

Hannigan et al. characterize the protein interactomes of four candidate ER ribosome-binding proteins, providing evidence that ER-bound ribosomes reside in distinct molecular environments. Their data link SEC62 to ER redox regulation and chaperone trafficking, and suggest a new role for LRRC59 in the regulation of mRNA translation.
Keywords: Protein synthesis, ribosomes, mass spectrometry, membranes, affinity proteomics, endoplasmic reticulum, protein translocation, quantitative proteomics, RNA binding protein
Graphical Abstract

Highlights
Identification of subcellular protein interactomes via proximity labeling and quantitative multiplexed proteomics.
SEC61β and RPN1 interactomes overlap with translocon-associated protein networks.
SEC62 interacts with redox-linked proteins and ER luminal chaperones.
LRRC59 directly interacts with mRNA translation factors and SRP machinery on the ER.
Abstract
Protein synthesis on the endoplasmic reticulum (ER) requires the dynamic coordination of numerous cellular components. Together, resident ER membrane proteins, cytoplasmic translation factors, and both integral membrane and cytosolic RNA-binding proteins operate in concert with membrane-associated ribosomes to facilitate ER-localized translation. Little is known, however, regarding the spatial organization of ER-localized translation. This question is of growing significance as it is now known that ER-bound ribosomes contribute to secretory, integral membrane, and cytosolic protein synthesis alike. To explore this question, we utilized quantitative proximity proteomics to identify neighboring protein networks for the candidate ribosome interactors SEC61β (subunit of the protein translocase), RPN1 (oligosaccharyltransferase subunit), SEC62 (translocation integral membrane protein), and LRRC59 (ribosome binding integral membrane protein). Biotin labeling time course studies of the four BioID reporters revealed distinct labeling patterns that intensified but only modestly diversified as a function of labeling time, suggesting that the ER membrane is organized into discrete protein interaction domains. Whereas SEC61β and RPN1 reporters identified translocon-associated networks, SEC62 and LRRC59 reporters revealed divergent protein interactomes. Notably, the SEC62 interactome is enriched in redox-linked proteins and ER luminal chaperones, with the latter likely representing proximity to an ER luminal chaperone reflux pathway. In contrast, the LRRC59 interactome is highly enriched in SRP pathway components, translation factors, and ER-localized RNA-binding proteins, uncovering a functional link between LRRC59 and mRNA translation regulation. Importantly, analysis of the LRRC59 interactome by native immunoprecipitation identified similar protein and functional enrichments. Moreover, [35S]-methionine incorporation assays revealed that siRNA silencing of LRRC59 expression reduced steady state translation levels on the ER by ca. 50%, and also impacted steady state translation levels in the cytosol compartment. Collectively, these data reveal a functional domain organization for the ER and identify a key role for LRRC59 in the organization and regulation of local translation.
RNA localization and accompanying local translation serve critical roles in the spatiotemporal regulation of post-transcriptional gene expression. Reflecting the importance of such regulation, localized mRNA translation requires the coordinate localization of numerous proteins, including aminoacyl-tRNA synthetases, translation factors, RNA-binding proteins (RBPs), molecular chaperones, enzymes/scaffolding proteins which act to modify the nascent polypeptide chain, as well as cis-encoded mRNA localization and trafficking information (1–14). At the endoplasmic reticulum (ER), the primary site for secretory and membrane protein synthesis, mRNA translation becomes more complex, requiring additional protein factors including proteins that facilitate ribosome association with the ER membrane, namely the protein translocation machinery, which participates in ribosome association, and newly discovered non-canonical integral membrane RNA-binding proteins (15–30). With understanding of the structural organization and regulation of ER-associated translation being largely derived from the classical canine pancreas rough microsome system, a largely unexplored question in the field concerns the cellular components and mechanisms regulating ER-localized translation in intact cells. A further level of complexity to ER-localized protein synthesis appears when considering the multiple lines of evidence supporting a transcriptome-wide role for the ER in proteome expression (17, 21, 24, 25, 28, 31–37). Notably, investigations of ER-localized mRNA composition in human cells, tissues, yeast, and fly revealed that all transcripts, not just those encoding secretory and membrane proteins, are translated on the ER (17, 21, 24, 25, 28, 31, 33, 35–39). Although landmark biochemical and structural studies have advanced our understanding of how secretory/membrane protein synthesis is coupled to protein translocation, it remains unclear how translation on the ER is compartmentalized to accommodate the coincident translation of both cytosolic and secretory/membrane protein-encoding mRNAs. One model proposes that an mRNA-wide role for the ER in proteome expression is achieved by translocon-independent modes of ribosome association with the ER membrane (25, 40–45). In this view, the SEC61 translocon serves a canonical role in secretory/membrane protein biogenesis by recruiting ribosomes engaged in the translation of this mRNA cohort, whereas other candidate ribosome interactors (e.g., p180, p34/LRRC59, SEC62) function as non-translocon ribosome binding sites. Ribosomes bound at these non-translocon sites may engage in the translation of both cytosolic and secretory/membrane protein-encoding transcripts. In the case of secretory/membrane polypeptides undergoing early elongation on non-translocon-associated ribosomes, signal sequence-bearing nascent chains might access translocons via lateral diffusion (21, 25, 31, 46, 47). A primary prediction of this model is that different ribosome interacting proteins would reside in distinct membrane protein environments, perhaps reflecting the degree to which their bound ribosomes are dedicated to secretory/membrane protein synthesis.
Using a BioID proximity-labeling approach, we recently reported that SEC61β, a translocon subunit, and the candidate ribosome-binding protein LRRC59 interact with populations of ribosomes engaged in the translation of divergent cohorts of mRNAs (34). In this communication, we extend these studies by investigating the proximal ER protein interactomes of the four previously engineered BioID reporters (SEC61β, RPN1, SEC62, and LRRC59) (34). In time course labeling studies, we observed that for each reporter, proximal interactome labeling intensified but only modestly diversified as a function of labeling time, a finding consistent with a functional domain organization of the ER. Unexpectedly, our data revealed that the previously reported ribosome receptor SEC62 interacts with functionally divergent protein networks, including those with roles in cell proliferation, signaling pathways, redox homeostasis, and cytoplasmic displaced ER luminal chaperones. In contrast, LRRC59 displays a highly SRP pathway-, translation-, and RNA-binding protein-enriched interactome. Both proximity proteomics and native immunoprecipitation studies found LRRC59 to interact almost exclusively with SRP machinery, non-canonical ER-RBPs, and translation initiation factors, suggesting a previously unappreciated role for LRRC59 in the organization and/or regulation of secretory/membrane protein synthesis on the ER. Consistent with this view, siRNA knockdown of LRRC59 expression substantially reduced protein synthesis levels in the cytosol and ER compartments.
EXPERIMENTAL PROCEDURES
Generation of BioID Chimera and Flp-InTM T-RexTM HEK293 Cell Lines
BirA-chimera constructs are described in (34). HEK293 Flp-InTM T-RExTM cell lines were generated according to the manufacturer's instructions (ThermoFisher Scientific). BirA-containing plasmids (0.4 µg), along with the pOGG4 (4 µg) plasmid were transfected into cells using 7.5 µL of Lipofectamine 3000 (ThermoFisher, L3000001). All transfections were performed in 6-well culture dishes at 80% confluency. Colonies were selected for between 48 hours and two weeks post-transfection using 100 μg/mL hygromycin (MediaTech, 30-240-CR, Manassas, VA) and 15 μg/mL blasticidin (ThermoFisher, R21001). A negative control cell line (“Empty Vector Control”) was generated by recombination of an empty vector pcDNA5-FRT/TO and antibiotic selection for an empty vector matched control.
Sequential Detergent Fractionation and Cell Lysis
Cells were washed twice with ice-cold PBS containing 50 μg/mL of cycloheximide (CHX) (VWR, 94271, Radnor, PA) for 3 minutes. To extract the cytosolic (C) fraction, cells were permeabilized for 5–10 minutes at 4°C in buffer containing 110 mM KOAc, 25 mM HEPES pH 7.2, 2.5 mM MgCl2, 0.03% digitonin (Calbiochem, 3004010), 1 mM DTT, 50 μg/mL CHX, 40 U/mL RNAseOUT (Invitrogen, 10777-019, Carlsbad, CA), and protease inhibitor complex (PIC) (Sigma Aldrich, P8340). Supernatants were collected as the cytosolic fraction, and cells were then rinsed with wash buffer (110 mM KOAc, 25 mM HEPES pH 7.2, 2.5 mM Mg(OAc)2, 0.004% to 0.008% digitonin, 1 mM DTT, 50 μg/mL CHX, 40 U/mL RNAseOUT, and PIC). To extract the membrane (M) fraction, the washed cells were then lysed either for 5 minutes at 4°C in lysis buffer 1 (400 mM KOAc, 25 mM HEPES pH 7.2, 15 mM MgCl2, 1% NP-40, 0.5% DOC, 1 mM DTT, 50 μg/mL CHX, 40 U/mL RNAseOUT, and PIC) or for 15 minutes at 4°C in lysis buffer 2 (200 mM KCl, 25 mM HEPES pH 7.2, 15 mM MgCl2, 1 mM DTT, 2% dodecylmaltoside (DDM), 50 µg/mL CHX, 40 U/ml RNaseOUT, and 1X protease inhibitor). Subcellular fractions were cleared by centrifugation (15,300 × g for 10 minutes). Total cell lysis was performed by incubating cells at 4°C for 10 minutes in membrane lysis buffer 1, followed by centrifugation at 15,300 × g for 10 minutes.
BirA Labeling of Microsomes
Canine pancreas rough microsomes (RM) (48) were adjusted to a concentration of 4 mg/mL in 500 μL of BirA reaction buffer (20 mM Tris pH 8, 5 mM CaCl2, 100 mM KCl2, 10 mM MgCl2, 3 mM ATP, 1.5 mM biotin, 5 mM phosphocreatine (Sigma-Aldrich, P7936), and 5 μg/mL of creatine kinase (Sigma-Aldrich, C3755)). Purified recombinant BirA*-GST fusion protein was added at a concentration of 10 μg/mL. Following 0, 1, 3, 6, and 18 hours, 100 μL of reaction was removed, flash frozen in a dry ice/ethanol bath, and stored at –80°C for subsequent analysis.
Immunoblotting
Protein lysate concentrations were determined using a Pierce BCA Protein Assay Kit (ThermoFisher, 23225). Proteins were resolved by SDS-PAGE using either 10% acrylamide gels or 12% acrylamide gels containing 0.5% trichloroethanol. Gels were UV irradiated for 5 minutes and imaged using an Amersham Imager 600 (GE Life Sciences). Gels or membranes were then equilibrated in Tris-glycine transfer buffer for 5-10 minutes and transferred onto nitrocellulose membranes. Membranes were blocked in 3% BSA and probed for BirA (Abcam, ab14002), streptavidin-RD680 (Li-Cor, P/N 925-68079; 1:20,000), TRAPα (49), GRP94 (50), or LRRC59 (Bethyl Laboratories, A305-076A). Membranes were incubated with isotype-matched secondary antibodies (Li-Cor, Lincoln, NE; 1:10,000), and imaged by infrared fluorescence detection using the Odyssey Clx (Li-Cor), where signal intensities were quantified by densitometry analyses. To examine total protein levels, immunoblots were stained with either India Ink or Ponceau S solution (Sigma-Aldrich).
Protease Protection Assay
The SEC62-BirA construct was expressed overnight as reported in (34). Cultures were then placed on ice, permeabilized in digitonin-supplemented cytosolic buffer (as described above), rinsed, and incubated with cytosolic buffer containing 0, 25, or 50 µg/mL of Proteinase K (Bioline) for 30 minutes at 4°C. Protease digestions were quenched by addition of 0.5 mM PMSF. Cell extracts were prepared and immunoblots performed as above.
siRNA Transfection
HEK293T cells were transfected at 60-80% confluency with either a non-targeting siRNA negative control (siCtl, ThermoFisher, 4390844) or siLRRC59 (ThermoFisher, s30851) using Lipofectamine RNAiMAX Transfection Reagent (ThermoFisher, 13778-150). At 72 hours post-transfection, cells were subjected to downstream analyses.
[35S]-Methionine Incorporation
Cells were washed twice in 1X PBS, then starved in methionine-free media for 15 minutes at 37°C. For experimental conditions, cells were incubated with labeling media (methionine-free DMEM, 50 µCi/ml [35S]-methionine) for 7.5 minutes at 37°C. Cell cultures were then washed twice with serum-free media containing 100 µg/mL CHX to inhibit translation. Cell cultures were subsequently washed twice with PBS containing 100 µg/mL CHX before proceeding to cell fractionation. For control conditions, cells were incubated in media containing 100 µg/mL CHX for 10 minutes at 37°C after initial methionine starvation. Cells were then incubated in labeling media (methionine-free DMEM, 50 µCi/mL [35S]-methionine, 100 µg/mL CHX) for 7.5 minutes at 37°C, followed by two washes in serum-free media containing 100 µg/mL CHX. As above, cells were washed two additional times in 1X PBS containing 100 µg/mL CHX before proceeding to cell fractionation. Experimental and control cells were subsequently lysed using sequential detergent-based fractionation methods (described above). Proteins from the cytosol and membrane lysates were precipitated in 10% TCA for 30 minutes on ice. Following centrifugation (14,000 rpm) for 10 minutes at 4°C, protein pellets were washed four times in ice cold 10% TCA, for 5 minutes each. Pellets were then washed two times in ice cold 100% acetone, dried at 95°C for 5 minutes, and resuspended in 10 µl of buffer containing 5% SDS and 0.5 M Tris, pH 10.5. Samples were then diluted 1:10 with water and assayed for radioactivity (CPM) using liquid scintillation counting.
TMT/Isobaric Tag Mass Spectrometry
Experimental Design and Statistical Rationale
Three biological replicates from each reporter cell line were divided evenly among two TMT 11-plex reporter sets. The biological replicates were split such that two replicates of a condition were on one 11-plex set and one replicate was on the other to avoid bias between the sets. Additionally, a Study Pool QC (SPQC) sample was created using equal amounts of each of the 15 samples, and this SPQC was analyzed with n = 3 on each 11-plex set. One channel was left empty as a negative control. Additional processing details are included in the relevant sections.
Sample Preparation and Proteolytic Digestion
Biological replicates from each reporter cell line were affinity isolated on streptavidin magnetic beads, eluted in 120 µL of biotin elution buffer (2% SDS, 20 mM biotin, 2 M thiourea, 0.5 M Tris unbuffered), and prepared according to the standard S-Trap digestion protocol (Protifi, Inc.; (51)). Briefly, each sample was loaded onto its respective S-Trap column, and washed four times with S-trap binding buffer (90% MeOH, 100 mM TEAB), and digested by adding 0.8 µg of sequencing grade trypsin to the top of each S-trap tip with incubation for one hour at 47°C. The peptides were eluted from the S-trap tip first with 50 mM TEAB, then with 0.2% aqueous formic acid, and finally with 50% acetonitrile in 0.2% aqueous formic acid. The peptide elutions were combined and dried via SpeedVac. Peptide yield from each sample was determined to be approximately 20 µg based on BCA Protein Assay (ThermoFisher Scientific).
TMT Labeling
Dried samples, determined to contain approximately 22 µg of digested peptide each, were brought to room temperature and resuspended in 70 µL 200 mM TEAB. An aliquot (20 µL) from each of the 15 samples was combined to make a Study Pool QC (SPQC). TMT reagents (TMT10Plex plus TMT11-131C, Product A37725) were dissolved in 45 µL acetonitrile for 5 minutes with vortexing. Labeling reagent (20 µl) was added to each sample for 2 hours at room temperature. Sample labeling was then quenched with 4 µL of 5% v/v hydroxylamine in 200 mM TEAB for 15 minutes. The TMT samples for each set were combined into a 1.5 mL Eppendorf tube, acidified to 1.0% formic acid, frozen, and lyophilized to dryness overnight.
Pre-fractionation
Each TMT labeled peptide set was fractionated to improve depth of proteome coverage using a Pierce High pH Reversed-Phase Peptide Fractionation Kit (ThermoFisher Scientific, Part 84868). The fractionation was performed according to the manufacturer's protocol and yielded 8 peptide fractions for analysis. Water/acetonitrile mixtures with 0.1% v/v triethylamine (TEA), pH 10, were used for reversed-phase fractionation. 5% v/v wash was used to remove excess TMT reagent, then fractions were collected at 10, 12.5, 15, 17.5, 20, 22.25, 25, and 50% v/v MeCN. These fractions were independently acidified to 1% formic acid and dried via SpeedVac. Samples were subsequently resuspended in 22 µL 1/2/97 v/v/v TFA/MeCN/water.
Liquid Chromatography – Tandem Mass Spectrometry
Approximately 1 µg of TMT-labeled peptide from each fraction was analyzed by nanoscale liquid chromatography – tandem mass spectrometry (LC-MS/MS) on a nanoAquity UPLC (Waters) coupled to an Orbitrap Fusion Lumos Tribrid mass spectrometer (ThermoFisher Scientific). Peptides were first trapped on a column at 99.9% water and 5 µL/min, followed by separation at 0.4 µL/min on an analytical column (Waters Corporation) with a gradient from 3 to 30% MeCN (0.1% formic acid) over 90 minutes. Column eluent was introduced to the MS via electrospray ionization (+2.1kV) and a source temperature of 275°C. Upon easy-IC internal mass calibration, tandem MS sequencing and quantification was performed using a full-scan spectrum at 120k resolution, followed by MS2 sequencing at 50k resolution with HCD fragmentation at 38 V. MS/MS was performed with an isolation width of 0.7 Da, a cycle time of 1 second until the next full scan spectrum, and 60 seconds dynamic exclusion. Raw data and *.mgf peaklist files for this study have been uploaded to the MASSive data repository and are available at: ftp://massive.ucsd.edu/MSV000085009/.
TMT-labeled MS Data Processing
Raw MS data was converted to *.mgf format using Proteome Discoverer v2.1 (ThermoFisher Scientific) and submitted to Mascot v2.5 (Matrix Sciences, Inc.) for database searching. Peptide matching included 5 ppm precursor and 0.02 Da product ion tolerance, fixed carbamidomethyl (C), along with variable modifications TMT10 (N-term, K) and deamidation (N, Q), and full trypsin specificity with one missed cleavage. Searches were performed against the curated human proteome (www.uniprot.org, downloaded Sept 2016, 20206 entries), plus common contaminant sequences such as ALBU_BOVIN, ADH1_YEAST, ENO1_YEAST, and BIRA_ECOLI. A reverse-sequence decoy database was appended for False Discovery Rate (FDR) determination. Scaffold Q+ v4.8.5 (Proteome Software, Inc.) was used to quantify TMT-label based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 50.0% probability by the Scaffold Local FDR algorithm, whereas protein identifications were accepted if they could be established at greater than 99.9% probability and contained at least 1 identified peptide. Protein probabilities were assigned by the Protein Prophet algorithm (52). TMT reporter ion channels were corrected based on isotopic purity in all samples according to the algorithm described in i-Tracker (53). Normalization was performed iteratively (across samples and spectra) on intensities, as described in (54). Spectra data were log-transformed, pruned of those matched to multiple proteins and those missing a reference value, and weighted by an adaptive intensity weighting algorithm. Relative protein abundance across the experiment was expressed as the log2 ratio to the reference (SPQC) channel average for all samples (supplemental File S1). Percent missing values were calculated at the protein level for the SPQC channels, as well as all channels. A p-value using a Student's t-test was then calculated comparing each biological group (n = 3) versus the SPQC (n = 6).
