

Abstract
Objective
Neurofilament light chain (NfL) has been established as a biomarker of axonal damage in many diseases of the central nervous system (CNS). Increased levels of serum NfL (sNfL) can derive as well from damage in the peripheral nervous system (PNS) as from CNS, but little is known about the quantities contributing to sNfL. Peripheral nerve damage may be reflected by an increase in sNfL levels, while the NfL CSF/serum ratio and NfL index decreases.
Methods
We collected serum and cerebrospinal fluid (CSF) from 21 Guillain‐Barré Syndrome (GBS) patients and measured NfL in serum and CSF and compared them with 19 neurologically healthy controls.
Results
In general, NfL in CSF and serum was significantly higher in GBS patients. Serum NfL was higher in GBS patients admitted to the intensive care unit (P = 0.02). Controls had a mean CSF/serum NfL ratio of 26.7 (ranging from 5.8 to 69.5) indicating a central origin of NfL. Three GBS patients had a similar range (23.9 to 42.7, mean 33.3) all of them with demyelinating pathology in the PNS. Eighteen GBS patients with axonal or mixed axonal‐demyelinating pathology showed significantly lower CSF/serum ratios (0.02–12.2, mean 4.4), indicative of a peripheral origin of NfL. When applying the NfL index subdivisions remain the same.
Interpretation
These results demonstrate that the PNS is a relevant contributor to sNfL levels and that the distribution can be identified by a lowered NfL CSF/serum ratio of NfL index. Furthermore, acute or subacute polyneuropathies are likely confounding factors in interpreting sNfL levels in CNS diseases.
Introduction
Neurofilaments are cytoskeletal proteins of exclusively neuronal origin. Neurofilament light chain (NfL) levels in CSF have been established as biomarker of neuronal damage in numerous neurological diseases of the CNS. 1 , 2 , 3
With the advent of the single molecule assay (SIMOA) technique NfL could also be quantitated reliably at low levels in serum, confirming its capacity to reflect disease activity in the CNS. 4 , 5 Implicitly, NfL in blood was considered of being of CNS origin when measured in terms of CNS disorders not taking into account the possibility of being derived from the PNS also. However, axons of the PNS hence release NfL upon damage. 6 Accordingly, elevated sNfL levels have already been shown in various vasculitic and hereditary neuropathies. 6 , 7 GBS separates from other neuropathies as it affects intrathecal and extrathecal parts of the peripheral nervous system. Accordingly, increased levels of NfL were found both in CSF, 8 , 9 and serum or plasma. 10 However, the question to which extent intrathecal/central and extrathecal/peripheral nerve pathology contribute to the neurofilament levels in serum has not been addressed although of upmost importance for correct interpretation of sNfL levels.
We hypothesized that increased sNfL in GBS results to a relevant degree from extrathecal axonal damage, which may be reflected by a decrease in the NfL CSF/serum ratio and the NfL index. Furthermore, these two parameters render possible to calculate the portions of NfL in every disease and in a consequence to assess the origin of the NfL levels.
