

Abstract
Recent advancements in T-cell biology and antibody engineering have opened doors to significant improvements in cancer immunotherapy. Initial success with monoclonal antibodies targeting key receptors that inhibit T-cell function such as cytotoxic T lymphocyte antigen 4 (CTLA-4) and programmed death-ligand 1 (PD-1) have demonstrated the potency of this new class of therapy, highlighted by long-term complete responses for metastatic cancers once thought incurable. However, only a subset of patients responds to checkpoint blockade because of a multitude of factors, including an immunosuppressive tumor microenvironment and the mutational burden of the cancer. Novel antibodies, as well as ligand-immunoglobulin fusion proteins that target costimulatory immune receptors, are being developed and tested in clinical trials to further enhance the anti-tumor immune response. Many of these costimulatory receptors are in the tumor necrosis factor receptor superfamily (TNFRSF) and are expressed on multiple immune cell types, including inhibitory cells. While TNFRSFs signal through common pathways, the outcome of targeting different receptors depends on the functional status of the cell types expressing the relevant receptors. In this review, we discuss the current state of targeted costimulatory immunotherapy.
1. Introduction
The generation of potent T-cell-mediated anti-tumor immunity relies on the provision of several critical signals, including T-cell receptor (TCR)-mediated recognition of peptide-major histocompatibility complex (MHC) molecules on antigen-presenting cells (APCs) along with appropriate costimulatory signals [1, 2]. Even in the presence of these optimal conditions, tumors utilize a myriad of mechanisms to evade and suppress the immune response. For example, tumors suppress recognition and visibility to the immune system by changing their microenvironment (TME) and reducing MHC class I presentation of tumor-associated antigens (TAAs). Changes to the TME can occur through secretion of immunosuppressive cytokines such as interleukin (IL)-10 and transforming growth factor (TGF)-β, which inhibit effector T-cell responses and promote regulatory FoxP3+ cluster of differentiation (CD)-4 T-cell regulatory (Treg) function [3] or through upregulation of programmed death-ligand 1 (PD-L1), which inhibits the TCR signaling pathway. PD-L1 binds to programmed cell death protein 1 (PD-1) expressed by activated or exhausted T cells, resulting in phosphatase activity that inhibits TCR kinase signaling [4, 5]. Importantly, tumors that are immunologically “silent,” which refers to low MHC I and high PD-L1 expression, often have little or no response to immunotherapy [6–9].
Thus, there is an urgent need to develop therapies that enhance tumor antigen presentation along with T-cell priming, activation, and differentiation to counteract tumor-induced immune evasion and suppression. One such approach is to enhance T-cell recognition of TAAs presented on MHC molecules through the provision of exogenous tumor-specific vaccines, known as therapeutic vaccination [10, 11]. However, therapeutic vaccines have achieved limited clinical success thus far. Another approach is the provision of monoclonal antibodies (mAb) that block inhibitory checkpoint molecules such as cytotoxic T lymphocyte antigen 4 (CTLA-4), PD-1, and PD-L1. In contrast to therapeutic vaccines, checkpoint blockade immunotherapy has demonstrated significant therapeutic benefit for patients with metastatic cancer, which has led to US FDA approval for a variety of tumor types [6, 7]. A third approach is the use of agonist mAbs that boost T-cell function by engaging costimulatory molecules such as OX40, 4–1BB, and CD40. In this review, we review checkpoint inhibitors and their impact on the current landscape of cancer immunotherapy, followed by a discussion of the current state of therapies that target T-cell costimulatory receptors with a focus on OX40, 4–1BB, glucocorticoid-induced tumor necrosis factor receptor (TNFR)-related protein (GITR), CD40, and inducible T-cell costimulator (ICOS) and their use in combination with checkpoint inhibitors.
2. Checkpoint Inhibitors (Anti-CTLA-4, Anti-PD-1) and the First Bispecific Antibody
The first T-cell-targeted immunotherapies came after the discovery of immune checkpoints that regulate activation and inhibition following TCR stimulation. One of the first inhibitory regulators of T-cell function to be cloned was CTLA-4 in 1987 [12]. CTLA-4 and the homologous receptor CD28 both bind to B7–1 and B7–2 ligands expressed on APCs [13–15]. When CD28 engages B7 ligands, it enhances the TCR signaling pathway through enhanced phosphoinositide 3 kinase (PI3K) activity. However, activated T cells downregulate expression of CD28 and upregulate CTLA-4, which inhibits T-cell signaling by engaging B7 ligands with 20-fold higher affinity than CD28, thereby blocking CD28-mediated costimulation. Additionally, CTLA-4 is constitutively expressed on Tregs. In mouse models of cancer, CTLA-4 blockade was shown to boost anti-tumor immunity by inhibiting Tregs and enhancing T-cell effector function [16, 17].
These data led to the clinical development of two blocking antibodies targeting human CTLA-4 (aCTLA-4): ipilimumab [18] and tremelimumab [19–21]. Clinical trials of ipilimumab for the treatment of metastatic melanoma showed promising results, which led to a successful phase III trial in 2009 for patients with high-grade unresectable melanoma. This trial showed a 10.1-month overall median survival when receiving ipilimumab alone compared with 6.4 months’ survival for patients receiving a gp100 vaccine alone. The addition of the gp100 vaccine to ipilimumab therapy had no additional impact on overall survival in these patients [22]. Tremelimumab was not as successful, perhaps because of its staggered treatment schedule of infusions every 3 months compared with every 3 weeks for ipilimumab. Ipilimumab was subsequently approved for metastatic melanoma in 2011. Tremelimumab is still being investigated for several other indications but has not seen clinical success for melanoma, small cell lung cancer, or mesothelioma [20].
The success of immunotherapies such as ipilimumab in 2011 and the vaccine-based therapy sipuleucel-T for hormone-refractory prostate cancer in 2010 led to the development of multiple T-cell-targeted immunotherapy agents (Table 1). Leading candidates to be targeted by the next wave of immunotherapies were T-cell checkpoints PD-1 and PD-L1, molecules that inhibit T-cell activity and function when engaged [23]. The PD-1/PD-L1 pathway is upregulated in many cancers and is an important mechanism of inhibiting activated tumor-reactive T cells [24]. Two PD-1-blocking antibodies (aPD-1), pembrolizumab and nivolumab, demonstrated clinical success and received FDA approval. Pembrolizumab was approved in 2014 following a successful phase III trial investigating its use following failed ipilimumab therapy for advanced metastatic melanoma [25]. Pembrolizumab was approved for metastatic non-small-cell lung cancer in 2015 [26], metastatic head and neck squamous cell carcinoma in 2016 [27], and DNA mismatch repair-deficient solid tumors in 2017. Nivolumab is currently FDA approved for metastatic melanoma, renal cell carcinoma, lung cancer, bladder cancer, and Hodgkin’s lymphoma [28]. Additionally, three antibodies targeting PD-L1 (aPD-L1) have also seen success in clinical trials, including FDA approval of atezolizumab for the treatment of metastatic non-small-cell lung cancer [29], durvalumab for the treatment of metastatic urothelial cancer [30], and avelumab for the treatment of non-small-cell lung cancer [31].
Table 1.