Identification of Interaction Networks
A combination of statistical prioritization, 2D clustering, and principal components analysis (PCA) was used to identify putative interaction networks from the dataset. The data were curated such that proteins only quantified in one TMT set, or missing in more than 4 of the total channels were excluded from consideration (86 of 1263 proteins). A p-value was then calculated using a Student's t-test between each BirA-fusion sample group (n = 3) and the SPQC group (n = 6) to determine whether a protein was statistically different in each biological group (BirA reporter) from the average of all groups (SPQC). Proteins that did not pass a Bonferroni-corrected p-value < 0.1 (raw p-value < 1e-4) were removed, yielding 353 proteins as putative interactors (supplemental File S2). Finally, putative interaction networks were identified using unbiased 2D hierarchical clustering (Robust, Ward's Method) in JMP 14.0 (SAS Institute, Cary NC). The clustering analysis only included the BirA interactome samples (not the SPQC samples) to reduce the potential for cluster mis-assignment.
Label-free Proteomic Analysis of BioID Proteomes
Sample Preparation
For single BioID reporter studies, reporter cell culturing, reporter expression, cell fractionation, detergent lysis, and affinity isolation of biotinylated proteins was performed as above. Samples were subjected to one dimensional SDS-PAGE. 25 µL of sample was combined with 5 µL of 100 mM DTT and 10 µL of NuPAGETM (ThermoFisher Scientific) 4× loading buffer, and samples were then heated to 70°C for ten minutes with shaking. SDS-PAGE separation was performed using 1.5 mm 4–12% Bis-Tris pre-cast polyacrylamide gels (Novex, ThermoFisher Scientific) and 1× MES SDS NuPAGETM Running Buffer (ThermoFisher Scientific), including NuPAGETM antioxidant. SDS-PAGE separation was performed at a constant 200V for five minutes, gels fixed for 10 minutes, stained for 3 hours, and destained overnight following manufacturer instructions.
Gel Band Isolation and Trypsin Digestion
Gel bands of interest were isolated using a sterile scalpel, transferred to protein LoBind tubes (Eppendorf), and minced. Gel pieces were washed with 500 µL of 40% LCMS grade acetonitrile (MeCN, ThermoFisher Scientific) in AmBic, with shaking at 30°C. Gel pieces were shrunk with LCMS grade MeCN, the solution discarded, and the gel pieces dried at 50°C for 3 min. Reduction of disulfides was performed using 100 µL of 10 mM DTT at 80°C for 30 min with shaking, followed by alkylation with 100 µL of 55 mM IAM at room temperature for 20 min. This liquid was aspirated from the samples and gel pieces were washed twice with 500 µL AmBic. LCMS grade MeCN was added to shrink the gel pieces in each sample, then samples were swelled in AmBic, and this process was repeated. The gel pieces were shrunk a final time by adding 200 µL of LCMS grade MeCN, and heating for 3 min at 50°C to promote evaporation. Trypsin digestion was performed by addition of 30 µL of 10 ng/µL sequencing grade trypsin (Promega, Madison, WI) in AmBic followed by 30 µL of additional AmBic. The samples were incubated overnight at 37°C with shaking at 750 rpm. Following overnight digestion, 60 µL of 1/2/97 v/v/v TFA/MeCN/water was added to each sample and incubated for 30 min at room temperature and 750 rpm to extract peptides, and the combined supernatant was transferred to an autosampler vial (Waters). Gel pieces were shrunk in 50 µL additional MeCN for 10 min to extract the maximum number of peptides, and combined with the previous supernatant. The samples were dried in the Vacufuge (Eppendorf) and stored at −80°C.
Qualitative Analysis of Gel Electrophoresis Samples
All gel band samples were resuspended in 20 µL of 1/2/97 v/v/v TFA/MeCN/water and analyzed by nanoLC-MS/MS using a Waters nanoAcquity LC interfaced to a Thermo Q-Exactive Plus via a nanoelectrospray ionization source. One microliter of each gel band sample was injected for analysis. Each sample was first trapped on a Symmetry C18, 300 μm × 180 mm trapping column (5 µL/min at 99.9/0.1 v/v H2O/MeCN for 5 minutes), after which the analytical separation was performed using a 1.7 μm ACQUITY HSS T3 C18 75 μm × 250 mm column (Waters). The peptides were eluted using a gradient of 5–40% MeCN with 0.1% formic acid at a flow rate of 400 nL/min with a column temperature of 55°C for 90 minutes. Data collection on the Q Exactive Plus mass spectrometer was performed with data dependent acquisition (DDA) MS/MS, using a 70,000 resolution precursor ion (MS1) scan followed by MS/MS (MS2) of the top 10 most abundant ions at 17,500 resolution. MS1 was performed using an automatic gain control target of 1e6 ions and maximum ion injection (max IT) time of 60 ms. MS2 used AGC target of 5e4 ions, 60 ms max IT time, 2.0 m/z isolation window, 27 V normalized collision energy, and 20 s dynamic exclusion.
Single Reporter MS Data Processing
Database searching was performed as described by TMT-labeled MS data processing. For single reporters, data was searched using trypsin enzyme cleavage rules and a maximum of 4 missed cleavages, fixed modification carbamidomethylated cysteine, variable modifications biotinylated lysine, deamidated asparagine and glutamic acid, and oxidized methionine. The peptide mass tolerance was set to +/− 5 ppm and the fragment mass tolerance was set to +/− 0.02 Da. False discovery rate control for peptide and protein identifications was performed using Scaffold v4 (Proteome Software, Inc.) (supplemental File S3).
Native LRRC59 Immunoprecipitation and Mass Spectrometry
Sample Preparation
Caco-2 cells were cultured according to ATCC recommendations and processed at ca. 90% confluence. Cell extracts were prepared by addition of 0.5 mL per 15 cm plate of NP-40 lysis buffer (1% NP-40, 100 mM KOAc, 50 mM HEPES pH 7.2, 2 mM Mg(OAc)2, PIC, 1 mM DTT). Lysates were maintained on ice for 20 minutes and cleared by centrifugation (10,000 × g, 10 minutes). The supernatant fractions were diluted 1:1 in dilution buffer (50 mM HEPES, 100 mM KOAc, 2 mM Mg(OAc)2, PIC, 1 mM DTT) and supplemented with 5 µg/mL of LRRC59 antibody (A305-076A, Bethyl Labs, Montgomery TX) or rabbit IgG (Sigma-Aldrich, St. Louis, MO). Samples were incubated with end-over-end rotation overnight at 4°C. Dynabead Protein G beads (ThermoFisher, Waltham MA) were added to a concentration of 30 µL/mL and rotated for 2 hours at 4°C. Beads were washed 3× in buffer 1 (0.1% NP-40, 100 mM KOAc, 50 mM HEPES pH 7.2, 2 mM Mg(OAc)2, PIC, 1 mM DTT), 1× in buffer 2 (0.1% NP-40, 500 mM KOAc, 50 mM HEPES pH 7.2, 2 mM Mg(OAc)2, PIC, 1 mM DTT), and 1× in PBS. Proteins were eluted in an equi-bead volume of 2× Laemmli buffer by heating at 70°C for 20 minutes and submitted for mass spectrometry analysis.
LRRC59-IP Data Analysis
Raw MS data (.sf3 files) were processed using Scaffold 4 Proteome Software (Proteome Software, Inc.) to obtain total spectral counts for each sample. Protein interactors of LRRC59 and IgG (control) were identified using CompPASS (55), an unbiased, comparative proteomics analysis suite. Any prey in each IP with a D-score greater than or equal to one was a high-confidence interacting protein (supplemental File S4).
Bioinformatic Analyses
Gene Ontology
GO analyses were performed using the Cytoscape application, BiNGO (56), with a Benjamini and Hochberg FDR correction (significance level of 0.05) to enrich for terms after multiple testing correction. A custom set of genes expressed in our multiplexed BioID experiment was used as background for examination of SEC62-BirA and LRRC59-BirA interactors, whereas the entire human annotation (provided within the application) was used as a reference background for LRRC59 interactors determined by native IP. Additional functional information (as depicted by the heatmaps/matrices and protein color-coding) was extracted by batch querying each set of protein interactors against the MGI (57–59) and STRING (60, 61) databases.
Protein-Protein Interaction Networks
Protein-protein interaction analyses of SEC62-BirA (n = 50) and LRRC59-BirA (n = 25) interactors were performed using the STRING database (60, 61). Only experimentally determined interactions and those reported from a curated database were considered.
Identification of Membrane Proteins
The list of SEC62-BirA interactors (n = 50) was intersected with a membrane protein annotation file downloaded from the MembranOME database (62, 63). Of the 50 SEC62-interacting proteins, 21 (42%) were identified as membrane proteins. Membrane protein classification was validated by manually searching each of the 50 proteins against the Human Protein Atlas (64, 65).
RESULTS
Evidence for Domain Organization of ER Membrane Protein Interactomes
In a recent study, we examined the spatial organization of mRNA translation on the endoplasmic reticulum via proximity proteomics, where BioID reporters of translocon-associated (SEC61β, RPN1) and candidate (SEC62, LRRC59) ribosome interacting proteins were used to biotin label proximal ribosomes in vivo. Together with RNA-seq analysis of mRNAs isolated from the biotin-tagged ribosome populations (34), these studies revealed that translation on the ER membrane is heterogeneous and that ER-bound ribosomes display local environment-specific enrichments in their associated mRNAs. The mechanism(s) responsible for this regional organization of translation, however, is unknown. Here, we used proximity proteomics and the previously utilized BioID reporters to test the hypothesis that ribosome-binding proteins reside in distinct protein networks or functional domains, as a potential mechanism to support higher order organization of mRNA translation on the ER.
In the experiments presented below, BioID reporters of known or proposed ribosome interacting proteins were used to map proximal ER membrane protein interactomes at previously identified mRNA translation sites (Fig. 1A) (34). BioID proximity labeling experiments are typically conducted over many hours (66–68) (e.g. 16–24 hours) because of the slow release kinetics of the reactive biotin-AMP catalytic intermediate from the BirA* active site (69). In context of this study, we considered that extended labeling times coupled with reporter diffusion in the ER membrane would confound identification of proximal-interacting vs. random-interacting proteins. In line with this consideration, we expected that for each reporter, the composition of biotin-tagged proteins would diversify as a function of labeling time (70). Although it has been previously demonstrated that neighboring interactomes can be distinguished from random interactors by their higher relative labeling over non-specific controls, we examined the biotin labeling patterns of BioID reporters as a function of labeling time (66, 70, 71). The results of these experiments are shown in Fig. 1B. Depicted are streptavidin blots of the cytosol (C) and membrane (M) protein fractions from the four BioID reporter cell lines, sampled over a labeling time course of 0–6 hours. Two observations are highlighted here. One, although the BirA domains are cytosolically disposed and would be expected to label both cytosolic and membrane proteins, biotin-tagging is strongly enriched for membrane proteins. Two, the major membrane protein biotin labeling patterns intensified but did not substantially diversify over the labeling time course (Fig. 1B). Densitometric analysis of the biotin labeling patterns revealed by SDS-PAGE are depicted in Fig. 1B, right panels, where it can be appreciated that the biotin labeling patterns were relatively constant over labeling time. These data suggest that the BioID interactomes of the tested reporters include largely stable membrane protein assemblies, rather than the randomizing interactomes expected of diffusion-based interactions (72–75). The data presented above (Fig. 1B) are consistent with a model where the local environments of the BioID reporters are constrained. Such spatial restriction may reflect organization of the ER via functional interactome networks, like the well documented observations regarding domain organization of the plasma membrane (73–75). We also considered that the distinctive labeling patterns of the different reporters could be influenced by ER dynamics and/or distribution biases of the reporters (e.g. tubules vs. lamellar regions of the ER). To examine these scenarios, we performed BirA* labeling time course experiments in vitro, by adding soluble, recombinant BirA*-GST fusion protein to canine pancreas rough microsomes (RM), which lack the native topology and dynamics of the ER (Fig. 1C). Using this experimental system, the reactive biotin-AMP intermediate was delivered in trans and accessible to the microsome surface by diffusion. The results of these experiments demonstrate that when accessible to RM proteins in trans, biotin labeling is pervasive, with RM proteins being broadly labeled and labeling intensities increasing as a function of labeling time (Fig. 1C, upper panel). A protein loading control for this experiment is depicted in Fig. 1C, lower panel. Combined, the distinct and temporally stable proximity labeling patterns identified for each BioID reporter cell line suggest that the BirA-chimeras reside in distinct protein interactome domains of the ER.
Fig. 1.

Identification of ER membrane protein interactomes by proximity proteomics. A, Schematic of known (SEC61 translocon, OST complex), and candidate (SEC62, LRRC59) ER-ribosome receptors. SEC61β (purple), a subunit of the SEC61 translocon, RPN1 (green), a subunit of the OST complex, SEC62 (orange), and LRRC59 (blue) are expressed as BioID chimeras, labeling interacting and near-neighbor proteins (indicated by starred ribosomes and proteins W, X, Y, and Z). B, Left panel: Streptavidin blots reporting on the subcellular distribution of biotin-labeled proteins from HEK293 cells following overnight induction of either the LRRC59-, SEC62-, SEC61β-, or RPN1-BirA reporter construct. Biotin labeling was performed over a time course spanning 0–6 hours and cytosol (C) and membrane (M) extracts prepared by detergent fractionation. Right panel: Densitometric quantifications of biotin labeling intensities for cytosolic and membrane fractions. C, Canine pancreas rough microsomes with (+BirA) or without (−BirA) the addition of recombinant, soluble BirA* in trans. Biotin labeling of proteins was conducted over 0–18 hours (top, left). Biotin labeling intensities were quantified using densitometric analyses (top, right). As a loading control, total protein lysate was analyzed by India ink staining (bottom, left) and quantified by densitometric analysis (bottom, right).
Investigation of Local Interactomes via TMT Quantitative Mass Spectrometry
To enable quantitative measurements of the protein interactomes depicted in Fig. 1, an isobaric-tagging mass spectrometry analytical approach was used (TMT, tandem mass tagging) (Fig. 2A). Isobaric labeling methods provide multiplexing and assist in the quantitative analysis of biological replicates. Two oligomeric protein complexes known to reside near sites of translation on the ER, the SEC61 translocon and the oligosaccharyltransferase (OST) complex, were used as spatial reference points, where labeling of their associated subunits serves as a test for proximity (Fig. 1A, 2A). Specifically, for the SEC61 translocon, a BioID reporter of its subunit, SEC61β, was used to map the interactome of this well-studied complex (15, 76–79). Similarly, ribophorin I (RPN1), a subunit of the OST complex that is transiently recruited to the SEC61 translocon during nascent glycoprotein translocation, served as an additional proximity labeling control for the local environments of ER translation sites (Fig. 1A, 2A) (41, 80–82). To expand our analysis to less studied ER environments, we examined LRRC59 as it was previously reported to reside proximal to ER-bound ribosomes, in vivo (34) and to function in ribosome binding, in vitro (45, 83) (Fig. 1A). We also investigated a second candidate ribosome-binding protein, SEC62 (34,66,67). Although LRRC59 and SEC62 have been shown to interact with ribosomes, their native protein interactomes are largely unstudied.
Fig. 2.
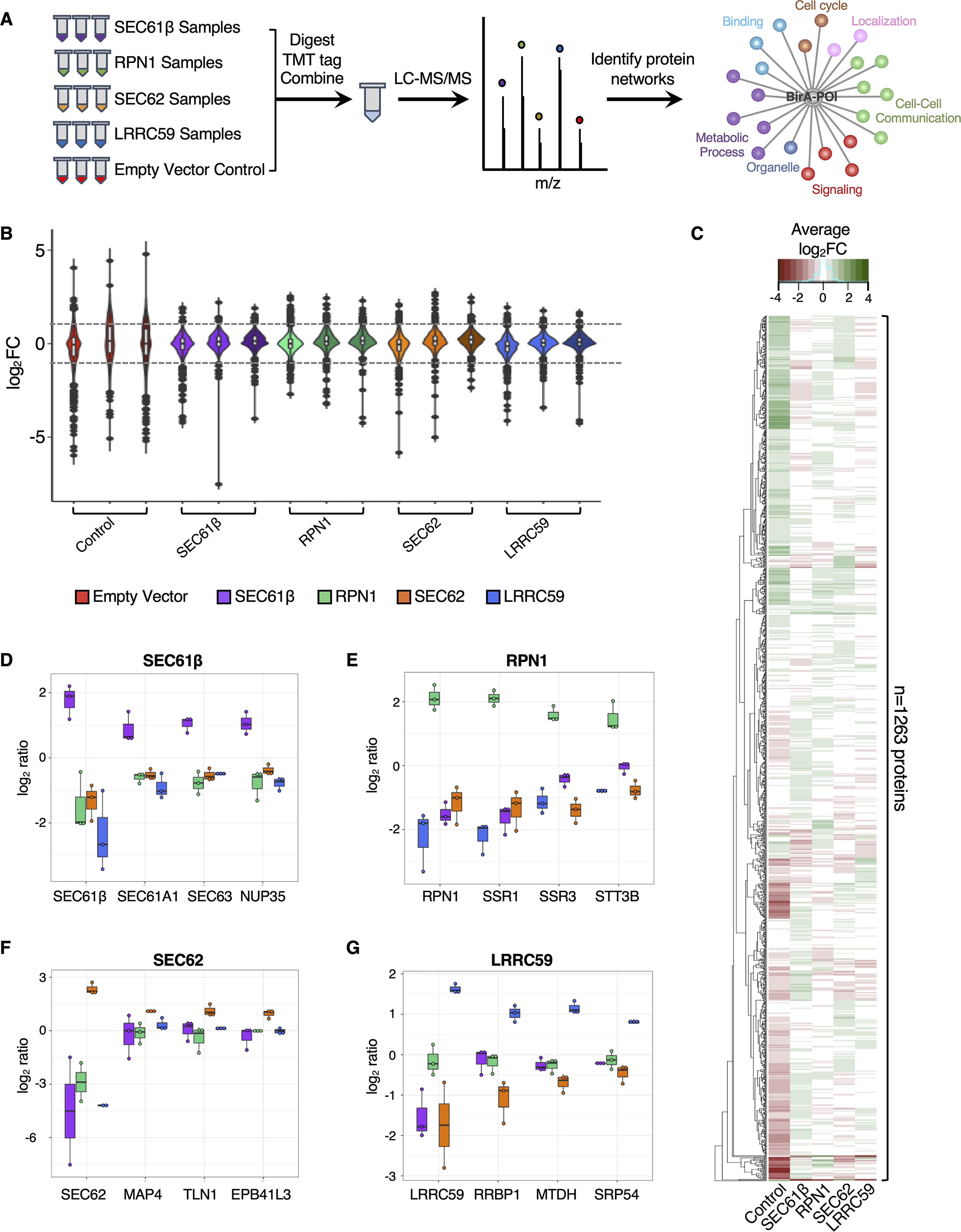
Proximity proteomics reveals unique interactomes for each of the four tested baits. A, Schematic of the experimental approach. BirA-reporters for known (SEC61β (purple), RPN1 (green)) and candidate (SEC62 (orange), LRRC59 (blue)) ER-resident ribosome receptors were expressed with biotin labeling (3 hours) conducted in biological triplicate. An empty vector negative control (red) was included. Samples were digested, tandem mass tag (TMT) labeled, and combined for liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. Enrichment analyses of biotin-labeled proteins identify protein-protein interactions and/or functional networks for each of the five baits. B, Violin plots of the protein abundance distributions for all biotin-labeled proteins (n = 1263) for each bait. C, Clustered heatmap showing the average log2FC (across biological replicates) for each of 1263 identified proteins per bait (green represents enriched protein abundance; red indicates decreased protein abundance). Boxplots depicting fold-enrichment values for high confidence prey proteins identified in the (D) SEC61β, (E) RPN1, (F) SEC62, and (G) LRRC59 BioID reporter studies. Each dot represents the log2 FC value per biological replicate.
