

Abstract
Indanyloxyacetic acid-94 (IAA-94), an intracellular chloride channel blocker, is shown to ablate cardioprotection rendered by ischemic preconditioning (IPC), N (6)-2-(4-aminophenyl) ethyladenosine or the PKC activator phorbol 12-myristate 13-acetate and cyclosporin A (CsA) in both ex-vivo and in-vivo ischemia-reperfusion (IR) injury. Thus signifying the role of the IAA-94 sensitive chloride channels in mediating cardio-protection upon IR injury. Although IAA-94 sensitive chloride currents are recorded in cardiac mitoplast, there is still a lack of understanding of the mechanism by which IAA-94 increases myocardial infarction (MI) by IR injury. Mitochondria are the key arbitrators of cell life and death pathways. Both oxidative stress and calcium overload in the mitochondria, elicit pathways resulting in the opening of mitochondrial permeability transition pore (mPTP) leading to cell death. Therefore, in this study we explored the role of IAA-94 in MI and in maintaining calcium retention capacity (CRC) of cardiac mitochondria after IR. IAA-94 inhibited the CRC of the isolated cardiac mitochondria in a concentration-dependent manner as measured spectrofluorimetrically using calcium green-5 N. Interestingly, IAA-94 did not change the mitochondrial membrane potential. Further, CsA a blocker of mPTP opening could not override the effect of IAA-94. We also showed for the first time that IAA-94 perfusion after ischemic event augments MI by reducing the CRC of mitochondria. To conclude, our results demonstrate that the mechanism of IAA-94 mediated cardio-deleterious effects is via modulating the mitochondria CRC, thereby playing a role in mPTP opening. These findings highlight new pharmacological targets, which can mediate cardioprotection from IR injury.
Keywords: Mitochondria, Chloride intracellular channel proteins, Myocardial infarction, Cardioprotection, Calcium retention capacity
1. Introduction
Ischemia-reperfusion (IR) causes immediate necrotic cell-death resulting in myocardial infarction (MI) [1]. Several studies have been performed with pharmacological agents that may either protect the myocardium or increase the MI from IR injury [2–6]. Majority of cations present in the cardiomyocytes are well characterized in regulating the MI from IR injury [7–9] but studies involving anions are inadequate. The major anion in cardiomyocytes is chloride (Cl−) and its concentration in myocardial cells ranges from 10 to 30 mmol L−1 [10], which is significantly higher from that of an expected equilibrium. Cl− is also a key anion involved in cell-volume regulation and during ischemic pre-conditioning (IPC); increase in cell-volume is accompanied by Cl− efflux [11]. Higher Cl− concentrations in cytoplasm results in activation of Cl− channels located at the plasma membrane and intracellular membranes.
Cl− channels present in cardiac cells play a profound role in the development of cardiac hypertrophy, MI due to IR injury and heart failure [5,10,12–14]. Expectedly, many Cl− channels blockers are known to abolish the cardioprotection rendered by ischemic or pharmacological preconditioning [15,16]. The commonly used intracellular Cl− channel blocker, Indanyloxyacetic acid 94 (IAA-94), was shown to ablate the cardioprotective effects of IPC and preconditioning with cyclosporin A (CsA) [17,18]. It is also known to block Cl− channel currents in cardiac mitoplast and planar bilayer systems [19–23].However, the precise roles and mechanisms involved in increasing MI from IR injury attributable to IAA-94 are unknown.
Since IAA-94 abrogates cardioprotective effects of IPC as well as of CsA, we focused on studying the role of IAA-94 on mitochondrial function, as mitochondria are the key targets for cardioprotection from IR injury. One of the major determinants of cell death in cardiomyocytes during IR is the formation of mitochondrial permeability transition pore (mPTP) [24–26]. MPTP is a pore formed in the mitochondria, which allows the passage of solutes of around 1.5 kDa disrupting the synthesis of ATP and further triggering the activation of cell death pathways [27]. It plays a vital role in the transition from reversible to irreversible damage of adult cardiomyocytes during IR [18,28]. We have recently established the role of IAA-94 in mitochondrial reactive oxygen species (ROS) generation [29]. Calcium overload along with oxidative stress increased phosphate concentration and adenine nucleotide depletion are the major determinants of mPTP formation [30,31]. CsA is a well-characterized inhibitor of mPTP [26,32,33] but is also known to stimulate Cl− efflux [18]. Since IAA-94 blocks CsA-mediated cardioprotection, it is plausible that IAA-94 prevents Cl− efflux facilitated by CsA in the heart.
In this study, we established that IAA-94 shows concentration-dependent inhibition of cardiac mitochondrial calcium retention capacity (CRC). Thus, implicating its role in the early onset of mPTP opening. Further, CsA could not rescue the effect of IAA-94 mediated reduction in CRC. IAA-94 does not affect mitochondrial membrane potential (ψmito). In addition, we showed IAA-94 increases MI in the rat model upon in vivo IR injury as observed in other species. CRC of mitochondria after IR upon IAA-94 treatment was also significantly reduced. Our results suggest that IAA-94 influences cardiac mitochondrial CRC, and therefore possibly play a role in the regulation of mPTP opening during ischemia.
2. Material and methods
All experiments were conducted in accordance with guidelines and approved by the Ohio State University, Drexel University and UT Health Science Center at San Antonio IACUC committees. Two months old Rattus norvegicus were purchased from Charles River (PA). Mitochondria isolation and CRC was measured as described [34,35].
2.1. Left anterior descending coronary artery occlusion and measurement of infarct size
Male Sprague-Dawley rats (250–300 g) were anesthetized with ketamine [80 mg kg−1, intraperitoneally (i.p.)] and xylazine (8 mg kg−1, i.p.). The rats were intubated and ventilated (CWE SAR-830/P). The hearts were exposed through a left thoracotomy in the fourth inter-costal space. The pericardium was opened, and a 5.0 Prolene polypropylene suture was tightened around the proximal left anterior descending coronary artery. Ischemia was confirmed by ST elevation in electrocardiograph. The heart was subjected to 45 min of ischemia, followed by 3 h of reperfusion, which was achieved by releasing the tension on the ligature. An IAA-94 (InformEx New Orleans) bolus [20 mg kg−1 body weight (final concentration: 50 μmol L−1)] was applied via the femoral vein 5 min prior to reperfusion. The same volume of phosphate buffer saline (PBS) was given in the control group. At the end of the experiment, the coronary artery at the same position was occluded again prior to injection of 2.5 ml of 1% (w/v) Evans blue dye into the femoral vein. This reocclusion would specifically delineate the myocardial ischemic area at risk (AAR) which, was identified as the region lacking blue staining. The ventricles of the hearts were sliced transversely into 2 mm thick slices. The slices were incubated in 1% (w/v) triphenyl tetrazolium chloride (TTC) at 37 °C for 15 min to identify the non-infarcted and infarcted areas. The infarcted area was displayed as the area unstained by TTC. Infarct size was expressed as a percentage of the AAR.
2.2. Ischemia-reperfusion injury model ex-vivo
2-month-old SD rats were anesthetized using isoflurane (2%). The hearts were rapidly excised, washed in ice-cold modified Krebs-Henseleit (KH, pH 7.4, concentrations in mmol L−1: 118 NaCl, 4.7 KCl, 1.2 KH2PO4, 1.2 MgSO4, 24 NaHCO3, 11.1 Glucose, 2 CaCl2, 1 sodium pyruvate) solution, mounted on a cannula and perfused with KH solution at 37 °C at a constant volume (2.5 mL/min). After 10 mins of perfusion to remove blood, hearts were subjected to 30 min of global ischemia and 30 min of reperfusion. In some of the group of rats prior to reperfusion hearts were treated with IAA-94 (50 μmol L−1) or DMSO as vehicle control for 10 min. Hearts without any ischemia were used as controls (sham). Mitochondria from the heart were isolated in the various experimental groups to assess calcium retention capacity.
2.3. Isolation of mitochondria
Mitochondria were isolated as described earlier [34,35]. Briefly, rat heart was finely minced and homogenized in ice-cold mitochondrial isolation buffer (in mmolesL−1, 70 sucrose, 210 mannitol, 1 EDTA-Na2, 50 Tris-HCl, pH 7.4) using a Potter-Elvejem homogenizer (10 rapid strokes). The homogenate was transferred into a 2.0 ml Eppendorf tube and centrifuged at 2000 ×g for 5 min. The supernatant was carefully transferred into a clean 1.5 ml Eppendorf tube and centrifuged at 12,000 ×g for 10 min. The pellet containing crude mitochondria was suspended in 55 μL of resuspension buffer (in mmol L−1, 70 sucrose, 210 mannitol, 0.1 EDTA-Na2, 50 Tris HCl, pH 7.4). 5 μl of the mitochondrial fraction was used for measuring protein concentration using DC™ protein assay reagent (Bio-Rad, Cat#5000111). The fluorescence values were normalized with the protein concentration.
