

Abstract
Objective
To describe clinical features and disease progression of Selenoprotein N‐related myopathy in a large multicenter cohort of patients.
Methods
Cross‐sectional multicenter data analysis of 60 patients (53 families) with Selenoprotein N‐related myopathy and single‐center retrospective longitudinal analysis of 25 patients (21 families) over a median period of 5.3 years.
Results
The majority of patients (46/60, 77%) presented before age 2 years with hypotonia, poor head/neck control, and developmental delay. At last assessment (median age 14 years; range 2.5 to 36 years), 10/60 patients had minimal or no ambulation. Ventilatory support was initiated in 50/60 patients at a mean Forced Vital Capacity (FVC) of 38% and at a median age of 13 years. Forty‐five/60 patients developed scoliosis (at median age 12.1 years) and 18 had scoliosis surgery at a median age of 13.6 years. Five children needed nasogastric feeds and/or gastrostomy. Longitudinal data analysis on 25 patients showed progressive decline of Hammersmith functional motor scores (estimated annual change −0.55 point), time to walk 10 meter, time standing from sitting, and from lying. Sixteen patients had weights < 2nd centile. The estimated change in FVC % per year was −2.04, with a 95% CI (−2.94, −1.14).
Conclusions
This comprehensive analysis of patients with Selenoprotein N‐related myopathy further describes the clinical course of this rare condition. The observed functional motor and respiratory data provide evidence of the slow decline patients experience over time which is useful when considering therapeutic intervention.
Introduction
Selenoprotein N‐related myopathies are recessive conditions caused by pathogenic variants in the SELENON gene (previously known as SEPN1). 1 , 2 , 3 The disease spectrum includes congenital muscular dystrophy (CMD) with rigid spine, multiminicore disease, desmin‐related myopathy with Mallory body‐like inclusions and congenital fiber‐type disproportion myopathy, 3 , 4 , 5 , 6 all sharing the primary features of early‐onset muscle weakness affecting axial muscles, leading to early respiratory insufficiency. 7 , 8 Selenoprotein N‐related myopathies (SELENON ‐RM) are estimated to account for 11.65% of CMDs 9 and 16% of congenital myopathies (CM) 10 in the UK. The international standards of care for CMD and CM 11 , 12 recommend multidisciplinary follow‐up for SELENON ‐RM and management of respiratory and orthopedic complications.
Selenoprotein N, the protein product of the SELENON gene, localizes in the endoplasmic/sarcoplasmic reticulum (ER/SR) and interacts with SERCA2, the SR/ER Ca2 + pump, to keep it active within the relatively oxidizing environment of the SR. 13 The absence of Selenoprotein N leads to a maladaptive ER stress response by oxidizing and inhibiting SERCA2, adversely affecting muscle function. 14 The use of ER stress inhibitors is being investigated as a possible treatment for SELENON‐RM and a clinical trial with N‐acetylcysteine is ongoing (NCT02505087). Improved understanding of the pathogenesis of this condition followed by early therapeutic trials, make it necessary to better define the natural history and identify clinical endpoints to support clinical trial readiness. Currently, no longitudinal studies on SELENON ‐RM have been reported.
This study aims to provide deep phenotypic analysis and description of the disease course in a large multicenter cohort of SELENON‐RM patients. In particular, we analyzed progression over time of motor abilities, respiratory function, and scoliosis, aiming to identify clinical parameters to guide clinicians on disease timeline, and provide information on prognosis, anticipatory care, and trajectories of functional changes, to help developing outcome measures for future clinical trials.
Methods
Sixty patients from (51 families) with SELENON‐RM were enrolled from three clinical sites: Site N1, Beggs Laboratory Congenital Myopathy Research Program, Boston Children’s Hospital (Boston Children’s Hospital IRB protocol: 03‐08‐128) (22 patients) Site N2, Hacettepe University Children’s Hospital, Turkey (13 patients); and Site N3, Dubowitz Neuromuscular center (DNC), London, England (25 patients). Twenty‐four patients were previously reported. 6 , 8 All patients had a molecularly confirmed diagnosis of SELENON‐RM with two pathogenic SELENON variants in trans, identified either by Sanger sequencing or next‐generation sequencing (Table S1).
Cross‐sectional data were collected for 60 patients. Clinical data for patients from site N1 and N2 were collected using a comprehensive medical questionnaire (Table S2). Clinical data for patients from Site N3 were available for more than one time‐point and thus the most recent value was used for cross‐sectional analysis, to make the information comparable among the three sites.