We established inducible Flp-InTM T-RexTM HEK293 cell lines for each of the BioID reporters and included an empty vector negative control for background characterization. By the rationale detailed above, cell lines were biotin-labeled for three hours to allow for significant labeling of intracellular membrane proteins (Fig. 1B). Biotin-tagged protein fractions were then affinity isolated from cell extracts, digested with trypsin, derivatized with isobaric mass tag reagents, combined, and analyzed by LC-MS/MS (Fig. 2A). To accomplish the analysis of three biological replicates of each of the five reporters, in addition to six study pool QC replicates, two TMT 10-plex reagent sets were utilized. Biological groups were divided between the TMT sets to avoid between-set bias, and the SPQC replicates were used to normalize between TMT sets.
Identification of ER Membrane Protein Interactomes
To identify the protein interactomes for each of the different BioID reporters, we analyzed the raw mass spectrometry data generated from the proximity proteomics experiments described above. Quantification and identification of TMT-labeled peptides were performed with Protein Discoverer 2.3 and Scaffold Q+ software. TMT signals were normalized to the total intensity within each channel, peptides derived from each protein summed to represent the protein abundance, and relative protein abundance was calculated as a log2 fold change (FC) relative to the mean of the SPQC reference channels, which represents the “biological average” of all samples in the experiment. In total, 1,263 proteins were identified across the entire sample set, with most proteins showing modest to no reporter-specific enrichment (Fig. 2B, supplemental File S1). Violin plots in Fig. 2B highlight the technical reproducibility of the approach; and for each reporter construct, a small subset of proteins appear to be specifically enriched for biotin labeling (log2FC > 1, dashed line). Despite SEC61β, RPN1, SEC62 and LRRC59 sharing similar overall log2FC distribution patterns (Fig. 2B), examination of the magnitude of biotin labeling at the protein level revealed that each reporter is associated with a unique set of prominent near-neighbor interactors, as summarized in the heatmap profile (Fig. 2C), and individual reporter representations (Fig. 2D–2G). As an example, the SEC61β reporter labeled other members of the SEC61 translocon, as well as a nuclear pore complex protein (Fig. 2D); the RPN1 reporter labeled subunits of the OST complex and other glycoproteins (Fig. 2E); the SEC62 interactome includes an array of proteins involved in redox regulation, cytoskeleton architecture, and the cell cycle (Fig. 2F); and the LRRC59 interactome included ribosome-binding proteins, RNA-binding proteins, and SRP pathway components (Fig. 2G). Importantly, all the bait proteins significantly labeled themselves, providing a quantitative index of relative proximity (Fig. 2D–2G). Because identification and quantification are not decoupled in isobaric tagging experiments (e.g. the identification and quantification come from the same spectrum, which is a mixture of all samples), we also performed BirA-reporter proteomic studies using label-free shotgun proteomics (not multiplexed). Although this approach did not have the proteome coverage of the TMT-tagging approach, we were able to independently verify the high-confidence interactors for each reporter. Specifically, we identified SEC61 subunits, members of the OST complex, factors related to redox homeostasis and the cytoskeleton, and an enrichment of SRP machinery, translation factors and RBPs in the SEC61β, RPN1, SEC62, and LRRC59 interactomes, respectively, using this approach (Table I, supplemental File S3). Combined, these data indicate that ER proteins can reside in discrete protein interactomes, which is consistent with a model where cohorts of functionally-related or interacting proteins comprise stable membrane domain interactomes, as previously reported for other membrane systems (84–87).
Table I. Classification of BioID-chimeras and their associated interactomes using a label-free shotgun proteomics approach. Subset of enriched, high-confidence interactors of BioID-SEC61β, -RPN1, -SEC62, or -LRRC59, as determined by single reporter, label-free shotgun mass spectrometry analyses.
| UniProt_ID | Gene ID | Molecular weight | Localization | Type | Normalized count data | Bait | GO Annotation |
|---|---|---|---|---|---|---|---|
| MSPD2_HUMAN | MOSPD2 | 60 kDa | Endoplasmic reticulum | Membrane | 2.38 | Sec61B | chemotaxis, integral component of membrane |
| TMX3_HUMAN | TMX3 | 52 kDa | Endoplasmic reticulum | Membrane | 1.19 | Sec61B | cell redox homeostasis, isomerase activity, endoplasmic reticulum |
| PMYT1_HUMAN | PKMYT1 | 55 kDa | Cytoplasm | Soluble | 1.92 | Sec61B | endoplasmic reticulum, kinase activity |
| S61A1_HUMAN | SEC61A1 | 52 kDa | Endoplasmic reticulum | Membrane | 0.95 | Sec61B | SRP-dependent cotranslational protein targeting to membrane, translocation, endoplasmic reticulum, ribosome binding |
| ITPR3_HUMAN | ITPR3 | 304 kDa | Endoplasmic reticulum | Membrane | 5.91 | Sec61B | endoplasmic reticulum, ion transport, positive regulation of cytosolic calcium ion concentration |
| GCP60_HUMAN | ACBD3 | 61 kDa | Cytoplasm | Soluble | 1.42 | Sec61B | Golgi apparatus, lipid metabolic process |
| NU153_HUMAN | NUP153 | 154 kDa | Nucleus | Membrane | 8.91 | Sec61B | protein binding, negative regulation of RNA export from nucleus, Ran GTPase binding |
| SC61B_HUMAN | SEC61B | 10 kDa | Endoplasmic reticulum | Membrane | 6.30 | Sec61B | SRP-dependent cotranslational protein targeting to membrane, translocation, endoplasmic reticulum, ribosome binding |
| SSRA_HUMAN | SSR1 | 32 kDa | Endoplasmic reticulum | Membrane | 3.08 | RPN1 | endoplasmic reticulum |
| STT3B_HUMAN | STT3B | 94 kDa | Endoplasmic reticulum | Membrane | 10.16 | RPN1 | co-translational protein modification, oligosaccharyl transferase activity, response to unfolded protein |
| RPN1_HUMAN | RPN1 | 69 kDa | Endoplasmic reticulum | Membrane | 101.37 | RPN1 | proteosome complex, protein glycosylation, endoplasmic reticulum |
| FKBP8_HUMAN | FKBP8 | 45 kDa | Mitochondrion | Membrane | 7.81 | RPN1 | isomerase activity, apoptosis, signaling pathway |
| DJC16_HUMAN | DNAJC16 | 91 kDa | Endoplasmic reticulum | Membrane | 7.72 | RPN1 | cell redox homeostasis |
| SEC62_HUMAN | SEC62 | 46 kDa | Endoplasmic reticulum | Membrane | 36.53 | Sec62 | posttranslational protein targeting to membrane, translocation, endoplasmic reticulum |
| PRDX4_HUMAN | PRDX4 | 31 kDa | Endoplasmic reticulum | Soluble | 5.03 | Sec62 | oxidoreductase activity, protein folding |
| PDIA3_HUMAN | PDIA3 | 57 kDa | Endoplasmic reticulum | Soluble | 4.93 | Sec62 | cell redox homeostasis, isomerase activity, endoplasmic reticulum |
| COR1B_HUMAN | CORO1B | 54 kDa | Cytoplasm | Soluble | 4.71 | Sec62 | cytoskeleton, protein localization |
| FLNA_HUMAN | FLNA | 281 kDa | Cytoplasm | Soluble | 27.94 | Sec62 | cytoskeleton, cell adhesion, GTPase activity |
| E41L3_HUMAN | EPB41L3 | 121 kDa | Nucleus | Soluble | 3.28 | Sec62 | cytoskeleton, protein localization, cell adhesin, cell growth |
| ATF6A_HUMAN | ATF6 | 75 kDa | Endoplasmic reticulum | Membrane | 2.55 | Sec62 | unfolded protein response |
| SRP68_HUMAN | SRP68 | 71 kDa | Cytoplasm | Membrane | 14.54 | LRRC59 | SRP-dependent cotranslational protein targeting to membrane, ribosome binding, 7S RNA binding |
| SRP54_HUMAN | SRP54 | 56 kDa | Cytoplasm | Soluble | 4.39 | LRRC59 | SRP-dependent cotranslational protein targeting to membrane, ribosome binding, 7S RNA binding, GTPase activity |
| LYRIC_HUMAN | MTDH | 64 kDa | Cell membrane | Membrane | 54.70 | LRRC59 | cell adhesion, signaling pathways |
| VIGLN_HUMAN | HDLBP | 141 kDa | Cytoplasm | Soluble | 3.80 | LRRC59 | ipid metabolic process, RNA binding |
| SRP72_HUMAN | SRP72 | 75 kDa | Nucleus | Soluble | 5.83 | LRRC59 | SRP-dependent cotranslational protein targeting to membrane, ribosome binding, 7S RNA binding |
| LRC59_HUMAN | LRRC59 | 35 kDa | Endoplasmic reticulum | Membrane | 56.07 | LRRC59 | endoplasmic reticulum |
| SND1_HUMAN | SND1 | 102 kDa | Cytoplasm | Soluble | 16.67 | LRRC59 | endonuclease activity, RISC complex, RNA binding |
| LSG1_HUMAN | LSG1 | 75 kDa | Cytoplasm | Soluble | 27.87 | LRRC59 | GTPase activity, protein transport |
| RRBP1_HUMAN | RRBP1 | 152 kDa | Endoplasmic reticulum | Membrane | 57.81 | LRRC59 | endoplasmic reticulum, protein transport |
| KTN1_HUMAN | KTN1 | 156 kDa | Cytoplasm | Membrane | 78.32 | LRRC59 | endoplasmic reticulum, kinesin binding |
Characterization of SEC61β and RPN1 Interactomes Using Proximity Proteomics
To further characterize the protein interactomes of the reporter baits, we combined statistical prioritization, 2D clustering, and principal components analysis. This integrative approach bypasses the somewhat arbitrary requirement of filtering against a specific fold-change value, and instead uses protein co-expression patterns to identify interaction networks and correct for variability in protein abundance across each of our reporter cell lines. This analysis identified 145, 13, 50, and 25 high-confidence protein interactors of SEC61β, RPN1, SEC62, and LRRC59, respectively (supplemental File S2). Because the interactomes of SEC61β and RPN1 are at least in part characterized, we first examined the protein networks of these two baits. In this analysis, SEC61β had the highest number of high-confidence interactors (n = 145 proteins) (Fig. 3A), making it the largest interactome captured by our study. Despite its large size, gene ontology (GO) analysis demonstrated that nearly all of SEC61β protein partners (either direct or proximal) are membrane proteins and/or have functions related to protein transport (Fig. 3B), which aligns with the known functions of SEC61 in ER targeting, membrane insertion, and translocation of newly synthesized polypeptides (88). Moreover, almost half (44%) of the identified protein interactors are annotated to physically interact with one another, suggesting that the SEC61β interactome is not only enriched for membrane/secretory proteins but that these high-confidence interactors comprise large protein-protein complexes/networks (Fig. 3C). Notably, our proteomics and protein-protein interaction (PPI) analyses revealed that SEC61β interacts with SEC61α (SEC61A1) and SEC63 (Figs. 2D, 3C), which is consistent with previous reports (18, 76, 89, 90) and further validates that the putative interactors identified by our approach are likely bona fide targets.
Fig. 3.
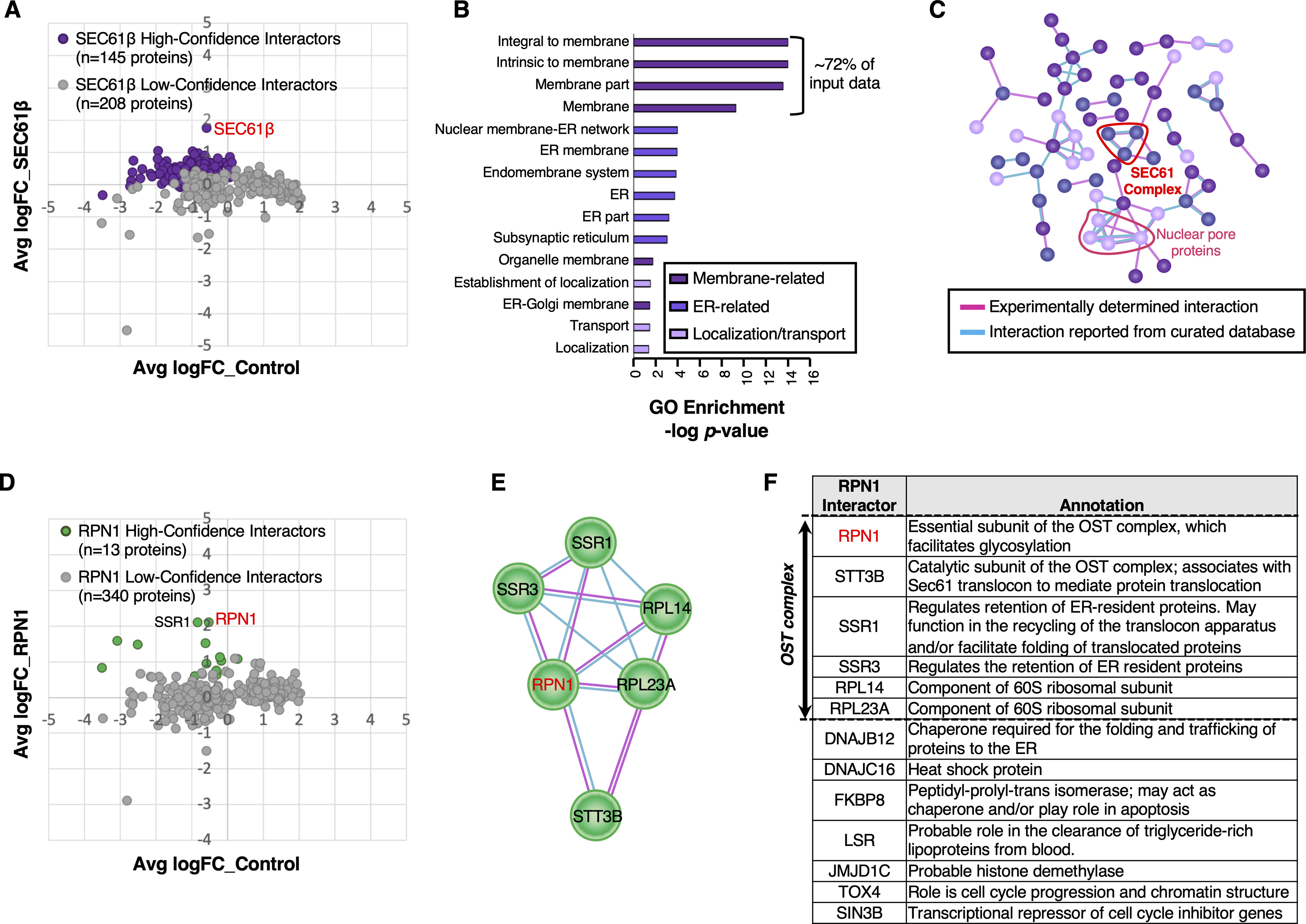
Characterization of protein networks for known ribosome interactors, SEC61β and RPN1. A, Comparison of protein abundance for the 353 identified putative interactors in SEC61β-BirA and empty vector control HEK293 cells. Purple dots represent enriched, high-confidence interactors. Gray dots represent identified proteins whose enrichments are below cutoff, and represent less likely bona fide interactors of SEC61β-BirA. B, Enriched Gene Ontology (GO) terms associated with high-confidence SEC61β-BirA interactors. Dark purple, purple, and light purple bars represent membrane-, endoplasmic reticulum (ER)-, and protein transport-related GO enriched terms, respectively. C, Protein-protein interactions (PPI) among high-confidence SEC61β-BirA interactors, based on STRING annotations. Pink and cyan edges indicate experimentally determined and curated interactions, respectively. D, Comparison of protein abundance for the identified 353 putative interactors in RPN1-BirA and empty vector control HEK293 cells. Green dots represent enriched, high-confidence proteins that interact with RPN1-BirA. Gray dots, as above, represent identified proteins that are less likely bona fide interactors of RPN1-BirA. E, PPI network among high-confidence RPN1-BirA interactors, based on STRING annotations. F, Functional comparison of all 13 high-confidence RPN1-BirA interactors, based on STRING annotations.
In contrast to the large number of proteins identified as SEC61β interactors, examination of the RPN1 interactome yielded the smallest number of interactors (n = 13 proteins) (Fig. 3D). Despite its small size, about one-third of the RPN1 interactome comprises members of the OST complex (Fig. 3E–3F), including STT3B, and the α and β subunits of the TRAP complex (SSR1, SSR3), as expected (81, 91). Additionally, our analysis revealed RPN1 to interact with 60S ribosomal proteins (RPL14, RPL23A), supporting a role for RPN1 in ribosome association (92). Collectively, our characterizations of the SEC61β and RPN1 interactomes parallel high-resolution structural analyses of the SEC61 translocon, which place the OST and TRAP complexes in close physical proximity to the SEC61 oligomer (81, 91).
