

Abstract
Background
CRIPTO is a multi-functional signaling protein that promotes stemness and oncogenesis. We previously developed a CRIPTO antagonist, ALK4L75A-Fc, and showed that it causes loss of the stem cell phenotype in normal mammary epithelia suggesting it may similarly inhibit CRIPTO-dependent plasticity in breast cancer cells.
Methods
We focused on two triple negative breast cancer cell lines (MDA-MB-231 and MDA-MB-468) to measure the effects of ALK4L75A-Fc on cancer cell behavior under nutrient deprivation and endoplasmic reticulum stress. We characterized the proliferation and migration of these cells in vitro using time-lapse microscopy and characterized stress-dependent changes in the levels and distribution of CRIPTO signaling mediators and cancer stem cell markers. We also assessed the effects of ALK4L75A-Fc on proliferation, EMT, and stem cell markers in vivo as well as on tumor growth and metastasis using inducible lentiviral delivery or systemic administration of purified ALK4L75A-Fc, which represents a candidate therapeutic approach.
Results
ALK4L75A-Fc inhibited adaptive responses of breast cancer cells under conditions of nutrient and ER stress and reduced their proliferation, migration, clonogenicity, and expression of EMT and cancer stem cell markers. ALK4L75A-Fc also inhibited proliferation of human breast cancer cells in stressed tumor microenvironments in xenografts and reduced both primary tumor size and metastatic burden.
Conclusions
Cancer cell adaptation to stresses such as nutrient deprivation, hypoxia, and chemotherapy can critically contribute to dormancy, metastasis, therapy resistance, and recurrence. Identifying mechanisms that govern cellular adaptation, plasticity, and the emergence of stem-like cancer cells may be key to effective anticancer therapies. Results presented here indicate that targeting CRIPTO with ALK4L75A-Fc may have potential as such a therapy since it inhibits breast cancer cell adaptation to microenvironmental challenges and associated stem-like and EMT phenotypes.
Keywords: Breast cancer, Plasticity, CRIPTO, Stress adaptation, Cancer stem cells
Background
The ability of tumor cells to reprogram their behavior in order to cope with environmental challenges is critical to tumor progression. Challenges include growth and survival in abnormal contexts during dissemination or at distant sites, or when normative signals are corrupted by tissue disruption, nutrient deprivation, or chemotherapy. Importantly, cellular and molecular mechanisms of plasticity that engender tumor cell persistence or even proliferation under microenvironmental-, therapeutic-, or metastasis-associated stress have begun to emerge as key therapeutic targets [1–4].
CRIPTO is a small GPI-anchored signaling protein that regulates normal stem cell activity and oncogenesis [5–8]. It is selectively upregulated in multiple human tumor types including ~ 80% of human breast cancers where it is associated with poor patient prognosis [9–11]. CRIPTO promotes anchorage-independent growth, migration, invasion, and EMT in mammary epithelial cells and breast cancer cell lines in vitro [11–13]. CRIPTO overexpression increases ductal branching, hyperplasia, and latent growth of tumors with an EMT-like phenotype in the mammary epithelium of mice [14–17]. In addition, CRIPTO knockdown inhibits proliferation of human MDA-MB-468 and SKBR3 breast cancer cell lines in vitro [18], while CRIPTO knockdown in a triple negative breast cancer (TNBC)-like transplantable mouse mammary tumor model blocks progression [19]. A CRIPTO mAb carrying a toxic payload has been shown to inhibit growth of MDA-MB-231 cells in a xenograft model [20]. CRIPTO has also been reported as a functional cancer stem cell marker in head and neck cancers and, more recently, in esophageal squamous cell carcinoma, hepatocellular carcinoma, and post-therapy, recurrent osteotropic prostate cancer [21, 22]. Several groups are seeking to develop CRIPTO as a biomarker of tumor aggressiveness and validate therapeutic modalities directed at blocking CRIPTO-induced oncogenesis [19, 23–25].
CRIPTO has multiple signaling functions. It is an essential co-receptor for certain TGF-β superfamily ligands including Nodal and attenuates signaling by others such as activin A, activin B, and TGF-β1 (reviewed in [5, 6]). CRIPTO also acts independently of its effects on TGF-β superfamily ligands as a soluble secreted factor that activates SRC and MAPK/PI3K pathways including activation of AKT [5, 6]. These signaling activities require CRIPTO binding to cell surface glucose regulated protein 78 kDa (GRP78), an endoplasmic reticulum chaperone that localizes to the cell surface under stress conditions and which, like CRIPTO, is overexpressed in breast cancers [26–32]. CRIPTO has also been reported to signal via WNT [33] and NOTCH [34] pathways providing additional mechanisms that may contribute to its roles in stem cells and oncogenesis.
We have developed a novel antagonist based on the CRIPTO binding properties of activin-like kinase 4 (ALK4), a transmembrane serine kinase receptor that mediates signaling by several TGF-β superfamily ligands including activins and Nodal [12]. This antagonist, ALK4L75A-Fc, is designed to have enhanced specificity for CRIPTO as it comprises a human Fc domain fused to a mutant ALK4 extracellular domain bearing a point mutation (L75A) that disrupts ALK4 binding to TGF-β superfamily ligands (i.e., activin and Nodal) without affecting its binding to CRIPTO [35]. We previously demonstrated that ALK4L75A-Fc specifically binds CRIPTO, inhibits CRIPTO signaling, and antagonizes CRIPTO-dependent maintenance of the normal mammary epithelial stem cell state [12]. In the present study, we demonstrate that ALK4L75A-Fc similarly inhibits stem cell phenotypes, plasticity, and adaptability of human breast cancer cells in vitro and in vivo, suggesting it may represent a viable therapeutic strategy for targeting facultative, tumorigenic, stem cell phenotypes that emerge as cancer cells respond to microenvironmental or therapy-associated stress.
Methods
Production of ALK4L75A-Fc
Purified ALK4L75A-Fc was produced as previously described [12] from 293T-conditioned media using sequential protein A and Flag affinity chromatography. For inducible ALK4L75A-Fc, the open reading frame reported in Spike et al. [12] was cloned into a custom 3rd generation lentiviral backbone (described in Supplemental Fig. 1). Lentiviral particles were packaged and concentrated according to the procedure described in Tiscornia et al. [36].
Cell lines
293T, NCCIT, MDA-MB-231, and MDA-MB-468 cell lines were acquired from ATCC. Cells were cultured in “Tumor media” (Dulbecco’s modified Eagle’s media (DMEM) with ciprofloxacin 10 μg/ml, and 10% fetal calf serum), except NCCIT cells which were cultured in “NCCIT media” (RPMI 1640 medium, ATCC modification; GIBCO with ciprofloxacin 10 μg/ml, and 10% fetal calf serum). Cells were grown in sterile humidified tissue culture incubators at 37 °C with 5% CO2 and ambient (~ 17–18%) O2. Cells were transduced with lentiviral concentrates in the presence of 7 μg/ml polybrene for up to 16 h with transduction efficiency (not shown) suggesting MOI < 1. Cells were subsequently sorted for green fluorescence on a FACSAria fluorescence activated cell sorter (BD Biosciences, Franklin Lakes, NJ), following at least 3 days of culture. ALK4L75A-Fc expression was confirmed by western blot of cell lysates and immune-precipitation from conditioned media with protein A agarose beads (Abcam, Cambridge, UK, ab193254) and anti-Flag antibodies (Cell Signaling Technology, Danvers, MA, 8146). Inducible short hairpin vectors were acquired from Dharmacon, Inc. (Lafayette, CO, Cat# V3SH11255-01EG6997).
Treatments
Low serum conditions were achieved by switching from each cell line’s standard media (above) to Opti-MEM (GIBCO, Waltham, MA) for 16 h. Cells were treated in vitro with purified ALK4L75A-Fc (10 μg/ml), recombinant mouse CRIPTO (0.3 μg/ml; R&D Systems, Minneapolis, MN), thapsigargin (Invitrogen, Carlsbad, CA) at 125 nM for MDA-MB-231 and 10 nM for MDA-MB-468 cells, 2-deoxy-d-glucose (4 mM; Sigma-Aldrich, St. Louis, MO), doxycycline (0.2–2 μg/ml; Sigma-Aldrich, St. Louis, MO), and LY290042 (10 μM; Sigma-Aldrich, St. Louis, MO) where indicated. A doxycycline dose of 1 μg/ml was used for in vitro cell treatments except where establishing doxycycline dose response (Supplement Fig. 1B).
