the tumors (Figures 2D–2G). We further confirmed oncogene expression and constitutive ALK activation in CHNBs and compared their expression to NB patient-derived xenografts (referred to as PDX NBs) with amplified MYCN (MYCNamp), with or without ALK mutation (ALKF1174L or ALKWT), and to GEMM NB tumors overexpressing MYCN and ALKF1174L (Th-MYCN+ALKF1174L, referred to as GEMM NBs; Berry et al., 2012; Figures 2K and S4E). CHNBs were highly proliferative, as indicated by the expression of the cell-cycle marker Ki67 (Figures 2H and S4D). Finally, CHNBs were positive for phosphorylated H2AX (γH2AX), a marker of DNA damage that indicates genome instability, which is typically associated with malignancy (Figure 2I). Thus, MYCN and mutated ALK-expressing hNCCs are capable of forming highly proliferative tumors in locations and distributions similar to those of human NB.
Figure 2. Human NB Formation in Mouse-Human Chimeras.
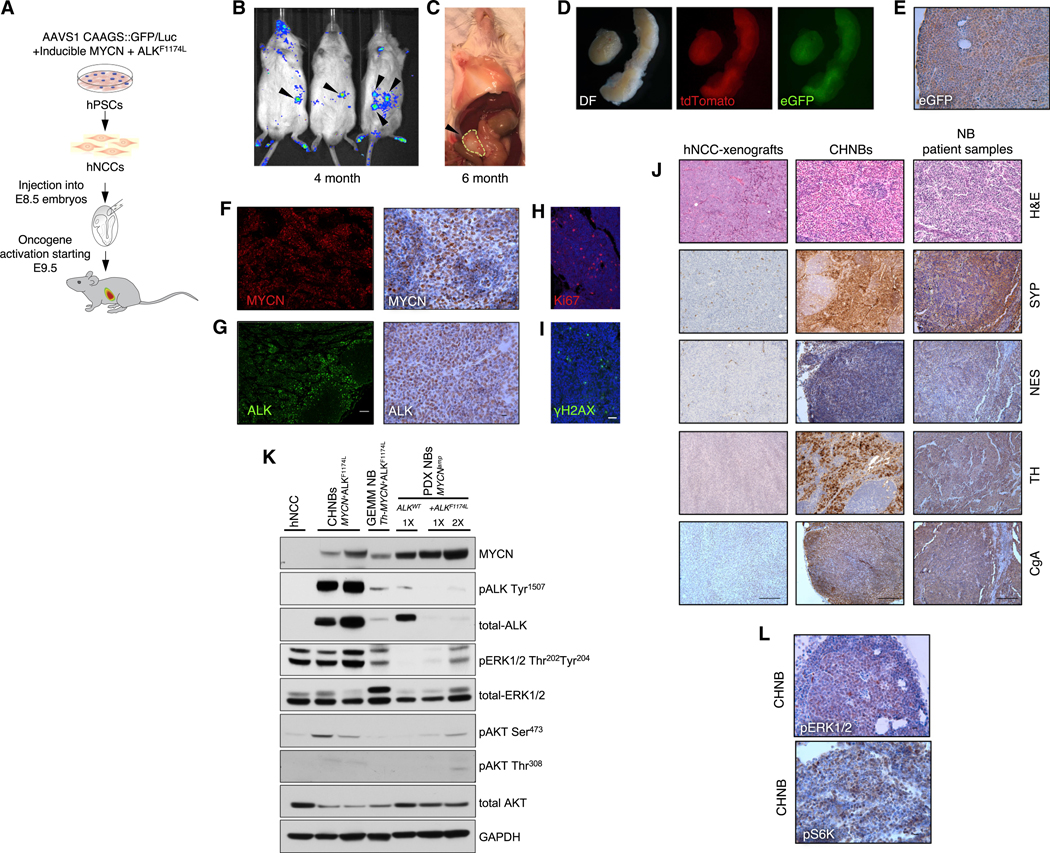
(A) Schematic representation of the experiment. E8.5 mouse embryos injected with hNCCs and monitored for tumor induction.
(B and C) Bioluminescent imaging shows human tumor formation at the age of 4 months (B) and macroscopic tumor growth at the age of 6 month (C).
(D) Tumors of chimeric mice dissected at P120 (left; DF, dark filed) express tdTomato (middle) and eGFP (right), indicating their origin from donor hNCCs.
(E) eGFP expression within tumors was confirmed by IHC (scale bar indicates 10 μm).
(F and G) The human tumors were found to express MYCN and ALK by IF (left; scale bar indicates 100 μm) and IHC staining (right; scale bar indicates 10 μm).
(H and I) CHNBs express the typical cancer hallmarks Ki67 (H) and γH2AX (I); scale bar indicates 20 μm.
(J) H&E and IHC show that CHNB express the typical NB markers synaptophysin (SYP), nestin (NES), tyrosine hydroxylase (TH), and chromogranin A (CgA) similarto NB patient samples. Dox-treated hNCCs, subcutaneously injected into immunocompromised mice, formed xenograft tumors (left column), which did not express these NB markers. See IHC quantifications in Figures S4F–S4I; scale bars indicate 100 μm.
(K) Western blot analysis of different NB models show expression of MYCN and ALK along with ALK (pALK Tyr1507) self-activation in CHNBs, in PDX NBs (NB-patient-derived xenografts) with amplified MYCN (MYCNamp), with and without mutation in ALK (ALKF1174L or ALKWT), and in mouse NB tumors of GEMM overexpressing MYCN and ALKF1174L (Th-MYCN+ALKF1174L). Activation of MAPK and PI3K/AKT pathways in CHNBs and other MYCN+ALKF1174L NB models was shown by phosphorylation of pERK1/2 Thr202 Tyr204, pAKT Ser473, and pAKT Thr308. Total ERK1/2, AKT, and GAPDH are presented for controls. 1× = 1 volume of loading; 2× = twice the loading volume.
(L) IHC staining for pERK1/2 and pS6K, confirming MAPK and PI3K/AKT activity in CHNB tumors (scale bar indicates 10 μm).
Figure 4. Mouse-Human Chimeras Develop Immune-Tolerance to Human Cells and Allow Human Tumor Growth.

(A) Fraction of mice with CHNB detected at necropsy at different ages. The detection rates of CHNB in mouse-human chimeras was similar in immunocompetent (green; n = 151) and in immunocompromised hosts (orange; n = 54), indicating that CHNB tumors develop in host mice regardless of immune state (p value = 0.45, χ2).
(B) To indirectly test the human cells’ tolerance in immunocompetent chimeric mice, a different group of mice were challenged with subcutaneous xenograft formation of Dox-induced hNCCs (see Figure 1G), and the fractions of mice that developed xenograft tumors rates was measured over time. Whereas xenograft tumors never grew in WT immunocompetent mice (gray line; n = 14), over 90% immunocompromised mice (red line; n = 25) and about a quarter of the chimeric mice (black; n = 39) developed xenograft tumors, which is similar to the previously reported chimera-formation rates of 30% (* p value < 0.05; *** p value < 0.0001, χ2).
(C) Representative IVIS images of luciferase-expressing hNCCs forming xenograft tumors in an immunocompromised host (right) and in an immunocompetent mouse-human chimera (left).
Histological examination of hematoxylin-eosin (H&E)-stained CHNB tumors revealed densely packed small, round, blue cells, with lobular patterns and occasional rosettes typically seen in human NB (Figures 2J and S4C). CHNB tumors consisted of cells expressing NB-associated markers, such as tyrosine hydroxylase (TH; 46.71% ± 18.29% of the cells), synaptophysin (SYP; 44.45% ± 10.95%), chromogranin A (CgA; 58.05% ± 16.20%), and nestin (NES; 37.38% ± 22.69%) in heterogeneous patterns (Figure 2J, middle and right, and Figures S4F–S4I). By contrast, hNCC xenografts generated subcutaneously in immunocompromised mice (Figure 1G) were homogeneous, appeared to resemble human NB samples less closely than CHNBs, and lacked cells expressing these markers (Figure 2J, left, and Figures S4F–S4I). CHNBs also showed phosphorylation of the MAPK and PI3K/AKT pathways, similar to that seen in human PDX NBs and in the GEMM NB (Th-MYCN+ALKF1174L) (Figures 2K and 2L; Berry et al., 2012; Moore et al., 2014). These results demonstrate that hNCCs expressing MYCN and ALKF1174L, when integrated into the developing mouse embryo, induce the growth of tumors that resemble primary human NB tumors, express NB markers, and activate tumor-relevant signaling pathways.