Functional Diversity Across the BioID-SEC62 Interactome
Following the statistical methodology described above, we interrogated the SEC62 interactome. As mentioned earlier, SEC62 has been demonstrated to interact with ribosomes and to facilitate mRNA translation and protein translocation on the ER (89, 93); however, a comprehensive understanding of the SEC62 interactome in mammalian cells has not been previously reported. As assessed by BioID proteomics, the SEC62 interactome of HEK293 cells is comprised of a large cohort of proteins (n = 50) (Fig. 4A). Consistent with our previous study (34), we did not identify significant interactions of the SEC62 reporter with ribosomes, indicating that SEC62 may participate in ER translation independent of ribosome binding, as postulated for the canine pancreas rough microsome system (94). Alternatively, the BioID reporter construct may occlude ribosome binding activity present in the native protein. To examine the functional significance of the BioID-SEC62 interactome, we performed GO analysis and database mining on the 50 identified proteins. This analysis revealed that SEC62 interacts with a wide range of proteins involved in biologically diverse functions, including roles in cell cycle and proliferation, cytoskeleton architecture, protein localization, signaling pathways, ER chaperones, and redox homeostasis (Fig. 4B–4C). Because SEC62-interacting proteins have overlapping cellular functions (Fig. 4C), we next asked if these proteins physically interact with one another to form protein complexes that may provide mechanistic insights into the biological functions of the SEC62 interactome. Protein-protein interaction analysis revealed that 54% of the SEC62 interactome physically interact with one another (Fig. 4D). Using literature-based searches, database mining, and informatic approaches, we assigned a primary function to each protein, as indicated by the color legend in Fig. 4B. In this depiction, the edges connecting interacting proteins were color coded to distinguish experimentally determined (pink) interactions (95) from those reported/curated in databases (cyan), as annotated by the STRING database (60, 61). Like the GO analysis, interrogation of PPI networks demonstrated heterogeneity in the functional assignment of interacting proteins. Notably, SEC62 was not reported in any of the six PPI networks, which illustrates the current lack of knowledge regarding SEC62 interactions in cells. Nonetheless, in seemingly unexpected interactions, such as with the ER-Golgi trafficking components STX5 and SCFD1 (Fig. 4D), an ER-centric element is evident. Thus, and for the three Golgi-related proteins depicted (SCFD1, STX5, and GOLGA5), all function in ER-Golgi/Golgi-Golgi vesicular transport, have been previously identified as co-interactors, and participate in ER-linked vesicular transport processes. Specifically, SCFD1 functions with the SNARE protein STX5 to regulate ER to Golgi transport (96, 97). Intriguingly, STX5 exists in two forms, long and short, where the long form both contains an ER retrieval sequence and is predominately localized to the ER (98).
Fig. 4.
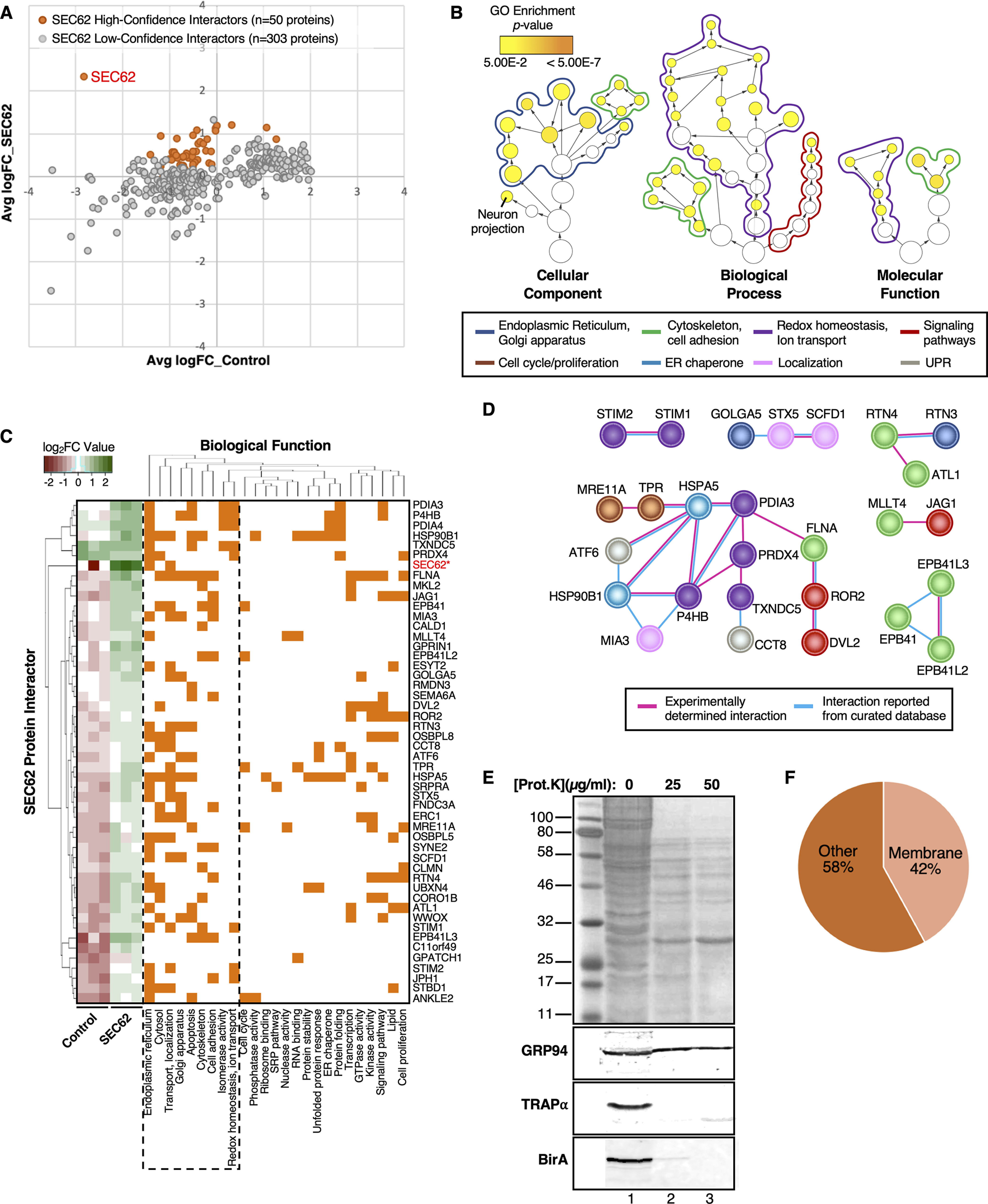
BioID-SEC62 labels functionally diverse proteins. A, Comparison of protein abundance for the 353 identified putative interactors in SEC62-BirA and empty vector control HEK293 cells. Orange dots represent enriched, high-confidence proteins that interact with SEC62-BirA. Gray dots represent identified proteins that are less likely bona fide interactors of SEC62-BirA. B, Hierarchical view of relationships for GO terms associated with SEC62 high-confidence protein interactors. GO term circles are outlined to match the colors assigned to each enriched GO category, as indicated beneath the panel. Circle sizes represent the number of genes in each enriched term, whereas circle color indicates the GO enrichment p-value. C, Clustering of SEC62 high-confidence interactors based on co-occurrence of functional annotations. The left-most heatmap represents protein abundance values across biological replicates in control and SEC62-BirA HEK293 cells. D, Protein-protein interactions (PPI) among high-confidence SEC62-BirA interactors, based on STRING annotations. Proteins are color-coded to match their functional assignment, as indicated above the panel. E, Topology analysis of SEC62-BirA reporter line. SEC62-BirA expressing cells were chilled on ice, permeabilized with a digitonin-supplemented cytosol buffer, and subjected to digestion with the indicated concentrations of Proteinase K for 30 min on ice. Cells were subsequently lysed and total protein was resolved via SDS-PAGE (top panel). Following transfer, membranes were probed for GRP94 (ER-luminal protein), TRAPα (ER-resident protein with cytosolically-disposed antibody epitope), and BirA (BioID-SEC62 reporter). Lanes 1, 2, and 3 represent digestions with 0, 25, and 50 μg/ml proteinase K, respectively. F, Distribution of membrane proteins identified within the SEC62-BirA interactome, based on membranOME annotations.
A particularly striking finding in the SEC62-BirA interactome was the presence of ER luminal-resident proteins, including PDIA3, PRDX4, and HSP90B1/GRP94. With the identification of ER luminal proteins limited to the SEC62-BirA reporter line, we initially presumed that the membrane topology of the SEC62-BirA reporter was inverted from the native protein, whose N- and C-termini are cytoplasmic, thereby placing the BirA domain in the ER lumen (93, 99). Alternatively, and given that the ER luminal proteins identified were present in biological triplicates and exceeded significance cutoffs, these data imply that SEC62 is functionally coupled with, or proximal to, the recently discovered ER luminal protein reflux pathway machinery (100). To distinguish between these two possibilities, we examined the membrane topology of the SEC62-BirA reporter by protease protection assays, performed on digitonin-permeabilized SEC62-BirA expressing cells (Fig. 4E). In this approach, cytosolic domains of ER membrane proteins are expected to be protease accessible, whereas ER lumen proteins are largely protected against protease digestion. GRP94 and TRAPα, both ER-resident proteins, were used as proteolysis topology controls. GRP94, an ER luminal protein was protected from proteinase K digestion (Fig. 4E). In contrast, TRAPα is digested completely at 25 µg/ml of proteinase K (the lowest concentration tested), with detection by a polyclonal antibody raised against the cytosolic domain (Fig. 4E). Similar to what we observed with TRAPα digestion, anti-BirA reactivity was lost at the lowest proteinase K concentration tested, demonstrating that the SEC62-BirA reporter assumes the membrane topology of the native protein (Fig. 4E). To further examine if there was an over-representation of ER luminal proteins in our SEC62-BioID dataset, we assessed the membrane vs. soluble distribution of all 50 interacting proteins using the membranOME database (62, 63) (Fig. 4F). Using this approach, we determined that only 42% of the SEC62 interactome is made up of membrane proteins (Fig. 4F). This suggests that although we did observe an enrichment of ER luminal proteins, the majority of the unique SEC62 interactors are indeed soluble proteins. Notably, this distribution between membrane vs. soluble protein interactors was mirrored in the set of high-confidence SEC62 interactors identified by the single BioID-reporter experiments, which also include ER luminal proteins (Table I, “Type” column). Together, these data further suggest that SEC62 may be proximal to and/or an interactor with an ER luminal protein reflux pathway. Further studies are needed to establish this putative functional link.
The LRRC59 Interactome is Enriched in the SRP Pathway, ER-resident RNA-binding Proteins, and Translation Factors
Previous in vitro studies have shown LRRC59 to interact with the 60S ribosomal subunit, however the biological importance of this interaction and the local LRRC59 membrane environment remains unknown. Following the methodology detailed above, we examined the BioID-LRRC59 interactome. As noted, our analysis identified 25 high-confidence LRRC59 interacting proteins (Fig. 5A). Unlike the SEC62-BirA interactome which is enriched for ER functions other than mRNA translation/protein biogenesis, proteins identified in the LRRC59-BirA dataset were highly enriched for functions related to mRNA translation (e.g. eIF2A, eIF5), the SRP pathway (e.g. SRP54, SRP72), and RNA binding (e.g. MTDH, SND1) (Fig. 5B–5D). Notably, the proteins that had the highest quantitative enrichments for LRRC59-BirA labeling include LRRC59, RRBP1 (p180, ribosome-binding protein), MTDH (AEG-1, an RNA-binding protein), SERBP1 (RNA-binding protein), and SRP72 (SRP protein) (Fig. 5C, leftmost heatmap).
Fig. 5.
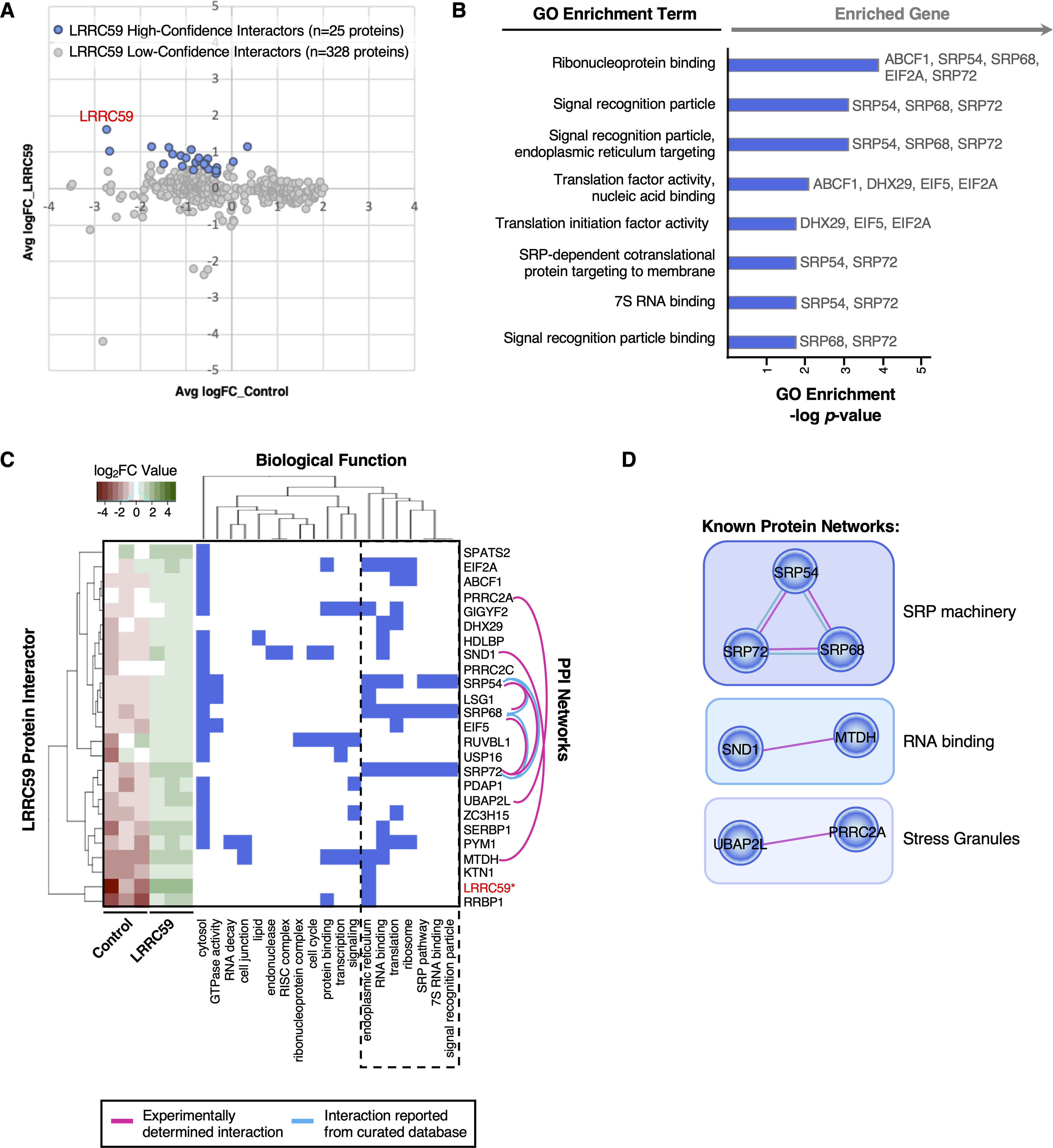
BioID-LRRC59 interacts with SRP pathway, translation machinery, and RNA-binding proteins. A, Comparison of protein abundance for the 353 identified putative interactors in LRRC59-BirA and empty vector control HEK293 cells. Blue dots represent enriched, high-confidence proteins that interact with LRRC59-BirA. Gray dots represent identified proteins that are less likely bona fide interactors of LRRC59-BirA. B, Enriched Gene Ontology (GO) terms associated with high-confidence LRRC59-BirA interactors. Genes assigned to each enriched GO term are listed on the right. C, Clustering of LRRC59 high-confidence interactors based on co-occurrence of functional annotations. The left-most heatmap represents protein abundance values across biological replicates in control and LRRC59-BirA HEK293 cells. Protein-protein interaction (PPI) networks among LRRC59 interacting proteins, as annotated by STRING, are visualized by pink and/or cyan edges. D, Alternative view of PPI networks among LRRC59 high-confidence interacting proteins, based on STRING annotations.
Similar to our previous interactome analyses, we generated a PPI network for the 25 LRRC59-interacting proteins, using STRING (60, 61) (Fig. 5C–5D). This analysis revealed three primary protein networks within the LRRC59 interactome: the RNA-binding proteins MTDH/AEG-1 and SND1 (101), the stress granule proteins UBAP2L and PRRC2A (102), and importantly the SRP subunit proteins SRP54, SRP68 and SRP72, indicating broad coverage of SRP (Fig. 5C–5D, depicted as pink and cyan edges linking interacting proteins). Recently, we reported that MTDH/AEG-1 is an ER-resident integral membrane RBP whose interactome is highly enriched for integral membrane protein-encoding transcripts (19). Importantly, our previous study implicated MTDH/AEG-1 in the localization of secretory and membrane protein-encoding mRNAs to the ER, suggesting that LRRC59 may also bind functionally-related mRNAs. SND1, which has been shown to interact with MTDH during overexpression studies in cancer models, is a tudor domain-containing protein that modulates the transcription, splicing, and stability of mRNAs related to cell proliferation, signaling pathways, and tumorigenesis (103, 104). These functional annotations are consistent with models where LRRC59 functions in a complex with MTDH/AEG-1 and SND1 to recruit/regulate mRNAs for translation on the ER membrane. In a similar vein, the BioID data identified LRRC59 as an SRP interactor. SRP is best characterized for its role in the signal sequence-dependent trafficking of ribosomes engaged in the translation of secretory/membrane proteins. Intriguingly, the C-terminus of LRRC59 (located in the ER lumen) shares overlapping sequence structure with the SR receptor (105), further implicating LRRC59 function in the SRP pathway and/or translation of secretory/membrane protein mRNAs on the ER. Additionally, the interaction of LRRC59 with the protein-protein network pair, UBAP2L-PRRC2C, may relate to mRNA regulation via stress granule assembly. Stress granules are membrane-less structures formed from non-translating mRNPs during stress (106, 107). Stress granules are typically composed of several RNA-binding proteins, along with factors involved in translation initiation and mRNA decay. Interestingly, the LRRC59 interactome is enriched for all three classes of factors. We also report PRRC2C, a known paralog of PRRC2A which is required for the efficient formation of stress granules (102), as an LRRC59 interacting protein, indicating that stress granule proteins may associate with ER-compartmentalized translation centers (e.g. LRRC59 interactome). Combined with our previous study, these data demonstrate that LRRC59 associates with ER-bound ribosomes and scaffolds a protein interactome highly enriched in SRP pathway machinery and RNA-binding proteins, suggesting a relationship between LRRC59 and stress granule formation. Experiments to test these hypotheses are currently ongoing.
