

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is the most lethal common malignancy, with little improvement in patient outcomes over the past decades. Recently, subtypes of pancreatic cancer with different prognoses have been elaborated, however the inability to model these subtypes has precluded mechanistic investigation of their origins. Here, we present a xenotransplantation model of PDAC in which neoplasms originate from patient-derived organoids injected directly into murine pancreatic ducts. Our model enables distinction of the two main PDAC subtypes: intraepithelial neoplasms from this model progress in an indolent or invasive manner representing the classical or basal-like subtypes of PDAC, respectively. Parameters that influence PDAC subtype specification in this intraductal model include cell plasticity and hyperactivation of the Ras pathway. Finally, through intratumoral dissection and the direct manipulation of RAS gene dosage, we identify a suite of RAS-regulated secreted and membrane bound proteins which may represent potential candidates for therapeutic intervention in PDAC patients.
Keywords: Pancreatic cancer, preclinical mouse model, intraductal neoplasia, molecular subtypes, cell plasticity, KRAS, patient-derived organoids
Introduction
Pancreatic ductal adenocarcinoma (PDAC) has one of the highest mortality rates among all types of cancers even when diagnosed at an early stage. For all stages combined, the 5-year survival rate is 9%. However, more than half of patients are diagnosed with metastatic disease, which is associated with a 5-year survival rate of only 3% (1). The development of early detection methods and effective therapies are needed to improve the outcome of PDAC patients (2). Accordingly, the recent revolution in cancer genomics has led to the characterization of the genetic landscape of PDAC, and clinical trials in which treatment is matched to underlying mutations are currently underway (3). A new perspective relevant to therapeutic development has emerged from the recent identification of two major transcriptional subtypes of PDAC with characteristic histopathological features and different prognoses (4-9). These include a “basal-like” (or squamous) subtype which is poorly differentiated and carries a worse prognosis when compared to the better-differentiated “classical” (or progenitor) subtype (4,7,10). Basal-like and classical subtypes have been shown to share features of the squamous and progenitor subtypes, respectively, as defined by an independent PDAC classification system (5,11,12), which also can predict response to chemotherapy (6,13-15). In addition, the basal-like subtype is associated with the activation of genes involved in the epithelial–mesenchymal transition (EMT), activation of the transcription factors MYC and TP63, and down-regulation of endodermal identity markers, such as HNF4A and GATA6 whose expression are therefore hallmarks of the classical subtype (4,6,7). In addition, expression of the ∆N isoform of TP63 (∆Np63) and GLI2 were recently shown to promote the basal-like identity in PDAC (16,17). Further characterization of the molecular phenotype of these transcriptional subtypes may lead to the identification of tailored therapies with improved efficacy.
Beyond the genetic mechanisms of subtype specification (18,19), epigenetic alterations have also been shown to dynamically regulate PDAC transcriptional subtypes. For example, paracrine communication from the tumor microenvironment can promote subtype specification (9,20-22). In addition, mutations in several epigenetic regulator genes, including KDM6A, KMT2C, and KMT2D, have been associated with the basal-like subtype (4,6,23). However, the relative importance of genetic and epigenetic aberrations to promote the stable or dynamic regulation of subtypes is still unknown. Moreover, the development of subtype-based therapeutic strategies remains challenging due to the complex heterogeneous features of PDAC and the lack of preclinical models that recapitulate these subtypes.
To address these limitations, we sought to establish an in vivo xenograft model that could recapitulate the basal-like and classical subtypes of PDAC. As the pancreatic ducts are the sites where preinvasive pancreatic neoplasms progress in humans, we reasoned that seeding the murine pancreatic ducts system with human neoplastic cells would offer new cellular and molecular insights into the mechanisms promoting progression of this malignancy. We transplanted a panel of human patient-derived PDAC organoids, which genetically and transcriptionally reflect the diversity of PDAC (24), into the pancreatic ductal system of immunodeficient mice. The intraepithelial neoplasms that formed following xenotransplantation followed one of two patterns of progression: aggressive or indolent, which reflected the basal-like or classical subtypes of PDAC, respectively. Through analysis of the two progression phenotypes observed in our model, we have identified roles for both epigenetic and KRAS-driven signaling in promoting the progression to a basal-like subtype.
Results
Establishing an in vivo model that recapitulates the progression from intraductal epithelial neoplasia to invasive PDAC
We previously developed a three-dimensional organoid culture system to propagate patient-derived PDAC tissue from surgically resected primary tumors (T) and metastases (M), or fine needle aspirates (F) (25). To generate in vivo models of PDAC, we previously injected these organoids directly into the pancreatic interstitium of immunodeficient mice (Fig. 1A) (25,26). This “orthotopically grafted organoid” (OGO) model initially develops high-grade, extra-ductal neoplasms that resemble advanced pancreatic intraepithelial neoplasms (PanINs) and progress morphologically over several weeks to months to form frankly invasive tumors (25), markedly faster than the timescale of PDAC progression hypothesized to occur in humans (27,28). While this model is well-suited for generating large PDAC tumors, these tumors arise within the interstitial space rather than within the pancreatic ductal system where human PDAC progression is believed to occur (29). We therefore sought to develop an intraductally grafted organoid (IGO) model that better recapitulates the progression from pre-invasive cells within the duct system to tissue-invasive extra-ductal neoplasms with surrounding desmoplasia. To evaluate how IGO-derived cells develop tumors we injected single-cell suspensions of organoids into the main pancreatic ducts of immunodeficient mice (Fig. 1A; Supplementary Fig. S1A) via a modified retrograde pancreatic intraductal injection method (30) and analyzed the location of cancer cells by immunofluorescence (IF) and three-dimensional imaging. At day 1 post-transplantation, organoids were detected focally along the main pancreatic duct (Supplementary Fig. S1B). Within 1 to 2 weeks post-transplantation, organoids were engrafted within the intercalated ducts (Fig. 1B and 1C, white arrow marks ducts labeled with CK19 staining) and the centro-acinar area (Fig. 1B and 1C, white dotted line marks the centro-acinar area; Supplementary Video S1 and S2; Supplementary Fig. S1C) indicating that organoid cells were ultimately able to seed the terminal end of the duct, resulting in intraductal neoplasms focally connected to the host murine pancreatic duct (Fig. 1D; Supplementary Fig.S1D). Two distinct histological features could be observed at 4 weeks following transplant: some regions derived from these transplants exhibited features of intraductal glandular prominence with columnar epithelium (Fig. 1E, white arrows), while other regions lacked glandular features and rather exhibited invasive features (Fig. 1E, white arrowheads; Supplementary Video S3 and S4).
Figure 1. Establishment and characterization of an intraductally grafted organoid (IGO) transplant model of pancreatic cancer.