US FDA-approved T-cell targeted immunotherapies
| Target | Mechanism | Name | Company |
|---|---|---|---|
| CTLA-4 | Treg depletion, enhanced CD28 costimulation | Ipilimumab | BMS |
| PD-1 | Block PD-1:PD-L1 interaction by targeting PD-1 on T cells, enhanced TCR signaling | Pembrolizumab | Merck |
| Nivolumab | BMS | ||
| PD-L1 | Block PD-1:PD-L1 interaction by blocking PD-L1 on tumor cells, enhanced TCR signaling | Durvalumab | AstraZeneca |
| Avelumab | Merck | ||
| Atezolizumab | Genentech | ||
| CD19 and CD3 | Link T cells to cancerous B cells, enhanced TCR signaling by activating CD3 | Blinatumomab | Amgen |
BMS Bristol-Myers Squibb, CD cluster of differentiation, CTLA cytotoxic T lymphocyte antigen, PD-1 programmed cell death protein 1, PD-L1 programmed death ligand 1, TCR T-cell receptor, Treg regulatory T cells
Among this next wave of agents was an alternate class of immunotherapeutic antibodies that simultaneously target two different receptors, known as Bispecific T-cell Engagers (BiTE) [32]. BiTEs are engineered antibodies that contain Fab (Fragment, antigen-binding) regions against different targets to link two different surface proteins together. If the targeted surface proteins are expressed on different cells, then the BiTE will link the two cells together. This rationale was used to develop one of the first BiTEs, blinatumomab [33]. This BiTE targets CD19 overexpressed on B-cell malignancies and CD3 expressed by T cells in order to bring the cancer cell and the T cell together, with the goal of simultaneously enhancing T-cell activity through CD3 binding [33]. Blinatumomab was FDA approved for acute lymphoblastic leukemia in 2014.
While these various immunotherapeutic agents have demonstrated marked success, particularly in a subset of cancers (melanoma, lung), several important issues remain. One of the primary challenges is that immunotherapy typically only benefits a subset of patients. Other issues include the onset of potentially severe adverse events (SAEs) associated with therapy that can be enhanced when these agents are given in combination, such as in the case of aCTLA-4/aPD-1 therapy [34]. Additional questions remain about treatment timing, dosage, administration route, and identification of patients who are most likely to respond to therapy. While ipilimumab, pembrolizumab, and nivolumab have proven that immune checkpoint blockade can lead to long-term responses in metastatic disease, immunotherapy agents in development are aimed at engaging T-cell costimulatory receptors to activate and enhance the T-cell-mediated anti-tumor response.
3. Targeting the TNFRSF, ICOS, and Combination Immunotherapy
Recent studies have increasingly focused on therapies targeting costimulatory receptors expressed on activated T cells and APCs (Fig. 1). TNFR superfamily (TNFRSF) members are a promising class of immune-modulating molecules that are under rapid development for cancer immunotherapy. TNFRSF members are typically homotrimeric transmembrane proteins with a cysteine-rich extracellular domain [35]. Intracellular signaling domains of TNFRSFs can induce pro-apoptotic or pro-survival and pro-inflammatory programs depending on the cell type and signaling domains expressed. TNFRSFs signal through TNFR-associated factor (TRAF) adaptor proteins. Activating TRAF proteins (TRAF1, TRAF2, and TRAF5) signal to activate the canonical nuclear factor (NF)-κB and c-Jun N-terminal kinase (JNK) pathway and activate the inhibitor of apoptosis (IAP) [36]. These signaling pathways enhance T-cell function and survival following TNFRSF engagement. However, some TNFRSFs, such as TNF-related apoptosis-inducing ligand (TRAIL) receptor and apoptosis antigen 1 (FAS) receptor, signal through death domains to initiate a caspase-mediated apoptosis program [37]. Multiple pro-survival and pro-inflammatory TNFRSFs are expressed on T cells following TCR activation, making them promising targets both as monotherapies and as complementary targets to the established checkpoint inhibitor immunotherapy agents. Thus, targeting TNFRSFs with agonistic antibodies and ligand-Fc fusion proteins has the potential to potently activate and enhance the anti-tumor T-cell response.
Fig. 1.
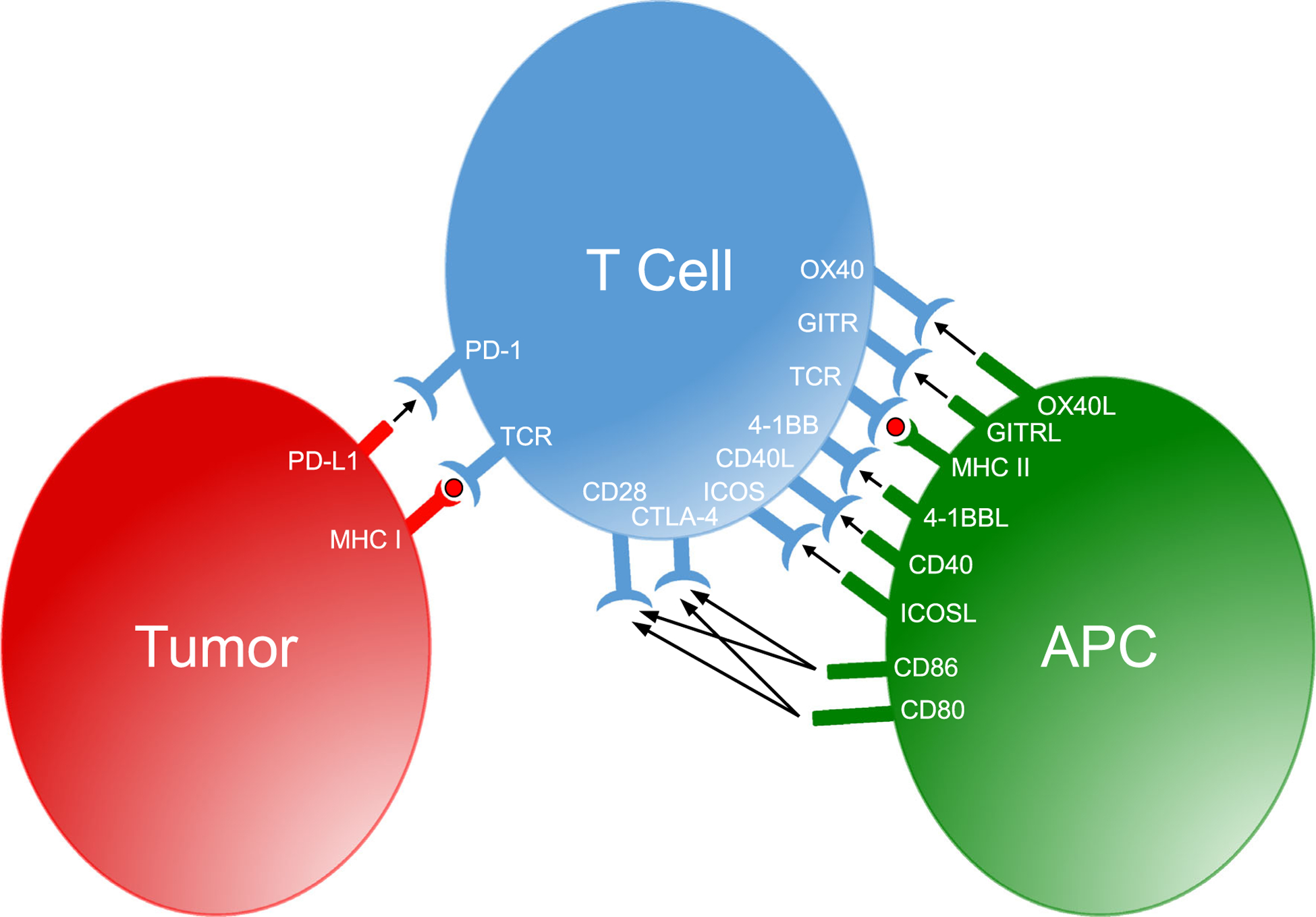
Inhibitory checkpoint and costimulatory receptors with their respective ligands expressed by antigen-presenting cells, T cells, and tumor cells. APC antigen-presenting cells, CD cluster of differentiation, CTLA cytotoxic T lymphocyte antigen, GITR glucocorticoid- induced tumor necrosis factor receptor -related protein, ICOS inducible T-cell costimulator, MHC major histocompatibility complex, PD programmed cell death protein, TCR T-cell receptor
Antibodies that target receptors can either be inhibitory (antagonist) or activating (agonist) and must be designed and engineered specifically to accomplish those signals. The most commonly used antibody isotype for immunotherapy is immunoglobulin G (IgG), but several subtypes of IgG influence its immune function depending on how they engage Fc γ receptors or activate the complement system cascade [38]. IgG1 strongly induces antibody-dependent cellular cytotoxicity (ADCC) by engaging Fc γ receptors, which lends itself well to targeting tumor-specific antigens (e.g., herceptin, which targets the Her2 receptor on breast cancer) or potentially depleting inhibitory immune cells, such as Tregs. However, nivolumab and pembrolizumab are IgG4 isotype antibodies, which only weakly engage Fc γ receptors and makes them amenable for receptor blockade without inducing ADCC.