2.4. Measurement of CRC
Extra mitochondrial calcium (Ca2+) was detected by Calcium green™-5N using a fluorescence spectrophotometer (Hitachi F-2710).2.5 μmol L−1 Calcium green™-5N, Hexapotassium Salt (Thermo-Fisher) and IAA-94 or DMSO (control) were added to the CRC Buffer (mmol L−1, 150 sucrose, 50 KCl, 2 KH2PO4, 5 succinic Acid, 20 Tris-HCl, pH 7.4) at 25 °C and the fluorescence was measured (excitation at 500 nm and emission at 530 nm). Different concentration of IAA-94/DMSO was added along with the calcium green™-5N. After 30 s, 50 μL of isolated mitochondria sample was added. After another 90 s, and subsequently at every 60 s thereafter, 5 μmol L−1 CaCl2 was added until a sudden increase in fluorescence indicated mitochondrial death. In some experiments, Cyclosporin A (1 nmol L−1) was added before the addition of mitochondria.
2.5. Measurement of mitochondrial membrane potential (MMP)
Rhodamine 123 was used to measure MMP by fluorescence spectrophotometer. Mitochondria were isolated as described earlier [36]. Briefly, rat heart was excised and homogenized in mitochondrial isolation buffer (mmol L−1, 220 mannitol, 70 sucrose, 20 HEPES, 0.4% BSA, 1 K-EDTA, 100 K-EGTA, pH 7.4). The homogenate was centrifuged at 2500 ×g for 5 min. The supernatant was carefully transferred into a clean 1.5 ml Eppendorf tube and centrifuged at 12,100 ×g for 10 min. The mitochondrial pellet was resuspended in mitochondrial resuspension buffer (mmol L−1, 250 sucrose, 20 HEPES, 1 K-EDTA, and 100 EGTA, pH 7.4). 50 nmol L−1 rhodamine 123 was added to 2 ml of mitochondrial membrane potential buffer (in mmol/L, 250 sucrose, 10 HEPES, 0.1 K-EGTA, 2 MgCl2, 0.1 ATP, 4 KH2PO4, pH 7.4) and the basal level fluorescence was measured at 503 nm and 527 nm. 25 μl of the mitochondria was added at 30 s. 90 s later mitochondria was energized with 3 mmol L−1 succinate followed by addition of IAA-94/vehicle control at 7 min. This was followed by addition of 40 nmol L−1 FCCP at 12 min. The fluorescence was measured for an additional 5 min.
2.6. Mitoplast patch clamp
For electrophysiological measurements, mitoplasts (mitochondria without outer membranes) patching was performed as described previously [37,38]. Briefly, mitoplasts were prepared from rat heart mitochondria placed in a hypotonic solution (mmol L−1 5 HEPES, 100 CaCl2, pH 7.2) to induce swelling and disruption of the outer membrane. To restore the sample to an isotonic condition (mmol L−1 150KCl, 10 HEPES, 0.1 CaCl2, pH 7.2) a hypertonic solution (mmol L−1 750 KCl, 30 HEPES, 0.1 CaCl2, pH 7.2) was added. The patch-clamp pipette was filled with an isotonic solution. Mitoplasts are easily recognizable due to their size, round shape, transparency, and presence of a “cap”, characteristics that distinguish these structures from the cellular debris that is also present in the preparation. The currents were low-pass filtered at 1 kHz and sampled at a frequency of 100 kHz (Axopatch 200B). The pipettes had a resistance of about 12–18 MΩ and were pulled using a PC-10 Narishige puller. An isotonic solution containing 100 mmol L−1 CaCl2 was used as the control solution for the presented data. The experiments to assess the channel activity were carried out in patch-clamp inside-out mode. Data were analyzed with pClamp software.
2.7. Immunolabeling
Ultra-pure cardiac mitochondria [7] were incubated with 200 nmol L−1 mitotracker (Mitotracker™ Red CMXRos) in mitochondrial isolation buffer (in mmol L−1, 70 sucrose, 210 mannitol, 1 EDTA-Na2, 50 Tris-HCl, pH 7.4) for 60 min at 4 °C on a rotator shaker. After loading, the mitochondria were seeded onto poly-l-lysine coated coverslips (0.17-mm thickness) for 2 h at 4 °C, followed by fixing and permeabilization [39]. The mitochondrial chloride intracellular channel (CLIC) proteins were used as a positive control. The permeabilized mitochondria were labeled with anti-CLIC4, -CLIC5 and L-type channel antibodies at 4 °C for 16 h. The samples were washed with PBS containing 0.1% (v/v) Triton-X 100, followed by incubation with corresponding secondary antibody conjugates Atto 647 N (1 μg mL−1 each of anti-mouse and anti-rabbit IgGs) or Alexa-488 (2 μg mL−1 anti-mouse IgG or anti-rabbit IgG) in 0.1% (v/v) Triton-X 100 in PBS, containing 1% (w/v) normal goat serum (NGS, G9023, Sigma Aldrich) at room temperature for 60 min. Samples were mounted for confocal microscopy with mowiol® 4–88 (Sigma Aldrich). Images were acquired with an Olympus confocal microscope IX 81 using a 60× oil immersion objective with 1.42 NA (PlanAppoN) and median filtered [7]. The percentage colocalization was quantified using image J.
2.8. Data analysis
Data are presented as mean ± SEM, n ≥ 3. Student’s two-tailed t-test and one way ANOVA followed by Newman Keuls comparison test were used to determine statistical significance. A p value of < 0.05 was considered significant.
3. Results
3.1. IAA-94 reduces mitochondrial CRC but does not affect ψmito
IAA-94 was shown to abrogate cardioprotection mediated by IPC and cyclosporin A (CsA) [17,18]. Our results (supplementary fig. 1), as well as previous studies, demonstrated the presence of IAA-94 sensitive channels in cardiac mitochondria. The channel current was inhibited by a relatively lower concentration of IAA-94 (10 μmol L−1) in cardiac mitoplast. The mechanism, by which IAA-94 sensitive channels play a role in cardioprotection in in vivo, as well as ex-vivo condition, is unknown. Mitochondrial Ca2+ is involved in mPTP opening which triggers cell death signaling pathways. Therefore, we elucidated the role of IAA-94 in CRC of the mitochondria. Interestingly, IAA-94 showed a concentration-dependent inhibition of CRC of the cardiac mitochondria as represented in Fig. 1. We have observed that the calcium uptake by the mitochondria for mPTP opening has significantly reduced to39.6 ± 8% (n = 4) and 84.8 ± 4.2% (n = 4) at 30 μmol L−1 and 100 μmol L−1 IAA-94, respectively. Moreover, there was no change in the ψmito in the presence of IAA-94 as measured spectrophotometrically using rhodamine 123 (Fig. 2A). Membrane potential was also measured in the presence of ATP (Fig. 2B). ATP is known to increase intracellular Ca2+, thereby inducing mitochondrial membrane depolarization. Therefore, we evaluated whether the blocking of CLICs affects the ATP-induced mitochondrial membrane depolarization. Our results show that ATP induces mitochondrial depolarization as indicated by the increased fluorescence but no change in ψmito was observed in the presence of 30 μmol L−1 IAA-94 (Fig. 2B).
Fig. 1.