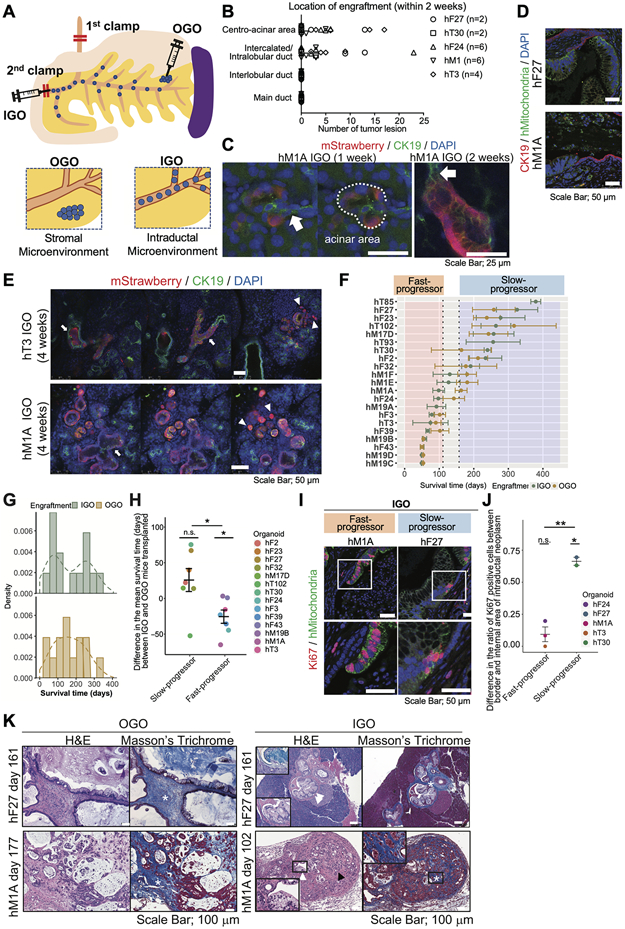
A) Diagram depicting the two organoid transplant models. In the intraductally grafted organoid (IGO) transplant model, two clamps (marked by red bars) are placed on the common bile duct between liver and pancreas, and on the papilla of Vater in order to block the backflow of cells. Organoids are injected at the papilla of Vater (injection site marked by syringe with double red bars). In the orthotopically grafted organoid (OGO) model, organoids are injected directly into the interstitial space in the tail of the pancreas. The relative locations of the injected cells are depicted in the bottom two panels.
B) Quantification of the numbers of lesions of engrafted tumor sites at the indicated sites 1-2 weeks after IGO transplantation. Presence of engrafted cells was confirmed by immunohistochemical (IHC) staining for human Mitochondria (hMitochondria). n, number of mice analyzed.
C) Representative immunofluorescent (IF) images of mStrawberry-hM1A IGO xenografts 1 to 2 weeks post-transplantation. White arrows, intercalated ducts. White dotted line, centro-acinar area. Scale bars, 25 μm.
D) Representative IF images of hF27 (top) and hM1A (bottom) IGO xenografts at endpoint stained with CK19 (red), hMitochondria (green), with DAPI counterstain (blue). Scale bars, 50 μm.
E) Representative IF images of mStrawberry-labeled hT3 (top) and hM1A (bottom) IGO xenografts 4 weeks after transplantation. Mouse ducts are stained with antibodies against CK19 (green). Engrafted organoids are stained with antibodies against mStrawberry (red). Nuclei are counterstained with DAPI (blue). White arrows, intraductal tumor. White arrowheads, invading tumor. Scale bars, 50 μm.
F) Mean survival times (days) of IGO mice (green) and OGO mice (yellow) transplanted with indicated organoid lines. Points and error bars, means and standard deviations of survival times are calculated using the actual survival times for uncensored mice and the imputed survival times for censored mice. Vertical dotted lines indicate the thresholds for Fast- and Slow-progressors.
G) Distribution of the mean survival times (days) of IGO and OGO mice. For each organoid line, the mean survival time is calculated using the actual survival times for uncensored mice and the imputed survival times for censored mice. Only organoid lines having at least 3 IGO and 3 OGO mice have been retained. The histogram is using density instead of count on y-axis and is overlaid with a density curve (dashed line).
H) Difference in the mean survival times (days) between IGO and OGO mice. Each point represents the difference in a specific organoid line. The mean and the standard error of the mean are shown for the Fast- and Slow-progressors. Paired Student’s t test has been performed on organoids grouped according to Fast and Slow-progressors to determine if the mean of the group is different from zero. In addition, a Welch’s test has been used to evaluate if there is a significant difference between the means of the Fast- and Slow-progressors.
I) Representative IF staining for hMitochondria (green) and Ki67 (red) of tumors generated from IGO transplants of hM1A (left) and hF27 (right). Scale bars, 50 μm. Bottom panels, magnified views of boxed areas in top panels.
J) Difference in the ratio of Ki67 positive cells between border and intraductal area for Fast- and Slow-progressors. Each point represents the difference in a specific organoid line. The mean and the standard error of the mean are shown for the Fast- and Slow-progressors. Paired Student’s t test has been done on organoids grouped according to Fast- and Slow-progressors to determine if the mean of the group is different from zero. In addition, a Welch's t test has been used to determine if there is a significant difference between the means of the Fast- and Slow-progressors.
K) Representative images of hematoxylin and eosin (H&E) and Masson’s Trichrome staining of OGO (left) and IGO xenografts (right) of hF27 or hM1A at endpoint. Scale bar, 100 μm. White asterisk, desmoplastic reaction. White arrowheads, representative intraductal lesion. Black arrowhead, invasive lesion. Insets, magnified images.
Thus, IGO-derived xenografts displayed a range of histological features, with a subset of organoid cultures remaining restricted to the intraductal region.
To comprehensively characterize the difference in the phenotypes between the IGO and OGO models, we transplanted 21 distinct organoid cultures including three KRAS wild-type lines (24), using both methods and analyzed differences in survival time of the transplant mice. All 21 organoids successfully engrafted in both models with variable efficiency (Supplementary Fig. S1E) except for two organoids (hF23, hF27), which exhibited a much lower engraftment rate (2/6 and 1/6) in the OGO model compared to that (7/8 and 7/7) in the IGO model. This result suggests that the ductal environment better supports survival of hF23 and hF27 compared to the extra-ductal environment.
A wide variation in survival times following PDAC organoid transplantation was observed (Fig. 1F). Interestingly, the survival times of the IGO transplants formed a bimodal distribution (Fig. 1G; Supplementary Table S1), while the survival of OGO transplants comprised a unimodal distribution (Fig 1G). By approximating sampled survival times with two normal distributions, we defined survival thresholds for fast-progressing lines (“Fast-progressors”) and slow-progressing lines (“Slow-progressors”) in the IGO model (Fig. 1F and 1G; Supplementary Fig. S1F; and SM1). Mice bearing Fast-progressors (10/21 transplanted organoid lines) had mean survival times of fewer than 111 days, whereas the mean survival times of mice bearing Slow-progressors (9/21 transplanted organoid lines) were greater than 157 days (Fig. 1F and 1G; Supplementary Fig. S1F; and SM1). Though xenograft growth rates of each organoid in the OGO model showed similar trends as seen in corresponding organoids in the IGO model, survival times of Fast-progressors and Slow-progressors in the IGO model were shorter and longer respectively, than survival times of the corresponding lines in the OGO model (Fig. 1H).
