

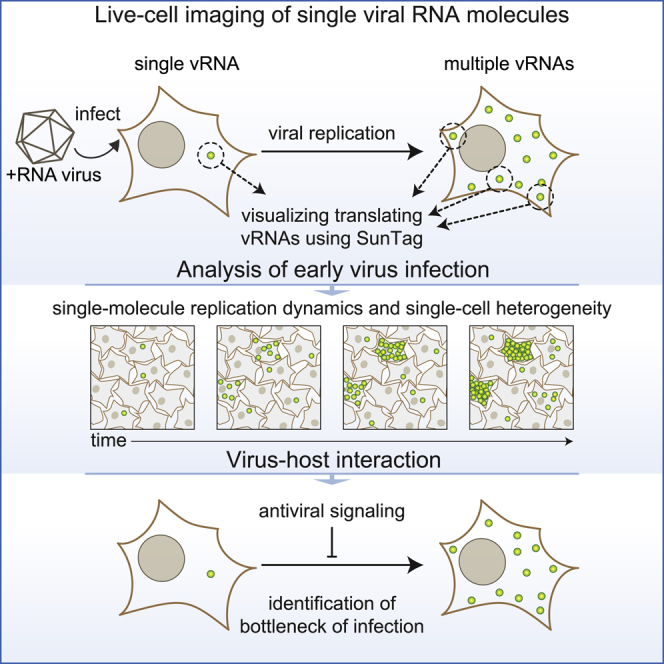
Summary
RNA viruses are among the most prevalent pathogens and are a major burden on society. Although RNA viruses have been studied extensively, little is known about the processes that occur during the first several hours of infection because of a lack of sensitive assays. Here we develop a single-molecule imaging assay, virus infection real-time imaging (VIRIM), to study translation and replication of individual RNA viruses in live cells. VIRIM uncovered a striking heterogeneity in replication dynamics between cells and revealed extensive coordination between translation and replication of single viral RNAs. Furthermore, using VIRIM, we identify the replication step of the incoming viral RNA as a major bottleneck of successful infection and identify host genes that are responsible for inhibition of early virus replication. Single-molecule imaging of virus infection is a powerful tool to study virus replication and virus-host interactions that may be broadly applicable to RNA viruses.
Keywords: +RNA virus, viral translation, viral replication, virus-host competition, single-molecule imaging, SunTag, VIRIM
Graphical Abstract
Highlights
-
•
Single-molecule imaging assay to study translation and replication of +RNA viruses
-
•
Early picornavirus infection occurs in five distinct phases
-
•
Heterogeneity in translation and replication of viral RNAs
-
•
Replication of incoming vRNA represents major target for host antiviral activity
Boersma et al. develop a single-molecule imaging assay (VIRIM) to study translation, replication, and virus-host interactions of +RNA viruses. They observe heterogeneity in translation and replication of single viruses, identify replication of the incoming viral genome as a bottleneck for successful infection, and identify host genes mediating this antiviral activity.
Introduction
The group of positive-strand RNA (+RNA) viruses comprises many virus families, including important pathogens of humans and animals such as Coronaviridae (e.g., Middle East respiratory syndrome coronavirus [MERS-CoV] and severe acute respiratory syndrome coronavirus 2 [SARS-CoV-2]), Flaviviridae (e.g., Zika virus, dengue virus, hepatitis C virus), Caliciviridae (e.g., norovirus), and Picornaviridae (e.g., poliovirus, coxsackievirus, rhinovirus, and other emerging enteroviruses such as EV-A71 and EV-D68). +RNA virus infections are a major health and economic burden on society, and very few treatment options currently exist for the majority of +RNA virus infections.
Most +RNA viruses contain a single-strand positive-sense RNA genome that can be directly translated into viral proteins upon release into the cytoplasm of a host cell. Upon synthesis, viral proteins execute various functions, such as viral RNA (vRNA) replication, modification of host cell processes to facilitate virus amplification, and repression of antiviral signaling in the host cell. After translation of the incoming vRNA (i.e., the vRNA that infected the host cell), the newly synthesized vRNA-dependent RNA polymerase (RdRp) generates negative-sense RNA (−RNA), which, in turn, is used as a template for synthesis of additional +RNAs. These new +RNAs can enter a new round of translation and replication or can be encapsulated to form new infectious virus particles (Baggen et al., 2018; Barrows et al., 2018; Thorne and Goodfellow, 2014; de Wit et al., 2016).
Because vRNA molecules can engage in multiple processes (translation, replication, and/or packaging), tightly controlled switching between these dynamic processes is likely important for virus reproduction. Even for the incoming vRNA, a translation-to-replication switch is essential to initiate virus replication in newly infected cells. Although some factors have been identified that may contribute to the switch from translation to replication (Ahlquist et al., 2003; Sean et al., 2009), currently there are few mechanistic insights into this switch.
Cells have sophisticated mechanisms to detect and counteract viral infection, including protein sensors that detect long double-stranded (viral) RNA (dsRNA), which is formed during replication of +RNA viruses. Upon detection of viral dsRNA, host cell signaling leads to rapid activation of innate antiviral pathways, such as the interferon (IFN) induction pathway. Subsequent IFN signaling leads to upregulation of IFN-induced genes (ISGs), which are critical to limit reproduction of the virus (Samuel, 2001; Schoggins et al., 2011; Stetson and Medzhitov, 2006). Viruses, in turn, actively counteract antiviral signaling pathways. For example, many picornaviruses produce proteases that target host dsRNA sensors or members of the IFN signaling cascade to prevent detection of the virus and concomitant production of antiviral signaling molecules (Wang et al., 2018). Moreover, many RNA viruses shut down host translation and transcription, which may also hamper the antiviral response (Chase and Semler, 2012; Walsh et al., 2013). Therefore, early virus detection may improve the likelihood of mounting an effective antiviral response in an infected cell. Thus, the outcome of a viral infection is likely determined by competition between viral translation/replication kinetics and host-cell antiviral signaling kinetics. Interestingly, substantial cell-to-cell heterogeneity has been observed for antiviral signaling, even in a homogeneous population of cells in culture (Doǧanay et al., 2017; Patil et al., 2015; Zawatzky et al., 1985), suggesting that cellular and/or viral heterogeneity may be an important aspect of virus-host competition.
For multiple reasons, currently available assays are suboptimal to study viral translation and replication dynamics or virus-host competition. First, most current assays (e.g., western blot, PCR, immunofluorescence, engineered GFP-expressing viruses, etc.) are not sufficiently sensitive to detect the virus during the first few hours of infection, when viral translation and replication and antiviral responses are initiated. It is particularly challenging to interrogate the incoming virus particle because it contains only a single vRNA molecule and can easily evade detection. Second, most assays require cell lysis or fixation and therefore do not provide real-time measurements of live, infected cells. As a result, it is difficult to correlate molecular events that occur early during infection with the eventual outcome of infection. Third, many cells are often required for a single measurement, which is particularly problematic for analysis of dynamic processes that are not synchronized in time. If multiple cells in a population are infected at different times, then an ensemble method is inadequate to study temporally defined events, such as replication of the incoming vRNA or initiation of antiviral signaling. Moreover, the highly heterogeneous response to viral infection is an additional problem for ensemble methods. Fourth, most assays assess only a single parameter of the viral infection (e.g., vRNA levels or viral protein levels). However, viral translation and replication are interconnected; a translation defect results in production of less polymerase, which may reduce the replication rate. Therefore, single-parameter assays have a limited ability to specifically uncover mechanistic insights into regulation of translation or replication. Finally, ensemble methods are also unable to assess spatial information of viral infection. Therefore, new tools are urgently required to provide real-time, spatially resolved, single-molecule measurements of viral translation and replication and virus-host interactions.
Results
Single-Molecule Analysis of Viral Translation and Replication
To analyze early events during virus infection, we aimed to develop a live-cell imaging assay to visualize individual vRNAs. We applied our previously developed SunTag fluorescence imaging system (Tanenbaum et al., 2014), which allows single-molecule detection in live cells. The SunTag system consists of an array of small peptides (SunTag peptides) and a fluorescently labeled intracellular single-chain variable fragment antibody (scFv-GFP; SunTag antibody [STAb]) that can bind to the SunTag peptides. We and others have shown previously that the SunTag system can be used to visualize translation of single mRNAs (Morisaki et al., 2016; Pichon et al., 2016; Wang et al., 2016; Wu et al., 2016; Yan et al., 2016); when multiple SunTag peptides are introduced at the N terminus of an open reading frame (ORF), STAbs can bind to the SunTag peptides co-translationally as soon as they emerge from the ribosome, fluorescently labeling the translating mRNA (Figure 1A). Because single mRNA molecules are generally translated simultaneously by multiple ribosomes, translating mRNAs are often associated with a high level of SunTag fluorescence and can be distinguished easily from single “mature” (i.e., ribosome-released) proteins based on fluorescence intensity. In contrast, single-molecule analysis of translation cannot be achieved using GFP encoded by the mRNA; fluorescent proteins like GFP have a relatively long “maturation time” (i.e., the time between synthesis and fluorescence) (Balleza et al., 2018), so most GFP molecules do not become fluorescent until after translation is completed and the GFP molecule has been released from the mRNA. In the latter scenario, GFP fluorescence is not associated with the mRNAs and does not directly report the translational status or localization of individual translating mRNAs.
Figure 1.

A Single-Molecule Imaging Assay to Study Translation and Replication of Individual RNA Viruses
(A) Cartoon of the SunTag-CVB3 single-molecule imaging assay.
(B–D) Representative images of STAb cells (B and D) and U2OS cells (C) 2 h after administration of SunTag-CVB3 and representative images of vRNA smFISH and STAb staining (C).
(E) Combined analysis of live-cell imaging and vRNA smFISH in the same cells. Every dot represents a single cell; a dashed line indicates linear fit.
(F) Mean number of translating vRNAs per cell over time, aligned at first detection of a vRNA with or without pre-treatment with 10 μM 3Dpol inhibitor (GPC-N114).
(G) Representative images of NLS-BFP STAb cells 1 h after SunTag-CVB3 administration.
(H) Difference in BFP fluorescence intensity between nucleoplasm and cytoplasm. Data are aligned at the start of phase 1 (dashed line) and normalized to the values of 3 min before the start of phase 1.
(I) Combined analysis of live-cell imaging and vRNA smFISH in the same cells. Every dot represents a single cell.
(J) Time projection of a single translating vRNA. Color indicates time in minutes since first detection of the vRNA; dotted lines indicate cell and nuclear outlines at the first time-point.
(K) Diffusion kinetics of translating vRNA or mRNA molecules.
Time in minutes since first detection of a translating vRNA (arrow head) is given in (B) and (G). Shaded areas in (F), (H), and (K) indicate SEM. Scale bars, 15 μm. See also Video S1 and Figure S1. The number of experimental repeats and cells analyzed per experiment are listed in Table S1.
We reasoned that the SunTag translation imaging system could also be employed to visualize translation of single vRNAs, allowing tracking of viral infections in space and time with single-molecule sensitivity (Figure 1A). We engineered coxsackievirus B3 (CVB3), a representative member of the Enterovirus genus, with 5 SunTag peptide repeats at the N terminus of the viral polyprotein (SunTag-CVB3) (Figure 1A). The SunTag array was stably maintained in the vRNA through multiple passages (Figure S1A), although it caused some reduction in overall vRNA levels, similar to other inserts in CVB3, such as GFP (Figures S1B–S1D; Andino et al., 1994; Feuer et al., 2002; Lanke et al., 2009)). Shortly after infection of human U2OS cells stably expressing the STAb (referred to as STAb cells) with SunTag-CVB3 at a low MOI (MOI = 0.25), one or more bright GFP foci could be observed in infected cells (Figure 1B). Single-molecule fluorescence in situ hybridization (smFISH) analysis showed that SunTag GFP foci co-localized with CVB3 +RNA (Figure 1C) and that GFP foci disappeared rapidly upon administration of the translation inhibitor puromycin (Figure 1D), confirming that GFP foci represent nascent polypeptides associated with translating vRNAs rather than mature proteins. Quantitative analysis of GFP focus intensities revealed that individual GFP foci correspond to ∼90 SunTag peptides (Figure S1E). Because a single ribosome translating the vRNA is associated with only 5 SunTag peptides, these results indicate that GFP foci represent vRNAs translated simultaneously by many ribosomes.
Figure S1.

A Single-Molecule Assay to Visualize and Analyze Translation and Replication of Individual CVB3 vRNAs, Related to Figure 1
(A) Agarose gel analysis of the SunTag insert in the wild-type (WT) or SunTag (ST) CVB3 genome after RNA isolation, cDNA synthesis and PCR amplification of the indicated region. (B-D) Virus growth curves of the indicated viruses in indicated cell lines. Similar MOIs were used in all experiments. (E) Number of nascent SunTag peptides per translating vRNA focus based on the vRNA GFP fluorescence intensities compared to the GFP fluorescence intensity of 24xSunTag arrays expressed in STAb cells. (F) Representative images of STAb cell infected with SunTag-CVB3, during live-cell imaging (left) or after fixation and smFISH against +CVB3. (G) The number of smFISH +CVB3 ‘background’ foci in cells not exposed to virus (left) or cells that were uninfected after incubation with SunTag-CVB3 (right). For each repeat, the mean number of smFISH foci in uninfected cells was used to correct the number of smFISH foci in infected cells in Figures 1E, 1I, and 2E. (H) Combined analysis of viral protein synthesis (based on 3Dpol protein immunofluorescence) and viral load (based on fluorescence intensity of +CVB3 smFISH) in the STAb cells infected with indicated virus. Dashed lines indicate linear fits. (I, J) Representative images (I) and quantification (J) of combined analysis of live-cell imaging and dsRNA immunofluorescence of the same STAb cells infected with SunTag-CVB3. Color of outline (I) indicates the time between first detection of a translating vRNA and fixation. Cells in which no translating vRNAs were observed are indicated by a white outline and were used to correct for background fluorescence. (K, L) Representative images (K) and quantification (L) of combined analysis of GFP fluorescence and dsRNA immunofluorescence in the same U2OS cells infected with eGFP-CVB3. (M) Violin and boxplots of diffusion kinetics of translating vRNAs in cells that contain the indicated number of translating vRNAs. (N) Images of representative time-lapse movie of a STAb U2OS cell infected with SunTag-CVB3. Zooms indicate areas with mobile (pink) or immobilized (blue) translating vRNAs. (O) Bar graph of the fraction of immobilized translating vRNAs per cell. Every dot (G, H, J, L) indicates a single cell. Statistics is based on Kruskal-Wallis test. Error bars indicate SEM. Scale bars, 15 μm. The number of experimental repeats and cells analyzed per experiment are listed in Table S1.
When cells were followed by time-lapse microscopy, we generally observed an increase in the number of GFP foci over time in individual cells (Figure 1B), indicative of virus replication. The number of GFP foci observed in live-cell imaging experiments correlated well with the number of vRNAs, as assessed by smFISH in the same cells after fixation (R2 = 0. 90; Figures 1E and S1F). The increase in GFP foci was strongly attenuated by an inhibitor of the RdRp, 3Dpol (GPC-N114; van der Linden et al., 2015), confirming that an increase in the number of GFP foci in single cells reflects vRNA replication (Figure 1F).
To assess whether SunTag-CVB3 replicated with kinetics similar to wild-type CVB3, we developed a live-cell sensor of infection by CVB3 that does not rely on the SunTag. Previous reports have shown that nuclear pore integrity becomes impaired upon infection with CVB3, resulting in increased exchange of nuclear and cytoplasmic components (Belov et al., 2000; Flather and Semler, 2015; Gustin and Sarnow, 2001). To test whether we could leverage impaired nuclear transport as a marker for infection, we generated STAb cells stably expressing BFP fused to a nuclear localization signal (NLS-BFP). We found that translocation of BFP from the nucleus to the cytoplasm occurs extremely rapidly after infection with SunTag-CVB3, within minutes of translation of the incoming vRNA (Figures 1G and 1H; Video S1). The start of BFP translocation can therefore be used as a proxy for the moment of infection. Using live-cell imaging of NLS-BFP localization (to assess the moment of infection) combined with smFISH of the same cells after fixation (to assess viral replication), we compared the number of vRNAs over time for SunTag and wild-type CVB3. Although the number of vRNAs over time was highly heterogeneous between cells for wild-type and SunTag-CVB3, the average number of vRNAs per cell over time was similar for both viruses, indicating that insertion of the SunTag into the viral genome does not affect early vRNA replication (Figure 1I). Furthermore, by combining smFISH with immunofluorescence, we found that the ratio of vRNAs to viral proteins is also similar for wild-type and SunTag CVB3 (Figure S1H), indicating that the SunTag also does not hamper viral translation.
Maximal intensity projection of 11 GFP Z-slices (green) and a single BFP Z-slice (magenta) of STAb cells stably expressing NLS-BFP infected with SunTag-CVB3. Images were acquired every 2 min. Time is indicated in hours:minutes since the start of the acquisition. The first translating vRNA is detected at 00:10. Note that images of the same cell are used in Figures 1G and 2A. Scale bar shown in the first image, 15 μm.
Quantitative comparison of SunTag-CVB3 with previously established methods to detect viral infection (staining of viral dsRNA and fluorescence generated by GFP-CVB3) confirmed that previous methods could reliably detect viral infection only 3–5 h after infection (Figures S1I–S1L), when the vRNA has already undergone (several rounds of) replication. These results show that SunTag-CVB3 faithfully recapitulates wild-type viral infection dynamics and uniquely reports early events during infection. We refer to this single-molecule virus imaging assay using SunTag as VIRIM (virus infection real-time imaging).