Orthogonal Validation Confirms the Direct Interaction of LRRC59 with mRNA Translation-related Factors
One limitation to a proximity proteomics approach is that the identified protein interactors cannot be distinguished as stable vs. transient interactors. To determine if LRRC59 stably interacts with SRP machinery, translation factors, and/or RNA-binding proteins, we performed LRRC59 native co-immunoprecipitation (co-IP) studies followed by mass spectrometry. In brief, Caco-2 cells were cultured, detergent extracts prepared, and LRRC59 captured via indirect immunoprecipitation, using an affinity purified anti-LRRC59 antisera. Following mass spectrometric analysis, raw data files were processed with Protein Discoverer and Scaffold to perform semi-quantitative analysis via total spectral counts for the identified proteins. High-confidence interacting proteins of LRRC59 were subsequently identified using CompPASS (55), which is an unbiased, comparative proteomics software platform. In total, 2,678 prey within each IP were identified (Fig. 6A), and of these proteins, 102 were determined to be high-confidence interacting proteins (HCIP) of LRRC59 (D-score ≥ 1) (Fig. 6B, supplemental File S4). Notably, 20% of the LRRC59 targets determined by BioID overlapped with these HCIPs (Fig. 6B). As expected, these shared LRRC59 targets include SERPB1, DHX29, PRRC2C, SRP68, and LRRC59 itself, which were among the most enriched biotin-labeled proteins within the LRRC59-BirA experiment. We also recovered the other highly enriched proteins SRP72, SRP54, and RRBP1 in the LRRC59 co-IP data (Fig. 6E); however, their D-scores (0.95, 0.92, and 0.90, respectively) were just below the conservative threshold. Given that SRP is itself a ribonucleoprotein complex, these data are consistent with SRP acting as a stable member of the LRCC59 interactome.
Fig. 6.
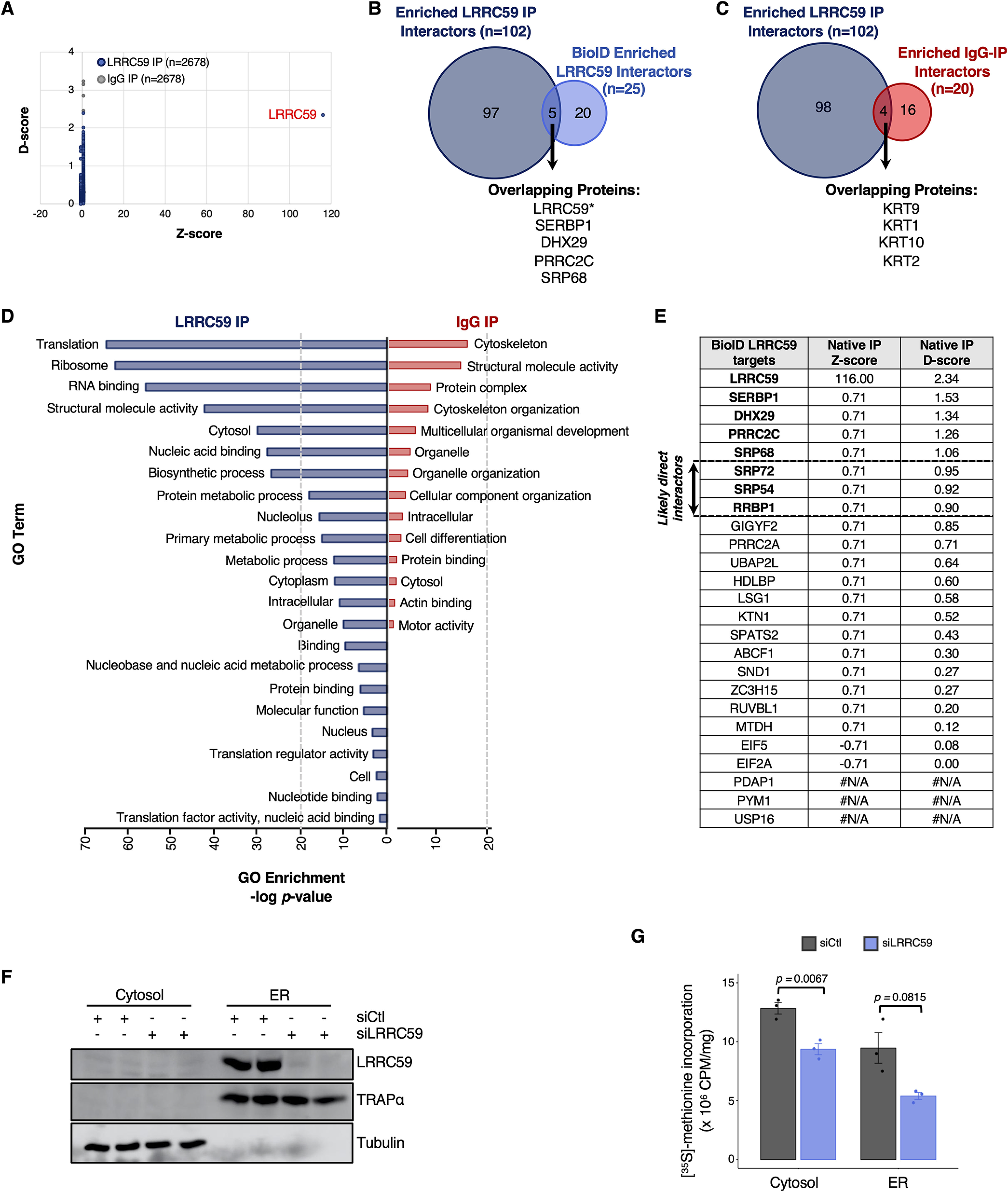
LRRC59 co-IP screen for direct interactions with SRP pathway, translation machinery, and RNA-binding proteins. A, Comparison of D- and Z-scores, as determined by CompPASS analysis, for all proteins identified to interact with LRRC59 (blue) and IgG (control; gray) via immunoprecipitation (IP). Each dot is one of the 2,678 proteins identified by mass spectrometry. B, Number and overlap of enriched, high-confidence interactors of LRRC59, as determined by co-IP (dark blue) or isobaric tagging (BioID; light blue) approaches. C, Number and overlap of high-confidence interactors (D-score ≥ 1) of LRRC59 (dark blue) or IgG (red), as determined by co-IP. D, Enriched Gene Ontology (GO) terms associated with high-confidence LRRC59 interactors (dark blue, left) or IgG interactors (red, right). E, Comparison of D- and Z-scores for each of the 25 LRRC59-interacting proteins, as determined by the BioID approach. F, Western blot analysis depicting LRRC59 expression in cells transfected with either control (scrambled) siRNA or LRRC59-targeted siRNA. Tubulin and TRAPα serve as controls for protein loading and subcellular fractionation, respectively. G, Bar plot showing normalized levels of [35S]-methionine incorporation in the cytosol or ER compartments of non-targeting siRNA (siCtl, negative control) or LRRC59-targeted siRNA transfected cells.
Because co-IP assays are generally accompanied by high background, we also co-immunoprecipitated non-specific IgG as a control. Importantly, analysis of our IgG-IP yielded only 20 HCIPs, which is a small fraction (19%) compared to the LRRC59 interactome (Fig. 6C). We did observe an enrichment of keratin proteins as HCIPs in both LRRC59 and IgG interactomes (Fig. 6C), and attribute this to common environmental contamination, as has been previously reported (108). Thus, our data suggests that the primary bona fide interactors of LRRC59 are uniquely enriched by co-IP.
To assess the biological functions of all the HCIPs that directly interact with LRRC59 (n = 102), we performed GO analysis. Consistent with our observations of the BioID-LRRC59 interactome, HCIPs determined by co-IP were also strongly enriched for proteins with functions related to mRNA translation and RNA binding (Fig. 6D, left side). In contrast, non-immune IgG high-confidence interactors were only mildly enriched in common background proteins (108) (Fig. 6D, right side). Therefore, our data collectively demonstrates that LRRC59 directly interacts with SRP machinery, translation initiation factors, and RNA-binding proteins.
Loss of LRRC59 Disrupts mRNA Translation
Given that the LRRC59 interactome is enriched for networks and individual proteins that strongly converge on mRNA translation regulation, we hypothesized that LRRC59 may function in local protein synthesis. To functionally test whether mRNA translation is impacted by the loss of LRRC59, we first altered levels of LRRC59 expression using siRNA transfection (Fig. 6F). At 72 hours post-transfection, we observed near-complete inhibition of LRRC59 expression in HEK293T cells (Fig. 6F). Using this timepoint, we next performed [35S]-methionine incorporation assays on cells transfected with a non-targeting siRNA (negative control, siCtl) or siLRRC59 to measure changes in protein synthesis. Strikingly, we observed that mRNA translation is reduced by nearly 50% on the ER membrane upon loss of LRRC59 (Fig. 6G). We also observed a significant decrease in mRNA translation occurring in the cytosolic compartment (Fig. 6G). Together, these data indicate that LRRC59 is critical for maintaining cellular mRNA translation.
DISCUSSION
Although the protein machinery involved in secretory and membrane biogenesis on the ER is well established, it remains unclear how mRNA translation on the ER, including translation of cytosolic protein-encoding mRNAs, is spatially organized. Moreover, our understanding of how the resident ER proteome contributes to mRNA localization, anchoring, and translational control is lacking. In this communication, we examine these questions by characterizing the protein interactomes of known and candidate ER-resident ribosome receptors in the mammalian cell line HEK293. Of interest, our data place LRRC59 in a functional nexus for secretory and membrane protein synthesis via interactions with SRP, translation initiation factors, and RNA-binding proteins. Combined, the results of this study reveal new modes of compartmentalized mRNA translation and expand upon the canonical understanding of the SRP pathway.
Functional Domain Organization of the ER Membrane
The endoplasmic reticulum is a structurally complex organelle known to serve multiple functions, including mRNA translation, protein translocation, protein folding, post-translational protein modifications, lipid biosynthesis, and calcium transport (85, 109). In addition, the ER contains specialized domains dedicated to interactions with other membrane organelles, such as the mitochondria and endosomes, and was recently demonstrated to participate in stress granule and processing body dynamics (110–113). With the regulation of dynamic ER morphology and organelle-organelle interactions under active investigation, insights into the spatial organization of the ER membrane and how this higher order is necessary to accommodate its wide-range of biological functions can be expected to provide molecular intersections between the two processes.
Using an unbiased, multiplexed proteomics approach to examine the protein neighborhoods of membrane-bound ribosomes, we identified over 200 proteins in the HEK293 reporter model, many of which clustered into discrete functional categories. Importantly, each of the four tested ER ribosome interactor-BioID reporters had unique sets of interacting proteins, which is consistent with proteins being enriched in functional domains of the ER membrane. In agreement with published structural data, our proximity proteomics study revealed SEC61β to interact with other members of the SEC61 translocon, including SEC61α and SEC63. Remarkably, we also discovered SEC61β to interact with 143 (n = 145 proteins, total) other proteins, making it the largest interactome identified by our study. Despite its large size, gene ontology analysis of the SEC61β interactome yielded a strong enrichment for membrane and transport proteins, which parallels SEC61β's primary role in secretory/membrane protein biogenesis. Although our list of SEC61β-BirA protein interactors have functions that converge on those expected of the translocon, our analysis also provides new candidate interacting proteins that may function alongside SEC61β; and by extension, suggests alternative mechanisms for mRNA translation via the SEC61 translocon.
Similarly, we characterized the protein interactome of the ER-resident protein, RPN1, a subunit of the OST complex and an accessory component of the translocon (40, 82, 114, 115). In contrast to SEC61β, the high-confidence RPN1 interactome was limited, comprising 13 proteins, making it the smallest interactome identified by our study. Nonetheless, we found RPN1 to interact with SSR1/TRAPα and SSR3/TRAPγ, which has been previously structurally validated (81, 91). Members of the OST complex, as well as 60S ribosomal proteins, were also among the list of RPN1 interactors, which is consistent with the spatial assignment of RPN1 and its function in N-linked glycosylation and ribosome binding, respectively. Although we did not pursue the direct functional relationship between these proteins and RPN1, our data provides a new platform for studying dynamic regulation of mRNA translation by the OST complex.
The SEC62 Interactome is Functionally Diverse
The ER-localized members of the SEC gene family have been extensively studied via genetic and biochemical approaches, revealing how Sec61p, Sec62p and Sec63p interact with one another and operate collectively to support translocation of membrane and secretory proteins (89, 99, 116). Although these studies have advanced our understanding of the SEC61 translocon and the biological functions of SEC62 and SEC63 in protein translocation, how these proteins interact with the translation machinery, particularly in mammalian cells, has only recently gained attention (93, 94). Our system identified SEC62 to interact with 50 proteins. Unexpectedly, the SEC62 interactome was enriched for ER luminal proteins, including BiP, GRP94, PDI, and PRDX4. Identifications of these interactions by both TMT-multiplexed and single reporter proteomics analyses was further corroborated by topological orientation studies, confirming that the SEC62-BirA reporter has the appropriate orientation at the ER membrane, with the BirA moiety displayed on the cytosolic domain of the ER. Moreover, noting that these interactions were not identified in the three other BioID reporters examined, we conclude that these are likely bona fide interactions. The existing literature on cytoplasmic and nuclear localizations of ER luminal chaperones such as calreticulin and BiP (117–120), along with the recent identification of an ER lumen protein reflux pathway (100), provide key evidence for a retrograde trafficking pathway for ER luminal proteins across the ER membrane and suggest that SEC62 may functionally intersect with such processes. Further study is needed, however, to understand the molecular basis for the observed SEC62-ER luminal protein interactions.
Surprisingly, the SEC62 interactome also includes proteins functioning in cell-cell adhesion, vesicle transport, signaling pathways, and cytoskeleton formation, indicating that SEC62 may have functions independent of protein biogenesis. For example, SEC62 may be important for ER tubule organization and protein transport to the Golgi apparatus. Interestingly, our data also suggests that SEC62 may have a critical role in multiple signaling pathways. To date, the best characterized ER signaling pathway is the Unfolded Protein Response (UPR). In the UPR, the accumulation of misfolded proteins at the ER triggers a signaling cascade that includes transcriptional (e.g. ATF6) upregulation of ER chaperones (e.g. BiP, protein disulfide isomerases (PDI), GRP94), and ERAD components – which are all represented in our list of interactors. We also found SEC62 to interact with proteins that function in the Wnt and Notch signaling pathways, which are less commonly studied in the context of ER regulation, though it has been reported that Wnt signaling proteins are retained in the ER because of inefficient secretion (121, 122). To this point, the ER-resident glycoprotein Oto regulates Wnt activity by binding Wnt1 and Wnt3a to facilitate its retention in the ER (123). Whether SEC62 acts as another ER-resident protein that binds Wnt-related factors to regulate the accumulation and burst of Wnt ligands remains to be determined. Similarly, our data suggests that SEC62 may play a role in the glycosylation of Notch proteins, thereby influencing Notch activation. Understanding how SEC62 may function in the UPR, Notch, and/or Wnt signaling pathways has the potential to shed new light on how defects in these signaling cascades at the ER contributes to genetic human disorders.
A Role for LRRC59 in the Spatial Organization of Protein Synthesis on the ER
Despite the discovery of LRRC59 decades ago, little is known about its biological function (34, 83, 105, 124–126). Early sequence analysis revealed that the cytoplasmic domain of LRRC59 contains a number of intriguing structural features, including leucine-rich repeats (LRR), which are known protein-protein interaction motifs, hydrophilic regions (KRE), and several regions of charged residues that could serve as sites for protein-protein interactions and ribosome binding activity (105, 124). Indeed, via proximity proteomics, we identified high-confidence interactions with 25 proteins. Importantly, these interactions are likely occurring on the cytosolic domain of LRRC59, as predicted, because the reporter construct places the BirA terminal to the LRR, KRE, and transmembrane-spanning domains (34). Prominent in the LRRC59 interactome were subunits of the SRP (e.g. SRP54, SRP72, SRP68), translation initiation factors (e.g. eIF2A, eIF5, DHX29), and other ER-RBPs (e.g. SERBP1, MTDH). The prevalence of these interactions link LRRC59 to the regulation of secretory and membrane protein synthesis on the ER. In support of this view, we recently demonstrated that LRRC59-BirA constructs robustly label ER-bound ribosomes (34), and previous in vitro studies demonstrated that LRRC59 binds the 60S ribosomal subunit (105, 124). Importantly, we functionally validated that LRRC59 is indeed necessary for mRNA translation. Our analysis of [35S]-labeled nascent proteins demonstrated, for the first time, that protein synthesis is negatively impacted when LRRC59 expression is ablated. However, our understanding of how LRRC59 is precisely regulating mRNA translation requires additional study, which we are currently pursuing.
Here, we propose six possible mechanisms by which LRRC59 may regulate mRNA translation on the ER membrane (Fig. 7). First, given the enrichment of SRP subunits in both our BioID and co-IP experiments, LRRC59 may directly interact with the SRP receptor (Fig. 7A) and/or SRP (Fig. 7B) to recruit mRNA/ribosome/nascent peptide complexes to the ER membrane for continued mRNA translation. Alternatively, LRRC59 may bind translationally active ribosomes via protein-protein interactions occurring on its large LRR- and KRE-containing cytoplasmic domain (Fig. 7C). Given the enrichment of translation initiation factors interacting with LRRC59, our data also suggests that LRRC59 may bind both 60S and 40S ribosomal subunits, as well as initiation factors in proximity to facilitate mRNA translation initiation on the ER membrane (Fig. 7D). Another possible mechanism for a LRRC59 function in mRNA translation is via directly binding mRNA (19) and/or indirectly targeting mRNAs through interactions with ER-localized RBPs (Fig. 7E). By anchoring localized mRNAs either directly or indirectly, LRRC59 may then recruit ribosomes (as previously postulated) for subsequent mRNA translation (Fig. 7E). Finally, our data also reveal LRRC59 to interact with proteins that associate with stress granules (e.g. UBAP2L, PRRC2A, PRRC2C). Therefore, we hypothesize that stress granules may reside proximal to LRRC59 as a mechanism to spatially and temporally fine-tune protein synthesis upon changes in cellular homeostasis (Fig. 7F). Although we have presented data supporting a role for LRRC59 in translational regulation on the ER membrane, further studies are necessary to provide detailed mechanistic characterization of this proposed function.
Fig. 7.