To determine whether the survival of mice bearing IGO transplants (IGO mice) reflected patient disease characteristics, clinical features of the patients from whom organoids were derived were compared to features of the IGO tumors. 86% (6/7) of Fast-progressors were derived from stage 3/4 patients, whereas only 44% of Slow-progressors (4/9) were derived from stage 3/4 patients (Supplementary Table S2). The common metastatic sites for both IGOs and OGOs were lung and liver, followed by peritoneum (Supplementary Table S3), similar to PDAC patients (31), and the metastatic rate of the Slow-progressor IGO models was less than the Fast-progressor IGOs, consistent with stage of the patients from whom the organoids were derived. We observed a slightly reduced metastatic frequency in the Fast-progressors in the IGO model compared to the same organoids engrafted in the OGO model (Supplementary Table S3), potentially reflecting the more rapid death of IGO mice from obstructive jaundice that temporally precluded the development of metastases (Supplementary Fig. S1G, left panel). At humane endpoint, 43% (16/37) of Slow-progressor IGOs (3 out of 9 organoid lines) developed cystic tumors (Supplementary Fig. S1G, right panel; Supplementary Fig. S1H), although all IGOs except one (hT102) showed evidence of tumor progression with invasion into adjacent pancreatic tissues and/or distant metastasis (Supplementary Fig. S1H and S1I).
Next, we sought to investigate whether differences in the murine microenvironment where the organoids were engrafted may affect the proliferation of cancer cells and shape the histological features between IGO and OGO tumors. Accordingly, the number of actively proliferating cells was assessed by immunofluorescence (IF) staining for Ki67 on Fast- and Slow-progressor IGO and OGO tumor sections, and organoid-derived cancer cells were identified by expression of a human mitochondrial protein. We found that the Ki67 content in intraductal lesions of four IGO-derived Slow-progressors (hF23, hF27, hT93, and hT102) was significantly lower than their corresponding OGO tumors, while proliferation in Fast-progressors was consistently comparable in both the IGO or OGO setting (Supplementary Fig. S1J and S1K). Notably, while many xenografted cells in the intraductal areas of Fast-progressors expressed Ki67, in Slow-progressors, only human PDAC cells adjacent to murine duct cells were positive (Fig. 1I and 1J). OGO transplants uniformly displayed abundant interactions between cancer and stromal cells as well as prominent collagen deposition in comparison to tumors derived from IGO transplants (Fig. 1K). Invasive tumors following the progression of IGO transplants were oftentimes contiguous with the duct and accompanied by an abundant stromal reaction (Fig. 1K). Thus, we observed differences in both the neoplastic histology and the grade of stromal reaction present in IGO compared to OGO transplants. Our results suggest that the pancreatic microenvironment engraftment site may dictate the resulting histological phenotypes.
Tumor microenvironment influences transcriptional subtypes of PDAC
We noted that a major difference between these two models was the microenvironment that the PDAC cells were engrafted in: while IGO transplants were exposed primarily to cues from the pancreatic ducts, OGO transplants were exposed to cues from the more varied tissue elements and cell types present in the interstitial space of pancreas, including blood vessels, nerves, connective tissue and pancreatic stellate cells - a resident fibroblast type in the pancreas. To evaluate whether the microenvironment in which organoids engraft influences the transcriptome of cancer cells, we performed RNA-sequencing (RNA-seq) using pairs of tumors derived from OGO- and IGO-transplants of 14 different organoid lines, taken at endpoint. We used the Callari et al (32) method to discriminate between human cancer cells and mouse stroma transcriptomes. Gene expression changes in cancer cells were determined by analyzing those reads that mapped specifically to the human reference genome. Gene set enrichment analysis (GSEA) using the Cancer Hallmarks database indicated that genes involved in EMT and interferon-ɑ/γ response were enriched in the OGO- relative to the IGO-xenografts (Fig. 2A). We confirmed that the EMT marker gene Vimentin was highly expressed in OGO tumors compared to IGO tumors in hF2 and hT93 tumors (Fig. 2B and 2C; Supplementary Fig. S2A-S2C). The previously described “Squamous PDAC” (17) and “Basal-like” (7) gene signatures were enriched in the OGO xenografts (Fig. 2D). Conversely, the previously described “Progenitor PDAC” (17) and “Classical” (7) signatures were enriched in IGO-derived tumors compared to OGO-derived tumors (17) (Fig. 2D). Moreover, the oxidative phosphorylation and fatty acid metabolism pathways, which have been associated with the PDAC progenitor/classical subtypes (4,33), were upregulated in cancer cells from IGO tumors (Fig. 2A). These results suggest that differences in the microenvironment to which engrafted cells are exposed can influence the metabolic phenotype as well as the molecular subtype of PDAC. Next, to evaluate whether xenografted cancer cells influence the transcriptome of mouse stromal cells in the microenvironment, gene expression changes in stromal cells were analyzed by reads mapped to the mouse reference genome. GSEA revealed the enrichment of genes involved in EMT, WNT, and TGF-β response in mouse stromal cells in the OGO- relative to the IGO-xenografts (Fig. 2E). The “ECM-rich” gene signature (9) and “Moffitt-Activated” signatures (7) were also found to be enriched in the mouse stromal cells in OGO xenografts (Fig. 2F). Contrastingly, genes involved in oxidative phosphorylation and xenobiotic metabolism as well as a “Moffitt-Normal” signature (7) were enriched in mouse stroma cells in IGO-derived tumors compared to OGO-derived tumors (Fig. 2F).
Figure 2. Molecular features of IGO model and OGO model.
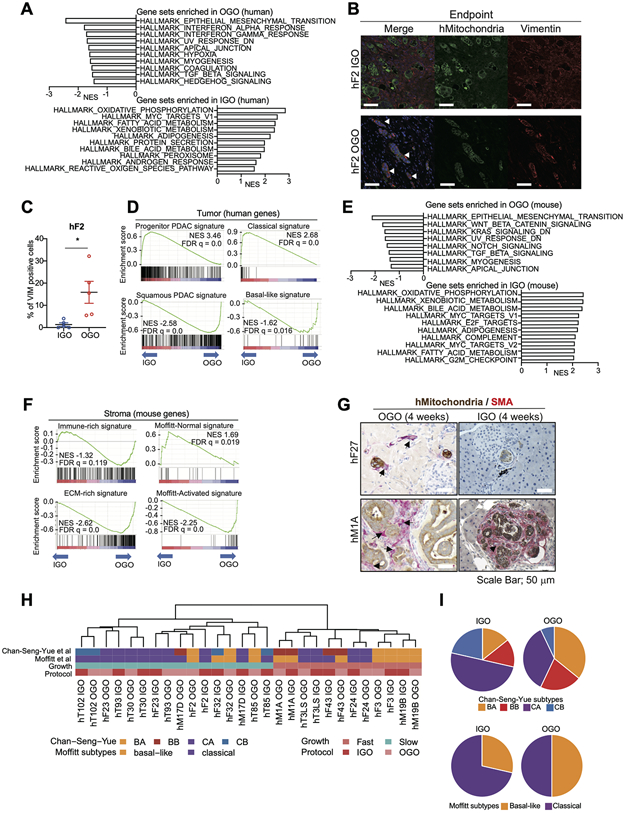
A) Hallmark pathways found significantly enriched by GSEA among genes differentially expressed in tumor cells of OGO tumors relative to IGO tumors. 10 gene sets with high Normalized Enrichment Score (NES) are shown. False Discovery Rate (FDR) q values of all gene sets are less than 0.25.