Gene-Expression Profiling of CHNBs Is Comparable to Human NBs and Other NB Models
To further characterize the identity of our CHNBs, we analyzed their gene expression profiles by RNA sequencing (RNA-seq; CHNBs MYCN+ALKF1174L; n = 8). Comparison of the expression of 20 genes frequently associated with NB revealed a significant correlation between CHNBs and human NBs (Figure 3A; Harenza et al., 2017). We performed unbiased comparison of the transcriptomes of CHNBs with patient-derived NBs and other NB models. We extracted RNAs from PDX NBs (MYCNamp+ALKWT PDX; n = 2 and MYCNamp+ALKF1174L PDX; n = 3), GEMM NBs (Th-MYCN+ALKF1174L; n = 3), NB cell lines (n = 2; Kelly, MYCNamp+ALKF1174L; SH-SY5Y, MYCNWT+ALKF1174L), hNCCs (wild type [WT] hNCCs; n = 2 and MYCN+ALKF1174L hNCCs with Dox; n = 2), and samples from hNCC xenografts (MYCN+ALKF1174L; n = 6). Because CHNBs are composed of both human and murine cells, these samples were compiled with human and mouse RNA-seq reads represented in a 50:50 ratio, and mouse genes (from CHNBs and GEMM NBs) were annotated with their human orthologs (see STAR Methods). Table S2 summarizes the samples that were processed with RNA-seq. We compared these RNA-seq profiles with publicly available expression profiles of a cohort of human NB tumors by assembling a NB meta-signature that represents the median expression levels of all genes across multiple samples of human NBRNA-seq datasets (n = 669 patients; RNA-seq samples collected from publicly available datasets: Zhang et al., 2015; and the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) initiative; see STAR Methods). We further subdivided this cohort to separately analyze NB meta-signatures for non-amplified MYCN tumors (n = 529; MYCNWT) and for NB samples with amplified MYCN (n = 125; MYCNamp). Principal component analysis (PCA) of the NB patient meta-signature against that of the various NB models listed above showed that individual samples clustered closely together but that the different NB models clustered separately (Figure 3B). Importantly, the CHNB samples clustered adjacent to the NB patient meta-signature and closer than the GEMM and PDX NB models. Similar results were seen in the PCA analysis comparing the meta-signatures of NB samples expressing MYCNWT and MYCNamp (Figure S5A). Although PCA uses the most variable genes for analyses, we further investigated similarities in the expression profiles of the different NB models when compared with the NB patient tumor meta-signature. We calculated the fraction of all genes that were similarly expressed in any of the models and the patient NB samples (Δ < 2-fold). This analysis revealed that all four models shared comparable expression levels, with 70% of the genes being similarly active when compared with genes expressed in patient tumors. The variation of expression profiles was slightly greater for CHNB samples, reflecting a higher degree of heterogeneity likely due to the greater and more variable dynamics of tumor initiation and progression in the interspecies chimeras as compared to GEMM, PDX, and established cell lines, which usually represent clonal populations (Figure 3C).
Figure 3. CHNB Tumors Present Gene Expression Profiles Similar to Human NB.

(A) RNA-seq analysis of the human genes expressed in CHNB show a significant correlation in the expression of a panel of key genes normally expressed in NB tumors (ABCC1, BIRC5, CAMTA1, CCND1, DDX1, ENOS, IGF1R, KIF1B, KRAS, MAX, NES, NGFR, NME1, NRAS, PHOX2B, RAF1, SNW1, TH, TP53, and VEGFA) with NB cell lines (Kelly and SH-SY5Y; p value = 0.011, linear regression).
(B) Principal component (PCA) analysis of CHNBs (purple), patient NB samples (black), PDX NB (orange), GEMM NB (red), NB cell lines (olive), hNCCs (blue), and Dox-induced hNCC xenografts (green). 669 RNA-seq samples of NB patients are presented as a meta-sample (in black), which represents the median expression levels of all genes across multiple samples.
(C) A direct comparison of the fraction of genes, which are similarly expressed in CHNBs and in NB models when compared to NB patients (CHNB, PDX, GEMM, or cell lines versus Patient NB; Δ < 2-fold) revealed that all four models show comparable fraction of shared genes. Bars presented as means; error bars represent SD; p values range = 0.75–0.64, t test.
NBs are typically associated with copy number variations (CNVs), such as gain of chromosome 17q (Ho et al., 2018). A previous study showed that gene expression profiles of RNA-seq data can be utilized for the analysis of chromosomal aberrations in hPSCs (Weissbein et al., 2016). To assess whether similar genomic changes occur in CHNBs, we compared the RNA-seq data from CHNBs, hNCCs (the cellular origins of the CHNBs), and PDX NBs. As a positive control we used the NB cell line SH-SY5Y carrying a CNV on 17q (17q21.33–17q25.3 gain; Yusuf et al., 2013). Figure S5B shows that gain of 17q was found in a PDX tumor and in one out of 8 CHNBs samples, but not in hNCCs. We conclude that a subset of CHNBs harbor chromosomal alterations that occur in human tumors.
Tumors Form in Immunocompetent Recipient Mice
In contrast to conventional xenograft models, hNCCs expressing MYCN and mutated ALK developed NBs in immunocompetent and immunocompromised hosts with similar tumor penetrance (weeks 30 to 70, detection by necropsy; Figure 4A). However, the onset of detectable tumor growth was observed at a slightly earlier time in immunocompromised hosts (weeks 10 to 30, early detection by IVIS; Figure S4B). The growth of human tumors in immunocompetent mice suggests that the hosts acquired tolerance to the human cells (Xing and Hogquist, 2012). To test tolerance in chimeric mice, we challenged 3 groups of mice with hNCC-xenograft formation; hNCCs expressing the two oncogenes, when injected into immunocompromised mice, formed hNCC-derived xenograft tumors (Figures 1G, 4B, in red, and 4C), whereas when injected into non-chimeric, immunocompetent mice, they never developed tumors due to xenogeneic rejection (Figure 4B, in gray). However, when the cells were injected into immunocompetent mice that had been derived from embryos injected at E8.5 with hNCCs, approximately 25% developed hNCC xenograft tumors (Figures 4B, in black, and 4C), which is similar to the fraction of mice that are chimeric after in utero injection (Cohen et al., 2016). We conclude that interspecies chimeras acquire tolerance to hNCCs and their malignant derivatives, thereby enabling the growth of human tumors in immunocompetent hosts.
To test for different types of immune tolerance in the context of tumor formation in mouse-human chimeras, we investigated the immune microenvironment of the human NBs in immunocompetent chimeric mice. The lack of xenograft rejection could be mediated by either central or peripheral tolerance. The induction of central tolerance would manifest by depletion of T cell clones reactive to human antigens from the repertoire during T cell development and, therefore, should not be able to infiltrate tumors. However, if tumor-induced T cell dysfunction would cause the lack of T cell-mediated rejection, T cells could be present in the tumor microenvironment but held in check by immune inhibitory pathways, as also frequently observed in human tumors. Immuno-histological analysis of CHNB tumors growing in adult chimeric mice showed significant infiltration by host (murine) CD3+ and cytotoxic CD8a+ T cells and F4/80+ macrophages, consistent with a significant host immune response (Figures 5A–5C). Furthermore, following the assessment of the immune score of the tumors (Galon et al., 2006), CHNB tumors displayed a high score, similar to patient-derived inflamed tumors (Figures 5E–5G). This is in sharp contrast to PDX NBs, which are formed in immunocompromised hosts, or to GEMM NBs (Th-MYCN, ALKF1174L; Berry et al., 2012) that lack T cell activation or immune cell infiltration, as indicated by the lack of CD3 and CD8 staining (Figures 5D–5G). Thus, tumors growing in chimeric mice appear to induce local immune evasion by inducing the expression of immune-suppressive checkpoint molecules within the tumor microenvironment.
Figure 5. Immune-Microenvironment of NB Tumors Formed in Mouse-Human Chimeras.

(A–C) Host T cell, CTL, and macrophage infiltration marked by CD3 (A), CD8 (B), and F4/80 (C) in NB tumors of chimeric mice.
(D) No CD3+ T cell infiltrate was observed in a NB of GEMM (Th-MYCN+ALKF1174L).
(E) Representative fields of IHC staining of CHNB and GEMM NB for CD8a, followed by software analysis for quantifications of IHC and immune scores (yellow, tissue area; dark yellow, nuclei staining; red, DAB staining for CD8a; see STAR Methods).
(F and G) Quantifications of IHC experiments for mouse-specific CD3 (F) and CD8a (G) for comparing immune cell infiltration in tumors of GEMM NB (n = 2) and CHNB (n = 6). Data presented as means; error bars represent SD; *** p value < 0.0001, t test.
Cytotoxic T Cells Recognize Oncogene-Related Antigens on Human NBs
To investigate the cross-talk between immune and tumor cells in the milieu of mouse-human chimeras, we isolated T cells from chimeric mice to test any direct interactions between the host immune system and the human tumor cells. Splenocytes of chimeric mice that had developed CHNB tumors were isolated and co-cultured with donor hNCCs to test T cell activation as a measure of specific recognition of the TCR and inducing epitope(s) (Figure 6A). Co-cultures with hNCCs that did not express the NB-related oncogenes did not stimulate T cell proliferation, similar to other human cell controls, such as human dermal fibroblast cells (lacking NB relevance but with high expression of the human leukocyte antigen class I [HLA-I] and genes related to antigen presentation; see Figure S6) and MYCN-amplified, ALKF1174L-mutated Kelly NB cells (expressing NB relevant epitopes but lacking HLA-I and antigen presentation; Pistoia et al., 2013; Figure S6). In contrast, when the T cells were co-cultured with hNCCs overexpressing MYCN and ALKF1174L, T cell proliferation was highly induced (Figures 6B and 6C), indicating specific recognition of antigens originating from the CHNB tumors (Passoni et al., 2002; Pistoia et al., 2012). To rule out any cross-presentation on antigen presenting cells and to further show that this specific recognition was driven by a direct interaction between T cells and antigens presented on human tumor cells, we repeated these experiments by pre-sorting CD8+ T cells from splenocytes of chimeric mice (Figure 6A). When CD8+ T cells were co-cultured with the different groups of human cells described above, we observed that the CD8+ T cells reacted only to specific human epitopes on oncogene-expressing hNCCs (Figure 6D). These results suggest that expression and presentation of tumor-specific epitopes on NB cells promote immune recognition by the host T cells. These tumor-reactive T cells are then prevented from rejecting tumor cells by local immune suppressive factors within the tumor microenvironment.
Figure 6. T Cells from Chimeric Mice Specifically Recognize Dox-Induced hNCCs.

(A) Schematic representation of the experiment. CFSE-labeled splenocytes or CD8+ T cells from CHNBs bearing chimeric mice were co-cultured with hNCCs expressing or not expressing the ALKF1174L and MYCN oncogenes with human dermal fibroblasts (hDF; for HLA-Ihigh control, see Figure S6) and with Kelly (NB cell line HLA-Ilow, see Figure S6). T cell proliferation was monitored after 7 days.
(B) CD8+ T cells from chimeric mice proliferated when exposed to oncogene-activated hNCCs but much less when stimulated by unrelated human NB cell lines or hNCCs not expressing the oncogenes. Anti-CD3 and -CD28 antibodies were used to stimulate T cell as a positive control (activated) and non-treated splenocytes as negative controls (NT).