Localization and Mobility of vRNAs during Early Infection
Using SunTag-CVB3, we first examined the localization and mobility of translating vRNAs. During the first 2–3 h, we did not observe any preferential localization of translating vRNAs in the cytoplasm. vRNAs moved rapidly throughout the cytoplasm and showed mobility that was similar to the mobility of host mRNAs of comparable length (Figures 1J, 1K, and S1M). At later stages during infection (2–4 h post-infection [p.i.]), we did observe a small subset of vRNAs that became immobilized in the vicinity of the nucleus (Figures S1N and S1O). All GFP foci (mobile and immobile) disappeared rapidly upon treatment with puromycin (Figure S2A), indicating that both types of foci represent translating vRNAs. We conclude that, during early infection, the majority of translating vRNAs are not localized to specific sub-cellular sites but, rather, diffuse freely through the cytoplasm.
Figure S2.

Live-Cell Imaging of Translating vRNAs Identifies Infection Phases, Related to Figure 2
(A) Representative images of SunTag-CVB3 infected NLS-BFP STAb cells in different infection phases 3 hr after administration of virus before (middle panels) and 5 minutes after (bottom panels) puromycin administration. NLS-BFP (top panel) is shown as well to indicate that cells have been infection, based on loss of nuclear NLS-BFP fluorescence. Arrow head indicates first translating vRNA during phase1. (B) Frequency of time-points during assigned phases during which indicated number of vRNAs were observed. Infection phase1p refers to phase1 pulses, indicating that only time-points assigned as a phase1 pulse were included in the analysis. (C) Combined analysis of the number of translating vRNAs and SunTag labeling stoichiometry in the same cells, normalized to the mean labeling stoichiometry in uninfected cells. SunTag labeling stoichiometry is based on GFP fluorescence intensity of 24xSunTag-peptide arrays expressed in the same cells. To illustrate corresponding phases, phase3, 4, and 5 are indicated based on the distribution of the number of translating vRNAs during phase4. (D) Fluorescence intensity of smFISH foci normalized to the mean of single vRNAs. Red dashed line indicates the intensity threshold of smFISH foci classified as replicating vRNAs. (E) Replicating vRNA intensity normalized to the mean single vRNA smFISH. (F, G) Combined analysis of the same cells of live-cell imaging SunTag-CVB3 infection in SunTag U2OS cells to determine start of infection and smFISH to analyze replicating vRNAs. Replicating vRNA intensities are normalized to mean smFISH intensity of single vRNAs (F). 3Dpolinhibitor: GPC-N114 (10 μM). ∗∗p < 0.01; ns, not significant, based on Kruskal-Wallis test (E) or unpaired Mann-Whitney test (F). Scale bars, 15 μm. The number of experimental repeats and cells analyzed per experiment are listed in Table S1.
Heterogeneity and Dynamics of Replication
Long-term time-lapse imaging of SunTag-CVB3-infected cells revealed a remarkable recurring pattern in viral replication, including five distinct phases (Figures 2A–2C; Video S1). Infection phase 1 starts with the appearance of a single GFP spot (at MOI < 1), representing translation of the incoming vRNA. Phase 1 is followed by a period without GFP foci, referred to as phase 2. Phase 2 may represent vRNA replication (synthesis of a −RNA and subsequent synthesis of multiple new +RNAs templated from the −RNA) because vRNA translation is shut down during replication (Barton et al., 1999; Gamarnik and Andino, 1998). Consistent with this, the average duration of phase 2 was in line with the expected time required to synthesize −RNA and +RNA based on in vitro measurements of replication speed (Arnold and Cameron, 2000; Barton and Flanegan, 1997). Phase 3 starts with re-appearance of a GFP spot, followed by a rapid increase in the number of GFP foci, likely because of translation of newly synthesized +RNAs. During phase 3, additional +RNAs are likely synthesized from the –RNA that was produced in phase 2. In phase 4, the number of GFP foci remains constant, indicating that the newly synthesized +RNAs are undergoing translation in preparation of a new round of replication. On average, 15–20 translating vRNAs were observed during phase 4, consistent with a previous observation that +RNAs typically outnumber –RNAs by approximately 20-fold (Dave et al., 2019). During phase 4, a subset of vRNAs is expected to undergo replication, yielding new –RNAs and subsequent new +RNAs. In phase 5, a second rapid increase in the number of foci is observed, likely reflecting synthesis and translation of the additional +RNAs. Phase 5 continues until the number of foci per cell exceeds the detection limit.
Figure 2.

Single-Cell Dynamics and Heterogeneity of Virus Replication
(A and B) Representative images (A) and example quantifications (B) of time-lapse movies of STAb cells infected with SunTag-CVB3. Example images from the same time-lapse movie are also used in Figure 1G and Video S1.
(C) Cartoon of infection phases in single cells.
(D) Representative images of vRNA smFISH and STAb staining.
(E and F) Combined analysis of live-cell imaging to determine infection phase and smFISH in the same cells to quantify smFISH foci (E) and replicating vRNAs (F) in STAb cells infected with SunTag-CVB3.
(G) Fraction of cells in each infection phase over time. Uninfected cells are excluded from quantification.
(H–K) Kaplan-Meier graphs showing durations of infection phases.
(L) Mean number of translating vRNAs during phase 4.
(M) Fraction of infected cells with successful phase 2, based on the plateau in the Kaplan-Meier curve of the duration of phase 2. Every dot indicates a repeat, and lines indicate mean.
(N) Kaplan-Meier survival curve.
(O and P) Representative images (O) and example quantifications (P) of representative time-lapse movie of STAb cells infected with SunTag-EMCV.
(Q) Violin plot of the combined timing of phases 1 and 2 or phases 3 and 4.
Colors (B, C, and P) illustrate infection phases. Data points during phases 0, 1, and 2 (B and P) are increased 3-fold to aid visualization of data that are very close to the x axes. Arrowheads (A and E) indicate the first translating vRNA; an arrow (D) indicates a replicating vRNA. Error bars (E, F, and L) indicate SD; circles (H–K and N) indicate the last analyzable time point for individual cells. Scale bars, 15 μm. See also Videos S1, S2, and S3 and Figures S2 and S3. The number of experimental repeats and cells analyzed per experiment are listed in Table S1.
Quantitative analysis revealed that focus calling, which underlies annotation of infection phases, was highly accurate, with only 0.4% false positives and 7.5% false negatives (Figure S2B). Moreover, GFP foci in all infection phases disappeared upon treatment with the translation inhibitor puromycin, confirming that the GFP foci exclusively represent translating vRNAs (Figure S2A). Note that, at the end of phase 5, GFP, which localized previously in the cytoplasm and nucleus, accumulated in the cytoplasm, likely reflecting an excess of mature SunTag protein in the cytoplasm, which sequestered the STAb (Figure 2A; Video S1). The excess of SunTag peptides over STAb molecules may cause a lower binding stoichiometry of the STAb to SunTag peptide arrays and interfere with quantitative interpretation of GFP focus intensities. However, we found that STAb binding stoichiometry was not affected during early infection, at least until ∼60 foci per cell were present (i.e., beyond phase 5) (Figure S2C), demonstrating that STAb binding stoichiometry was constant through phases 1–5.
To confirm that replication of the incoming vRNA occurred during phases 2 and 3, we combined VIRIM with CVB3 +RNA smFISH analysis of the same cells (Figures S1F and 2D). This analysis revealed that the number of smFISH foci increased starting from phase 3 onward (Figure 2E), consistent with vRNA replication in phases 2 and 3. Interestingly, close inspection of the smFISH foci revealed that two types of smFISH foci could be distinguished in infected cells based on the smFISH intensity (Figures 2D and S2D); most smFISH foci showed a uniform low intensity, whereas a small subset of foci was much brighter (>2.5-fold). During replication, multiple polymerases can simultaneously use the –RNA as a template, resulting in multiple co-localizating nascent +RNAs, suggesting that these bright foci may represent replicating vRNAs. The numbers and fluorescence intensities of the bright foci was indeed reduced in cells treated with the 3Dpol inhibitor (Figures S2F and S2G), and the bright foci rarely co-localized with STAb fluorescence (1.2%, n = 329 bright foci, 2 repeats; Figure 2D), indicating that the bright smFISH foci indeed represent replicating vRNAs. Quantitative analysis revealed that these replicating vRNAs were not detected during phase 1 but became visible from phase 2 onward (Figure 2F), further confirming that replication initiates during phase 2.
Although the five infection phases were observed in most infected cells, the duration of the individual phases, as well as the number of translating vRNAs during each phase was heterogeneous between cells (Figure 2B). In particular, substantial heterogeneity was observed in the duration of phase 1, ranging from 12 min to more than 4 h. As a result, cells in all infection phases co-exist within a population of cells at any moment (Figure 2G). Such diverse cells are pooled and averaged in ensemble measurements, highlighting the importance of a real-time single-molecule imaging approach, like VIRIM. Similar infection phases were observed in HeLa cells as well as in cells derived from the airway epithelium (A549 cancer cells) and gastrointestinal tract (primary human intestinal organoids), which represent natural targets of CVB3 (Figures 2H–2M and Figures S3A-S3J; Baggen et al., 2018). These results suggest that the infection phases and their timing may be a universal phenomenon for CVB3.
Figure S3.

Reproducibility of Single-Cell Dynamics and Heterogeneity of Viral Replication, Related to Figure 2
(A-D) Kaplan-Meier graphs showing durations of infection phases of all repeats combined (black) or separated into 6 datasets (colors). Circles indicate last analyzable time-point for individual cells. Data in black is replotted from Figure 2H–2K for comparison. (E-J) Representative images (E, G, I) and example quantifications (F, H, J) of time-lapse movies of indicated STAb cells infected with SunTag-CVB3. (E, G, I) Arrow head indicates the first translating vRNA. Asterisk indicates a background spot. (F, H, J) Colors illustrate infection phases. Note that data points during phase0, 1, and 2 are enhanced 3-fold to aid visual inspection of data. (K) Number of nascent SunTag peptides per translating vRNA focus based on the vRNA GFP fluorescence intensities compared to the GFP fluorescence intensity of 24xSunTag arrays expressed in STAb cells. Data plotted in green is replotted from Figure S1E for comparison. (L) Frequency of time-points during assigned phases during which indicated number of vRNAs were observed. Infection phase1p refers to phase1 pulses, indicating that only time-points assigned as a phase1 pulse were included in the analysis (See STAR Methods). (M) Kaplan-Meier graphs of the fraction of uninfected cells remaining after incubation with SunTag-CVB3. Data is corrected for the fraction of cells that were infected before the start of the movies, as indicated by the gap at the start of each graph. (N) Representative example quantifications of STAb cells infected by multiple viruses after incubation with SunTag-CVB3 at MOI 1.25, based on observing multiple vRNAs during phase1. As phase1 and2 of individual infections could not be distinguished, the duration between first detection of a translating vRNA and start of phase3 was determined as proxy for phase1+2 duration. Scale bars, 15 μm (E, G) or 10 μm and 25 μm (I). The number of experimental repeats and cells analyzed per experiment are listed in Table S1.
When analyzing the replication phases in more detail, we found that 15%–20% of SunTag-CVB3-infected cells were arrested during phase 2 (Figures 2I and 2M; Video S2), indicating that the incoming vRNA did not undergo replication to produce new vRNAs in those cells. In all cases where phase 2 was completed successfully, subsequent phases were also successful, indicating that phase 2 is the most vulnerable phase of the virus life cycle and is key for successful infection. Notably, most cells in which no detectable virus replication occurred still died eventually, albeit slightly slower than cells with regular replication (Figure 2N; Video S3). Cells containing unsuccessfully replicated viruses may have an important role in antiviral immunity in vivo; for example, through cytokine production or release of viral antigens without release of viral progeny.
Maximal intensity projection of 11 GFP Z-slices (green) and a single BFP Z-slice (magenta) of STAb cells stably expressing NLS-BFP infected with SunTag-CVB3. Images were acquired every 2 min. Time is indicated in hours:minutes since the start of the acquisition. The first translating vRNA in the middle cell is detected at 00:22, and does not result in successful viral replication (phase2 block). Scale bar shown in the first image, 15 μm.
Maximal intensity projection of 4 GFP Z-slices (green) and a single BFP Z-slice (magenta) of STAb cells stably expressing NLS-BFP infected with SunTag-CVB3 and imaged with propidium iodide (cyan). Images were acquired every 10 min. Time is indicated in hours:minutes since the start of the movie. (A) The first translating vRNA in the middle cell is detected at 03:00 and results in a successful infection. (B) The first translating vRNA in the middle cell is detected at 03:10, and does not result in a successful infection (block in phase2).Scale bar shown in the first images, 15 μm.
To determine whether we could similarly assess virus replication kinetics of other viruses, we employed VIRIM to encephalomyocarditis virus (EMCV), a member of the Cardiovirus genus in the picornavirus family (Baggen et al., 2018). SunTag-EMCV was viable and could be stably propagated in human cells. After infection of STAb-U2OS cells with SunTag-EMCV, we observed rapidly diffusing GFP foci, similar to SunTag-CVB3. SunTag-EMCV foci were, on average, ∼3-fold dimmer than SunTag-CVB3 foci (Figure S3K). Although EMCV GFP foci were more difficult to detect, GFP focus calling could still be performed with high accuracy (Figure S3L). Dimmer translating vRNA foci suggest that the translation efficiency of EMCV is lower than that of CVB3 (the vRNA is occupied by fewer translating ribosomes). Nonetheless, analysis of the number of translating vRNAs over time revealed that EMCV replication followed a similar pattern as CVB3 replication (Figures 2O and 2P). The infection phases may thus be a general phenomenon of picornaviruses. These results show that VIRIM may be widely applicable to study translation and replication dynamics of +RNA viruses.
The Translation-to-Replication Switch
During phase 1, a virus translates its genome in preparation of vRNA replication (phases 2/3). Similarly, newly synthesized vRNAs are translated in phases 3 and 4 in preparation for replication, which occurs during phases 4/5. Interestingly, we found that the median duration of phases 1 and 2 (translation and replication of incoming vRNA) was similar to the median duration of phases 3 and 4 (translation and replication of daughter vRNAs) (60 versus 72 min; Figure 2Q), even though far more viral proteins and vRNAs are present in the cell during phases 3 and 4. These results suggest that the timing of vRNA replication is not determined by the number of vRNAs or viral proteins in the cell but, rather, intrinsic to individual vRNAs (i.e., controlled in cis). Consistent with this, a similar median duration of phases 1 and 2 was observed when using a higher MOI (Figure 2Q) that resulted in a substantial number of cells (∼35%) infected by multiple viruses (STAR Methods; Figures S3M and S3N).
Because replication of an incoming vRNA depends on newly translated viral polymerase, we wondered whether the duration of phase 1 (which determines the number of viral polymerase proteins produced) is predictive of the success of replication during phase 2. We found that cells with unsuccessful replication (phase 2 arrest) generally did have an extended phase 1 duration (Figure 3A). In most cells, phase 1 consists of an uninterrupted period during which only a single translating vRNA is observed (Figures 2C and 3B). However, in a subset of cells (15%–20%), we observed a single translating vRNA, followed by a period (>12 min) without detectable GFP foci during phase 1, followed by another period with only a single translating vRNA (we refer to these uninterrupted periods of a single GFP spot as translation “pulses”; Figure 3B). Although multiple translation pulses were rare in cells with successful replication, they were more frequently observed in cells with unsuccessful replication (Figure 3C). Reappearance of GFP foci after their initial disappearance was unlikely to be due to a second infection of the same cell (STAR Methods; Figure S4A). Instead, we speculated that reinitiation of translation occurs as result of a failure in replication. Indeed, inhibition of virus replication using the 3Dpol inhibitor resulted in an extended phase 1 with additional translation pulses (Figures 3B and S4B). Similar effects were observed with rupintrivir (3Cpro inhibitor), which prevents proper processing of the viral polymerase and also acts as a replication inhibitor (Figures 3C, S4B, and S4C). The infection rate was not affected by replication inhibitors, excluding secondary infections as a major cause for the observed increase in translation pulses (Figure S4D). Notably, in the majority (∼65%) of untreated infected cells that underwent a second translation pulse, replication occurred successfully, indicating that the virus strategy to reinitiate translation upon failed replication frequently results in successful replication.
Figure 3.

Coordination between vRNA Translation and Replication
(A) Kaplan-Meier graphs showing the duration of infection phase 1.
(B) Examples of phase 1 pulses in single cells. Colors illustrate individual infection phases, pulses, and breaks.
(C) Frequency of pulses. 3Dpol inhibitor, GPC-N114 (10 μM); 3Cpro inhibitor, rupintrivir (10 μM).
(D) Kaplan-Meier graphs showing the duration of first pulses.
(E) Violin plots of fluorescence intensities of translating vRNAs, normalized to mean of the wild-type (WT) IRES virus.
(F and G) Kaplan-Meier graphs showing the duration of phase 1 (F) and phase 2 (G). Data plotted in black are replotted from Figures 2J and 2K for comparison.
Circles (A, D, F, and G) indicate the last analyzable time point for individual cells. ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; n.s., not significant (based on two-tailed unpaired Student’s t test). Scale bar, 15 μm. See also Figure S4. The number of experimental repeats and cells analyzed per experiment are listed in Table S1.
Figure S4.

Timing of Translation-to-Replication Transition, Related to Figure 3
(A) Frequency of multiple infections or phase1 pulses in infected cells. (B) Kaplan-Meier graphs showing durations of infection phase1 (sum of all pulses and breaks; indicated with arrow in Figures 3B and S3C). (C) Quantification of a representative example cell of the number of translating vRNAs over time after 10 μM Rupintrivir treatment. (D) Kaplan Meier graphs of the fraction of uninfected cells remaining after incubation with SunTag-CVB3. Data is corrected for the fraction of cells that were infected before the start of the movies, as indicated by the gap at the start of each graph. (E) Frequency at which indicated number of consecutive time-points without a translating vRNA (gaps) occurred in the analysis of time-lapse movies of STAb cells infected with SunTag-CVB3. (F-K) Kaplan-Meier graphs showing the duration of individual phase1 pulses (F-H) or individual phase1 breaks (I-K). The data plotted in gray (F-K) represents the combination of all data. Datasets with less than six cells were not plotted, but are included in the combined datasets (gray). Data plotted in orange (F-H) is replotted from Figure 3D, for comparison. (L, M) Kaplan-Meier graphs showing the duration of phase3 (J) and phase4 (K). Data plotted in black is replotted from Figure 2J, K, for comparison. The number of experimental repeats and cells analyzed per experiment are listed in Table S1.