Model depicting LRRC59 interactions with ER localized mRNA translation. Proximity proteomics revealed LRRC59 to significantly interact with SRP factors, translation machinery (including the ribosome), RNA-binding proteins (RBPs), and proteins associated with stress granules. As depicted, LRRC59 may interact with the (A) SRP receptor or (B) SRP to recruit translationally-engaged ribosomes to the ER membrane for continued mRNA translation. C, LRRC59 recruits mRNA/ribosome/nascent peptide complexes by directly interacting with associated translation factors and/or RBPs, independent of the SRP pathway. D, LRRC59 interacts with the 40S and 60S ribosomal subunits, along with translation factors, RBPs, and mRNA to facilitate translation initiation. E, LRRC59 may anchor mRNAs on the ER membrane via direct RNA binding activity and/or through interactions with other mRNA-bound RBPs, thereby recruiting nearby ribosomes for subsequent mRNA translation. F, LRRC59 may interact with stress granules to fine-tune the activity of translating ribosomes in response to alterations in cellular homeostasis. The depicted modes of mRNA regulation by LRRC59 are not mutually exclusive.
In summary, we demonstrate that ER-resident proteins proximal to bound ribosomes are organized via protein network interactions. We provide evidence that SEC61β interacts with proteins important for secretory and membrane translocation; demonstrate that RPN1 interacts with OST complex subunits and ribosomal proteins; and propose a new function for LRRC59 in regulating mRNA translation of secretory/membrane-encoding proteins via the SRP pathway. These data also reveal an array of possible biological functions that SEC62 may facilitate, including signaling pathways, redox homeostasis, and protein folding. Together, these data offer important insights into subcellular translation regulation, and provide an opportunity to unravel connections between ribosome/protein organization and human physiology.
Data Availability
The quantitative mass spectrometry data has been made publicly available using MassIVE at: ftp://massive.ucsd.edu/MSV000085009/.
Supplementary Material
Acknowledgments
We thank the Duke University School of Medicine Proteomics and Metabolomics Shared Resource for their invaluable proteomics services, along with members of the Nicchitta laboratory, in particular Qiang Chen, Jessica Child, Jason Arne, and JohnCarlo Kristofich, for their helpful feedback on this project.
Footnotes
This article contains supplemental data.
Funding and additional information—This work was supported by NIH grant GM101533 to C.V.N. and GM139254 to M.M.H. We would like to acknowledge NIH Shared Instrumentation grant S10OD024999 (MA Moseley) for analytical support of this study.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
Abbreviations—The abbreviations used are:
- RBP
- RNA-binding proteins
- ER
- endoplasmic reticulum
- FC
- fold change.
REFERENCES
- 1. Gunkel N., Yano T., Markussen F. H., Olsen L. C., and Ephrussi A. (1998) Localization-dependent translation requires a functional interaction between the 5' and 3' ends of oskar mRNA. Genes Dev. 12, 1652–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Smibert C. A., Lie Y. S., Shillinglaw W., Henzel W. J., and Macdonald P. M. (1999) Smaug, a novel and conserved protein, contributes to repression of nanos mRNA translation in vitro. RNA 5, 1535–1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Micklem D. R., Adams J., Grunert S., and St Johnston D. (2000) Distinct roles of two conserved Staufen domains in oskar mRNA localization and translation. EMBO J. 19, 1366–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tiruchinapalli D. M., Oleynikov Y., Kelic S., Shenoy S. M., Hartley A., Stanton P. K., Singer R. H., and Bassell G. J. (2003) Activity-dependent trafficking and dynamic localization of zipcode binding protein 1 and beta-actin mRNA in dendrites and spines of hippocampal neurons. J. Neurosci. 23, 3251–3261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gu W., Deng Y., Zenklusen D., and Singer R. H. (2004) A new yeast PUF family protein, Puf6p, represses ASH1 mRNA translation and is required for its localization. Genes Dev. 18, 1452–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huttelmaier S., Zenklusen D., Lederer M., Dictenberg J., Lorenz M., Meng X., Bassell G. J., Condeelis J., and Singer R. H. (2005) Spatial regulation of beta-actin translation by Src-dependent phosphorylation of ZBP1. Nature 438, 512–515 [DOI] [PubMed] [Google Scholar]
- 7. Paquin N., Menade M., Poirier G., Donato D., Drouet E., and Chartrand P. (2007) Local activation of yeast ASH1 mRNA translation through phosphorylation of Khd1p by the casein kinase Yck1p. Mol. Cell 26, 795–809 [DOI] [PubMed] [Google Scholar]
- 8. Willett M., Brocard M., Davide A., and Morley S. J. (2011) Translation initiation factors and active sites of protein synthesis co-localize at the leading edge of migrating fibroblasts. Biochem. J. 438, 217–227 [DOI] [PubMed] [Google Scholar]
- 9. Yasuda K., Kotani T., and Yamashita M. (2013) A cis-acting element in the coding region of cyclin B1 mRNA couples subcellular localization to translational timing. Developmental Biol. 382, 517–529 [DOI] [PubMed] [Google Scholar]
- 10. Zhang Q., Meng X., Li D., Chen S., Luo J., Zhu L., Singer R. H., and Gu W. (2017) Binding of DEAD-box helicase Dhh1 to the 5'-untranslated region of ASH1 mRNA represses localized translation of ASH1 in yeast cells. J. Biol. Chem. 292, 9787–9800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bellon A., Iyer A., Bridi S., Lee F. C. Y., Ovando-Vazquez C., Corradi E., Longhi S., Roccuzzo M., Strohbuecker S., Naik S., Sarkies P., Miska E., Abreu-Goodger C., Holt C. E., and Baudet M. L. (2017) miR-182 regulates Slit2-mediated axon guidance by modulating the local translation of a specific mRNA. Cell Reports 18, 1171–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vidaki M., Drees F., Saxena T., Lanslots E., Taliaferro M. J., Tatarakis A., Burge C. B., Wang E. T., and Gertler F. B. (2017) A requirement for Mena, an actin regulator, in local mRNA translation in developing neurons. Neuron 95, 608–622.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Debard S., Bader G., De Craene J. O., Enkler L., Bar S., Laporte D., Hammann P., Myslinski E., Senger B., Friant S., and Becker H. D. (2017) Nonconventional localizations of cytosolic aminoacyl-tRNA synthetases in yeast and human cells. Methods 113, 91–104 [DOI] [PubMed] [Google Scholar]
- 14. Koppers M., Cagnetta R., Shigeoka T., Wunderlich L. C., Vallejo-Ramirez P., Qiaojin Lin J., Zhao S., Jakobs M. A., Dwivedy A., Minett M. S., Bellon A., Kaminski C. F., Harris W. A., Flanagan J. G., and Holt C. E. (2019) Receptor-specific interactome as a hub for rapid cue-induced selective translation in axons. eLife 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beckmann R., Spahn C. M., Eswar N., Helmers J., Penczek P. A., Sali A., Frank J., and Blobel G. (2001) Architecture of the protein-conducting channel associated with the translating 80S ribosome. Cell 107, 361–372 [DOI] [PubMed] [Google Scholar]
- 16. Berkovits B. D., and Mayr C. (2015) Alternative 3' UTRs act as scaffolds to regulate membrane protein localization. Nature 522, 363–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cui X. A., Zhang H., and Palazzo A. F. (2012) p180 promotes the ribosome-independent localization of a subset of mRNA to the endoplasmic reticulum. PLoS Biol 10, e1001336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gorlich D., Prehn S., Hartmann E., Kalies K. U., and Rapoport T. A. (1992) A mammalian homolog of SEC61p and SECYp is associated with ribosomes and nascent polypeptides during translocation. Cell 71, 489–503 [DOI] [PubMed] [Google Scholar]
- 19. Hsu J. C., Reid D. W., Hoffman A. M., Sarkar D., and Nicchitta C. V. (2018) Oncoprotein AEG-1 is an endoplasmic reticulum RNA-binding protein whose interactome is enriched in organelle resident protein-encoding mRNAs. RNA 24, 688–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jagannathan S., Hsu J. C., Reid D. W., Chen Q., Thompson W. J., Moseley A. M., and Nicchitta C. V. (2014) Multifunctional Roles for the Protein Translocation Machinery in RNA Anchoring to the Endoplasmic Reticulum. J. Biol. Chem. 289, 25907–25924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jan C. H., Williams C. C., and Weissman J. S. (2014) Principles of ER cotranslational translocation revealed by proximity-specific ribosome profiling. Science 346, 1257521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Johnson A. E., and van Waes M. A. (1999) The translocon: a dynamic gateway at the ER membrane. Annu. Rev. Cell Dev. Biol. 15, 799–842 [DOI] [PubMed] [Google Scholar]
- 23. Rapoport T. A. (2007) Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 450, 663–669 [DOI] [PubMed] [Google Scholar]
- 24. Reid D. W., and Nicchitta C. V. (2012) Primary role for endoplasmic reticulum-bound ribosomes in cellular translation identified by ribosome profiling. J. Biol. Chem. 287, 5518–5527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reid D. W., and Nicchitta C. V. (2015) Diversity and selectivity in mRNA translation on the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 16, 221–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Simsek D., Tiu G. C., Flynn R. A., Byeon G. W., Leppek K., Xu A. F., Chang H. Y., and Barna M. (2017) The mammalian Ribo-interactome reveals ribosome functional diversity and heterogeneity. Cell 169, 1051–1065.e1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stephens S. B., Dodd R. D., Brewer J. W., Lager P. J., Keene J. D., and Nicchitta C. V. (2005) Stable ribosome binding to the endoplasmic reticulum enables compartment-specific regulation of mRNA translation. Mol. Biol. Cell 16, 5819–5831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Voigt F., Zhang H., Cui X. A., Triebold D., Liu A. X., Eglinger J., Lee E. S., Chao J. A., and Palazzo A. F. (2017) Single-molecule quantification of translation-dependent association of mRNAs with the endoplasmic reticulum. Cell Reports 21, 3740–3753 [DOI] [PubMed] [Google Scholar]
- 29. Walter P., and Blobel G. (1981) Translocation of proteins across the endoplasmic reticulum. II. Signal recognition protein (SRP) mediates the selective binding to microsomal membranes of in vitro-assembled polysomes synthesizing secretory proteins. J. Cell Biol. 91, 551–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Walter P., and Blobel G. (1981) Translocation of proteins across the endoplasmic reticulum. III. Signal recognition protein (SRP) causes signal sequence dependent and site-specific arrest of chain elongation that is released by microsomal membranes. J. Cell Biol. 91, 557–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chartron J. W., Hunt K. C., and Frydman J. (2016) Cotranslational signal-independent SRP preloading during membrane targeting. Nature 536, 224–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Diehn M., Bhattacharya R., Botstein D., and Brown P. O. (2006) Genome-scale identification of membrane-associated human mRNAs. PLoS Genet. 2, e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Diehn M., Eisen M. B., Botstein D., and Brown P. O. (2000) Large-scale identification of secreted and membrane-associated gene products using DNA microarrays. Nat. Genet. 25, 58–62 [DOI] [PubMed] [Google Scholar]
- 34. Hoffman A. M., Chen Q., Zheng T., and Nicchitta C. V. (2019) Heterogeneous translational landscape of the endoplasmic reticulum revealed by ribosome proximity labeling and transcriptome analysis. J. Biol. Chem. 294, 8942–8958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lerner R. S., Seiser R. M., Zheng T., Lager P. J., Reedy M. C., Keene J. D., and Nicchitta C. V. (2003) Partitioning and translation of mRNAs encoding soluble proteins on membrane-bound ribosomes. Rna 9, 1123–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mueckler M. M., and Pitot H. C. (1981) Structure and function of rat liver polysome populations. I. Complexity, frequency distribution, and degree of uniqueness of free and membrane-bound polysomal polyadenylate-containing RNA populations. J. Cell Biol. 90, 495–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mueckler M. M., and Pitot H. C. (1982) Structure and function of rat liver polysome populations. II. Characterization of polyadenylate-containing mRNA associated with subpopulations of membrane-bound particles. J. Cell Biol. 94, 297–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen Q., Jagannathan S., Reid D. W., Zheng T., and Nicchitta C. V. (2011) Hierarchical regulation of mRNA partitioning between the cytoplasm and the endoplasmic reticulum of mammalian cells. Mol. Biol. Cell 22, 2646–2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kopczynski C. C., Noordermeer J. N., Serano T. L., Chen W. Y., Pendleton J. D., Lewis S., Goodman C. S., and Rubin G. M. (1998) A high throughput screen to identify secreted and transmembrane proteins involved in Drosophila embryogenesis. Proc. Natl. Acad. Sci. U S A 95, 9973–9978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Harada Y., Li H., Li H., and Lennarz W. J. (2009) Oligosaccharyltransferase directly binds to ribosome at a location near the translocon-binding site. Proc. Natl. Acad. Sci. U S A 106, 6945–6949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kreibich G., Freienstein C. M., Pereyra B. N., Ulrich B. L., and Sabatini D. D. (1978) Proteins of rough microsomal membranes related to ribosome binding. II. Cross-linking of bound ribosomes to specific membrane proteins exposed at the binding sites. J. Cell Biol. 77, 488–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Levy R., Wiedmann M., and Kreibich G. (2001) In vitro binding of ribosomes to the beta subunit of the Sec61p protein translocation complex. J. Biol. Chem. 276, 2340–2346 [DOI] [PubMed] [Google Scholar]
- 43. Muller M., and Blobel G. (1984) In vitro translocation of bacterial proteins across the plasma membrane of Escherichia coli. Proc. Natl. Acad. Sci. U S A 81, 7421–7425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Savitz A. J., and Meyer D. I. (1993) 180-kD ribosome receptor is essential for both ribosome binding and protein translocation. J. Cell Biol. 120, 853–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tazawa S., Unuma M., Tondokoro N., Asano Y., Ohsumi T., Ichimura T., and Sugano H. (1991) Identification of a membrane protein responsible for ribosome binding in rough microsomal membranes. J. Biochem. Tokyo. 109, 89–98 [PubMed] [Google Scholar]
- 46. Jan C. H., Williams C. C., and Weissman J. S. (2015) LOCAL TRANSLATION. Response to comment on “Principles of ER cotranslational translocation revealed by proximity-specific ribosome profiling. Science 348, 1217. [DOI] [PubMed] [Google Scholar]
- 47. Reid D. W., and Nicchitta C. V. (2015) LOCAL TRANSLATION. Comment on “Principles of ER cotranslational translocation revealed by proximity-specific ribosome profiling. Science 348, 1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Walter P., and Blobel G. (1980) Purification of a membrane-associated protein complex required for protein translocation across the endoplasmic reticulum. Proc. Natl. Acad. Sci. U S A 77, 7112–7116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Migliaccio G., Nicchitta C. V., and Blobel G. (1992) The signal sequence receptor, unlike the signal recognition particle receptor, is not essential for protein translocation. J. Cell Biol. 117, 15–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jagannathan S., Nwosu C., and Nicchitta C. V. (2011) Analyzing mRNA localization to the endoplasmic reticulum via cell fractionation. Methods Mol. Biol. 714, 301–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang Y., Anderson E., and Zhang S. (2018) Evaluation of six sample preparation procedures for qualitative and quantitative proteomics analysis of milk fat globule membrane. Electrophoresis. 39, 2332–2339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nesvizhskii A. I., Keller A., Kolker E., and Aebersold R. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 53. Shadforth I. P., Dunkley T. P., Lilley K. S., and Bessant C. (2005) i-Tracker: for quantitative proteomics using iTRAQ. BMC Genomics 6, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Oberg A. L., Mahoney D. W., Eckel-Passow J. E., Malone C. J., Wolfinger R. D., Hill E. G., Cooper L. T., Onuma O. K., Spiro C., Therneau T. M., and Bergen H. R. 3rd. (2008) Statistical analysis of relative labeled mass spectrometry data from complex samples using ANOVA. J. Proteome Res. 7, 225–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sowa M. E., Bennett E. J., Gygi S. P., and Harper J. W. (2009) Defining the human deubiquitinating enzyme interaction landscape. Cell 138, 389–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Maere S., Heymans K., and Kuiper M. (2005) BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21, 3448–3449 [DOI] [PubMed] [Google Scholar]