Retrospective longitudinal clinical data were collected for patients from Site N3 only for the following parameters: weight, motor function, respiratory function, and Cobb angle. Motor function tests included The Hammersmith standardized functional motor scores (HFMS), 15 timed 10 meters walk, timed raising from sitting, and lying. Respiratory function measurements included spirometry (forced vital capacity, FVC), performed in a pediatric lung function laboratory by experienced operators. The Global Lung Initiative dataset was used for reference. 16 Sleep studies were performed in a pediatric sleep laboratory. Sleep‐disordered breathing (SDB) was scored according to a standardized scoring system 17 .
The project followed Caldicott Guardian regulations and only anonymized de‐identified aggregated patient data were shared between the three sites.
Statistical Methods
Summaries are presented at the latest follow‐up visit with mean and standard deviation for normally distributed data, and median and interquartile range for skewed data. For the cross‐sectional cohort, Kaplan–Meier estimates were calculated to describe the median time to scoliosis and ventilation. Frequency data are presented along with proportions and comparisons between groups, for example, diagnosis before or after 2 years of age were made using a chi‐squared test. For the longitudinal cohort trajectories for functional motor score, respiratory function, and weight centiles over time were modeled using mixed‐effects regression, accounting for the repeated measures within patients. Estimated slopes are presented along with 95% confidence intervals. All statistical analyses were conducted in Stata v 15 (StataCorp. 2017. Stata Statistical Software: Release 15. College Station, TX: StataCorp LLC) and a p value < 0.05 was deemed significant.
Results
Patients’ data and data availability are presented in Table S1 and Figure 1.
Figure 1.
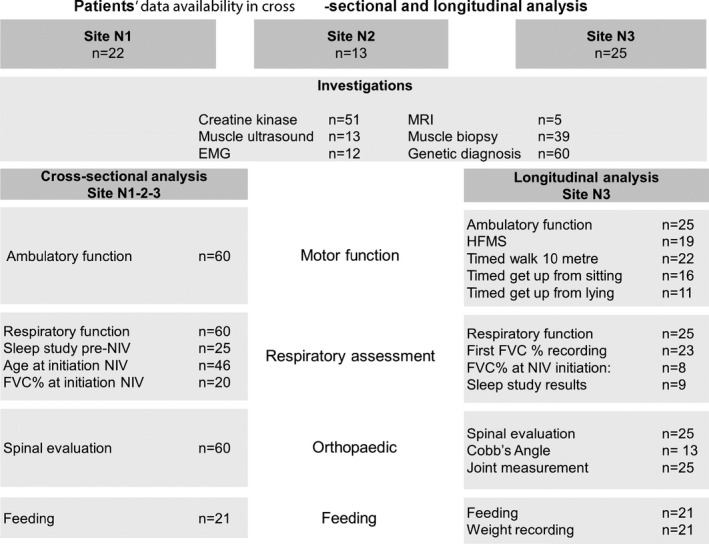
Patients’ data availability in cross‐sectional and longitudinal analysis. Flow diagram showing the number of patients for whom cross‐sectional and longitudinal data is available for different investigations and assessment. Sites are indicated on the top. n: number; EMG electromyography; MRI muscle magnetic resonance imaging; NIV noninvasive ventilation; FVC Forced vital capacity; FVC% forced vital capacity percentage predicted; HFMS Hammersmith Functional motor score.
Cross‐sectional data analysis
The age range of patients at last assessment was 2–58 years (median 15 years, mean 16.5 years) with a male:female ratio of 29:31.
Onset
Age at onset ranged from birth to 13 years (mean 2.3 years, median 9 months). Twenty‐six of 60 patients (43%) had onset ≤ 6 months with hypotonia and/or poor head/neck control. Twenty patients (33%) presented between 6 months and 2 years with gross motor delay, failure to thrive, and frequent respiratory tract infections; 14 patients (23%) presented> 2 years of age with motor difficulties, scoliosis and/or failure to thrive.
Ambulatory function
Fifty of 60 patients (83%) were ambulant at last assessment (median age at last evaluation: 14 years; range 2.5 to 36 years). Four patients (two aged 13 and 15 years; age unknown for further two) were only able to take steps indoors holding furniture or walk with assistance. Eight patients lost ambulation. Age at loss of ambulation was known for four and was 6.6, 15, 16, and 18 years, respectively (Fig. 2A).