Interestingly, the average duration of each translation pulse was similar for untreated cells and cells treated with replication inhibitors (Figures 3D and S4F–S4K). Replication itself is thus not required to shut down translation. Instead, these findings suggest that an independent phase 1 “timer” exists that regulates the translation-to-replication switch. During phase 1, the replication machinery (including 3Dpol) that is essential for vRNA replication during phase 2 is synthesized. We hypothesized that the phase 1 “timer” may reflect production of a threshold amount of viral protein. To test this hypothesis, we reduced the viral translation rate by introduction of a point mutation in the viral internal ribosome entry site (IRES). Introduction of an IRES mutation (Sabin-like 3 mutation) reduced viral translation by ∼30% (Figure 3E), consistent with previous studies (Ben M’hadheb-Gharbi et al., 2006; Svitkin et al., 1985, 1990). Surprisingly, however, the IRES mutation did not alter the phase 1 “timer” that regulates the translation-to-replication switch (Figures 3F, S4L, and S4M), indicating that translation shutdown occurs at a set time, independent of the amount of viral protein that has been synthesized. Interestingly, IRES mutant viruses did show a defect in virus replication during phase 2 (Figure 3G), suggesting that entry into the replication phase with a reduced level of viral proteins impairs replication.
Host versus Viral Translation
During infection, many RNA viruses shut down host translation, although the specific mechanism varies between viruses (Walsh et al., 2013). Enteroviruses inhibit host translation through cleavage of the translation initiation factor eIF4G by the viral 2Apro protein, which has been suggested to stimulate viral translation (Hambidge and Sarnow, 1992; Kräusslich et al., 1987; Lamphear et al., 1995). To study the coordination between host and viral protein synthesis directly, we examined viral translation rates along with host translation efficiency over time in single cells. Viral translation rates were assessed based on GFP focus intensity, whereas host protein synthesis was determined using a fluorescently labeled methionine analog to label newly synthesized proteins (Estell et al., 2017; Figure S5A; STAR Methods). Strikingly, global host protein synthesis was already substantially reduced during infection phase 1, which occurs before replication of the incoming vRNA (Figures 4A–4C and S5B). The rapid global reduction in protein synthesis rates most likely reflects inhibition of translation rather than transcription because a global decrease in protein synthesis rates would be difficult to achieve so quickly through transcription inhibition. To assess how the timing of host translation shutdown relates to the kinetics of eIF4G cleavage, we developed a live-cell eIF4G cleavage biosensor and examined the timing of eIF4G cleavage during early infection. In this biosensor, a fusion of mCherry-eIF4G-BFP was tethered to the outer mitochondrial membrane (Figure 4D). Cleavage of eIF4G results in dissociation of BFP fluorescence from mCherry-labeled mitochondria. Potent eIF4G cleavage was observed during viral infection (Figures 4E and S5C; Video S4), with half of the eIF4G biosensor cleaved ∼1.5 h after the start of phase 1 (Figures 4F and S5D). These results show that host translation is shut down rapidly after infection. In contrast, viral translation efficiency was similar during all phases of infection (Figures 4G and 4H), including before and after host translation shutdown (Figures 4I and 4J; STAR Methods). Therefore, despite accumulation of cleaved eIF4G and host translation shutdown, viral translation remained constant (Figure 4K). Our kinetics analysis also revealed that host shutdown occurs before (substantial) dsRNA is present in the cell and therefore could pre-empt production of antiviral proteins.
Figure S5.

Inhibition of Host Cell Translation, Related to Figure 4
(A) Fluorescence intensity of the labeled methionine analog either in the cytoplasm or in the nucleus after indicated treated measured in single cells. Data are normalized to the mean cytoplasmic intensity. Every dot indicates an experimental repeat; lines indicate mean values. (B) Violin and boxplots of the cytoplasmic fluorescence intensity of the methionine analog in single cells, after indicated treatments. All datasets were normalized to the mean of uninfected (set to 1) and puromycin treated (set to 0) cells. For comparison, the data on uninfected cells in A is replotted from Figures 3B and 3C (in blue). (C) western blots of STAb cells expressing the eIF4G cleavage reporter after indicated incubation times with CVB3. (D) STAb cells expressing the eIF4G cleavage reporter were imaged after infection with SunTag-CVB3. BFP fluorescence intensity differences between mitochondria and cytoplasm over time for infected cells with either successful or unsuccessful replication of the incoming vRNA. The data in black is replotted from Figure 4F. Shaded areas indicate SEM. The number of experimental repeats and cells analyzed per experiment are listed in Table S1.
Figure 4.

Viral Translation Efficiency Is Unaffected by Shutdown of Host Cell Translation
(A) Representative images of uninfected (white outline) and infected (green outline) NLS-BFP STAb cells stained for an incorporated methionine analog to indicate global translation rates.
(B and C) Fluorescence intensities of the methionine analog normalized to the mean of uninfected cells (blue, set to 1) from the same sample and to a puromycin-treated control (dotted line, set to 0). Data in pink (cells with a single translating vRNA) are replotted in (C). Every dot represents a single infected cell.
(D) Cartoon of the eIF4G cleavage reporter.
(E) Representative images of STAb cells expressing the eIF4G cleavage reporter at the indicated time (minutes) since first detection of a translating vRNA (arrowhead).
(F) BFP fluorescence intensity difference between mitochondria and cytoplasm. Intensity difference traces are aligned to the start of phase 1 (dashed line) and normalized to the values of the 15 min before start of phase 1.
(G and H) Comparison of GFP fluorescence intensity of translating vRNA in infected cells with a single or multiple translating vRNAs.
(I and J) GFP fluorescence intensity of single translating vRNAs over the course of phase 1 (I) or after administration of 3 μM harringtonine (J). Intensity traces (J) are aligned to the moment of drug administration (dashed line) and normalized to the mean intensity over 2 min before drug treatment.
(K) Comparison of viral translation and host cell protein production based on the moving averages from (B) and (H).
Error bars and shaded areas indicate SD (B, C, I, and J) or SEM (F). Statistics are based on two-tailed unpaired Student’s t test (G) or paired Wilcoxon test (I). Scale bars, 15 μm. See also Video S4 and Figure S5. The number of experimental repeats and cells analyzed per experiment are listed in Table S1.
Maximal intensity projection of 11 GFP Z-slices (green) and single Z-slice of BFP (blue) and mCherry (red) of STAb cells expressing the eIF4G cleavage reporter infected with SunTag-CVB3. Images were acquired every 5 min. Time is indicated in hours:minutes since the start of the acquisition. The first translating vRNA is detected at 01:35. Of note, image acquisition is started 12 h after induction of expression of the eIF4G cleavage reporter and the expression is still increasing throughout the movie. Therefore, only GFP, but not BFP and mCherry, is corrected for photo bleaching. Scale bar shown in the first image, 15 μm.
Virus-Host Interactions
Innate antiviral responses, most notably IFN signaling, play a key role in repressing the spread of most RNA viruses (Belkowski and Sen, 1987; Samuel, 2001; Schoggins et al., 2011; Stetson and Medzhitov, 2006). Although the effects of IFN signaling on gene expression have been well documented (Schneider et al., 2014; Schoggins, 2018), its effects on early viral infection are largely unknown. We stimulated the innate antiviral state by treating cells with IFN α2 (referred to as IFN) and assessed the timing, efficiency, and heterogeneity of infection. IFN signaling did not alter the fraction of cells over time that became infected (i.e., showed at least 1 GFP spot; Figure 5A) but led to a striking increase in cells in which the incoming vRNA was arrested during phase 2 (replication phase) (Figures 5B–5E). Surprisingly, the subset of cells (∼40%) in which the incoming vRNA did undergo successful replication progressed normally through the later infection phases (Figures 5C–5F), albeit with a slight reduction in the number of vRNAs in phase 4 (Figure 5G). Together, these results show that the IFN-induced antiviral state predominantly acts to prevent replication of the incoming vRNA with little effect on other aspects of early infection.
Figure 5.

Potent IFN-Induced Inhibition of Replication of the Incoming vRNA
(A) Kaplan-Meier graphs of the fraction of uninfected cells remaining after incubation with SunTag-CVB3 with or without IFNα2 pretreatment. Data are corrected for the fraction of cells that were infected before the start of the time-lapse movies, as indicated by the gap at the start of each graph. Data plotted in black are replotted from Figure S3M.
(B–E) Kaplan-Meier graphs showing durations of infection phases. Data in black are replotted from Figures 2H–2K for comparison.
(F) Kaplan-Meier graphs of the fraction of surviving cells that were infected or uninfected after the indicated treatment. Groups were further subdivided based on whether infection resulted in successful replication during phase 2. For comparison, data plotted with dark colors (untreated control) are replotted from Figure 2H.
(G) Violin and boxplots of the mean number of translating vRNAs per infected cell during phase 4. Data plotted in black are replotted from Figure 2L. ∗∗p < 0.01 based on unpaired Student’s t test.
Circles (B–F) indicate the last analyzable time point for individual cells. The number of experimental repeats and cells analyzed per experiment are listed in Table S1.
Because IFN promotes an antiviral state through upregulation of ISGs (Schneider et al., 2014; Schoggins, 2018), we set out to identify ISG(s) responsible for suppression of replication of the incoming vRNA. Using RNA sequencing (RNA-seq), we identified 37 genes that were upregulated more than 3-fold upon IFN treatment in U2OS cells (Figure 6A), which included many well-known antiviral genes (Figure S6A). We adapted the VIRIM assay to facilitate screening of many experimental conditions (STAR Methods; the adapted VIRIM assay lacks single-molecule detection sensitivity) and tested the involvement of 28 of these ISGs in suppressing replication of the incoming virus. We identified 6 ISGs (IFIT1, OAS1, OAS3, STAT1, HELZ2, and C19orf66) whose depletion partially neutralized repression of vRNA replication by IFN (Figures 6B and S6B). Identification of STAT1, the transcription factor of ISGs, validated our screen (Schneider et al., 2014). Combined knockdown of multiple ISGs relieved phase 2 arrest even more (up to ∼60%; Figures 6C and S6B–S6G), demonstrating that multiple antiviral mechanisms act in parallel to block replication of the incoming vRNA. Notably, the well-known ISG protein kinase R (PKR) was present in our screen but did not inhibit replication of the incoming vRNA (Figures 6B, S6H, and S6I). Although all proteins identified in our screen have been implicated previously in antiviral signaling (Fusco et al., 2017; Kumar et al., 2014; Li et al., 2016; Suzuki et al., 2016), none of these proteins have been implicated in inhibition of early vRNA replication, illustrating the potential of VIRIM in analyzing antiviral mechanisms.
Figure 6.

IFN Acts through Multiple Parallel Mechanisms to Block Replication of the Incoming vRNA
(A) Scatterplot of gene expression changes after IFNα2 treatment. Every dot indicates a single gene, and red dots indicate genes with at least 3-fold increased expression upon IFNα2 treatment in both repeats.
(B and C) Fraction of infected cells with successfully replicating viruses after transfection with the indicated siRNAs and/or treatment with IFNα2. Every dot represents an independent experiment, and black lines indicate the means. Grey control data are based on experiments without siRNA transfections; blue control data are based on non-targeting siRNAs. Dashed lines represent the means of controls.
(D and E) Kaplan-Meier graphs showing the duration of infection phase 2 (D) or phase 1 (E). Data in black and red are replotted from Figures 5B and 5C for comparison.
(F) Frequency of pulses. Control data are replotted from Figure 3C.
(G) Kaplan-Meier graphs showing the duration of phase 1 pulses. Data in black are replotted from Figure S3D for comparison.
(H) Kaplan Meier graphs showing the duration of infection phase 2. Data in black and red are replotted from Figure 5C for comparison.
Circles (D, E, G, and I) indicate the last analyzable time point for individual cells. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.01, p < 0.001, ∗∗∗∗p < 0.0001; Dunnett’s multiple comparisons test (B and C) or Gehan-Breslow-Wilcoxon test (D and H). † indicates non-significant genes that were included in the follow-up analysis. See also Figure S6. The number of experimental repeats and cells analyzed per experiment are listed in Table S1.
Figure S6.

The IFN-Induced Antiviral State Represses Replication in Phase 2, Related to Figure 6
(A) Selection of the top gene ontology terms and the corresponding p values for genes with increased expression upon IFNα2 treatment. (B) Fraction of infected cells with successfully replicating virus after transfection with the indicated siRNAs, and/or treatment with IFNα2. (C-G) qPCR analysis of expression levels of indicated genes, relative to GADPH expression levels, normalized to untreated controls. (H) Fraction of infected cells with successfully replicating virus after transfection with the indicated siRNA, and/or treatment with IFNα2. (I) qPCR analysis of expression levels of PKR, relative to GADPH expression levels, normalized to untreated controls. (J) Fraction of infected cells with successfully replicating virus after transfection with the indicated siRNA, and/or treatment with IFNα2. (K-M) qPCR analysis of expression levels of indicated genes, relative to GADPH expression levels, normalized to untreated controls. (N) An example single-cell trace of pulses and breaks in an IFNα2-treated cell. (O) Fraction of infected cells with successfully replicating virus after transfection with the indicated siRNAs, and/or treatment with IFNα2. Every dot (B, H, J, O) represents an independent experiment, black lines indicate the mean, and dashed lines indicate the mean of the untreated controls. Error bars indicate SD. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.01; p < 0.001; ∗∗∗∗p < 0.0001; ns, not significant, based on Dunnett’s multiple comparisons tests (B, H, J, O) or two-tailed unpaired Student’s t test compared to no siRNA conditions (C-G, I, K-M). The number of experimental repeats and cells analyzed per experiment are listed in Table S1.
The observed phase 2 arrest upon IFN treatment could be caused by inhibition of vRNA replication or could result from RNA decay of the incoming vRNA (thus preventing its replication). To explore the role of RNA decay in phase 2 arrest, we first depleted the 5′-to-3′ exonuclease Xrn1 as well Dis3L, an essential subunit of the exosome (the major 3′-to-5′ exonuclease complex). Neither Xrn1 nor Dis3L depletion affected the phase 2 arrest induced by IFN (Figures S6J–S6L). In our screen, we did identify 2′-5′-oligoadenylate synthetase 1 and 3 (OAS1 and OAS3; Figures 6B and S6B), whose activity is required for activation of the well-known antiviral gene RNase L (Li et al., 2016), and RNase L is known to stimulate RNA decay through endonucleolytic cleavage during viral infection (Chakrabarti et al., 2011; Silverman, 2007). Therefore, we tested the role of RNase L in IFN-induced phase 2 arrest. Indeed, knockdown of RNase L also mitigated phase 2 arrest induced by IFN, as assessed by VIRIM and the adapted VIRIM assay (Figures 6C, 6D, S6J, and S6M), suggesting that RNase L-mediated RNA decay may be important for IFN-induced phase 2 arrest. However, RNase L depletion did not decrease the duration of phase 1 (Figure 6E), suggesting that RNase L-induced RNA decay is limited to phase 2 and, thus, likely triggered by vRNA replication.
Interestingly, in addition to the strong phase 2 arrest upon IFN treatment, we observed a modest extension of phase 1 (Figure 5B). Closer examination revealed that the phase 1 extension was caused by an increase in the number of translation pulses during phase 1 (Figures 6F, 6G, and S6N), indicative of replication failure (Figures 3A–3C). Thus, IFN likely acts through multiple mechanisms to suppress viral replication, including vRNA degradation and inhibition of replication.
Finally, we further explored the role of one of the identified ISGs, IFN-induced protein with tetratricopeptide repeats 1 (IFIT1). IFIT1 is a repressor of viral infection and is thought to inhibit viral translation through binding to triphosphate-containing 5′ viral ends (Daffis et al., 2010; Kumar et al., 2014; Pichlmair et al., 2011). However, enteroviruses lack a triphosphate group at the 5′ end of their genome (Baggen et al., 2018). Recently, an additional translation-independent function was proposed for IFIT1, although the mechanistic details remain unknown (Mears et al., 2019). We reproduced the small interfering RNA (siRNA) phenotype of IFIT1 using an additional IFIT1 siRNA (Figure S6O), confirming the specificity of IFIT1 knockdown. Next we performed live-cell analysis using VIRIM of early infection in IFN-treated and IFIT1-depleted cells, revealing a reduction in phase 2 arrest without substantial changes to the other infection phases (Figure 6H). These results reveal that the replication phase of the incoming vRNA is the key point of repression by IFN signaling and that IFN likely acts through multiple parallel mechanisms.
Discussion
Insights into Viral Replication
Using VIRIM, we found that early CVB3 infection consists of five phases, each reflecting a distinct set of molecular events in the enteroviral life cycle. Similar infection phases were observed in various cell types and for the cardiovirus EMCV and may represent a universal phenomenon for picornaviruses. Through detailed analysis of these infection phases, we were able to provide multiple insights into viral translation and replication. (1) We found that the transition from translation to replication is controlled by a “timer” that likely acts in cis on each vRNA individually (Figure 2Q). (2) Translation shutdown is induced independent of vRNA replication because shutdown occurred with identical kinetics in cells treated with replication inhibitors (Figure 3D). (3) Upon failed replication, vRNAs frequently reinitiate translation for another attempt to replicate (Figures 3B and 3C). Because our results suggest that replication inhibition of the incoming vRNA is a major point of attack of antiviral signaling (see below), reinitiation of translation followed by a second attempt at replication may represent a novel mechanism by which viruses combat the antiviral response. Interestingly, this second “pulse” of translation shows kinetics similar to the first one, indicating that the translation-to-replication timer is reset after every replication attempt. (4) The onset of the translation-to-replication switch is not dependent on the amount of viral protein that has been produced because CVB3 with a mutated IRES that shows reduced translation initiates replication with similar kinetics as wild-type CVB3 (Figure 3F). However, the efficiency of virus replication is lower for IRES mutant viruses, suggesting that they enter the replication phase with insufficient viral protein to execute replication with high fidelity (Figure 3G). We speculate that the timing of the translation-to-replication switch (i.e., the duration of phase 1) has evolved to occur as soon as possible, but only after sufficient levels of viral proteins have been produced to ensure productive replication.