- 57. Krupke D. M., Begley D. A., Sundberg J. P., Richardson J. E., Neuhauser S. B., and Bult C. J. (2017) The Mouse Tumor Biology Database: A Comprehensive Resource for Mouse Models of Human Cancer. Cancer Res. 77, e67–e70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bult C. J., Blake J. A., Smith C. L., Kadin J. A., Richardson J. E., and Mouse Genome Database, G. (2019) Mouse Genome Database (MGD) 2019. Nucleic Acids Res. 47, D801–D806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Smith C. M., Hayamizu T. F., Finger J. H., Bello S. M., McCright I. J., Xu J., Baldarelli R. M., Beal J. S., Campbell J., Corbani L. E., Frost P. J., Lewis J. R., Giannatto S. C., Miers D., Shaw D. R., Kadin J. A., Richardson J. E., Smith C. L., and Ringwald M. (2019) The mouse Gene Expression Database (GXD): 2019 update. Nucleic Acids Res. 47, D774–D779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Szklarczyk D., Morris J. H., Cook H., Kuhn M., Wyder S., Simonovic M., Santos A., Doncheva N. T., Roth A., Bork P., Jensen L. J., and von Mering C. (2017) The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 45, D362–D368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Szklarczyk D., Gable A. L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., Simonovic M., Doncheva N. T., Morris J. H., Bork P., Jensen L. J., and Mering C. V. (2019) STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lomize A. L., Lomize M. A., Krolicki S. R., and Pogozheva I. D. (2017) Membranome: a database for proteome-wide analysis of single-pass membrane proteins. Nucleic Acids Res. 45, D250–D255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lomize A. L., Hage J. M., and Pogozheva I. D. (2018) Membranome 2.0: database for proteome-wide profiling of bitopic proteins and their dimers. Bioinformatics 34, 1061–1062 [DOI] [PubMed] [Google Scholar]
- 64. Uhlen M., Oksvold P., Fagerberg L., Lundberg E., Jonasson K., Forsberg M., Zwahlen M., Kampf C., Wester K., Hober S., Wernerus H., Bjorling L., and Ponten F. (2010) Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol. 28, 1248–1250 [DOI] [PubMed] [Google Scholar]
- 65. Uhlen M., Fagerberg L., Hallstrom B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson A., Kampf C., Sjostedt E., Asplund A., Olsson I., Edlund K., Lundberg E., Navani S., Szigyarto C. A., Odeberg J., Djureinovic D., Takanen J. O., Hober S., Alm T., Edqvist P. H., Berling H., Tegel H., Mulder J., Rockberg J., Nilsson P., Schwenk J. M., Hamsten M., von Feilitzen K., Forsberg M., Persson L., Johansson F., Zwahlen M., von Heijne G., Nielsen J., and Ponten F. (2015) Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- 66. Roux K. J., Kim D. I., Raida M., and Burke B. (2012) A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196, 801–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Varnaite R., and MacNeill S. A. (2016) Meet the neighbors: Mapping local protein interactomes by proximity-dependent labeling with BioID. Proteomics 16, 2503–2518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sears R. M., May D. G., and Roux K. J. (2019) BioID as a Tool for Protein-Proximity Labeling in Living Cells. Methods Mol. Biol. 2012, 299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kwon K., and Beckett D. (2000) Function of a conserved sequence motif in biotin holoenzyme synthetases. Protein Sci 9, 1530–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rees J. S., Li X. W., Perrett S., Lilley K. S., and Jackson A. P. (2015) Protein Neighbors and Proximity Proteomics. Mol. Cell. Proteomics 14, 2848–2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kim D. I., Birendra K. C., Zhu W., Motamedchaboki K., Doye V., and Roux K. J. (2014) Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc. Natl. Acad. Sci. U S A 111, E2453–E2461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Singer S. J., and Nicolson G. L. (1972) The fluid mosaic model of the structure of cell membranes. Science 175, 720–731 [DOI] [PubMed] [Google Scholar]
- 73. Kusumi A., Suzuki K. G., Kasai R. S., Ritchie K., and Fujiwara T. K. (2011) Hierarchical mesoscale domain organization of the plasma membrane. Trends Biochem. Sci. 36, 604–615 [DOI] [PubMed] [Google Scholar]
- 74. Kusumi A., Fujiwara T. K., Chadda R., Xie M., Tsunoyama T. A., Kalay Z., Kasai R. S., and Suzuki K. G. (2012) Dynamic organizing principles of the plasma membrane that regulate signal transduction: commemorating the fortieth anniversary of Singer and Nicolson's fluid-mosaic model. Annu. Rev. Cell Dev. Biol. 28, 215–250 [DOI] [PubMed] [Google Scholar]
- 75. Goyette J., and Gaus K. (2017) Mechanisms of protein nanoscale clustering. Curr. Opin. Cell Biol. 44, 86–92 [DOI] [PubMed] [Google Scholar]
- 76. Becker T., Bhushan S., Jarasch A., Armache J. P., Funes S., Jossinet F., Gumbart J., Mielke T., Berninghausen O., Schulten K., Westhof E., Gilmore R., Mandon E. C., and Beckmann R. (2009) Structure of monomeric yeast and mammalian Sec61 complexes interacting with the translating ribosome. Science 326, 1369–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Dejgaard K., Theberge J. F., Heath-Engel H., Chevet E., Tremblay M. L., and Thomas D. Y. (2010) Organization of the Sec61 translocon, studied by high resolution native electrophoresis. J. Proteome Res. 9, 1763–1771 [DOI] [PubMed] [Google Scholar]
- 78. Pfeffer S., Burbaum L., Unverdorben P., Pech M., Chen Y., Zimmermann R., Beckmann R., and Forster F. (2015) Structure of the native Sec61 protein-conducting channel. Nat. Commun. 6, 8403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Voorhees R. M., Fernandez I. S., Scheres S. H., and Hegde R. S. (2014) Structure of the mammalian ribosome-Sec61 complex to 3.4 A resolution. Cell 157, 1632–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kelleher D. J., Kreibich G., and and Gilmore R. (1992) Oligosaccharyltransferase activity is associated with a protein complex composed of ribophorins I and II and a 48 kD protein. Cell 69, 55–65 [DOI] [PubMed] [Google Scholar]
- 81. Nilsson I., Kelleher D. J., Miao Y., Shao Y., Kreibich G., Gilmore R., von Heijne G., and Johnson A. E. (2003) Photocross-linking of nascent chains to the STT3 subunit of the oligosaccharyltransferase complex. J. Cell Biol. 161, 715–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wild R., Kowal J., Eyring J., Ngwa E. M., Aebi M., and Locher K. P. (2018) Structure of the yeast oligosaccharyltransferase complex gives insight into eukaryotic N-glycosylation. Science 359, 545–550 [DOI] [PubMed] [Google Scholar]
- 83. Ichimura T., Shindo Y., Uda Y., Ohsumi T., Omata S., and Sugano H. (1993) Anti-(p34 protein) antibodies inhibit ribosome binding to and protein translocation across the rough microsomal membrane. FEBS Lett. 326, 241–245 [DOI] [PubMed] [Google Scholar]
- 84. de Brito O. M., and Scorrano L. (2010) An intimate liaison: spatial organization of the endoplasmic reticulum-mitochondria relationship. EMBO J. 29, 2715–2723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. English A. R., and Voeltz G. K. (2013) Endoplasmic reticulum structure and interconnections with other organelles. Cold Spring Harb. Perspect. Biol. 5, a013227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Helle S. C., Kanfer G., Kolar K., Lang A., Michel A. H., and Kornmann B. (2013) Organization and function of membrane contact sites. Biochim. Biophys. Acta 1833, 2526–2541 [DOI] [PubMed] [Google Scholar]
- 87. Hung V., Lam S. S., Udeshi N. D., Svinkina T., Guzman G., Mootha V. K., Carr S. A., and Ting A. Y. (2017) Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. eLife 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lang S., Pfeffer S., Lee P. H., Cavalie A., Helms V., Forster F., and Zimmermann R. (2017) An update on Sec61 channel functions, mechanisms, and related diseases. Front. Physiol. 8, 887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lang S., Benedix J., Fedeles S. V., Schorr S., Schirra C., Schauble N., Jalal C., Greiner M., Hassdenteufel S., Tatzelt J., Kreutzer B., Edelmann L., Krause E., Rettig J., Somlo S., Zimmermann R., and Dudek J. (2012) Different effects of Sec61alpha, Sec62 and Sec63 depletion on transport of polypeptides into the endoplasmic reticulum of mammalian cells. J. Cell Sci. 125, 1958–1969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hartmann E., Sommer T., Prehn S., Gorlich D., Jentsch S., and Rapoport T. A. (1994) Evolutionary conservation of components of the protein translocation complex. Nature 367, 654–657 [DOI] [PubMed] [Google Scholar]
- 91. Pfeffer S., Dudek J., Gogala M., Schorr S., Linxweiler J., Lang S., Becker T., Beckmann R., Zimmermann R., and Forster F. (2014) Structure of the mammalian oligosaccharyl-transferase complex in the native ER protein translocon. Nat. Commun. 5, 3072. [DOI] [PubMed] [Google Scholar]
- 92. Braunger K., Pfeffer S., Shrimal S., Gilmore R., Berninghausen O., Mandon E. C., Becker T., Forster F., and Beckmann R. (2018) Structural basis for coupling protein transport and N-glycosylation at the mammalian endoplasmic reticulum. Science 360, 215–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Muller L., de Escauriaza M. D., Lajoie P., Theis M., Jung M., Muller A., Burgard C., Greiner M., Snapp E. L., Dudek J., and Zimmermann R. (2010) Evolutionary gain of function for the ER membrane protein Sec62 from yeast to humans. Mol. Biol. Cell 21, 691–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Jadhav B., McKenna M., Johnson N., High S., Sinning I., and Pool M. R. (2015) Mammalian SRP receptor switches the Sec61 translocase from Sec62 to SRP-dependent translocation. Nat. Commun. 6, 10133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Klionsky D. J., Abdalla F. C., Abeliovich H., Abraham R. T., Acevedo-Arozena A., Adeli K., Agholme L., Agnello M., Agostinis P., Aguirre-Ghiso J. A., Ahn H. J., Ait-Mohamed O., Ait-Si-Ali S., Akematsu T., Akira S., Al-Younes H. M., Al-Zeer M. A., Albert M. L., Albin R. L., Alegre-Abarrategui J., Aleo M. F., Alirezaei M., Almasan A., Almonte-Becerril M., Amano A., Amaravadi R., Amarnath S., Amer A. O., Andrieu-Abadie N., Anantharam V., Ann D. K., Anoopkumar-Dukie S., Aoki H., Apostolova N., Arancia G., Aris J. P., Asanuma K., Asare N. Y., Ashida H., Askanas V., Askew D. S., Auberger P., Baba M., Backues S. K., Baehrecke E. H., Bahr B. A., Bai X. Y., Bailly Y., Baiocchi R., Baldini G., Balduini W., Ballabio A., Bamber B. A., Bampton E. T., Banhegyi G., Bartholomew C. R., Bassham D. C., Bast R. C. Jr., Batoko H., Bay B. H., Beau I., Bechet D. M., Begley T. J., Behl C., Behrends C., Bekri S., Bellaire B., Bendall L. J., Benetti L., Berliocchi L., Bernardi H., Bernassola F., Besteiro S., Bhatia-Kissova I., Bi X., Biard-Piechaczyk M., Blum J. S., Boise L. H., Bonaldo P., Boone D. L., Bornhauser B. C., Bortoluci K. R., Bossis I., Bost F., Bourquin J. P., Boya P., Boyer-Guittaut M., Bozhkov P. V., Brady N. R., Brancolini C., Brech A., Brenman J. E., Brennand A., Bresnick E. H., Brest P., Bridges D., Bristol M. L., Brookes P. S., Brown E. J., Brumell J. H., Brunetti-Pierri N., Brunk U. T., Bulman D. E., Bultman S. J., Bultynck G., Burbulla L. F., Bursch W., Butchar J. P., Buzgariu W., Bydlowski S. P., Cadwell K., Cahova M., Cai D., Cai J., Cai Q., Calabretta B., Calvo-Garrido J., Camougrand N., Campanella M., Campos-Salinas J., Candi E., Cao L., Caplan A. B., Carding S. R., Cardoso S. M., Carew J. S., Carlin C. R., Carmignac V., Carneiro L. A., Carra S., Caruso R. A., Casari G., Casas C., Castino R., Cebollero E., Cecconi F., Celli J., Chaachouay H., Chae H. J., Chai C. Y., Chan D. C., Chan E. Y., Chang R. C., Che C. M., Chen C. C., Chen G. C., Chen G. Q., Chen M., Chen Q., Chen S. S., Chen W., Chen X., Chen X., Chen X., Chen Y. G., Chen Y., Chen Y., Chen Y. J., Chen Z., Cheng A., Cheng C. H., Cheng Y., Cheong H., Cheong J. H., Cherry S., Chess-Williams R., Cheung Z. H., Chevet E., Chiang H. L., Chiarelli R., Chiba T., Chin L. S., Chiou S. H., Chisari F. V., Cho C. H., Cho D. H., Choi A. M., Choi D., Choi K. S., Choi M. E., Chouaib S., Choubey D., Choubey V., Chu C. T., Chuang T. H., Chueh S. H., Chun T., Chwae Y. J., Chye M. L., Ciarcia R., Ciriolo M. R., Clague M. J., Clark R. S., Clarke P. G., Clarke R., Codogno P., Coller H. A., Colombo M. I., Comincini S., Condello M., Condorelli F., Cookson M. R., Coombs G. H., Coppens I., Corbalan R., Cossart P., Costelli P., Costes S., Coto-Montes A., Couve E., Coxon F. P., Cregg J. M., Crespo J. L., Cronje M. J., Cuervo A. M., Cullen J. J., Czaja M. J., D'Amelio M., Darfeuille-Michaud A., Davids L. M., Davies F. E., De Felici M., de Groot J. F., de Haan C. A., De Martino L., De Milito A., De Tata V., Debnath J., Degterev A., Dehay B., Delbridge L. M., Demarchi F., Deng Y. Z., Dengjel J., Dent P., Denton D., Deretic V., Desai S. D., Devenish R. J., Di Gioacchino M., Di Paolo G., Di Pietro C., Diaz-Araya G., Diaz-Laviada I., Diaz-Meco M. T., Diaz-Nido J., Dikic I., Dinesh-Kumar S. P., Ding W. X., Distelhorst C. W., Diwan A., Djavaheri-Mergny M., Dokudovskaya S., Dong Z., Dorsey F. C., Dosenko V., Dowling J. J., Doxsey S., Dreux M., Drew M. E., Duan Q., Duchosal M. A., Duff K., Dugail I., Durbeej M., Duszenko M., Edelstein C. L., Edinger A. L., Egea G., Eichinger L., Eissa N. T., Ekmekcioglu S., El-Deiry W. S., Elazar Z., Elgendy M., Ellerby L. M., Eng K. E., Engelbrecht A. M., Engelender S., Erenpreisa J., Escalante R., Esclatine A., Eskelinen E. L., Espert L., Espina V., Fan H., Fan J., Fan Q. W., Fan Z., Fang S., Fang Y., Fanto M., Fanzani A., Farkas T., Farre J. C., Faure M., Fechheimer M., Feng C. G., Feng J., Feng Q., Feng Y., Fesus L., Feuer R., Figueiredo-Pereira M. E., Fimia G. M., Fingar D. C., Finkbeiner S., Finkel T., Finley K. D., Fiorito F., Fisher E. A., Fisher P. B., Flajolet M., Florez-McClure M. L., Florio S., Fon E. A., Fornai F., Fortunato F., Fotedar R., Fowler D. H., Fox H. S., Franco R., Frankel L. B., Fransen M., Fuentes J. M., Fueyo J., Fujii J., Fujisaki K., Fujita E., Fukuda M., Furukawa R. H., Gaestel M., Gailly P., Gajewska M., Galliot B., Galy V., Ganesh S., Ganetzky B., Ganley I. G., Gao F. B., Gao G. F., Gao J., Garcia L., Garcia-Manero G., Garcia-Marcos M., Garmyn M., Gartel A. L., Gatti E., Gautel M., Gawriluk T. R., Gegg M. E., Geng J., Germain M., Gestwicki J. E., Gewirtz D. A., Ghavami S., Ghosh P., Giammarioli A. M., Giatromanolaki A. N., Gibson S. B., Gilkerson R. W., Ginger M. L., Ginsberg H. N., Golab J., Goligorsky M. S., Golstein P., Gomez-Manzano C., Goncu E., Gongora C., Gonzalez C. D., Gonzalez R., Gonzalez-Estevez C., Gonzalez-Polo R. A., Gonzalez-Rey E., Gorbunov N. V., Gorski S., Goruppi S., Gottlieb R. A., Gozuacik D., Granato G. E., Grant G. D., Green K. N., Gregorc A., Gros F., Grose C., Grunt T. W., Gual P., Guan J. L., Guan K. L., Guichard S. M., Gukovskaya A. S., Gukovsky I., Gunst J., Gustafsson A. B., Halayko A. J., Hale A. N., Halonen S. K., Hamasaki M., Han F., Han T., Hancock M. K., Hansen M., Harada H., Harada M., Hardt S. E., Harper J. W., Harris A. L., Harris J., Harris S. D., Hashimoto M., Haspel J. A., Hayashi S., Hazelhurst L. A., He C., He Y. W., Hebert M. J., Heidenreich K. A., Helfrich M. H., Helgason G. V., Henske E. P., Herman B., Herman P. K., Hetz C., Hilfiker S., Hill J. A., Hocking L. J., Hofman P., Hofmann T. G., Hohfeld J., Holyoake T. L., Hong M. H., Hood D. A., Hotamisligil G. S., Houwerzijl E. J., Hoyer-Hansen M., Hu B., Hu C. A., Hu H. M., Hua Y., Huang C., Huang J., Huang S., Huang W. P., Huber T. B., Huh W. K., Hung T. H., Hupp T. R., Hur G. M., Hurley J. B., Hussain S. N., Hussey P. J., Hwang J. J., Hwang S., Ichihara A., Ilkhanizadeh S., Inoki K., Into T., Iovane V., Iovanna J. L., Ip N. Y., Isaka Y., Ishida H., Isidoro C., Isobe K., Iwasaki A., Izquierdo M., Izumi Y., Jaakkola P. M., Jaattela M., Jackson G. R., Jackson W. T., Janji B., Jendrach M., Jeon J. H., Jeung E. B., Jiang H., Jiang H., Jiang J. X., Jiang M., Jiang Q., Jiang X., Jiang X., Jimenez A., Jin M., Jin S., Joe C. O., Johansen T., Johnson D. E., Johnson G. V., Jones N. L., Joseph B., Joseph S. K., Joubert A. M., Juhasz G., Juillerat-Jeanneret L., Jung C. H., Jung Y. K., Kaarniranta K., Kaasik A., Kabuta T., Kadowaki M., Kagedal K., Kamada Y., Kaminskyy V. O., Kampinga H. H., Kanamori H., Kang C., Kang K. B., Kang K. I., Kang R., Kang Y. A., Kanki T., Kanneganti T. D., Kanno H., Kanthasamy A. G., Kanthasamy A., Karantza V., Kaushal G. P., Kaushik S., Kawazoe Y., Ke P. Y., Kehrl J. H., Kelekar A., Kerkhoff C., Kessel D. H., Khalil H., Kiel J. A., Kiger A. A., Kihara A., Kim D. R., Kim D. H., Kim D. H., Kim E. K., Kim H. R., Kim J. S., Kim J. H., Kim J. C., Kim J. K., Kim P. K., Kim S. W., Kim Y. S., Kim Y., Kimchi A., Kimmelman A. C., King J. S., Kinsella T. J., Kirkin