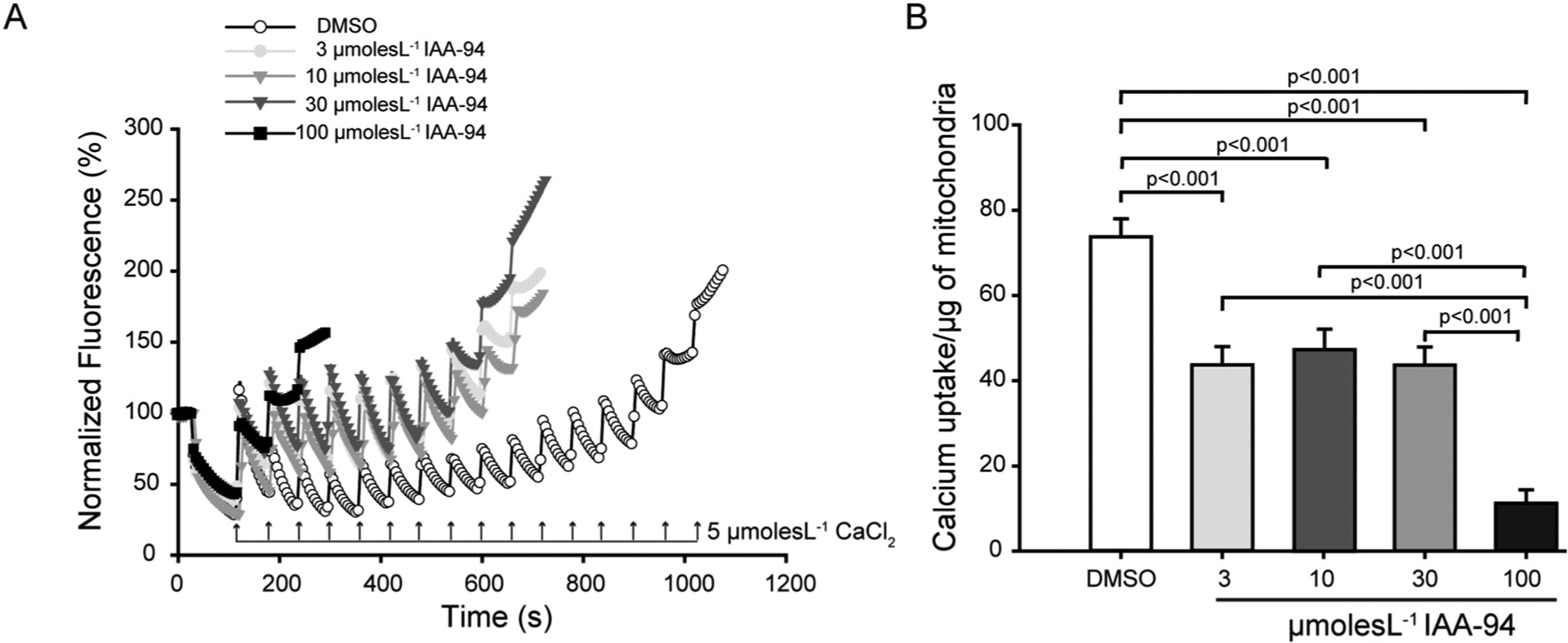
IAA-94 treatment inhibits calcium retention capacity (CRC) of the cardiac mitochondria. Extramitochondrial calcium was labeled with calcium green 5 N and the uptake of calcium was measured in the presence and absence of different concentration of IAA-94 (3–100 μmol L−1), by the addition of 5 μmol L−1 calcium chloride (CaCl2) pulses. After each addition of CaCl2, a fast uptake was observed followed by a state where the equilibrium between the influx and efflux of Ca2+ is reached. The equilibrium is disrupted and the Ca2+ is released when the maximum threshold of Ca2+ is reached within mitochondria. (A), in the representative figure, 14 pulses of 5 μmol L−1 CaCl2 are required to induce mPTP opening in case of control (DMSO), whereas the varying concentration of IAA-94 show decreased CRC of the cardiac mitochondria. (B), bar graph representing average pulses of CaCl2 required for mPTP opening in control as well as varying concentration of IAA-94 (3–100 μmol L−1) in n = 4. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 2.
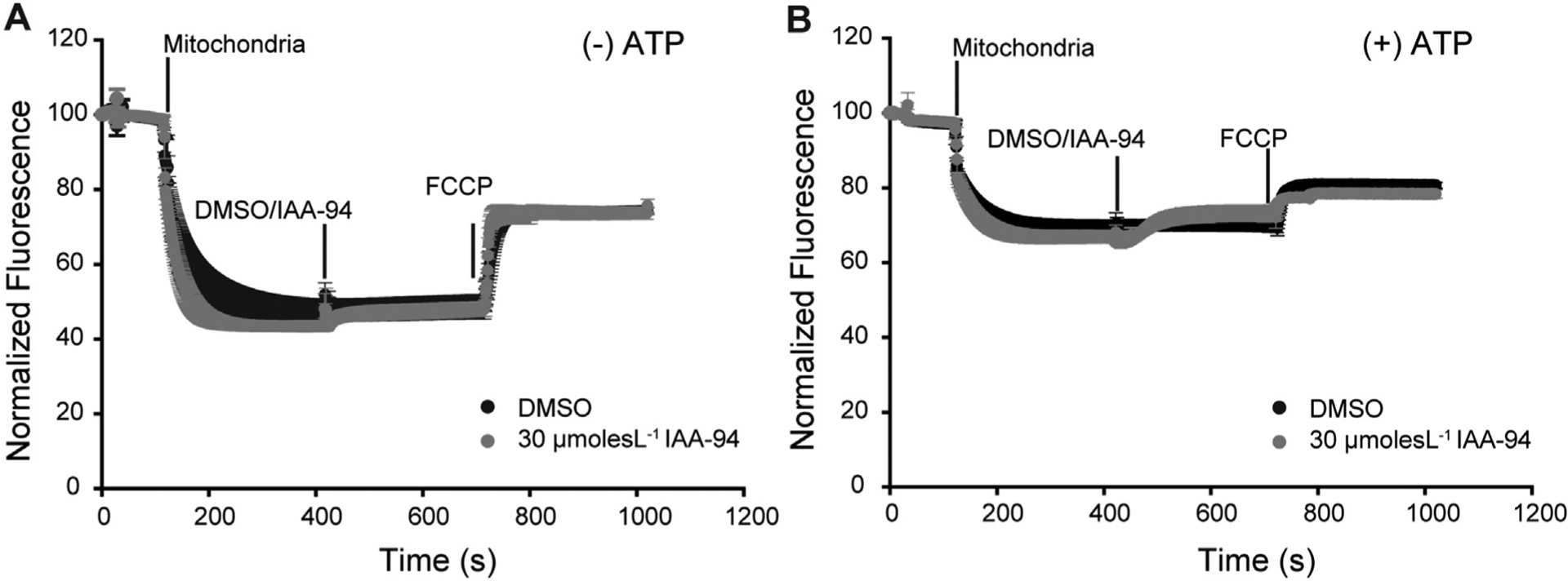
IAA-94 treatment does not alter mitochondrial membrane potential (Δψmito). Cardiac mitochondria were loaded with Rhod-123 followed by exposure to either DMSO or 30 μmol L−1 of IAA-94 at 7 min in the (A) absence and (B) presence of 1mmolesL−1 ATP. FCCP, an uncoupling agent was added at 12th min and the fluorescence intensities were recorded for another 5 min. The y-axis represents percentage relative fluorescence intensities (F/Fi). Presence of ATP depolarizes the mitochondria but there was no change in the Δψmito in the presence or absence of ATP.
3.2. Effect of IAA-94 on cyclosporin A-induced CRC
Cyclophilin D is a mitochondrial matrix protein and a predominant mediator of mPTP opening [40]. The activity of this protein is inhibited by an immunosuppressive drug cyclosporin A (CsA) [41–44]. Previous results have shown that IAA-94 prevented the cardioprotection as well as cellular protection mediated by CsA [18]. Therefore, we tested the effect of IAA-94 on CRC of the cardiac mitochondria in the presence of CsA to understand the mechanism of action of IAA-94. As expected, CsA increased the calcium threshold of cardiac mitochondria by 29.69 ± 2.34% (n = 4) (Fig. 3). However, in the presence of IAA-94 (30 μmol L−1), CsA could not increase the CRC of cardiac mitochondria as shown in Fig. 3A&B. These results indicate that the effect of IAA-94 on mitochondria cannot be reverted by CsA.
Fig. 3.