Flow cytometry
Single cell suspensions of cells were stained with antibodies to CD44 (AF647-anti-CD44, IM7; BioLegend, San Diego, CA) and surface GRP78 (AF488-anti-HSPA5, polyclonal rabbit; Novus Biologicals, Centennial, CO), and with DAPI. Viable (i.e., DAPI-negative) cells were gated for singlets, and the proportion of cells beyond a uniform threshold were enumerated under equivalent staining and voltages across samples. Aldefluor™ kit (StemCell Technologies, Vancouver, Canada) staining was conducted according to the manufacturer’s recommendation.
Surface GRP78 detection
Surface GRP78 was detected in intact adherent cells by incubating cells on ice with primary GRP78-directed antibodies (N-20, Santa Cruz Biotechnology, Inc., Dallas, TX), followed by cold wash in PBS and incubation with 4% paraformaldehyde, followed by subsequent washing and mounting in the presence of DAPI. Cells were imaged on an SP8 white light confocal microscope (Leica Biosystems, Wetzlar, Germany).
Cell proliferation and migration
To quantitatively measure proliferation, cells were equivalently seeded at low confluency and imaged every 2 h for the times indicated in an automated multi-well incubator/imaging apparatus Incucyte™ (Essen BioSciences, Ann Arbor, MI). Alternatively, cells were plated at density (50,000 cells per 96-well well), and scratch wounds were generated to allow longitudinal imaging in the Incucyte™. System intrinsic automated quantification of proliferation and wound filling was used. Cells were treated with Mitomycin C (Sigma-Aldrich, St. Louis, MO) at 40 μg/ml for MDA-MB-231 and 1 μg/ml for MDA-MB-468 cells.
Methylcellulose cultures
Cells were suspended in 1% methylcellulose (R&D Systems, Minneapolis, MN) in tumor media and allowed to grow 10 days before analysis.
Organoid serial passaging
MDA-MB-468 cells transfected with either the ALKL75A-Fc inducible lentiviral vector or a mock control were seeded in a 24-well low adhesion dish (Corning Inc., Corning, NY) at 5000 cells/well in tumor media supplemented with 10% phenol-free/growth factor reduced Matrigel (Corning Inc., Corning, NY). Cultures were treated with 1 μg/ml doxycycline from the beginning of organoid culture or only subsequent to passaging, as indicated. On day 9, cells were washed with cold PBS and pelleted. Organoids were dissociated with subsequent treatments of Dispase (1 U/ml) (Stem Cell Technologies, Vancouver, Canada) and TrypLE (Thermo Fisher Scientific, Waltham, MA). Cell pellets were then plated in a 24-well low adhesion dish in phenol-free tumor media supplemented with 5% phenol-free/growth factor reduced Matrigel. Images were collected on day 6 following passaging.
Mice and tumor studies
Athymic female nude mice (for NCCIT grafts) and 10–12-week-old SCID beige female mice (for orthotopic breast cancer xenografts) were obtained from Charles River Laboratories (Wilmington, MA). All animal care and procedures were approved and monitored by an Institutional Animal Care and Use Committee. 1 × 106 NCCIT cells (ATCC, Manassas, VA, CRL-2073) in phosphate-buffered saline were injected into each flank of nude mice and allowed to graft for 1 week (Fig. 1d) or 2 weeks (Fig. 1e) as indicated, prior to first treatment with soluble ALK4L75A-Fc or vehicle. As indicated, mice were treated with ALK4L75A-Fc daily for 5 days prior to overt tumor formation followed by bi-weekly injection or were treated bi-weekly after appearance of measurable tumor growth at 2 weeks. Tumor growth was measured with calipers. Cell suspensions of 1 × 105 (Fig. 6a, c) or 2.5 × 105 cells (Fig. 6b) for MDA-MB-231 and 1 × 104 cells for MDA-MB-468 (Fig. 6e) in 40 μl of 50% Matrigel (50% media) were injected transdermally into the #4 fat pad of recipient mice, and tumors were allowed to form and grow to 1 cm or until mice were moribund or tissue was required for analysis. Following euthanasia, tumor tissue and other internal organs were harvested for processing. The lungs were isolated and imaged immediately on a M165 Fc fluorescent stereomicroscope (Leica Biosystems, Wetzlar, Germany) using Leica software. Metastases were identified as fluorescent green foci of varying sizes and were enumerated from static images. Mice were administered doxycycline ad libitum in drinking water (doxycycline hyclate, 0.8 mg/ml).
Fig. 1.

Recombinant purified ALK4L75A-Fc inhibits CRIPTO-dependent tumor cell growth in vitro and in vivo. a Confluency of NCCIT cells grown in vitro for 6 days in the presence (10 μg/ml) or absence of purified recombinant ALK4L75A-Fc. Standard deviation across triplicates is shown. b Effect of rmCRIPTO on the growth inhibitory effect of ALK4L75A-Fc in NCCIT cell cultures. Standard deviation across triplicates is shown. c Representative immunofluorescent anti-human IgG staining of tumor tissue from mice bearing NCCIT xenografts that were treated with ALK4L75A-Fc (5 mg/kg, I.P.) or vehicle (inset). A, acellular region; V, presumptive vasculature. Scale bar = 100 μm. d, e Effect of ALK4L75A-Fc injection compared to saline on growth of NCCIT xenografted tumors at the indicated doses and over the indicated time periods
Fig. 6.

Expression of ALK4L75A-Fc in vivo inhibits growth and metastasis of breast cancer xenografts. a Orthotopic MDA-MB-231 xenograft growth in the presence or absence of doxycycline induction of ALK4L75A-Fc. Student’s t test, p < 0.05 (across each of the final four measurements). n = 15 per group. b Bright field and fluorescent images of representative lung tissue samples from MDA-MB-231 xenografted mice in the absence (LV-S14-Mock; n = 12) or presence (LV-S14-ALK4L75A-Fc; n = 8) of ALK4L75A-Fc expression. c Number of macroscopic fluorescent metastatic foci in the lungs of mice bearing control or ALK4L75A-Fc-expressing tumor cells. Fisher’s exact test. d Size of primary MDA-MB-231 orthotopic xenografts at 5 weeks post-inoculation in mice injected intraperitoneally with ALK4L75A-Fc (10 mg/kg; n = 6) or vehicle (n = 4). e Size of MDA-MB-468 orthotopic xenografts at 5 weeks in the presence (n = 6) or absence (n = 4) of ALK4L75A-Fc expression
Tumor sections
Freshly dissected tumor tissue was manually sectioned into slices < 4 mm in diameter and fixed overnight at 4 °C in 10% neutral buffered formalin. Fixed tissues were transferred to 70% ethanol for storage. Processing for histology followed standard protocols including paraffin embedding, sectioning at 5 μm thickness, baking for 1 h at 55 °C, and deparaffinizing with Histochoice™ Clearing agent (Amresco Inc., Solon, OH) or CitriSolv (Decon Labs, King of Prussia, PA) (Fig. 5e, Supplemental Fig. 3B only), and rehydration through graded alcohol/water washes. Antigen retrieval was achieved by boiling samples in citrate buffer (10 mM citric acid, 0.05% Tween 20, pH 6.0). Tumor sections were subjected to histological analysis and immunostaining with antibodies to E-cadherin (24E10, Cell Signaling Technology, Danvers, MA), MKI67 (SP6, Abcam, Cambridge, UK), HIF1α (S46-16, BioLegend, San Diego, CA), cleaved caspase-3 (5A1E, Cell Signaling Technology, Danvers, MA), CD31-PE (MEC13.3, BioLegend, San Diego, CA), GRP78 (IH11-1H7, Thermo Fisher Scientific, Waltham, MA), CRIPTO (ab19917, Abcam, Cambridge, UK), phalloidin (ab176757, Abcam, Cambridge, UK), donkey anti-rabbit IgG AlexaFluor 647 (ab150075, Abcam, Cambridge, UK), goat anti-mouse IgG PE/Cy5.5 (130784, Abcam, Cambridge, UK), and DAPI (Thermo Fisher Scientific, Waltham, MA). GRP78/CRIPTO/phalloidin samples were also blocked with mouse Fc receptor blocking reagent (Miltenyi Biotec, Bergisch Gladbach, Germany). Stained slides were imaged on a Nikon Eclipse Ni microscope with DSRi2 camera (Nikon, Tokyo, Japan) or an SP8 white light confocal microscope (Leica Biosystems, Wetzlar, Germany). For quantification of proliferation in stressed microenvironments, 12 high-powered images were captured from 7 mock and 5 ALK4L75A-Fc-expressing tumors by scanning for HIF1α positivity without other fluorescent channels visible. Images were subsequently captured across all relevant fluorescent channels to observe costaining with HIF1α and MKI67 antibodies (and DAPI). These were manually scored blind (i.e., unlabeled) and then assigned to their appropriate tumor type, post hoc. Pimonidazole was injected intraperitoneally at 60 mg/kg according to the recommendations in the Hypoxiprobe™ kit (Hypoxyprobe, Burlington, MA) and detection of signal was carried out according to the manufacturer’s recommendations.