B) Representative IF staining for hMitochondria (green) and Vimentin (red) of tumors generated from IGO (top panels) and OGO (bottom panels) transplants of hF2. Scale bars, 100 μm.
C) Quantification of the percent of Vimentin-positive cells in the IF stains of h2 IGO and OGO transplants as described in (B). Data are means SEMs (n = 5 fields of view). P value, unpaired Student’s t test.
D) Enrichment of Progenitor PDAC signature (top left), Classical signature (top right), Squamous PDAC signature (bottom left) and Basal-like signature (bottom right) by GSEA among genes more highly expressed in tumor cells of IGO tumors and OGO tumors, respectively. NESs and FDR q values are shown.
E) Hallmark pathways found significantly enriched by GSEA among genes differentially expressed in stromal cells of OGO tumors relative to IGO tumors. Gene sets with high NES are shown. FDR q values of all gene sets are less than 0.25.
F) Enrichment of Progenitor PDAC signature (top left), Classical signature (top right), Squamous PDAC signature (bottom left) and Basal-like signature (bottom right) by GSEA among genes more highly expressed in stromal cells of IGO tumors and OGO-tumors, respectively. NESs and FDR q values are shown.
G) Co-IHC staining for hMitochondria (brown) and α-SMA (red) in pancreata of mice transplanted with hF27 or hM1A OGO (left panels) or IGO (right panels) xenografts, 4 weeks after transplantation. Black arrows, fibroblasts. Black arrow head, desmoplastic reaction. Scale bars, 50 μm.
H) Heatmap showing Chan-Seng-Yue (top bar) and Moffitt (second top bar) transcriptional subtypes in IGO and OGO transplants of the indicated organoid lines. BA, Basal-like-A; BB, Basal-like-B: CA, Classical-A; CB, Classical-B. Pink and blue horizontal bar (second bottom) indicates Fast- and Slow-progressor organoids, respectively. Red and pink horizontal bar (bottom) indicates IGO and OGO transplants, respectively.
I) Pie charts showing the proportions of the Chan-Seng-Yue (top) and Moffitt (bottom) transcriptional subtypes in IGO and OGO transplants.
We then sought to examine how the microenvironment influences tumor progression in the IGO model. At four to eight weeks following the establishment of an OGO model, the PDAC tumors had formed extra-ductal neoplasms with infiltration and activation of stromal cells (Fig. 2G; Supplementary Fig. S2D). In contrast, four weeks after IGO transplant, some organoids, such as hM1A, demonstrated invasion through the duct and generated lesions with abundant desmoplastic reaction, while other organoids, such as hF27, expanded intraductally and lacked an associated desmoplastic reaction (Fig. 2G; Supplementary Fig. S2D). Previous studies have shown that the microenvironment can influence the heterogeneity and aggressiveness of PDAC lesions through up-regulation of signaling pathways including the TGF-β pathway, a major inducer of EMT (7,22,34,35). As shown above, EMT and TGF-β pathways were upregulated in both the cancer cells and the stromal cells of OGO tumors. To assess the extent of TGF-β signaling in tumor development in IGO and OGO tumors, we analyzed the difference between IGO and OGO tumors at an early time point. To do so, we collected neoplastic cells from hM1A and hF24 xenograft tumors 4 weeks after transplant by either the IGO or OGO method, using human EpCam-coated magnetic beads and RNA was isolated for qPCR analysis. The hF24 organoid line is SMAD4-null (Supplementary Fig. S2E) and therefore lacks SMAD4-dependent TGF-β signaling. Accordingly, the TGF-β effectors SERPINE1 and SMAD7 were undetectable in both OGO- and IGO-derived transplants of hF24 (Supplementary Fig. S2F). Consistent with our hypothesis, SERPINE1 and SMAD7 were more highly expressed in the neoplastic cells arising from OGO transplants of hM1A compared to those from IGO transplants (Supplementary Fig. S2F), suggesting that TGF-β signaling is accentuated by the microenvironment present in the OGO model, compared to that of the IGO model. To confirm which cells express TGF-β ligands, we performed qPCR for the isolated tumor cells 4 weeks after transplantation (Supplementary Fig. S2G) and RNA-ISH for Tgfb1 and Tgfb2 in stromal cells using mouse specific probes (Supplementary Fig. S2H). Both TGFB1 and TGFB2 were more highly expressed in cancer cells in OGO tumors compared to IGO tumors (Supplementary Fig. S2G). Moreover, increased expression of Tgfb1 and Tgfb2 was observed in stromal cells in OGO tumors compared to IGO tumors (Supplementary Fig. S2H). These results suggested that active autocrine and paracrine TGF-β signaling in both tumor and stromal cells may play a major role in tumor progression in OGO tumors.
To confirm that the transplantation method can influence the molecular subtype of the resulting tumor, we assessed OGO- and IGO-derived hM1A cells from tumors 4 weeks post-transplantation for levels of molecular subtype markers (Supplementary Fig. S2I). S100A2, FAM83A, IGTA3, KRT5, and C16orf74, markers of squamous/basal-like PDAC (4,7,12,17) were up-regulated, while TFF2, AGR2, REG4, LGALS4, ANXA10, and DDC, markers of progenitor/classical PDAC (4,7,12,17)were down-regulated in hM1A OGO tumors compared to IGO tumors (Supplementary Fig. S2I) supporting our RNA-seq findings (Fig. 2D). In hF24 which is SMAD4-null, ITGA3 (squamous/basal-like marker) was upregulated and DDC (progenitor/classical marker) was downregulated in OGO tumors compared to IGO tumors (Supplementary Fig. S2I). Collectively, our results show that the microenvironment in which organoids are engrafted influences molecular characteristics of the cancer cells.