Figure 2.
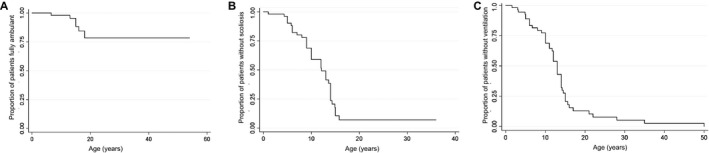
Cross‐sectional data analysis for ambulation, scoliosis, and ventilation in SELENON‐RM patients. Kaplan–Meier estimates for minimal/noambulation (A), scoliosis onset (Cobbs Angle> 10°) (B), and initiation of ventilation (C) from birth.
Orthopedic complications
Scoliosis was reported in 45 of 60 patients (75%). The median age at onset of scoliosis was 12.1 years (95% CI (9,14)) (Fig. 2B). Eighteen patients had spinal surgery at the median age of 13.6 years (range 9–17.6 years). Cobb’s angle range at the time of surgery, known for seven patients, was 50–77°. Six patients with Cobb’s angle range 15–72° were conservatively managed with spinal bracing.
Respiratory function
Fifty of 60 patients (83%) required ventilatory support from the median age 13 years (95% CI (11, 14), age range 1.6–50 years) (Fig. 1C). SDB prior to initiation of ventilation was documented for 25/50 patients. Four patients developed respiratory insufficiency following recurrent chest infections (defined as> 3/year), aspiration pneumonia, or general anesthesia leading to initiation of respiratory support. Forty‐eight of 50 patients were on noninvasive ventilation (NIV), 46 on bilevel positive airway pressure (BiPAP), and two on continuous positive airway pressure ventilation (CPAP). Forty‐four patients were on nocturnal NIV only, two needed additional support during respiratory illness and further two during respiratory illness and daytime as well. Two further patients had tracheostomy ventilation following acute respiratory failure. Age at the start of ventilation was known for 46 patients. Thirty‐five of 50 patients started ventilation before age 15 years (70%): 5 patients < 5 years, 7 between 5–10 years, and 23 patients between 10–15 years. The mean FVC percentage predicted (FVC%) at initiation of ventilation was 38.3% (range 19–58%).
Three patients were recommended ventilator support, but this was refused by the family. Two of these three patients died at 10 and 18 years, respectively. The third patient was last seen at age 7 years. Further seven patients were not recommended ventilatory support at the time of their more recent follow‐up, based on their last respiratory assessment (median age at last follow‐up 7.3 years, range 2.5‐14 years).
Feeding
Ten patients had swallowing or chewing difficulty. Three children had video‐fluoroscopy that showed delayed swallow initiation in two patients. Two patients had naso‐gastric and one naso‐jejunal feeds, and additional two patients underwent gastrostomy tube placement. A further patient was awaiting gastrostomy for persistent weight loss at the time of the last assessment at 7.4 years.
Other features
Nasal intonation of voice was reported in 22/60 patients. Facial weakness was observed in 20/60 patients, ptosis in seven, and ophthalmoplegia in one individual.
II. Longitudinal data analysis
Twenty‐five patients from Site N3 (male:female ratio 12:13), aged 2.5–24 years at the last follow‐up (median 14 years), were assessed at more than one time point, for a median period of 5.3 years (range 1.6–17.8 years). Median time from onset of symptoms to genetic diagnosis ranged from 5 months to 17.4 years.
Motor ability
Estimated annual change in the HMFS was −0.55 (95% CI (−1.05, −0.05)) (Fig. 3A, Fig S1), of timed 10 meter walk 0.16 sec (95 % CI (−0.03, 0.36)), of timed rise from floor sitting 0.86 sec (95 % CI (0.16, 1.55)), and of timed rise from floor lying 0.87 sec (95 % CI (0.06, 1.68)) (Table S3). Seven children had post spinal surgery motor function follow‐up data for a median of 8 months (range 0.4 months–9 years) and all remained ambulant after surgery.
Figure 3.