Fig. 5.

ALK4L75A-Fc reduces aggressive breast cancer cell phenotypes and proliferation of stressed cancer cells in vivo. a Expression changes in select stem cell, cancer stem cell, and EMT/MET associated transcripts in a contralateral pair of mock and ALK4L75A-Fc MDA-MB-231 tumors following 96 h of in vivo doxycycline treatment. b Representative images of mock and ALK4L75A-Fc-expressing MDA-MB-231 xenografts (n = 3 of each type). H&E, hematoxylin/eosin staining. E-Cadherin at the periphery of advanced tumors (2nd column). d = mammary duct. MKI67 staining of tumors in highly cellular areas and adjacent to acellular regions, A (right 4 panels). Scale = 250 μM. c Representative images of the overlap of MKI67 (green) and HIF1a (red) staining in MDA-MB-468 tumor sections with and without ALK4L75A-Fc expression. Four classes of cells could be identified, I–IV. d Blinded quantification of the four cell classes indicated in c across a set of 12 images (7 mock and 5 ALK4L75A-Fc) collected from 2 MDA-MB-468 mock and 2 MDA-MB-468 ALK4L75A-Fc tumors. *p < 0.05; Student’s t test. e Representative (n = 2) costaining for CRIPTO (green), GRP78 (red), phalloidin (blue), and DAPI (gray). Bracket = acellular region-adjacent CRIPTO staining cells. Scale bars = 100 μM (i) and 25 μM (ii), insets show colocalization of CRIPTO and GRP78 (yellow) at the cell periphery in bracketed regions
Western blots
Protein lysates from cultured cells and immune precipitates from conditioned media were suspended in RIPA buffer (50 mM Tris, pH 7.4, 1% Nonidet P-40 Substitute (Sigma-Aldrich, St. Louis, MO), 0.5% sodium deoxycholate, 0.1% SDS supplemented with protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO #P8340), and phosphatase inhibitor cocktail (Thermo Fisher Scientific, Waltham, MA, #78420)). Samples were boiled in LDS loading buffer prior to loading and were run on pre-cast 4–12% gradient Bis-Tris, 1.5 mm × 15 well gels (Invitrogen, Carlsbad, CA, #NP0336BOX). Proteins were transferred on an iBlot2 Dry Blotting System (Thermo Fisher Scientific, Waltham, MA) to PVDF (Invitrogen, Carlsbad, CA, #IB24002) or nitrocellulose (Invitrogen, Carlsbad, CA, #IB23002) membranes for protein detection. Membranes were blocked in Intercept Blocking buffer (Li-Cor Biosciences, Lincoln, NE) and probed with the following: mouse anti-human AKT (40D4, Cell Signaling Technology, Danvers, MA #2920, 1:2000), rabbit anti-human phospho-AKT (Ser473) (D9E, Cell Signaling Technology, Danvers, MA #4060, 1:2000), and rat anti-human β-actin (W16197A, BioLegend #664802, 1:10,000). The following secondary antibodies were used to detect unconjugated primary antibodies: goat anti-mouse IgG H&L, AF790 (Thermo Fisher Scientific, Waltham, MA, #A11375, 1:20,000); goat anti-rabbit IgG H&L, AF680 (Thermo Fisher Scientific, Waltham, MA, #A21076, 1:1000); and donkey anti-rat IgG H&L, Dylight 800 (Thermo Fisher Scientific, Waltham, MA, #SA5-10032, 1:10,000).
RT-PCR
Mouse and human TDGF1 (forward: TCCTTTTGTGCCTGCCCTC; reverse: CACAGGGAACACTTCTTGG) or commercially available primers for ACTB (Integrated DNA Technologies, Coralville, IA, IDT-PrimeTime Std qPCR Assay Hs.PT.39a.22214847) were included in standard PCR or quantitative real-time RT-PCR using the Power SYBR Green Cells-to-CT kit (Thermo Fisher Scientific, Waltham, MA, #4402954) according to the manufacturer’s instructions. Real-time PCR was carried out on a lightCycler 480 (Roche, Basel, Switzerland), and comparisons were made using the ΔΔCt method with β-actin as a control transcript.
RNA sequencing
A mouse bearing one pair of contralateral LV-S14-Mock and LV-S14-ALK4L75A-Fc tumors of approximately equal size (D = ~ 0.66 cm) was administered doxycycline ad libitum in water for 96 h prior to tumor harvest. We generated sequencing libraries from several snap frozen, cold pulverized 2-mm fragments of tumors corresponding to a spherical wedge of approximately 1/3 of the total tumor volume and extending from the center to the periphery immediately after isolation. We employed Truseq™ RNA preparation kits (Illumina, San Diego, CA), according to the manufacturer’s instructions. Libraries were validated using an Agilent Technologies bioanalyzer 2100 and Quant-iT™ HS dsDNA assay (Life Technologies, Carlsbad, CA). One hundred base, paired end sequencing was carried out on a Hiseq2000 (Illumina, San Diego, CA). Reads were mapped to the human genome (hg38) using STAR [37]. We quantified gene expression as FPKM (fragments per kilobase of gene model per million reads) using Cufflinks [38].
Results
ALK4L75A-Fc treatment inhibits CRIPTO-dependent tumor cell growth in vitro and in vivo
Embryonal carcinoma-derived NCCIT cells provide a model tumor cell system to test CRIPTO inhibitors as these cells have stem cell properties and express high levels of CRIPTO protein. We previously reported that growth of NCCIT cells in vitro is inhibited by CRIPTO knockdown or treatment with a neutralizing GRP78 antibody that disrupts CRIPTO/GRP78 binding [26]. Here, we show that treatment of NCCIT cells with ALK4L75A-Fc similarly inhibits NCCIT cell growth (Fig. 1a) and that this effect is rescued by addition of soluble recombinant CRIPTO protein (Fig. 1b). Next, we sought to test the effects of ALK4L75A-Fc protein on NCCIT xenografts grown in the flanks of nude mice by injecting the reagent intraperitoneally. ALK4L75A-Fc accumulated in NCCIT tumors since high immuno-reactivity was visible in occasional cells distributed throughout the tumors, in structures resembling vessels (V) and in cells distal to the presumptive vasculature at the boundary between intact tumor tissue and acellular zones (A) (Fig. 1c, Supplemental Fig. 3). Critically, treating mice with ALK4L75A-Fc slowed the growth of these very aggressive NCCIT cell tumors when injected either before (Fig. 1d) or after (Fig. 1e) measurable tumors were detectable. No adverse effects were observed when mice were injected with ALK4L75A-Fc at the highest dose tested of 10 mg/kg (Fig. 1d, and data not shown).
CRIPTO signaling is regulated by stress in breast cancer cells
MDA-MB-231 is an aggressive, poorly differentiated, TNBC cell line that has been reported to express CRIPTO [39]. While we found that expression of CRIPTO transcripts was barely detectable in these cells under standard growth conditions, CRIPTO levels increased when these cells were grown overnight in medium that had reduced glucose levels and lacked serum-derived growth factors (Opti-MEM, GIBCO) (Fig. 2a, b). Culturing MDA-MB-231 cells under these conditions also resulted in increased levels of phospho-AKT (Ser473), which could be inhibited by treating cells with ALK4L75A-Fc or the PI3K-inhibitor LY294002 (Fig. 2c). Expression of a short hairpin targeting CRIPTO similarly reduced AKT phosphorylation in MDA-MB-231 cells (Fig. 2d, Supplemental Fig. 1). Cell surface GRP78 levels also increased under these growth conditions to an even greater extent than was observed following treatment with thapsigargin, which is known to strongly increase the expression and cell surface localization of GRP78 (Fig. 2e). Finally, and consistent with previous results [40], both thapsigargin and the glycolysis inhibitor 2-deoxyglucose (2-DG) increased cell surface GRP78 levels relative to untreated controls in both MDA-MB-231 cells and a second TNBC cell line, MDA-MB-468 (Fig. 2f). Together, these results are consistent with CRIPTO/GRP78 signaling being stress responsive in breast cancer cells.