(C) Summary of multiple experiments presenting the normalized proliferation rates of the gated CFSElow/CD4+ (bottom) and CFSElow/CD8+ (top) T cells subpopulations after co-cultures in vitro with the different groups.
(D) CD8+ T cells were pre-sorted from splenocytes of chimeric mice with CHNBs and co-cultured with similar groups of human cells as described above. CD8+ T cell proliferation was induced when co-cultured with hNCCs overexpressing MYCN and ALKF1174L, but not with the control cells. Data presented as means, error bars represent SD, * p value < 0.05; ** p value < 0.01, t test.
Modeling Immune-Evasion of Human NBs
Because CD8+ cytotoxic T lymphocytes (CTLs) are the main effectors of tumor cell destruction in various cancer types, we investigated potential mechanisms of immune escape in CHNB tumors. Figure 7 shows that the CHNBs recruited regulatory T cells (Tregs) expressing FoxP3 and Il2ra, consistent with the induction of an anti-tumor immune response (Figures 7A, 7B, S7A, and S7D). In addition, the tumor-infiltrating T cells showed signs of T cell dysfunction. as indicated by murine-specific Tim-3 expression in host T cells (Figures 7C, S7B, S7D, and S7F), whereas the tumor cells express PD-L1 (the ligand of PD-1) and CD47 (Figures 7D, 7E, S7C, and S7E). This suggests that human tumors in the chimeric model escape immune distruction using the same immune checkpoints as described in patient samples (Woo et al., 2001; Dong et al., 2002; Jaiswal et al., 2009; Fourcade et al., 2010). Gene expression profiling of the host murine microenvironment confirmed a gene signature typical of activated T cells and gene set enrichment nalysis (GSEA) indicated significant upregulation in genes involved in tumor inflammation (Figures 7F and 7G). Finally, as an outcome of the immune evasion, the majority of the CTLs in the CHNBs were dysfunctional, as indicated by the lack of IFNγ signals in CD8+ T cells (Figure S7G). Thus, our results suggest that human tumors growing in chimeric mice evade a strong anti-tumor immune response by escape mechanisms similar to those seen in primary human tumors.
Figure 7. Immune-Evasion and Immune-Suppressed T Cells in NB Tumors from Mouse-Human Chimeras.

(A and B) Human NB tumor of chimeric mice shows immune tolerance marked by infiltration of Tregs (A, stained for FoxP3) and activated T cells (B, Il2ra).
(C–E) T cell exhaustion was observed in NB chimeric mice, indicated by the expression of immune checkpoint signals, such as the mouse-specific Tim-3 on the hosts T cells (C) and human-specific PD-L1 (D, the ligand for PD1 expresses on T cells; IHC scale bars indicate 100 mm) and human-specific CD47 on human tumor cell (E, activating “don’t eat me” signals on macrophages; IF scale bar indicates 50 μm).
(F) RNA-seq analysis for the murine genes expressed in CHNB tumor (represent the tumor microenvironment) show a gene expression signature with enrichment for genes associated with T cell immune activation and exhaustion.
(G) Similarly, the enrichment plot of gene set enrichment analysis (GSEA) of the tumor microenvironment-associated genes (mouse reads) from CHNBs show an expression signature significantly associated with cancer-linked inflammation response.
(H) Schematic representation of the experiment. T cells from chimeric mice bearing CHNBs and CD3+ T cells from WT mice were transferred to recipient immunocompromised mice, which were challenged with subcutaneous xenograft injection of hNCCs overexpressing MYCN and ALK (see Figure 1G). Xenografts’ growth were monitored for their size following the T cells transfers (TCT) as measurement for T cell acclivity.
(I) hNCCs xenografts in immune-compromised mice with no TCT kept growing rapidly (red). Xenograft tumors in mice injected with CD3+ cells from CHNB-bearing mice grew significantly slower than in mice transferred with WT T cells (n = 10 per group; solid lines represent means; error bars represent SD, ** p value < 0.001; *** p value < 0.0001, repeated-measures ANOVA).
To functionally assess the suppressed immune state in chimeric mice harboring CHNBs, we isolated the T cells and tested their ability to inhibit tumor xenograft formation. hNCCs expressing the two oncogenes were injected subcutaneously into the flanks of immune compromised mice to form hNCC xenografts as previously described (Figure 1G; n = 10 for each experimental group). For immune compatibility, we used immune-compromised mice from a previously generated strain, isogenic to the chimeric mice (KitW-sh; Rag2−/−; Il2rγ−/−; Bakthavatchalu et al., 2018). Twenty days post hNCC injection, 1×106 CD3+ T cells isolated from WT or chimeric CHNB-bearing mice were injected into the immune-compromised mice, and the hNCC xenograft volume was measured as a gauge for tumor growth or regression (Figure 7H). hNCC xenografts in immune-compromised mice that had never received CD3+ T cells continued to grow rapidly (Figure 7I, in red), whereas xenograft tumors in mice that were injected with WT T cells showed tumor regression that declined over time, likely due to rejection of the xenograft by the T cells (Figure 7I, in black). However, mice that were given CD3+ cells from CHNB tumor-bearing chimeric mice revealed significantly slower regression rates (Figure 7I, in blue). This suggests that CD3+ T cells from chimeric animals can be suppressed via mechanisms of T cell exhaustion and is consistent with the CHNB tumor microenvironment in the chimeric mice posing an immune suppressive context. The local immune suppression functionally affects T cells and facilitates the immune-evasion of human NBs. Our data suggest that the human-mouse CHNB-bearing animals experience both partial central tolerance and immune evasion via local immune suppression within the tumor microenvironment.
DISCUSSION
We demonstrate that the human-mouse chimeric platform presented here enables the modeling of human NB tumors in an animal system. The MYCN+ALKF1174L-induced CHNBs closely resembled primary human NBs expressing the same oncogenes, as judged by sites of tumor formation, histology, expression of specific markers, and genetic and transcriptomic profiles. We show that hNCCs expressing oncogenes known to initiate human disease can result in multifactorial disease phenotypes in an animal model when placed in the relevant developmental niche in the mouse embryo. By contrast, similar hNCCs transplanted into an ectopic niche in immunocompromised mice formed homogeneous outgrowths that lacked the expression of NB-specific markers and genes typically expressed in human tumor cells. Others have shown that mouse NCCs (JoMa1 cell line) overexpressing the same oncogenes formed mouse xenograft tumors that resembled primary human NBs (Schulte et al., 2013). Our results suggest that the integration of tumor-initiating hPSC-derived hNCCs into tissues of the early embryo enables faithful development of human tumor models in mice and recapitulation of human disease.
Similar to the Th-MYCN+ALKF1174L GEMM NBs (Berry et al., 2012), CHNBs were formed in the host’s retroperitoneal space and did not develop metastases. However, although more aggressive NB mouse models exist (Weiss et al., 1997; Berry et al., 2012; Althoff et al., 2015), the CHNBs were less invasive, grew slower, and did not lead to a significantly decreased survival of chimeric mice and, hence, might reflect a low risk, less aggressive disease. RNA-seq of CHNBs revealed similarities to the expression profiles with the other NBs models and, more importantly, to patient NBs, though the CHNBs presented with high expression profile heterogeneity. This heterogeneity may be related to greater and more variable dynamics of tumor formation and progression in interspecies chimeras, as opposed to those seen with GEMM, PDX, and cell lines that usually represent clonal populations. The heterogeneity and lower levels of aggressiveness of CHNB may be also related to early immune response.
Our CHNBs differ from primary human NBs expressing MYCN and ALKF1174L in exhibiting a vigorous host anti-tumor response as indicated by massive tumor infiltrationwithCD8+CTLs. This immune response was blunted by recruitment of Tregs and the activation of check point inhibitors, such as mouse Tim-3, human PD-L1, and CD47, allowing progressive tumor growth. MYCN-amplified NBs are frequently described as “cold” non-T cell-inflamed tumors and do not usually evoke an immune response. It was suggested that, similar to the role of MYC in suppressing immune surveillance in hematological cancers (Casey et al., 2016), MYCN is also associated with the repression of cellular immunity in NB (Zhang et al., 2017; Wei et al., 2018). Since the CHNB tumors present high levels of CTLs infiltrate with moderate-to-high levels of immune evasion strategies, it is possible that the intermediate expression level of MYCN in our current model captures the dynamics of tumor development along with the relevant immune reaction and the tumor evasion response.
Interspecies chimeras allow growth of the human oncogenic donor cells. We observed a similar incidence of tumor formation regardless of the immune-state of the host, whereas the delay of tumor outgrowth in immunocompetent chimeras may be due to the active immune response. Immune tolerance may develop because of central thymic tolerance or because of peripheral tolerance (Mathis and Benoist, 2004; Xing and Hogquist, 2012). In this study, we present evidence for the presence of peripheral immune tolerance in the CHNB microenvironment. We previously showed that WT hNCCs can contribute to the pigment system of adult immunocompetent chimeras (Cohen et al., 2016), but we do not know whether normal hNCCs can induce tolerance to xenografts in these mice. Interspecies chimeric mice may develop thymic tolerance to human donor cells during embryonic development of the chimeric host when exposed to human NC cells. Indeed, in a similar chimeric system, induction of central immune tolerance to human donor cells was observed in immunocompetent mice following injection of human colorectal cancer cells into mouse blastocysts (Chen et al., 2015). Further investigation is needed to determine whether thymic tolerance is achieved by the direct contribution of human cells to the mouse thymic development or by cross-antigen presentation of human antigens by dendritic cells in the developing thymus of chimeras. Although the thymus epithelium has been suggested to originate from the NC, there is no clear evidence for the involvement of NC-derived cells in the T cell education processes within the thymus (Foster et al., 2008). Hence, the nature of interspecies tolerance in post-implantation chimeric systems remains to be investigated.