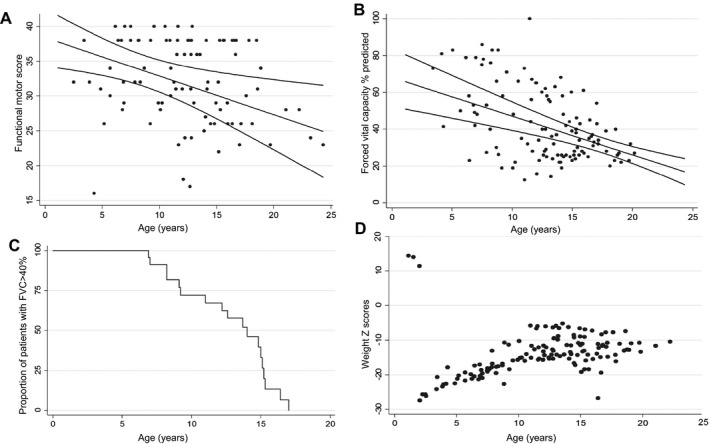
Longitudinal analysis of functional motor score, respiratory function, and feeding in SELENON‐RM patients. (A) population average model for the relationship between Hammersmith functional motor score and age along with 95% confidence intervals. Dots represent multiple visits per patient (n = 19). (B) Population average model for the relationship between Force Vital Capacity (FVC)% and age along with 95% confidence intervals. Dots represent multiple visits per patient (n = 23). (C) Kaplan–Meier estimates for time to FVC < 40%. (D) Line charts representing the progression of weight Z‐Scores. Dots represent multiple visits per each patient (n = 21).
Skeletal complications
Spinal stiffness preceding scoliosis was described in 21 of 25 patients (84%), from a median age of 10 years (range 2.0–15.6 years). The estimated change/year in Cobb’s angle was + 7.39 ° (95 % CI (1.70, 13.07)). Joint contractures were reported in 20 of 25 patients (80%), from a median age of 12 years (range 0–16.5 years). Achilles tendons contractures were observed in 18 patients (75%), of elbows in six (24%), and of knees in five (20%).
Respiratory function
Median age at first FVC% recording was 12 years (range 5–15 years). Seventeen patients had FVC < 60% at first recording. Estimated change in FVC%/year was −2.04 (95% CI (−2.94, −1.14); Fig. 3B). Fourteen patients had FVC < 40% (predictor of nocturnal hypoventilation) 18 at a median age of 14.8 years (95% CI (9.1–14.8)) (Fig. 3C). After initiation of NIV, yearly change in FVC% was −1.15 (95% CI (−2.12, −1.17); P value 0.02). FVC% decline following scoliosis surgery was estimated at −0.95 (95% (CI −1.5, 1.5)).
Recurrent chest infections were documented for 12 of 25 patients (48%) from a median age of 5 years (3 months–14 years) and 10 of these patients needed NIV from a median age of 6 years (1.6–14 years). Three patients had recurrent chest infections, whereas on NIV and two of the three needed daytime ventilator support as well. The cough assist machine was used by 3/25 children.
Results of polysomnography were available for nine of 25 patients. One patient had normal polysomnography at age 7.8 years, whereas eight showed hypoventilation from a mean age of 9.7 years (range 1.6–15 years). Sleep study data for five patients prior to BiPAP initiation showed elevated Apnea–Hypopnea Index (AHI); post NIV initiation follow‐up studies showed a reduction in AHI. Initial BIPAP pressures were adequate in two patients to overcome nocturnal hypoventilation with no need for further titration of pressures. Four patients required increments on inspiratory positive airway pressure during follow‐up sleep studies to overcome nocturnal hypoventilation. The increment on expiratory positive airway pressure was required in one patient. Down titration on BiPAP pressures over the observation period was possible in two patients, both initiated on NIV at very young ages (3.2 years and 6.7 years) (Fig S2).
Based on our data we estimated the number of patients required to run future clinical trials in which FVC is one of the endpoints. A clinical trial, in which further progression of respiratory decline is completely abolished over a 12‐month period, would require 50 patients for 80% power. On the other hand, a therapeutic intervention that could decrease disease progression by 75%, that is, reducing the average yearly decline of 2% to 0.5%, would require about 80 patients for 80% power (Fig. 4).
Figure 4.