Methods
Cohorts
We performed a retrospective analysis of CSF and serum samples from 21 patients suffering from GBS and 19 non‐neurological disease controls who underwent lumbar puncture (LP) to exclude diseases such as cerebral haemorrhage or neuroborreliosis and without signs of polyneuropathy or neurodegeneration having normal CSF parameters. At every lumbar puncture CSF and serum were drawn in one session. We included 21 GBS patients who had been diagnosed and treated at the Department of Neurology, University Hospital Magdeburg, Germany. Six patients had clinical follow‐up data with longitudinally sampled CSF and serum available. GBS was diagnosed according to the criteria presenting with at least bilateral or flaccid weakness of limbs accompanied by decreased or absent tendon reflexes, CSF cell count under 50 cells/µl, monophasic course and time between onset‐nadir of 12 h to 28 days. 11
This study included GBS patients with Brighton collaboration criteria 2 with some GBS patients fulfilling Brighton Criteria 1. 12 Assessment of disease severity was performed with the GBS Disability Scale (GBSDS). 13
Laboratory procedures
Patients were tested for presence of ganglioside antibodies using a standard immunoblot (EUROLINE, DL‐1130‐1601‐2 G/M comprising Anti‐Hu, Anti‐Ri, Anti‐Yo, Anti‐Zic4, Anti‐DNER, Anti‐Recoverin, Anti‐GAD65, Anti‐SOX1, Anti‐Titin, Anti‐Ma2, Anti‐Amphiphysin, EUROIMMUN, Lübeck, Germany), had a CSF analysis. Every patient received a standard electrophysiological procedure including measuring the motor and sensory nerve conduction velocities and f‐wave latency measurements in at least N. tibialis and N. peroneaus in one leg. Additionally, most patients received the same program on N. medianus and N. ulnaris at least at one arm. Furthermore, a minimum of one electromyographic measurement from the M. Tibialis anterior was added in each patient. Axonal motor GBS subtype required next to pathological nerve conduction velocities present f‐waves without pathological latency. 11 Electrophysiological classification (axonal, demyelinating, or mixed type neuropathy) was performed in all GBS patients. We used the electrophysiological GBS criteria according to Hadden 14 before 2015 and then changed to the electrophysiological criteria according to Rajabally. 15
This diagnostical workup enabled us to categorize according to the Brighton Criteria.
NfL measurements
sNfl measurements were performed with SIMOA at the University Hospital Basel, Switzerland, by an in‐house assay as described. 4 NfL in CSF was measured with the ELISA using the similar antibody pair (Umandiagnostics®, Umeå, Sweden) at the University Hospital Magdeburg.
Statistics
Statistical analyses were performed with GraphPad Prism. We used a one‐way ANOVA with Bonferroni correction, for correlation analysis Spearman rho, and considered P < 0.05 as significant. We calculated an NfL index using CSF‐NfL/sNfL divided by the Albumin ratio.
The study was approved by the ethics committee of the University Hospital Magdeburg, Germany (approval number 22/19). Every patient gave written informed consent.
Results
Clinical presentation, standard CSF evaluation, and electrophysiological evaluation of GBS patients
Table 1 shows that in the mean all patients demonstrated an increased albumin ratio; 24% (5/21) had serum anti‐ganglioside antibodies. Guillain‐Barré Disability Score (GBSDS) ranged from 2 to 5 (Fig. 1a).
Table 1.
Statistical values and analyses from patients and related measurements of NfL and albumin levels
| GBS‐ratiolow | GBS‐ratiohigh | Controls | |
|---|---|---|---|
| n | 18 | 3 | 19 |
| Age (f:m) | 58.4 (6:12) | 60,7 (0:3) | 64.7 (9:10) |
| Time to LP [days] | 17.1 | 3,7 | n/a |
| Mean CSF cell count | 2.2 | 1 | 1.2 |
| CSF/serum Albumin ratio (Qalb) | 17.1 | 13.8 | 6.1 |
| Mean serum NfL [pg/mL] | 2871.6 | 397.4 | 50.7 |
| Mean CSF NfL [pg/mL] | 5939.5 | 15854 | 1115 |
| NfL CSF/serum ratio | 3.96 | 33.32 | 26.7 |
| NfL‐index | 0.4 | 3.15 | 4.97 |
| Ganglioside antibody positive | 4 | 1 | n/a |
| GBS disablility score | 3.6 | 3.1 | n/a |
| Electrophysiology: axonal/deymelinating | 16/14 | 0/3 | n/a |
| Electrophysiology: F‐waves measurable? | 0/18 | 1/3 | n/a |
| sNfL/CSF‐NfL | sNfL/CSF‐NfL | ||
|---|---|---|---|
| ANOVA NfL levels versus controls | 0.003*/0.07 | 0.005*/0.004* | n/a |
| Spearman correlation GBSDS P‐value | 0.023*/0.048* | 0.333/0.333 | n/a |
| Spearman correlation Qalb P‐value | 0.099/0.443 | <0.001*/<0.001* | n/a |
| Spearman correlation CSF/Serum ratio P‐value | 0.581/0.003* | 0.666/0.666 | n/a |
Figure 1.