Antibodies used for clinical applications are typically produced in three forms: murine, humanized, and fully human. Murine antibodies produced by mouse hybridomas are seen as foreign and can thus only be dosed for a short time before the human immune system mounts an adaptive immune response to clear the foreign (murine) antibody. Humanized antibodies are modified murine antibodies engineered such that the Fc regions of the Fab domain are comprised of the human antibody sequence [39]. Humanized antibodies can be dosed without inducing an adaptive immune response [40]. An alternative to antibody targeting is to generate ligand-Fc fusion proteins, which express the natural ligand bound to the Fc region of the antibody rather than engineering a human antibody with different binding kinetics and regions of interaction compared with the natural ligand [41]. This may provide an advantage over conventional antibodies for targeting TNFRSFs, which are homotrimeric and bind homotrimeric ligands. The natural homotrimeric TNFRSF ligand binds the TNFRSF molecules in a way that may be different than the complementary-binding region of an antibody to produce superior engagement.
3.1. OX40
OX40 (CD134; TNFRSF4) is expressed by CD4 and CD8 T cells following TCR ligation [42, 43]. Murine Tregs also express OX40, although high OX40 expression on human Tregs is only seen following activation [44–46]. OX40 is transiently expressed 24–72 h after T-cell activation, which creates a critical window for engagement with its ligand, OX40 ligand (OX40L; CD252). OX40L is also transiently expressed on APCs, with particularly high expression on CD40-licensed dendritic cells (DC), which are important for priming CD8 T-cell responses [47]. The costimulatory activity of OX40 was initially discovered in 1987, when it was shown that agonistic antibody targeting of OX40-enhanced CD4 T-cell proliferation in vitro [48]. Following this, the ligand for OX40 was discovered to be a previously known glycoprotein expressed in human T-cell lymphoma/leukemia virus-1 infected cells, formerly known as gp34 [49].
Early studies on the role of OX40-OX40L interaction in experimental autoimmune encephalomyelitis (EAE) and murine models of arthritis demonstrated that blocking antibodies against either OX40 or OX40L effectively decreased autoimmunity [50]. Building upon these data, it was shown that T cells isolated from the TME expressed OX40 and subsequently hypothesized that these were tumor-reactive T cells. Additional studies revealed that treatment with an agonist aOX40 mAb or OX40L-IgG fusion protein significantly enhanced tumor-free survival across four different murine tumor models [51]. Depletion of CD4 or CD8 T cells demonstrated that the efficacy of aOX40 therapy was T cell-dependent. To investigate whether OX40 expression on CD8 T cells was necessary for treatment efficacy, OT-I TCR transgenic mice, which express a TCR specific for the SIINFEKL peptide of ovalbumin protein, were crossed with OX40-deficient mice, to create OX40−/− OT-Is. In this antigen-specific model, OX40−/− OT-I T cells exhibited reduced CD8 T-cell expansion and survival as compared with wild-type OT-I T cells, highlighting the critical role that OX40 signaling plays in regulating CD8 T-cell activation and survival [52]. Moreover, direct ligation of an OX40 agonist to antigen-specific CD8 T cells significantly enhanced expression of the effector molecule, granzyme B, leading to enhanced tumor regression and long-term survival of tumor-bearing mice [53].
These preclinical studies showed the efficacy of agonist aOX40 therapy and formed a rationale for further evaluation in clinical trials. An initial phase I clinical trial used a murine IgG anti-human OX40 mAb for the treatment of patients with metastatic carcinoma, lymphoma, or sarcoma (NCT01644968). After three doses of aOX40, 12 of 30 patients saw a reduction in at least one metastatic lesion [54]. However, the study was limited to three sequential doses given within 5 days because a murine antibody was used. Despite this use of a murine antibody, the trial still demonstrated that aOX40 therapy led to immunological effects (T-cell proliferation) and supported further clinical development. Currently, several phase I and II clinical trials are ongoing to evaluate aOX40 therapy across multiple cancer types, including head and neck (MEDI6469; NCT02274155), colorectal neoplasia (MEDI6469; NCT02559024), metastatic prostate (MEDI6469; NCT01303705), renal cell carcinoma (PF-04518600; NCT03092856), and solid tumors (INCAGN01949; NCT02923349) [Table 2]. Additionally, multiple phase I clinical trials are exploring aOX40 in combination with other immunotherapies, including tremelimumab (aCTLA-4) and durvalumab (aPD-L1) for the treatment of advanced solid tumors (MEDI0562; NCT02705482, and NCT02205333), in combination with pembrolizumab alone (GSK3174998; NCT02528357), or the combination of pembrolizumab and nivolumab for advanced cancers (BMS-986178; NCT02737475), and in combination with a Toll-like receptor 9 (TLR9) agonist for lymphomas (BMS-986178; NCT03410901). Results from these clinical trials are pending, but results from a phase I dose-escalation trial for the treatment of advanced solid malignancies using a combination of aOX40 and aPD-L1 (MOXR0916; NCT02410512) showed no high-grade adverse effects at the higher dosage, indicating that this combination was well tolerated. Agonist aOX40 therapy is a promising and exciting treatment that may lend itself well to combinations with checkpoint inhibitors such as aPD-1 and aCTLA-4.
Table 2.