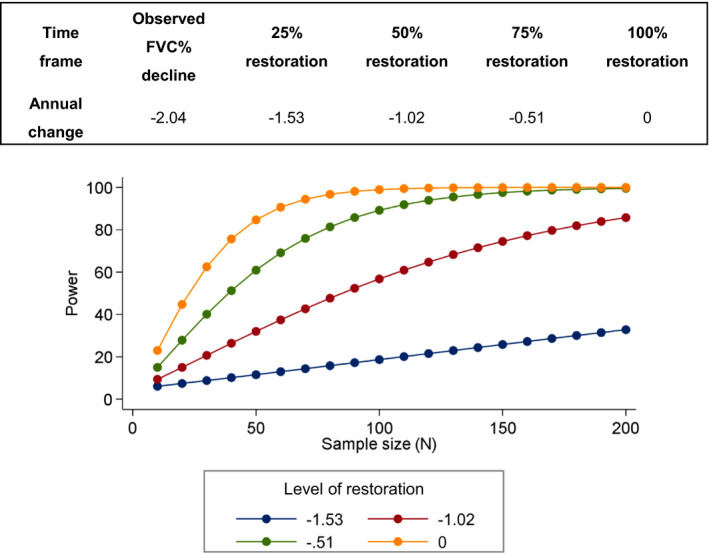
Estimated statistical power and sample sizes to detect restoration in decline of Forced Vital Capacity % predicted in SELENON‐RM cohort.
Weight
At first assessment, 10 children (aged 1–15 years, mean 10.5 years) had weights below the 2nd centile. By the last follow‐up, 16 of 21 patients (76%) had weight below the 2nd centile. Eighteen patients had weight < 50th centile (Fig. 3D). Fall in weight centiles coincided in some patients with respiratory decline (abnormal sleep study, chest infections and/or NIV initiation), progressive scoliosis, or spinal surgery. The population average model graph showed an average annual change in weight z scores of 0.65 (P = 0.008). Two children, after an initial period of low weight centiles, presented weight increased> 91st centile, in parallel to a reduction of their motor abilities.
III. Investigations
Creatine kinase (CK) levels were within normal limits in 39 patients (76%) and moderately elevated in 12 (median 477 U/L, range 236–1505 U/L). One patient with CK of 2459 U/L was a heterozygous female carrier of a nonsense DMD gene variant (c.2348A> C; Ref sequence LRG_199t1).
Muscle ultrasound evidenced increased echogenicity in quadriceps femoris in all investigated patients (not shown). Electromyography showed myopathic changes in ten patients. Five patients had muscle MRI of lower limbs evidencing selective sartorius muscle involvement (not shown). Muscle biopsies revealing both dystrophic and myopathic changes, often with accompanying minicores or core‐like areas. One patient’s muscle exhibited Mallory body inclusions (Table S1).
Twenty‐four patients were compound heterozygous and 36 homozygous for SELENON gene pathogenic/likely pathogenic variants. Thirty‐two individuals had missense variants (in homo or compound heterozygosity), 20 a loss of function variant (frameshift, stop or splice site) and eight a combination of the two. The c.943G> A p.(Gly315Ser) missense change was the most common variant, present in homozygosity in seven patients and in compound heterozygosity in 10.
IV. Genotype and phenotype correlation analysis
We compared phenotypes of patients with either two loss of function mutations, two missense variants or a combination of one loss of function and one missense mutation. No clear association was seen between the type of mutation and age at onset of symptoms, ambulation, frequency of scoliosis, and NIV or median age at start of ventilation (data not shown).
We then subdivided the entire cohort of patients based on age at onset ≤ 2 and> 2 years. Patients presenting ≤ 2 years had onset of scoliosis at a median age of 10 year versus 14.3 years in the> 2 year group (Fig. 5A). Loss of ambulation occurred earlier in patients with onset ≤ 2 years. Among these, six children with minimal or no ambulation had symptom onset < 1 year. No difference was observed in the age at the start of ventilation between the two groups (Fig. 5B). No statistical analysis was performed due to the limited numbers of patients.
Figure 5.
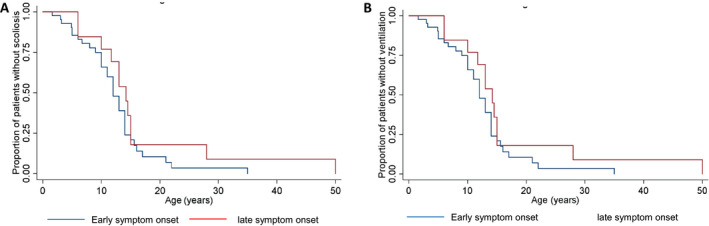
Time to event analysis for scoliosis and ventilation need according to onset. Survival curves for scoliosis (A) and ventilation free probability (B) of patients with onset of symptoms ≤ 2 years (early symptom onset, blue line) and> 2 years (late symptom onset, red line).