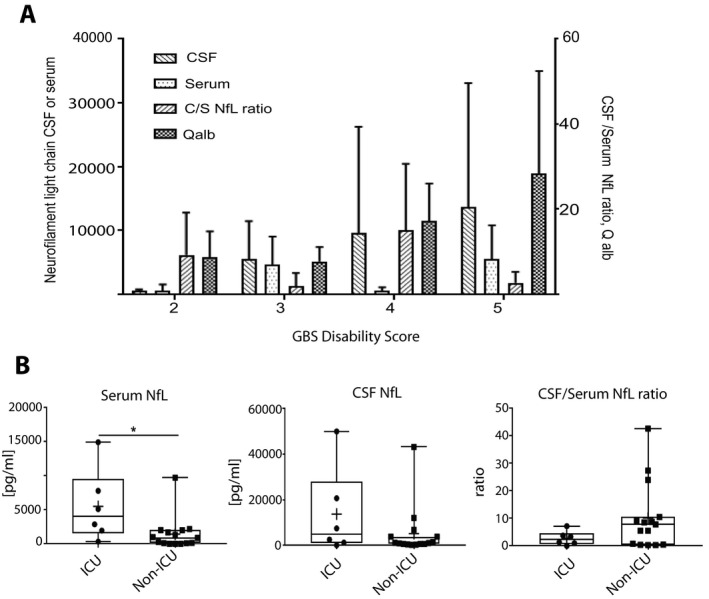
(A) Split up of the GBS patients per GBS Disability score 2 to 5 showing NFL CSF/serum ratio, Serum‐NfL and CSF‐NfL levels, and Albumin quotient per score. (B) CSF‐NfL, Serum‐NfL and its ratio separated for GBS patients who had been admitted to the ICU and not. Significant difference is marked with *.
Based on electrophysiological work‐up, 67% (14/21) of patients showed a mixed neuropathy pattern; 9% (2/21) were classified as purely axonal and 24% (5/21) as purely demyelinating neuropathies. The electrophysiological workup was applied according to standardized methods. All patients were treated with either intravenous immunoglobulins or with plasma exchange at first. Six patients had to be admitted to the Intensive Care Unit (ICU).
NfL levels in CSF and serum: biomarker for GBS and clinical course
Serum NfL levels were significantly higher in every GBS patient (mean = 2603, 25.1 to 14935 pg/mL) than in controls (mean = 50.7, 11.1 to 84.9 pg/mL, P < 0.01, Table 1). Accordingly, CSF‐NfL levels (mean = 7623, 149–50,000 pg/mL) were ranging more than 5‐fold higher versus CSF‐Controls (mean = 1114, 545 to 1957 pg/mL, P = 0.02, Table 1). Serum‐NfL, but not CSF‐NfL was significantly higher in patients who were admitted to ICU compared to patients who remained on the ward (P = 0.02 vs. P = 0.16, Fig. 1B).
NfL levels in CSF and serum: categorisation into high vs low CSF/serum ratio groups
The NfL CSF/serum ratio in controls with NfL originating predominantly from CNS structures ranged from 5.9 to 69.8 (mean = 26.7) The margin to its lowest quartile (12.8) was used as a cut‐off value indicating the intra‐ or extrathecal origin of sNfL, respectively. This allowed us to categorize patients into a NfL‐ratiohigh (n = 3, mean = 33.23, range 24.00–42.65) versus a NfL‐ratiolow group (n = 18, mean = 3.95, 0.03–10.52, Fig. 2A).
Figure 2.