Insights into Virus-Host Competition
To combat viral infection, host cells must rapidly detect and inhibit a virus upon infection. However, viruses have evolved various mechanisms to evade detection and/or counteract antiviral pathways (Wang et al., 2018). Our study reveals that this competition between picornaviruses and their host is initiated very early during infection. Advanced technologies, like VIRIM or the recently developed assay to identify host proteins interacting with vRNAs (Kim et al., 2020), are ideally suited to study early viral replication. Here we applied VIRIM to study virus-host interactions during the first hours of infection. We find that CVB3-induced host cell modifications occur extremely rapidly upon infection; eIF4G cleavage, host cell translation shutdown, and impairment of nuclear transport are initiated within minutes of initial infection, at the start of phase 1, and generally reach completion 1–3 h after initial infection (Figures 1G, 1H, 4B, and 4C). Very few (<100) viral proteins have been synthesized when host cell modifications initiate, highlighting the remarkable potency of enteroviral proteases (which drive many of the abovementioned host cell modifications). The swift host shutdown may provide a key advantage to the virus; substantial host cell inhibition is already induced before replication of the incoming vRNA occurs, and maximal host inhibition is generally achieved when the larger burst of replication occurs (phases 4/5). Because formation of dsRNA during viral replication is a key trigger of an antiviral response, shutdown of host protein synthesis before formation of dsRNA may be an effective virus strategy to limit production of antiviral signaling molecules. These findings regarding virus-host interaction kinetics may explain in part why only a small subset of infected cells mount a strong IFN response (Doǧanay et al., 2017; Patil et al., 2015; Zawatzky et al., 1985).
Many RNA viruses specifically target the host cell translation machinery (Chase and Semler, 2012; Walsh et al., 2013). As discussed above, it is possible that host translation shutdown counteracts antiviral signaling. An additional consequence of host cell translation shutdown is that the translation machinery (e.g., ribosomes) becomes available exclusively for vRNA translation, possibly boosting vRNA translation. Previous reports have suggested that eIF4G cleavage stimulates vRNA translation (Hambidge and Sarnow, 1992; Kräusslich et al., 1987; Lamphear et al., 1995). Here we examined the relationship between eIF4G cleavage and host translation shutdown with viral translation rates using VIRIM. We found that vRNA translation rates are similar before and after host translation shutdown and eIF4G cleavage (Figure 4K), indicating that viral translation is not directly affected by these processes. Our results do not exclude the possibility that host cell translation shutdown boosts viral translation at later stages during infection, when the number of vRNAs is substantially larger and translation machinery may become limiting.
A key aspect of antiviral signaling is production of IFN, which induces expression of a large set of ISGs and strongly represses virus spreading. Although the set of genes that is upregulated upon IFN signaling has been well documented, the function of the majority of ISGs in combatting enterovirus infection is largely unknown. Using VIRIM, we find that IFN signaling causes strong and specific inhibition of phase 2; i.e., replication of the incoming vRNA (Figure 5C). In all cases where the incoming vRNA replicated successfully, substantial virus replication was observed during the subsequent infection phases (Figures 5D–5G). These findings indicate that replication of the incoming vRNA is a major point of attack for antiviral signaling. To provide insights into the mechanism of this attack, we screened for ISGs that inhibit early enteroviral replication. We provide evidence that multiple mechanisms, including replication-triggered RNA decay through OAS-RNase L as well as inhibition of vRNA replication, have important roles in mediating the effects of IFN on early viral replication. These results uncover a major target of antiviral signaling (replication of the incoming vRNA) and identify multiple genes involved in this process. More broadly, this study shows how VIRIM can be used to dissect antiviral signaling. Whether replication of the incoming vRNA is also a bottleneck in infection of other +RNA viruses is an important topic for future research. VIRIM may also be a useful tool to dissect the mechanisms of antiviral drugs and potentially screen for novel drugs with specific modes of action during early infection.
Success Rate of Infection
Previous work has shown that multiple viral particles (in some cases even hundreds) are needed for (detectable) infection of a cell (Klasse, 2015), suggesting that productive infection by individual virus particles fails in the majority of cases. Little is known about the limiting steps in the virus life cycle that are responsible for this bottleneck. Using VIRIM, we discovered that, in 15%–20% of cells, replication of the incoming vRNA fails (Figures 2I and 2N), resulting in elimination of the viral infection. This number dramatically increased upon activation of the IFN signaling pathway (Figure 5C). In addition, we found that ∼40% of vRNAs did not undergo translation (Figure 1E). It is possible that vRNAs cycle between a translated and non-translated state. Alternatively, a subset of vRNAs may be defective in translation, which could also contribute to the failure of some virus particles in host cell infection. VIRIM will be a valuable tool to study the success rate of different steps in the life cycle of many different viruses, in different cell types, or in distinct cell states (e.g., stimulated with different signaling molecules).
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-dsRNA | English & Scientific Consulting | Cat #J2 |
| Mouse polyclonal anti-3Dpol | Oh et al., 2009 | N/A |
| Mouse monoclonal anti-tubulin | Sigma-Aldrich | Cat# T9026 |
| Rabbit polyclonal anti-eIF4GI | Bethyl Laboratories | Cat# A300-502A |
| Mouse monoclonal anti-mCherry | Invitrogen | Cat# MA5-32977 |
| Donkey polyclonal anti-mouse-Cy5 | Jackson ImmunoResearch | Cat# 715-175-151 |
| Goat polyclonal anti-mouse IRDye680 | LI-COR | Cat# 926-68070 |
| Goat polyclonal anti-rabbit IRDye800 | LI-COR | Cat# 926-32211 |
| Virus strains | ||
| CVB3 | Wessels et al., 2006 | N/A |
| EMCV | Duke and Palmenberg, 1989 | N/A |
| eGFP-CVB3 | Lanke et al., 2009 | N/A |
| SunTag-EMCV | This study | N/A |
| SunTag-CVB3 | This study | N/A |
| SunTag-CVB3Sabin-like1 | This study | N/A |
| SunTag-CVB3Sabin-like2 | This study | N/A |
| SunTag-CVB3Sabin-like3 | This study | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM | GIBCO | Cat# 31966021 |
| Leibovitz’s L15 medium | GIBCO | Cat# 21083-027 |
| Opti-MEM | Sigma-Aldrich | Cat# 11058-021 |
| Methionine-free DMEM | GIBCO | Cat# 21013024 |
| Advanced DMEM/F12 | Thermo Fisher scientific | Cat# 12634-010 |
| TryplE | Thermo Fisher scientific | Cat# 12605010 |
| Fetal Bovine Serum (FBS) | Sigma-Aldrich | Cat# F7524 |
| Penicillin-Streptomycin | GIBCO | Cat# 15140-122 |
| Glutamine | Sigma-Aldrich | Cat# G6392-1VL |
| Cysteine | Sigma-Aldrich | Cat# C7352-10MG |
| FuGENE 6 | Promega | Cat# E231A |
| Lipofectamine 2000 | ThermoFisher | Cat #11668019 |
| Lipofectamine RNAiMAX | Invitrogen | Cat# 13778-075 |
| Polyethylenimine | Polysciences Inc | Cat# 23966 |
| Polybrene | Santa Cruz Biotechnology, Inc | Cat# sc-134220 |
| Propidium Iodide | Sigma-Aldrich | Cat# P4170 |
| Doxycycline | Sigma-Aldrich | Cat# D9891-1G |
| Zeozin | Invitrogen | Cat# R25001 |
| Puromycin | ThermoFisher Scientific | Cat# 12122530 |
| Harringtonine | Cayman Chemical | Cat# 15361 |
| GPC-N114 | van der Linden et al., 2015 | N/A |
| Rupitrivir | Sigma-Aldrich | Cat# PZ0315 |
| Interferon α2 | Sigma-Aldrich | Cat# IF007 |
| scFv-sfGFP-StrepII | This study | N/A |
| Glucose oxidase | Sigma-Aldrich | Cat# G2133-10KU |
| Catalase | Sigma-Aldrich | Cat# C3515-10MG |
| Cultrex Basement Membrane Extract (BME), Growth Factor Reduced, Type 2 | R&D Systems, Bio-Techne | Cat# 3533-001-02 |
| Protease inhibitor cocktail | Roche | Cat# 11697498001 |
| Rho kinase inhibitor | Calbiochem | Cat# 555550 |
| TRIsure | Bioline | Cat# 38033 |
| Atto633-NHS | Atto-Tec | Cat# AD 633-31 |
| Cy5-azide | Lumiprobe | Cat# A3030 |
| Amino-11-ddUTP | Lumiprobe | Cat# 15040 |
| L-Homopropargylglycine | Sigma-Aldrich | Cat# 900893-100MG |
| Desthiobiotin | IBA Life Science | Cat# 2-1000-001 |
| Digitonine | Sigma-Aldrich | Cat# D141 |
| Paraformaldehyde | Aurion | Cat# 15710 |
| Formamide | ThermoFischer | Cat# AM9342 |
| Critical commercial assays | ||
| T7 RiboMax | Promega | Cat# P1320 |
| HiScribe | New England Biolabs | Cat# E2040S |
| Superscript III reverse transcriptase | Invitrogen | Cat# 18080093 |
| Tetro reverse transcriptase | Bioline | Cat# BIO-65050 |
| Terminal deoxynucleotidyl Transferase | ThermoFisher | Cat# EP0162 |
| iQ SYBR Green SuperMix | Bio-Rad | Cat# 1708885 |
| Nucleospin RNA | Macherey-Nagel | Cat# 740990.50 |
| Zymo RNA cleanup | ZymoResearch | Cat# R2061 |
| Deposited Data | ||
| Raw and analyzed RNA sequencing -/+ IFN | Gene expression omnibus https://www.ncbi.nlm.nih.gov/geo/ | GEO: GSE159280 |
| Raw data of imaging experiments | Mendeley data | https://doi.org/10.17632/9sxbk6cvn9.1 |
| Experimental Models: Cell Lines | ||
| U2OS cells | Tanenbaum lab | Cat# HTB-96 |
| A549 cells | Van Kuppeveld lab | Cat# CCL-185 |
| BHK-21 cells | Van Kuppeveld lab | Cat# CCL-10 |
| HeLa cells | Tanenbaum lab | Cat# CCL-2 |
| RPE1 cells | Tanenbaum lab | Cat# CRL-4000 |
| HEK293T cells | Tanenbaum lab & Van Kuppeveld lab | Cat# CRL-3216 |
| Oligonucleotides | ||
| See Table S2 for all sequences of smFISH probes, siRNAs, or qPCR oligos | This study | N/A |
| Recombinant DNA | ||
| Plasmids used in this study | Tanenbaum lab | https://www.tanenbaumlab.org/tools |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Micromanager | Micro-Manager 1.4.22 | https://micro-manager.org |
| NIS-Elements Imaging software | Nikon | https://www.microscope.healthcare.nikon.com/en_EU/products/software |
| Graphpad Prism 8 | GraphPad Software Inc | https://www.graphpad.com:443/scientific-software/prism/ |
| Other | ||
| StrepTactin Sepharose beads | IBA life sciences | Cat# 2-1201-002 |
| Zeba desalting column | VWR | Cat# GE17-0851-01 |
| 96-well glass bottom imaging plates-(Matriplates) | Brooks Life Science Systems | Cat# MGB096-1-2-LG-L |
Resource Availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Marvin Tanenbaum (M.Tanenbaum@hubrecht.eu).
Materials availability
The unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
The RNA sequencing data of this study has been deposited in the Gene Expression Omnibus (GEO) under accession code GEO: GSE159280. A selection of raw imaging data is made available through Mendeley data: https://doi.org/10.17632/9sxbk6cvn9.1.
Experimental Model and Subject Details
Cell lines
Human U2OS, HeLa, RPE1, HEK293T cells used for imaging, lentivirus production, and CVB3 production were grown in DMEM (4.5 g/L glucose, GIBCO) supplemented with 5% fetal bovine serum (Sigma-Aldrich) and 1% penicillin/streptomycin (GIBCO). Human A549 cells and Hamster BHK-21 for imaging and EMCV production were cultured in in DMEM (4.5 g/L glucose, GIBCO) supplemented with 10% fetal bovine serum (Sigma-Aldrich) and 1% penicillin/streptomycin (GIBCO). All cells were grown with 5% CO2 at 37°C. Cells were confirmed to be mycoplasma negative.
Intestinal organoids
Tissue from the human small intestine was obtained from the UMC Utrecht with informed consent of the patient. The patient was diagnosed with small intestinal cancer that was resected. A sample from non-transformed, normal mucosa was obtained for organoid culture used in the study. The study was approved by the UMC Utrecht ethical committee (Utrecht, the Netherlands) and was in accordance with the Declaration of Helsinki and according to Dutch law. This study is compliant with all relevant ethical regulations regarding research involving human participants. Human small intestinal cells were isolated, processed and cultured as described previously (Sato et al., 2011).
Method Details
Plasmids
The sequences of plasmids used in this study can be found via https://www.tanenbaumlab.org/tools. The eIF4G1 coding sequence was amplified from Addgene #45640.
Cell line generation
For generation of cell lines stably expressing transgenes, lentiviral transduction was used. To produce lentivirus, HEK293T cells were transfected using Polyethylenimine (PEI) with the lentiviral plasmid of interest and packaging vectors psPax and pMD2. The cell culture medium was refreshed 1 day after transfection. The supernatant containing the lentivirus was collected 3 days after transfection. To make stable cell lines, cells (U2OS, HeLa, or A549) were seeded at ∼35% confluency one day before infection. To infect cells, the viral supernatant was added to the cells along with Polybrene (10 mg/ml) (Santa Cruz Biotechnology Inc) and the cells were spin-infected for 90-120 minutes at 2000 rpm at 25°C. After spin-infection, the medium was refreshed and cells were cultured for minimally 2 days before further analysis was performed. To isolate cells that express the fluorescent transgenes, cells were FACS-sorted. To generate U2OS and HeLa cell lines expressing STAb in which all cells expressed the STAb at similar levels, single cells were sorted into 96-wells plates to generate monoclonal cell lines. To generate a cell line expressing nuclear BFP, a previously-generated monoclonal cell line stably expressing TetR, STAb, and PP7-2xmCherry-CAAX (Yan et al., 2016; hereafter referred to as STAb U2OS cells) was infected with lentivirus encoding nuclear BFP. A polyclonal population of BFP-positive cells was sorted that expressed BFP at similar levels. To generate an A549 cell line expressing STAb, cells were infected with the STAb lentivirus and a polyclonal population of GFP positive cells was sorted. Cells that had similar GFP expression levels as those in the STAb-U2OS cells were selected.
To generate a cell line expressing the eIF4G cleavage reporter, we could not use lentiviral transduction, as the eIF4G cleavage reporter is too large for efficient lentivirus production. Instead, the cell line expressing the eIF4G cleavage reporter was generated by transfecting (Fugene 6; Promega) the STAb-U2OS cells with a plasmid encoding the eIF4G cleavage reporter. One day after the transfection, the medium was refreshed and selection for stable integration of the eIF4G cleavage reporter was initiated (0.4 mg/ml Zeocin (Invitrogen)). Selection was performed for 14 days during which the Zeocin-containing medium was refreshed every 5 days. Note that eIF4G cleavage reporter was expressed from an inducible promoter to prevent possible toxicity due to long-term expression. Expression of the eIF4G cleavage reporter was induced by incubating cells with doxycycline (1 μg/ml; Sigma-Aldrich) for 12-24 hr before an experiment.
Generation of STAb intestinal organoids
For generation of intestinal organoids stably expressing STAb, lentiviral transduction was used, as described previously (Koo et al., 2011). To generate a polyclonal population of STAb expressing intestinal cells, organoids were dissociated by incubating 5 min with TrypLE (TryplE Express; Life Technologies) and GFP positive cells were FACS-sorted. Only cells with GFP fluorescence comparable to STAb U2OS were sorted. After sorting, cells were cultured for three days in the presence of a Rho kinase inhibitor (Rho kinase inhibitor, 10μM, Calbiochem) before further analysis was performed.
CVB3 and EMCV design and production
The CVB3 used in this study was derived from the pRibCVB3/T7 plasmid, which contains the cDNA of CVB3 strain Nancy driven by a T7 RNA polymerase promoter (Wessels et al., 2006). The EMCV used in this study was derived from the pM16.1 plasmid (Duke and Palmenberg, 1989). Mutations in the IRES were introduced by site-directed mutagenesis of the pRIB infectious clone. To make SunTag-CVB3 or SunTag-EMCV, 5 copies of the SunTag-coding sequence were introduced in-frame upstream of the viral coding region, at a location that was previously successfully used for insertions in recombinant CVB3 (Lanke et al., 2009); see Figure S1A and plasmid sequence). A cleavage site for the viral 3C protease was included between the SunTag and the rest of the viral polyprotein (ALFQG for CVB3, VFETQG for EMCV) to enable removal of SunTag from the viral protein VP4 (CVB3) or L protein (EMCV) and to prevent possible SunTag interference during virus particle assembly.
Virus stocks were made as described previously (Lanke et al., 2009; Wessels et al., 2006). In brief, the infectious clones were linearized using the MluI (CVB3) or BamHI (EMCV) restriction enzymes and used as template for in vitro transcription (T7 RiboMAX; Promega or HiScribe; New England Biology). RNA was purified (Nucleospin RNA; Machery-Nagel or ZymoResearch) and transfected into HEK293T or BHK-21 cells using Lipofectamine 2000 (ThermoFisher). After 2-3 days, when complete CPE (cytopathic effect) was observed, cells and supernatant were collected and freeze-thawed three times. The supernatant containing CVB3 was aliquoted and stored at −80°C. Virus titers were determined by endpoint titration, as described previously (Reed and Munech, 1938).