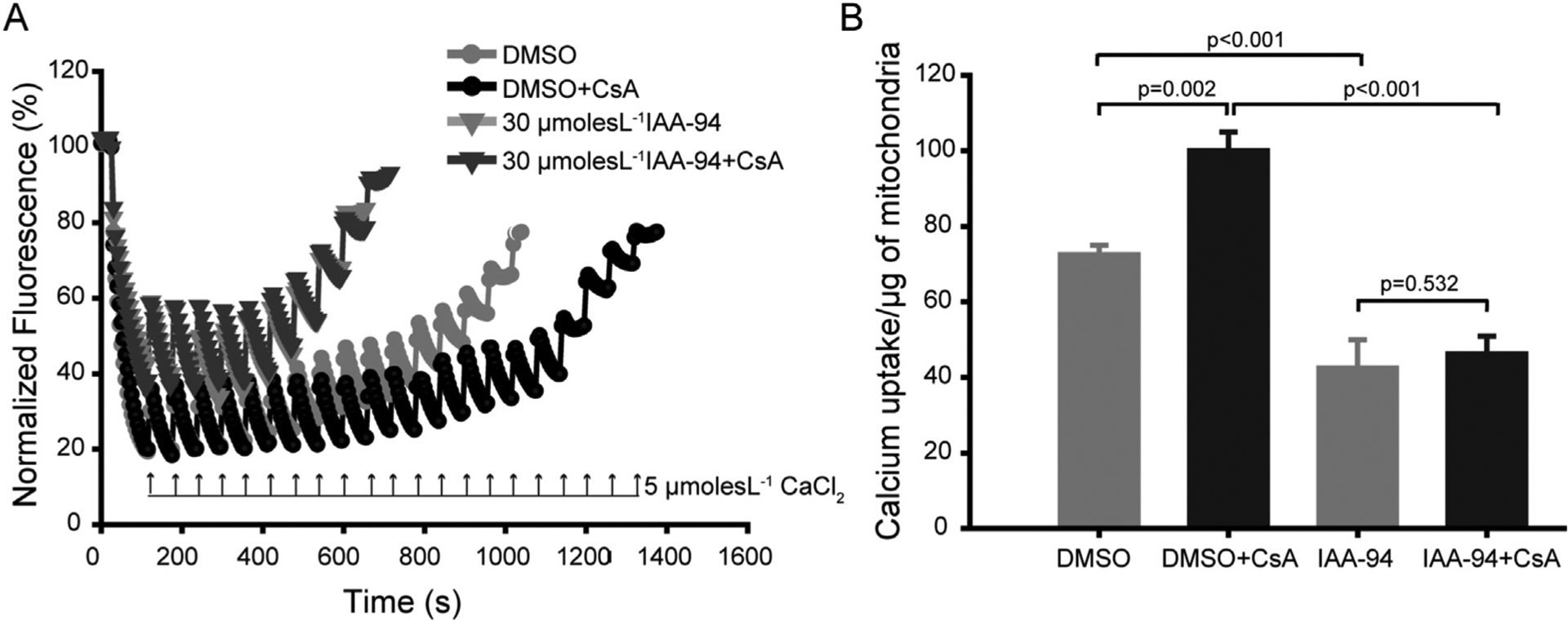
IAA-94 inhibits the cyclosporin A-mediated increase in calcium retention capacity (CRC) of cardiac mitochondria. Extramitochondrial calcium was labeled with calcium green 5 N as described previously and the uptake of the calcium was measured in the presence and absence of both IAA-94 (30 μmol L−1) and cyclosporin A (CsA, 1 nmol L−1). (A) In this representative figure, CsA increased the Ca2+ pulses required to induce mPTP opening to 21 pulses from 14 pulses (absence of CsA) of 5 μmol L−1 CaCl2. IAA-94 treatment inhibited the CsA mediated delayed onset of mPTP opening. (B), bar graphs representing average pulses of CaCl2 required for mPTP opening in both the presence and absence of CsA and IAA-94 in n = 4. Statistical significance values were determined by one way ANOVA test, n.s. is not significant. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3. IAA-94 increases myocardial infarction
IAA-94 is known to prevent cardioprotection due to IPC, and increase MI in rabbit hearts [16,17]. In this study, we examined the effect of IAA-94 (50 μmol L−1) in the heart subjected to IR injury using an in vivo rat model by performing left anterior descending coronary artery occlusion. The area at risk (AAR) to left ventricle (LV) ratio was similar in both groups [54.8 ± 5% in control (n = 5) vs. 54.6 ± 5% in IAA-94 (n = 5)], indicating that the two groups were subjected to a comparable degree of ischemic risk (Fig. 4 A, B). However, the infarct size (IS) was significantly higher in the IAA-94-treated group compared to control; the ratio of IS to AAR was 58.1 ± 7% in IAA-94 vs. 33.5 ± 6% in control (p = 0.01). These results in accordance with the previous data [11,16,17] pharmacologically implicate the significance of IAA-94-sensitive Cl− channels in cardioprotection.
Fig. 4.
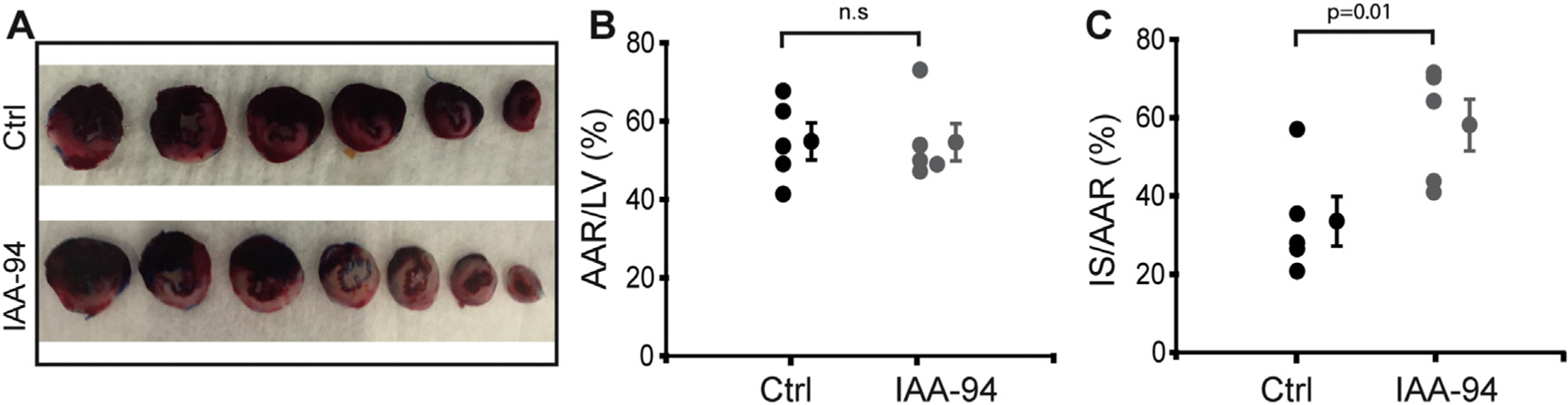
IAA-94 administration increases MI size in in vivo IR in a rat model. Adult male rat hearts were subjected to the left coronary artery occlusion for 45 min followed by 3 h of reperfusion. A single intravenous bolus of phosphate buffered saline (PBS) (control) or IAA-94 (20 mg/kg body weight) was administered 5 min before reperfusion. (A) Cardiac sections showing acute post-ischemic IAA-94-induced increase in MI size compared to control. (B) The percentage ratio of area at risk (AAR) normalized by left ventricle (LV) showed no significant difference in control vs IAA-94 treated group. Administration of IAA-94 significantly increased infarct size (IS) as demonstrated by percentage ratio IS normalized to AAR (C). Values are mean ± SEM. Statistical significance values were determined by one way ANOVA test, p = 0.01in IAA-94 vs. control, n.s. is not significant.
3.4. IAA-94 decreases CRC after ex-vivo ischemia-reperfusion
IAA-94 modulated the CRC of isolated mitochondria (Fig. 1) and also increased the myocardial infarction upon left anterior descending coronary artery occlusion (Fig. 4). We further determined the mechanism of increased infarction upon IAA-94 treatment during IR injury. CRC was measured in isolated mitochondria from heart subjected to ex-vivo ischemia-reperfusion (30 min of global ischemia and reperfusion). In some of the experimental groups hearts were treated with IAA-94 (50 μmol L−1) or DMSO (vehicle control) 10 mins prior to reperfusion. As expected we observed a decrease in CRC upon IR injury [7,45] (Fig. 5). Interestingly, upon IAA-94 post-conditioning, the CRC of mitochondria was significantly decreased (43.1 ± 1.6) vs. DMSO group (56.7 ± 1.4) (n ≥ 3, p < 0.001). Thus, indicating that during IR injury, IAA-94 increases infarction by modulating CRC and thereby affecting mPTP opening.
Fig. 5.