Fig. 2.

Stress response of the CRIPTO signaling pathway in breast cancer cell lines. a RT-PCR for TDGF1 (CRIPTO) in MDA-MB-231 under serum starvation. b Confirmation of TDGF1 sequence of the PCR product from a. c Western blot indicating that ALK4L75A-Fc blocks starvation-induced AKT activation in MDA-MB-231 cells. d AKT activation in MDA-MB-231 cells harboring a doxycycline-inducible shCRIPTO construct. e Flow cytometric analysis of surface GRP78 under thapsigargin treatment or serum starvation in MDA-MB-231. The percentage of cells brighter than the control midpoint are given from one of two independent experiments. f Immunofluorescent detection (prior to fixation) of surface GRP78 in 2-deoxyglucose (2-DG)- or thapsigargin-treated MDA-MB-231 and MDA-MB-468 cells. Representative images from one of three independent experiments are shown. Scale bar = 50 μm
ALK4L75A-Fc inhibits adaptation of breast cancer cells to glycolytic stress
Next, we tested the effect of ALK4L75A-Fc on cellular phenotypes related to EMT and stemness in the context of stress adaptation in these TNBC cell lines. Notably, treatment of MDA-MB-231 cells with ALK4L75A-Fc had little effect on cell migration in standard, high glucose culture conditions (4.5 g/l), but inhibited migration under conditions of glucose deprivation (1 g/l) (Fig. 3a, b). In order to provide an alternative method of delivering ALK4L75A-Fc and facilitate parallel in vitro and in vivo studies, we generated a lentiviral vector (LV-S14) that allows doxycycline (Dox)-inducible expression of secreted ALK4L75A-Fc while also providing luminescent and fluorescent tracking capability (Supplemental Fig. 1). The effect of ALK4L75A-Fc using this delivery method was highly similar to that resulting from treatment of MDA-MB-231 cells with purified ALK4L75A-Fc protein. Thus, Dox treatment of LV-ALK4L75A-Fc MDA-MB-231 cells caused reduced migration specifically under low glucose conditions while Dox treatment of LV-Mock MDA-MB-231 (negative control) had no effect (Fig. 3c, d). Similarly, Dox-induced ALK4L75A-Fc had little effect on the overall proliferation of MDA-MB-231 cells in standard (high glucose) media but increased sensitivity to growth inhibition by 2-DG (Fig. 3e, f). Strikingly, when 2-DG was removed from the media after 48 h, control (mock) cells grew faster than ALK4L75A-Fc-expressing cells and untreated control cells, consistent with a role for CRIPTO in adaptive cellular growth responses under glucose deprivation in vitro (Fig. 3e, f). Targeting CRIPTO expression with a Dox-inducible shRNA similarly reduced the rate of growth of MDA-MB-231 cells challenged by or released from 2-DG treatment (Fig. 3g).
Fig. 3.

ALK4L75A-Fc reduces migration and proliferation in stressed breast cancer cells, Scale bar = 300m. a, b In vitro scratch wound filling by MDA-MB-231 cells treated with soluble ALK4L75A-Fc protein under glucose restriction. Representative images from among 5 replicates per treatment are shown. c, d In vitro scratch wound filling of ALK4L75A-Fc-expressing and mock, LVS14-transduced MDA-MB-231 cell cultures under glucose restriction. Representative images from among 5 replicates per treatment are shown. e ALK4L75A-Fc inhibits MDA-MB-231 human breast cancer cells’ growth recovery from 2-DG treatment. Replicates = 3 *Student’s t test. f Maximum slope values from the graph in e. g Growth rate of MDA-MB-231 cells expressing shCRIPTO under 2-DG challenge and release, represented as maximum slope. All time-lapse studies were conducted with 3–5 replicates and were repeated 2–3 times
ALK4L75A-Fc also inhibited properties associated with cancer stem cells (CSC) and breast cancer cell aggressiveness in the context of experimental stress (Fig. 4). For example, 2-DG increased the percentage of cultured MDA-MB-231 cells that were positive for the stem cell marker Aldefluor and this effect was inhibited by treatment with ALK4L75A-Fc (Fig. 4a). Similarly, ALK4L75A-Fc reduced cell surface levels of the cancer stem cell-associated marker CD44 in MDA-MB-231 and MDA-MB-468 cells treated with 2-DG or thapsigargin, while increasing expression of the CSC contraindicator CD24 in MDA-MB-231 cells (Fig. 4b, c) [41]. MDA-MB-468 cells express naturally high levels of CD24 even prior to treatment (Fig. 4c). Colony growth under non-adherent conditions, such as methylcellulose and Matrigel cultures, provides a measure of cancer stem cell function and aggressiveness. MDA-MB-468 cells formed discrete colonies in methylcellulose, and the size of these colonies was not significantly altered by 2-DG treatment, although some large colonies were noted in 2-DG-treated cultures (Fig. 4d, e). Whereas ALK4L75A-Fc treatment did not affect the size of colonies grown in the absence of 2-DG, it strongly inhibited colony growth in the presence of 2-DG (Fig. 4d, e). We also examined the growth of colonies from ALK4L75A-Fc- and mock-transduced MDA-MB-468 cells plated in organoid culture conditions containing Matrigel. Similar to what was observed in methyl-cellulose cultures (Fig. 4d, e), Dox induction of ALK4L75A-Fc in MDA-MD-468 cells grown in organoid culturing conditions had no effect on initial organoid growth but resulted in almost no organoids in secondary cultures compared to uninduced primary cultures and mock controls, which produced many secondary organoids (Fig. 4f, Supplemental Fig. 2). These results suggest that the stress of 2-DG and reseeding renders MDA-MB-468 cells sensitive to CRIPTO blockade by ALK4L75A-Fc.
Fig. 4.

The CRIPTO blocking agent, ALK4L75A-Fc, reduces cancer stem cell properties in stressed breast cancer cells. a Stress and ALK4L75A-Fc associated changes in Aldefluor markers. b, c Effects of ALK4L75A-Fc on CD44 expression in thapsigargin- or 2-DG-treated MDA-MB-231 (b) and MDA-MB-468 cells (c). *Student’s t test, p < 0.05. d Changes in suspension colony frequency and growth of LVS14-Mock- and LVS14-ALKL75A-Fc-transduced MDA-MB-468 cells following stress. Duplicate wells were examined, and the experiment was carried out twice. e Quantification of suspension colony growth from the experiment shown in d and using multiple images. *Fisher’s exact test, p < 0.05. f Serial passage rates of MDA-MB-468 cells (in triplicate) induced to express ALKL75A-Fc. *Kruskal-Wallis, p = 0.0152
CRIPTO antagonism with ALK4L75A-Fc alters breast cancer stem cell, EMT, and stress response properties in vivo
Next, we examined the effect of ALK4L75A-Fc on breast cancer growth and metastasis in vivo. We transplanted stably transduced LV-S14-Mock and LV-S14-ALK4L75A-Fc MDA-MB-231 cells orthotopically into mammary fat pads of SCID mice, allowed tumors to grow to a size of 60–70 mm in diameter, and then administered Dox for 96 h to induce ALK4L75A-Fc. We compared the expression patterns of several previously reported EMT and stem cell markers (Fig. 5a). Expression levels of known cancer stem cell markers declined under ALK4L75A-Fc treatment, including the majority of aldehyde dehydrogenase (ALDH) transcripts that have been shown to contribute to positivity for the cancer stem cell-associated Aldefluor reagent and the CSC-associated isoforms of CD44 (CD44v3, CD44v6) [41, 42]. Markers associated with normal stem cells such as POU5F1, SOX2, and NANOG were mixed in the directionality of their change. The majority of transcripts for known EMT markers (e.g., VIM, ACTA2, ZEB1, ZEB2, SNAI2, TWIST1, TWIST2) also declined under ALK4L75A-Fc induction although SNAI1 was slightly elevated (Fig. 5a). Conversely, markers of epithelial fate and mesenchymal to epithelial transition (MET) (e.g., EPCAM, Cytokeratins (avg.), CLDN4, and the MaSC-associated marker CD24 (also a putative CSC contraindicator)) increased under ALK4L75A-Fc treatment (Fig. 5a). Together, these data suggest that ALK4L75A-Fc promotes a transition from a stem cell-like, mesenchymal phenotype to a more differentiated, epithelial phenotype in vivo.