To confirm that recombinant viruses contained the correct inserts, viral RNA was isolated from the virus stocks (Nucleospin viral RNA; Machery-Nagel) and cDNA was synthesized using Superscript III reverse transcriptase (Invitrogen) according to manufacturer’s protocol. Next, the region of interest was amplified by PCR (see Figure S1A), and the correct size of the PCR product was verified by agarose gel electrophoresis and the sequence of the SunTag insert and IRES mutations were verified by Sanger sequencing.
siRNA transfections
All siRNAs used in this study were ordered as SMARTpool ONTargetPlus reagents from Dharmacon, except for: siXrn1 (AGAUGAACUUACCGUAGAAUU; Hoek et al., 2019), siDs3L (CCAUGUAACCGUAAGAAUA; Staals et al., 2010); siRNASEL_#2 (SantaCruz sc-45966), siIFIT1_#2 (Ambion s7150), siIFIT1_#3 (CCAGACAAUGGAUAUAUUAAG; John et al., 2018). STAb U2OS cells or NLS-BFP STAb U2OS cells were reverse-transfected with siRNAs at a final concentration of 10 nM siRNA using RNAiMAX transfection reagent (Invitrogen) and seeded in 48-well plates. After 24-48 hr, the cells were trypsinized and re-plated on either glass or plastic plates for imaging or for qPCR analysis, respectively. One day after re-plating, cells were imaged or harvested for qPCR. Where indicated, cells were treated with IFN (1000 U/ml IFNα2 (Sigma)) approximately 24 hr before analysis.
Quantitative RT-PCR (qPCR)
Virus replication
To compare replication kinetics between CVB3, GFP-CVB3, and SunTag-CVB3 viruses in either HeLa, U2OS, or STAb U2OS cells, the amount of viral RNA was determined over time after infection. Cells were plated in 384-well plate 1 day before infecting with the indicated recombinant viruses at an MOI (multiplicity of infection) of 1. 1 Hr after inoculation, the virus-containing medium was removed, and cells were washed 3 times with PBS. At the indicated time points, cells were harvested and RNA was extracted using Nucleospin RNA (Machery-Nagel) according to manufactor’s guidelines. qPCR was used to assess viral load over time.
siRNA knockdown efficiency
To determine the knock-down efficiency of siRNAs, siRNA-treated STAb cells were harvested and RNA was isolated using TRIsure (Bioline). Next, cDNA was synthesized using Random hexamers and Tetro Reverse Transcriptase (Bioline). qPCRs were performed using SYBR-Green Supermix (Bio-Rad) on a Bio-Rad Real-time PCR machines (CFX Connect Real-Time PCR Detection System). mRNA levels were determined by qPCR. If the Cq (quantitation cycle) of a sample was higher than the Cq of a water control, the sample was excluded from analysis. See Table S2 for sequence details of oligonucleotides used for qPCR. All RNA levels were normalized to GAPDH mRNA levels.
smFISH
Labeling of smFISH probes
Single-molecule Fluorescence In Situ Hybridization (smFISH) was performed as described previously (Lyubimova et al., 2013; Raj et al., 2008). Custom-made oligonucleotide probes (96) targeting the positive RNA strand of CVB3 were designed using the website https://www.biosearchtech.com/ (See Table S2 for sequence of the probes). Probes were labeled with Atto633-NHS (Atto-Tec), as described previously (Gaspar et al., 2018). In brief, Atto633-NHS was dissolved in DMSO and mixed with NaHCO3 (final concentration 0.05 M; pH 8.4) and Amino-11-ddUTP (5 mM; Lumiprobe). The oligonucleotides were labeled by mixing 200 μM of each oligo with Terminal deoxynucleotidyl Transferase (TdT) buffer, 10 mM dye solution, and TdT (ThermoFischer) and incubating at 37þC overnight. Next, the labeled probes were precipitated using 100% ethanol, washed with 80% ethanol to remove free dye, and resuspended in nuclease-free water to a final concentration of 30 μM. Prior to hybridization, probes were diluted in TE to a working stock concentration of 1 μM.
Probe hybridization
STAb cells were plated on glass and incubated with SunTag-CVB3 as described in the section ‘Live-cell microscopy; cell culture before imaging’. Two additional PBS washing steps were performed after virus incubation to remove SunTag-CVB3 adhering to the outside of cells. 2-4 Hr after inoculation with virus, live-cell imaging was terminated and cells were washed with PBS and fixed with 3.7% paraformaldehyde in PBS for 5 minutes at room temperature. After fixation, cells were washed twice with PBS and permeabilized with 100% ice-cold ethanol at 4°C for 30 minutes, cells were then washed twice for 15 minutes with smFISH wash buffer (2X SSC, 10 % formamide in DEPC-treated water). smFISH probes were incubated in hybridization buffer (10 nM probe concentration; 1% dextran sulfate, 2x SSC, 10% formamide in DEPC-treated water) at 37°C. After overnight incubation with smFISH probes, cells were washed twice for 1 hr at 37°C and for 15 min at room temperature with smFISH wash buffer at room temperature. The wash buffer was replaced with imaging buffer (10 mM Tris, pH 8; 2x SCC; 0.63 % glucose, supplemented with glucose oxidase (Sigma) and catalase (Sigma)) and samples were stored at 4°C until imaging.
Combined smFISH and immunofluorescence
To combine smFISH and immunofluorescence, only a single wash was performed after hybridization of the smFISH probes. Then, the immunofluorescence staining was performed as described in the section ‘Immunofluorescence of dsRNA and 3Dpolymerase’, starting with blocking until washing away the secondary antibody. Then, a final wash step using smFISH wash buffer was performed and the wash buffer was replaced with imaging buffer and stored at 4°C until imaging.
Immunofluorescence of dsRNA and 3Dpolymerase
Cells infected with SunTag-CVB3 virus were first followed by time-lapse microscopy for 3-7 hr. and then cells were fixed with 3.7% paraformaldehyde for 30 minutes and permeabilized using PBS + 0.1% Triton X-100 for 5 minutes. Blocking was performed using PBS + 2% BSA + 50 mM NH4Cl for 45 minutes. Anti-dsRNA antibody (J2, English & Scientific Consulting) or anti-3Dpolymerase (Oh et al., 2009) diluted 1:1000 in blocking buffer was incubated for 45 minutes. After incubation with the primary antibody, three wash steps with block buffer were performed. Donkey anti-mouse Cy5 (Jackson lab) was used as a secondary antibody (1:200 in blocking buffer) and incubated for 45 minutes. After washing away the secondary antibody once with PBS, samples were kept in PBS at 4°C, until imaging.
Immunofluorescence with STAb
Purification of STAb
To purify STAb, RPE1 cells stably expressing STAb-StrepII were harvested, resuspended in lysis buffer (50 mM HEPES/KOH, pH 7.4, 200 mM KCl, 0.5 % Triton X-100, 1 mM PMSF, and EDTA free protease inhibitor cocktail (Sigma Aldrich)) on ice, and sonicated. To remove cell debris, the sample was centrifuged at 20000 g for 1 hr at 4°C and the supernatant was collected. To purify STAb-StrepII, the supernatant was incubated for 1 hr at 4°C with StrepTactin Sepharose beads (IBA life sciences) that were pre-equilibrated with wash buffer (20 mM HEPES/KOH, pH 7.4, 200 mM KCl) and HEPES/KOH, 200 mM KCl, 0.5 % Triton X-100. After washing with wash buffer, the STAb-StrepII was eluted from the beads using elution buffer (20 mM HEPES/KOH, pH 7.4, 200 mM KCl, 5 mM desthiobiotin (IBA life Sciences)). The eluate was buffer exchanged using a Zeba desalting column (ThermoFisher) into storage buffer (20 mM HEPES/KOH, 200 mM KCl, 5 % glycerol). The purified protein was stored at −80°C.
STAb staining
Immunofluorescence staining of SunTag using purified STAb was combined with smFISH as described in the section ‘Combined smFISH and immunofluorescence’, using 1:100 STAb dilution in blocking buffer. As the purified STAb is already fluorescently labeled, no secondary antibody incubation step is required. Instead, immunofluorescence staining of SunTag was finalized by six wash steps with blocking buffer after primary antibody incubation.
Analysis of global translation efficiency
Global translation efficiency was determined by labeling newly-synthesized proteins using a methionine analog, followed by fluorescence labeling of the methionine analog based on an adapted protocol of a previously established assay (Estell et al., 2017). NLS-BFP STAb cells were seeded in 96-well glass-bottom plates. To deplete methionine, regular cell culture medium was replaced with methionine-free medium ((Met)-free DMEM (GIBCO) supplemented with 2 mM glutamine (Sigma Aldrich), 0.02 mg/ml cysteine (Sigma Aldrich), 5% fetal bovine serum (Sigma Aldrich) and 1% penicillin/streptomycin (GIBCO)). SunTag-CVB3 was then added to cells at indicated time-points and the methionine analog, L-Homopropargylglycine (HPG, 1 μM, Sigma) was added to cells 15 minutes before fixation. Several controls were taken along in these experiments: 1) cells that were not incubated with virus, 2) cells that were not treated with the HPG, or 3) cells that were treated with translation inhibitor puromycin (ThermoFisher Scientific; 0.1 mg/ml) starting 1 minute before incubation with HPG. HPG was washed away with ice-cold wash buffer (10mM HEPES/KOH pH7.4; 10mM NaCl; 5mM MgCl2; 300mM sucrose) and cells were fixed in wash buffer with 3.7% PFA for 30 minutes at room temperature. After fixation, the cells were permeabilized with 0.015% digitonine (Sigma) in wash buffer for 5 minutes on ice. A click reaction was performed to fluorescently label HPG with Cy5 in freshly prepared click reaction buffer (100 mM Tris pH 8.8; 100 mM ascorbic acid; 1 mM CuSO4; 10 μM Cy5-azide (Lumiprobe)) for 15 minutes at room temperature. The click reaction buffer was washed away 5 times and replaced with PBS and samples were stored on ice, until imaging (which occurred within 12 hr after finalizing the click reaction). Note that the protocol described above results in nuclear background fluorescence (Figure S5A), which was not observed in a previous study, as in the earlier study permeabilization is performed prior to fixation (Estell et al., 2017). To prevent loss of cytoplasmic signal during permeabilization before fixation, cells were fixed prior to permeabilization in our experiments.
Western blot
STAb cells with inducible expression of the eIF4G cleavage reporter were seeded at 75% confluency in 6 cm dishes. One day after seeding, wild-type CVB3 virus (MOI = 2) was added to each well. At the indicated time points post infection, cells were released by trypsin treatment, and subsequently lysed on ice in lysis buffer (40 mM Tris-HCl pH7.4, 150 mM NaCl, 10 mM EDTA, 1% NP-40, protease inhibitors [Roche]) for 30 min. For each sample, 100 μg of total protein content in 1xLSB was heated to 95°C for 5 min, separated on a 7.5 % SDS-PAGE gel, and transferred to nitrocellulose membranes by wet transfer. Membranes were blocked in block buffer (PBS + 0.1% Tween-20 + 2% BSA) for 45 min, and then probed using primary antibodies mouse anti-tubulin (Sigma-Aldrich), rabbit anti-eIF4GI (Bethyl Laboratories), and mouse anti-mCherry (Invitrogen), used at 1:1000 dilution in block buffer for 1 hr. After three wash steps in wash buffer (PBS + 0.1% Tween-20), membranes were incubated in secondary antibodies goat anti-mouse IRDye680 (LI-COR) and goat anti-rabbit IRDye800 (LI-COR), used at 1:10,000 dilution in block buffer for 45 min. Membranes were washed twice in wash buffer and membranes were imaged using the Odyssey Imager (LI-COR).
RNA sequencing
STAb cells were seeded at 20% confluency in a 12-wells plate. One day after seeding, cells were incubated with 1000 U/ml IFN for 24h. As control, cells without IFN treatment were processed in parallel. Cells were harvested and RNA was isolated using TRISure (Bioline), according to manufacturer’s guidelines. Further processing and sequencing was performed by Single Cell Discoveries, based on the CEL-seq2 protocol (Hashimshony et al., 2016; Muraro et al., 2016).
Live-cell microscopy
Microscopes
Imaging experiments were performed using either a 1) Nikon TI inverted microscope with NIS Element Software (Nikon), equipped with a perfect focus system, a Yokagawa CSU-X1 spinning disc, an iXon Ultra 897 EM-CCD camera (Andor), or 2) a Nikon TI2 inverted microscope with NIS Element Software, equipped with a perfect focus system, a Yokagawa CSU-X1 spinning disc and an Prime 95B sCMOS camera (Photometrics). Both microscopes were equipped with a temperature-controlled hood. For experiments involving long-term time-lapse analysis (> 3 hr) a 60x 1.40 NA oil-immersion objective was used, while a 100x 1.49 NA oil-immersion objective was used for short-term analyses. A 100x 1.49 NA oil-immersion objective was used for experiments using 2D organoid cells. The siRNA screen (Figure 6B, 6C, S6B, S6H, S6J, and S6O) was performed using a 40x 0.95 NA air objective.
Image acquisition
For long-term (> 3 hr) time-lapse imaging to determine infection phase durations, x,y positions for imaging were selected randomly. Images were acquired every 1, 2, 3, or 5 minutes for 3-12 hours, using 50-70 ms exposure times. Multiple Z-slices (∼10-15 planes with 0.8 μm steps) were imaged for GFP to ensure that the entire cell was imaged. If relevant, a single Z-slice in the middle of the cell was also imaged for BFP and/or mCherry. To determine cell survival, images were acquired every 10 minutes for 36 hr, using similar laser and Z-slice settings. To quantify success-rate of infection after siRNA-mediated knockdown of IFN-induced genes, a large-image comprised of 6-8 to 6-8 regular field-of-views (FOVs) was generated for each condition. Every large-image was acquired every 15-30minutes for 10-12h. Images of one Z-plane were acquired for BFP, and mCherry and 4 Z-slices (1.5 μm steps) were imaged for GFP.
For short-term live-cell imaging, x,y positions for imaging were selected based on the presence of translating vRNAs. For harringtonine run-off experiments, cells were selected based on the number of translating vRNAs (either 1 or 20-50) per cell and images were acquired every 10 s for 20-30 minutes, using 50 ms exposure and 10-15 Z-slices with 0.8 μm steps. To determine the intensity of translating vRNAs, images were acquired for a single time-point using 50-100 ms exposure times. Multiple Z-slices (12-20 with 0.5 μm steps) were imaged for GFP to ensure that the entire cell was imaged. Compared to the long-term imaging experiments, 2-fold higher GFP laser power was used.
To determine diffusion speeds of translating vRNAs, cells were selected based on the number of translating vRNAs. A single GFP Z-slice was acquired in the middle of cells, using maximal laser power with 20 ms exposure for 100 frames at ∼40 ms interval (Figure S1M). In a separate assay (Figures 1J and 1K), the diffusion speed of translating vRNAs during infection phase1 was measured in randomly-selected cells and imaged every 1 minute for 4 hr using using 50-70 ms exposure times. Multiple Z-slices (12 planes with 0.8 μm steps) were imaged for GFP and a single Z-slice was imaged for BFP and mCherry. To determine the diffusion speed of translating cellular mRNAs, cells transfected with an mRNA reporter plasmid were selected that contained less than 10 translating reporter mRNAs per cells. Images of 10-15 Z-slices with 0.5 μm steps were acquired every 10 s using 50 ms exposure time.
Cell culture for imaging
Unless noted otherwise, live-cell imaging was performed by seeding STAb cells (U2OS, HeLa, or A549) or NLS-BFP STAb cells 1 day before imaging in a 96-well glass-bottom plate (Matriplates, Brooks Life Science Systems) at ∼40%–45% confluency. An MOI of ∼0.25 was used for infection using SunTag-CVB3 virus and cells were incubated with virus-containing medium for ∼30 minutes. After incubation with virus-containing medium, the virus-containing medium was replaced with imaging medium (pre-warmed CO2-independent Leibovitz’s-15 medium (GIBCO) containing 5% fetal bovine serum (Sigma-Aldrich) and 1% penicillin/streptomycin (GIBCO)). Live-cell imaging was initiated 15-30 minutes after removal of virus-containing medium.
For experiments in which the diffusion of translating mRNA molecules was assessed, U2OS cells stably expressing TetR, and STAb were seeded at 45% confluency and reverse transfected with a plasmid expressing the reporter mRNAs using Fugene (Promega) 1 day before imaging. For experiments in which the GFP fluorescence intensity of individual 24xSunTag arrays was measure, STAb U2OS cells were seeded at 45% confluency and reverse transfected using Fugene (Promege) 1 day before imaging with a plasmid encoding the 24xSunTag-CAAX protein. One hour before the start of imaging, the cell culture medium was replaced with imaging medium. All live-cell imaging experiments were performed at 37°C.
2D-culture of organoid cells for live-cell imaging
To enable imaging of single translating vRNAs in intestinal organoid cells, organoid cells were cultured in 2D. 96-well glass-bottom plates (Matriplates, Brooks Life Science Systems) were coated for 30 minutes at 37°C with 100 μL coating mix (5% Basement Membrane Extract (Type 2, Cultrex reduced growth factor, R&D systems) dissolved in Advanced DMEM (Thermofisher Scientific)). The coating mix was removed and the wells were dried for 15 minutes at 37°C. Organoids grown in 3D were dissociated into single cells by incubating the organoids for 5 minutes with TrypLE (TryplE Express; Life Technologies). Approximately 5000 cells were plated per well in expansion medium (see Sato et al., 2011). Virus incubation was performed as described in the section ‘Cell culture for imaging’. Expansion medium was replaced with imaging medium (pre-warmed CO2-independent Leibovitz’s-15 medium (GIBCO) containing 5% fetal bovine serum (Sigma-Aldrich) and 1% penicillin/streptomycin (GIBCO)) 15-30 minutes before the start of live-cell imaging.