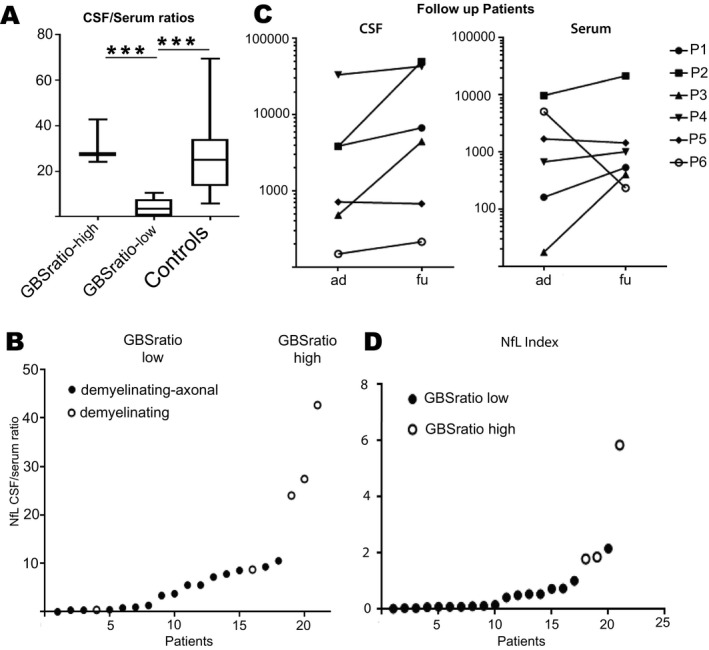
(A) CSF/serum NfL ratio are separated in groups with high and low ratio. High NfL ratios can be found in patients with predominant CNS affection and in control. (B) NfL CSF/serum ratios in all GBS patients, full circle = mixed demyelinating axonal affection in nerve conduction velocity, hollow circle = demyelinating lesions only in nerve conduction velocity. (C) NfL indices of all GBS patients. Full circle meaning NfL‐ratiolow and hollow circle representing NfL‐ratiohigh. (D) CSF‐NfL (left) and Serum NfL (right) levels in follow‐up GBS patients (ad = at diagnose, fu = follow up) Significant difference is marked with *.
Serum‐NfL and CSF/serum NfL ratio were strongly correlated (P < 0.001), but not CSF‐NfL with the CSF/serum NfL ratio (P = 0.44). The GBSDS correlated well with CSF‐NfL (P = 0.005), sNfL (P = 0.02), and the Qalb (P = 0.04), but not with the NfL CSF/serum ratio (P = 0.41, Table 1).
Clinical data show particular differences between both cohorts
Nerve conduction studies showed either axonal or mixed patterns in the NfL‐ratiolow, while in the NfL‐ratiohigh cohorts only demyelinating patterns were found (Fig. 2B). GBSDS was slightly higher in the NfL‐ratiolow group (mean = 3.6, range 2–5) than in the NfL‐ratiohigh group (mean = 3.3, range from 2 to 4). The mean albumin ratio scored slightly higher in the NfL‐ratiolow (17.11 pg/mL) than in NfL‐ratiohigh (13.77 pg/mL) group, but this difference did not reach levels of significance (P = 0.74).
A major difference was time from disease onset to lumbar puncture with NfL‐ratiohigh at 3.7 days and in the NfL‐ratiolow at 17.1 days.
Ratios in follow‐up patients
Six GBS patients had clinical follow‐up data. The mean time interval between lumbar punctures was 8.8 days for five patients and one patient having a second spinal tap 127 days after disease onset. Serum and CSF NfL levels increased in most patients with CSF/serum NfL ratios staying within the low or high ratio groups irrespective of GBSDS (Fig. 2C).
Applying the NfL index results in almost similar results
Albumin ratio and sNfL did not correlate in GBS patients overall (P = 0.76) and not in the NfL‐ratiolow group (P = 0.44) but in the group NfL‐ratiohigh (P < 0.01). CSF‐NfL and Qalb in NfL‐ratiohigh also correlated well (P < 0.01) but not in the NfL‐ratiolow (Table 1).