We reviewed age at onset, presentation, and disease course in the nine sib pairs of this cohort. Age at onset was dissimilar in one sib pair, with the older child presenting at birth and the younger at age 4.5 years. In two further sib pairs, BiPAP was initiated at an earlier age in the younger siblings. In view of the small number of families, no statistical analysis was performed on these findings.
Discussion
In this study, we comprehensively describe cross‐sectional and longitudinal clinical and functional data in a large cohort of SELENON‐RM patients. The cross‐sectional study not only confirms early onset with hypotonia, with high frequency of poor neck/head control, spinal rigidity, progressive scoliosis, and need for early respiratory support in SELENON‐RM patients, but also describes the frequency of additional complications and features such as swallowing difficulties and ptosis. the higher frequency of patients with minimal or no ability to walk (17%) contrasts with earlier reports, which focused on function group of patients with better motor function, with in particular only 2/41 (5%) nonambulant patients reported in one of the previous studies 7 , 8 , 19 . We provide for the first time longitudinal functional data analysis and disease course for SELENON‐RM patients, focusing on respiratory, and functional scores over time. Previous studies suggested that age at onset, clinical severity, and course of disease were independent variables. 7 , 8 However, our observation of earlier loss of ambulation and onset of scoliosis in patients with earlier age at onset suggests a correlation between these variables (Fig. 5A).
Our estimate of yearly decline in FVC% (−2.04 before NIV, −1.15 after initiation of ventilation), differs from previous reports, indicating relative stability of respiratory function over time. 7 , 20 Our estimates were calculated over a longer period of time and this might explain the observed differences. A 5% change/year in FVC% is defined as a minimally clinically significant change in the DMD population. 21 Extrapolating this to our SELENON‐RM cohort, we can estimate an average decline of 5% in FVC% after 2.5 years. Considering the severity of respiratory involvement in SELENON‐RM patients, longitudinal assessments of FVC could represent an effective outcome measure to demonstrate progression over time, in children where it can be reliably performed. We also provide for the first time power analysis for future clinical trials in which FVC would be used as one of the endpoints (Fig. 4).
This large cohort analysis confirms survival into adulthood for the majority of patients provided with adequate respiratory and orthopedic management and treatments. 22
Previous reports indicate a relative stability in motor function over time in SELENON‐RM patients. 7 , 8 In absence of a motor scoring scale validated for SELENON‐RM, we used the HFMS previously validated in pediatric neuromuscular conditions. 15 Our results highlight a modest annual decline in HFMS (−0.55 points/year in children> 5 years). A similar modest, but clear decline was also noted for timed tests. Overall, the motor decline appears to parallel scoliosis progression, weight fluctuations, and respiratory decline.
Failure to thrive, reported in up to 57% of reported SELENON‐RM patients 3 , 7 , 8 , 12 . was also frequently observed in this cohort, with 76% of patients with weights < 2nd centile at the last follow‐up. Interestingly, while dietary supplementation helped to increase weight centiles, in two children steep weight gain was observed in parallel with the decline in motor abilities, one of the two being at the more severe end of the phenotypic spectrum. The European Society for Paediatric Gastroenterology, Hepatology and Nutrition consensus statement on diagnosis and management of gastrointestinal and nutritional complications in children with neurological impairment 23 suggests that weight, height, body mass index, height‐for‐age, weight‐for‐age are poor predictors of body composition and lead to misinterpretation of low body mass index values and/or low weight z‐scores leading to overfeeding. Further research is needed to better evaluate body composition in SELENON‐RM patients (including whole body Dual‐energy X‐ray absorptiometry analysis) and to assess nutritional status to develop a disease‐specific growth curve.
In the patients studied, we did not observe any obvious genotype–phenotype correlations, confirming previous observations. 7 , 8
Our current study has some limitations. Patients were assessed in three clinical sites and although we followed the International standards of care guidelines, different clinical protocols were used and standards of care have evolved over time. This study presents a mix of cross‐sectional and longitudinal data analysis, with limited longitudinal observations. Our real‐world study also stresses the importance to collect data according to standardized assessment tools and clinically meaningful end points and outcome measures to guide clinicians and researchers on disease monitoring and planning of therapeutic clinical trials.
In conclusion, our study expands the knowledge on the clinical course of patients with SELENON‐RM and provides longitudinal data for patients affected by this condition. The observed functional motor and FVC trend data provide evidence for a slow decline over time. Our results emphasize the importance of robust and consistent multidisciplinary management of SELENON‐RM patients to improve outcomes and inform clinical trials.