Our results show a clear anti-tumor immune response of the host against human NB tumors. This normally involves infiltration of immune cells and activation of IFN-γ signaling and of genes associated with an effective inflammation and immune response (Spranger, 2016). CD8+ T lymphocytes play a key role in immunity to cancer due to their capacity to kill tumor cells upon recognition by the T cell receptor of specific antigenic peptides presented on the surface of target cells. Our data show that the host CD8+ T cells infiltrate the human NB tumors and specifically react against oncogene-related antigens when co-cultured with relevant cells. This may suggest that specific tumor-associated antigens produced by the human donor hNCCs elicit recognition by sub-clones of T cells. A typical productive cytotoxic T cell infiltrate is accompanied by the upregulation of PD-L1 on the cancer cells (Spranger et al., 2013), similar to our observations in the chimeric model. The immune reaction against CHNBs resembles the characteristics typical of anti-tumor immunity seen in patients, including a T cell-inflamed microenvironment signature, suggesting the formation of an equilibrium between the tumor and the anti-tumor immune response. The tumor ultimately escapes this equilibrium by the induction of immune evasion mechanisms, such as upregulation of PD-L1 and recruitment of Tregs, leading to T cell exhaustion in the tumor microenvironment, marked by Pd-1, Havcr2 (Lag3), Ctla4, and Tim-3 (Spranger et al., 2013; Wherry and Kurachi, 2015). Several studies have provided evidence that immune evasion via T cell dysfunction is a consequence of an initially productive anti-tumor immune response frequently driven by IFN-γ (Spranger et al., 2013; Taube et al., 2012).
A characteristic feature of NB is its clinical diversity, ranging from spontaneous regression to highly metastatic and poor prognosis disease. The mechanisms underlying such disparate behavior are unclear, although recent evidence suggests that host immunity plays an important role in the evolution of NBs during development (Brodeur and Bagatell, 2014). Current GEMM, PDX, and cell-line models of NB offer limited insights into the tumor immune microenvironment. Yet, the NB-bearing mouse-human chimera platform presented here provides a highly defined model for studying NB origins, the initiating early events in the disease, the evolution and progression of NB, and its tumor immune-microenvironment and the immune–tumor interactions which govern immune evasion. We note, however, that the reaction of immune cells against interspecies cells is complex and that a number of issues germane to the mechanism of the immune response in the interspecies chimeras, such as the exact nature of the relevant antigens and the role of central versus peripheral immune tolerance, remain to be clarified. Nevertheless, the ability to grow clinically derived human NB tumor cells in immune-competent hosts serves as a starting point for facilitating the study of interactions between tumor cells and the immune system, specifically, the mechanisms governing immune tolerance and evasion. Ultimately, the chimeric model system may prove to be an optimal platform for enabling the evaluation of different combinations of anti-oncogenic and immune-based therapies.
Interspecies chimeras are thought to be of great value for modeling human diseases in animals and for the eventual production of human organs in animals for transplantation therapy (Wu et al., 2016; Suchy and Nakauchi, 2017). Although the injection of rat PSCs into mouse blastocysts created interspecies chimeras (Kobayashi et al., 2010), generation of chimeras with PSCs from more distant species, such as injection of human donor PSCs into mouse or pig blastocysts, has been highly inefficient with functional integration of human donor cells into later embryos not having been convincingly demonstrated (Wu et al., 2017). The hourglass model of development postulates that the basic body plans of vertebrates are established at the most conserved embryonic or “phylotypic” period, which occurs at gastrulation (Irie and Kuratani, 2014). In contrast to gastrulation, vertebrate species differ fundamentally at the pre- and post-gastrulation stages, consistent with the possibility that integration of multipotent cells into gastrulation embryos may be the more efficient approach to generate interspecies chimeras with functional donor cell integration than injection of pluripotent cells into the blastocyst. We have previously shown that matching the developmental stage of both donor cell and host embryo (isochronic injection) is an important parameter for successful engraftment of pluripotent or multipotent cells (Cohen et al., 2018). Our current study presents the first example for the successful application of this approach in modeling the development and progression of a human disease in chimeric animals using hPSC-derived donor cells.
STAR*METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Rudolf Jaenisch (jaenisch@wi.mit.edu). Plasmids generated in this study have been deposited to Addgene. All other reagents are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal
C57BL/6, Nod scid gamma (NSG), and KitW-sh mice were obtained from the Jackson Laboratory. CD1-Elite mice, NSG and NU/NU (Crl:NU-Foxn1nu) were obtained from Charles River Laboratories. Mice were maintained in the WIBR animal facility. GEMM NB mice (Berry et al., 2012) and PDX mice were maintained in the Dana Farber Cancer Institute (DFCI) animal facility. GEMM Th-MYCN; ALKF1174L mice were generated by breeding of Th-MYCN and Th-ALKF1174L mice maintained in 100% 129X1/SvJ and a mixed C57BL/6J/129X1/SvJ (50%/50%) background respectively. Animal procedures were performed following the National Institute of Health (NIH) guidelines. All animal experiments were approved by the Committee on Animal Care (CAC) and the Department of Comparative Medicine (DCM) at the Massachusetts Institute of Technology (MIT) and/or the Institutional Animal Care and Use Committee (IACUC) of DFCI.
Embryonic hNCCs microinjection
Microinjections of hNCCs were performed as previously described (Cohen et al., 2016). Briefly, pregnant females at E8.5 were anesthetized via an intraperitoneal injection of avertin. Laparotomy was performed and the uterus was exposed and held with forceps during the injection of each embryo. NCCs were drawn into a glass micropipette, and the tip of the glass micropipette was inserted into the distal third of the decidual swelling. Approximately 2–5 × 103 cells (suspended in 0.2–0.8μl of cell culture media) were injected per embryo.
Neural crest cell contribution
Pregnant female mice were sacrificed between E9.5 to E15.5 of gestation following institutional guidelines. Embryos were harvested by dissection from the uterus and the placenta. NCC contribution to the embryos was determined by the presence of a fluorescent protein signal. Embryos containing a fluorescent signal were imaged using a stereomicroscope (Nikon SMZ18) and fixed for IF. Whole mount IF for E9.5 mouse embryos was performed as previously described (Ahnfelt-Rønne et al., 2007), and larger embryos were frozen-sectioned following IF.
Oncogene activation
ALKF1174L and MYCN genes were cloned into a lentiviral backbone under the doxyclycline (Dox)-inducible Tet-ON (TetO) promoter. hESCs or hiPSCs carrying the tetracycline transactivator (M2rtTA) were infected with virus encoding ALKF1174L and MYCN transgenes followed by Dox administration to activate the oncogenes (2 μg/mL in culture media; 2 mg/mL in mouse drinking water; MilliporeSigma).
Xenograft formation assay
1.5×106 hNCCs resuspended in 200 μl Matrigel/DMEM/F12 (50%, v/v) were injected into the flanks of different mice as indicated. To activate the oncogenes, Dox was administrated in drinking water (2 mg/mL). Mice were monitored daily for any signs of adverse health and/or tumor growth. For monitoring xenograft growth and/or regression we routinely measured the xenograft size every 4 days (maximal high x perpendicular width) using a caliper.
Monitoring xenograft and tumor growth by optical imaging
For luciferase imaging mice were anesthetized with Isofluorane (1.5%–2.0%) and injected intaperitoneally with D-luciferin (30 mg/mL, Perkin Elmer), 5 μl/gr of body weight, using a 26-gauge needle. Mice were imaged using Caliper IVIS Spectrum.
Generation of PDX NBs
MYCNamp; ALKWT PDXs (SJNBL046148-x1, SJNBL013763_x1) were obtained from St. Jude Children’s Research Hospital and propagated once at DFCI in NSG 6–9-week-old female mice. MYCNamp; ALKF1174L PDXs (COG-N-415X-p4, COG-N-453X-p5) were obtained from Children’s Oncology Group and propagated once at DFCI in NU/NU 6–9-week-old female mice. Presence of ALK mutation in the PDXs was confirmed by sequencing the ALK cDNA.
T cells isolation and in vivo transfer
For in vivo T cells transfers into immunosuppressed mice, CD3+ cells were isolated from spleens of wt or chimeric mice carrying CHNB tumors following the manufacturer protocol (Pan T cells isolation kit II, mouse, 130–095-130, MACS, Miltenyi Biotech). Cells were resuspended in Saline (15×106 cells/mL) and injected via retro-orbital administration into the anesthetized immunocompromised mice (100ul per animal).
Cells
PSCs (WIBR#3 (X,X) and hiPSC line AA#1 (X,X)) were cultured in DMEM/F12 medium supplemented with 15% FBS and 5% KOSR (Thermo Fisher Scientific) and differentiated to hNCCs as previously described (Cohen et al., 2016). Briefly, cells were cultured in KOSR medium (Thermo Fisher Scientific) supplemented with 10μM SB431542 and 500nM LDN193189 and 3 μM CHIR99021 (all from Stemgent) for the first 72 h after plating hESCs/hiPSCs at a density of 2–3×105 cells per cm2 on Fibronectin + Laminin precoated dishes. Subsequently, a gradual switch from KOSR to N2B27 medium (Thermo Fisher Scientific), supplemented with 3 μM CHIR99021 was used until day 11. At day 12 cell cultures were enriched with hNCCs and cultured in N2B27 supplemented with bFGF and EGF (both 20ng/mL, Peprotech) and 3 μM CHIR99021. Kelly (X,X), SH-SY5Y (X,X), IMR-32 (X,Y), MCF7 (X,X), and Colo-829 (X,Y) cell lines were received as gifts form Susan Lindquist’s and Bob Weinberg’s labs at WIBR.
METHOD DETAILS
Soft agar colony formation assay
hNC single-cells were resuspended and plated in 0.3% agar (m/v) in N2B27 hNCCs media, on top of a layer of 0.5% agar (m/v) in 1:1 mix of autoclaved ddH2O and N2B27. After the cells/agar mixture solidified, cells were topped with hNCCs media and were cultured for 15 days with, or without Dox, to allow cells to form colonies. 0.5ml of medium were added every 3 days. To visualize the colonies, cells were stained over night by nitroblue tetrazolium chloride solution (NBT, 1mg/mL; VWR), and colonies of the different culture conditions were counted.