Agonist antibody and Fc-ligands in clinical trials as mono and combination immunotherapies
| Target | Mechanism | Name | Company | Clinical Trial |
|---|---|---|---|---|
| 0X40 | Enhanced T cell NF-κB signaling | MEDI6469 | MedImmune | NCT02274155 |
| NCT02559024 | ||||
| NCT01303705 | ||||
| PF-04518600 | Pfizer | NCT03092856 | ||
| INCAGN01949 | Incyte | NCT02923349 | ||
| MEDI0562 | MedImmune | NCT02705482 | ||
| BMS-986178 | BMS | NCT02205333 | ||
| GSK3174998 | GlaxoSmithKline | NCT02737475 | ||
| MOXR0916 | Genentech | NCT03410901 | ||
| NCT02528357 | ||||
| NCT02410512 | ||||
| CD40 | Enhanced DC cross-presentation to CD8 T cells | CP-870,893 | Pfizer | NCT01103635 |
| CDX-1140 | Celldex Therapeutics | NCT03329950 | ||
| SGN-40 | Seattle Genetics | NCT00435916 | ||
| HCD122 | Novartis | NCT01275209 | ||
| NCT00670592 | ||||
| APX005 M | Apexigen | NCT02482168 | ||
| NCT02706353 | ||||
| NCT03123783 | ||||
| NCT03389802 | ||||
| NCT03165994 | ||||
| NCT03214250 | ||||
| ADC1013 | Alligator Bioscience | NCT02379741 | ||
| Chi Lob 4/7 | University of Southampton | NCT01561911 | ||
| 4–1BB | Enhanced NK cell ADCC | Utomilumab | Pfizer | NCT02554812 |
| Urelumab | BMS | NCT02315066 | ||
| NCT02444793 | ||||
| NCT02179918 | ||||
| NCT02845323 | ||||
| NCT02253992 | ||||
| NCT02534506 | ||||
| GITR | Intratumoral Treg depletion | MK-4166 | Merck | NCT02132754 |
| NCT02553499 | ||||
| BMS-986156 | BMS | NCT02598960 | ||
| NCT03335540 | ||||
| MEDI1873 | MedImmune | NCT02583165 | ||
| INCAGN01876 | Incyte | NCT03277352 | ||
| GWN323 | Novartis | NCT03126110 | ||
| TRX518 | Leap Therapeutics | NCT02697591 | ||
| OMP-336B11 | OncoMed Pharmaceuticals | NCT02740270 | ||
| NCT01239134 | ||||
| NCT03295942 | ||||
| ICOS | Enhanced downstream TCR signaling following aCTLA-4 therapy | MEDI570 | MedImmune | NCT02520791 |
| GSK3359609 | GlaxoSmithKline | NCT02723955 | ||
| JTX-2011 | Jounce Therapeutics | NCT02904226 |
aCTLA-4 antibodies targeting cytotoxic T lymphocyte antigen 4, ADCC antibody-dependent cell-mediated cytotoxicity, BMS Bristol-Myers Squibb, CD cluster of differentiation, DC dendritic cell, GITR glucocorticoid-induced tumor necrosis factor receptor-related protein, ICOS inducible T-cell costimulator, NF nuclear factor, NK natural killer, TCR T-cell receptor, Treg regulatory T cell
3.2. CD40
CD40L (CD154; TNFSF5) is expressed primarily on activated CD4 T cells, along with B cells, monocytes, natural killer (NK) cells, basophils, and mast cells [55]. This ligand plays an important role in binding CD40 (TNFRSF5) expressed on APCs and B cells and acting as an important signal to induce activation [56]. The interaction between CD40 and CD40L is critical for developing an adaptive immune response that is highly context dependent on the cell types and cytokines involved. Activated CD4 T-cell CD40L engagement of CD40 on B cells results in TRAF adaptor protein induction of NF-κB, mitogen-activated protein kinase (MAPK), PI3K, and phospholipase γ pathways that activate B cells [57, 58]. Activated B cells form germinal centers, undergo antibody isotype switching, and differentiate into plasma cells to produce antibodies [59]. Signaling through CD40 on DCs is a critical step for DC “licensing,” which enables DCs to prime CD8 T-cell responses effectively through cross-presentation [60–63]. Enhancing APC activation and DC licensing is the main rationale behind agonist aCD40 therapy and the rationale behind its use in combination therapy [64]. Studies of agonistic aCD40 antibodies in murine models of cancer have shown significant therapeutic efficacy. In a mesothelioma model, aCD40 was effective in inhibiting tumor growth in a dose-dependent manner [65]. Interestingly, one group discovered that aCD40 therapy was more effective at clearing tumors in a lymphoma model when treatment was delayed [66]. It was speculated that the larger tumor burden of delayed treatment may increase the available tumor antigens that can be taken up and cross-presented by DCs.
An initial clinical trial using recombinant human CD40L for non-Hodgkin’s lymphoma showed some promise when one of 32 patients had a complete response and another had a partial response in the absence of major toxicity [67]. Several CD40 agonists have moved forward into clinical trials, including the humanized aCD40 CP-870,893, which led to a partial response in 14% of patients with melanoma (NCT01103635) [68]. Studies investigating SGN-40 have not induced any clinical responses to date, but SGN-40 is being used in a clinical trial for lymphomas (NCT00435916) and in combination with rituximab (NCT00655837). Other agents in clinical trials include HCD122 (lucatumumab) for lymphomas (NCT01275209, NCT00670592) [69], CDX-1140 for solid cancers (NCT03329950), and APX005 M for solid tumors (NCT02482168), central nervous system tumors (NCT03389802), and esophageal cancer (NCT03165994). Additional monotherapy trials include ADC-1013 for solid tumors (NCT02379741) and Chi Lob 4/7 for advanced malignancies (NCT01561911). Combination therapy trials include APX005 M in combination with pembrolizumab for metastatic melanoma (NCT02706353) and in combination with nivolumab for metastatic lung cancer, melanoma, and pancreatic cancer (NCT03123783, NCT03214250).
While initial clinical trial data suggest that agonistic CD40 agents may not be effective as monotherapies, CD40 agonists may be effective agents in combination with other immunotherapies. Agonistic CD40 agents may also synergize well with chemotherapies and radiation, which release tumor antigens to aid in the antigen-presentation process. Different antibodies have seen varying levels of SAEs, which suggests that a clear understanding of the optimal timing and dosage for these drugs will be critical to their development and future success.
3.3. 4–1BB
4–1BB (CD137; TNFRSF9) is a glycosylated costimulatory molecule expressed transiently on activated T cells, NK cells, and DCs, as well as expressed constitutively on Tregs [70]. Engagement of 4–1BB by its ligand 4–1BBL (CD137L; TNFSF9) induces strong T-cell activation and survival, particularly in activated CD8 T cells [71, 72]. The role of 4–1BB in cancer immunology has been evaluated in 4–1BBL-deficient mice, which develop spontaneous B-cell lymphomas, and in 4–1BB−/− mice, which develop systemic lupus erythematosus likely due to tumor-suppressive activity of 4–1BBL for B cells [73, 74]. Initial studies of 4–1BB agonists in cancer models of sarcoma and mastocytoma showed enhanced numbers of tumor-specific CD8 T cells and improved T-cell memory against tumor re-challenge [75]. 4–1BB agonists have since been shown to have efficacy across multiple tumor models by inducing a population of tumor-specific cytotoxic CD8 T cells that produce potent pro-inflammatory cytokines and effector molecules, such as granzymes [76–79].
4–1BB also plays an important role in activating and enhancing NK cell function through Fc γ receptors [80]. Based on this finding, it was hypothesized that 4–1BB stimulation of NK cells aids in NK-mediated antibody-dependent cell-mediated cytotoxicity (ADCC). This would make agonistic 4–1BB therapy a candidate for use in combination with tumor antigen-targeted antibodies such as rituximab, which targets CD20 for the treatment of non-Hodgkin’s lymphoma and chronic lymphocytic leukemia. In murine models of lymphoma, an agonist a4–1BB showed strong efficacy when given after rituximab treatment [81].
A phase I clinical trial of utomilumab (a4–1BB) in combination with rituximab for patients with B-cell lymphomas resulted in two complete responses in follicular lymphoma that lasted beyond 2 years (NCT01307267). An increase in memory T cells and activated NK cells was also observed. No significant SAEs were reported, with no patients stopping treatment due to toxicity in the utomilumab study [82]. Utomilumab is also being tested in multiple clinical trials with other agents such as avelumab (aPD-L1; NCT02554812), aOX40 (NCT02315066), mogamulizumab (an aCCR4 antibody; NCT02444793), and pembrolizumab (NCT02179918). Another phase I/II study of an a4–1BB agonist (urelumab; NCT02253992) resulted in dose-dependent adverse effects and a 50% objective response rate in patients with advanced metastatic melanoma when given with aPD-1 (nivolumab); it is also being tested in combination with nivolumab for bladder cancer and other malignant tumors (NCT02845323, NCT02534506). Clearly, 4–1BB agonists are an agent of great interest both as a monotherapy and in combination with other immunotherapies.