We did not observe major morphological changes in MDA-MB-231 tumor cells following 96 h of ALK4L75A-Fc induction in vivo (data not shown). However, examination of a contralateral pair of tumors induced with Dox for 96 h suggested early gene expression changes toward a more epithelial, less mesenchymal, less CSC-like state when comparing the ALK4L75A-Fc-expressing tumor to its mock counterpart (Fig. 5a). Nevertheless, even when tumor cells were induced to express ALK4L75A-Fc for 6 weeks, few differences in histopathological features were noted (Fig. 5b, left panels). Both treated and untreated tumors retained an overall carcinomatous histopathology and had regions of apparent necrosis surrounded by regions of high cellularity (see also Supplemental Fig. 3). However, in some regions, ALK4L75A-Fc-expressing cells were slightly larger than their mock-transduced counterparts (Mock 2-4 rbc vs ALK4L75A-Fc 6-8 rbc) (Fig. 5b and data not shown). We noted very few pyknotic nuclei in the highly cellular zones of either mock or ALK4L75A-Fc-expressing tumors. Consistent with this, the highly cellular regions of both mock and ALK4L75A-Fc tumors only rarely showed cells positive for the apoptosis marker cleaved caspase 3, though the boundary with acellular zones was strongly positive for this marker in both cases (Supplemental Fig. 3). Though treated and untreated tumor cells appeared grossly similar and E-cadherin transcript levels (CDH1) did not appear to be acutely modulated by ALK4L75A-Fc in MDA-MB-231 xenografts (Fig. 5a), end stage tumors expressing ALK4L75A-Fc stained more strongly for E-cadherin protein (compared for instance to endogenous mammary ducts (d)), consistent with ongoing reversal of EMT and promotion of a more differentiated phenotype over the longer term (Fig. 5b).
Although we found that ALK4L75A-Fc expression had no obvious effect on cellular proliferation rates in highly cellular regions of tumors as judged by mitotic counts (not shown) and MKI67 positivity (Fig. 5b, right panels), ALKL75A-Fc-expressing tumors showed occasional abnormal mitoses and a marked reduction in MKI67 staining specifically in regions adjacent to necrotic areas when compared to mock controls (Fig. 5b, far right panels). Acellular regions in tumors frequently represent avascular regions where nutrient supply is insufficient to support cellular survival, and the cells adjacent to these regions are expected to be under nutrient deprivation stress. Indeed, we found regions adjacent to these acellular zones to stain positive for pimonidazole adducts indicating that they are hypoxic (Supplemental Fig. 3). Therefore, we quantified Ki67 positivity in cells exhibiting HIF1α stabilization to compare proliferation rates of stressed cells in tissue sections from control and ALK4L75A-Fc-expressing tumors (Fig. 5c, d). We were able to identify all four classes (I–IV) of DAPI-stained nuclei in tissue sections from LV-S14-Mock and LV-S14-ALK4L75A-Fc tumors (Fig. 5c). Enumeration of these four classes across multiple micrographs per tumor type revealed a statistically significant reduction in double-positive (MKI67+,HIF1a+) cells in ALK4L75A-Fc-expressing tumors (Fig. 5d). Consistent with our observations in vitro, these in vivo data indicate that CRIPTO signaling is required for tumor cell proliferation under conditions of stress and that this stress-adaptation signaling can be blocked with ALK4L75A-Fc. Indeed, immune detection of CRIPTO and GRP78 in tumor xenografts showed robust staining for both CRIPTO and GRP78 at the cell periphery specifically in regions we previously identified as nutrient deprived (Fig. 5e, Supplemental Fig. 3). CRIPTO and GRP78 were often colocalized at the cell surface in these regions (Fig. 5e, ii insets). These regions also exhibited qualitative differences in phospho-SMAD2/3 and phospho-AKT staining (Supplemental Fig. 4).
CRIPTO blockade with ALK4L75A-Fc inhibits growth and metastasis of breast cancer xenografts
We conducted a larger tumor study to examine the overall impact of ALK4L75A-Fc on breast cancer growth and spread. LV-S14-Mock and LV-S14-ALK4L75A-Fc MDA-MB-231 cells were transplanted orthotopically into nude mice, and then, Dox was administered 2 weeks later to induce expression of ALK4L75A-Fc. Tumors arising from cells that carry Dox-inducible ALK4L75A-Fc grew more slowly than those arising from LV-S14-Mock cells or with either cell type in the absence of Dox (Fig. 6a). Furthermore, at the endpoint of the study, gross fluorescent visualization of lung tissue from mice with MDA-MB-231 tumors lacking ALK4L75A-Fc expression revealed numerous macro- and micro-metastatic lesions (Fig. 6b). By contrast, of the 8 mice with ALK4L75A-Fc-expressing tumors, only 1 with a single macro-metastasis was observed (Fig. 6c). Critically, these anticancer effects of ALK4L75A-Fc were not restricted to overexpression of ALK4L75A-Fc in MDA-MB-231 cells since injection of purified ALK4L75A-Fc (10 mg/kg; I.P.) also inhibited MDA-MB-231 tumor growth (Fig. 6d), and similar to what we observed in MDA-MB-231 cells, ALK4L75A-Fc induction in a second TNBC model (MDA-MB-468 cells) following transplantation also inhibited growth of resulting tumors (Fig. 6e).
Discussion
Phenotypic plasticity allows cells to adapt to stresses such as nutrient deprivation, hypoxia, and chemotherapy and likely represents a key contributor to tumor cell heterogeneity and the major clinical problems of dormancy, metastasis, therapy resistance, and recurrence. Our studies indicate that CRIPTO plays critical roles in plasticity and the adaptive process. We show that the CRIPTO directed antagonist ALK4L75A-Fc is active against poorly differentiated TNBC cells in vitro and in vivo, and that it blocks their ability to adapt to stresses such as nutrient deprivation that are commonly encountered by tumors. Specifically, ALK4L75A-Fc decreased the expression of EMT and CSC markers that are hallmarks of cancer aggressiveness, blocked proliferation of cells in stressed microenvironments in vivo, and also reduced tumor burden especially at metastatic sites.
We focused on two models of TNBC due to the unmet need for molecular targets in this subtype of breast cancer and because of their tractability as tumor xenografts for modeling tumor growth and metastasis. Our study establishes proof of principal for ALK4L75A-Fc in treating these tumors. It remains to be determined whether increases in surface CRIPTO or surface GRP78 are more important in rendering the CRIPTO/GRP78 signaling axis sensitive to stress. However, we note that CRIPTO is overexpressed in a range of cancers including over 80% of breast cancers, and accordingly, some preliminary studies have indicated CRIPTO effects in both ER+ and Her2+ breast cancers [18]. Using similar approaches to those described here, it should be possible to identify additional tumor types susceptible to ALK4L75A-Fc using patient-derived organoid and xenograft models (PDO and PDX, respectively) derived from diverse breast cancer patients and representing the broader intertumoral heterogeneity of breast cancer.
CRIPTO’s effects on breast cancer cells are likely mediated by its joint modulation of the TGF-β pathway and growth factor-like signaling (i.e., activation of SRC/MAPK/PI3K pathways), and there may also be important roles for other pathways regulated by CRIPTO including WNT and NOTCH [33, 34]. We previously showed that ALK4L75A-Fc acts as a “ligand trap” that inhibits CRIPTO growth factor-like signaling, including AKT activation, in primary mouse mammary epithelial cells and a non-transformed human mammary epithelial cell line (MCF10A) [12]. The AKT pathway is a critical regulator of tumor cell growth and stem cell-like properties, and our results suggest that blocking this aspect of CRIPTO signaling plays a key role in inhibiting maintenance of the stem cell phenotype and oncogenesis. While there currently is no evidence supporting such a role, it remains formally possible that ALK4L75A-Fc also has additional unanticipated targets besides CRIPTO that contribute to its anti-tumor effects.
CRIPTO binding to GRP78 at the cell surface is thought to be critical for CRIPTO signaling function [12, 26, 27]. Although GRP78’s best defined roles in cellular stress responses involve its ER/UPR activities, it is now widely recognized that a portion of GRP78 re-localizes to non-ER locations including the cell surface in response to a variety of cellular stresses such as ER stress and nutrient deprivation. Cell surface GRP78 is proposed to mediate additional cellular stress responses such as those demonstrated for CRIPTO previously and in the present study [5, 29]. Notably, nutrient stress was reported to promote GRP78-dependent AKT phosphorylation in a variety of tumor cell lines including breast cancer cells [43]. Recently, GRP78 was also implicated in the maintenance/reestablishment of MEK/ERK signaling in therapy-resistant BRAF mutant melanomas, and in maintaining stemness in pancreatic cancer cells in response to therapy-associated oxidative and protein stress [44, 45]. Similarly, another recent report has indicted that cell surface GRP78 also marks cells with enhanced tumor- and metastasis-initiating capacities in breast cancer [46]. Based on the findings presented here, we propose that re-localization of GRP78 to the cell surface during stressful conditions enhances CRIPTO signaling and thereby promotes cellular plasticity, adaptation to stress, and tumor progression (Fig. 7).