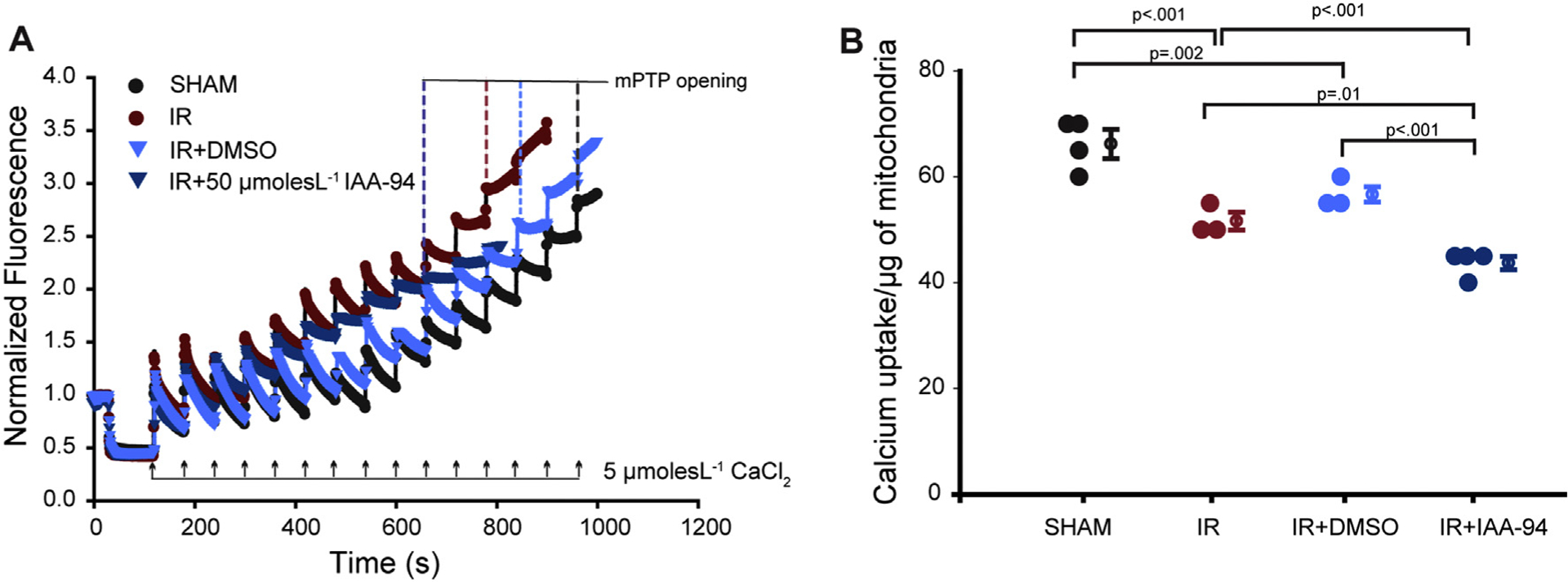
IAA-94 treatment reduces CRC of the cardiac mitochondria during IR injury. Excised hearts were subjected to global ischemia/reperfusion (30 min each) in a modified Langendorff perfusion system. Hearts were treated with IAA-94/DMSO 10 mins prior to reperfusion. CRC was determined by labeling extramitochondrial calcium with calcium green 5 N and the uptake of calcium was measured in the different experimental groups, by the addition of 5 μmol L−1 calcium chloride (CaCl2) pulses. (A), in the representative figure, 14 pulses of 5 μmol L−1 CaCl2 are required to induce mPTP opening in case of sham group, whereas IR group requires 11 pulses of 5 μmol L−1 CaCl2 to open the mPTP. IAA-94 treatment further decreased the CRC upon IR injury (9 pulses). (B), average pulses of CaCl2 required for mPTP opening in different experimental groups (n ≥ 3). Statistical significance values were determined by one way ANOVA test. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
4. Discussion
Coronary heart diseases (CHD) are the major leading cause of morbidity and mortality worldwide [46,47]. The effects of CHD are attributed to the detrimental effects of acute myocardial IR injury [31]. IR injury is accompanied by rapid changes in the ionic equilibrium of myocytes and thereby leading to cell death. Ion channels present in the plasma membrane and intracellular membranes play a huge part in maintaining cardiac physiology as they regulate action potential, and mutations in them are often associated with many cardiac-related diseases [48]. In addition, activation of mitochondrial cation channels like small conductance calcium-activated potassium channels (SKCa) [49,50], large-conductance calcium-activated potassium channels (BKCa) [7,8] and KATP [51–53] have been implicated in mediating cardioprotection from IR injury by modulating the mitochondrial bioenergetics.
Cl− channels and transporters are equally important as they play a role in modulating the volume decrease in response to the increase in cell volume during ischemia, hypoxia, and hypertrophy and thereby protecting cardiomyocytes from cell death [15]. Some of the Cl− channels very well implicated in cardioprotection from IR injury are volume-regulated chloride channels (VRCCs) [15] and cystic fibrosis transmembrane conductance regulator (CFTR) [54,55]. VRCCs are known to regulate redox signaling pathway through interaction with NADPH oxidase (Nox) and/or as a superoxide anion transporter to enhance myocyte viability as well as modulate anti-apoptotic cell signaling pathways during myocardial damage [10]. CFTR was found to be one of the key player mediating cardioprotective effects of IPC and post conditioning (POC).
The role of Cl− channels on cardioprotection is mostly deciphered using their pharmacological inhibitors due to lack of information on their molecular identity. Studies by Diaz et al. demonstrated that Cl− channel blocker like Indanyloxyacetic acid 94 (IAA-94) prevented the IPC and hypo-osmotic stress-mediated protection of rabbit hearts from IR injury [17]. Thus, establishing the involvement of IAA-94 sensitive Cl− channels in IPC. Blocking these channels with IAA-94 also abrogated the cardioprotective effect of pharmacological conditioning using adenosine receptor agonist [2-chloro-N6-cyclopentyladenosine (CCPA)/N6–2-(4-aminophenyl) ethyl adenosine (APNEA)] and the PKC activator (phorbol 12-myristate 13-acetate (PMA) [16]. Unlike IAA-94, blocking with non-selective anion channel blocker 4,4′-Diisothiocyanostilbene-2,2′-disulfonic acid (DIDS) resulted in cardioprotection from IR injury [5]. This differential effect could be attributed to the different targets of these inhibitors.
IAA-94 was used to affinity purify the chloride intracellular channel (CLIC) proteins from bovine kidney lysates [56,57]. Some of the para-logs of mammalian CLICs, namely, CLIC4 and CLIC5 were also established to be present in the cardiac mitochondria [22,29,34]. Similar to previous studies [21], we observed IAA-94 sensitive Cl− channel activity in cardiac mitoplast isolated from R. norvegicus at very low concentration (10 μmol / L) of IAA-94 further confirming the presence of IAA-94 sensitive channels in mitochondria (supplementary fig. 1). Although these sub-conductance states of IAA-94 sensitive mitochondrial chloride channels are attributed to the presence of CLICs in cardiac mitochondria [23], it still needs to be deciphered using CLIC specific genetic knock out mouse.
In agreement with the study by Diaz et al., [17], we showed that blocking Cl− channels with IAA-94, 5 min prior to reperfusion increased myocardial infarction (Fig. 4) suggesting activation of IAA-94 sensitive Cl− channels are necessary during ischemia for cardioprotection against IR injury. Although the role of IAA-94 sensitive Cl− channels in cardioprotection has been very well studied [16,17], the mechanism by which they mediate cardioprotection is not very clear.
In this study, we show that IAA-94 sensitive Cl− channels modulate the CRC of the isolated cardiac mitochondria. IAA-94 showed a concentration-dependent reduction in the CRC of cardiac mitochondria (Fig. 1). CRC of mitochondria is known to determine its viability. It is a measure of the calcium threshold in the mitochondria required to open the mPTP [27]. We observed a very low concentration of IAA-94 (3 μmol L−1) was able to reduce the CRC by ~40% (Fig. 1). A higher concentration of IAA-94 (100 μmol L−1) further reduced the CRC by ~85% as shown in Fig. 1. This suggests the role of IAA-94 sensitive Cl− chloride channels in modulating the mPTP opening via regulating the calcium homeostasis in the cardiomyocyte.
Remarkably, IAA-94 (30 μmol L−1) could not alter the mitochondrial membrane potential both in the presence and absence of ATP (Fig. 2) as measured by rhodamine 123 suggesting that inhibiting Cl− channels does not alter the mitochondrial membrane potential. This is in accordance with the previous studies wherein it was shown that cyclosporin A (CsA), a prototypic inhibitor of mPTP opening disrupted the mitochondrial membrane potential in cardiomyocytes but IAA-94 could not [18]. The mechanism of how IAA-94 treatment results in mPTP opening but does not cause any change in membrane potential is intriguing and needs to be investigated further.
