

We report on an engineered yeast strain that allows for the facile isolation of the proteasome and associated proteasome substrates after rapid inactivation. Nano-flow liquid chromatography tandem mass spectrometry was performed, and the identification of every proteasome resident was confirmed. When inactivated, known and new proteasome substrates were identified, including an ER-resident enzyme in the sterol biosynthetic pathway. Modification of the developed tools described in this report can be used for a variety of future applications.
Keywords: Protein degradation, protein folding, mass spectrometry, yeast, protein complex analysis, ubiquitin, ERAD, proteasome
Graphical Abstract

Highlights
Epitope-tagging of a proteasome subunit allows for facile immuno-isolation.
An engineered yeast strain permits capture of proteasome-associated substrates.
MS/MS identified all 33 resident proteasome subunits in the 20S and 19S particles.
Analysis of associated proteins and characterization of newly identified ERAD substrate.
Abstract
Studies in the yeast Saccharomyces cerevisiae have helped define mechanisms underlying the activity of the ubiquitin–proteasome system (UPS), uncover the proteasome assembly pathway, and link the UPS to the maintenance of cellular homeostasis. However, the spectrum of UPS substrates is incompletely defined, even though multiple techniques—including MS—have been used. Therefore, we developed a substrate trapping proteomics workflow to identify previously unknown UPS substrates. We first generated a yeast strain with an epitope tagged proteasome subunit to which a proteasome inhibitor could be applied. Parallel experiments utilized inhibitor insensitive strains or strains lacking the tagged subunit. After affinity isolation, enriched proteins were resolved, in-gel digested, and analyzed by high resolution liquid chromatography-tandem MS. A total of 149 proteasome partners were identified, including all 33 proteasome subunits. When we next compared data between inhibitor sensitive and resistant cells, 27 proteasome partners were significantly enriched. Among these proteins were known UPS substrates and proteins that escort ubiquitinated substrates to the proteasome. We also detected Erg25 as a high-confidence partner. Erg25 is a methyl oxidase that converts dimethylzymosterol to zymosterol, a precursor of the plasma membrane sterol, ergosterol. Because Erg25 is a resident of the endoplasmic reticulum (ER) and had not previously been directly characterized as a UPS substrate, we asked whether Erg25 is a target of the ER associated degradation (ERAD) pathway, which most commonly mediates proteasome-dependent destruction of aberrant proteins. As anticipated, Erg25 was ubiquitinated and associated with stalled proteasomes. Further, Erg25 degradation depended on ERAD-associated ubiquitin ligases and was regulated by sterol synthesis. These data expand the cohort of lipid biosynthetic enzymes targeted for ERAD, highlight the role of the UPS in maintaining ER function, and provide a novel tool to uncover other UPS substrates via manipulations of our engineered strain.
The 26S proteasome, a multi-catalytic cytosolic protease, serves as the major proteolytic factory in eukaryotes. The 26S particle is formed by two species, a 20S core particle, which houses pairs of trypsin, chymotrypsin, and caspase-like enzymes, and two 19S “caps” (or PA700 particles) that abut each end of the core particle (1, 2). Based on its abundance and robust activity, the targeting and destruction of substrates to this protease are tightly controlled because the majority of proteasome substrates are covalently modified by an isopeptide ubiquitin (Ub) linkage. Ub is added onto a target via a cascade of Ub activating, conjugating, and ligating enzymes, and covalent Ub appendages are formed between the C terminus of Ub and most commonly the ε amino group on a Lys in a protein substrate or on a previously attached Ub (3). Although numerous varieties of poly-Ub “chains” have been described—due to the presence of seven internal Lys residues in Ub as well as the N terminus—the most common poly-Ub linkage recognized by the proteasome is formed by the sequential addition of the C terminus of Ub onto a Lys in Ub at position 48 (4–6). Early results indicated that a poly-Ub chain of at least four Ub substituents is required for proteasome recognition (7).
As a result of fine-tuned mechanisms underlying the recognition and Ub-tagging of substrates, the Ub-proteasome system (UPS) plays a critical role in myriad cellular processes, which include the cell cycle, gene expression, immune system function, signal transduction, and cell fate (8). The UPS also plays a critical role in protein “quality control,” which mediates the destruction of damaged, mutated, unassembled, and improperly folded proteins in the cell. Perhaps not surprisingly, the UPS is linked to an ever-growing number of human diseases.
One prominent protein quality control pathway is known as endoplasmic reticulum (ER) associated degradation, or ERAD. Approximately one-third of all proteins in eukaryotes are synthesized at the ER, including all membrane and the majority of secreted proteins (9). The maintenance of ER homeostasis requires the high fidelity folding, post-translational modification, and assembly of proteins in the ER (10–13). During ERAD, proteins are first selected by molecular chaperones and chaperone-like proteins that reside in or on the cytosolic face of the ER. After recognition, the proteins are polyubiquitinated. However, because the Ub machinery is absent from the ER lumen, many ERAD substrates—such as soluble proteins that reside completely within the ER—must be extracted or “retrotranslocated” prior to Ub tagging (14, 15). In contrast, integral membrane proteins in the ER contain domains facing the cytosol that can be polyubiquitinated before retrotranslocation and proteasome-mediated destruction. Based on its central role in maintaining ER and secretory protein homeostasis, the ERAD pathway has also been linked to numerous human diseases (16).
Initially, ERAD substrates were identified by the analysis of mutated secreted proteins, primarily in yeast, along with disease-causing proteins in human cells (17–21). Select ERAD substrates were also identified via MS. For example, a transporter assembly factor, CD147, was uncovered as an ERAD substrate in human cells by isolating partners of an ER lectin in cells lacking a downstream component (22). In another example, Stable Isotope Labeling by/with Amino acids in Cell culture (SILAC) of yeast containing or lacking components of the ER-associated ubiquitination machinery led to the isolation of Erg1, an enzyme in the ergosterol pathway, as a regulated ERAD substrate (23). A homolog of Erg1 that acts in the sterol biosynthetic pathway in higher cells was targeted by homologous components of the ubiquitination machinery in human cells, validating the power of the yeast-based screen. Moreover, the capture of ubiquitinated proteins after treating human cells with a retrotranslocation inhibitor and SILAC analysis identified other ERAD substrates (24). Many of these more recently identified substrates are enzymes whose steady-state levels are controlled by ERAD(25–27). Nevertheless, there are undoubtedly undiscovered ERAD substrates because >7,000 proteins in a human cell associate at some point during their biogenesis with the ER (28). In addition, due to the expansion of genome datasets, a plethora of mutations and polymorphisms have been identified in secreted and membranes proteins, and many of these variants may also be targeted for ERAD. Together, there is a growing appreciation that metabolic pathway components residing in the ER as well as aberrant proteins are subject to ERAD.
In this study, we wished to identify previously uncharacterized UPS substrates using an approach that, to our knowledge, had not previously been pursued in yeast. First, we created a yeast strain that lacked a drug efflux pump so a specific inhibitor of the proteasome could be applied. Next, a proteasome subunit was tagged with an epitope and the gene encoding the modified subunit was integrated into the chromosome in the drug-sensitive yeast strain. We then performed a series of pulldown assays using experimental and control strains treated with a proteasome inhibitor, and label-free differential MS was used to compare isolated proteins from each condition. Data obtained in the presence of a proteasome inhibitor identified known UPS substrates along with an ER resident protein, Erg25, which had not previously been directly characterized as a UPS target. Subsequent biochemical and genetic analyses confirmed that Erg25 associates with the proteasome, is polyubiquitinated, and is stabilized in a yeast strain lacking ERAD-associated E3 ligases, thereby establishing Erg25 as a bona fide ERAD substrate. Because this enzyme plays a critical role in the biosynthesis of a yeast sterol (29), our data expand the number of regulated, WT enzymes directed to the ERAD pathway and highlight the ER as a dynamic regulator of both protein synthesis and degradation.
EXPERIMENTAL PROCEDURES
Experimental Design and Statistical Rationale
Six biological replicates per condition were analyzed by GFP-affinity purification. MaxQuant software suite (version 1.6.6.0) (30, 31) was used to analyze the MS raw files. The statistical significances of the differences were determined using a two-sample Student's t test on the log2-transformed intensity values. A combination of statistical significance and fold change differences were used to select significant candidates.
Yeast Genetics
A yeast strain expressing GFP-tagged Pre8 from its endogenous promoter was obtained from the Yeast GFP Clone Collection (32) (Thermo Fisher Scientific, Waltham, MA). The parental WT strain used for the construction of the GFP clones was obtained from ATCC (ATCC 201388: MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0). Homologous recombination was used to disrupt the PDR5 locus in these two yeast backgrounds. In brief, a PDR5 deletion cassette was generated by PCR amplifying the NATMX cassette from the pFA6a–NAT-MX6 cassette, with primers containing homology to the 5' and 3' regions of the PDR5 locus (33). The resulting PCR product was purified and transformed into the yeast strain described above and plated on media containing the antibiotic, nourseothricin (NAT; clonNAT, Werner BioAgents, Jena, Germany) to select for integration of the NATMX cassette at the PDR5 locus. The genotypes of the resulting strains were confirmed by PCR amplification of the NAT cassette at the PDR5 locus. The Δpdr5 phenotype was confirmed by assessing the inhibition of the degradation of a known proteasome substrate, NBD2*, in response to MG132 treatment, as described (34). A plasmid containing a GFP-tagged control protein, SZ*, was used as described (35). Unless indicated otherwise, all yeast manipulations and growth conditions employed standard methods (36).