Drug treatment
In experiments in which cells were treated with IFN, 1000 U/ml IFNα2 (Sigma) was added approximately 24 hr before imaging. Virus-containing medium and imaging medium did not contain IFN. Where indicated, cells were treated with the inhibitors 3Ci: Rupitrivir (Sigma, 10 μM) or 3Di: GPC-N114; van der Linden et al., 2015; 10 μM) 30-60 minutes before addition of virus-containing medium and treatment of these drugs continued during incubation with virus-containing medium and during live-cell imaging.
To determine cell survival during time-lapse imaging, Propidium Iodide (PI, 20 μg/ml; Sigma) was added to the imaging medium.
To investigate viral translation dynamics, translation inhibitors puromycin (0.1 mg/ml; ThermoFischer Scientific) or harringtonine (3 μg/ml; Cayman Chemical) were added to the imaging medium at indicated time-points.
Image acquisition of fixed cells
Unless noted otherwise, imaging of fixed cells was performed using the 60x 1.40 NA objective. The x,y positions for imaging were selected either randomly or, in the cases where smFISH and/or immunofluorescence were performed after live-cell imaging, the same x,y positions were used as those imaged during live-cell imaging. Images were acquired using 70 ms exposure. For GFP, Cy5, and Atto633, 15-20 Z-slices with 0.5 μm steps were acquired. For cells expressing NLS-BFP, a single Z-slice focused in the middle of the cell was imaged for BFP.
The 100x 1.49 NA objective was used to image fixed cells in which the methionine analog was stained. The x,y positions were selected randomly. Large-images of 6 to 6 (control conditions) or 9 to 9 (all other conditions) regular FOVs were generated. For BFP, GFP, and Cy5, images of 10-15 Z-slices with 0.8 μm steps were acquired using 100 ms exposure.
Quantification and Statistical Analysis
Post-acquisition processing of microscopy data
For all acquired Z-slices, maximal intensity projections were generated using NIS elements software. All downstream analyses were performed on the maximal intensity projections. For experiments in which fluorescence intensities were measured, correction for photobleaching was performed, unless noted otherwise, for each color individually, using the ImageJ plugin Bleach correction, using the Exponential fit option. In brief, the fluorescence intensity over time of the entire FOV was determined and fit with an exponential decay distribution. The bleaching rate was then calculated based on the exponential decay fit and was used to correct the fluorescence intensities of images at all time-points.
Counting the number of translating vRNAs per cell
In live-cell imaging experiments in which the duration of infection phases was determined, cells were selected for analysis based on the following criteria:
-
1)
Only cells that were completely in the FOV for the entire duration of the analysis were included.
-
2)
Maximally 5 cells per FOV were analyzed.
-
3)
Cells with multiple translating vRNAs in the first time point of the movie were excluded from analysis.
-
4)
If a cell undergoes mitosis during the movie, the cell is excluded from the analysis.
The number of translating vRNAs per cell was determined manually for each time point, starting from the first time point that a translating vRNA could be observed, until either individual vRNAs could no longer be distinguished anymore due to the large number of vRNAs in the cell or until the number of translating vRNAs exceeded 50 vRNAs/cell. All bright GFP spots were considered individual translating vRNAs. However, the following additional criteria were taken into account during counting of translating vRNAs:
-
1)
Translating vRNAs are highly mobile (Figures 1K and S1M) and a single vRNAs can therefore often be observed in multiple slices of a Z stack at different x,y coordinates, resulting in two spots close together in the maximal intensity projection. To prevent double counting of the same translating vRNA in the maximal projection, two spots positioned very close together were counted as a single translating vRNA.
-
2)
In many STAb-expressing cells, we also observed one or two distinct GFP foci adjacent to the nucleus, most likely indicating that the STAb bound with weak affinity to the centrosomes. These foci were independent of virus infection, as similar foci could also be observed in uninfected cells. These foci were excluded from all analyses.
-
3)
Some cells contain GFP foci that were larger and far less mobile than translating vRNAs. These foci were observed in both infected and uninfected cells, and thus represent background foci. These background foci often co-localized with mCherry positive foci, indicative of autofluorescence. A combination of morphology, mobility, and dual positivity with mCherry was used to exclude these background foci during counting of translating vRNAs.
Of note, in experiments in which the GFP fluorescence intensity of translating vRNAs were measured, images were acquired for only a single time-point. The mobility of background foci could therefore not be taken into account to exclude background foci from analysis. However, we found that the morphology, size, mCherry fluorescence co-localization and subcellular location of such background foci was still sufficient to reliably distinguish the background foci from translating vRNAs.
Infection phases
Annotation of infection phases
Based on the number of translating vRNAs over time, infection phases 1 to 5 were called. Phase1 is defined as the period from the first detection of a translating vRNA until the disappearance of the translating vRNAs; phase2 is the period from disappearance of the first translating vRNA until the appearance of multiple translating vRNAs. The period during which the number of translating vRNAs increased after phase2 is referred to as phase3. Once the number of vRNAs plateaus, phase4 is called. Then, a second increase in the number of translating vRNAs is observed, which is referred to as phase5.
Of note, in some cases we observed a single translating vRNAs, which disappeared after some time, followed by the re-appearance of a single translating vRNA (rather than the appearance of multiple vRNAs, characteristic of phase2 followed by phase3). Such disappearance and reappearance of a single translating vRNA could be observed multiple times in rare cases (called phase1 pulses and breaks; see for example Figures 3B and S3C). In these cases, phase1 is defined as the entire period from the first detection of a translating vRNA until the end of the last phase1 pulse (i.e., the last pulse before phase2), as indicated with an arrow in Figures 3B and S3C. In analyses specifically focusing on these phase1 pulses, the timing of individual phase1 pulses and breaks was analyzed.
Since a small amount of noise (i.e., variations in the number of detected foci between adjacent time-points) was observed in the calling of the number of translating vRNAs per time-point (likely due to imperfect detection of vRNAs), the following additional criteria were taken into account for phase annotation:
-
1)
Phase1, phase1 pulses, phase1 breaks, and phase2 were each called only if their duration was at least 12.5 minutes. Depending on the timing interval, 3-12 consecutive time points are needed to fulfill the 12.5 minutes requirement. A gap in phase 1 (i.e., one or multiple consecutive time-points without detection of a translating vRNA) could be the result of noise in counting of translating vRNAs or a phase1 break. To test whether a 12.5 minutes threshold is sufficient to distinguish ‘counting noise’ from true biological changes in the infection phase, we analyzed the frequency and the number of consecutive time-points without a translating vRNA during phase1. For each cell, the frequency and duration of gaps of less than 12.5 minutes was determined (Figure S3E). The majority of gaps consisted of a single time-point, indicating noise in translating vRNA counting. Gaps of 12.5 minutes or longer due to noise would not be expected based on the distributions of gap duration, but instead reflect a biological change in the number of GFP foci. A threshold of 12.5 minutes can thus be used to adequately annotate phase1, phase2, phase1 pulses, and phase1 breaks.
-
2)
A moving average of the number of translating vRNAs over time was used to approximate both the end of phase3 and the start of phase5. Then, the raw data on the number of translating vRNAs over time was used to determine the exact first or last time-point. The end of phase3 is the last time-point that an increase relative to the previous time-point was observed. The start of phase5 is the first time-point that an increase relative to the previous time-point was observed.
-
3)
Phase4 consists of the time-points between the last time-point of phase3 and the first time-point of phase5.
Analysis of infection phases
For each infected cell, the duration of each infection phase was determined. Additionally, we determined whether the infection phase was complete (i.e., the start and end of an infection phase were observed during the movie). The distributions of infection phase durations are plotted as Kaplan-Meier graphs, which allows inclusion of both complete and incomplete infection phases (for example Figures 2H–2K). In these Kaplan-Meier graphs, the timing of an incomplete infection phase is plotted as a circle. For phase 3, 4, and 5 additional infection phase parameters were determined; the number of translating vRNAs per cell over time was plotted, and, for phases3 and 5, linear regression analysis of performed to determine the rate of vRNA synthesis. The mean number of translating vRNAs during phase4 was calculated as the average of all time points during phase4.
Independent repeats
Analyses of the durations infection phases were performed based on many (2-21) independent experiments. However, the number of cells per experiment varied greatly. Therefore, we did not display the mean values of the individual repeats, as experiments with a low number of analyzed cells make an inappropriately large contribution to this mean value. Instead, all analyzed cells were combined into one Kaplan-Meier graph. To verify that individual cells from individual repeats could be combined into one dataset, we confirmed the reproducibility of the infection phases by comparing the combined datasets to individual repeats. As some repeats contained a low number of analyzed cells, some repeats were combined into datasets of at least 30 cells (Figures S2A–S2D).
Analysis of cells with multiple infections
Unless stated otherwise, experiments were performed using a low MOI (0.25) to ensure that single viruses infected individual cells. However, even at a low MOI, double infections (i.e., two viruses infecting a single cell) were observed in a small number of cells (∼10.6 %). A double infection was called when a single cell contained two GFP foci for at least 12.5 min. To simplify analysis of infection phases, double infected cells were excluded from all analyses, except from the experiments using a high (1.25) MOI (Figures 2Q, S3M, and S3N). For high MOI experiments double infections could be observed in many cells. To determine the duration between initial infection and translation of newly synthesized vRNAs, the time between first detection of a translating vRNA and rapid increase in translating vRNAs was determined (arrows in Figure S3N). This time is referred to as combined duration of phase1+2.
Analysis to exclude that phase1 pulses are a result of double infections
Even though most of our experiments were performed using a low MOI (0.25), limiting the fraction of cells with double infections, in some infected cells we did observe double infections (i.e., multiple translating vRNAs present simultaneously during phase1; also see section ‘Analysis of cells with multiple infections’). In addition to double infected cells, phase1 pulses were observed in some infected cells (Figures 3C and S3C). In theory, two translation pulses in a single cell could represent either a single vRNA turning translation off and on again, or pulses could be the result of two sequential infections in a single cell. To examine whether phase1 pulses are likely to be due to double infections, we calculated the fraction of cells expected to be infected by multiple viruses (Ellis and Delbrück, 1939), the fraction of cells observed to be infected by multiple viruses, and the fraction of cells with translation pulses. If for example, 10% of cells are expected to contain double infections, but no double infections are called and 10% of infections are called as containing translation pulses, it is likely that such pulses in fact represent double infections. In contrast, if 10% of cells are expected to contain double infection and 10% of infections are called as double infections, while in addition another 10% of infections show pulses, we conclude that such pulses likely do not represent double infections, as we conclude that we are calling the expected number of double infections, and can thus accurately identify double infections.
The expected number of double infections can be calculated using the following equation (Ellis and Delbrück, 1939):
P(n) is the expected fraction of cells infected by n viruses using an MOI of m. With an MOI of 0.25, 77.9%, 19.5% and 2.6% of all cells is expected to be uninfected, infected by a single virus, or infected by > 1 virus respectively. Thus, 11.7% ( = 2.6/(19.5+2.6)) of all infected cells is expected to be infected by > 1 virus (Figure S4A). We called double infections in 10.6% of the infected cells (Figure S4A), a value very similar to the expected value. In addition, we called translation pulses in 11.4% of cells. This analysis thus strongly suggests that double infections are not primarily responsible for the observed translation pulses.
Correlation between durations of different infection phases
To investigate whether successful replication during phase2 (i.e., transition from phase2 to phase3) correlated with the duration of phase1 (Figure 3A), all infected cells were subdivided based on the duration of phase2. Cells with successful replication were defined as cells in which phase2 is equal to or less than 60 minutes. If the end of phase2 was not observed, the infection was categorized as unsuccessful replication. We excluded cells for which we could not determine whether phase2 was successful; for example, if phase2 was incomplete and shorter than 60 minutes (for example, due to the end of the movie).
For analyses in which the duration of phase1+2 was compared to the duration of phase3+4 (Figure 2Q), only cells were included in which both relevant infectious phases were completely observed (i.e., start and end observed). As a result, cells for which the end of phase2 was not observed (i.e., mostly cells with unsuccessful replication) are excluded from this analysis. The comparison between the combined duration of phase1+2 and phase3+4 was made based on the median of the distributions, instead of the average of the distributions, as the phase1+2 duration has a long tail toward longer times. This long tail is the result of the larger heterogeneity in phase1 duration (Figure 2Q). During phase1+2, there is only a single vRNA that can undergo replication, so a delay for that vRNA molecule causes an increased translation+replication time. In contrast, during phase3+4 multiple newly synthesized vRNAs are translated and replicated. If only a single one of those vRNAs replicates successfully, new vRNAs are observed and phase3+4 is completed (start of phase5 is defined as the appearance of additional GFP foci). Thus, even if one vRNA is delayed in replication, another vRNA can still replication resulting in the completion of phase3+4. As a result, the shape of the distribution of phase3+4 duration is narrower compared to phase1+2 duration. Since we wanted to examine the duration of translation+replication of a ‘typical’ vRNA in phase1+2 with a vRNA in phase3+4, we chose the median in this analysis to prevent artificial skewing of the value by the outliers present only when a single vRNA is present in the cell.
Quantifying the number of smFISH foci per cell
Single vRNAs
To determine the number of vRNAs in single infected cells (Figures 1E, 1I, 2E, S1F, S1G, and S2D), the number of CVB3 smFISH spots in infected cells was determined. Loss of nuclear NLS-BFP was used as a readout of cell infection. Spots with an intensity of over 2.5-fold of the median intensity of all spots were excluded from the analyses, as they represent sites of replication and do not represent single vRNAs (See section ‘Quantifying the number of smFISH foci per cell-Replicating vRNAs’). For each replicate experiment the average number of CVB3 smFISH spots in uninfected cells (i.e., cells without loss of nuclear NLS-BFP) was determined (range 2 – 7 spots; see Figure S1G) and this number was subtracted from the spot count of infected cells. Note that these ‘background foci’ are not observed if no virus incubation step was performed (Figure S1F). These background foci therefore likely represent (intact) CVB3 particles adhering to the outside of the cell or in endosomes.
Replicating vRNAs
Based on the fluorescence intensity of smFISH foci, two types of smFISH foci were defined: 1) single vRNAs with a relatively low and homogeneous distribution in fluorescence intensity, and 2) replicating vRNAs with > 2.5x the fluorescence intensity of single vRNAs (Figure S2D). The fluorescence intensity of single vRNA spots and replicating vRNAs was normalized to the mean of three smFISH foci in each cell.
Calculating mobility of translating RNAs
For analysis of the mobility of translating RNAs (either vRNAs or mRNAs), the ImageJ plugin TrackMate (v3.7.0) was used. Manual Tracking was used to generate tracks of single RNAs in Figures 1J and 1K. Based on these tracks, the mean square displacement (MSD) was calculated for each RNA.
In a separate experiment, the mobility of translating vRNAs was determined in cells with more than one translating vRNA (Figure S1M). The tracks were generated semi-automatically, using the LoG detector option of TrackMate. Spots were identified after applying a Gaussian filter and the intensity threshold was set manually. Next, tracks were generated using the LAP tracker option, allowing a maximal linking distance of 1 μm and up to 5 frame gaps in the track. All generated tracks were manually inspected. Tracks with less than 10 time-points were excluded from further analysis. Finally, the diffusion coefficient of each trace was calculated by fitting the first 10 time-points of the MSD curve.
Determining the fraction of translating immobilized vRNAs
To classify translating vRNAs based on their mobility, we visually determined whether a translating vRNA was mobile or immobile in 5 consecutive time points with 2 min interval. If a translating vRNA was visible in each time point at the same location, the vRNA was classified as immobile. The fraction of immobile vRNAs per cell was then calculated based on the number of immobilized sites and the total number of vRNAs for each cell as determined by the mean number of vRNAs counted over the 5 time points. If no immobilized vRNAs were observed, the fraction of immobilized vRNAs in that cell was classified as ‘0’. All cells, with and without immobilized vRNAs, were combined to calculate the mean fraction of translating immobilized vRNAs (Figure S1O).
Translating vRNA intensity and vRNA translation efficiency
Fluorescence intensity of a translating vRNA
To analyze the translation efficiency of vRNAs over the course of infection, fluorescence intensities of translating vRNAs were measured manually in different cells with variable number of translating vRNAs. The fluorescence intensity was measured in an ROI of 8x8 pixels centered on the translating vRNA. In cells with less than 10 translating vRNAs, the intensity of all vRNAs was measured. In cells with more than 10 translating vRNAs, the intensity of 10 vRNAs was measured by pre-selecting a region of the cell and measuring all vRNAs within this region. For each translating vRNA, a local background subtraction was performed based on the mean of the intensity of 3 random ROIs near the vRNA.
Analyzing GFP foci intensity
The fluorescence intensity of a translating vRNA depends on the number of nascent SunTag peptides (and thus the number of ribosomes) associated with the vRNA. To determine the number of nascent SunTag peptides per translating vRNA, we aimed to determine the precise number of GFP molecules associated with each GFP spot. To this end, the GFP fluorescence intensity of translating vRNAs was compared to the GFP fluorescence intensity of a SunTag ‘stoichiometry reporter’ with a known number of SunTag peptides. As it is difficult to unambiguously detect SunTag proteins with few SunTag peptides, a 24xSunTag-peptide array was selected, for which single molecules can be readily observed after expression in STAb cells. To further facilitate observation of the stoichiometry reporter, a C-terminal prenylation (CAAX) sequence was added to the 24xSunTag-peptie array to tether the proteins to the plasma membrane. Membrane tethering reduces mobility and limits localization to a 2D plane (when focusing on the bottom of the cell), facilitating intensity quantification. High expression of the SunTag-peptide arrays can interfere with unambiguous identification of single SunTag peptide arrays. Therefore, the highly repressive Emi 5′UTR (Tanenbaum et al., 2015) was used to ensure low expression of the stoichiometry reporter and only cells with low expression of the 24xSunTag-CAAX (< 100 foci visible on bottom plasma membrane) were included in the analyses. Using identical imaging parameters as used to determine translating vRNA intensity, short time-lapse movie (< 10 time-points; 25ms interval) were acquired of selected cells focused on the bottom of the cells. Only spots that were visible during 5 consecutive time-points were included in the analysis. The 24xSunTag-CAAX intensity was measured in an ROI of 8x8 pixels centered on the spot in the first time-point of the time-lapse. For each focus, a local background subtraction was performed based on the mean of the intensity of 3 random ROIs near the spot. The relative fluorescence intensity corresponding to a single SunTag peptide was calculated by dividing the mean fluorescence intensity of 24xSunTag-CAAX by 24. The fluorescence intensity of single translating vRNAs was measured as described in section ‘Fluorescence intensity of a translating vRNA’. The number of nascent SunTag peptides associated with each vRNA was then determined based on the fluorescence intensity of a 1x SunTag and of the translating vRNA.