To evaluate which model better represents the diversity of transcriptional subtypes of human PDAC, we compared the proportion of subtypes in IGO and OGO tumors with those published in the COMPASS database (19), in which authors reported that previously reported “basal-like” and “Classical” subtypes were split into “Basal-like-A (BA)” and “Basal-like-B (BB)”, and “Classical-A (CA) and “Classical-B (CB)”, respectively. Genes from four signatures (19) were used for non-biased hierarchical clustering (Fig. 2H) and we found that the IGO tumors revealed a predominantly comprised CA and CB subtypes, whereas the OGO tumors revealed the predominantly comprised BA and BB subtypes. The distribution of subtypes in IGO tumors best reflected the relative distribution of subtypes reported in COMPASS database (Fig. 2I) To further confirm if IGO models represent human PDAC, we performed IHC analyses for S100A2 (basal-like marker), TP63 (squamous marker), and GATA6 (progenitor marker) in human PDAC sections. The fine needle aspiration sample from which the hF27 organoid was derived contained high GATA6 staining and low S100A2 and TP63 staining, whereas the original tumor from which the hM19B organoid was derived contained high S100A2 and low GATA6 staining (Supplementary Fig. S2J). IGO transplants of hM19B were classified as basal-like (Fig. 2H), consistent with the IHC results of subtype markers for the limited number of available samples. Additionally, the IHC staining of subtype markers for IGO transplants of hM19B and hF27 was similar to the IHC results for their corresponding human samples (Supplementary Fig. S2K). Collectively, these results suggest that the IGO model can recapitulate the transcriptomic subtypes of human PDAC.
Indolent and invasive tumor phenotypes correlate with molecular subtypes
In the IGO model, organoids gave rise to distinct intraductal and invasive phenotypes that resemble the different histopathological phenotypes observed in human PDAC progression (25). We sought to investigate whether observed differences in tumor kinetics were a function of intrinsic features of organoids observed in vitro. In vitro proliferation rates of 7 organoids were not significantly different and did not correlate with the in vivo tumor growth kinetics (Supplementary Fig. S3A and S3B). However, consistent with the findings of another group (36), hM1A and hF3 organoids (Fast-progressors) exhibited a dense, filled-in spherical morphology compared to the hollow cystic organoids formed by hF2 and hF27 (Slow-progressors) (Supplementary Fig. S3C).
To gain insight into the molecular mechanisms underlying the biological differences between Fast- and Slow-progressor IGO transplants, we performed global mRNA sequencing on 6 Fast- and 9 Slow-progressors. Differentially expressed gene (DEG) analysis identified 231 and 335 genes significantly up-regulated in Fast- and Slow-progressors, respectively (“Fast-signature” and “Slow-signature,” respectively, Fig. 3A). To determine whether there was a correlation between our progression phenotype and previously identified transcriptional subtypes, we compared genes up-regulated in Fast- and Slow-progressors with “Squamous”, “Basal-like” “Progenitor” and “Classical” gene sets. By GSEA, EMT and proliferation/cell cycle-related genes (i.e. MYC-targets, E2F-targets and G2M checkpoint signatures) were enriched in Fast-progressors (Fig. 3B). Additionally, KRAS and mTORC1 signaling genes, which have been associated with PDAC progression (37), were also enriched in the Fast-progressors (Fig. 3B), while bile acid metabolism and fatty acid metabolism signaling genes were enriched in the Slow-progressors (Fig. 3B). We also observed that the progenitor-type PDAC signature and classical signatures were significantly enriched in Slow-progressors, whereas the Squamous PDAC signature and basal-like signature were significantly enriched in Fast-progressors (Fig. 3C). To confirm these results, we examined the expression levels of known progenitor/classical and squamous/basal-like marker genes by qPCR (Supplementary Fig. S3D). Among the progenitor/classical markers (4,7,17), LYZ, AGR2 and FOXA2 were significantly more highly expressed and GATA6 was modestly increased in Slow- compared to Fast-progressors, while CAV1 and S100A2 were more highly expressed among the squamous/basal-like markers (4,7,17) in Fast-progressors (Supplementary Fig. S3D). Moreover, IHC staining of classical markers AGR2 and GATA6, and basal-like marker, S100A2 confirmed these results (Fig. 3D and 3E; Supplementary Fig. S3E-S3G)
Figure 3. Fast progression to invasive carcinoma in the IGO model is correlated with a basal-like phenotype.
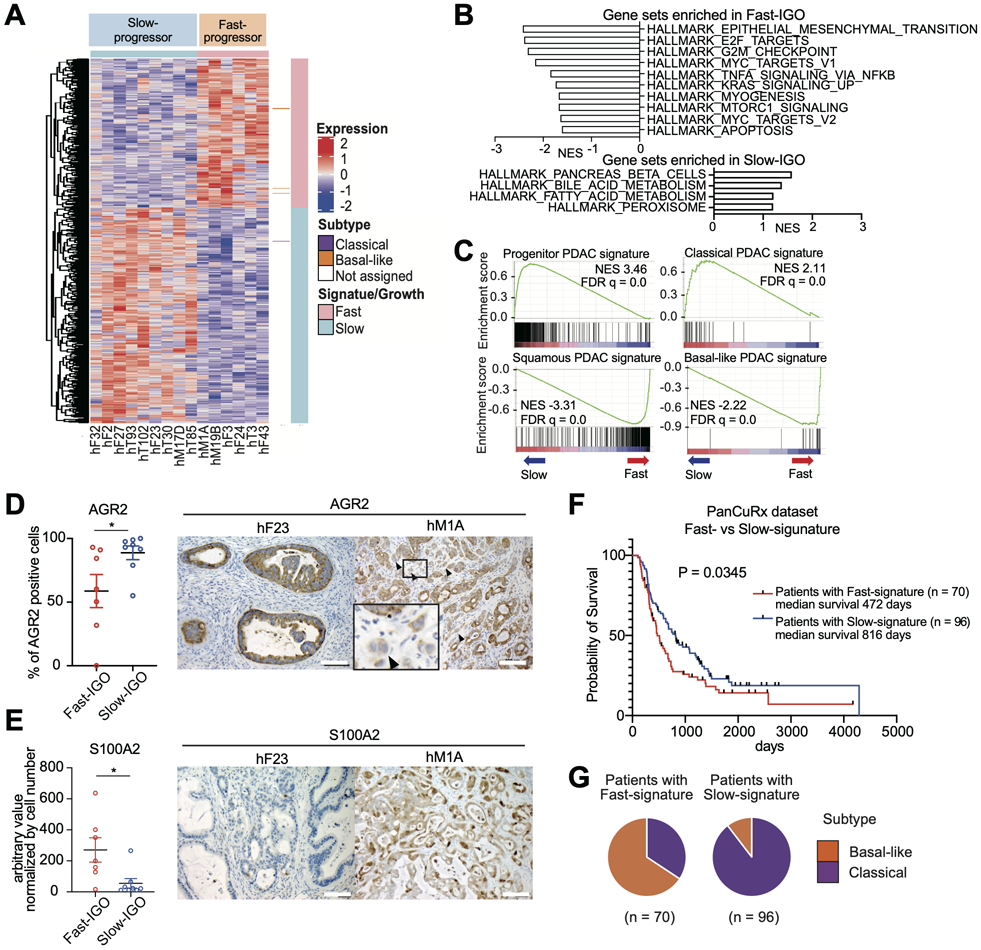
A) Heatmap of unbiased hierarchical clustering of expression levels of differentially expressed genes in Fast- relative to Slow-progressor IGO transplants of the indicated organoid lines. Pink and blue horizontal bar at the top indicates Fast- and Slow-progressor organoids, respectively. Pink and blue vertical bar indicates Fast- and Slow-progressor gene signatures, respectively. Purple and orange marks to the right of the heatmap indicate known classical and basal-like genes, respectively.