Pre8 Affinity Purification
The following yeast strains were used for the MS analysis: (1) those containing a WT copy of PDR5 and an untagged proteasome subunit (PRE8 cells), (2) those containing the proteasome-tagged subunit as well as the WT copy of PDR5 (PRE8:GFP cells), and (3) those lacking PDR5 but harboring the tagged proteasome subunit (PRE8:GFP Δpdr5; Fig. 1A). All three strains also expressed NBD2* (see above) from an introduced vector. The strains were grown to late log phase in selective media at 30 °C and MG132 (Selleckchem, Houston, TX) was added to a final concentration of 100 μm to all cultures for 1 h prior to harvesting. Approximately 20 OD600 units of cells were used for each GFP affinity purification. After incubation, cells were harvested by centrifugation at 2000 × g for 5 min and the cell pellets were washed twice with 5 ml of ice-cold PBS. Cell lysis was carried out in 1 ml of ice-cold lysis buffer (10 mM TrisCl, pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 5 mM MgCl2, 10 mM ATP, 0.5% Nonidet P-40, and an ATP regeneration mix (37)) in the presence of freshly added protease and phosphatase inhibitors (Thermo Fisher Scientific) using the FastPrep24 instrument and Lysing Matrix C (MP Biomedical, LLC, Santa Ana, CA). The disruption was achieved with 3 rounds of vigorous Vortex mixing at a speed setting of 6 meters/sec for 60 s. After centrifugation for 5 min at 4 °C at 16000 × g, the supernatants were transferred to new Eppendorf tubes containing 20 μl GFP-Trap®_A bead slurry (ChromoTek GmbH, 82152 Planegg-Martinsried, Germany) pre-washed with washing buffer (10 mM TrisCl, pH 7.5, 150 mM NaCl, 0.5 mM EDTA). After 2 h of end-over-end rotation at 4 °C, the GFP-Trap®_A beads were collected by centrifugation at 2000 × g for 2 min and then washed twice with 500 µL of ice-cold washing buffer. Bound proteins were eluted at 95 °C in 25 µL 2 × NuPAGE™ LDS Sample Buffer (Thermo Fisher Scientific) for 10 min. All 18 GFP affinity purifications (6 purifications per transformed cell type; Fig. 1A) were performed simultaneously on the same day.
Fig. 1.

Synthesis and characterization of yeast strains used in this study. A, Models of the yeast strains constructed for this study. As described under Experimental Procedures, where indicated a GFP tag was inserted at the 5′ end of the PRE8 locus and the PDR5 gene was replaced with a drug resistance cassette, NATMX. B, To confirm expression of a GFP tagged proteasome subunit (Pre8:GFP), yeast lysates from the indicated strains were subject to SDS-PAGE and Western blotting. Blots were probed with either anti-GFP or anti-Sec61 antibody, which was used as a loading control. C, Cycloheximide chase analyses were performed as described under Experimental Procedures using the PRE8:GFP Δpdr5 strain transformed with a known ERAD substrate, NBD2*, in the presence or absence of 50 μm MG132. Protein lysates were prepared and immunoblotted with anti-HA (NBD2*) or anti-G6P (anti-glucose-6-phosphate dehydrogenase), which was used as a loading control. Data represent the means of 8-10 experiments, +/− S.D. **, p < 0.01.
In-Gel Trypsin Digestion
In-gel trypsin digestion was carried out as previously described (38). In brief, eluates from the affinity purification were resolved by SDS-PAGE (NuPAGE™ 4–12% Bis-Tris Protein Gels, 1.0 mm, 10-well; Thermo Fisher Scientific) at 150 V for 10 min and stained with SimplyBlueTM SafeStain (Thermo Fisher Scientific). The stained region was excised, washed with HPLC water, and destained with 50% acetonitrile (ACN)/25 mM ammonium bicarbonate until no visible staining remained. Gel slices were dehydrated with 100% ACN and reduced with 10 mM DTT (DTT) at 56 °C for 1 h, which was followed by alkylation with 55 mM iodoacetamide (IAA) at room temperature for 45 min in the dark. The gel pieces were then dehydrated with 100% ACN to remove excess DTT and IAA. Next, 50 µL of 20 ng/µL trypsin in 25 mM ammonium bicarbonate was added for overnight digestion at 37 °C. The resulting tryptic peptides were extracted with 70% ACN/5% formic acid (FA), vacuum dried, and reconstituted in 18 µL 0.1% FA. A pooled instrument control sample was generated by combining equal volumes of the individual digests and analyzed repeatedly to monitor nLC-MS/MS performance throughout the duration of the experiment. To minimize systemic bias, a randomized block design was used to balance the sample analysis order by sample type (supplemental Table S1).
Nano-Flow Liquid Chromatography Tandem Mass Spectrometry
Tryptic peptides were analyzed with a nLC-MS/MS system consisting of a nanoACQUITY (Waters Corporation) nano-flow HPLC coupled to an Orbitrap Velos Pro hybrid ion trap mass spectrometer (Thermo Fisher Scientific, Inc.). For each nLC-MS/MS analysis, 1 µL of each peptide digest was injected onto a 0.075 × 250 mm PicoChipTM column (New Objective, Inc.) packed with 3 μm 120 Å Reprosil C18 chromatography media and terminated with an integrated 15 μm electrospray emitter. Reverse-phase separation was achieved with a 66 min linear gradient composition of 2–35% ACN/0.1% formic acid and a flow rate of 300 nL/min. A positive electrospray ionization voltage of 1.9 kV and a capillary temperature of 275 °C was used to ionize and desolvate the eluted peptides, respectively. The mass spectrometer was operated in the data-dependent acquisition mode that records one high-resolution mass spectrum (MS1) followed by low-resolution tandem mass spectra (MS2) for each of the 13 most abundant precursor ions detected in the MS1. All high-resolution MS1 spectra were acquired with a m/z range of 375–1800 Da, an Orbitrap resolution setting of 60,000, and an automatic gain control (AGC) target value of 1.0E06. Low resolution MS2 spectra were acquired using collision-induced dissociation (CID) and an AGC target value of 5.0E06. The acquisition of replicate MS2 spectra was minimized using a dynamic exclusion time of 90 s.
Label-Free Differential Mass Spectrometry
The nLC-MS/MS data were analyzed with the MaxQuant software suite (version 1.6.6.0) (30, 31). The Andromeda protein identification search engine and SwissProt Saccharomyces cerevisiae protein database (downloaded June, 2019 with 6,721 entries) was utilized with default settings for Orbitrap instruments unless specified. Briefly, the parameters used included a precursor mass tolerance of 20 ppm for the first search and 4.5 ppm for the main search, a product ion mass tolerance of 0.8 Da, and a minimum peptide length of 7 amino acids. Trypsin was set as the proteolytic enzyme with a maximum of two missed cleavages allowed. The enzyme specifically cleaves peptide bonds C-terminal of Arginine (R) and Lysine (K) if they are not followed by Proline (P). Carbamidomethylation of Cysteine (C) was set as a fixed modification. Oxidation of Methionine (M), deamination of Asparagine (N) and Glutamine (Q), GlyGly modification of Lysine (K), and acetylation of the protein N terminus were set as variable modification. Both isotope time correlation and theoretical isotope correlation were set at 0.4. Advanced peak splitting was turned on. The alignment time window was set at 20 min and match time window at 1.5 min. A 1% false discovery rate (FDR) was used to filter the peptide identification results. Quantification of the MS1 peptide intensity was performed for all identified peptides following retention time alignment and peak matching across samples, and these values are provided in supplemental Tables S2, S3, and S4 (protein groups with single peptide identification were excluded). The MS proteomics data have been deposited to the ProteomXchange (39) consortium via the MassIVE partner repository (also see “Data Availability” section, below).
In general, MaxQuant-calculated protein and peptide intensity values, statistical tests, fold-change cutoffs, and practical considerations were used to select Pre8 interacting partners, including proteasome particles, proteasome partners, and UPS substrates from nonspecific contaminants. Proteins with a single identified peptide were excluded from further data analysis. First, log2-transformed protein intensity values were subject to a Student's t test and a p-value cut-off were used to evaluate “protein-level” expression differences between experimental and control samples. Next, proteins with a protein-intensity fold-change greater than 2 when comparing PRE8:GFP or PRE8:GFP Δpdr5 to PRE8 protein intensity values were selected for additional analysis. Proteins with a protein-intensity fold-change greater than 2 and p value less than 0.01 when comparing the PRE8:GFP or PRE8:GFP Δpdr5 to Pre8 protein intensity values were selected for additional analysis. A protein was considered a bona fide Pre8 interacting partner if two or more peptides had p-values < 0.01 and fold-change > 2 based on peptide intensity values. To select putative UPS substrates, protein intensity values of Pre8-interacting partners in each sample were normalized by its Pre8 protein intensity, and a Student's t test on log2-transformed Pre8-adjusted protein intensity values was used to compare PRE8:GFP samples and PRE8:GFP Δpdr5 samples. A p-value cut-off < 0.05 and fold change greater than 1.5 was used for UPS substrate selection.