Conflicts of Interest
Dr. Beggs serves on the scientific advisory boards of Audentes Therapeutics Inc and Dynacure Inc., has received sponsored research support from Audentes Therapeutics Inc, receives sponsored research support from Alexion Pharmaceuticals, Inc., is a paid consultant for F. Hoffmann‐La Roche, performs occasional paid consulting for Guidepoint Global Advisors and Gerson Lehrman Group, Inc., and receives research support from the US National Institutes of Health and the Muscular Dystrophy Association. There are no disclosures from other authors.
Supporting information
Figure S1. Changes over time in Hammersmith Functional motor score. Individual functional motor score pattern over time. The maximum functional motor score being 40/40. Lines represent individual patients (n = 19).
Figure S2. Changes over time in noninvasive nocturnal bilevel positive airway pressures. Data for seven patients are presented. Intervals between cardiorespiratory sleep studies on the x‐axis differed between patients. Required levels of expiratory airway pressure (EPAP) and inspiratory airway pressure (IPAP) are displayed on the y‐axis..
Table S1. Characteristics and progression of patients with SELENON‐RM.
Table S2. Data collection form for site N1 and N2.
Table S3. Yearly Progression of timed rise from floor sitting and lying, in second.
Acknowledgments
Special thanks and appreciation to Dr Nigel Clarke (two of the patients in the cohort were previously reported by his group), who unfortunately passed away during the course of this study. We acknowledge the contribution of the Highly Specialised Service for Congenital muscular dystrophies and congenital myopathies at the Dubowitz Neuromuscular Centre and in particular Dr Lucy Feng for her assistance. We thank Dr Stephanie Robb, Dr Ros Quinlivan, and Dr Pinki Munot and the Neuromuscular Physiotherapy team for the clinical assessment of our patients at DNC and the Great Ormond Street Lung physiology team for spirometry data.
Funding Information
This work was supported in part by The Muscular Dystrophy UK Central Grant to the DNC and by grant MDA602235 from the Muscular Dystrophy Association (USA) and a generous gift from the Lee and Penny Anderson Family Foundation. Diagnostic DNA sequencing of patients in the Boston cohort utilized the resources of the Boston Children’s Hospital IDDRC Molecular Genetics Core Laboratory funded by U54HD090255 from the US National Institutes of Health. This research was also supported by the NIHR Great Ormond Street Hospital Biomedical Research Centre. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health.
Funding Statement
This work was funded by Muscular Dystrophy Association grant MDA602235; US National Institutes of Health grant U54HD090255; NIHR Great Ormond Street Hospital Biomedical Research Centre grant .
References
- 1. Dubowitz V. Rigid spine syndrome: a muscle syndrome in search of a name. Proc R Soc Med 1973;66:219–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Moghadaszadeh B, Desguerre I, Topaloglu H, et al. Identification of a new locus for a peculiar form of congenital muscular dystrophy with early rigidity of the spine, on chromosome 1p35–36. Am J Hum Genet 1998;62:1439–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moghadaszadeh B, Petit N, Jaillard C, et al. Mutations in SEPN1 cause congenital muscular dystrophy with spinal rigidity and restrictive respiratory syndrome. Nat Genet 2001;29:17–18. [DOI] [PubMed] [Google Scholar]
- 4. Ferreiro A, Quijano‐Roy S, Pichereau C, et al. Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early onset myopathies. Am J Hum Genet 2002;71:739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ferreiro A, Ceuterick‐de Groote C, Marks JJ, et al. Desmin‐related myopathy with Mallory body‐like inclusions is caused by mutations of the selenoprotein N gene. Ann Neurol 2004;55:676–686. [DOI] [PubMed] [Google Scholar]
- 6. Clarke NF, Kidson W, Quijano‐Roy S, et al. SEPN1: associated with congenital fiber‐type disproportion and insulin resistance. Ann Neurol 2006;59:546–552. [DOI] [PubMed] [Google Scholar]
- 7. Schara U, Kress W, Bonnemann CG, et al. The phenotype and long‐term follow‐up in 11 patients with juvenile selenoprotein N1‐related myopathy. Eur J Paediatr Neurol 2008;12:224–230. [DOI] [PubMed] [Google Scholar]