B) Hallmark pathways found significantly enriched by GSEA among genes differentially expressed in Fast- relative to Slow-progressor IGO tumors. Gene sets with high NES are shown. FDR q values of all gene sets are less than 0.25.
C) Enrichment of Progenitor PDAC signature (top left), Classical signature (top right), Squamous PDAC signature (bottom left) and Basal-like signature (bottom right) by GSEA among genes more highly expressed in Slow-progressors and Fast-progressors, respectively. NESs and FDR q values are shown.
D) Quantification of the percent of AGR2 positive cells in the IHC stains of Fast- and Slow-IGO transplants. Representative IHC images of IGO transplants of hF23 (left) and hM1A (right) at endpoint. Scale bars, 100 μm. Representative Data are means SEMs (n ≧ 3 fields of view). P value, unpaired Student’s t test.
E) Quantification of the percent of S100A2 positive cells in the IHC stains of Fast- and Slow-IGO transplants. Representative IHC images of IGO transplants of hF23 (left) and hM1A (right) at endpoint. Scale bars, 100 μm. Representative Data are means SEMs (n ≧ 3 fields of view). P value, unpaired Student’s t test.
F) Kaplan-Meier curve of percent overall survival based on PanCuRx data for patients found to express either the Fast-signature (red) or Slow-signature (blue). Median survival and log-rank (Mantel-Cox) test P value are shown.
G) Pie charts showing the percentage of PanCuRx patients whose tumors were classified as either classical (purple) or basal-like subtype (orange) for patients whose tumors expressed the Fast-signature (left) or Slow-signature (right).
To determine whether our Fast- and Slow-progressor gene signatures are relevant to human PDAC, we performed unbiased, hierarchical clustering of the PanCuRx Translational Research Initiative dataset of locally advanced PDAC patients (38) based on expression of our Fast- and Slow-signature genes (Supplementary Fig. S3H). Interestingly, the Fast- and Slow-signature genes divided patients into two groups which we classified as “Fast-Signature” patients and “Slow-Signature” patients (Supplementary Fig. S3H). Fast-Signature patients exhibited significantly worse prognosis compared to Slow-Signature patients (Fig. 3F). Moreover, tumors from 48 out of 70 (65.7%) Fast-Signature patients were classified as belonging to the basal-like subtype, and 86 out of 96 (89.6%) Slow-Signature patients were classified as the classical subtype (Fig. 3G). Taken together, these results show that our IGO-defined Fast- and Slow-signatures correlate with the basal-like and classical transcriptional subtypes of human PDAC, respectively, and with patient prognosis. Moreover, our results suggest that the growth kinetics of tumors derived from IGO transplants are predictive of PDAC transcriptional subtypes.
Transcriptional shift is governed by tumor cell plasticity in vivo
Other groups have reported that transcriptional subtypes correlate with histological manifestations and immunohistochemical staining for common squamous markers such as CK5, CK6 and TP63 (15,18). We performed IHC for TP63 and S100A2, to distinguish basal-like and classical cell types in IGO-tumors. We found that the immunostaining pattern of TP63 and S100S2 was heterogeneous (Supplementary Fig. S4A and S3F), suggesting that the squamous differentiation program may be dynamic. Previously, we reported that based on the expression of genes in PDAC organoids cultured in vitro, organoids can be classified into classical and basal-like subtypes (24). Interestingly, the previously identified in vitro transcriptional subtypes were not always concordant with the in vivo transcriptional subtypes of IGO transplants (Supplementary Fig. S4B). The Fast-progressor organoids hF3 and hM19B were found to have a basal-like subtype both in vitro and in IGO transplants (Supplementary Fig. S4B). Accordingly, hF3 expressed the basal-like markers S100A2 and TP63 in vitro (Supplementary Fig. S4C). In contrast, the Fast-progressor organoid hM1A was classified as the classical subtype in vitro but demonstrated a basal-like phenotype in vivo (i.e. a “hybrid” phenotype, Supplementary Fig. S4B). hF24 was classified as classical subtype both in vitro and in vivo and did not express the basal-like markers S100A2 and TP63 in vitro, however, hF24-IGO tumors contained S100A2 and TP63 positive cells in vivo (Supplementary Fig. S3F and S4A). The ability of the same organoid to manifest different subtypes depending on context suggests that transcriptional subtypes may be affected by microenvironmental cues.
Recent single-cell analyses have demonstrated the coexistence of basal-like and classical subtypes within a single tumor with the relative proportions of these cells determining the overarching subtype at the bulk transcriptome level (19,39). In another recent study, evolutionary analysis of mixed phenotype tumors, comprising classical and basal-like features, from autopsy patients revealed that the basal-like subtype represented a subclone contained within an otherwise classical tumor (18). We questioned whether the hybrid phenotype of organoids in the IGO xenograft model might result from clonal selection or, alternatively, cellular plasticity driven by exposure to different environmental cues. To distinguish between these two possibilities, we labeled organoids with the Autobow system, which facilitates lineage tracing (Supplementary Fig. S4D) (40). In this system, three fluorescent proteins with loxP sites and Cre are combined in a single cassette, and Cre activation results in the random and irreversible activation of various combinations of the three fluorescent proteins. We used the organoids hM1A and hF24 for these studies because they displayed a hybrid phenotype. Following introduction of the Autobow cassette to hM1A and hF24 and Cre activation in vitro, we confirmed that the system was working by detection of multicolor expression in cultured organoids (Supplementary Fig. S4E). Next, single-cell suspensions of these Autobow-expressing hM1A and hF24 organoids (hM1A-, hF24-Autobow) were transplanted by the IGO-method. Four weeks post-transplantation of hM1A-Autobow, each lesion expressed a single fluorescent protein, indicating that each individual lesion likely arose from a single engrafted cell (Supplementary Fig. S4F). At a later timepoint (8 weeks post-transplantation) of hM1A-Autobow, multi-focal and multichromatic tumors were observed (Supplementary Fig. S4G), indicating merging of individual clonal tumors. We observed both inter-and intra-clonal differences in histological grade, suggesting that individual clones retain the capacity to generate different histopathological phenotypes. Notably, we observed intra-clonal differences in the expression of basal-like and classical markers by IHC staining: the intraductal region of a monoclonal lesion expressed the classical marker GATA6, while an invasive region derived from the same clone expressed the basal-like marker S100A2 (Fig. 4A). These data suggest that the subtype identity of a transplanted single clone is not fixed and can be determined by the tumor microenvironment in vivo.
Figure 4. Cellular plasticity contributes to shifting molecular subtypes.