Immunoprecipitation Assays
The GFP-tagged Pre8 strain lacking PDR5, described above, was transformed with pIU2593 (2 μ URA PADH1-ERG25-HA) (40), a kind gift from the laboratory of Drs. Jerry Kaplan and Diane Ward (University of Utah), or a vector control (pRS416) (41). As a control, a Δpdr5 strain lacking GFP-tagged Pre8 was transformed with pIU2593. Overnight yeast cultures were grown at 30 °C in selective media overnight, diluted into 30 ml of media the next morning, grown for 2–3 h and then incubated for 1 h with 50 μm MG132 (to stabilize the substrate) prior to harvesting ∼30 OD600 of log phase cells by centrifugation (see above). Cell pellets were resuspended in 500 µL of buffer (50 mM Tris-Cl, pH 7, 150 mM NaCl, 1% TX-100, plus Roche Complete EDTA-Free PI mixture and 10 mM N-Ethylmaleimide (NEM)) and disrupted by the addition of glass beads and agitation on a Vortex mixer four times for 1 min. The lysate was cleared by centrifugation at ∼16,000 × g for 10 min at 4 °C in microfuge, and the supernatant was incubated with 25 µL anti-HA affinity matrix (Roche) overnight on a rotator at 4 °C. The anti-HA affinity matrix was washed three times with buffer, and the immunoprecipitated proteins were eluted with SDS-PAGE sample buffer plus NEM and sequentially incubated at 50 °C (for anti-HA) and then at 90 °C (for anti-GFP). Protein elution was followed by SDS-PAGE and Western blot analysis. Erg25 was detected with anti-HA HRP-linked antibody (Roche) and Pre8 was detected with anti-GFP and anti-mouse IgG HRP linked secondary antibody (Cell Signaling). Image analysis was performed using a BioRAD Universal Hood II Imager, and signals were quantified using ImageJ 1.51h software (National Institutes of Health).
Assays to detect the levels of ubiquitinated Erg25 were performed essentially as described (42). A yeast strain lacking Pdr5 (Δpdr5; Open Biosystems, Thermo Fisher) was transformed with pIU2593 (Erg25-HA; see above) and pRS423mycUbiqutin (2 µ HIS3 PCUP1-mycUb) (43). Overnight yeast cultures were grown in selective media at 30 °C and diluted in the morning. After continued growth, log phase cultures were treated with either 50 μm MG132 (to stabilize the substrate) or an equivalent volume of DMSO for 1 h, and ∼30 OD600 of log phase cells were harvested by centrifugation and the cell pellet was washed once with ice-cold ddH2O. Cell pellets were resuspended in 500 µL of denaturing RIPA buffer (10 mM Tris-Cl pH 8.0, 140 mM NaCl, 1 mM EDTA, 1% Triton-× 100, 0.1% sodium deoxycholate, 0.1% SDS) plus a protease inhibitor tablet (Roche) and 10 mM NEM. The cells were disrupted with glass beads, as outlined above, and the lysate was cleared by centrifugation, also as described above. The protein input for immunoprecipitations was normalized based on an A280 reading in 2% SDS, and the normalized lysates were incubated with 25 µL anti-HA affinity matrix (Roche) overnight on a rotator at 4 °C. The matrix was then washed three times with denaturing RIPA buffer plus protease inhibitors and 10 mM NEM before proteins were eluted with SDS-PAGE sample buffer plus 10 mM NEM and heated to 50 °C for 10 min prior to analysis by SDS-PAGE and western blotting. Erg25 was detected as described above, and ubiquitin was detected by immersing the blot in a boiling water bath for 10 min prior to blocking in a milk solution and incubation with anti-ubiquitin antibody (P4D1, Santa Cruz Biotechnology). Image analysis and quantification were performed as described above.
Cycloheximide Chase Protein Degradation Assays
The following yeast strains were used to measure the turnover of specific proteins in yeast cells: HRD1/DOA10, Δhrd1Δdoa10, Δpdr5, and Δpdr5Δpep4 (44, 45), BY4742, Δerg2, and Δerg3 (Open Biosystems, Thermo Scientific), and CDC48 and cdc48-2 (45). Yeast strains were transformed with pIU2593 (see above), NBD2* (34) (also see above) or left untransformed, and cultures were grown in the appropriate media at 30 °C before cycloheximide chases were performed as described (46). The CDC48 and cdc48-2 strains were grown at room temperature and then incubated at 39 °C degrees for 2 h prior to addition of cycloheximide to induce the thermosensitive phenotype. The Δpdr5 and isogenic WT strains were pretreated with 50 μm MG132 or DMSO for 1 h or, where indicated, with 40 μm fluconazole (Sigma) for 2 h prior to the addition of cycloheximide. Yeast cell lysates were generated using alkaline lysis followed by trichloroacetic acid precipitation (47). Protein pellets were resuspended in SDS-PAGE sample buffer, heated at 50 °C for 10 min, and subject to SDS-PAGE and western blotting. Western blot detection of Erg25-HA, imaging and quantitation were performed as described above. Native Erg25 was detected using anti-Erg25 immunosera (a kind gift from Dr. Jerry Kaplan and Diane Ward at the University of Utah), and an anti-rabbit IgG (IgG) HRP-conjugated secondary antibody (Cell Signaling Technology).
RESULTS
The goal of this study was to develop an alternate approach to identify UPS substrates in yeast that might have been missed through other methods. This pursuit is warranted because each attempt to identify UPS substrates—using a variety of methods—has provided a list of nonoverlapping substrates compared with prior attempts. Thus, each experimental regimen most likely exhibits unique attributes and, in turn, suffers from deficiencies. We therefore developed a new substrate capture assay in yeast in which proteasome particles, associated partners, and UPS substrates could all be isolated when the proteasome was either active or inactive. We reasoned that substrates would be rapidly degraded when the proteasome was active, but these substrates would instead associate with the proteasome when it was inactive. In contrast, core proteasome components would be isolated regardless of whether the proteasome was active or inactive. As a negative control, a pulldown was also conducted in which an epitope on a proteasome subunit for antibody-based isolation was absent.
We first utilized a strain in which the PRE8 chromosomal locus in a WT yeast strain was replaced with Pre8-GFP to allow for detection and isolation (48). PRE8 encodes an α subunit in the proteasome core particle and resides near a component in the 19S cap that stabilizes the holoenzyme, i.e. the 26S particle (49). Next, we deleted the PDR5 gene in this strain (see Experimental Procedures) since loss of the encoded drug efflux pump allows for controlled proteasome inhibition when a specific inhibitor is added, such as MG132 (50, 51). A WT strain in which WT PRE8 and PDR5 loci were maintained in the chromosome was used as a negative control (Fig. 1A).
Next, we confirmed that the constructed strains behaved as expected. First, we noted that each yeast strain grew identically (data not shown), and we detected the GFP-tagged Pre8 subunit in yeast in which this modified subunit was integrated (Fig. 1B, lane 1). The specificity of the signal was confirmed by introducing a GFP-tagged protein into a WT strain (lane 3) (35). In contrast, cells containing the endogenous, unaltered copy of Pre8 lacked the GFP signal (lane 2). Second, we confirmed that the degradation of a known UPS substrate, NBD2* (34), was slowed in cells lacking the Pdr5 drug efflux pump after a short pre-incubation with MG132, as expected (Fig. 1C).
After confirming the expected behavior of the strains, we followed the protocol outlined in Fig. 2A in which the indicated three strains were treated with MG132 for 1 h and the proteasome was immunoprecipitated via GFP-affinity capture (see Experimental Procedures). NBD2* was also expressed from a transformed vector in all three strains to ensure quality control. Mass spectrometry identified 4166 peptides belonging to 1039 proteins, with 575 of them with a minimum of 2 peptides (supplemental Table S2, S3, and S4). We found 84 and 144 proteins significantly enriched in the pulldown samples from PRE8:GFP and PRE8:GFP Δpdr5 cells respectively, resulting in a combined total of 149 proteins as Pre8-interacting partners (supplemental Table S5).
Fig. 2.