3.4. GITR
GITR (CD357; TNFRSF18) is expressed on activated T cells, constitutively expressed on Tregs, and moderately expressed on memory T cells [83, 84]. Once activated, T cells transiently express GITR 24 h after stimulation. GITR expression is regulated by the FoxP3 transcription factor in Tregs and by canonical NF-κB signaling in activated T cells [85]. GITR signaling in activated T cells lowers the threshold for CD28 co-stimulation and results in NF-κB, MAPK, and JNK signal pathway activation through TRAF adaptor proteins [86, 87]. However, the rationale for targeting GITR with mAbs relies on its high constitutive expression on Tregs and costimulatory signaling in CD4 and CD8 T cells. GITR was originally thought to be a unique marker of Tregs before later being found on other cell types [88]. Anti-GITR antibodies seem to be well tolerated without inducing significant autoimmunity. Targeting GITR with IgG1 antibodies has been effective at depleting tumor-infiltrating Tregs from the TME, but not in the periphery in a B16 mouse model of melanoma [89]. Using a FoxP3-GFP transgenic mouse, it was discovered that aGITR was effective in depleting Tregs in B16 tumor-bearing mice, resulting in tumor clearance [90]. On activated CD4 and CD8 T cells, GITR is upregulated following TCR stimulation and signals through activating TRAF molecules to enhance proliferation, pro-inflammatory cytokine production, and resistance to Treg-mediated suppression [91–93].
Based on the high expression of GITR on tumor-infiltrating Tregs, aGITR therapy is expected to be more effective in cancers with high levels of infiltrating Tregs such as cervical, renal cell, hepatocellular, lung, and melanoma [94, 95]. Therefore, it is hypothesized that aGITR therapy would synergize well with other immunotherapies; however, the potential for more severe SAEs resulting from Treg depletion will need to be taken into consideration. Multiple clinical trials are underway testing both aGITR mAbs and GITRL-Fc fusion proteins, including trials as monotherapy (INCAGN0187; NCT02697591), (GWN323; NCT02740270), (TRX518; NCT01239134), and (OMP-336B11; NCT03295942). Anti-GITR mAbs are also being tested in combination with pembrolizumab (MK-4166; NCT02132754, NCT02553499), (INCAGN0187; NCT03277352, NCT03126110) and nivolumab (BMS-986156; NCT02598960, NCT03335540). MEDI1873 is a novel hexameric GITRL-Fc fusion protein aimed at enhancing activated T-cell function, rather than depleting Tregs, and has shown superior activity compared with aGITR mAbs in vivo (NCT02583165) [96].
3.5. ICOS
ICOS (CD278) is an immunoglobulin superfamily receptor expressed on activated T cells [97]. Its ligand, ICOSL (CD275; B7-H2) is expressed on both B cells and DCs [98]. ICOS, CD28, and CTLA-4 all share a homologous proline-rich motif that facilitates their binding to B7 ligands. While CD28 and CTLA-4 can bind B7–1, B7–2, and B7-H2, ICOS is only known to bind B7-H2, although in a different position from CD28 and CTLA-4 and at much higher affinity [99, 100]. ICOS stimulation by ICOSL induces PI3K and AKT pathway signaling, which aides in CD4 T-cell differentiation into follicular T helper cells (TFH), Th1, and Th2 T cells [101]. The effect of ICOS signaling on activated CD4 T cells appears to be context dependent, although signaling likely drives IL-4 and IL-10 production for Th2 differentiation in the absence of additional stimulation [102].
Interestingly, ICOS is upregulated on CD4 T cells following aCTLA-4 therapy, suggesting that ICOS expression is linked to CTLA-4 and may play a compensatory role to CTLA-4 when CTLA-4 is blocked therapeutically [103]. Additionally, therapeutic synergy was observed when both pathways were targeted [103–105]. In the B16 melanoma model, CTLA-4 blockade and an agonist aICOS mAb synergized to provide protection that the monotherapies did not [103]. Clinical trials for aICOS include MEDI-570 for various lymphomas (NCT02520791), GSK3359609 in combination with pembrolizumab for solid tumors (NCT02723955), and JTX-2001 in combination with nivolumab for solid tumors (NCT02904226). Future clinical trials may focus on aICOS in combination with aCTLA-4 agents to determine whether this combination induces additional therapeutic benefit over either therapy alone.
4. Conclusions
Immunotherapy has shown great promise in recent years, leading to durable responses and even cures for a subset of patients with metastatic cancer. While targeting immune checkpoints such as CTLA-4 and PD-1 has proven to be a viable therapy, it has not been a universal success. Targeting inhibitory receptors with a single agent may not be enough to augment the anti-cancer response and overcome cancer-mediated immune suppression. New antibodies and ligand-Fc fusion proteins targeting costimulatory receptors such as OX40, 4–1BB, GITR, and ICOS might provide T cells with a stimulus that is otherwise lacking in the TME. Other costimulatory therapies are targeting alternative (non-T cell) cell types such as DCs (aCD40 therapy) and NK cells (a4–1BB). Moving forward, there is a critical need to understand how to rationally combine these agents, including balancing increased efficacy with the potential for increased toxicity. As the field moves into combination therapies of inhibitory and activating antibodies, sequencing of these agents will also be critical for eliciting potent anti-cancer responses. However, synergistic combination therapy may have the added benefit of working effectively at lower doses, leading to less severe adverse events. A great deal of effort is also focusing on identifying predictive biomarkers of response. For example, tumor-specific PD-L1 expression has already proven to be a predictive marker for aPD-1 therapy in some instances; however, additional immunological analysis may be critical for deciding which immunotherapies to use in combination. In conclusion, immunotherapy targeting of costimulatory receptors, particularly of the TNFRSF, are a promising addition to the growing list of immunotherapy agents being tested in clinical trials.
Key Points.
Immune checkpoint blockade releases the brakes on effector T cells and can induce clinical responses in a subset of patients with metastatic cancer.
Antibodies capable of activating costimulatory receptors on T cells are a promising new class of immunotherapy drugs being evaluated alone or in combination with other immunotherapeutics for patients with advanced cancer.
Acknowledgements
The authors thank Annah Rolig for her careful review of this manuscript.
Funding This work was supported by the Providence Portland Medical Foundation, Susan G. Komen Grant Career Catalyst Research Grant (CCR15329664), and NIH R21CA190790.
Footnotes
Conflicts of interest DE has no competing interests. WLR has received commercial research grants, consulting fees, and/or royalties from MedImmune, Bristol-Myers Squibb, Merck, Galectin Therapeutics, Nektar Therapeutics, Aeglea Biotherapeutics, IRX Therapeutics, Tesaro, Shire, and Shimadzu.