- 8. Scoto M, Cirak S, Mein R, et al. SEPN1‐related myopathies: clinical course in a large cohort of patients. Neurology 2011;76:2073–2078. [DOI] [PubMed] [Google Scholar]
- 9. Sframeli M, Sarkozy A, Bertoli M, et al. Congenital muscular dystrophies in the UK population: Clinical and molecular spectrum of a large cohort diagnosed over a 12‐year period. Neuromuscul Disord 2017;27:793–803. [DOI] [PubMed] [Google Scholar]
- 10. Maggi L, Scoto M, Cirak S, et al. Congenital myopathies‐clinical features and frequency of individual subtypes diagnosed over a 5‐year period in the United Kingdom. Neuromuscul Disord 2013;23:195–205. [DOI] [PubMed] [Google Scholar]
- 11. Wang CH, Bonnemann CG, Rutkowski A, et al. Consensus statement on standard of care for congenital muscular dystrophies. J Child Neurol 2010;25:1559–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang CH, Dowling JJ, North K, et al. Consensus statement on standard of care for congenital myopathies. J Child Neurol 2012;27:363–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Marino M, Stoilova T, Giorgi C, et al. SEPN1, an endoplasmic reticulum‐localized selenoprotein linked to skeletal muscle pathology, counteracts hyper‐oxidation by means of redox‐regulating SERCA2 pump activity. Hum Mol Genet 2015;24:1843–1855. [DOI] [PubMed] [Google Scholar]
- 14. Pozzer D, Varone E, Chernorudskiy A, et al. A maladaptive ER stress response triggers dysfunction in highly active muscles of mice with SELENON loss. Redox Biol 2018;20:354–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Scott OM, Hyde SA, Goddard C, Dubowitz V. Quantitation of muscle function in children: a prospective study in duchenne muscular dystrophy. Muscle Nerve 1982;5:291–301. [DOI] [PubMed] [Google Scholar]
- 16. Quanjer PH, Stanojevic S, Cole TJ, et al. Multi‐ethnic reference values for spirometry for the 3–95‐yr age range: the global lung function 2012 equations. Eur Respir J 2012;40:1324–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Berry RB, Brooks R, Gamaldo CE, et al. The AASM Manual for the Scoring of Sleep and Associated Events: Rules, Terminology and Technical Specifications, Version 2.0.3.www.aasmnet.org, Darien, Illinois: American Academy of Sleep Medicine 2014.
- 18. Ragette R, Mellies U, Schwake C, Voit T, Teschler H. Patterns and predictors of sleep disordered breathing in primary myopathies. Thorax 2002;57:724–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ardissone A, Bragato C, Blasevich F, et al. SEPN1‐related myopathy in three patients: novel mutations and diagnostic clues. Eur J Pediatr 2016;175:1113–1138. [DOI] [PubMed] [Google Scholar]
- 20. Caggiano S, Khirani S, Dabaj I, et al. Diaphragmatic dysfunction in SEPN1‐related myopathy. Neuromuscul Disord 2017;27:747–755. [DOI] [PubMed] [Google Scholar]
- 21. McDonald CM, Gordish‐Dressman H, Henricson EK, et al. Longitudinal pulmonary function testing outcome measures in Duchenne muscular dystrophy: long‐term natural history with and without glucocorticoids. Neuromuscul Disord 2018;28:897–909. [DOI] [PubMed] [Google Scholar]
- 22. Sponholz S, von der Hagen M, Hahn G, et al. Selenoprotein N muscular dystrophy: differential diagnosis for early‐onset limited mobility of the spine. J Child Neurol 2006;21:316–20. [DOI] [PubMed] [Google Scholar]
- 23. Romano C, van Wynckel M, Hulst J, et al. European Society for Paediatric Gastroenterology, Hepatology and Nutrition guidelines for the evaluation and treatment of gastrointestinal and nutritional complications in children with neurological impairment. J Pediatr Gastroenterol Nutr 2017;65:242–64. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Changes over time in Hammersmith Functional motor score. Individual functional motor score pattern over time. The maximum functional motor score being 40/40. Lines represent individual patients (n = 19).
Figure S2. Changes over time in noninvasive nocturnal bilevel positive airway pressures. Data for seven patients are presented. Intervals between cardiorespiratory sleep studies on the x‐axis differed between patients. Required levels of expiratory airway pressure (EPAP) and inspiratory airway pressure (IPAP) are displayed on the y‐axis..
Table S1. Characteristics and progression of patients with SELENON‐RM.
Table S2. Data collection form for site N1 and N2.
Table S3. Yearly Progression of timed rise from floor sitting and lying, in second.