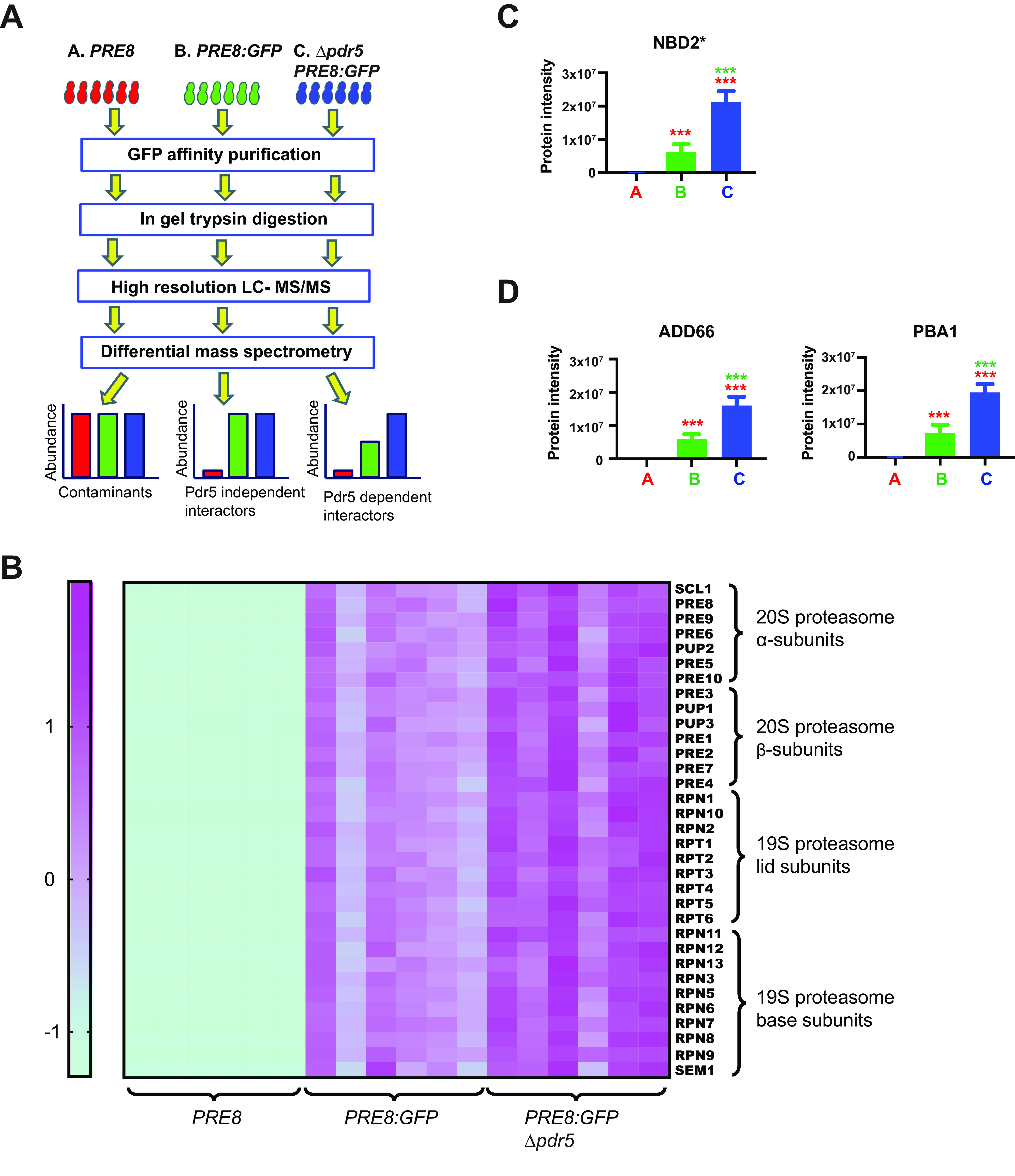
Validation of a new proteasome trapping proteomics workflow to identify proteasome substrates. A, Diagram of the substrate trapping proteomics workflow used to identify UPS substrates, as described under Experimental Procedures. B, A heat map illustrating the abundance of all 33 proteasome subunits identified in the Pre8-GFP immunoprecipitation. The values for protein intensity, used as a surrogate metric for protein abundance, were standardized using Z-transformation. Higher abundance is illustrated as purple and lower abundance is shown in green for each of the 6 replicate pull-downs. C, The three yeast strains described in part (A) were transformed with a plasmid to express NBD2*, a UPS substrate, and subject to the workflow illustrated in (A). NBD2* serves as a positive control, further validating our methods. D, The proteasome assembly factors Add66 and Pba1 were also enriched in the Pre8-GFP immunoprecipitation and are enriched further when the proteasome is inhibited. For (C) and (D) n = 6, +/-S.D. ***p < 0.001, A versus B (red), B versus C (green).
The robustness of GFP-affinity capture in isolating proteasomes was confirmed by the identification of all 33 proteasome subunits, including the 14 core subunits as well as the entire proteasome lid and base (8). In addition, high resolution MS based quantification indicated that these subunits were significantly enriched in samples expressing GFP-tagged Pre8 compared with untagged-Pre8 (Fig. 2B). These results validate the efficacy of the pulldown protocol and mass spec detection. We further validated the experimental design by asking whether the known UPS substrate, NBD2*, was enriched in the presence of MG132 in the PRE8:GFP tagged strain lacking the Pdr5 drug efflux pump. As shown in Fig. 2C, the intensity of NBD2* rose about 2.5-fold when the proteasome was inhibited. We also detected two known proteasome assembly chaperones as being stabilized in the presence of MG132 in the Pre8/GFP tagged strains. These proteins, Add66 (Pba2) and Pba1, assist the formation of the 20S core particle and are degraded after aiding in the assembly process (Fig. 2D) (52–54). Together, these data support our development of a new method to isolate previously unknown proteasome substrates.
We next more closely examined the list of proteins that were statistically enriched in replicate samples from PRE8:GFP Δpdr5 cells compared with PRE8:GFP cells that had been treated with MG132. As presented in Table I, a total of 27 proteins were identified, which in principle represent potential proteasome substrates. One protein, which had not previously been identified as a UPS substrate but exhibited a >5-fold enrichment and the third lowest p-value was Erg25. Erg25 is a known ER resident protein that converts dimethylzymosterol to zymosterol (29). Zymosterol is an ergosterol precursor (see below), and ergosterol is the primary sterol in the yeast plasma membrane where it plays an analogous role to cholesterol in higher eukaryotic cells. Because other enzymes required for the synthesis of ergosterol were identified as ERAD substrates (23, 55–57), we focused our subsequent efforts on Erg25 and asked whether this enzyme is also an ERAD substrate.
Table I. Ubiquitin-proteasome substrates identified using a proteasome trapping proteomics work-flow. Proteins are ranked by lowest p-value. The selection was based on Student's t-test p value < 0.05 and fold change >1.5 comparing PRE8:GFP Δpdr5 to PRE8:GFP samples. Proteins with only one identified peptide are excluded.
| Gene | Protein Name | Fold Change | # of Peptides Identified | p value | UniProt Accession |
|---|---|---|---|---|---|
| ECM29 | Proteasome component ECM29 | 3.8 | 72 | 3.19E-09 | P38737 |
| Uncharacterized RING finger protein YBR062C | 9.0 | 6 | 1.83E-08 | P38239 | |
| ERG25 | Methylsterol monooxygenase | 5.1 | 6 | 2.30E-08 | P53045 |
| BLM10 | Proteasome activator BLM10 | 2.4 | 116 | 8.35E-08 | P43583 |
| HSP26 | Heat shock protein 26 | 6.2 | 6 | 4.57E-07 | P15992 |
| PBA1 | Proteasome chaperone 1 | 1.9 | 16 | 2.37E-05 | Q05778 |
| CDC48 | Cell division control protein 48 | 1.8 | 23 | 2.62E-05 | P25694 |
| ADD66 | Proteasome assembly chaperone 2 | 1.8 | 11 | 5.65E-05 | P36040 |
| RAD23 | UV excision repair protein RAD23 | 1.7 | 12 | 1.27E-04 | P32628 |
| ZRT1 | Zinc-regulated transporter 1 | 2.3 | 7 | 1.61E-04 | P32804 |
| STE6 | Alpha-factor-transporting ATPase | 2.3 | 18 | 1.85E-04 | P12866 |
| UBP6 | Ubiquitin carboxyl-terminal hydrolase 6 | 1.6 | 37 | 8.28E-04 | P43593 |
| VPH1 | V-type proton ATPase subunit a, vacuolar isoform | 2.2 | 10 | 9.27E-04 | P32563 |
| ZPS1 | Protein ZPS1 | 2.0 | 6 | 2.03E-03 | Q12512 |
| SSH1 | Sec61 protein homolog | 1.6 | 4 | 3.22E-03 | P38353 |
| SCS2 | Vesicle-associated membrane protein-associated protein SCS2 | 1.6 | 9 | 3.35E-03 | P40075 |
| HSP82 | ATP-dependent molecular chaperone HSP82 | 2.0 | 4 | 4.47E-03 | P02829 |
| DSK2 | Ubiquitin domain-containing protein DSK2 | 1.9 | 3 | 6.44E-03 | P48510 |
| RPL23B RPL23A | 60S ribosomal protein L23-B; 60S ribosomal protein L23-A | 1.7 | 3 | 1.01E-02 | P0CX42 P0CX41 |
| RPL22A | 60S ribosomal protein L22-A | 1.5 | 2 | 1.35E-02 | P05749 |
| SSA2 | Heat shock protein SSA2 | 1.6 | 31 | 1.89E-02 | P10592 P09435 |
| LYS20 LYS21 | Homocitrate synthase, cytosolic isozyme; Homocitrate synthase, mitochondrial | 2.0 | 4 | 2.02E-02 | P48570 Q12122 |
| PHO88 | Inorganic phosphate transport protein PHO88 | 2.5 | 3 | 2.04E-02 | P38264 |
| SSA1 | Heat shock protein SSA1 | 1.9 | 7 | 2.48E-02 | P10591 |
| SSE1 | Heat shock protein homolog SSE1 | 1.6 | 10 | 3.68E-02 | P32589 P32590 |
| CHO2 | Phosphatidylethanolamine N-methyltransferase | 5.1 | 3 | 3.80E-02 | P05374 |
| URA3 | Orotidine 5-phosphate decarboxylase | 1.7 | 11 | 4.73E-02 | P03962 |
We first wished to confirm that Erg25 is proteasome-associated and therefore performed a coimmunoprecipitation experiment in PRE8:GFP Δpdr5 yeast expressing an HA epitope-tagged form of Erg25. When Erg25 was immunoprecipitated using anti-HA antibody, GFP tagged Pre8 coprecipitated under nondenaturing conditions after treatment with MG132 (Fig. 3A, lane 3). In contrast, coprecipitation was absent in cells lacking Pre8-GFP or Erg25-HA (lanes 1 and 2). These results confirmed proteasome-Erg25 interaction, as suggested by the mass spec analysis.