References
- 1.Heath WR, Carbone FR. Cross-presentation, dendritic cells, tolerance and immunity. Annu Rev Immunol. 2001;19:47–64. [DOI] [PubMed] [Google Scholar]
- 2.Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol. 1996;14:233–58. [DOI] [PubMed] [Google Scholar]
- 3.Burkholder B, Huang RY, Burgess R, Luo S, Jones VS, Zhang W, et al. Tumor-induced perturbations of cytokines and immune cell networks. Biochim Biophys Acta. 2014;1845(2):182–201. [DOI] [PubMed] [Google Scholar]
- 4.Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. 2015;14(4):847–56. [DOI] [PubMed] [Google Scholar]
- 5.Hicklin DJ, Marincola FM, Ferrone S. HLA class I antigen downregulation in human cancers: T-cell immunotherapy revives an old story. Mol Med Today. 1999;5(4):178–86. [DOI] [PubMed] [Google Scholar]
- 6.Emens LA, Ascierto PA, Darcy PK, Demaria S, Eggermont AMM, Redmond WL, et al. Cancer immunotherapy: opportunities and challenges in the rapidly evolving clinical landscape. Eur J Cancer. 2017;81:116–29. [DOI] [PubMed] [Google Scholar]
- 7.Hellmann MD, Friedman CF, Wolchok JD. Combinatorial cancer immunotherapies. Adv Immunol. 2016;130:251–77. [DOI] [PubMed] [Google Scholar]
- 8.Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol. 2018;18(3):153–67. [DOI] [PubMed] [Google Scholar]
- 9.Garrido F, Aptsiauri N, Doorduijn EM, Garcia Lora AM, van Hall T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol. 2016;39:44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tagliamonte M, Petrizzo A, Tornesello ML, Buonaguro FM, Buonaguro L. Antigen-specific vaccines for cancer treatment. Hum Vaccin Immunother. 2014;10(11):3332–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJ. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat Rev Cancer. 2016;16(4):219–33. [DOI] [PubMed] [Google Scholar]
- 12.Brunet JF, Denizot F, Luciani MF, Roux-Dosseto M, Suzan M, Mattei MG, et al. A new member of the immunoglobulin superfamily-CTLA-4. Nature. 1987;328(6127):267–70. [DOI] [PubMed] [Google Scholar]
- 13.Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 core-ceptor expression and signal transduction. Immunol Rev. 2009;229(1):12–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–48. [DOI] [PubMed] [Google Scholar]
- 15.Inman BA, Frigola X, Dong H, Kwon ED. Costimulation, coinhibition and cancer. Curr Cancer Drug Targets. 2007;7(1):15–30. [DOI] [PubMed] [Google Scholar]
- 16.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–6. [DOI] [PubMed] [Google Scholar]
- 17.Kuhns MS, Epshteyn V, Sobel RA, Allison JP. Cytotoxic T lymphocyte antigen-4 (CTLA-4) regulates the size, reactivity, and function of a primed pool of CD4 + T cells. Proc Natl Acad Sci U S A. 2000;97(23):12711–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolchok JD, Hodi FS, Weber JS, Allison JP, Urba WJ, Robert C, et al. Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann N Y Acad Sci. 2013;1291:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tremelimumab. Drugs R D. 2010;10(2):123–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ribas A, Kefford R, Marshall MA, Punt CJ, Haanen JB, Marmol M, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol. 2013;31(5):616–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Callahan MK, Wolchok JD, Allison JP. Anti-CTLA-4 antibody therapy: immune monitoring during clinical development of a novel immunotherapy. Semin Oncol. 2010;37(5):473–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol. 2013;14(12):1212–8. [DOI] [PubMed] [Google Scholar]
- 24.Homet Moreno B, Ribas A. Anti-programmed cell death protein-1/ligand-1 therapy in different cancers. Br J Cancer. 2015;112(9):1421–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kang SP, Gergich K, Lubiniecki GM, de Alwis DP, Chen C, Tice MAB, et al. Pembrolizumab KEYNOTE-001: an adaptive study leading to accelerated approval for two indications and a companion diagnostic. Ann Oncol. 2017;28(6):1388–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375(19):1823–33. [DOI] [PubMed] [Google Scholar]
- 27.Bauml J, Seiwert TY, Pfister DG, Worden F, Liu SV, Gilbert J, et al. Pembrolizumab for platinum- and cetuximab-refractory head and neck cancer: results from a single-arm, phase II study. J Clin Oncol. 2017;35(14):1542–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deeks ED. Nivolumab: a review of its use in patients with malignant melanoma. Drugs. 2014;74(11):1233–9. [DOI] [PubMed] [Google Scholar]
- 29.Santini FC, Rudin CM. Atezolizumab for the treatment of non-small cell lung cancer. Expert Rev Clin Pharmacol. 2017;10(9):935–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Powles T, O’Donnell PH, Massard C, Arkenau HT, Friedlander TW, Hoimes CJ, et al. Efficacy and safety of durvalumab in locally advanced or metastatic urothelial carcinoma: updated results from a phase 1/2 open-label study. JAMA Oncol. 2017;3(9):e172411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gulley JL, Rajan A, Spigel DR, Iannotti N, Chandler J, Wong DJL, et al. Avelumab for patients with previously treated metastatic or recurrent non-small-cell lung cancer (JAVELIN Solid Tumor): dose-expansion cohort of a multicentre, open-label, phase 1b trial. Lancet Oncol. 2017;18(5):599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu S, Li A, Liu Q, Yuan X, Xu H, Jiao D, et al. Recent advances of bispecific antibodies in solid tumors. J Hematol Oncol. 2017;10(1):155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu J, Fu J, Zhang M, Liu D. Blinatumomab: a bispecific T cell engager (BiTE) antibody against CD19/CD3 for refractory acute lymphoid leukemia. J Hematol Oncol. 2015;04(8):104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372(21):2006–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song Y, Buchwald P. TNF superfamily protein-protein interactions: feasibility of small- molecule modulation. Curr Drug Targets. 2015;16(4):393–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie P TRAF molecules in cell signaling and in human diseases. J Mol Signal. 2013;8(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walczak H Death receptor-ligand systems in cancer, cell death, and inflammation. Cold Spring Harb Perspect Biol. 2013;5(5):a008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kretschmer A, Schwanbeck R, Valerius T, Rosner T. Antibody Isotypes for Tumor Immunotherapy. Transfus Med Hemother. 2017;44(5):320–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chiu ML, Gilliland GL. Engineering antibody therapeutics. Curr Opin Struct Biol. 2016;38:163–73. [DOI] [PubMed] [Google Scholar]
- 40.Bruggemann M, Osborn MJ, Ma B, Hayre J, Avis S, Lundstrom B, et al. Human antibody production in transgenic animals. Arch Immunol Ther Exp (Warsz). 2015;63(2):101–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Czajkowsky DM, Hu J, Shao Z, Pleass RJ. Fc-fusion proteins: new developments and future perspectives. EMBO Mol Med. 2012;4(10):1015–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bansal-Pakala P, Halteman BS, Cheng MHY, Croft M. Costimulation of CD8 T Cell responses by OX40. J Immunol. 2004;172(8):4821–5. [DOI] [PubMed] [Google Scholar]