SunTag labeling stoichiometry during infection
The fluorescence intensity of a translating vRNA depends on the number of nascent peptides per vRNA and the intracellular concentration of unbound STAb. During infection of a SunTag-encoding virus, many SunTag peptides are synthesized, which recruit STAb. The amount of ’mature’ SunTag peptides may therefore affect the unbound cellular STAb concentration and thereby the translating vRNA intensity. Reduced brightness of vRNA foci due to limiting STAb availability may hamper analysis of the number of translating vRNAs and may complicate the analysis of translating vRNA intensities. To examine whether changes in the concentration of available STAb occur during early infection phases, the SunTag labeling stoichiometry in infected cells was determined. If the concentration of available STAb drops below a threshold, the recruitment of STAb to the SunTag peptide array is reduced, leading to an altered fluorescence intensity of the SunTag peptide array. To this end, the fluorescence intensity of the stoichiometry reporter (24xSunTag-CAAX; see section ‘Analyzing GFP foci intensity’) was measured in infected cells, as described in ‘Analyzing GFP foci intensity’. In the same cells, the number of translating vRNAs was quantified. The fluorescence intensity of 24xSunTag-CAAX was normalized to the mean intensity in uninfected cells.
Translation elongation rates
To compare the viral translation elongation rate at different time-points during infection, we performed harringtonine run-off experiments (Yan et al., 2016). The fluorescence intensity of single translated vRNAs was measured manually in an ROI of 5x5 pixel on the vRNA. For each vRNA a local background correction was performed by subtracting the local background fluorescence intensity. Translating vRNAs that showed an increasing intensity prior to harringtonine administration were excluded from analysis, as these translating vRNAs were likely not yet in steady-state translation. Where possible, translating vRNAs under the nucleus were selected for intensity measurements, as these vRNAs have low mobility and are thus more easily tracked over time. The intensity was measured for at least 10 minutes after harringtonine administration. If a vRNA could not be detected anymore, the ROI was positioned at the location where the translating RNA was last detected. The fluorescence intensity of each translating vRNA was normalized to the mean intensity of the same vRNA in the 2 minutes before harringtonine administration.
Measuring intensities of fluorescence reporters
eIF4G cleavage reporter
Cleavage of the eIF4G reporter was determined by the dissociation of BFP from mitochondria (as assessed by the mitochondrially-localized mCherry signal). The cleavage of the eIF4G cleavage reporter was quantified only in cells in which the start of phase 1 could be observed. In each of these cells, the BFP fluorescence intensity was measured in 3 ROIs (of 5x5 pixels) in the cytoplasm and in 3 ROIs on the mitochondria. In experiments with relatively short expression of the eIF4G cleavage reporter (12 hr doxycycline induction), the fluorescence intensities of BFP and mCherry increased over time, suggesting that expression of the eIF4G cleavage reporter had not yet reached steady state. To correct for variations in expression of the reporter over time, the BFP intensities were normalized based on the expression of mCherry. We first determined the fluorescence change in mCherry signal over time by calculating the mean mCherry intensity of 3 ROIs (5x5 pixels) on the mitochondria. The (auto fluorescence) background - the mean mCherry intensity in the cytoplasm was also measured in cells that did not express the eIF4G cleavage reporter – was determined and subtracted. Second, a correction factor for expression of the reporter was calculated for each time-point by dividing the mCherry intensity of that time-point by the mean mCherry intensity of the first five time-points of the movie. Third, the BFP fluorescence intensities of each time-point were then corrected by dividing the measured BFP intensities by the time-point-specific correction factor. Then, the difference between the mitochondrial and cytoplasmic BFP intensities (after correction) was calculated by subtracting the cytoplasmic intensity from the mitochondrial intensity. Finally, the difference in BFP intensity over time was normalized to the mean difference in BFP intensity of the three time-points before the start of phase 1. The difference in BFP intensity between mitochondria and cytoplasm over time was also calculated for cells that were not infected (i.e., no translating vRNAs were observed). As the start of phase 1 could not be assigned in these cells, the difference in BFP intensity could not be normalized based on the start of phase 1. Instead, the difference in BFP intensity of control cells were aligned and normalized to the start of the movie.
Nuclear leakage of NLS-BFP
Loss of nuclear NLS-BFP during infection was analyzed in cells in which the start of phase 1 could be observed. The BFP fluorescence intensity in 3 ROIs (of 5x5 pixels) in the nucleoplasm and 3 ROIs in the cytoplasm was measured. Then, the mean cytoplasmic BFP intensity was subtracted from the mean nuclear BFP intensity. Finally, the difference in BFP intensity over time was normalized to the mean difference in BFP intensity of the three time-points before the start of phase 1. The difference in BFP intensity over time was also calculated for cells that were not infected (i.e., no translating vRNAs were observed). As the start of phase 1 could not be assigned in these cells, the difference in BFP intensity could not be normalized based on the start of phase 1. Instead, the difference in BFP intensity of control cells were aligned and normalized to the start of the movie.
Measurements of global host translation
All cells with translating vRNAs were included in the analysis. Additionally, at least 20 uninfected cells from the same images were included as controls. Mitotic cells were excluded from the analysis. The HPG-associated Cy5 fluorescence intensity (referred to as ‘methionine fluorescence’) of each cell was determined by calculating the mean intensity of 3 20x20 pixel ROIs in the cytoplasm of cells. If a cell crossed the boundaries of the large-image stich, the stitching area was excluded from measuring fluorescence intensities, as intensities were generally lower in these regions. Fluorescence background was measured in cells that were not subjected to HPG treatment. The methionine fluorescence intensities of infected cells were normalized to the mean intensity of uninfected cells (which was set to 1) and the mean intensity of puromycin treated cells (set to 0).
Determining the moment of infection
To determine the fraction of infected cells over time, the moment of infection was determined, based on the start of phase1. In some experiments, like when wild-type CVB3 was used, the first appearance of a translating vRNA could not be used to determine the start of phase1. In those instances, the moment of loss of nuclear NLS-BFP was used to determine the start of infection, because the start of phase 1 coincides with the loss of nuclear NLS-BFP (Figures 1G and 1H; Videos S1, S2, and S3). The time between inoculation with virus-containing medium and the first detection of loss of NLS-BFP was used. Cells that already displayed (partial) nuclear leakage at the first time-point of the movie were excluded from analysis, as these cells were already infected before the start of the movie. In a separate analysis, the fraction of cells with nuclear leakage in the first time-point was determined to calculate the fraction off pre-imaging infected cells infected before the start of the movie. The Kaplan-Meier graph with the fraction of uninfected cells over time was then corrected for pre-imaging infection by using the fraction of pre-imaging infected cells as starting point of the Kaplan-Meier graph.
Determining the moment of cell death
Movies acquired with propidium iodide in the imaging medium were analyzed to determine cell survival after infection. Per FOV, maximally 10 cells were selected randomly for analysis. Only cells that were not yet infected at the start of the movie were selected. Cells that went through mitosis during the movie were excluded from analysis. For each cell we scored whether the cell became infected or not during the movie, based on nuclear leakage of NLS-BFP. Then, we determined whether the infection lead to successful replication (i.e., phase3) within 6 hr of the start of phase1. If an infected cell moved out of the FOV within 6 hr after infection, the cell was excluded from analysis, as we could not determine whether or not the infection led to successful replication. Third, for each cell the moment of cell death was determined based on PI-positivity. If a cell did not die, or moved out of the FOV before dying, the cell was classified as survivor and the last time-point that the cell could be observed was used to determine the survival time. Finally, the fraction of surviving cells over time since infection was plotted in a Kaplan-Meier graph. For control cells that did not get infected, the survival was plotted relative to the start of the movie.
siRNA screen
To facilitate high-throughput analysis of viral replication success, we designed a rapid analysis pipeline. If an infection leads to successful replication, many new vRNAs are produced and translated, resulting in the synthesis of many more viral proteins and SunTag peptides (in case of SunTag-CVB3 virus). The SunTag peptides are located in the cytoplasm, while the STAb is located in both the nucleus and the cytoplasm. Therefore, production of high levels of viral SunTag protein results in recruitment of all STAb from the nucleus to the cytoplasm. During normal infection, this occurs in cells around ∼2 hr after initiation of phase3 (See for example Videos S1 and S3). Importantly, nuclear depletion of STAb is not observed in infected cells with unsuccessful replication (i.e., arrest during phase2; see Videos S2 and S3). Loss of nuclear GFP signal can be readily observed during time-lapse imaging, even when using a low magnification imaging objective, and is a robust readout for successful vRNA replication. Usage of a low magnification objective allows for imaging of many more conditions simultaneously, and thus is especially useful for a quick screen.
All cells that were infected in the first 2 hr of a movie, as determined by the leakage of NLS-BFP from the nucleus, were included in the analysis. For each infected cells, we determined whether the infection led to successful replication, as determined by depletion of nuclear STAb, within 6 hr after the start of phase1. The fraction of infected cells with successful replication was then calculated by dividing the number of infected cells with successful replication by the total number of infected cells.
To assess the accuracy of using STAb nuclear loss as a readout for successful viral replication, we determined the fraction of infected cells with successful replication with or without 24 hr IFN pre-treatment (data plotted in black in Figure 6B). The fraction of infected cells with successful replication is 0.78 ± 0.05 (mean ± SD; n = 7) and 0.31 ± 0.05 (mean ± SD; n = 5) in the absence or presence of IFN treatment, respectively. The obtained values for the fraction of successful viral replication based on either loss of nuclear STAb or on single-molecule analysis of infection phases was comparable: without treatment 0.78 versus 0.85 (no IFN) or 0.31 versus 0.41 (with IFN). Note that the rapid analysis pipeline resulted in a slightly lower fraction of successful replication than the single-molecule VIRIM analysis, possibly due to too stringent scoring of loss of nuclear STAb. Thus, the analysis pipeline based on nuclear loss of STAb can be used to screen various conditions.
Analysis of RNA sequencing
To identify IFN-induced genes, cells with or without 24 hr IFN pre-treatment were compared. For each sample, paired-end reads from illumina sequencing were aligned to the human transcriptome, as described previously (Muraro et al., 2016). Genes with less than 10 reads in the datasets from the cells without IFN treatment were excluded from analysis. To enable comparison between the different samples, the read-count per gene was normalized to the average of the total read count of all samples (mean total read count per sample: 4535455). To determine the fold-induction of gene expression after IFN treatment, the ratio in gene expression in untreated and IFN pre-treated cells was calculated. All genes with at least 3-fold increase in expression upon IFN treatment in both experiments were selected (37 genes). To simplify the siRNA screen, a further selection of genes was made by excluding genes with well-described antiviral functions that are unrelated to inhibition of viral replication (9 genes excluded) and by excluding genes for which no SMARTpool ONTargetPlus reagents were available (1 gene excluded). As a result, 28 genes were included in the siRNA screen (for details on sequencing results and fold increase in expression after IFN treatment per gene see online data).
Statistical analyses and generation of graphs
All statistical analyses graphs were performed and generated using Prism GraphPad (v8.2.1). Statistical tests are explained in figure legends.
To facilitate visual inspection of general trends in heterogeneous data, moving averages were included in Figures 1I, 4B, 4C, 4H, and 4K. Moving averages were generated as locally weighted least-squares (LOWESS) using the ‘fit spline/lowess’ function in GraphPad and using a 5 point smoothening window.
To determine the mean duration of phase1 and phase2, the distribution of phase1 and phase2 durations were fit to a one-component exponential decay distribution. During phase annotations, a minimal threshold of 12.5 minutes was used to annotate phase1 and phase2. Therefore, additional constrains (y = 1 and x = 0-12) were taken into account to generate the one-component exponential decay fits of phase1 and phase2.
A fraction of cells did not complete phase2, which was referred to as the fraction of infected cells with ‘unsuccessful replication’. To determine the fraction of cells with unsuccessful replication, the one-component exponential decay fit of phase2 was generated and the plateau of the fit was used as the fraction of cells with unsuccessful replication.
Acknowledgments
We thank members of the Tanenbaum and Van Kuppeveld labs for helpful discussions. We also thank Ive Logister, Stijn Sonneveld, and Rupa Banerjee for help with experiments. This work was financially supported by an ERC starting grant to M.E.T. (EU/ERC-677936 RNAREG), the Howard Hughes Medical Institute through an international research scholar grant to M.E.T. (HHMI/IRS 55008747), and a NWO VICI grant to F.J.M.v.K. (91812628). S.B., H.H.R., L.J.M.B., T.P., J.B., H.C., and M.E.T. were supported by the Oncode Institute, which is partly funded by the Dutch Cancer Society (KWF).
Author Contributions
Conceptualization, S.B., H.H.R., F.J.M.v.K., and M.E.T.; Methodology, S.B., H.H.R., L.J.M.B., F.J.M.v.K., and M.E.T.; Validation, S.B. and H.H.R.; Formal Analysis, S.B.; Investigation, S.B., H.H.R., L.J.M.B., T.P., and A.L.W.v.V.; Resources, J.B. and H.C.; Data Curation, S.B., H.H.R., and L.J.M.B.; Writing – Original Draft, S.B. and M.E.T.; Writing – Review & Editing, all authors; Visualization, S.B.; Supervision, F.J.M.v.K. and M.E.T.; Funding Acquisition, F.J.M.v.K. and M.E.T.
Declaration of Interests
H.C. is the inventor of several patents related to organoid technology; his full disclosure is given at https://www.uu.nl/staff/JCClevers/.
Published: November 13, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.10.019.
Contributor Information
Frank J.M. van Kuppeveld, Email: f.j.m.vankuppeveld@uu.nl.
Marvin E. Tanenbaum, Email: m.tanenbaum@hubrecht.eu.
Supplemental Information
Overview of the number of repeats per experiment, and of the number of cells and foci analyzed per experiment. Some datasets are plotted multiple times. If relevant replotting of the same datasets is indicated in the last column and in the corresponding figure legends.