Fig. 7.
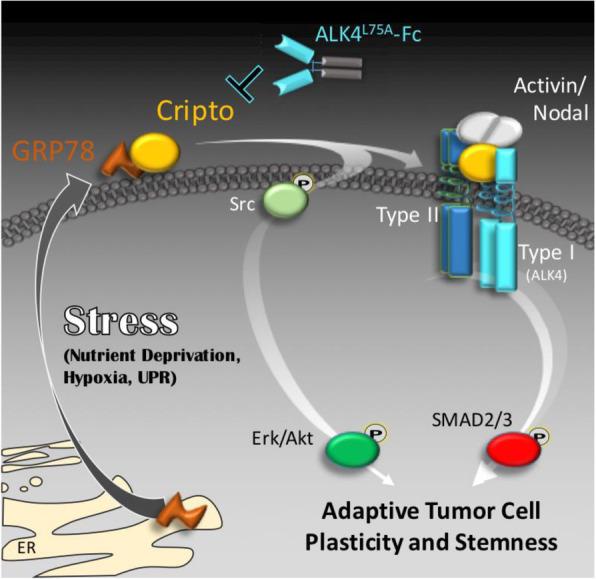
Graphical rendering of GRP78-dependent CRIPTO signaling driving stem-like tumor cell phenotypes. Cellular stresses that promote GRP78 re-localization to the cell surface could render such cells susceptible to CRIPTO-mediated signaling that balances growth factor-like signaling with TGF-beta pathway signaling to promote growth and plasticity
Here, we specifically examined the effects of ALK4L75A-Fc under conditions of growth factor restriction, glucose deprivation, and ER stress, challenges that are regularly encountered by tumor cells in vivo. Although the effects of chemotherapeutic and molecularly targeted therapies were not examined in this study, these treatments elicit similar stresses to those examined here. Our in vitro studies indicate that while blocking CRIPTO/GRP78 signaling does not inhibit oncogenic behaviors such as cell migration in the absence of stresses such as nutrient deprivation, it sustains these behaviors in the presence of such stresses. Similarly, we find that targeting CRIPTO signaling with ALK4L75A-Fc specifically impacts cell proliferation in stressed microenvironments in tumors and inhibits metastasis, a process that requires tumor cells to adapt to multiple novel and stressful environments. Thus, cellular dependencies on CRIPTO and perhaps other stem cell/plasticity promoting pathways are likely to be dependent on contexts that call for cellular adaptation, including therapy response.
We have shown that in addition to compromising cellular stress adaptation, blocking CRIPTO signaling inhibits EMT and CSC phenotypes, the acquisition of which are both plastic/reversible processes [12]. EMT is associated with invasive phenotypes that may contribute to tumor cell dissemination, and its reversibility is critical for metastatic outgrowth [47]. The EMT phenotype has been shown to emerge in tumor cells under therapeutic challenge and to engender drug resistance [48]. Similarly, CSC activity (i.e., enhanced tumor propagating capacity of subpopulations of tumor cells) has been shown to be a metastable phenotype associated with specific biomarkers in breast and other cancers [49, 50]. Importantly, breast CSC phenotypes may be augmented by microenvironmental stresses [51]. In a notable example of this, Wicha and colleagues showed that certain stem cell populations (i.e., ALDH+ cells) reside in stressed tumor microdomains and increase following antiangiogenic, hypoxia-inducing therapy (Avastin) [52]. It remains to be determined if other critical cell processes like metabolism or trafficking, which may also be under CRIPTO control [53, 54], are critical contributors to overall cellular plasticity and adaptability.
Conclusions
We conclude that TNBCs, and possibly many other types of cancers, critically rely on stress adaptive programs involving the CRIPTO/GRP78 signaling axis and that these can be inhibited by ALK4L75A-Fc. In a wide variety of cancers including breast cancers, subpopulations of cells exhibit specialized tumorigenic properties that contribute to disease progression and therapy resistance [55]. Elucidation of mechanisms governing tumor cell adaptation to stress has led to the identification of therapeutic targets such as CRIPTO for inhibiting plasticity and the emergence of subpopulations of cancer cells with facultative stem cell-like properties. While reagents such as ALK4L75A-Fc that block the function of these targets might show modest effects as single agents in some tumor types where many cells have secured ample nutrient supply, they are predicted to complement cytostatic and cytotoxic treatments by undercutting cells surviving debulking therapies that are capable of adapting and emerging as therapy-resistant clones following chemotherapy, recurring after dormancy, and disseminating through novel stress-inducing environments during metastasis.
Supplementary Information
Additional file 1: Supplemental Fig. 1. A) Schematic of a lentiviral expression vector (S14) with fluorescent luminescent markers and doxycycline-inducible ALK4L75A-Fc. (B,C) Western blot demonstrating doxycycline-dependent expression of FLAG-tagged ALK4L75A-Fc in S14-transduced MDA-MB-231 cells, B and secretion into conditioned media, C. (D) Amplification curves for TDGF1 (ie CRIPTO) from equivalent starting material in MDA-MB231 cells transduced with a Dox dependent shCRIPTO vector. Supplemental Fig. 2. Morphology of organoids seeded into secondary cultures following treatment with Dox to induce ALK4L75A-Fc expression during primary organoid out growth. Supplemental Fig. 3. A) a panoramic view of a hematoxylin/Eosin stained MDA-MB-468 tumor section by which position and morphology can be used to assign regional and structural information. P=periphery, v=presumptive vasculature, C=cellular region, S =stressed zone (see B), A = acellular zone. No gross morphological differences apart from average size were noted for ALK4L75A-Fc expressing tumors relative to mock controls. (B) High magnification images of presumptive vasculature in MDA-MB-468 xenografts containing obvious red blood cells (left panels). Lack of robust CD31 immunoreactivity in cellular regions of xenografts (second column). Cleaved caspase 3 staining in proximal acellular regions (third column). Identification of regions of stress in vivo via detection of Pimonidazole adducts with Hypoxyprobe antibodies at the junction between cellular and acellular zones. All images are representative of multiple tumors assayed for each genotype. No notable differences were seen between ALK4L75A-Fc expressing tumors and controls for these characteristics. Supplemental Fig. 4. Altered signaling in ALK4L75A-Fc expressing xenografts. A diminution of phospho-AKT signaling can be discerned in ALK4L75A-Fc expressing tumors relative to mock tumors in both Hypoxyprobe positive and negative cellular regions (top row). Hypoxic regions in Mock tumors had generally diminished SMAD2/3 phosphorylation whereas ALK4L75A-Fc tumors often exhibited SMAD2/3 phosphorylation in hypoxic zones especially as these abut the acellular zones. All images are representative of three tumors assayed for each genotype. Scale bar= 50μm.
Acknowledgements
We would like to thank M. Van Brocklin for constructive comments on the manuscript, and D. Lum and K. Modzelewska for valuable technical assistance.
Authors’ contributions
Concept and writing: PCG and BTS; experimental design: OB, REH, PCG, and BTS; experimentation: OB, REH, DWF, BLG, EB, HTD, SR, KB, BMH, MF, PCG, and BTS; analysis and interpretation: OB, REH, DWF, BLG, BMH, ML, EMM, PCG, and BTS; editorial revision: OB, REH, DWF, BLG, and BTS. The authors read and approved the final manuscript.
Funding
Portions of this work were supported by Huntsman Cancer Foundation, The Clayton Foundation, the Department of Defense BCRP, Susan G. Komen, and NIH CCSG support grant(s): P30 CA42014 (HCI) and P30 CA014195 (Salk).
Availability of data and materials
All data generated or analyzed during this study are included in this published article, or available upon reasonable request from the corresponding author.
Ethics approval and consent to participate
All animal studies were performed ethically in accordance with protocols approved by IACUC oversite committees at HCI and Salk.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s13058-020-01361-z.