NfL index in controls was 4.97 (ranged from 0.78 to 18.27) with lowest quartile at 2.8. Using the NfL index elicited slightly different subdivisions depending on the cut‐off used. Applying the lowest quartile from the NfL index obtained from controls would leave only one patient in the intrathecal NfL group. Simply using the NfL index produced a gap (Fig. 2D) separating four GBS patients from the other (NfL index range from 1.77 to 5.83) comprising the four patients with the highest CSF/Serum NfL ratio (ratio ranging from 10.95 to 42.65) and having a mixed demyelinating‐axonal PNS pathology being inside the high NfL index group (Fig. 2D). The parameter “time disease onset to LP” would remain highly different between NfL index high versus NfL index low group (t = 6.25 days to t = 17.8 days).
Mean NfL index in NfL‐ratiohigh was at 3.15 (1.78–4.83) and not different from that of controls (P = 0.34). In NfL‐ratiolow mean NfL index was 0.4 (ranging from 0.00581 to 2.14) with significant difference to the controls (P < 0.01). Comparison of the interquartile range for the C/S NfL ratio or the NfL index with the controls also showed highly significant differences (Kolmogorov‐Smirnoff P < 0.001, Fig. 3).
Figure 3.
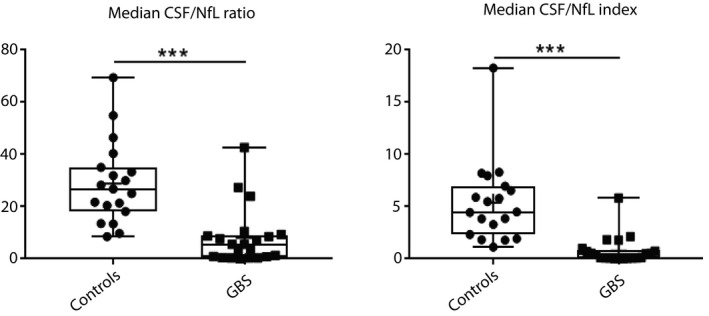
Comparison of the interquartile ranges of the C/S ratio and NfL indices between controls and GBS patients. Significant difference with P < 0.001 is marked with ***.
Discussion
NfL ratio and NfL index are markers to determine the origin of NfL. This is important to assess the contribution of the different NfL sources to the sNfl level. We could show in this study that NfL of peripheral origin can be a significant contributor of overall sNfL levels. In general, sNfL is a biomarker for GBS and is also a marker for disease severity as seen before for CSF‐NfL. 8
Guillain‐Barré Syndrome is a dynamic, acute polyneuropathy causing damage in PNS with intrathecal structures involved. 13 Edema in the nerve root 16 , 17 , 18 are probably causing the phenomena of cyto‐albumin dissociation because proteins with at least the size of 70 kDa as albumin cannot dissociate toward the blood compartment. 17 If this hypothesis is true, NfL having a molecular weight of 68 kDa may also not dissociate from and in the blood compartment explaining the high NfL levels in the CSF and serum in follow‐up examinations. The retrograde neurodegeneration involving the long peripheral nerves during the course of the disease may also contribute to the disproportionate increase in sNfL but this mechanism comes into significance later on. Parallel to this phenomenon axonal damage of the peripheral nerves including at the neuromuscular junction according to the underlying pathology of the GBS may cause an “NfL flood” into the serum releasing NfL into the blood compartment probably keeping the Nfl ratio constant at first. These mechanisms may explain the difference in both groups in time from disease onset to lumbar puncture and may explain the increase in total amount of NfL in CSF and serum over time in the follow‐up cohort. One more mechanism may also contribute to the increase of sNfL. Due to the damaged blood–nerve‐barrier at the nerve root NfL passes into the serum. This diffusion may be hampered due to the edema in the nerve root. The contribution of this mechanism to the overall sNFL and vice versa is not quantifiable with our data. In total several mechanisms are possibly contributing to the results as seen here.