- 43.Gramaglia I, Weinberg AD, Lemon M, Croft M. Ox-40 ligand: a potent costimulatory molecule for sustaining primary CD4 T cell responses. J Immunol. 1998;161(12):6510–7. [PubMed] [Google Scholar]
- 44.Bulliard Y, Jolicoeur R, Zhang J, Dranoff G, Wilson NS, Brogdon JL. OX40 engagement depletes intratumoral Tregs via activating FcgammaRs, leading to antitumor efficacy. Immunol Cell Biol. 2014;92(6):475–80. [DOI] [PubMed] [Google Scholar]
- 45.So T, Croft M. Cutting Edge: OX40 Inhibits TGF-β and antigen-driven conversion of Naive CD4 T Cells into CD25 + Foxp3 + T cells. J Immunol. 2007;179(3):1427–30. [DOI] [PubMed] [Google Scholar]
- 46.Vu MD, Xiao X, Gao W, Degauque N, Chen M, Kroemer A, et al. OX40 costimulation turns off Foxp3 + Tregs. Blood. 2007;110(7):2501–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murata K, Ishii N, Takano H, Miura S, Ndhlovu LC, Nose M, et al. Impairment of antigen-presenting cell function in mice lacking expression of OX40 ligand. J Exp Med. 2000;191(2):365–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paterson DJ, Jefferies WA, Green JR, Brandon MR, Corthesy P, Puklavec M, et al. Antigens of activated rat T lymphocytes including a molecule of 50,000 Mr detected only on CD4 positive T blasts. Mol Immunol. 1987;24(12):1281–90. [DOI] [PubMed] [Google Scholar]
- 49.Tanaka Y, Inoi T, Tozawa H, Yamamoto N, Hinuma Y. A glycoprotein antigen detected with new monoclonal antibodies on the surface of human lymphocytes infected with human T-cell leukemia virus type-I (HTLV-I). Int J Cancer. 1985;36(5):549–55. [DOI] [PubMed] [Google Scholar]
- 50.Weinberg AD, Wegmann KW, Funatake C, Whitham RH. Blocking OX-40/OX-40 ligand interaction in vitro and in vivo leads to decreased T cell function and amelioration of experimental allergic encephalomyelitis. J Immunol. 1999;162(3):1818–26. [PubMed] [Google Scholar]
- 51.Weinberg AD, Rivera MM, Prell R, Morris A, Ramstad T, Vetto JT, et al. Engagement of the OX-40 receptor in vivo enhances antitumor immunity. J Immunol. 2000;164(4):2160–9. [DOI] [PubMed] [Google Scholar]
- 52.Salek-Ardakani S, Moutaftsi M, Crotty S, Sette A, Croft M. OX40 drives protective vaccinia virus-specific CD8 T cells. J Immunol. 2008;181(11):7969–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Redmond WL, Gough MJ, Charbonneau B, Ratliff TL, Weinberg AD. Defects in the acquisition of CD8 T cell effector function after priming with tumor or soluble antigen can be overcome by the addition of an OX40 agonist. J Immunol. 2007;179(11):7244–53. [DOI] [PubMed] [Google Scholar]
- 54.Curti BD, Kovacsovics-Bankowski M, Morris N, Walker E, Chisholm L, Floyd K, et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Can Res. 2013;73(24):7189–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carbone E, Ruggiero G, Terrazzano G, Palomba C, Manzo C, Fontana S, et al. A new mechanism of NK cell cytotoxicity activation: the CD40-CD40 ligand interaction. J Exp Med. 1997;185(12):2053–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Quezada SA, Jarvinen LZ, Lind EF, Noelle RJ. CD40/CD154 interactions at the interface of tolerance and immunity. Annu Rev Immunol. 2004;22:307–28. [DOI] [PubMed] [Google Scholar]
- 57.Haxhinasto SA, Bishop GA. Synergistic B cell activation by CD40 and the B cell antigen receptor: role of B lymphocyte antigen receptor-mediated kinase activation and tumor necrosis factor receptor-associated factor regulation. J Biol Chem. 2004;279(4):2575–82. [DOI] [PubMed] [Google Scholar]
- 58.Zhu N, Ramirez LM, Lee RL, Magnuson NS, Bishop GA, Gold MR. CD40 signaling in B cells regulates the expression of the pim-1 kinase via the NF- B pathway. J Immunol. 2002;168(2):744–54. [DOI] [PubMed] [Google Scholar]
- 59.Danese S The CD40/CD40L costimulatory pathway in inflammatory bowel disease. Gut. 2004;53(7):1035–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smith CM, Wilson NS, Waithman J, Villadangos JA, Carbone FR, Heath WR, et al. Cognate CD4(+) T cell licensing of dendritic cells in CD8(?) T cell immunity. Nat Immunol. 2004;5(11):1143–8. [DOI] [PubMed] [Google Scholar]
- 61.Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393(6684):480–3. [DOI] [PubMed] [Google Scholar]
- 62.Sigal LJ, Crotty S, Andino R, Rock KL. Cytotoxic T-cell immunity to virus-infected non-haematopoietic cells requires presentation of exogenous antigen. Nature. 1999;398(6722):77–80. [DOI] [PubMed] [Google Scholar]
- 63.Guermonprez P, Saveanu L, Kleijmeer M, Davoust J, Van Endert P, Amigorena S. ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature. 2003;425(6956):397–402. [DOI] [PubMed] [Google Scholar]
- 64.Vonderheide RH, Glennie MJ. Agonistic CD40 antibodies and cancer therapy. Clin Cancer Res. 2013;19(5):1035–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Khong A, Brown MD, Vivian JB, Robinson BW, Currie AJ. Agonistic anti-CD40 antibody therapy is effective against postoperative cancer recurrence and metastasis in a murine tumor model. J Immunother. 2013;36(7):365–72. [DOI] [PubMed] [Google Scholar]
- 66.Tutt AL, O’Brien L, Hussain A, Crowther GR, French RR, Glennie MJ. T cell immunity to lymphoma following treatment with anti-cd40 monoclonal antibody. J Immunol. 2002;168(6):2720–8. [DOI] [PubMed] [Google Scholar]
- 67.Vonderheide RH, Dutcher JP, Anderson JE, Eckhardt SG, Stephans KF, Razvillas B, et al. Phase I study of recombinant human CD40 ligand in cancer patients. J Clin Oncol. 2001;19(13):3280–7. [DOI] [PubMed] [Google Scholar]
- 68.Vonderheide RH, Flaherty KT, Khalil M, Stumacher MS, Bajor DL, Hutnick NA, et al. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J Clin Oncol. 2007;25(7):876–83. [DOI] [PubMed] [Google Scholar]
- 69.Byrd JC, Kipps TJ, Flinn IW, Cooper M, Odenike O, Bendiske J, et al. Phase I study of the anti-CD40 humanized monoclonal antibody lucatumumab (HCD122) in relapsed chronic lymphocytic leukemia. Leuk Lymphoma. 2012;53(11):2136–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vinay DS, Kwon BS. 4–1BB signaling beyond T cells. Cell Mol Immunol. 2011;8(4):281–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dawicki W, Watts TH. Expression and function of 4–1BB during CD4 versus CD8 T cell responses in vivo. Eur J Immunol. 2004;34(3):743–51. [DOI] [PubMed] [Google Scholar]
- 72.Vinay DS, Kwon BS. Immunotherapy of cancer with 4–1BB. Mol Cancer Ther. 2012;11(5):1062–70. [DOI] [PubMed] [Google Scholar]
- 73.Middendorp S, Xiao Y, Song JY, Peperzak V, Krijger PH, Jacobs H, et al. Mice deficient for CD137 ligand are predisposed to develop germinal center-derived B-cell lymphoma. Blood. 2009;114(11):2280–9. [DOI] [PubMed] [Google Scholar]
- 74.Vinay DS, Choi JH, Kim JD, Choi BK, Kwon BS. Role of endogenous 4–1BB in the development of systemic lupus erythematosus. Immunology. 2007;122(3):394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellstrom KE, et al. Monoclonal antibodies against the 4–1BB T-cell activation molecule eradicate established tumors. Nat Med. 1997;3(6):682–5. [DOI] [PubMed] [Google Scholar]