In addition, we observed CRC of mitochondria was significantly reduced in hearts treated with IAA-94 prior to reperfusion during ex vivo IR injury (Fig. 5). This suggests that during ischemia targets of IAA-94 are involved in regulating mitochondrial calcium levels which are required for maintaining the integrity of mPTP. In this study, we provide direct evidence for the underlying mechanism of IAA-94 mediated increased myocardial infarction is via modulating the CRC of mitochondria.
It was shown earlier that blocking chloride channels with IAA-94 abrogated the cardioprotection mediated by CsA [18] in ex vivo studies. Concluding that cardioprotective effects of CsA are not only attributed to their role in inhibiting mPTP opening but also by enhancing the cell volume regulatory mechanisms during ischemia. Further, we investigated the role of IAA-94 sensitive channels on mPTP opening in the presence of CsA. As expected CsA treatment enhanced the CRC of the cardiac mitochondria (Fig. 3) delaying the mPTP opening. Intriguingly, upon blocking Cl− channels with IAA-94, CsA could not salvage the early onset of mPTP opening (Fig. 3).
Previous studies have also shown that CsA could not rescue the peak cell swelling of cardiomyocyte in the presence of IAA-94. In our experiments, IAA-94 treatment in cardiac mitochondria probably leads to mitochondrial swelling. This initial cascade of events is enough to activate the mPTP opening and hence could not be reverted in the presence of CsA. In support, we also show the presence of IAA-94 sensitive Cl− channel conductance in cardiac mitochondria (supplementary fig.1) which could probably play a part in Cl− efflux during volume regulation. Our results corroborated earlier findings that IAA-94 sensitive Cl channels are present in mitochondria [21,23].
All pharmacological approaches have limitations such as non-specific effects due to targeting other proteins or cellular components. IAA-94 is known to inhibit L type calcium channels non-specifically [58]. We isolated mitochondria, loaded them mitotracker and labeled with specific antibodies against L-type calcium channels, CLIC4 and CLIC5. This method of direct visualization of protein in isolated mitochondria provides an opportunity to test the presence (or absence) of target proteins [35,59–62]. In our study, we did not detect L-type calcium channels in cardiac mitochondria isolated from rat hearts (supplementary fig. 2). In the same preparation, IAA-94 sensitive mitochondrial anion channel proteins, CLIC4 (47 ± 5%) and CLIC5 (53 ± 7%) localize very well to cardiac mitochondria [34]. This suggests that the abrogative effects of IAA-94 are associated with the chloride channels and not any other non-specific targets of IAA-94 in mitochondria at the concentration used in this study. Further, these findings do not rule out the possibility of non-mitochondrial chloride channels in cardiomyocyte that could modulate the mPTP opening and thereby play a role in cardioprotection.
To conclude, in this study we show that IAA-94-sensitive chloride channels play a key role in mediating cardioprotection from in vivo IR injury. The IAA-94 mediated cardio deleterious effect probably is governed by regulation of mitochondrial Ca2+ retention capacity by modulating mPTP opening (Fig. 6). In support, we also show the presence of IAA-94 sensitive chloride channels in cardiac mitochondria, which could be putative targets of IAA-94 in mitochondria.
Fig. 6.
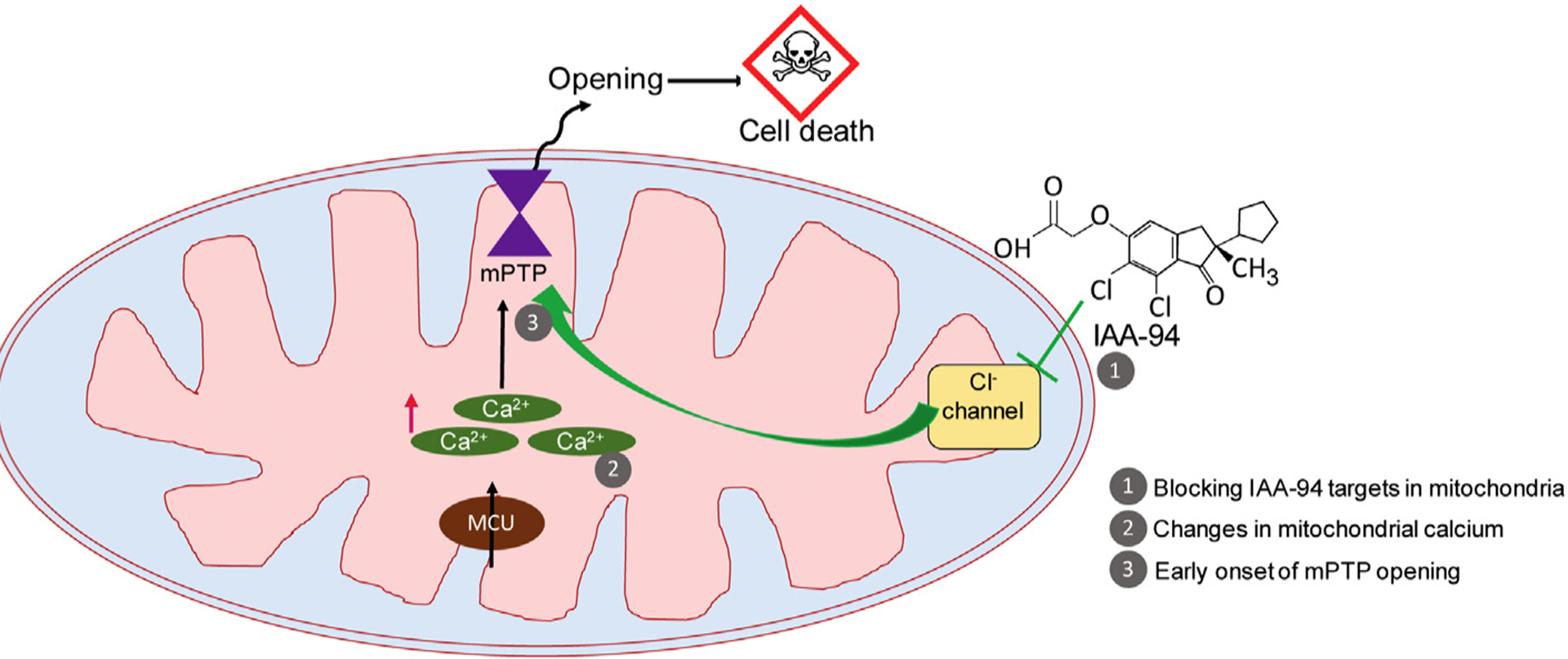
Working hypothesis. During ischemia-reperfusion injury, Calcium overload in cardiac mitochondria leads to mPTP opening resulting in cell death. When IAA-94 targets (Cl− channel) are blocked during IR injury, the mitochondrial calcium is probably increased further resulting in decreased CRC of cardiac mitochondria leading to early onset of mPTP opening.
Supplementary Material
Acknowledgments
We thank Dr. Patrick Osei-Owusu (Drexel University) for initial help and discussion with the IAA-94 studies, and Prof. Olimpia Meucci (Drexel University) for help with rats. This work is supported by American Heart Association Grant in Aid [16GRNT29430000 (HS)], American Heart Association Scientist Development Grant [17SDG33100000 (JCB)], American Heart Association Post-doc fellowship [17POST33670360 (DP)], InstitutesNational Heart, Lung, and Blood Institute [HL133050-01 (HS), HL138093 (JCB), HL136232 (MK)], and National Institute of Neurological Disorders and Stroke [P30NS104177].
Abbreviations:
- APNEA
N6-2-(4-aminophenyl) ethyladenosine
- AAR
area at risk
- ATP
adenosine tri phosphate
- CCPA
2-chloro-N6-cyclopentyladenosine
- CRC
calcium retention capacity
- CLIC
chloride intracellular channel
- (CsA)
cyclosporin A
- DIDS
4,4′-diisothiocyanostilbene-2, 2′-disulfonic acid
- IgGs
immunoglobulins
- IAA-94
R(+)-[(6,7-Dichloro-2-cyclopentyl-2,3-dihydro-2-methyl-1-oxo-1H-inden-5-yl)-oxy]acetic acid/indanyloxyacetic acid 94
- IS
infarct size
- IPC
ischemic preconditioning
- IR
ischemia-reperfusion
- LV
left ventricle
- mPTP
mitochondrial permeability transition pore
- MI
myocardial infarction
- NGS
normal goat serum
- PKC
protein kinase C
- PMA
phorbol 12-myristate 13-acetate
- POC
post-conditioning
- ROS
reactive oxygen species
- TTC
triphenyltetrazolium chloride
Footnotes
Declaration of competing interest
The authors declare that there is no conflict of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.lfs.2019.116841.