References
- 1.Saito Y, Li L, Coyaud E, Luna A, Sander C, Raught B, et al. LLGL2 rescues nutrient stress by promoting leucine uptake in ER(+) breast cancer. Nature. 2019;569(7755):275–279. doi: 10.1038/s41586-019-1126-2. [DOI] [PubMed] [Google Scholar]
- 2.Echeverria GV, Ge Z, Seth S, Zhang X, Jeter-Jones S, Zhou X, et al. Resistance to neoadjuvant chemotherapy in triple-negative breast cancer mediated by a reversible drug-tolerant state. Sci Transl Med. 2019;11(488):eaav0936. 10.1126/scitranslmed.aav0936. PMID: 30996079. [DOI] [PMC free article] [PubMed]
- 3.Wellenstein MD, Coffelt SB, Duits DEM, van Miltenburg MH, Slagter M, de Rink I, et al. Loss of p53 triggers WNT-dependent systemic inflammation to drive breast cancer metastasis. Nature. 2019;572(7770):538–542. doi: 10.1038/s41586-019-1450-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Janiszewska M, Tabassum DP, Castano Z, Cristea S, Yamamoto KN, Kingston NL, et al. Subclonal cooperation drives metastasis by modulating local and systemic immune microenvironments. Nat Cell Biol. 2019;21(7):879–888. doi: 10.1038/s41556-019-0346-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gray PC, Vale W. Cripto/GRP78 modulation of the TGF-beta pathway in development and oncogenesis. FEBS Lett. 2012;586(14):1836–1845. doi: 10.1016/j.febslet.2012.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klauzinska M, Castro NP, Rangel MC, Spike BT, Gray PC, Bertolette D, et al. The multifaceted role of the embryonic gene Cripto-1 in cancer, stem cells and epithelial-mesenchymal transition. Semin Cancer Biol. 2014;29:51–58. doi: 10.1016/j.semcancer.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagaoka T, Karasawa H, Castro NP, Rangel MC, Salomon DS, Bianco C. An evolving web of signaling networks regulated by Cripto-1. Growth Factors. 2012;30(1):13–21. doi: 10.3109/08977194.2011.641962. [DOI] [PubMed] [Google Scholar]
- 8.Rangel MC, Karasawa H, Castro NP, Nagaoka T, Salomon DS, Bianco C. Role of Cripto-1 during epithelial-to-mesenchymal transition in development and cancer. Am J Pathol. 2012;180(6):2188–2200. doi: 10.1016/j.ajpath.2012.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dublin E, Bobrow L, Barnes D, Gullick W. Amphiregulin and cripto overexpression in breast-cancer - relationship with prognosis and clinical and molecular-variables. Int J Oncol. 1995;7(3):617–622. doi: 10.3892/ijo.7.3.617. [DOI] [PubMed] [Google Scholar]
- 10.Gong YP, Yarrow PM, Carmalt HL, Kwun SY, Kennedy CW, Lin BP, et al. Overexpression of Cripto and its prognostic significance in breast cancer: a study with long-term survival. Eur J Surg Oncol. 2007;33(4):438–443. doi: 10.1016/j.ejso.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 11.Normanno N, Kim N, Wen D, Smith K, Harris AL, Plowman G, et al. Expression of messenger RNA for amphiregulin, heregulin, and cripto-1, three new members of the epidermal growth factor family, in human breast carcinomas. Breast Cancer Res Treat. 1995;35(3):293–297. doi: 10.1007/BF00665981. [DOI] [PubMed] [Google Scholar]
- 12.Spike BT, Kelber JA, Booker E, Kalathur M, Rodewald R, Lipianskaya J, et al. CRIPTO/GRP78 signaling maintains fetal and adult mammary stem cells ex vivo. Stem Cell Reports. 2014;2(4):427–439. doi: 10.1016/j.stemcr.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.di Bari MG, Ginsburg E, Plant J, Strizzi L, Salomon DS, Vonderhaar BK. Msx2 induces epithelial-mesenchymal transition in mouse mammary epithelial cells through upregulation of Cripto-1. J Cell Physiol. 2009;219(3):659–666. doi: 10.1002/jcp.21712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strizzi L, Bianco C, Normanno N, Seno M, Wechselberger C, Wallace-Jones B, et al. Epithelial mesenchymal transition is a characteristic of hyperplasias and tumors in mammary gland from MMTV-Cripto-1 transgenic mice. J Cell Physiol. 2004;201(2):266–276. doi: 10.1002/jcp.20062. [DOI] [PubMed] [Google Scholar]
- 15.Sun Y, Strizzi L, Raafat A, Hirota M, Bianco C, Feigenbaum L, et al. Overexpression of human Cripto-1 in transgenic mice delays mammary gland development and differentiation and induces mammary tumorigenesis. Am J Pathol. 2005;167(2):585–597. doi: 10.1016/S0002-9440(10)63000-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wechselberger C, Ebert AD, Bianco C, Khan NI, Sun Y, Wallace-Jones B, et al. Cripto-1 enhances migration and branching morphogenesis of mouse mammary epithelial cells. Exp Cell Res. 2001;266(1):95–105. doi: 10.1006/excr.2001.5195. [DOI] [PubMed] [Google Scholar]
- 17.Wechselberger C, Strizzi L, Kenney N, Hirota M, Sun Y, Ebert A, et al. Human Cripto-1 overexpression in the mouse mammary gland results in the development of hyperplasia and adenocarcinoma. Oncogene. 2005;24(25):4094–4105. doi: 10.1038/sj.onc.1208417. [DOI] [PubMed] [Google Scholar]
- 18.De Luca A, Casamassimi A, Selvam MP, Losito S, Ciardiello F, Agrawal S, et al. EGF-related peptides are involved in the proliferation and survival of MDA-MB-468 human breast carcinoma cells. Int J Cancer. 1999;80(4):589–594. doi: 10.1002/(SICI)1097-0215(19990209)80:4<589::AID-IJC17>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 19.Castro NP, Fedorova-Abrams ND, Merchant AS, Rangel MC, Nagaoka T, Karasawa H, et al. Cripto-1 as a novel therapeutic target for triple negative breast cancer. Oncotarget. 2015;6(14):11910–11929. doi: 10.18632/oncotarget.4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelly RK, Olson DL, Sun Y, Wen D, Wortham KA, Antognetti G, et al. An antibody-cytotoxic conjugate, BIIB015, is a new targeted therapy for Cripto positive tumours. Eur J Cancer. 2011;47(11):1736–1746. doi: 10.1016/j.ejca.2011.02.023. [DOI] [PubMed] [Google Scholar]
- 21.Lo RC, Leung CO, Chan KK, Ho DW, Wong CM, Lee TK, et al. Cripto-1 contributes to stemness in hepatocellular carcinoma by stabilizing Dishevelled-3 and activating Wnt/beta-catenin pathway. Cell Death Differ. 2018;25(8):1426–1441. doi: 10.1038/s41418-018-0059-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zoni E, Chen L, Karkampouna S, Granchi Z, Verhoef EI, La Manna F, et al. CRIPTO and its signaling partner GRP78 drive the metastatic phenotype in human osteotropic prostate cancer. Oncogene. 2017;36(33):4739–4749. doi: 10.1038/onc.2017.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu CH, Wang Y, Qian LH, Yu LK, Zhang XW, Wang QB. Serum Cripto-1 is a novel biomarker for non-small cell lung cancer diagnosis and prognosis. Clin Respir J. 2017;11(6):765–771. doi: 10.1111/crj.12414. [DOI] [PubMed] [Google Scholar]
- 24.Foca G, Iaccarino E, Foca A, Sanguigno L, Untiveros G, Cuevas-Nunez M, et al. Development of conformational antibodies targeting Cripto-1 with neutralizing effects in vitro. Biochimie. 2019;158:246–256. doi: 10.1016/j.biochi.2019.01.016. [DOI] [PubMed] [Google Scholar]
- 25.Castro NP, Rangel MC, Merchant AS, MacKinnon G, Cuttitta F, Salomon DS, et al. Sulforaphane suppresses the growth of triple-negative breast cancer stem-like cells in vitro and in vivo. Cancer Prev Res (Phila) 2019;12(3):147–158. doi: 10.1158/1940-6207.CAPR-18-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelber JA, Panopoulos AD, Shani G, Booker EC, Belmonte JC, Vale WW, et al. Blockade of Cripto binding to cell surface GRP78 inhibits oncogenic Cripto signaling via MAPK/PI3K and Smad2/3 pathways. Oncogene. 2009;28(24):2324–2336. doi: 10.1038/onc.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shani G, Fischer WH, Justice NJ, Kelber JA, Vale W, Gray PC. GRP78 and Cripto form a complex at the cell surface and collaborate to inhibit transforming growth factor beta signaling and enhance cell growth. Mol Cell Biol. 2008;28(2):666–677. doi: 10.1128/MCB.01716-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem J. 2011;434(2):181–188. doi: 10.1042/BJ20101569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee AS. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer. 2014;14(4):263–276. doi: 10.1038/nrc3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dong D, Ni M, Li J, Xiong S, Ye W, Virrey JJ, et al. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 2008;68(2):498–505. doi: 10.1158/0008-5472.CAN-07-2950. [DOI] [PubMed] [Google Scholar]