Fig. 3.

Erg25 is a proteasome substrate. A, Cell lysates were prepared from Pre8:GFP Δpdr5 or Δpdr5 yeast strains expressing C-terminally HA epitope tagged Erg25 (Erg25-HA) or harboring a vector control. Immunoprecipitations (IP) in the presence of MG132 were performed with anti-HA affinity matrix as described under Experimental Procedures. Immunoblots (IB) after precipitation of cell lysates with anti-HA or anti-GFP are indicated. *nonspecific band. B, Cell lysates from Δpdr5 yeast expressing Erg25-HA and myc-ubiquitin grown in the presence or absence of 50 μm MG132 were subjected to lysis under denaturing conditions (to preclude the isolation of nonspecific partners) and immunoprecipitated with anti-HA affinity matrix. Precipitated proteins were immunoblotted with anti-HA (Erg25) and anti-ubiquitin. Data represent the means of 5 experiments, ± S.D. *, p < 0.05. C, Cycloheximide chase assays were performed as described under Experimental Procedures in untransformed Δpdr5 yeast strains incubated in the absence or presence of MG132 and immunoblots were probed with anti-Erg25 antisera. Data represent the means of 4-6 experiments, +/-S.D. *, p ≤ 0.05.
Based on these data, we predicted that Erg25 is ubiquitinated and that this modification directs the enzyme to the proteasome. To this end, we precipitated HA-tagged Erg25 species in the presence or absence of MG132 in cells lacking the Pdr5 drug efflux pump. The cells also contained a plasmid that increased the steady-state levels of Ub, which aids in the detection of this transient modification (43). After immunoprecipitating Erg25, we examined the isolated proteins for the presence of a polyubiquitin chain by Western blot analysis. As shown in Fig. 3B, the level of ubiquitinated Erg25 increased when proteasome function was impaired, i.e. when cultures were incubated with MG132 prior to cell lysis (lane 3). Next, to more directly determine whether Erg25 is degraded in a proteasome-dependent manner, we measured Erg25 stability by performing a cycloheximide chase analysis when the proteasome was inhibited in a pdr5Δ background in the presence of MG132. As shown in Fig. 3C, Erg25 degradation was slowed in the presence of MG132. Combined with the fact that Erg25 is an ER-resident protein (29), these data establish Erg25 as an ERAD substrate.
Next, because ERAD substrates are ubiquitinated by ER-associated E3 ubiquitin ligases, we measured the rate of Erg25 turnover in a cycloheximide chase assay in the absence of one or both of the canonical ERAD-associated E3 ligases, Hrd1 and Doa10 (Fig. 4A) (19, 58). Based on a mass spec analysis, Christiano et al. previously found that deletion of Hrd1 somewhat increased Erg25 half-life (59); however in our hands, the rate of Erg25 degradation was not significantly affected in either of the single E3 mutants (Δhrd1 or Δdoa10). In contrast, deletion of both Hrd1 and Doa10 almost completely stabilized Erg25 (Fig. 4A). Most commonly, Doa10 and Hrd1 target distinct groups of ERAD substrates for degradation, which depends on where the misfolded lesion resides (60), yet other cases exist in which both enzymes are required to target a specific membrane protein for ERAD (46). The difference between our data and the previous data might reflect the unique strain backgrounds used or methodology. Regardless, these data further support the targeting of Erg25 to the ERAD pathway.
Fig. 4.

Erg25 is an ERAD substrate. Cycloheximide chase assays were performed as described under Experimental Procedures using the indicated yeast strains expressing Erg25-HA. Protein lysates were prepared and immunoblotted with anti-HA (Erg25) and anti-G6P as a loading control. Data represent the means of 4-6 experiments, +/-S.D. *, p ≤ 0.05; **, p < 0.01; ***, p < 0.001.
After ubiquitination of ERAD substrates at the ER membrane, Cdc48, a AAA+-ATPase, provides the energy required to retrotranslocate ERAD substrates from the ER to the cytosol. Cycloheximide chases performed in either WT (CDC48) yeast or yeast containing a temperature sensitive Cdc48 allele, cdc48-2, revealed that Erg25 degradation was significantly inhibited in the cdc48-2 strain when cells had been pre-shifted to a nonpermissive temperature (Fig. 4B). These data strengthen our conclusion that Erg25 is a proteasome dependent, ERAD substrate.
Previous work established that the ERAD of two other enzymes in the ergosterol biosynthetic pathway, Hmg2 and Erg1 (see Fig. 5A), are regulated by the level of biosynthetic intermediates (23, 61, 62). For example, the accumulation of lanosterol triggers Erg1 degradation, therefore blocking the synthesis of lanosterol precursors (23). To ask whether the degradation of Erg25 is also regulated by biosynthetic intermediates, we measured Erg25 stability in the presence or absence of fluconazole, which inhibits Erg11 (Fig. 5A) (63). As shown by cycloheximide chase analysis, pretreatment of yeast with fluconazole slowed Erg25 turnover (Fig. 5B). To confirm that the regulation of Erg25 stability is impacted by ergosterol pathway intermediates, we assessed Erg25 degradation in yeast lacking either Erg2 or Erg3. Unlike fluconazole, Erg2 and Erg3 act downstream of Erg25 in the ergosterol pathway. Surprisingly, deletion of either Erg2 or Erg3 also modestly stabilized Erg25 (Fig. 5B), implying that accumulation of downstream intermediates positively affects Erg25 stability. Because prior work reported that deletion of Erg6 has no effect on proteasome-dependent degradation (50), we do not believe that inhibiting the ergosterol pathway negatively affects ERAD generally. It is also possible that these perturbations affect ER homeostasis, stabilizing Erg25. Future studies will better characterize how the Erg25 degradation pathway is regulated. Nevertheless, these data indicate that Erg25 is a regulated ERAD substrate, that responds to the levels of sterol intermediates in the ER membrane.
Fig. 5.

Inhibition of ergosterol synthesis stabilizes Erg25. A, Schematic representation of select steps in the mevalonate/ergosterol pathway. Feedback inhibition by lanosterol is represented by red lines. B, Cycloheximide chase assays were performed as described under Experimental Procedures in yeast expressing Erg25-HA in the presence or absence of 40 μm fluconazole. Protein lysates were prepared and immunoblotted with anti-HA (Erg25) or anti-G6P as a loading control. C, Cycloheximide chases were performed in the indicated yeast strains, as above. Data represent the means of 4-8 experiments, +/-S.D. *, p <0.05; **p < 0.01, ***, p < 0.001.
DISCUSSION
MS has been used extensively to identify proteasome components along with the spectrum of UPS targets (64–66). The experimental system outlined in the current study was designed so that core components of the proteasome as well as UPS targets could be identified in parallel by MS and so that nonspecific partners could be readily excluded.
Among the 27 proteasome-associated proteins enriched in the PRE8:GFP Δpdr5 cells compared with the PRE8:GFP cells treated with MG132 were known UPS substrates. For example, Pba1 and Add66 are yeast homologs of a proteasome assembly chaperone complex that are degraded after aiding 20S core particle assembly (52, 53). Interesting, Rad23 and Dsk2, were also isolated as putative UPS substrates in our analysis. Dsk2 and Rad23 function redundantly to deliver ubiquitinated substrates to the proteasome during ERAD (67, 68), but Rad23 is known to escape degradation because it lacks a proteasome initiation region (69). We propose that their enrichment as proteasome partners when degradation is disabled reflects a build-up of ferried substrates to the proteasome. Cdc48 is another factor that escorts ubiquitinated substrates to the proteasome (70). Cdc48 was similarly enriched in the MG132-sensitive cells (Table I). Other enriched proteins include Blm10 and Ecm29. These known proteasome-associated proteins respectively open the aperture to the proteolytic chamber in the 20S core particle, which augments substrate entry (71, 72), and inhibit the ATPase and proteolytic activity of the proteasome (73). A structural analysis of Blm10 bound to the proteasome suggests that it favors peptide entry and proteolysis in the 20S core (74, 75). Our data suggest, under specific conditions, that it might also become a proteasome substrate. In turn, Ecm29 binds more avidly to the 26S particle than to dissociated subunits (76, 77). Because small molecule proteasome inhibitors, such as MG132, stabilize reconstituted 26S particles (76), the enrichment of Ecm29 with stalled proteasomes hints that this protein could similarly be a proteasome substrate. Finally, we note that Vph1 was identified as a high-confidence proteasome-targeted substrate. Cooper and colleagues previously reported that Vph1 is a robust ERAD substrate when a specific assembly chaperone is absent (78). The data in our paper suggest that even when the chaperone (Vma22) is present, a portion of this topologically complex membrane protein is targeted for ERAD. These data highlight the fact that membrane protein folding in the ER is faulty, as observed for numerous other substrates (16).