References
- [1].Bulluck H, Hausenloy DJ, Ischaemic conditioning: are we there yet? Heart 101 (2015) 1067–1077. [DOI] [PubMed] [Google Scholar]
- [2].Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N, Elbelghiti R, Cung TT, Bonnefoy E, Angoulvant D, Macia C, Raczka F, Sportouch C,Gahide G, Finet G, Andre-Fouet X, Revel D, Kirkorian G, Monassier JP,Derumeaux G, Ovize M, Effect of cyclosporine on reperfusion injury in acute myocardial infarction, N. Engl. J. Med 359 (2008) 473–481. [DOI] [PubMed] [Google Scholar]
- [3].Rupprecht HJ, vom Dahl J, Terres W, Seyfarth KM, Richardt G,Schultheibeta HP, Buerke M, Sheehan FH, Drexler H, Cardioprotective effects of the Na(+)/H(+) exchange inhibitor cariporide in patients with acute anterior myocardial infarction undergoing direct PTCA, Circulation 101 (2000) 2902–2908. [DOI] [PubMed] [Google Scholar]
- [4].Yamada K, Matsui K, Satoh K, Kitano M, Yamamoto S, Ohashi N, Reduction of myocardial infarct size by SM-20550, a novel Na(+)/H(+) exchange inhibitor, in rabbits, Eur. J. Pharmacol 404 (2000) 201–212. [DOI] [PubMed] [Google Scholar]
- [5].Wang X, Cao Y, Shen M, Wang B, Zhang W, Liu Y, He X, Wang L, Xia Y,Ding M, Xu X, Ren J, DIDS reduces ischemia/reperfusion-induced myocardial injury in rats, Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology 35 (2015) 676–688. [DOI] [PubMed] [Google Scholar]
- [6].Pasdois P, Quinlan CL, Rissa A, Tariosse L, Vinassa B, Costa AD, Pierre SV,Dos Santos P, Garlid KD, Ouabain protects rat hearts against ischemia-reperfusion injury via pathway involving src kinase, mitoKATP, and ROS, Am. J. Physiol. Heart Circ. Physiol 292 (2007) H1470–H1478. [DOI] [PubMed] [Google Scholar]
- [7].Singh H, Lu R, Bopassa JC, Meredith AL, Stefani E, Toro L, MitoBK(ca) is encoded by the Kcnma1 gene, and a splicing sequence defines its mitochondrial location, Proc. Natl. Acad. Sci. U. S. A 110 (2013) 10836–10841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Frankenreiter S, Bednarczyk P, Kniess A, Bork NI, Straubinger J, Koprowski P,Wrzosek A, Mohr E, Logan A, Murphy MP, Gawaz M, Krieg T, Szewczyk A,Nikolaev VO, Ruth P, Lukowski R, cGMP-elevating compounds and ischemic conditioning provide Cardioprotection against ischemia and reperfusion injury via cardiomyocyte-specific BK channels, Circulation 136 (2017) 2337–2355. [DOI] [PubMed] [Google Scholar]
- [9].Frankenreiter S, Groneberg D, Kuret A, Krieg T, Ruth P, Friebe A, Lukowski R, Cardioprotection by ischemic postconditioning and cyclic guanosine monophosphate-elevating agents involves cardiomyocyte nitric oxide-sensitive guanylyl cyclase, Cardiovasc. Res 114 (2018) 822–829. [DOI] [PubMed] [Google Scholar]
- [10].Duan DD, Phenomics of cardiac chloride channels, Comprehensive Physiology 3 (2013) 667–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Diaz RJ, Hinek A, Wilson GJ, Direct evidence of chloride ion efflux in ischaemic and pharmacological preconditioning of cultured cardiomyocytes, Cardiovasc. Res 87 (2010) 545–551. [DOI] [PubMed] [Google Scholar]
- [12].Li P, Sun X, Volume regulated anion channel and ischemia/reperfusion injury of myocardium, Sheng wu yi xue gong cheng xue za zhi = Journal of biomedical engineering = Shengwu yixue gongchengxue zazhi 25 (2008) 980–983. [PubMed] [Google Scholar]
- [13].Zheng XB, Wang R, Yang HL, Sun XL, Effects of chloride ion channel and its blockers on myocardial ischemia reperfusion arrhythmias in rabbits, Zhonghua Yi Xue Za Zhi 93 (2013) 1168–1173. [PubMed] [Google Scholar]
- [14].Ye L, Zhu W, Backx PH, Cortez MA, Wu J, Chow YH, McKerlie C, Wang A,Tsui LC, Gross GJ, Hu J, Arrhythmia and sudden death associated with elevated cardiac chloride channel activity, J. Cell. Mol. Med 15 (2011) 2307–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bozeat ND, Xiang SY, Ye LL, Yao TY, Duan ML, Burkin DJ, Lamb FS, Duan DD, Activation of volume regulated chloride channels protects myocardium from ischemia/reperfusion damage in second-window ischemic preconditioning, Cell. Physiol. Biochem 28 (2011) 1265–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Batthish M, Diaz RJ, Zeng HP, Backx PH, Wilson GJ, Pharmacological preconditioning in rabbit myocardium is blocked by chloride channel inhibition, Cardiovasc. Res 55 (2002) 660–671. [DOI] [PubMed] [Google Scholar]
- [17].Diaz RJ, Losito VA, Mao GD, Ford MK, Backx PH, Wilson GJ, Chloride channel inhibition blocks the protection of ischemic preconditioning and hypo-osmotic stress in rabbit ventricular myocardium, Circ. Res 84 (1999) 763–775. [DOI] [PubMed] [Google Scholar]
- [18].Diaz RJ, Fernandes K, Lytvyn Y, Hawrylyshyn K, Harvey K, Hossain T,Hinek A, Wilson GJ, Enhanced cell-volume regulation in cyclosporin a cardioprotection, Cardiovasc. Res 98 (2013) 411–419. [DOI] [PubMed] [Google Scholar]
- [19].Tulk BM, Schlesinger PH, Kapadia SA, Edwards JC, CLIC-1 functions as a chloride channel when expressed and purified from bacteria, J. Biol. Chem 275 (2000) 26986–26993. [DOI] [PubMed] [Google Scholar]
- [20].Singh H, Ashley RH, Redox regulation of CLIC1 by cysteine residues associated with the putative channel pore, Biophys. J 90 (2006) 1628–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Misak A, Grman M, Malekova L, Novotova M, Markova J, Krizanova O,Ondrias K, Tomaskova Z, Mitochondrial chloride channels: electrophysiological characterization and pH induction of channel pore dilation, European biophysics journal: EBJ 42 (2013) 709–720. [DOI] [PubMed] [Google Scholar]
- [22].Gururaja Rao S, Ponnalagu D, Patel NJ, Singh H, Three decades of chloride intracellular channel proteins: from organelle to organ physiology, Curr Protoc Pharmacol 80 (2018) 11 21 11–11 21 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tomasek M, Misak A, Grman M, Tomaskova Z, Subconductance states of mitochondrial chloride channels: implication for functionally-coupled tetramers, FEBS Lett. 591 (2017) 2251–2260. [DOI] [PubMed] [Google Scholar]
- [24].Morciano G, Bonora M, Campo G, Aquila G, Rizzo P, Giorgi C, Wieckowski MR,Pinton P, Mechanistic role of mPTP in ischemia-reperfusion injury, Adv. Exp. Med. Biol 982 (2017) 169–189. [DOI] [PubMed] [Google Scholar]
- [25].Hausenloy DJ, Yellon DM, The mitochondrial permeability transition pore: its fundamental role in mediating cell death during ischaemia and reperfusion, J. Mol. Cell. Cardiol 35 (2003) 339–341. [DOI] [PubMed] [Google Scholar]
- [26].Heusch G, Boengler K, Schulz R, Inhibition of mitochondrial permeability transition pore opening: the holy grail of cardioprotection, Basic Res. Cardiol 105 (2010) 151–154. [DOI] [PubMed] [Google Scholar]
- [27].Bernardi P, Di Lisa F, The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection, J. Mol. Cell. Cardiol 78 (2015) 100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Baines CP, The mitochondrial permeability transition pore and the cardiac necrotic program, Pediatr. Cardiol 32 (2011) 258–262. [DOI] [PubMed] [Google Scholar]
- [29].Ponnalagu D, Singh H, Anion channels of mitochondria, Handbook of Experimental Pharmacology, vol. 240, 2017, pp. 71–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Griffiths EJ, Halestrap AP, Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion, Biochem. J, 307 ( Pt 1) (1995) 93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hausenloy DJ, Yellon DM, Myocardial ischemia-reperfusion injury: a neglected therapeutic target, J. Clin. Invest 123 (2013) 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lim SY, Davidson SM, Hausenloy DJ, Yellon DM, Preconditioning and postconditioning: the essential role of the mitochondrial permeability transition pore, Cardiovasc. Res 75 (2007) 530–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Skyschally A, Schulz R, Heusch G, Cyclosporine a at reperfusion reduces infarct size in pigs, Cardiovasc. Drugs Ther. 24 (2010) 85–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ponnalagu D, Gururaja Rao S, Farber J, Xin W, Hussain AT, Shah K, Tanda S,Berryman M, Edwards JC, Singh H, Molecular identity of cardiac mitochondrial chloride intracellular channel proteins, Mitochondrion 27 (2016) 6–14. [DOI] [PubMed] [Google Scholar]