- 31.Fernandez PM, Tabbara SO, Jacobs LK, Manning FC, Tsangaris TN, Schwartz AM, et al. Overexpression of the glucose-regulated stress gene GRP78 in malignant but not benign human breast lesions. Breast Cancer Res Treat. 2000;59(1):15–26. doi: 10.1023/A:1006332011207. [DOI] [PubMed] [Google Scholar]
- 32.Lee WS, Sung MS, Lee EG, Yoo HG, Cheon YH, Chae HJ, et al. A pathogenic role for ER stress-induced autophagy and ER chaperone GRP78/BiP in T lymphocyte systemic lupus erythematosus. J Leukoc Biol. 2015;97(2):425–433. doi: 10.1189/jlb.6A0214-097R. [DOI] [PubMed] [Google Scholar]
- 33.Nagaoka T, Karasawa H, Turbyville T, Rangel MC, Castro NP, Gonzales M, et al. Cripto-1 enhances the canonical Wnt/beta-catenin signaling pathway by binding to LRP5 and LRP6 co-receptors. Cell Signal. 2013;25(1):178–189. doi: 10.1016/j.cellsig.2012.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Watanabe K, Nagaoka T, Lee JM, Bianco C, Gonzales M, Castro NP, et al. Enhancement of Notch receptor maturation and signaling sensitivity by Cripto-1. J Cell Biol. 2009;187(3):343–353. doi: 10.1083/jcb.200905105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kelber JA, Shani G, Booker EC, Vale WW, Gray PC. Cripto is a noncompetitive activin antagonist that forms analogous signaling complexes with activin and nodal. J Biol Chem. 2008;283(8):4490–4500. doi: 10.1074/jbc.M704960200. [DOI] [PubMed] [Google Scholar]
- 36.Tiscornia G, Singer O, Verma IM. Production and purification of lentiviral vectors. Nat Protoc. 2006;1(1):241–245. doi: 10.1038/nprot.2006.37. [DOI] [PubMed] [Google Scholar]
- 37.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quail DF, Zhang G, Walsh LA, Siegers GM, Dieters-Castator DZ, Findlay SD, et al. Embryonic morphogen nodal promotes breast cancer growth and progression. PLoS One. 2012;7(11):e48237. doi: 10.1371/journal.pone.0048237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsai YL, Zhang Y, Tseng CC, Stanciauskas R, Pinaud F, Lee AS. Characterization and mechanism of stress-induced translocation of 78-kilodalton glucose-regulated protein (GRP78) to the cell surface. J Biol Chem. 2015;290(13):8049–8064. doi: 10.1074/jbc.M114.618736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dontu G, Al-Hajj M, Abdallah WM, Clarke MF, Wicha MS. Stem cells in normal breast development and breast cancer. Cell Prolif. 2003;36(Suppl 1):59–72. doi: 10.1046/j.1365-2184.36.s.1.6.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1(5):555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao M, Liang J, Lu Y, Guo H, German P, Bai S, et al. Site-specific activation of AKT protects cells from death induced by glucose deprivation. Oncogene. 2014;33(6):745–755. doi: 10.1038/onc.2013.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ojha R, Leli NM, Onorati A, Piao S, Verginadis II, Tameire F, et al. ER translocation of the MAPK pathway drives therapy resistance in BRAF-mutant melanoma. Cancer Discov. 2019;9(3):396–415. doi: 10.1158/2159-8290.CD-18-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dauer P, Sharma NS, Gupta VK, Durden B, Hadad R, Banerjee S, et al. ER stress sensor, glucose regulatory protein 78 (GRP78) regulates redox status in pancreatic cancer thereby maintaining “stemness”. Cell Death Dis. 2019;10(2):132. doi: 10.1038/s41419-019-1408-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Conner C, Lager TW, Guldner IH, Min-Zu Wu YH, Hishida T, Ruiz S, Yamasaki AE, Gilson RC, Belmonte JCI, Gray PC, Kelber JA, Zhang S, Panopoulos AD. GRP78 promotes stemness in normal and neoplastic cells. Sci Rep. 2020;10(1):3474. 10.1038/s41598-020-60269-y. [DOI] [PMC free article] [PubMed]
- 47.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14(6):818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 48.Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14(10):611–629. doi: 10.1038/nrclinonc.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146(4):633–644. doi: 10.1016/j.cell.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 51.Spike BT. Breast cancer stem cells and the move toward high resolution stem cell systems. In: Liu H, Lathia J, editors. Cancer Stem Cells: Elsevier; 2016. p. 121–148. ISBN:978-0-12-803892-5.
- 52.Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci U S A. 2012;109(8):2784–2789. doi: 10.1073/pnas.1018866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fiorenzano A, Pascale E, D'Aniello C, Acampora D, Bassalert C, Russo F, et al. Cripto is essential to capture mouse epiblast stem cell and human embryonic stem cell pluripotency. Nat Commun. 2016;7:12589. doi: 10.1038/ncomms12589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoover M, Runa F, Booker E, Diedrich JK, Duell E, Williams B, et al. Identification of myosin II as a cripto binding protein and regulator of cripto function in stem cells and tissue regeneration. Biochem Biophys Res Commun. 2019;509(1):69–75. doi: 10.1016/j.bbrc.2018.12.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu H, Lathia JD. Cancer stem cells : targeting the roots of cancer, seeds of metastasis, and sources of therapy resistance. Available from: ScienceDirect. Restricted to UCB, UCD, UCI, UCLA, UCM, UCR, UCSC, UCSD, and UCSF http://uclibs.org/PID/284116.. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Supplemental Fig. 1. A) Schematic of a lentiviral expression vector (S14) with fluorescent luminescent markers and doxycycline-inducible ALK4L75A-Fc. (B,C) Western blot demonstrating doxycycline-dependent expression of FLAG-tagged ALK4L75A-Fc in S14-transduced MDA-MB-231 cells, B and secretion into conditioned media, C. (D) Amplification curves for TDGF1 (ie CRIPTO) from equivalent starting material in MDA-MB231 cells transduced with a Dox dependent shCRIPTO vector. Supplemental Fig. 2. Morphology of organoids seeded into secondary cultures following treatment with Dox to induce ALK4L75A-Fc expression during primary organoid out growth. Supplemental Fig. 3. A) a panoramic view of a hematoxylin/Eosin stained MDA-MB-468 tumor section by which position and morphology can be used to assign regional and structural information. P=periphery, v=presumptive vasculature, C=cellular region, S =stressed zone (see B), A = acellular zone. No gross morphological differences apart from average size were noted for ALK4L75A-Fc expressing tumors relative to mock controls. (B) High magnification images of presumptive vasculature in MDA-MB-468 xenografts containing obvious red blood cells (left panels). Lack of robust CD31 immunoreactivity in cellular regions of xenografts (second column). Cleaved caspase 3 staining in proximal acellular regions (third column). Identification of regions of stress in vivo via detection of Pimonidazole adducts with Hypoxyprobe antibodies at the junction between cellular and acellular zones. All images are representative of multiple tumors assayed for each genotype. No notable differences were seen between ALK4L75A-Fc expressing tumors and controls for these characteristics. Supplemental Fig. 4. Altered signaling in ALK4L75A-Fc expressing xenografts. A diminution of phospho-AKT signaling can be discerned in ALK4L75A-Fc expressing tumors relative to mock tumors in both Hypoxyprobe positive and negative cellular regions (top row). Hypoxic regions in Mock tumors had generally diminished SMAD2/3 phosphorylation whereas ALK4L75A-Fc tumors often exhibited SMAD2/3 phosphorylation in hypoxic zones especially as these abut the acellular zones. All images are representative of three tumors assayed for each genotype. Scale bar= 50μm.
Data Availability Statement
All data generated or analyzed during this study are included in this published article, or available upon reasonable request from the corresponding author.