The strain used for these studies, which allows for treatment with a proteasome inhibitor and the facile isolation of proteasomes, will prove valuable for future work in the field. For example, genes encoding specific proteins that target ubiquitinated substrates to the proteasome can be deleted in our strain. A comparison of isolated proteins, which takes advantage of the Pre8:GFP chimera, might identify specific substrates of these targeting factors (79). In addition, numerous post-translational modifications have been identified on proteasome substrates that regulate their degradation (80). The isolation of proteasome partners in strains deleted for genes encoding factors that add or remove these modifications may also identify new clients. Finally, the isolation of proteasomes after subcellular fractionation, e.g. of mitochondria, could identify new UPS substrates that are compartment-specific.
Prior to our work, Erg25 had not been characterized as an ERAD substrate. This is somewhat surprising given the range of MS studies performed to identify ERAD substrates and the fact that several other components of the ergosterol biosynthetic pathway are regulated by ERAD. An analysis of the stability of the human Erg25 homolog (81), which is 38% identical to the S. cerevisiae isoform, is now warranted. Interestingly, yeast Erg25 was identified in three other proteomic studies. First, the isolation of organelle membranes and immunoprecipitation from yeast expressing an epitope-tagged ubiquitin species in an ERAD mutant uncovered putative membrane-associated ERAD substrates (57). Erg25 was absent from the list of 83 putative substrates, but a ubiquitinated Lys in the protein (IPSAK*EQLYCLK) was detected in the larger pool of modified proteins. Other proteins in the ergosterol pathway, Erg1, Erg9, and Erg27, were also detected in this analysis, suggesting that these enzymes may also be ERAD substrates. Indeed, Erg1 was later confirmed as a regulated ERAD substrate by Carvalho and colleagues (23). Additional experiments—such as those performed in the present study—are needed to determine whether Erg9 and Erg27 are targeted for ERAD. Second, Erg25 was detected in a SILAC analysis comparing yeast containing or lacking components of the ubiquitin conjugation machinery. However, Erg25 failed to meet the criteria required to classify the protein as a bona fide ERAD client (55). Finally, Erg25, as well as Erg1, Erg3, Erg5, and ERg11 were classified as proteins with short half-lives relative to the proteome, and that the Erg25, Erg3 and ERg5 half-lives increased in a Hrd1 deletion strain (59). However, in that study, the role of the ERAD pathway in targeting Erg25 was not directly tested, nor was the potential role of sterol synthesis examined. Overall, these and other data are consistent with the fact that each MS protocol exhibits unique strengths regarding its ability to isolate intended targets (also see Introduction). We therefore suggest that an identification of the spectrum of ERAD and UPS substrates has and will continue to benefit from diverse approaches.
DATA AVAILABILITY
All spectra matched to yeast peptides have been deposited to MS-Viewer, accessible through at the following URL: http://prospector2.ucsf.edu/prospector/cgi-bin/msform.cgi?form=msviewer, with the search key: n0y0xqrhg6. Raw MS data files, together with all MaxQuant exported files and Saccharomyces cerevisiae SwissProt database used, have also been deposited to the ProteomXchange consortium via the MassIVE partner repository with the data set identifier PXD020736.
Supplementary Material
Acknowledgments
We thank Jerry Kaplan and Diane Ward for reagents and technical assistance. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
This article contains supplemental data.
Funding and additional information—This work was supported by National Institutes of Health grants R35 GM131732 and P30 DK 079307 to J.L.B., grant R01 DK109024 to T.M.B., and by grant P30CA047904 (The Hillman Cancer Center Proteomics Facility).
Conflict of interest —The authors declare that they have no conflicts of interest with the contents of this article.
Abbreviations—The abbreviations used are:
- MS
- Mass spectrometry
- nLC-MS/MS
- nanoflow liquid chromatography-tandem mass spectrometry
- Ub
- Ubiquitin
- UPS
- ubiquitin-proteasome system
- ER
- Endoplasmic reticulum
- ERAD
- Endoplasmic reticulum-associated degradation
- SILAC
- Stable Isotope Labeling by/with Amino acids in Cell culture.
REFERENCES
- 1. Bard J. A. M., Goodall E. A., Greene E. R., Jonsson E., Dong K. C., and Martin A. (2018) Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 87, 697–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Voges D., Zwickl P., and Baumeister W. (1999) The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 68, 1015–1068 [DOI] [PubMed] [Google Scholar]
- 3. Varshavsky A. (2008) Discovery of cellular regulation by protein degradation. J. Biol. Chem. 283, 34469–34489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yau R., and Rape M. (2016) The increasing complexity of the ubiquitin code. Nat. Cell Biol. 18, 579–586 [DOI] [PubMed] [Google Scholar]
- 5. Stolz A., and Dikic I. (2018) Heterotypic Ubiquitin Chains: Seeing is Believing. Trends Cell Biol. 28, 1–3 [DOI] [PubMed] [Google Scholar]
- 6. Kulathu Y., and Komander D. (2012) Atypical ubiquitylation - the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat. Rev. Mol. Cell Biol. 13, 508–523 [DOI] [PubMed] [Google Scholar]
- 7. Thrower J. S., Hoffman L., Rechsteiner M., and Pickart C. M. (2000) Recognition of the polyubiquitin proteolytic signal. EMBO J. 19, 94–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Finley D., Ulrich H. D., Sommer T., and Kaiser P. (2012) The ubiquitin-proteasome system of Saccharomyces cerevisiae. Genetics 192, 319–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ghaemmaghami S., Huh W. K., Bower K., Howson R. W., Belle A., Dephoure N., O'Shea E. K., and Weissman J. S. (2003) Global analysis of protein expression in yeast. Nature 425, 737–741 [DOI] [PubMed] [Google Scholar]
- 10. Braakman I., and Bulleid N. J. (2011) Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 80, 71–99 [DOI] [PubMed] [Google Scholar]
- 11. Brodsky J. L., and Skach W. R. (2011) Protein folding and quality control in the endoplasmic reticulum: Recent lessons from yeast and mammalian cell systems. Curr. Opin. Cell Biol. 23, 464–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smith M. H., Ploegh H. L., and Weissman J. S. (2011) Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science 334, 1086–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Berner N., Reutter K. R., and Wolf D. H. (2018) Protein quality control of the endoplasmic reticulum and ubiquitin-proteasome-triggered degradation of aberrant proteins: yeast pioneers the path. Annu. Rev. Biochem. 87, 751–782 [DOI] [PubMed] [Google Scholar]
- 14. Zattas D., and Hochstrasser M. (2015) Ubiquitin-dependent protein degradation at the yeast endoplasmic reticulum and nuclear envelope. Crit. Rev. Biochem. Mol. Biol. 50, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Christianson J. C., and Ye Y. (2014) Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat. Struct. Mol. Biol. 21, 325–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Needham P. G., Guerriero C. J., and Brodsky J. L. (2019) Chaperoning endoplasmic reticulum-associated degradation (ERAD) and protein conformational diseases. Cold Spring Harb. Perspect. Biol. 11, a033928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McCracken A. A., and Brodsky J. L. (1996) Assembly of ER-associated protein degradation in vitro: dependence on cytosol, calnexin, and ATP. J. Cell Biol. 132, 291–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hiller M. M., Finger A., Schweiger M., and Wolf D. H. (1996) ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science 273, 1725–1728 [DOI] [PubMed] [Google Scholar]
- 19. Hampton R. Y., Gardner R. G., and Rine J. (1996) Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol. Biol. Cell 7, 2029–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ward C. L., Omura S., and Kopito R. R. (1995) Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 83, 121–127 [DOI] [PubMed] [Google Scholar]
- 21. Jensen T. J., Loo M. A., Pind S., Williams D. B., Goldberg A. L., and Riordan J. R. (1995) Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 83, 129–135 [DOI] [PubMed] [Google Scholar]