V., Kirshenbaum L. A., Kitamoto K., Kitazato K., Klein L., Klimecki W. T., Klucken J., Knecht E., Ko B. C., Koch J. C., Koga H., Koh J. Y., Koh Y. H., Koike M., Komatsu M., Kominami E., Kong H. J., Kong W. J., Korolchuk V. I., Kotake Y., Koukourakis M. I., Kouri Flores J. B., Kovacs A. L., Kraft C., Krainc D., Kramer H., Kretz-Remy C., Krichevsky A. M., Kroemer G., Kruger R., Krut O., Ktistakis N. T., Kuan C. Y., Kucharczyk R., Kumar A., Kumar R., Kumar S., Kundu M., Kung H. J., Kurz T., Kwon H. J., La Spada A. R., Lafont F., Lamark T., Landry J., Lane J. D., Lapaquette P., Laporte J. F., Laszlo L., Lavandero S., Lavoie J. N., Layfield R., Lazo P. A., Le W., Le Cam L., Ledbetter D. J., Lee A. J., Lee B. W., Lee G. M., Lee J., Lee J. H., Lee M., Lee M. S., Lee S. H., Leeuwenburgh C., Legembre P., Legouis R., Lehmann M., Lei H. Y., Lei Q. Y., Leib D. A., Leiro J., Lemasters J. J., Lemoine A., Lesniak M. S., Lev D., Levenson V. V., Levine B., Levy E., Li F., Li J. L., Li L., Li S., Li W., Li X. J., Li Y. B., Li Y. P., Liang C., Liang Q., Liao Y. F., Liberski P. P., Lieberman A., Lim H. J., Lim K. L., Lim K., Lin C. F., Lin F. C., Lin J., Lin J. D., Lin K., Lin W. W., Lin W. C., Lin Y. L., Linden R., Lingor P., Lippincott-Schwartz J., Lisanti M. P., Liton P. B., Liu B., Liu C. F., Liu K., Liu L., Liu Q. A., Liu W., Liu Y. C., Liu Y., Lockshin R. A., Lok C. N., Lonial S., Loos B., Lopez-Berestein G., Lopez-Otin C., Lossi L., Lotze M. T., Low P., Lu B., Lu B., Lu B., Lu Z., Luciano F., Lukacs N. W., Lund A. H., Lynch-Day M. A., Ma Y., Macian F., MacKeigan J. P., Macleod K. F., Madeo F., Maiuri L., Maiuri M. C., Malagoli D., Malicdan M. C., Malorni W., Man N., Mandelkow E. M., Manon S., Manov I., Mao K., Mao X., Mao Z., Marambaud P., Marazziti D., Marcel Y. L., Marchbank K., Marchetti P., Marciniak S. J., Marcondes M., Mardi M., Marfe G., Marino G., Markaki M., Marten M. R., Martin S. J., Martinand-Mari C., Martinet W., Martinez-Vicente M., Masini M., Matarrese P., Matsuo S., Matteoni R., Mayer A., Mazure N. M., McConkey D. J., McConnell M. J., McDermott C., McDonald C., McInerney G. M., McKenna S. L., McLaughlin B., McLean P. J., McMaster C. R., McQuibban G. A., Meijer A. J., Meisler M. H., Melendez A., Melia T. J., Melino G., Mena M. A., Menendez J. A., Menna-Barreto R. F., Menon M. B., Menzies F. M., Mercer C. A., Merighi A., Merry D. E., Meschini S., Meyer C. G., Meyer T. F., Miao C. Y., Miao J. Y., Michels P. A., Michiels C., Mijaljica D., Milojkovic A., Minucci S., Miracco C., Miranti C. K., Mitroulis I., Miyazawa K., Mizushima N., Mograbi B., Mohseni S., Molero X., Mollereau B., Mollinedo F., Momoi T., Monastyrska I., Monick M. M., Monteiro M. J., Moore M. N., Mora R., Moreau K., Moreira P. I., Moriyasu Y., Moscat J., Mostowy S., Mottram J. C., Motyl T., Moussa C. E., Muller S., Muller S., Munger K., Munz C., Murphy L. O., Murphy M. E., Musaro A., Mysorekar I., Nagata E., Nagata K., Nahimana A., Nair U., Nakagawa T., Nakahira K., Nakano H., Nakatogawa H., Nanjundan M., Naqvi N. I., Narendra D. P., Narita M., Navarro M., Nawrocki S. T., Nazarko T. Y., Nemchenko A., Netea M. G., Neufeld T. P., Ney P. A., Nezis I. P., Nguyen H. P., Nie D., Nishino I., Nislow C., Nixon R. A., Noda T., Noegel A. A., Nogalska A., Noguchi S., Notterpek L., Novak I., Nozaki T., Nukina N., Nurnberger T., Nyfeler B., Obara K., Oberley T. D., Oddo S., Ogawa M., Ohashi T., Okamoto K., Oleinick N. L., Oliver F. J., Olsen L. J., Olsson S., Opota O., Osborne T. F., Ostrander G. K., Otsu K., Ou J. H., Ouimet M., Overholtzer M., Ozpolat B., Paganetti P., Pagnini U., Pallet N., Palmer G. E., Palumbo C., Pan T., Panaretakis T., Pandey U. B., Papackova Z., Papassideri I., Paris I., Park J., Park O. K., Parys J. B., Parzych K. R., Patschan S., Patterson C., Pattingre S., Pawelek J. M., Peng J., Perlmutter D. H., Perrotta I., Perry G., Pervaiz S., Peter M., Peters G. J., Petersen M., Petrovski G., Phang J. M., Piacentini M., Pierre P., Pierrefite-Carle V., Pierron G., Pinkas-Kramarski R., Piras A., Piri N., Platanias L. C., Poggeler S., Poirot M., Poletti A., Pous C., Pozuelo-Rubio M., Praetorius-Ibba M., Prasad A., Prescott M., Priault M., Produit-Zengaffinen N., Progulske-Fox A., Proikas-Cezanne T., Przedborski S., Przyklenk K., Puertollano R., Puyal J., Qian S. B., Qin L., Qin Z. H., Quaggin S. E., Raben N., Rabinowich H., Rabkin S. W., Rahman I., Rami A., Ramm G., Randall G., Randow F., Rao V. A., Rathmell J. C., Ravikumar B., Ray S. K., Reed B. H., Reed J. C., Reggiori F., Regnier-Vigouroux A., Reichert A. S., Reiners J. J. Jr., Reiter R. J., Ren J., Revuelta J. L., Rhodes C. J., Ritis K., Rizzo E., Robbins J., Roberge M., Roca H., Roccheri M. C., Rocchi S., Rodemann H. P., Rodriguez de Cordoba S., Rohrer B., Roninson I. B., Rosen K., Rost-Roszkowska M. M., Rouis M., Rouschop K. M., Rovetta F., Rubin B. P., Rubinsztein D. C., Ruckdeschel K., Rucker E. B. 3rd, Rudich A., Rudolf E., Ruiz-Opazo N., Russo R., Rusten T. E., Ryan K. M., Ryter S. W., Sabatini D. M., Sadoshima J., Saha T., Saitoh T., Sakagami H., Sakai Y., Salekdeh G. H., Salomoni P., Salvaterra P. M., Salvesen G., Salvioli R., Sanchez A. M., Sanchez-Alcazar J. A., Sanchez-Prieto R., Sandri M., Sankar U., Sansanwal P., Santambrogio L., Saran S., Sarkar S., Sarwal M., Sasakawa C., Sasnauskiene A., Sass M., Sato K., Sato M., Schapira A. H., Scharl M., Schatzl H. M., Scheper W., Schiaffino S., Schneider C., Schneider M. E., Schneider-Stock R., Schoenlein P. V., Schorderet D. F., Schuller C., Schwartz G. K., Scorrano L., Sealy L., Seglen P. O., Segura-Aguilar J., Seiliez I., Seleverstov O., Sell C., Seo J. B., Separovic D., Setaluri V., Setoguchi T., Settembre C., Shacka J. J., Shanmugam M., Shapiro I. M., Shaulian E., Shaw R. J., Shelhamer J. H., Shen H. M., Shen W. C., Sheng Z. H., Shi Y., Shibuya K., Shidoji Y., Shieh J. J., Shih C. M., Shimada Y., Shimizu S., Shintani T., Shirihai O. S., Shore G. C., Sibirny A. A., Sidhu S. B., Sikorska B., Silva-Zacarin E. C., Simmons A., Simon A. K., Simon H. U., Simone C., Simonsen A., Sinclair D. A., Singh R., Sinha D., Sinicrope F. A., Sirko A., Siu P. M., Sivridis E., Skop V., Skulachev V. P., Slack R. S., Smaili S. S., Smith D. R., Soengas M. S., Soldati T., Song X., Sood A. K., Soong T. W., Sotgia F., Spector S. A., Spies C. D., Springer W., Srinivasula S. M., Stefanis L., Steffan J. S., Stendel R., Stenmark H., Stephanou A., Stern S. T., Sternberg C., Stork B., Stralfors P., Subauste C. S., Sui X., Sulzer D., Sun J., Sun S. Y., Sun Z. J., Sung J. J., Suzuki K., Suzuki T., Swanson M. S., Swanton C., Sweeney S. T., Sy L. K., Szabadkai G., Tabas I., Taegtmeyer H., Tafani M., Takacs-Vellai K., Takano Y., Takegawa K., Takemura G., Takeshita F., Talbot N. J., Tan K. S., Tanaka K., Tanaka K., Tang D., Tang D., Tanida I., Tannous B. A., Tavernarakis N., Taylor G. S., Taylor G. A., Taylor J. P., Terada L. S., Terman A., Tettamanti G., Thevissen K., Thompson C. B., Thorburn A., Thumm M., Tian F., Tian Y., Tocchini-Valentini G., Tolkovsky A. M., Tomino Y., Tonges L., Tooze S. A., Tournier C., Tower J., Towns R., Trajkovic V., Travassos L. H., Tsai T. F., Tschan M. P., Tsubata T., Tsung A., Turk B., Turner L. S., Tyagi S. C., Uchiyama Y., Ueno T., Umekawa M., Umemiya-Shirafuji R., Unni V. K., Vaccaro M. I., Valente E. M., Van den Berghe G., van der Klei I. J., van Doorn W., van Dyk L. F., van Egmond M., van Grunsven L. A., Vandenabeele P., Vandenberghe W. P., Vanhorebeek I., Vaquero E. C., Velasco G., Vellai T., Vicencio J. M., Vierstra R. D., Vila M., Vindis C., Viola G., Viscomi M. T., Voitsekhovskaja O. V., von Haefen C., Votruba M., Wada K., Wade-Martins R., Walker C. L., Walsh C. M., Walter J., Wan X. B., Wang A., Wang C., Wang D., Wang F., Wang F., Wang G., Wang H., Wang H. G., Wang H. D., Wang J., Wang K., Wang M., Wang R. C., Wang X., Wang X., Wang Y. J., Wang Y., Wang Z., Wang Z. C., Wang Z., Wansink D. G., Ward D. M., Watada H., Waters S. L., Webster P., Wei L., Weihl C. C., Weiss W. A., Welford S. M., Wen L. P., Whitehouse C. A., Whitton J. L., Whitworth A. J., Wileman T., Wiley J. W., Wilkinson S., Willbold D., Williams R. L., Williamson P. R., Wouters B. G., Wu C., Wu D. C., Wu W. K., Wyttenbach A., Xavier R. J., Xi Z., Xia P., Xiao G., Xie Z., Xie Z., Xu D. Z., Xu J., Xu L., Xu X., Yamamoto A., Yamamoto A., Yamashina S., Yamashita M., Yan X., Yanagida M., Yang D. S., Yang E., Yang J. M., Yang S. Y., Yang W., Yang W. Y., Yang Z., Yao M. C., Yao T. P., Yeganeh B., Yen W. L., Yin J. J., Yin X. M., Yoo O. J., Yoon G., Yoon S. Y., Yorimitsu T., Yoshikawa Y., Yoshimori T., Yoshimoto K., You H. J., Youle R. J., Younes A., Yu L., Yu L., Yu S. W., Yu W. H., Yuan Z. M., Yue Z., Yun C. H., Yuzaki M., Zabirnyk O., Silva-Zacarin E., Zacks D., Zacksenhaus E., Zaffaroni N., Zakeri Z., Zeh H. J. 3rd, Zeitlin S. O., Zhang H., Zhang H. L., Zhang J., Zhang J. P., Zhang L., Zhang L., Zhang M. Y., Zhang X. D., Zhao M., Zhao Y. F., Zhao Y., Zhao Z. J., Zheng X., Zhivotovsky B., Zhong Q., Zhou C. Z., Zhu C., Zhu W. G., Zhu X. F., Zhu X., Zhu Y., Zoladek T., Zong W. X., Zorzano A., Zschocke J., and Zuckerbraun B. (2012) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8, 445–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Hou N., Yang Y., Scott I. C., and Lou X. (2017) The Sec domain protein Scfd1 facilitates trafficking of ECM components during chondrogenesis. Developmental Biol. 421, 8–15 [DOI] [PubMed] [Google Scholar]
- 97. Linders P. T., Horst C. V., Beest M. T., and van den Bogaart G. (2019) Stx5-mediated ER-golgi transport in mammals and yeast. Cells 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hui N., Nakamura N., Sonnichsen B., Shima D. T., Nilsson T., and Warren G. (1997) An isoform of the Golgi t-SNARE, syntaxin 5, with an endoplasmic reticulum retrieval signal. Mol. Biol. Cell 8, 1777–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Linxweiler M., Schick B., and Zimmermann R. (2017) Let's talk about Secs: Sec61, Sec62 and Sec63 in signal transduction, oncology and personalized medicine. Signal Transduct. Target Ther. 2, 17002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Igbaria A., Merksamer P. I., Trusina A., Tilahun F., Johnson J. R., Brandman O., Krogan N. J., Weissman J. S., and Papa F. R. (2019) Chaperone-mediated reflux of secretory proteins to the cytosol during endoplasmic reticulum stress. Proc. Natl. Acad. Sci. U S A 116, 11291–11298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Sarkar D. (2013) AEG-1/MTDH/LYRIC in liver cancer. Adv. Cancer Res. 120, 193–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Youn J. Y., Dunham W. H., Hong S. J., Knight J. D. R., Bashkurov M., Chen G. I., Bagci H., Rathod B., MacLeod G., Eng S. W. M., Angers S., Morris Q., Fabian M., Cote J. F., and Gingras A. C. (2018) High-density proximity mapping reveals the subcellular organization of mRNA-associated granules and bodies. Mol. Cell 69, 517–532.e11 [DOI] [PubMed] [Google Scholar]
- 103. Guo F., Wan L., Zheng A., Stanevich V., Wei Y., Satyshur K. A., Shen M., Lee W., Kang Y., and Xing Y. (2014) Structural insights into the tumor-promoting function of the MTDH-SND1 complex. Cell Reports 8, 1704–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Li C. L., Yang W. Z., Chen Y. P., and Yuan H. S. (2008) Structural and functional insights into human Tudor-SN, a key component linking RNA interference and editing. Nucleic Acids Res. 36, 3579–3589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Ohsumi T., Ichimura T., Sugano H., Omata S., Isobe T., and Kuwano R. (1993) Ribosome-binding protein p34 is a member of the leucine-rich-repeat-protein superfamily. Biochem. J. 294, 465–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Protter D. S. W., and Parker R. (2016) Principles and properties of stress granules. Trends Cell Biol. 26, 668–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Khong A., Matheny T., Jain S., Mitchell S. F., Wheeler J. R., and Parker R. (2017) The stress granule transcriptome reveals principles of mRNA accumulation in stress granules. Mol Cell 68, 808–820.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Mellacheruvu D., Wright Z., Couzens A. L., Lambert J. P., St-Denis N. A., Li T., Miteva Y. V., Hauri S., Sardiu M. E., Low T. Y., Halim V. A., Bagshaw R. D., Hubner N. C., Al-Hakim A., Bouchard A., Faubert D., Fermin D., Dunham W. H., Goudreault M., Lin Z. Y., Badillo B. G., Pawson T., Durocher D., Coulombe B., Aebersold R., Superti-Furga G., Colinge J., Heck A. J., Choi H., Gstaiger M., Mohammed S., Cristea I. M., Bennett K. L., Washburn M. P., Raught B., Ewing R. M., Gingras A. C., and Nesvizhskii A. I. (2013) The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 10, 730–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Schwarz D. S., and Blower M. D. (2016) The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol. Life Sci. 73, 79–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Murley A., and Nunnari J. (2016) The Emerging Network of Mitochondria-Organelle Contacts. Mol. Cell 61, 648–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Wu H., Carvalho P., and Voeltz G. K. (2018) Here, there, and everywhere: The importance of ER membrane contact sites. Science 361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Cohen S., Valm A. M., and Lippincott-Schwartz J. (2018) Interacting organelles. Curr. Opin. Cell Biol. 53, 84–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Lee J. E., Cathey P. I., Wu H., Parker R., and Voeltz G. K. (2020) Endoplasmic reticulum contact sites regulate the dynamics of membraneless organelles. Science 367, eaay7108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Kreibich G., Ulrich B. L., and Sabatini D. D. (1978) Proteins of rough microsomal membranes related to ribosome binding. I. Identification of ribophorins I and II, membrane proteins characteristics of rough microsomes. J. Cell Biol. 77, 464–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Nilsson I. M., and and von Heinje G. (1993) Determination of the distance between the oligosaccharyl transferase ative stie and the endoplasmic reticulum membrane. J. Biol. Chem. 268, 5798–5801 [PubMed] [Google Scholar]
- 116. Deshaies R. J., Sanders S. L., Feldheim D. A., and Schekman R. (1991) Assembly of yeast Sec proteins involved in translocation into the endoplasmic reticulum into a membrane-bound multisubunit complex. Nature 349, 806–808 [DOI] [PubMed] [Google Scholar]
- 117. Afshar N., Black B. E., and Paschal B. M. (2005) Retrotranslocation of the chaperone calreticulin from the endoplasmic reticulum lumen to the cytosol. Mol. Cell. Biol. 25, 8844–8853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Duriez M., Rossignol J. M., and Sitterlin D. (2008) The hepatitis B virus precore protein is retrotransported from endoplasmic reticulum (ER) to cytosol through the ER-associated degradation pathway. J. Biol. Chem. 283, 32352–32360 [DOI] [PubMed] [Google Scholar]
- 119. Halperin L., Jung J., and Michalak M. (2014) The many functions of the endoplasmic reticulum chaperones and folding enzymes. IUBMB Life 66, 318–326 [DOI] [PubMed] [Google Scholar]
- 120. Shaffer K. L., Sharma A., Snapp E. L., and Hegde R. S. (2005) Regulation of protein compartmentalization expands the diversity of protein function. Dev. Cell 9, 545–554 [DOI] [PubMed] [Google Scholar]
- 121. Burrus L. W., and McMahon A. P. (1995) Biochemical analysis of murine Wnt proteins reveals both shared and distinct properties. Exp. Cell Res. 220, 363–373 [DOI] [PubMed] [Google Scholar]
- 122. Moti N., Yu J., Boncompain G., Perez F., and Virshup D. M. (2019) Wnt traffic from endoplasmic reticulum to filopodia. PloS One 14, e0212711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Zoltewicz J. S., Ashique A. M., Choe Y., Lee G., Taylor S., Phamluong K., Solloway M., and Peterson A. S. (2009) Wnt signaling is regulated by endoplasmic reticulum retention. PloS One 4, e6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Ichimura T., Ohsumi T., Shindo Y., Ohwada T., Yagame H., Momose Y., Omata S., and Sugano H. (1992) Isolation and some properties of a 34kD membrane protein that may be essential for ribosome binding in rat liver rough microsomes. FEBS Lett. 296, 7–10 [DOI] [PubMed] [Google Scholar]
- 125. Tatematsu M., Funami K., Ishii N., Seya T., Obuse C., and Matsumoto M. (2015) LRRC59 regulates trafficking of nucleic acid-sensing TLRs from the endoplasmic reticulum via association with UNC93B1. J. Immunol. 195, 4933–4942 [DOI] [PubMed] [Google Scholar]
- 126. Xian H., Yang S., Jin S., Zhang Y., and Cui J. (2020) LRRC59 modulates type I interferon signaling by restraining the SQSTM1/p62-mediated autophagic degradation of pattern recognition receptor DDX58/RIG-I. Autophagy 16, 408–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The quantitative mass spectrometry data has been made publicly available using MassIVE at: ftp://massive.ucsd.edu/MSV000085009/.