- [35].Singh H, Lu R, Rodriguez PF, Wu Y, Bopassa JC, Stefani E, Toro L, Visualization and quantification of cardiac mitochondrial protein clusters with STED microscopy, Mitochondrion 12 (2012) 230–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Baracca A, Sgarbi G, Solaini G, Lenaz G, Rhodamine 123 as a probe of mitochondrial membrane potential: evaluation of proton flux through F(0) during ATP synthesis, Biochim. Biophys. Acta 1606 (2003) 137–146. [DOI] [PubMed] [Google Scholar]
- [37].Bednarczyk P, Wieckowski MR, Broszkiewicz M, Skowronek K, Siemen D,Szewczyk A, Putative structural and functional coupling of the mitochondrial BKCa Channel to the respiratory chain, PLoS One 8 (2013) e68125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gururaja Rao S, Bednarczyk P, Towheed A, Shah K, Karekar P, Ponnalagu D, Jensen HN, Addya S, Reyes BAS, Van Bockstaele EJ, Szewczyk A,Wallace DC, Singh H, BKCa (Slo) channel regulates mitochondrial function and lifespan in Drosophila melanogaster, Cells 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ponnalagu D, Rao SG, Farber J, Xin W, Hussain AT, Shah K, Tanda S,Berryman MA, Edwards JC, Singh H, Data supporting characterization of CLIC1, CLIC4, CLIC5 and DmCLIC antibodies and localization of CLICs in endoplasmic reticulum of cardiomyocytes, Data in brief 7 (2016) 1038–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ, Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia, Proc. Natl. Acad. Sci. U. S. A 102 (2005) 12005–12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Fournier N, Ducet G, Crevat A, Action of cyclosporine on mitochondrial calcium fluxes, J. Bioenerg. Biomembr 19 (1987) 297–303. [DOI] [PubMed] [Google Scholar]
- [42].Crompton M, Ellinger H, Costi A, Inhibition by cyclosporin a of a Ca2+−dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress, The Biochemical journal 255 (1988) 357–360. [PMC free article] [PubMed] [Google Scholar]
- [43].Broekemeier KM, Dempsey ME, Pfeiffer DR, Cyclosporin a is a potent inhibitor of the inner membrane permeability transition in liver mitochondria, J. Biol. Chem 264 (1989) 7826–7830. [PubMed] [Google Scholar]
- [44].Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P, Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D, J. Biol. Chem 280 (2005) 18558–18561. [DOI] [PubMed] [Google Scholar]
- [45].Gedik N, Heusch G, Skyschally A, Infarct size reduction by cyclosporine a at reperfusion involves inhibition of the mitochondrial permeability transition pore but does not improve mitochondrial respiration, Arch. Med. Sci 9 (2013) 968–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, de Ferranti SD, Ferguson JF, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Lutsey PL, Mackey JS, Matchar DB, Matsushita K, Mussolino ME, Nasir K, O’Flaherty M, Palaniappan LP,Pandey A, Pandey DK, Reeves MJ, Ritchey MD, Rodriguez CJ, Roth GA, Rosamond WD, Sampson UKA, Satou GM, Shah SH, Spartano NL, Tirschwell DL, Tsao CW, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, American E Heart Association Council on, C. Prevention Statistics, S. Stroke Statistics, Heart Disease and Stroke Statistics-2018 Update: A Report from the American Heart Association, Circulation, 137 (2018) e67–e492. [DOI] [PubMed] [Google Scholar]
- [47].Foley JR, Plein S, Greenwood JP, Assessment of stable coronary artery disease by cardiovascular magnetic resonance imaging: current and emerging techniques, World J. Cardiol 9 (2017) 92–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Amin AS, Tan HL, Wilde AA, Cardiac ion channels in health and disease, Heart rhythm: the official journal of the Heart Rhythm Society 7 (2010) 117–126. [DOI] [PubMed] [Google Scholar]
- [49].Stowe DF, Gadicherla AK, Zhou Y, Aldakkak M, Cheng Q, Kwok WM,Jiang MT, Heisner JS, Yang M, Camara AK, Protection against cardiac injury by small Ca(2+)-sensitive K(+) channels identified in guinea pig cardiac inner mitochondrial membrane, Biochim. Biophys. Acta 1828 (2013) 427–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Tano JY, Gollasch M, Calcium-activated potassium channels in ischemia reperfusion: a brief update, Front. Physiol 5 (2014) 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gross GJ, Auchampach JA, Blockade of ATP-sensitive potassium channels prevents myocardial preconditioning in dogs, Circ. Res 70 (1992) 223–233. [DOI] [PubMed] [Google Scholar]
- [52].Bu HM, Yang CY, Wang ML, Ma HJ, Sun H, Zhang Y, K(ATP) channels and MPTP are involved in the cardioprotection bestowed by chronic intermittent hypobaric hypoxia in the developing rat, J. Physiol. Sci 65 (2015) 367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Paggio A, Checchetto V, Campo A, Menabo R, Di Marco G, Di Lisa F, Szabo I,Rizzuto R, De Stefani D, Identification of an ATP-sensitive potassium channel in mitochondria, Nature 572 (2019) 609–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Xiang SY, Ye LL, Duan LL, Liu LH, Ge ZD, Auchampach JA, Gross GJ, Duan DD, Characterization of a critical role for CFTR chloride channels in cardioprotection against ischemia/reperfusion injury, Acta Pharmacol. Sin 32 (2011) 824–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Chen H, Liu LL, Ye LL, McGuckin C, Tamowski S, Scowen P, Tian H,Murray K, Hatton WJ, Duan D, Targeted inactivation of cystic fibrosis transmembrane conductance regulator chloride channel gene prevents ischemic preconditioning in isolated mouse heart, Circulation 110 (2004) 700–704. [DOI] [PubMed] [Google Scholar]
- [56].Landry DW, Akabas MH, Redhead C, Edelman A, Cragoe EJ Jr., Al-Awqati Q, Purification and reconstitution of chloride channels from kidney and trachea, Science 244 (1989) 1469–1472. [DOI] [PubMed] [Google Scholar]
- [57].Singh H, Two decades with dimorphic chloride intracellular channels (CLICs), FEBS Lett. 584 (2010) 2112–2121. [DOI] [PubMed] [Google Scholar]
- [58].Doughty JM, Miller AL, Langton PD, Non-specificity of chloride channel blockers in rat cerebral arteries: block of the L-type calcium channel, J. Physiol, 507 ( Pt 2) (1998) 433–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Martin LJ, Lau E, Singh H, Vergnes L, Tarling EJ, Mehrabian M, Mungrue I,Xiao S, Shih D, Castellani L, Ping P, Reue K, Stefani E, Drake TA, Bostrom K, Lusis AJ, ABCC6 localizes to the mitochondria-associated membrane, Circ. Res 111 (2012) 516–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Singh H, Lu R, Bopassa JC, Meredith AL, Stefani E, Toro L, mitoBKCa is encoded by the Kcnma1 gene, and a splicing sequence defines its mitochondrial location, Proc. Natl. Acad. Sci. U. S. A 110 (2013) 10836–10841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Singh H, Li M, Hall L, Chen S, Sukur S, Lu R, Caputo A, Meredith AL,Stefani E, Toro L, MaxiK channel interactome reveals its interaction with GABA transporter 3 and heat shock protein 60 in the mammalian brain, Neuroscience 317 (2016) 76–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Zhang J, Li M, Zhang Z, Zhu R, Olcese R, Stefani E, Toro L, The mitochondrial BKCa channel cardiac interactome reveals BKCa association with the mitochondrial import receptor subunit Tom22, and the adenine nucleotide translocator, Mitochondrion 33 (2017) 84–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.