- 22. Tyler R. E., Pearce M. M., Shaler T. A., Olzmann J. A., Greenblatt E. J., and Kopito R. R. (2012) Unassembled CD147 is an endogenous endoplasmic reticulum-associated degradation substrate. Mol. Biol. Cell 23, 4668–4678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Foresti O., Ruggiano A., Hannibal-Bach H. K., Ejsing C. S., and Carvalho P. (2013) Sterol homeostasis requires regulated degradation of squalene monooxygenase by the ubiquitin ligase Doa10/Teb4. Elife. 2, e00953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huang E. Y., To M., Tran E., Dionisio L. T. A., Cho H. J., Baney K. L. M., Pataki C. I., and Olzmann J. A. (2018) A VCP inhibitor substrate trapping approach (VISTA) enables proteomic profiling of endogenous ERAD substrates. Mol. Biol. Cell 29, 1021–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stevenson J., Huang E. Y., and Olzmann J. A. (2016) Endoplasmic Reticulum-Associated Degradation and Lipid Homeostasis. Annu. Rev. Nutr. 36, 511–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Printsev I., Curiel D., and Carraway K. L. 3rd (2017) Membrane Protein Quantity Control at the Endoplasmic Reticulum. J. Membr. Biol. 250, 379–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goder V., Alanis-Dominguez E., and Bustamante-Sequeiros M. (2020) Lipids and their (un)known effects on ER-associated protein degradation (ERAD). Biochim Biophys Acta Mol. Cell Biol. Lipids 1865(1), 158488. [DOI] [PubMed] [Google Scholar]
- 28. Venter J. C., Adams M. D., Myers E. W., Li P. W., Mural R. J., Sutton G. G., Smith H. O., Yandell M., Evans C. A., Holt R. A., Gocayne J. D., Amanatides P., Ballew R. M., Huson D. H., Wortman J. R., Zhang Q., Kodira C. D., Zheng X. H., Chen L., Skupski M., Subramanian G., Thomas P. D., Zhang J., Gabor Miklos G. L., Nelson C., Broder S., Clark A. G., Nadeau J., McKusick V. A., Zinder N., Levine A. J., Roberts R. J., Simon M., Slayman C., Hunkapiller M., Bolanos R., Delcher A., Dew I., Fasulo D., Flanigan M., Florea L., Halpern A., Hannenhalli S., Kravitz S., Levy S., Mobarry C., Reinert K., Remington K., Abu-Threideh J., Beasley E., Biddick K., Bonazzi V., Brandon R., Cargill M., Chandramouliswaran I., Charlab R., Chaturvedi K., Deng Z., Francesco V. D., Dunn P., Eilbeck K., Evangelista C., Gabrielian A. E., Gan W., Ge W., Gong F., Gu Z., Guan P., Heiman T. J., Higgins M. E., Ji R.-R., Ke Z., Ketchum K. A., Lai Z., Lei Y., Li Z., Li J., Liang Y., Lin X., Lu F., Merkulov G. V., Milshina N., Moore H. M., Naik A. K., Narayan V. A., Neelam B., Nusskern D., Rusch D. B., Salzberg S., Shao W., Shue B., Sun J., Wang Z. Y., Wang A., Wang X., Wang J., Wei M.-H., Wides R., Xiao C., Yan C., Yao A., Ye J., Zhan M., Zhang W., Zhang H., Zhao Q., Zheng L., Zhong F., Zhong W., Zhu S. C., Zhao S., Gilbert D., Baumhueter S., Spier G., Carter C., Cravchik A., Woodage T., Ali F., An H., Awe A., Baldwin D., Baden H., Barnstead M., Barrow I., Beeson K., Busam D., Carver A., Center A., Cheng M. L., Curry L., Danaher S., Davenport L., Desilets R., Dietz S., Dodson K., Doup L., Ferriera S., Garg N., Gluecksmann A., Hart B., Haynes J., Haynes C., Heiner C., Hladun S., Hostin D., Houck J., Howland T., Ibegwam C., Johnson J., Kalush F., Kline L., Koduru S., Love A., Mann F., May D., McCawley S., McIntosh T., McMullen I., Moy M., Moy L., Murphy B., Nelson K., Pfannkoch C., Pratts E., Puri V., Qureshi H., Reardon M., Rodriguez R., Rogers Y.-H., Romblad D., Ruhfel B., Scott R., Sitter C., Smallwood M., Stewart E., Strong R., Suh E., Thomas R., Tint N. N., Tse S., Vech C., Wang G., Wetter J., Williams S., Williams M., Windsor S., Winn-Deen E., Wolfe K., Zaveri J., Zaveri K., Abril J. F., Guigó R., Campbell M. J., Sjolander K. V., Karlak B., Kejariwal A., Mi H., Lazareva B., Hatton T., Narechania A., Diemer K., Muruganujan A., Guo N., Sato S., Bafna V., Istrail S., Lippert R., Schwartz R., Walenz B., Yooseph S., Allen D., Basu A., Baxendale J., Blick L., Caminha M., Carnes-Stine J., Caulk P., Chiang Y.-H., Coyne M., Dahlke C., Mays A. D., Dombroski M., Donnelly M., Ely D., Esparham S., Fosler C., Gire H., Glanowski S., Glasser K., Glodek A., Gorokhov M., Graham K., Gropman B., Harris M., Heil J., Henderson S., Hoover J., Jennings D., Jordan C., Jordan J., Kasha J., Kagan L., Kraft C., Levitsky A., Lewis M., Liu X., Lopez J., Ma D., Majoros W., McDaniel J., Murphy S., Newman M., Nguyen T., Nguyen N., Nodell M., Pan S., Peck J., Peterson M., Rowe W., Sanders R., Scott J., Simpson M., Smith T., Sprague A., Stockwell T., Turner R., Venter E., Wang M., Wen M., Wu D., Wu M., Xia A., Zandieh A., and Zhu X. (2001) The sequence of the human genome. Science 291, 1304–1351 [DOI] [PubMed] [Google Scholar]
- 29. Bard M., Bruner D. A., Pierson C. A., Lees N. D., Biermann B., Frye L., Koegel C., and Barbuch R. (1996) Cloning and characterization of ERG25, the Saccharomyces cerevisiae gene encoding C-4 sterol methyl oxidase. Proc. Natl. Acad. Sci. U S A 93, 186–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 31. Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M. Y., Geiger T., Mann M., and Cox J. (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 [DOI] [PubMed] [Google Scholar]
- 32. Huh W. K., Falvo J. V., Gerke L. C., Carroll A. S., Howson R. W., Weissman J. S., and O'Shea E. K. (2003) Global analysis of protein localization in budding yeast. Nature 425, 686–691 [DOI] [PubMed] [Google Scholar]
- 33. Hecht K. A., Wytiaz V. A., Ast T., Schuldiner M., and Brodsky J. L. (2013) Characterization of an M28 metalloprotease family member residing in the yeast vacuole. FEMS Yeast Res. 13, 471–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guerriero C. J., Weiberth K. F., and Brodsky J. L. (2013) Hsp70 targets a cytoplasmic quality control substrate to the San1p ubiquitin ligase. J. Biol. Chem. 288, 18506–18520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sun Z., and Brodsky J. L. (2018) The degradation pathway of a model misfolded protein is determined by aggregation propensity. Mol Biol Cell 12, 1422–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Adams A., Gottschling D. E., Kaiser C. A., and Stearns T. (1997) Methods in Yeast Genetics, Cold Spring Harbor Laboratory Press [Google Scholar]
- 37. Brodsky J. L., Hamamoto S., Feldheim D., and Schekman R. (1993) Reconstitution of protein translocation from solubilized yeast membranes reveals topologically distinct roles for BiP and cytosolic Hsc70. J. Cell Biol. 120, 95–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Braganza A., Li J., Zeng X., Yates N. A., Dey N. B., Andrews J., Clark J., Zamani L., Wang X. H., St Croix C., O'Sullivan R., Garcia-Exposito L., Brodsky J. L., and Sobol R. W. (2017) UBE3B is a calmodulin-regulated, mitochondrion-associated E3 ubiquitin ligase. J. Biol. Chem. 292, 2470–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vizcaino J. A., Deutsch E. W., Wang R., Csordas A., Reisinger F., Rios D., Dianes J. A., Sun Z., Farrah T., Bandeira N., Binz P. A., Xenarios I., Eisenacher M., Mayer G., Gatto L., Campos A., Chalkley R. J., Kraus H. J., Albar J. P., Martinez-Bartolome S., Apweiler R., Omenn G. S., Martens L., Jones A. R., and Hermjakob H. (2014) ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ward D. M., Chen O. S., Li L., Kaplan J., Bhuiyan S. A., Natarajan S. K., Bard M., and Cox J. E. (2018) Altered sterol metabolism in budding yeast affects mitochondrial iron-sulfur (Fe-S) cluster synthesis. J. Biol. Chem. 293, 10782–10795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sikorski R. S., and Hieter P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Preston G. M., Guerriero C. J., Metzger M. B., Michaelis S., and Brodsky J. L. (2018) Substrate insolubility dictates Hsp104-dependent endoplasmic-reticulum-associated degradation. Mol. Cell. 70, 242–253.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nakatsukasa K., Huyer G., Michaelis S., and Brodsky J. L. (2008) Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell 132, 101–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pagant S., Kung L., Dorrington M., Lee M. C., and Miller E. A. (2007) Inhibiting endoplasmic reticulum (ER)-associated degradation of misfolded Yor1p does not permit ER export despite the presence of a diacidic sorting signal. Mol. Biol. Cell 18, 3398–3413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guerriero C. J., Reutter K. R., Augustine A. A., Preston G. M., Weiberth K. F., Mackie T. D., Cleveland-Rubeor H. C., Bethel N. P., Callenberg K. M., Nakatsukasa K., Grabe M., and Brodsky J. L. (2017) Transmembrane helix hydrophobicity is an energetic barrier during the retrotranslocation of integral membrane ERAD substrates. Mol Biol Cell 28, 2076–2090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Buck T. M., Kolb A. R., Boyd C. R., Kleyman T. R., and Brodsky J. L. (2010) The endoplasmic reticulum-associated degradation of the epithelial sodium channel requires a unique complement of molecular chaperones. Mol. Biol. Cell 21, 1047–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang Y., Michaelis S., and Brodsky J. L. (2002) CFTR expression and ER-associated degradation in yeast. Methods Mol. Med. 70, 257–265 [DOI] [PubMed] [Google Scholar]