Western Blot
Cells were lysed by RIPA buffer with proteinase inhibitor (Thermo Fisher).Tumors were homogenized in NP40 lysis buffer (Thermo Fisher) containing 1 × complete protease inhibitor and a cocktail of phosphatase inhibitors (Roche). Protein concentration was measured by the DC Protein Assay (Bio-Rad) or Qubit (Thermo Fisher). 100ug total protein was denatured in LDS sample buffer, separated on pre-cast 4%–12% Bis-Tris gels (Thermo Fisher) and transferred to nitrocellulose membranes. Membranes were incubated in a blocking buffer (5% non-fat dry milk in TBS with 0.2% Tween-20) for 1 h, and then incubated in the primary antibody in blocking buffer overnight at 4 °C. Chemiluminescent detection was performed with appropriate HRP-conjugated secondary antibodies and visualized using enhanced chemiluminescence reagents (Thermo Fisher 34580). Following antibodies: N-MYC (51705), ALK (3333), pALK Y1604 (3341), AKT (4691), pAKT-T308 (9275), pAKT-S473 (9271), ERK (4695), pERK1/2 (9106), GAPDH (2118), and anti-mouse IgG-HRP (7076) all from CST, and pALK-Y1507 (ab73996, Abcam), and anti-rabbit IgG-HRP (sc-2357, SCB; 40139, MilliporeSigma).
H&E (Hematoxylin and Eosin) IF (immunofluorescence) and IHC (immunohistochemistry) staining
Cells, tumors or tissues were collected and fixed in 4% paraformaldehyde in PBS (MilliporeSigma). Patient-derived NB tissue microarray were obtained from US Biomax (26 patients’ samples, in duplicates). For IHC, samples were embedded in paraffin and sectioned into 4μm/slide. Following deparaffinizing in xylene and alcohol gradient, antigen retrieval was performed in sodium citrate or EDTA buffers (Vectashield). For immunostainings, samples were blocked with 2% BSA and incubated with primary antibodies including Rabbit anti-ALK (1:200, Cell Signaling technology (CST), 3633), Rabbit anti-MYCN (1:50 84406; 1:100 51705, CST), Rabbit anti-CD3 (1:300, RM9107S0, Thermo Fisher), Rabbit anti-CD8a (1:300, 98941, CST), Rabbit anti-FoxP3 (1:75, MAB8214, R&D), Mouse anti-Il2ra (CD25, 1:100, NB600–564, Novus), Rabbit anti mouse Tim-3 (1:200, 83882, CST), Rabbit anti-human-PD-L1 (1:300, 13684, CST), Sheep anti-human-CD47 (1:100, AF4670SP, R&D), Mouse anti- IFNγ (1:200, DB-1, BioLegend), Rat anti-mouse F4/80 (1:100, MF48000, Thermo Fisher), Rabbit anti-γH2AX (1:300, ab2893, Abcam), Rabbit anti-human-Ki67 (1:20, PA516785, Thermo Fisher), Rabbit anti-Chromogranin A (1:500, NB120–15160, Novus), Rabbit anti-Synaptophysin (1:200, 36406, CST), Mouse anti-human-Nestin (1:300, ab176571, Abcam), Rabbit anti-TH (1:500, P40101–150, Pel-Freez), Rabbit anti-pERK1/2 (1:400, 4370, SCT), Rabbit anti-p-p70 S6 (1:300, 9205, CST), Rabbit anti-Peripherin (1:500, ab4666, Abcam), Rabbit anti-human neurofilament (1:100, 160kD, ab92539, Abcam), anti-eGFP (1:1000, GFP-1020 Aves Labs; 1:300 AF647-Alpaca nanobody; 1:5000, ab290, Abcam), mouse anti-HNK-1 (1:50, C6680, MilliporeSigma) and rabbit anti-p75 (1:175, G3231, Promega) overnight at 4°C followed by appropriate secondary antibody incubation for 1–2 h (for IF: A31571, A21235, A21202, A11001, A21206, A11008, A31573, A21244, A11015 or A21209, Thermo Fisher; 703–545-155, 115–585-044, Jax ILR; for IHC: Rabbit-on-Rodent and Mouse-on-Mouse HRP-Polymer, RMR622; MM620, Biocare, or Biotinylated anti-rabbit IgG, BA-1000 Vector). IF samples were imaged using Zeiss LSM 710 confocal microscope. For H&E staining, deparaffinized sections were stained in hematoxylin, incubated with 1% acid alcohol (1% HCl in 70% EtOH) and ammonia water, and stained with 1% Eosin.
Splenocyte and T cells isolation for in vitro co-cultures
Total splenocytes or CD8+ isolated T cells (CD8a+ isolation kit, mouse, 130–104-075, Miltenyi Biotech) were collected from spleens of chimeric mice harboring CHNBs. At a density of 5×106 cells /mL splenocytes/ CD8+ T cells were labeled with CFSE (2μM in PBS, Thermo Fisher) and plated in 96-well U-bottom plates (2×105 cells/well splenocytes and 5×104 for CD8+ cells). As positive controls, T cells were activated in vitro by pre-coating the wells with CD3 (1:100, 100302 BioLegend) and CD28 (1:100, 102102 BioLegend) and incubated at 37°C for 2 h, or by culture with or without ionomycin (1ug/mL) and PMA (100ng/mL, both from MilliporeSigma). For cell co-cultures, 2×104 mitotically inactivated (mitomycin-C, 10μg/mL for 2 h, MilliporeSigma) human dermal fibroblasts (hDF), Kelly cells or hNCCs were co-plated with splenocytes or CD8+ T cells. Cultures were maintained at 37°C, 5% CO2 in RPMI/10% (v/v) FBS, supplemented with β-ME (50μM; MilliporeSigma) and HEPES (10mM; Thermo Fisher Scientific). After 7 days of incubation, cells were collected and analyzed by flow cytometer.
LacZ staining
Cell were fixed shortly with 2% formaldehyde and 0.5% glutaraldehyde in PBS, and incubated in staining-solution containing 1 mg/mL X-Gal, 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide and 2mM MgCl2, in PBS (All from MilliporeSigma) at 37°C, over-night.
Southern Blot
10μg of DNA isolated from hPSCs was digested overnight with the appropriate enzymes and run on a 0.8% agarose gel. DNA was transferred to a nylon membrane (Amersham) and hybridized with 32P-labeled random primer (Stratagene) probes.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA isolation, RNA-seq and RT-qPCR
Cells and tumor samples were homogenized, and RNA was extracted (RNeasy,QIAGEN, or mirVana miRNA isolation kit, Thermo Fisher, See Table S2). Poly(A) mRNA capture and construction of stranded mRNA-Seq libraries were made using KAPA mRNA HyperPrep Kit according to instructions (Roche). Sequencing was performed at the Whitehead Genome Technology Core (HiSeq 2500, Illumina). For RNA-seq analysis a genome index was created with STAR, using the human (hg38) and mouse (mm10) genomes and Ensembl 91 gene annotations. Reads were mapped to this human/mouse genome STAR, using default parameters, and gene counts were obtained with featureCounts (with option ‘-s 2’), using the same Ensembl 91 gene annotations. Counts from technical replicates were summed. Raw counts were converted to FPKM with DESeq2, using gene lengths provided by featureCounts. We obtained Ensembl human and mouse genes with 1-to-1 orthology (n = 16781), using these human genes for human samples and orthologous mouse genes for mouse samples. Human neuroblastoma samples (with expression linked to 15103 gene symbols; n = 669) were merged from these sources: TARGET RNA-seq BCCA, summarized by FPKM (n = 10; from https://ocg.cancer.gov/programs/target/data-matrix; TARGET RNA-seq NCI-Khan, summarized by FPKM (n = 161), from https://ocg.cancer.gov/programs/target/data-matrix; GSE49711 RNA-seq summarized by FPKM (n= 498) (Zhang et al., 2015). We merged our samples (n= 29) with public databases of NB samples by matching gene symbols, producing a profile of 13,101 genes in 705 samples, which were then batch-corrected with ComBat. To make CHNB samples more comparable to human tumor samples, they were subsequently represented by a combination of human and mouse profiles in equal proportions. Gene levels were log2-transformed, and the 500 most variable genes were used for PCA.
For RT-qPCR, total RNA was isolated (RNeasy, QIAGEN) and reversed transcribed (Superscript III First Strand Synthesis kit, Invitrogen). qRT-PCR analysis was performed in triplicate (See list of primers in Table S3). Gene expression was normalized to GAPDH expression. Error bars represent the standard deviation (SD) of triplicate reactions.
Quantitative Image Analysis
IHC images were processed into separate channels representing nuclei staining (hematoxylin) and IHC staining (DAB) using Fiji. Images were then analyzed using the open-source CellProfiler software (https://cellprofiler.org; Kamentsky et al., 2011) for cell segmentation and staining characterization. Briefly, cell nuclei and IHC staining were segmented based on Gaussian blurred images in both hematoxylin and DAB channels respectively using size and intensity thresholds. Cells were then filtered based on the channels overlapping rate, and those which exhibited over 60% overlapping rates, representing cells expressing the tested markers, were scored and quantified. The software pipeline detection was tested to be highly correlated with visual detection.
Flow cytometry
Single-cell suspensions were prepared and splenocytes were incubated with APC-conjugated anti-CD4 (0.25μg/106 cells in 100μl volume, 100412 BioLegend) and Perp-Cy5.5-conjugated anti-CD8 (1μg/106 cells in 100μl volume, 100733 BioLegend) for 30 min on ice. For HLA class I analysis, cells were counted and 2×106 cells were incubated with mouse anti-HLA class I antibody (1:200, 311402 BioLegend) for 30 min followed by secondary antibody (anti-mouse IgG AF647, Thermo Fisher) incubation for 30min. Cells were analyzed by flow cytometry on LSRII or LSRFortessa device with BDFacs DIVA software. The data were analyzed by Flow Jo software (TreeStar).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using the Prism 8 software (GraphPad). Statistical parameters including the exact value of n and measures (mean ± SD) and statistical significance are reported in the Figures and/or the Figure Legends. Data are judged to be statistically significant when p-value < 0.05 by linear regression, Student’s T-Test, x2 test, two-way or repeated-measures ANOVA, where appropriate.