References
- Ahlquist P., Noueiry A.O., Lee W.-M., Kushner D.B., Dye B.T. Host factors in positive-strand RNA virus genome replication. J. Virol. 2003;77:8181–8186. doi: 10.1128/JVI.77.15.8181-8186.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andino R., Silvera D., Suggett S.D., Achacoso P.L., Miller C.J., Baltimore D., Feinberg M.B. Engineering poliovirus as a vaccine vector for the expression of diverse antigens. Science. 1994;265:1448–1451. doi: 10.1126/science.8073288. [DOI] [PubMed] [Google Scholar]
- Arnold J.J., Cameron C.E. Poliovirus RNA-dependent RNA polymerase (3D(pol)). Assembly of stable, elongation-competent complexes by using a symmetrical primer-template substrate (sym/sub) J. Biol. Chem. 2000;275:5329–5336. doi: 10.1074/jbc.275.8.5329. [DOI] [PubMed] [Google Scholar]
- Baggen J., Thibaut H.J., Strating J.R.P.M., van Kuppeveld F.J.M. The life cycle of non-polio enteroviruses and how to target it. Nat. Rev. Microbiol. 2018;16:368–381. doi: 10.1038/s41579-018-0005-4. [DOI] [PubMed] [Google Scholar]
- Balleza E., Kim J.M., Cluzel P. Systematic characterization of maturation time of fluorescent proteins in living cells. Nat. Methods. 2018;15:47–51. doi: 10.1038/nmeth.4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrows N.J., Campos R.K., Liao K.C., Prasanth K.R., Soto-Acosta R., Yeh S.C., Schott-Lerner G., Pompon J., Sessions O.M., Bradrick S.S., Garcia-Blanco M.A. Biochemistry and Molecular Biology of Flaviviruses. Chem. Rev. 2018;118:4448–4482. doi: 10.1021/acs.chemrev.7b00719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton D.J., Flanegan J.B. Synchronous replication of poliovirus RNA: initiation of negative-strand RNA synthesis requires the guanidine-inhibited activity of protein 2C. J. Virol. 1997;71:8482–8489. doi: 10.1128/jvi.71.11.8482-8489.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton D.J., Morasco B.J., Flanegan J.B. Translating ribosomes inhibit poliovirus negative-strand RNA synthesis. J. Virol. 1999;73:10104–10112. doi: 10.1128/jvi.73.12.10104-10112.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkowski L.S., Sen G.C. Inhibition of vesicular stomatitis viral mRNA synthesis by interferons. J. Virol. 1987;61:653–660. doi: 10.1128/jvi.61.3.653-660.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov G.A., Evstafieva A.G., Rubtsov Y.P., Mikitas O.V., Vartapetian A.B., Agol V.I. Early alteration of nucleocytoplasmic traffic induced by some RNA viruses. Virology. 2000;275:244–248. doi: 10.1006/viro.2000.0427. [DOI] [PubMed] [Google Scholar]
- Ben M’hadheb-Gharbi M., Gharbi J., Paulous S., Brocard M., Komaromva A., Aouni M., Kean K.M. Effects of the Sabin-like mutations in domain V of the internal ribosome entry segment on translational efficiency of the Coxsackievirus B3. Mol. Genet. Genomics. 2006;276:402–412. doi: 10.1007/s00438-006-0155-3. [DOI] [PubMed] [Google Scholar]
- Chakrabarti A., Jha B.K., Silverman R.H. New insights into the role of RNase L in innate immunity. J. Interferon Cytokine Res. 2011;31:49–57. doi: 10.1089/jir.2010.0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase A.J., Semler B.L. Viral subversion of host functions for picornavirus translation and RNA replication. Future Virol. 2012;7:179–191. doi: 10.2217/fvl.12.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daffis S., Szretter K.J., Schriewer J., Li J., Youn S., Errett J., Lin T.Y., Schneller S., Zust R., Dong H. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature. 2010;468:452–456. doi: 10.1038/nature09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave P., George B., Balakrishnan S., Sharma D.K., Raheja H., Dixit N.M., Das S. Strand-specific affinity of host factor hnRNP C1/C2 guides positive to negative-strand ratio in Coxsackievirus B3 infection. RNA Biol. 2019;16:1286–1299. doi: 10.1080/15476286.2019.1629208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit E., van Doremalen N., Falzarano D., Munster V.J. SARS and MERS: recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016;14:523–534. doi: 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doğanay S., Lee M.Y., Baum A., Peh J., Hwang S.Y., Yoo J.Y., Hergenrother P.J., García-Sastre A., Myong S., Ha T. Single-cell analysis of early antiviral gene expression reveals a determinant of stochastic IFNB1 expression. Integr. Biol. 2017;9:857–867. doi: 10.1039/c7ib00146k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duke G.M., Palmenberg A.C. Cloning and synthesis of infectious cardiovirus RNAs containing short, discrete poly(C) tracts. J. Virol. 1989;63:1822–1826. doi: 10.1128/jvi.63.4.1822-1826.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis E.L., Delbrück M. The growth of bacteriophage. J. Gen. Physiol. 1939;22:365–384. doi: 10.1085/jgp.22.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estell C., Stamatidou E., El-Messeiry S., Hamilton A. In situ imaging of mitochondrial translation shows weak correlation with nucleoid DNA intensity and no suppression during mitosis. J. Cell Sci. 2017;130:4193–4199. doi: 10.1242/jcs.206714. [DOI] [PubMed] [Google Scholar]
- Feuer R., Mena I., Pagarigan R., Slifka M.K., Whitton J.L. Cell cycle status affects coxsackievirus replication, persistence, and reactivation in vitro. J. Virol. 2002;76:4430–4440. doi: 10.1128/JVI.76.9.4430-4440.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flather D., Semler B.L. Picornaviruses and nuclear functions: targeting a cellular compartment distinct from the replication site of a positive-strand RNA virus. Front. Microbiol. 2015;6:594. doi: 10.3389/fmicb.2015.00594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco D.N., Pratt H., Kandilas S., Cheon S.S.Y., Lin W., Cronkite D.A., Basavappa M., Jeffrey K.L., Anselmo A., Sadreyev R. HELZ2 is an IFN effector mediating suppression of dengue virus. Front. Microbiol. 2017;8:240. doi: 10.3389/fmicb.2017.00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamarnik A.V., Andino R. Switch from translation to RNA replication in a positive-stranded RNA virus. Genes Dev. 1998;12:2293–2304. doi: 10.1101/gad.12.15.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar I., Wippich F., Ephrussi A., Gáspár I., Wippich F., Ephrussi A. Terminal Deoxynucleotidyl Transferase Mediated Production of Labeled Probes for Single-molecule FISH or RNA Capture. Bio Protoc. 2018;8 doi: 10.21769/BioProtoc.2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustin K.E., Sarnow P. Effects of poliovirus infection on nucleo-cytoplasmic trafficking and nuclear pore complex composition. EMBO J. 2001;20:240–249. doi: 10.1093/emboj/20.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambidge S.J., Sarnow P. Translational enhancement of the poliovirus 5′ noncoding region mediated by virus-encoded polypeptide 2A. Proc. Natl. Acad. Sci. USA. 1992;89:10272–10276. doi: 10.1073/pnas.89.21.10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimshony T., Senderovich N., Avital G., Klochendler A., de Leeuw Y., Anavy L., Gennert D., Li S., Livak K.J., Rozenblatt-Rosen O. CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 2016;17:77. doi: 10.1186/s13059-016-0938-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoek T.A., Khuperkar D., Lindeboom R.G.H., Sonneveld S., Verhagen B.M.P., Boersma S., Vermeulen M., Tanenbaum M.E. Single-Molecule Imaging Uncovers Rules Governing Nonsense-Mediated mRNA Decay. Mol. Cell. 2019;75:324–339.e11. doi: 10.1016/j.molcel.2019.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John S.P., Sun J., Carlson R.J., Cao B., Bradfield C.J., Song J., Smelkinson M., Fraser I.D.C. IFIT1 Exerts Opposing Regulatory Effects on the Inflammatory and Interferon Gene Programs in LPS-Activated Human Macrophages. Cell Rep. 2018;25:95–106.e6. doi: 10.1016/j.celrep.2018.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B., Arcos S., Rothamel K., Jian J., Rose K.L., McDonald W.H., Bian Y., Reasoner S., Barrows N.J., Bradrick S. Discovery of Widespread Host Protein Interactions with the Pre-replicated Genome of CHIKV Using VIR-CLASP. Mol. Cell. 2020;78:624–640.e7. doi: 10.1016/j.molcel.2020.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klasse P.J. Molecular determinants of the ratio of inert to infectious virus particles. Prog. Mol. Biol. Transl. Sci. 2015;129:285–326. doi: 10.1016/bs.pmbts.2014.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo B.K., Stange D.E., Sato T., Karthaus W., Farin H.F., Huch M., van Es J.H., Clevers H. Controlled gene expression in primary Lgr5 organoid cultures. Nat. Methods. 2011;9:81–83. doi: 10.1038/nmeth.1802. [DOI] [PubMed] [Google Scholar]
- Kräusslich H.G., Nicklin M.J., Toyoda H., Etchison D., Wimmer E. Poliovirus proteinase 2A induces cleavage of eucaryotic initiation factor 4F polypeptide p220. J. Virol. 1987;61:2711–2718. doi: 10.1128/jvi.61.9.2711-2718.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P., Sweeney T.R., Skabkin M.A., Skabkina O.V., Hellen C.U.T., Pestova T.V. Inhibition of translation by IFIT family members is determined by their ability to interact selectively with the 5′-terminal regions of cap0-, cap1- and 5′ppp- mRNAs. Nucleic Acids Res. 2014;42:3228–3245. doi: 10.1093/nar/gkt1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamphear B.J., Kirchweger R., Skern T., Rhoads R.E. Mapping of functional domains in eukaryotic protein synthesis initiation factor 4G (eIF4G) with picornaviral proteases. Implications for cap-dependent and cap-independent translational initiation. J. Biol. Chem. 1995;270:21975–21983. doi: 10.1074/jbc.270.37.21975. [DOI] [PubMed] [Google Scholar]
- Lanke K.H.W., van der Schaar H.M., Belov G.A., Feng Q., Duijsings D., Jackson C.L., Ehrenfeld E., van Kuppeveld F.J.M. GBF1, a guanine nucleotide exchange factor for Arf, is crucial for coxsackievirus B3 RNA replication. J. Virol. 2009;83:11940–11949. doi: 10.1128/JVI.01244-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Banerjee S., Wang Y., Goldstein S.A., Dong B., Gaughan C., Silverman R.H., Weiss S.R. Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. Proc. Natl. Acad. Sci. USA. 2016;113:2241–2246. doi: 10.1073/pnas.1519657113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyubimova A., Itzkovitz S., Junker J.P., Fan Z.P., Wu X., van Oudenaarden A. Single-molecule mRNA detection and counting in mammalian tissue. Nat. Protoc. 2013;8:1743–1758. doi: 10.1038/nprot.2013.109. [DOI] [PubMed] [Google Scholar]
- Mears H.V., Emmott E., Chaudhry Y., Hosmillo M., Goodfellow I.G., Sweeney T.R. Ifit1 regulates norovirus infection and enhances the interferon response in murine macrophage-like cells. Wellcome Open Res. 2019;4:82. doi: 10.12688/wellcomeopenres.15223.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisaki T., Lyon K., DeLuca K.F., DeLuca J.G., English B.P., Zhang Z., Lavis L.D., Grimm J.B., Viswanathan S., Looger L.L. Real-time quantification of single RNA translation dynamics in living cells. Science. 2016;352:1425–1429. doi: 10.1126/science.aaf0899. [DOI] [PubMed] [Google Scholar]
- Muraro M.J., Dharmadhikari G., Grün D., Groen N., Dielen T., Jansen E., van Gurp L., Engelse M.A., Carlotti F., de Koning E.J.P., van Oudenaarden A. A Single-Cell Transcriptome Atlas of the Human Pancreas. Cell Syst. 2016;3:385–394.e3. doi: 10.1016/j.cels.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh H.S., Pathak H.B., Goodfellow I.G., Arnold J.J., Cameron C.E. Insight into poliovirus genome replication and encapsidation obtained from studies of 3B-3C cleavage site mutants. J. Virol. 2009;83:9370–9387. doi: 10.1128/JVI.02076-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil S., Fribourg M., Ge Y., Batish M., Tyagi S., Hayot F., Sealfon S.C. Single-cell analysis shows that paracrine signaling by first responder cells shapes the interferon-β response to viral infection. Sci. Signal. 2015;8:ra16. doi: 10.1126/scisignal.2005728. [DOI] [PubMed] [Google Scholar]
- Pichlmair A., Lassnig C., Eberle C.A., Górna M.W., Baumann C.L., Burkard T.R., Bürckstümmer T., Stefanovic A., Krieger S., Bennett K.L. IFIT1 is an antiviral protein that recognizes 5¢-triphosphate RNA. Nat. Immunol. 2011;12:624–630. doi: 10.1038/ni.2048. [DOI] [PubMed] [Google Scholar]
- Pichon X., Bastide A., Safieddine A., Chouaib R., Samacoits A., Basyuk E., Peter M., Mueller F., Bertrand E. Visualization of single endogenous polysomes reveals the dynamics of translation in live human cells. J. Cell Biol. 2016;214:769–781. doi: 10.1083/jcb.201605024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A., van den Bogaard P., Rifkin S.A., van Oudenaarden A., Tyagi S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods. 2008;5:877–879. doi: 10.1038/nmeth.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed L.J., Munech H. A simple method of estimating fifty percent endpoints. Am. J. Epidemiol. 1938;27:493–497. [Google Scholar]
- Samuel C.E. Antiviral actions of interferons. Clin. Microbiol. Rev. 2001;14:778–809. doi: 10.1128/CMR.14.4.778-809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T., Stange D.E., Ferrante M., Vries R.G.J., Van Es J.H., Van den Brink S., Van Houdt W.J., Pronk A., Van Gorp J., Siersema P.D., Clevers H. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology. 2011;141:1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- Schneider W.M., Chevillotte M.D., Rice C.M. Interferon-stimulated genes: a complex web of host defenses. Annu. Rev. Immunol. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins J.W. Recent advances in antiviral interferon-stimulated gene biology. F1000Res. 2018;7:309. doi: 10.12688/f1000research.12450.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins J.W., Wilson S.J., Panis M., Murphy M.Y., Jones C.T., Bieniasz P., Rice C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sean P., Nguyen J.H.C., Semler B.L. Altered interactions between stem-loop IV within the 5′ noncoding region of coxsackievirus RNA and poly(rC) binding protein 2: effects on IRES-mediated translation and viral infectivity. Virology. 2009;389:45–58. doi: 10.1016/j.virol.2009.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman R.H. Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol. 2007;81:12720–12729. doi: 10.1128/JVI.01471-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staals R.H.J., Bronkhorst A.W., Schilders G., Slomovic S., Schuster G., Heck A.J.R., Raijmakers R., Pruijn G.J.M. Dis3-like 1: a novel exoribonuclease associated with the human exosome. EMBO J. 2010;29:2358–2367. doi: 10.1038/emboj.2010.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson D.B., Medzhitov R. Type I interferons in host defense. Immunity. 2006;25:373–381. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Suzuki Y., Chin W.-X., Han Q., Ichiyama K., Lee C.H., Eyo Z.W., Ebina H., Takahashi H., Takahashi C., Tan B.H. Characterization of RyDEN (C19orf66) as an Interferon-Stimulated Cellular Inhibitor against Dengue Virus Replication. PLoS Pathog. 2016;12:e1005357. doi: 10.1371/journal.ppat.1005357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkin Y.V., Maslova S.V., Agol V.I. The genomes of attenuated and virulent poliovirus strains differ in their in vitro translation efficiencies. Virology. 1985;147:243–252. doi: 10.1016/0042-6822(85)90127-8. [DOI] [PubMed] [Google Scholar]
- Svitkin Y.V., Cammack N., Minor P.D., Almond J.W. Translation deficiency of the Sabin type 3 poliovirus genome: association with an attenuating mutation C472----U. Virology. 1990;175:103–109. doi: 10.1016/0042-6822(90)90190-3. [DOI] [PubMed] [Google Scholar]
- Tanenbaum M.E., Gilbert L.A., Qi L.S., Weissman J.S., Vale R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell. 2014;159:635–646. doi: 10.1016/j.cell.2014.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanenbaum M.E., Stern-Ginossar N., Weissman J.S., Vale R.D. Regulation of mRNA translation during mitosis. eLife. 2015;4:e07957. doi: 10.7554/eLife.07957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne L.G., Goodfellow I.G. Norovirus gene expression and replication. J. Gen. Virol. 2014;95:278–291. doi: 10.1099/vir.0.059634-0. [DOI] [PubMed] [Google Scholar]
- van der Linden L., Vives-Adrián L., Selisko B., Ferrer-Orta C., Liu X., Lanke K., Ulferts R., De Palma A.M., Tanchis F., Goris N. The RNA template channel of the RNA-dependent RNA polymerase as a target for development of antiviral therapy of multiple genera within a virus family. PLoS Pathog. 2015;11:e1004733. doi: 10.1371/journal.ppat.1004733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D., Mathews M.B., Mohr I. Tinkering with translation: protein synthesis in virus-infected cells. Cold Spring Harb. Perspect. Biol. 2013;5:a012351. doi: 10.1101/cshperspect.a012351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Han B., Zhou R., Zhuang X. Real-Time Imaging of Translation on Single mRNA Transcripts in Live Cells. Cell. 2016;165:990–1001. doi: 10.1016/j.cell.2016.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Ma L., Stipkovits L., Szathmary S., Li X., Liu Y. The strategy of picornavirus evading host antiviral responses: Nonstructural proteins suppress the production of IFNs. Front. Microbiol. 2018;9:2943. doi: 10.3389/fmicb.2018.02943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessels E., Duijsings D., Niu T.K., Neumann S., Oorschot V.M., de Lange F., Lanke K.H.W., Klumperman J., Henke A., Jackson C.L. A viral protein that blocks Arf1-mediated COP-I assembly by inhibiting the guanine nucleotide exchange factor GBF1. Dev. Cell. 2006;11:191–201. doi: 10.1016/j.devcel.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Wu B., Eliscovich C., Yoon Y.J., Singer R.H. Translation dynamics of single mRNAs in live cells and neurons. Science. 2016;352:1430–1435. doi: 10.1126/science.aaf1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X., Hoek T.A., Vale R.D., Tanenbaum M.E. Dynamics of Translation of Single mRNA Molecules In Vivo. Cell. 2016;165:976–989. doi: 10.1016/j.cell.2016.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawatzky R., De Maeyer E., De Maeyer-Guignard J. Identification of individual interferon-producing cells by in situ hybridization. Proc. Natl. Acad. Sci. USA. 1985;82:1136–1140. doi: 10.1073/pnas.82.4.1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Maximal intensity projection of 11 GFP Z-slices (green) and a single BFP Z-slice (magenta) of STAb cells stably expressing NLS-BFP infected with SunTag-CVB3. Images were acquired every 2 min. Time is indicated in hours:minutes since the start of the acquisition. The first translating vRNA is detected at 00:10. Note that images of the same cell are used in Figures 1G and 2A. Scale bar shown in the first image, 15 μm.
Maximal intensity projection of 11 GFP Z-slices (green) and a single BFP Z-slice (magenta) of STAb cells stably expressing NLS-BFP infected with SunTag-CVB3. Images were acquired every 2 min. Time is indicated in hours:minutes since the start of the acquisition. The first translating vRNA in the middle cell is detected at 00:22, and does not result in successful viral replication (phase2 block). Scale bar shown in the first image, 15 μm.
Maximal intensity projection of 4 GFP Z-slices (green) and a single BFP Z-slice (magenta) of STAb cells stably expressing NLS-BFP infected with SunTag-CVB3 and imaged with propidium iodide (cyan). Images were acquired every 10 min. Time is indicated in hours:minutes since the start of the movie. (A) The first translating vRNA in the middle cell is detected at 03:00 and results in a successful infection. (B) The first translating vRNA in the middle cell is detected at 03:10, and does not result in a successful infection (block in phase2).Scale bar shown in the first images, 15 μm.
Maximal intensity projection of 11 GFP Z-slices (green) and single Z-slice of BFP (blue) and mCherry (red) of STAb cells expressing the eIF4G cleavage reporter infected with SunTag-CVB3. Images were acquired every 5 min. Time is indicated in hours:minutes since the start of the acquisition. The first translating vRNA is detected at 01:35. Of note, image acquisition is started 12 h after induction of expression of the eIF4G cleavage reporter and the expression is still increasing throughout the movie. Therefore, only GFP, but not BFP and mCherry, is corrected for photo bleaching. Scale bar shown in the first image, 15 μm.
Overview of the number of repeats per experiment, and of the number of cells and foci analyzed per experiment. Some datasets are plotted multiple times. If relevant replotting of the same datasets is indicated in the last column and in the corresponding figure legends.
Data Availability Statement
The RNA sequencing data of this study has been deposited in the Gene Expression Omnibus (GEO) under accession code GEO: GSE159280. A selection of raw imaging data is made available through Mendeley data: https://doi.org/10.17632/9sxbk6cvn9.1.