The precise half‐life of NfL in serum is unknown but could be another explanation for the increase of NfL levels overall. Due to its resistance to proteases NfL has an estimated half‐life of several months in blood. This resistance could result in an additive effect increasing Nfl levels over time. 18
The albumin ratio in the NfL‐ratiolow group was slightly higher than in the NfL‐ratiohigh group. In favor of this, serum and CSF levels of NfL and Qalb correlated in the NfL‐ratiohigh group (P < 0.001), while this was not the case for the NfL‐ratiolow group (Table 1).
Applying the NfL index produces highly significant differences between controls and GBS patients pointing at a peripheral origin of sNfL in most GBS patients but not in controls. The NfL‐ratiohigh patients showed a NfL index similar to that of controls again suggesting that the larger part of NfL should derive from within the CSF space. In contrast, the NfL‐ratiolow group had a mean NfL index far lower than in controls, suggesting that a major quantity is released from extrathecal nerve tissue.
Another factor in favor for sNfL as a biomarker is its correlation with the severity of the disease, which is very heterogeneous and dynamic in GBS. Here, we do find a correlation with GBSDS meaning that the higher the GBSDS the more NfL is released, predominantly from PNS into the serum. Furthermore, sNfL and not CSF‐NfL is higher in patients admitted to the ICU confirming the biomarker status at least for sNfL.
Conclusion
The existence of NfL with peripheral origin in blood changes the way we look at sNfL. One suggestion is to measure the NfL ratio or index to assure the biomarker quality of sNfL for a CNS disease. Another suggestion is to rule out every patient having subacute or acute damage to peripheral nerves due to, for example, contact sports, medication with potentially neurotoxic agents or diabetic and other inflammatory polyneuropathies.
Author Contributions
PK contributed to design and conceptualization of the study, major role in the acquisition of data, analysis or interpretation of the data, drafting or revising the manuscript for intellectual content. JK contributed to analysis or interpretation of the data drafting or revising the manuscript for intellectual content major role in the acquisition of data. ED contributed to drafting or revising the manuscript for intellectual content. SV, CS, AH, FL, and DR contributed to major role in the acquisition of data, drafting or revising the manuscript for intellectual content. BS contributed to drafting or revising the manuscript for intellectual content. DL and AG contributed to major role in the acquisition of data, drafting or revising the manuscript for intellectual content, analysis or interpretation of the data.
Conflict of Interest
Jens Kuhle reports grants from Biogen, Novartis, Roche, Teva, the Swiss MS Society, Genzyme, the University of Basel Swiss National Research Foundation, Bayer Vital, Merck, and Celgene outside the submitted work. David Leppert has been an employee of Novartis until January 2019. Frank Leypoldt reports speaker honoraria from Bayer, Roche, Novartis, Fresenius, travel funding from Merck, Grifols, and Bayer and serving on advisory boards for Roche, Biogen and Alexion. Peter Körtvélyessy, Emrah Düzel, Stefan Vielhaber, Christian Schmidt, Annika Heinius, Burkhart Schraven, Dirk Reinhold, and Alexander Goihl report no conflict of interests.
Acknowledgments
The authors thank Jeannette Witzke and Kerstin Kaiser at the CSF lab in Magdeburg and Prof. Hansotto Reiber from Göttingen for his scientific input. Open access funding enabled and organized by Projekt DEAL.
Funding Information
No funding information provided.