- 76.Gauttier V, Judor JP, Le Guen V, Cany J, Ferry N, Conchon S. Agonistic anti-CD137 antibody treatment leads to antitumor response in mice with liver cancer. Int J Cancer. 2014;135(12):2857–67. [DOI] [PubMed] [Google Scholar]
- 77.Li B, Lin J, Vanroey M, Jure-Kunkel M, Jooss K. Established B16 tumors are rejected following treatment with GM-CSF-secreting tumor cell immunotherapy in combination with anti-4–1BB mAb. Clin Immunol. 2007;125(1):76–87. [DOI] [PubMed] [Google Scholar]
- 78.Li Q, Carr A, Ito F, Teitz-Tennenbaum S, Chang AE. Polarization effects of 4–1BB during CD28 costimulation in generating tumor-reactive T cells for cancer immunotherapy. Cancer Res. 2003;63(10):2546–52. [PubMed] [Google Scholar]
- 79.Morales-Kastresana A, Catalan E, Hervas-Stubbs S, Palazon A, Azpilikueta A, Bolanos E, et al. Essential complicity of perforingranzyme and FAS-L mechanisms to achieve tumor rejection following treatment with anti-CD137 mAb. J Immunother Cancer. 2013;1:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lin W, Voskens CJ, Zhang X, Schindler DG, Wood A, Burch E, et al. Fc-dependent expression of CD137 on human NK cells: insights into “agonistic” effects of anti-CD137 monoclonal antibodies. Blood. 2008;112(3):699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kohrt HE, Houot R, Goldstein MJ, Weiskopf K, Alizadeh AA, Brody J, et al. CD137 stimulation enhances the antilymphoma activity of anti-CD20 antibodies. Blood. 2011;117(8):2423–32. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 82.Tolcher AW, Sznol M, Hu-Lieskovan S, Papadopoulos KP, Patnaik A, Rasco DW, et al. Phase Ib study of utomilumab (PF-05082566), a 4–1BB/CD137 agonist, in combination with pembrolizumab (MK-3475) in patients with advanced solid tumors. Clin Cancer Res. 2017;23(18):5349–57. [DOI] [PubMed] [Google Scholar]
- 83.Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol. 2002;3(2):135–42. [DOI] [PubMed] [Google Scholar]
- 84.McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, et al. CD4(+)CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16(2):311–23. [DOI] [PubMed] [Google Scholar]
- 85.Tone Y, Kidani Y, Ogawa C, Yamamoto K, Tsuda M, Peter C, et al. Gene expression in the Gitr locus is regulated by NF-kappaB and Foxp3 through an enhancer. J Immunol. 2014;192(8):3915–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Snell LM, McPherson AJ, Lin GH, Sakaguchi S, Pandolfi PP, Riccardi C, et al. CD8 T cell-intrinsic GITR is required for T cell clonal expansion and mouse survival following severe influenza infection. J Immunol. 2010;185(12):7223–34. [DOI] [PubMed] [Google Scholar]
- 87.Esparza EM, Lindsten T, Stockhausen JM, Arch RH. Tumor necrosis factor receptor (TNFR)-associated factor 5 is a critical intermediate of costimulatory signaling pathways triggered by glucocorticoid-induced TNFR in T cells. J Biol Chem. 2006;281(13):8559–64. [DOI] [PubMed] [Google Scholar]
- 88.Ronchetti S, Ricci E, Petrillo MG, Cari L, Migliorati G, Nocentini G, et al. Glucocorticoid-induced tumour necrosis factor receptor-related protein: a key marker of functional regulatory T cells. J Immunol Res. 2015;2015:171520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schaer DA, Budhu S, Liu C, Bryson C, Malandro N, Cohen A, et al. GITR pathway activation abrogates tumor immune suppression through loss of regulatory T cell lineage stability. Cancer Immunol Res. 2013;1(5):320–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cohen AD, Schaer DA, Liu C, Li Y, Hirschhorn-Cymmerman D, Kim SC, et al. Agonist anti-GITR monoclonal antibody induces melanoma tumor immunity in mice by altering regulatory T cell stability and intra-tumor accumulation. PLoS One. 2010;5(5):e10436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tone M, Tone Y, Adams E, Yates SF, Frewin MR, Cobbold SP, et al. Mouse glucocorticoid-induced tumor necrosis factor receptor ligand is costimulatory for T cells. Proc Natl Acad Sci USA. 2003;100(25):15059–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stephens GL, McHugh RS, Whitters MJ, Young DA, Luxenberg D, Carreno BM, et al. Engagement of glucocorticoid-induced TNFR family-related receptor on effector T cells by its ligand mediates resistance to suppression by CD4 + CD25 + T cells. J Immunol. 2004;173(8):5008–20. [DOI] [PubMed] [Google Scholar]
- 93.Ronchetti S, Zollo O, Bruscoli S, Agostini M, Bianchini R, Nocentini G, et al. GITR, a member of the TNF receptor superfamily, is costimulatory to mouse T lymphocyte subpopulations. Eur J Immunol. 2004;34(3):613–22. [DOI] [PubMed] [Google Scholar]
- 94.Liu C, Workman CJ, Vignali DA. Targeting regulatory T cells in tumors. FEBS J. 2016;283(14):2731–48. [DOI] [PubMed] [Google Scholar]
- 95.Shang B, Liu Y, Jiang SJ, Liu Y. Prognostic value of tumor-infiltrating FoxP3 + regulatory T cells in cancers: a systematic review and meta-analysis. Sci Rep. 2015;14(5):15179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tigue NJ, Bamber L, Andrews J, Ireland S, Hair J, Carter E, et al. MEDI1873, a potent, stabilized hexameric agonist of human GITR with regulatory T-cell targeting potential. Oncoimmunology. 2017;6(3):e1280645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Kraft R, Anagnostopoulos I, et al. ICOS is an inducible T-cell costimulator structurally and functionally related to CD28. Nature. 1999;397(6716):263–6. [DOI] [PubMed] [Google Scholar]
- 98.Yao S, Zhu Y, Zhu G, Augustine M, Zheng L, Goode DJ, et al. B7-h2 is a costimulatory ligand for CD28 in human. Immunity. 2011;34(5):729–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang S, Zhu G, Chapoval AI, Dong H, Tamada K, Ni J, et al. Costimulation of T cells by B7-H2, a B7-like molecule that binds ICOS. Blood. 2000;96(8):2808–13. [PubMed] [Google Scholar]
- 100.Yoshinaga SK, Whoriskey JS, Khare SD, Sarmiento U, Guo J, Horan T, et al. T-cell co-stimulation through B7RP-1 and ICOS. Nature. 1999;402(6763):827–32. [DOI] [PubMed] [Google Scholar]
- 101.So L, Fruman DA. PI3K signalling in B- and T-lymphocytes: new developments and therapeutic advances. Biochem J. 2012;442(3):465–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Löhning M, Hutloff A, Kallinich T, Mages HW, Bonhagen K, Radbruch A, et al. Expression of ICOS in vivo defines CD4 + effector t cells with high inflammatory potential and a strong bias for secretion of interleukin 10. J Exp Med. 2003;197(2):181–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fan X, Quezada SA, Sepulveda MA, Sharma P, Allison JP. Engagement of the ICOS pathway markedly enhances efficacy of CTLA-4 blockade in cancer immunotherapy. J Exp Med. 2014;211(4):715–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen H, Fu T, Suh WK, Tsavachidou D, Wen S, Gao J, et al. CD4 T cells require ICOS-mediated PI3K signaling to increase T-Bet expression in the setting of anti-CTLA-4 therapy. Cancer Immunol Res. 2014;2(2):167–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fu T, He Q, Sharma P. The ICOS/ICOSL pathway is required for optimal antitumor responses mediated by anti-CTLA-4 therapy. Can Res. 2011;71(16):5445–54. [DOI] [PubMed] [Google Scholar]