DATA AND CODE AVAILABILITY
The RNA-seq datasets generated during this study are available at GEO: GSE144366.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti N-MYC | Cell Signaling Technology | Cat#51705; RRID:AB_2799400 |
| Rabbit anti N-MYC | Cell Signaling Technology | Cat#84406; RRID:AB_2800038 |
| Rabbit anti ALK | Cell Signaling Technology | Cat#3333; RRID:AB_836862 |
| Rabbit anti ALK | Cell Signaling Technology | Cat#3633; RRID:AB_11127207 |
| Rabbit anti Phospho-ALK (Y1507) | Abcam | Cat#ab73996; RRID:AB_2226754 |
| Rabbit anti Phospho-ALK (Y1604) | Cell Signaling Technology | Cat#3341; RRID:AB_331047 |
| Rabbit anti AKT | Cell Signaling Technology | Cat#4691; RRID:AB_915783 |
| Rabbit anti Phospho-AKT (T308) | Cell Signaling Technology | Cat#9275; RRID:AB_329828 |
| Rabbit anti Phospho-AKT (S473) | Cell Signaling Technology | Cat#9271; RRID:AB_329825 |
| Rabbit anti ERK1/2 (p44/42 MAPK) | Cell Signaling Technology | Cat#4695; RRID:AB_390779 |
| Mouse anti Phospho-ERK1/2 (p44/42 MAPK; T202/Y204) | Cell Signaling Technology | Cat#4370; RRID:AB_2315112 |
| Mouse anti Phospho-ERK1/2 (p44/42 MAPK; T202/Y204) | Cell Signaling Technology | Cat#9106; RRID:AB_331768 |
| Rabbit anti Phospho-p70 S6 Kinase (T389) | Cell Signaling Technology | Cat#9205; RRID:AB_330944 |
| Rabbit anti GAPDH | Cell Signaling Technology | Cat#2118; RRID:AB_561053 |
| Rabbit anti CD3 | Thermo Fischer/ Lab Vision | Cat#RM-9107-S0; RRID:AB_149923 |
| Armenian hamster anti CD3e | BioLegend | Cat#100302; RRID:AB_312667 |
| Mouse anti rat IFN-γ | BioLegend | Cat#507801; RRID:AB_315492 |
| Armenian hamster anti CD3e | BioLegend | Cat#100302; RRID:AB_312667 |
| Armenian hamster anti CD28 | BioLegend | Cat#102102; RRID:AB_312867 |
| Rat anti-mouse CD4, APC | BioLegend | Cat#100412; RRID:AB_312697 |
| Rat anti-mouse CD8a, PerCP/Cyanine5.5 | BioLegend | Cat#100733; RRID:AB_2075239 |
| Rabbit anti CD8 | Cell Signaling Technology | Cat#98941; RRID:AB_2756376 |
| Rabbit anti FoxP3 | R&D Systems | Cat#MAB8214 |
| Mouse anti IL2 Receptor alpha | Novus | Cat#NB600-564; RRID:AB_10002565 |
| Rabbit anti Tim3 | Cell Signaling Technology | Cat#83882; RRID:AB_2800033 |
| Rabbit anti PD-L1 | Cell Signaling Technology | Cat#13684; RRID:AB_2687655 |
| Sheep anti Human Cd47 | R&D Systems | Cat#AF4670; RRID:AB_1026196 |
| Rat anti Mouse F4 / 80 | Thermo Fischer/ Innovative Research | Cat#MF48000; RRID:AB_1500089 |
| Rabbit anti gamma H2A.X (phospho S139) | Abcam | Cat#ab2893; RRID:AB_303388 |
| Rabbit anti Human Ki67 | Thermo Fischer | Cat#PA5-16785; RRID:AB_11000602 |
| Rabbit anti Chromogranin A | Novus | Cat#NB120-15160; RRID:AB_789299 |
| Rabbit anti Synaptophysin | Cell Signaling Technology | Cat#36406; RRID:AB_2799098 |
| Rabbit anti Nestin | Abcam | Cat#ab176571 |
| Rabbit anti Tyrosine Hydroxylase (TH) | Pel-Freez Biologicals | Cat#P40101; RRID:AB_2313713 |
| Rabbit anti Peripherin | Abcam | Cat#ab4666; RRID:AB_449340 |
| Rabbit anti 160 kD Neurofilament | Abcam | Cat#ab92539; RRID:AB_10562619 |
| Mouse anti HNK-1 (CD57) | Millipore-Sigma | Cat#C6680; RRID:AB_1078474 |
| Rabbit anti p75 | Promega | Cat#G3231; RRID:AB_430853 |
| Mouse anti human HLA-A,B,C | BioLegend | Cat# 311402; RRID:AB_314871 |
| Chicken anti Green Fluorescent Protein (GFP) | Aves Labs/Antibodies Incorporated | Cat#GFP-1020; RRID:AB_10000240 |
| Rabbit anti Green Fluorescent Protein (GFP) | Abcam | Cat#ab290; RRID:AB_303395 |
| Alpaca anti Green Fluorescent Protein (GFP), Alexa Fluor 647 | H. Ploegh lab/WIBR | N/A |
| Anti mouse IgG, HRP | Cell Signaling Technology | Cat#7076; RRID:AB_330924 |
| Anti rabbit IgG, HRP | Santa Cruz Biotechnology | Cat#sc-2357; RRID:AB_628497 |
| Anti rabbit IgG, HRP | Millipore-Sigma | Cat#401393-2ML; RRID:AB_437797 |
| Anti rabbit IgG, Biotinylated | Vector Laboratories | Cat#BA-1000; RRID:AB_2313606 |
| Anti mouse IgG, Alexa Fluor 647 | Thermo Fischer/ Molecular Probes | Cat#A-31571; RRID:AB_162542 |
| Anti mouse IgG, Alexa Fluor 647 | Thermo Fischer/ Molecular Probes | Cat#A-21235; RRID:AB_141693 |
| Anti mouse IgG, Alexa Fluor 488 | Thermo Fischer/ Molecular Probes | Cat#A-21202; RRID:AB_141607 |
| Anti mouse IgG, Alexa Fluor 488 | Thermo Fischer/ Molecular Probes | Cat#A-11001; RRID:AB_2534069 |
| Anti rabbit IgG, Alexa Fluor 647 | Thermo Fischer/ Molecular Probes | Cat# A-31573; RRID:AB_2536183 |
| Anti rabbit IgG, Alexa Fluor 647 | Thermo Fischer/ Molecular Probes | Cat#A-21244; RRID:AB_141663 |
| Anti rabbit IgG, Alexa Fluor 488 | Thermo Fischer/ Molecular Probes | Cat#A-21206; RRID:AB_141708 |
| Anti rabbit IgG, Alexa Fluor 488 | Thermo Fischer/ Molecular Probes | Cat#A-11008; RRID:AB_143165 |
| Anti sheep IgG, Alexa Fluor 488 | Thermo Fischer/ Molecular Probes | Cat#A-11015; RRID:AB_141362 |
| Anti rat IgG, Alexa Fluor 488 | Thermo Fischer/ Molecular Probes | Cat#A-21209; RRID:AB_2535795 |
| Anti chicken IgY, Alexa Fluor 488 | Jackson ImmunoResearch Labs | Cat#703-545-155; RRID:AB_2340375 |
| Anti mouse IgG+IgM, Alexa Fluor 594 | Jackson ImmunoResearch Labs | Cat#115-585-044; RRID:AB_2338875 |
| Biological Samples | ||
| hNCCs Xenografts (MYCN+ALK F1174L) | This paper | N/A |
| Human NBs from Chimeric mice CHNB (MYCN+ALK F1174L) | This paper | N/A |
| Human NB PDX (MYCN amp, ALK WT) | St. Jude Children’s Research Hospital | Cat#SJNBL046148 |
| Human NB PDX (MYCN amp, ALK WT) | St. Jude Children’s Research Hospital | Cat#SJNBL013763 |
| Human NB PDX (MYCN amp, ALK F1174L) | Children’s Oncology Group | Cat#COG-N-415X |
| Human NB PDX (MYCN amp, ALK F1174L) | Children’s Oncology Group | Cat#COG-N-453X |
| Mouse NBs from GEMM (Th:MYCN+ALK F1174L) | Berry et al., 2012 | N/A |
| Human NBs tissue microarray | US Biomax | Cat#NB642 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Doxycycline hyclate ≥98% | Sigma Moilipore | Cat#D9891 |
| CFSE | Life Technologies | Cat#65-0850-84 |
| D-luciferin | Perkin Elmer | Cat#122799 |
| SB431542 | Stemgent | Cat#04-0010-10 |
| CHIR99021 | Stemgent | Cat#04-0004-10 |
| LDN193189 | Stemgent | Cat#04-0074-10 |
| bFGF | Peprotech | Cat#100-18B |
| Recombinant human HGF | Peprotech | Cat#AF-100-15 |
| Laminin | Millipore-Sigma | Cat#L2020 |
| Fibronectin | ThermoFischer | Cat#FC0105MG |
| Critical Commercial Assays | ||
| RNeasy Mini Kit | QIAGEN | Cat#74104 |
| mirVana™ miRNA Isolation Kit | ThermoFischer | Cat#AM1560 |
| KAPA mRNA HyperPrep Kit | Roche | Cat#08098115702 |
| SuperScript III First-Strand Synthesis SuperMix | ThermoFischer | Cat#18080400 |
| Fast SYBR Green Master Mix | ThermoFischer | Cat#4385618 |
| Pan T cells mouse kit isolation kit II | Miltenyi Biotec | Cat#130-095-130 |
| CD8a+ isolation kit, mouse | Miltenyi Biotec | Cat#130-104-075 |
| Rabbit-on-Rodent HRP-Polymer | Biocare Medical | Cat#RMR622 |
| Mouse-on-Mouse HRP-Polymer | Biocare Medical | Cat#MM620 |
| Deposited Data | ||
| Raw and processed data | This paper | GEO: GSE144366 |
| Experimental Models: Cell Lines | ||
| hESCs WIBR#3 AAVS1-Sa-Puro CAGGs::eGFP | Hockemeyer et al., 2009 | RRID: CVCL_9767 |
| hiPSC AA#1 AAVS1-Sa-Puro CAGGs::eGFP | Cohen et al., 2016 | N/A |
| hESCs WIBR#3 AAVS1-Sa-Puro CAGGs::Luc-T2A-Lac | This paper | N/A |
| hESCs WIBR#3 AAVS1-Sa-Puro CAGGs::Luc-T2A-Lac Teto MYCN+ALKF1174L | This paper | N/A |
| hiPSC AA#1 AAVS1-Sa-Puro CAGGs::eGFP Teto MYCN+ALKF1174L | This paper | N/A |
| Kelly | ECACC | Cat#92110411; RRID: CVCL_2092 |
| SH-SY5Y | ATCC | Cat#CRL-2266; RRID: CVCL_0019 |
| IMR-32 | ATCC | Cat#CCL-127; RRID: CVCL_0346 |
| MCF-7 | ATCC | Cat#HTB-22; RRID: CVCL_0031 |
| Colo-829 | ATCC | Cat#CRL-1974; RRID: CVCL_1137 |
| Human dermal fibroblasts | ATCC | Cat#PCS-201-12 |
| Experimental Models: Organisms/Strains | ||
| Mouse: NSG (NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ) | Jackson Laboratory | Cat#5557; RRID:IMSR_JAX:005557 |
| Mouse: C57BL/6 | Jackson Laboratory | Cat#664; RRID:IMSR_JAX:000664 |
| Mouse: Kit W-sh (B6.Cg-KitW-sh/HNihrJaeBsmGlliJ) | Jackson Laboratory | Cat#12861; RRID:IMSR_JAX:012861 |
| Mouse: NU/NU (Crl:NU-Foxn1nu) | Charles River | Cat#88; RRID:IMSR_CRL:088 |
| Mouse: NOD SCID (NOD.CB17-Prkdcscid/NCrCrl) | Charles River | Cat#394; RRID:IMSR_CRL:394 |
| Mouse: CD-1-IGS (Crl:CD1(ICR)) | Charles River | Cat#22; RRID:IMSR_CRL:022 |
| Mouse: Kit W-sh; Rag2−/− ; Il2rγ−/− | Bakthavatchalu et al., 2018 | N/A |
| Oligonucleotides | ||