References
- 1. Bridel C, Van Wieringen WN, Zetterberg H, et al. Diagnostic value of cerebrospinal fluid neurofilament light protein in neurology: a systematic review and meta‐analysis. JAMA Neurol 2019;76(9):1035–1048. 10.1001/jamaneurol.2019.1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Körtvelyessy P, Heinze HJ, Prudlo J, Bittner D. CSF biomarkers of neurodegeneration in progressive non‐fluent aphasia and other forms of frontotemporal dementia: Clues for pathomechanisms? Front Neurol 2018;9:504 10.3389/fneur.2018.00504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Steinacker P, Feneberg E, Weishaupt J, et al. Neurofilaments in the diagnosis of motoneuron diseases: a prospective study on 455 patients. J Neurol Neurosurg Psychiatry 2016;87:12–20. [DOI] [PubMed] [Google Scholar]
- 4. Barro C, Benkert P, Disanto G, et al. Serum neurofilament as a predictor of disease worsening and brain and spinal cord atrophy in multiple sclerosis. Brain 2018;141:2382–2391. Available from: https://academic.oup.com/brain/article/141/8/2382/5025690 [DOI] [PubMed] [Google Scholar]
- 5. Mattsson N, Cullen NC, Andreasson U, et al. Association between longitudinal plasma neurofilament light and neurodegeneration in patients with Alzheimer disease. JAMA Neurol 2019;76:791–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bischoff A, Manigold T, Barro C, et al. Serum neurofilament light chain: a biomarker of neuronal injury in vasculitic neuropathy. Annu Rheum Dis 2018;8(C):1093–1094. [DOI] [PubMed] [Google Scholar]
- 7. Maia LF, Maceski A, Conceição I, et al. Plasma neurofilament light chain: an early biomarker for hereditary ATTR amyloid polyneuropathy. Amyloid 2020;27(2):97–102. 10.1080/13506129.2019.1708716 [DOI] [PubMed] [Google Scholar]
- 8. Petzold A, Hinds N, Murray NMF, et al. CSF neurofilament levels: a potential prognostic marker in Guillain‐Barré syndrome. Neurology 2006;67:1071–1073. [DOI] [PubMed] [Google Scholar]
- 9. Petzold A, Brettschneider J, Jin K, et al. CSF protein biomarkers for proximal axonal damage improve prognostic accuracy in the acute phase of Guillain‐Barré syndrome. Muscle Nerve 2009;40:42–49. [DOI] [PubMed] [Google Scholar]
- 10. Gaiottino J, Norgren N, Dobson R, et al. Increased neurofilament light chain blood levels in neurodegenerative neurological diseases. PLoS One 2013;8:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Willison HJ, Jacobs BC, van Doorn PA. Guillain‐Barré syndrome. Lancet 2016;388:717–727. [DOI] [PubMed] [Google Scholar]
- 12. Fokke C, van den Berg B, Drenthen J, et al. Diagnosis of Guillain‐Barre syndrome and validation of Brighton criteria. Brain 2013;137:33–43. [DOI] [PubMed] [Google Scholar]
- 13. Kieseier BC, Mathey EK, Sommer C, Hartung H‐P. Immune‐mediated neuropathies [Internet]. Nat Rev Dis Prim 2018;4:31 Available from: http://www.nature.com/articles/s41572‐018‐0027‐2 [DOI] [PubMed] [Google Scholar]
- 14. Hadden RD, Cornblath DR, Hughes RA, et al. Electrophysiological classification of Guillain‐Barré syndrome: clinical associations and outcome. Plasma Exchange/Sandoglobulin Guillain‐Barré Syndrome Trial Group. Ann Neurol 1998;44(5):780–788. 10.1002/ana.410440512 [DOI] [PubMed] [Google Scholar]
- 15. Rajabally YA, Durand MC, Mitchell J, et al. Electrophysiological diagnosis of Guillain‐Barré syndrome subtype: could a single study suffice? J Neurol Neurosurg Psychiatry 2015;86:115–119. [DOI] [PubMed] [Google Scholar]
- 16. Alkan O, Yildirim T, Tokmak N, Tan M. Spinal MRI findings of Guillain‐Barré syndrome. J Radiol Case Rep 2009;3(3):25–28. 10.3941/jrcr.v3i3.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mulkey SB, Glasier CM, El‐Nabbout B, et al. Nerve root enhancement on spinal MRI in pediatric Guillain‐Barré syndrome. Pediatr Neurol 2010;43(4):263–269. 10.1016/j.pediatrneurol.2010.05.011 [DOI] [PubMed] [Google Scholar]
- 18. Byun WM, Park WK, Park BH, et al. Guillain‐Barre syndrome: MR imaging findings of the spine in eight patients. Radiology 1998;208(1):137–141. 10.1148/radiology.208.1.9646804 [DOI] [PubMed] [Google Scholar]