- 48. Chong Y. T., Cox M. J., and Andrews B. (2012) Proteome-wide screens in Saccharomyces cerevisiae using the yeast GFP collection. Adv. Exp. Med. Biol. 736, 169–178 [DOI] [PubMed] [Google Scholar]
- 49. Pathare G. R., Nagy I., Bohn S., Unverdorben P., Hubert A., Korner R., Nickell S., Lasker K., Sali A., Tamura T., Nishioka T., Forster F., Baumeister W., and Bracher A. (2012) The proteasomal subunit Rpn6 is a molecular clamp holding the core and regulatory subcomplexes together. Proc. Natl. Acad. Sci. U S A 109, 149–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lee D. H., and Goldberg A. L. (1996) Selective inhibitors of the proteasome-dependent and vacuolar pathways of protein degradation in Saccharomyces cerevisiae. J. Biol. Chem. 271, 27280–27284 [DOI] [PubMed] [Google Scholar]
- 51. Fleming J. A., Lightcap E. S., Sadis S., Thoroddsen V., Bulawa C. E., and Blackman R. K. (2002) Complementary whole-genome technologies reveal the cellular response to proteasome inhibition by PS-341. Proc. Natl. Acad. Sci. U S A 99, 1461–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Scott C. M., Kruse K. B., Schmidt B. Z., Perlmutter D. H., McCracken A. A., and Brodsky J. L. (2007) ADD66, a gene involved in the endoplasmic reticulum-associated degradation of alpha-1-antitrypsin-Z in yeast, facilitates proteasome activity and assembly. Mol. Biol. Cell 18, 3776–3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li X., Kusmierczyk A. R., Wong P., Emili A., and Hochstrasser M. (2007) beta-Subunit appendages promote 20S proteasome assembly by overcoming an Ump1-dependent checkpoint. EMBO J. 26, 2339–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hirano Y., Hendil K. B., Yashiroda H., Iemura S., Nagane R., Hioki Y., Natsume T., Tanaka K., and Murata S. (2005) A heterodimeric complex that promotes the assembly of mammalian 20S proteasomes. Nature 437, 1381–1385 [DOI] [PubMed] [Google Scholar]
- 55. Foresti O., Rodriguez-Vaello V., Funaya C., and Carvalho P. (2014) Quality control of inner nuclear membrane proteins by the Asi complex. Science 346, 751–755 [DOI] [PubMed] [Google Scholar]
- 56. Peng B., Plan M. R., Chrysanthopoulos P., Hodson M. P., Nielsen L. K., and Vickers C. E. (2017) A squalene synthase protein degradation method for improved sesquiterpene production in Saccharomyces cerevisiae. Metab. Eng. 39, 209–219 [DOI] [PubMed] [Google Scholar]
- 57. Hitchcock A. L., Auld K., Gygi S. P., and Silver P. A. (2003) A subset of membrane-associated proteins is ubiquitinated in response to mutations in the endoplasmic reticulum degradation machinery. Proc. Natl. Acad. Sci. U S A 100, 12735–12740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ravid T., Kreft S. G., and Hochstrasser M. (2006) Membrane and soluble substrates of the Doa10 ubiquitin ligase are degraded by distinct pathways. EMBO J. 25, 533–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Christiano R., Nagaraj N., Frohlich F., and Walther T. C. (2014) Global proteome turnover analyses of the Yeasts S. cerevisiae and S. Cell Rep. 9, 1959–1965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vashist S., and Ng D. T. (2004) Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J. Cell Biol. 165, 41–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bays N. W., Gardner R. G., Seelig L. P., Joazeiro C. A., and Hampton R. Y. (2001) Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat. Cell Biol. 3, 24–29 [DOI] [PubMed] [Google Scholar]
- 62. Hampton R. Y. (2002) ER-associated degradation in protein quality control and cellular regulation. Curr. Opin. Cell Biol. 14, 476–482 [DOI] [PubMed] [Google Scholar]
- 63. Anderson J. B., Sirjusingh C., Parsons A. B., Boone C., Wickens C., Cowen L. E., and Kohn L. M. (2003) Mode of selection and experimental evolution of antifungal drug resistance in Saccharomyces cerevisiae. Genetics 163, 1287–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wolf-Levy H., Javitt A., Eisenberg-Lerner A., Kacen A., Ulman A., Sheban D., Dassa B., Fishbain-Yoskovitz V., Carmona-Rivera C., Kramer M. P., Nudel N., Regev I., Zahavi L., Elinger D., Kaplan M. J., Morgenstern D., Levin Y., and Merbl Y. (2018) Revealing the cellular degradome by mass spectrometry analysis of proteasome-cleaved peptides. Nat. Biotechnol. 36, 1110–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kim W., Bennett E. J., Huttlin E. L., Guo A., Li J., Possemato A., Sowa M. E., Rad R., Rush J., Comb M. J., Harper J. W., and Gygi S. P. (2011) Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell. 44, 325–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rose A., and Mayor T. (2018) Exploring the Rampant Expansion of Ubiquitin Proteomics. Methods Mol. Biol. 1844, 345–362 [DOI] [PubMed] [Google Scholar]
- 67. Garza R. M., Sato B. K., and Hampton R. Y. (2009) In vitro analysis of Hrd1p-mediated retrotranslocation of its multispanning membrane substrate 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase. J. Biol. Chem. 284, 14710–14722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Medicherla B., Kostova Z., Schaefer A., and Wolf D. H. (2004) A genomic screen identifies Dsk2p and Rad23p as essential components of ER-associated degradation. EMBO Rep. 5, 692–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fishbain S., Prakash S., Herrig A., Elsasser S., and Matouschek A. (2011) Rad23 escapes degradation because it lacks a proteasome initiation region. Nat. Commun. 2, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Jentsch S., and Rumpf S. (2007) Cdc48 (p97): a 'molecular gearbox' in the ubiquitin pathway? Trends Biochem. Sci. 32, 6–11 [DOI] [PubMed] [Google Scholar]
- 71. Dange T., Smith D., Noy T., Rommel P. C., Jurzitza L., Cordero R. J., Legendre A., Finley D., Goldberg A. L., and Schmidt M. (2011) Blm10 protein promotes proteasomal substrate turnover by an active gating mechanism. J. Biol. Chem. 286, 42830–42839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lehmann A., Jechow K., and Enenkel C. (2008) Blm10 binds to pre-activated proteasome core particles with open gate conformation. EMBO Rep. 9, 1237–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. De La Mota-Peynado A., Lee S. Y., Pierce B. M., Wani P., Singh C. R., and Roelofs J. (2013) The proteasome-associated protein Ecm29 inhibits proteasomal ATPase activity and in vivo protein degradation by the proteasome. J. Biol. Chem. 288, 29467–29481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sadre-Bazzaz K., Whitby F. G., Robinson H., Formosa T., and Hill C. P. (2010) Structure of a Blm10 complex reveals common mechanisms for proteasome binding and gate opening. Mol. Cell. 37, 728–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Stadtmueller B. M., and Hill C. P. (2011) Proteasome activators. Mol. Cell. 41, 8–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kleijnen M. F., Roelofs J., Park S., Hathaway N. A., Glickman M., King R. W., and Finley D. (2007) Stability of the proteasome can be regulated allosterically through engagement of its proteolytic active sites. Nat. Struct. Mol. Biol. 14, 1180–1188 [DOI] [PubMed] [Google Scholar]
- 77. Leggett D. S., Hanna J., Borodovsky A., Crosas B., Schmidt M., Baker R. T., Walz T., Ploegh H., and Finley D. (2002) Multiple associated proteins regulate proteasome structure and function. Mol. Cell. 10, 495–507 [DOI] [PubMed] [Google Scholar]
- 78. Hill K., and Cooper A. A. (2000) Degradation of unassembled Vph1p reveals novel aspects of the yeast ER quality control system. EMBO J. 19, 550–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Raasi S., and Wolf D. H. (2007) Ubiquitin receptors and ERAD: a network of pathways to the proteasome. Semin. Cell Dev. Biol. 18, 780–791 [DOI] [PubMed] [Google Scholar]
- 80. Lucas X., and Ciulli A. (2017) Recognition of substrate degrons by E3 ubiquitin ligases and modulation by small-molecule mimicry strategies. Curr. Opin. Struct. Biol. 44, 101–110 [DOI] [PubMed] [Google Scholar]
- 81. Kennedy M. A., Johnson T. A., Lees N. D., Barbuch R., Eckstein J. A., and Bard M. (2000) Cloning and sequencing of the Candida albicans C-4 sterol methyl oxidase gene (ERG25) and expression of an ERG25 conditional lethal mutation in Saccharomyces cerevisiae. Lipids 35, 257–262 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All spectra matched to yeast peptides have been deposited to MS-Viewer, accessible through at the following URL: http://prospector2.ucsf.edu/prospector/cgi-bin/msform.cgi?form=msviewer, with the search key: n0y0xqrhg6. Raw MS data files, together with all MaxQuant exported files and Saccharomyces cerevisiae SwissProt database used, have also been deposited to the ProteomXchange consortium via the MassIVE partner repository with the data set identifier PXD020736.