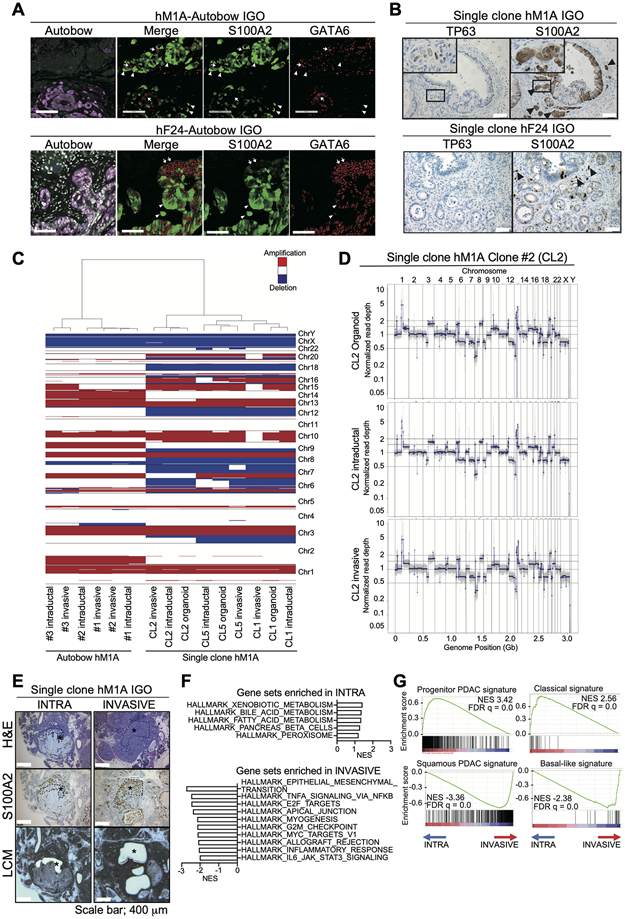
A) Representative images of IF staining for Autobow-colors (mCerulean, PhiYFP, and mKate2) (rightmost panel), IF staining for Merge, S100A2, and GATA6 (right three panels). Stainings were performed on sequential sections of tumors generated from IGO transplant of hM1A- (top) and hF24-Autobow (bottom) at endpoint. For IF, blue color, Cerulean; green color, Phi-YFP; red color, mKate and green color, S100A2; red color, GATA6. White arrow heads, cells with strong S100A2 and weak GATA6 staining. White arrows, cells with strong GATA6 and weak S100A2 staining. Scale bars, 100 μm.
B) Representative IHC staining for S100A2 and TP63 on sequential sections of tumors generated from IGO transplant of a clonal hM1A (top panels) and a clonal hF24 (bottom panels). Black arrow heads, invading cells with strong staining. Scale bars, 100 μm. Insets, magnified images.
C) Copy number variations in intraductal and invasive lesions of IGO transplants of Autobow hM1A organoid (Area #1, #2, and #3), in the clonal hM1A organoids (clone #1, #2, and #5 (CL1, −2, and −5)), and intraductal and invasive lesions of IGO transplants of corresponding clonal hM1A organoids. Hierarchical clustering of organoids and LCM samples indicated at a resolution of 20,000 bins (150 kbp per bin). Red indicates amplification, whereas blue indicates deletion.
D) Representative copy number profiles from DNA of clonal hM1A organoid clone #2 and DNA of IGO transplants of clonal hM1A organoid clone #2 excised by LCM in (C).
E) Representative H&E staining (top), S100A2 IHC staining (middle), and images following LCM (bottom) of tumors generated from IGO transplant of a clonal hM1A line. Intraductal lesions that are negative (left panel, INTRA) and invasive lesions (right panel, INVASIVE) were excised by LCM. Black asterisks and black dotted lines, excised area. Scale bars, 400 μm.
F) Hallmark pathways found significantly enriched by GSEA among genes differentially expressed in tumor cells of “INVASIVE” relative to “INTRA” lesions. Gene sets with high NES are shown. FDR q values of all gene sets are less than 0.25.
G) Enrichment of Progenitor PDAC signature (top left), Classical signature (top right), Squamous PDAC signature (bottom left) and Basal-like signature (bottom right) by GSEA among genes more highly expressed in “INVASIVE” and “INTRA” lesions, respectively. NESs and FDR q values are shown.
To exclude the possibility that different clones with the same color give rise to a single lesion by chance, hM1A and hF24 clonal organoids were derived from single cells in vitro and transplanted intraductally (Fig. S4H). hM1A and hF24 clones formed tumors with varied intratumor histology consisting of invasive lesions that expressed the basal-like markers S100A2 and TP63 as well as intraductal lesions negative for these markers (Fig. 4B). These results demonstrate that shifts in PDAC subtype during the progression of tumors formed from hybrid organoids are likely due to cancer cell plasticity rather than altered clonality. To further determine if these organoids are clonal, we performed laser capture microdissection (LCM) to isolate intra-ductal (S100A2-negative, lacking stromal interactions) and invasive (S100A2-positive, maintaining stromal interactions) lesions of IGO transplants of hM1A clones for DNA-seq analysis (Fig. 4C, Supplementary Fig. S5A). hM1A clones #1, #2, and #5 (CL1, −2, and −5) clustered together with LCM samples from both intraductal and invasive lesions of IGO tumors (Fig. 4C). We demonstrated that copy number profile of the clonal hM1A CL2 organoid was similar to the copy number profiles of intraductal and invasive lesions of its corresponding IGO transplants (Fig. 4D), suggesting that a single clone could indeed give rise to both intraductal (S100A2 negative) and invasive (S100A2 positive) lesions. CL1 and CL5 generated nearly identical copy number profiles in vitro and in vivo, with minor focal differences that can be attributed to noise or slight differences in data analysis methods used for organoids and IGO tumors (Supplementary Fig. S5B). We also performed LCM to isolate intra-ductal and invasive lesions from IGO transplants of Autobow-hM1A clones for DNA-seq analysis (Fig. 4C, Supplementary Fig. S5C). Autobow-hM1A lesion #1, #2, and #3 clustered separately from clonal hM1Al organoids, whereas intraductal and invasive lesions from same area clustered closely (Fig. 4C; Supplementary Fig. S5D) confirming that they likely originated from the same clone. We observed identical copy number profiles between intraductal lesions and invasive lesions of IGO tumors of Autobow-hM1A (Supplementary Fig. S5D). Collectively, the presence of intraductal and invasive lesions derived from a single clone suggests that cell plasticity drives histologically distinct phenotypes.
Tumor-stromal interaction contributes to tumor heterogeneity (22) and may influence the dynamic shift of molecular subtypes which are associated with histological phenotypes. In order to assess whether stromal interaction correlates with molecular subtype, we performed RNA-seq on LCM intra-ductal and invasive lesions of IGO transplants of hM1A clones (Fig. 4E). GSEA identified expression of EMT, TNFɑ signaling, and cell cycle-related gene sets to be enriched in invasive compared to intra-ductal lesions (Fig. 4F). EMT and proliferation signaling were previously found to be induced in cancer cells by interaction with stroma (22). Expression of the Squamous PDAC signature and the Basal-like signature were also enriched in invasive lesions, while the Progenitor PDAC and Classical signatures were enriched in intraductal lesions (Fig. 4G). Thus, IGO tumors derived from some PDAC organoids can simultaneously display squamous/basal-like and progenitor/classical phenotypes in a lesion-dependent manner, and the shift of subtypes is likely influenced by the tumor microenvironment.