| Primers for qPCR and sequencing, See Table S3 | This paper | N/A |
| Recombinant DNA | ||
| FUW-TetO-ALKF1174L-t2A-tdTomato | This paper | N/A |
| FUW-TetO-MYCN-3×2A-NeoR | This paper | N/A |
| AAVS1 CAGGS-Luciferase-T2A-LacZ | This paper | N/A |
| Software and Algorithms | ||
| Fiji | fiji.sc | N/A |
| FlowJo | BD Biosciences | N/A |
| BDFacs DIVA | BD Biosciences | N/A |
| DESeq2 | 10.18129/B9.bioc.DESeq2 | N/A |
| STAR | https://github.com/alexdobin/STAR | N/A |
| ComBat | http://www.bu.edu/jlab/wp-assets/ComBat | N/A |
| featureCounts | http://subread.sourceforge.net | N/A |
| CellProfiler | https://cellprofiler.org | N/A |
| Prism 8 | https://www.graphpad.com | N/A |
| BioRender | https://biorender.com | N/A |
Highlights.
A new platform to study human neuroblastoma using mouse-human neural crest chimeras
Chimeras are created by injecting hNCCs with relevant oncogenes into E8.5 mouse embryos
The chimeras developed tumors that resemble primary neuroblastomas seen in patients
The chimeric mice mount a strong immune response that is blunted by the tumor
ACKNOWLEDGMENTS
We thank Frank Soldner, Yonatan Stelzer, , Yuelin Song, and Shawn Liu for advice; Wendy Salmon (W. M. Keck Biological Imaging Facility, WI), Sumeet Gupta (Genome Technology Core, WI), Kathleen Cormier (Hope Babette Tang Histology Facility, KI, MIT), V. Spanoudaki (Animal Imaging & Preclinical Testing Facility, KI, MIT), Raaji Alagappan, Carrie Garrett-Engele, Stella Markoulaki, Ruthie Flannery, and Dongdong Fu for experimental assistance. We thank Robert Weinberg (WIBR, MIT), Jianzhu Chen, Tyler Jacks (the Koch Institute for Integrative Cancer Research at MIT), Paul Trainor (Stowers Institute), and their labs for advice and materials. This work was supported by grants from the Emerald Foundation, United Kingdom (R.J., M.A.C.), the LEO Foundation, Denmark (grant L18015 to R.J. and M.A.C.), the Melanoma Research Foundation, United States (grant 589136 to R.J.), the St. Baldrick’s Foundation, United States (R.E.G., R.J., S. Sengupta, M.A.C.), and by the NIH grants R37HD045022, 1R01-NS088538, and 5R01-MH104610 (to R.J.).
Footnotes
DECLARATION OF INTERESTS
R.J. is a cofounder of Fate Therapeutics, Fulcrum Therapeutics, and Omega Therapeutics and an advisor to Dewpoint Therapeutics.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.stem.2020.02.001.
REFERENCES
- Ahnfelt-Rønne J, Jørgensen MC, Hald J, Madsen OD, Serup P, and Hecksher-Sørensen J. (2007). An improved method for three-dimensional reconstruction of protein expression patterns in intact mouse and chicken embryos and organs. J. Histochem. Cytochem 55, 925–930. [DOI] [PubMed] [Google Scholar]
- Althoff K, Beckers A, Bell E, Nortmeyer M, Thor T, Sprüssel A, Lindner S, De Preter K, Florin A, Heukamp LC, et al. (2015). A Cre-conditional MYCN-driven neuroblastoma mouse model as an improved tool for preclinical studies. Oncogene 34, 3357–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakthavatchalu V, Wert KJ, Feng Y, Mannion A, Ge Z, Garcia A, Scott KE, Caron TJ, Madden CM, Jacobsen JT, et al. (2018). Cytotoxic Escherichia coli strains encoding colibactin isolated from immunocompromised mice with urosepsis and meningitis. PLoS ONE 13, e0194443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry T, Luther W, Bhatnagar N, Jamin Y, Poon E, Sanda T, Pei D, Sharma B, Vetharoy WR, Hallsworth A, et al. (2012). The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell 22, 117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodeur GM, and Bagatell R. (2014). Mechanisms of neuroblastoma regression. Nat. Rev. Clin. Oncol 11, 704–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodeur GM, Seeger RC, Schwab M, Varmus HE, and Bishop JM (1984). Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 224, 1121–1124. [DOI] [PubMed] [Google Scholar]
- Bronner ME, and LeDouarin NM (2012). Development and evolution of the neural crest: an overview. Dev. Biol. 366, 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, Gouw AM, Baylot V, Gütgemann I, Eilers M, and Felsher DW (2016). MYC regulates the antitumor immune response through CD47 and PD-L1. Science 352, 227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HJ, Sun J, Huang Z, Hou H Jr., Arcilla M, Rakhilin N, Joe DJ, Choi J, Gadamsetty P, Milsom J, et al. (2015). Comprehensive models of human primary and metastatic colorectal tumors in immunodeficient and immunocompetent mice by chemokine targeting. Nat. Biotechnol. 33, 656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, Wang L, Soda M, Kikuchi A, Igarashi T, et al. (2008). Oncogenic mutations of ALK kinase in neuroblastoma. Nature 455, 971–974. [DOI] [PubMed] [Google Scholar]
- Cheung NK, and Dyer MA (2013). Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat. Rev. Cancer 13, 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MA, Markoulaki S, and Jaenisch R. (2018). Matched Developmental Timing of Donor Cells with the Host Is Crucial for Chimera Formation. Stem Cell Reports 10, 1445–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MA, Wert KJ, Goldmann J, Markoulaki S, Buganim Y, Fu D, and Jaenisch R. (2016). Human neural crest cells contribute to coat pigmentation in interspecies chimeras after in utero injection into mouse embryos. Proc. Natl. Acad. Sci. USA 113, 1570–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. (2002). Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat. Med. 8, 793–800. [DOI] [PubMed] [Google Scholar]
- Espuny-Camacho I, Arranz AM, Fiers M, Snellinx A, Ando K, Munck S, Bonnefont J, Lambot L, Corthout N, Omodho L, et al. (2017). Hallmarks of Alzheimer’s Disease in Stem-Cell-Derived Human Neurons Transplanted into Mouse Brain. Neuron 93, 1066–1081.e18. [DOI] [PubMed] [Google Scholar]
- Fattahi F, Steinbeck JA, Kriks S, Tchieu J, Zimmer B, Kishinevsky S, Zeltner N, Mica Y, El-Nachef W, Zhao H, et al. (2016). Deriving human ENS lineages for cell therapy and drug discovery in Hirschsprung disease. Nature 531, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster K, Sheridan J, Veiga-Fernandes H, Roderick K, Pachnis V, Adams R, Blackburn C, Kioussis D, and Coles M. (2008). Contribution of neural crest-derived cells in the embryonic and adult thymus. J. Immunol. 180, 3183–3189. [DOI] [PubMed] [Google Scholar]
- Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, Kirkwood JM, Kuchroo V, and Zarour HM (2010). Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med 207, 2175–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gafni O, Weinberger L, Mansour AA, Manor YS, Chomsky E, Ben-Yosef D, Kalma Y, Viukov S, Maza I, Zviran A, et al. (2013). Derivation of novel human ground state naive pluripotent stem cells. Nature 504, 282–286. [DOI] [PubMed] [Google Scholar]
- Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Tosolini M, Camus M, Berger A, Wind P, et al. (2006). Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313, 1960–1964. [DOI] [PubMed] [Google Scholar]
- George RE, Sanda T, Hanna M, Fröhling S, Luther W 2nd, Zhang J, Ahn Y, Zhou W, London WB, McGrady P, et al. (2008). Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 455, 975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Chen M, Wang F, Windrem M, Wang S, Shanz S, Xu Q, Oberheim NA, Bekar L, Betstadt S, et al. (2013). Forebrain engraftment by human glial progenitor cells enhances synaptic plasticity and learning in adult mice. Cell Stem Cell 12, 342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harenza JL, Diamond MA, Adams RN, Song MM, Davidson HL, Hart LS, Dent MH, Fortina P, Reynolds CP, and Maris JM (2017). Transcriptomic profiling of 39 commonly-used neuroblastoma cell lines. Sci. Data 4, 170033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho N, Peng H, Mayoh C, Liu PY, Atmadibrata B, Marshall GM, Li J, and Liu T. (2018). Delineation of the frequency and boundary of chromosomal copy number variations in paediatric neuroblastoma. Cell Cycle 17, 749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver RC, Katibah GE, Amora R, Boydston EA, Zeitler B, et al. (2009). Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 27, 851–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Miller ML, McHenry LK, Zheng T, Zhen Q, Ilkhanizadeh S, Conklin BR, Bronner ME, and Weiss WA (2016). Generating trunk neural crest from human pluripotent stem cells. Sci. Rep. 6, 19727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huszar D, Sharpe A, and Jaenisch R. (1991). Migration and proliferation of cultured neural crest cells in W mutant neural crest chimeras. Development 112, 131–141. [DOI] [PubMed] [Google Scholar]
- Irie N, and Kuratani S. (2014). The developmental hourglass model: a predictor of the basic body plan? Development 141, 4649–4655. [DOI] [PubMed] [Google Scholar]
- Jaenisch R. (1985). Mammalian neural crest cells participate in normal embryonic development on microinjection into post-implantation mouse embryos. Nature 318, 181–183. [DOI] [PubMed] [Google Scholar]
- Jaiswal S, Jamieson CH, Pang WW, Park CY, Chao MP, Majeti R, Traver D, van Rooijen N, and Weissman IL (2009). CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 138, 271–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janoueix-Lerosey I, Lequin D, Brugières L, Ribeiro A, de Pontual L, Combaret V, Raynal V, Puisieux A, Schleiermacher G, Pierron G, et al. (2008). Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 455, 967–970. [DOI] [PubMed] [Google Scholar]
- Kamentsky L, Jones TR, Fraser A, Bray MA, Logan DJ, Madden KL, Ljosa V, Rueden C, Eliceiri KW, and Carpenter AE (2011). Improved structure, function and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinformatics 27, 1179–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Yamaguchi T, Hamanaka S, Kato-Itoh M, Yamazaki Y, Ibata M, Sato H, Lee YS, Usui J, Knisely AS, et al. (2010). Generation of rat pancreas in mouse by interspecific blastocyst injection of pluripotent stem cells. Cell 142, 787–799. [DOI] [PubMed] [Google Scholar]
- Ma H, Wert KJ, Shvartsman D, Melton DA, and Jaenisch R. (2018). Establishment of human pluripotent stem cell-derived pancreatic β-like cells in the mouse pancreas. Proc. Natl. Acad. Sci. USA 115, 3924–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathis D, and Benoist C. (2004). Back to central tolerance. Immunity 20, 509–516. [DOI] [PubMed] [Google Scholar]
- Moore NF, Azarova AM, Bhatnagar N, Ross KN, Drake LE, Frumm S, Liu QS, Christie AL, Sanda T, Chesler L, et al. (2014). Molecular rationale for the use of PI3K/AKT/mTOR pathway inhibitors in combination with crizotinib in ALK-mutated neuroblastoma. Oncotarget 5, 8737–8749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossé YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, et al. (2008). Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 455, 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passoni L, Scardino A, Bertazzoli C, Gallo B, Coluccia AM, Lemonnier FA, Kosmatopoulos K, and Gambacorti-Passerini C. (2002). ALK as a novel lymphoma-associated tumor antigen: identification of 2 HLA-A2.1-restricted CD8+ T-cell epitopes. Blood 99, 2100–2106. [DOI] [PubMed] [Google Scholar]
- Pistoia V, Morandi F, Bianchi G, Pezzolo A, Prigione I, and Raffaghello L. (2013). Immunosuppressive microenvironment in neuroblastoma. Front. Oncol. 3, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistoia V, Morandi F, Pezzolo A, Raffaghello L, and Prigione I. (2012). MYCN: from oncoprotein to tumor-associated antigen. Front. Oncol. 2, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, and Wolchok JD (2018). Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte JH, Lindner S, Bohrer A, Maurer J, De Preter K, Lefever S, Heukamp L, Schulte S, Molenaar J, Versteeg R, et al. (2013). MYCN and ALKF1174L are sufficient to drive neuroblastoma development from neural crest progenitor cells. Oncogene 32, 1059–1065. [DOI] [PubMed] [Google Scholar]
- Seeger RC, Brodeur GM, Sather H, Dalton A, Siegel SE, Wong KY, and Hammond D. (1985). Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N. Engl. J. Med 313, 1111–1116. [DOI] [PubMed] [Google Scholar]
- Soldner F, and Jaenisch R. (2018). Stem Cells, Genome Editing, and the Path to Translational Medicine. Cell 175, 615–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S. (2016). Mechanisms of tumor escape in the context of the T-cellinflamed and the non-T-cell-inflamed tumor microenvironment. Int. Immunol.28, 383–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, and Gajewski TF (2013). Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl. Med 5, 200ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchy F, and Nakauchi H. (2017). Lessons from Interspecies Mammalian Chimeras. Annu. Rev. Cell Dev. Biol 33, 203–217. [DOI] [PubMed] [Google Scholar]
- Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL, and Chen L. (2012). Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci. Transl. Med 4, 127ra37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theunissen TW, Friedli M, He Y, Planet E, O’Neil RC, Markoulaki S, Pontis J, Wang H, Iouranova A, Imbeault M, et al. (2016). Molecular Criteria for Defining the Naive Human Pluripotent State. Cell Stem Cell 19, 502–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trainor PA (2005). Specification of neural crest cell formation and migration in mouse embryos. Semin. Cell Dev. Biol 16, 683–693. [DOI] [PubMed] [Google Scholar]
- Vega-Lopez GA, Cerrizuela S, Tribulo C, and Aybar MJ (2018). Neurocristopathies: New insights 150 years after the neural crest discovery. Dev. Biol. 444 (Suppl 1), S110–S143. [DOI] [PubMed] [Google Scholar]
- Wei JS, Kuznetsov IB, Zhang S, Song YK, Asgharzadeh S, Sindiri S, Wen X, Patidar R, Najaraj S, Walton A, et al. (2018). Clinically Relevant Cytotoxic Immune Cell Signatures and Clonal Expansion of T-Cell Receptors in High-Risk MYCN-Not-Amplified Human Neuroblastoma. Clin. Cancer Res. 24, 5673–5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, and Bishop JM (1997). Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 16, 2985–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissbein U, Schachter M, Egli D, and Benvenisty N. (2016). Analysis of chromosomal aberrations and recombination by allelic bias in RNA-Seq. Nat. Commun. 7, 12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, and Kurachi M. (2015). Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 15, 486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windrem MS, Osipovitch M, Liu Z, Bates J, Chandler-Militello D, Zou L, Munir J, Schanz S, McCoy K, Miller RH, et al. (2017). Human iPSC Glial Mouse Chimeras Reveal Glial Contributions to Schizophrenia. Cell Stem Cell 21, 195–208.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo EY, Chu CS, Goletz TJ, Schlienger K, Yeh H, Coukos G, Rubin SC, Kaiser LR, and June CH (2001). Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and latestage ovarian cancer. Cancer Res. 61, 4766–4772. [PubMed] [Google Scholar]
- Wu J, Greely HT, Jaenisch R, Nakauchi H, Rossant J, and Belmonte JC (2016). Stem cells and interspecies chimaeras. Nature 540, 51–59. [DOI] [PubMed] [Google Scholar]
- Wu J, Platero-Luengo A, Sakurai M, Sugawara A, Gil MA, Yamauchi T, Suzuki K, Bogliotti YS, Cuello C, Morales Valencia M, et al. (2017). Interspecies Chimerism with Mammalian Pluripotent Stem Cells. Cell 168, 473–486.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Y, and Hogquist KA (2012). T-cell tolerance: central and peripheral. Cold Spring Harb. Perspect. Biol. 4, a006957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Liu B, Xu J, Wang J, Wu J, Shi C, Xu Y, Dong J, Wang C, Lai W, et al. (2017). Derivation of Pluripotent Stem Cells with In Vivo Embryonic and Extraembryonic Potency. Cell 169, 243–257.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusuf M, Leung K, Morris KJ, and Volpi EV (2013). Comprehensive cytogenomic profile of the in vitro neuronal model SH-SY5Y. Neurogenetics 14, 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Wu X, Basu M, Dong C, Zheng P, Liu Y, and Sandler AD (2017). MYCN Amplification Is Associated with Repressed Cellular Immunity in Neuroblastoma: An In Silico Immunological Analysis of TARGET Database. Front. Immunol. 8, 1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Yu Y, Hertwig F, Thierry-Mieg J, Zhang W, Thierry-Mieg D, Wang J, Furlanello C, Devanarayan V, Cheng J, et al. (2015). Comparison of RNA-seq and microarray-based models for clinical endpoint prediction. Genome Biol. 16, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq datasets generated during this study are available at GEO: GSE144366.