Induction of a basal-like phenotype in invasive areas of IGO tumors is associated with activation of the RAS pathway
We next sought to understand the molecular mechanisms underlying the dynamic nature of the molecular subtypes observed in IGO-derived tumors. Allelic imbalance and high expression of mutant KRAS have been associated with aggressive undifferentiated histological phenotypes in PDAC (19,41). Therefore, we hypothesized that levels of KRAS signaling were higher in invasive compared to intraductal regions of IGO-derived lesions. To determine whether KRAS signaling levels varied in different lesions within IGO-derived tumors, we evaluated the levels of phosphorylated ERK (p-ERK), a downstream effector of KRAS signaling, in Fast- and Slow-progressor IGO tumors. Elevated levels of p-ERK were observed in Fast-progressor compared to Slow-progressor tumors (Supplementary Fig S6A and S6B). Next, we sought to correlate the levels of p-ERK with molecular subtypes and histology in IGO-derived tumors of the Slow-progressor organoids, hF2 and hF27. While tumors derived from these organoids formed mostly well-differentiated intraductal lesions, smaller invasive lesions that express the basal-like marker S100A2 could also be detected (Fig. 5A; Supplementary Fig. S1I). Consistent with a role for KRAS signaling in promoting invasiveness, levels of p-ERK or phosphorylation of MAPK downstream target S6 (p-S6) were higher in invasive areas compared to intraductal areas in hF2 tumors and correlated with areas of high expression of S100A2 (Fig. 5A-5C; Supplementary Fig S6C and S6D). Similarly, phospho-ERK or p-S6 levels were modestly up-regulated in invasive lesions of hF27 tumors, which is consistent with the observed high expression of S100A2 (Fig. 5A-5C; Supplementary Fig S6C and S6D) and Ki67 staining (Supplementary Fig. 1I and 1J). These results suggest that differential activation of the RAS pathway in intraductal and invasive lesions may contribute to the dynamic shifting of molecular subtypes in Slow-progressors.
Figure 5. Induction of a basal-like phenotype in invasive areas of tumors derived from IGO transplants of Slow-progressor organoids is associated with activation of the RAS pathway.
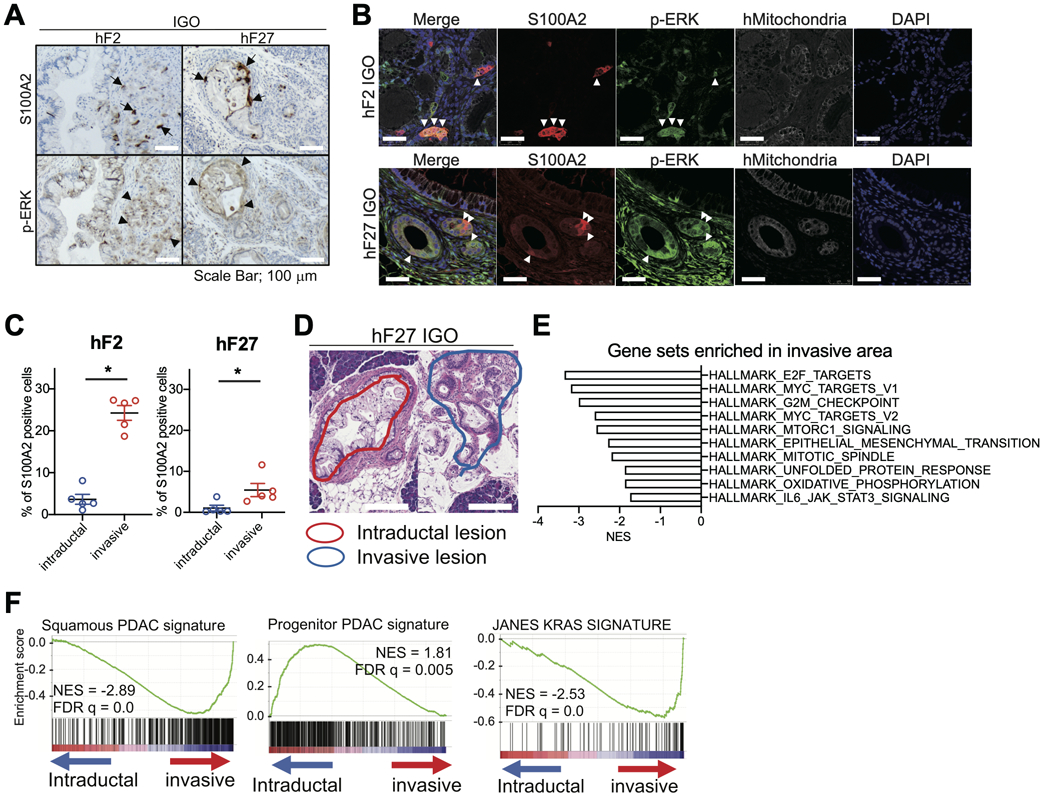
A) Representative IHC stainings for S100A2 (top) and p-ERK (bottom) in tumors derived from IGO transplants of hF2 or hF27 organoids. Black arrows, S100A2-positive cells. Black arrowheads, p-ERK-positive cells. Scale bars, 100 μm
B) Representative IF staining for hMitochondria (white), S100A2 (red), and p-ERK (green), with DAPI counterstain (blue) on section of tumor derived from IGO transplant of hF2 (top panels) and hF27 (bottom panels). Scale bars, 100 μm.
C) Quantification of the percent of S100A2-positive cells in the IHC stains described in (A). Data are means SEMs (n=5). P value, unpaired Student’s t test.
D) Representative H&E staining showing intraductal (red) and invasive (blue) regions excised by LCM from a tissue section of a tumor derived from an IGO transplant of hF27.
E) Hallmark pathways significantly enriched by GSEA among genes more highly expressed in invasive relative to intraductal lesions of IGO transplants of Slow-progressor organoids. Gene sets with high NES are shown. FDR q values of all gene sets are less than 0.25.
F) GSEA showing enrichment of Squamous PDAC signature, Progenitor PDAC signature, and JANES KRAS signature (42) among genes more highly expressed in invasive compared to intraductal lesions of IGO transplants of Slow-progressor organoids. NESs and FDR q values are shown.
To identify transcriptional features enriched specifically in invasive areas, we performed LCM and RNA-seq on intraductal and invasive regions of tumors derived from IGO transplants of the Slow-progressor organoids hF2 and hF27 (Fig. 5D and Supplementary Fig S6E). GSEA using the Cancer Hallmarks database identified the E2F target genes, G2M checkpoint genes, and EMT genes as enriched among genes more highly expressed in invasive areas (Fig. 5E), consistent with the presence of a basal-like phenotype in the invasive regions of tumors generated from IGO transplants of Slow-progressor organoids. Accordingly, the classes of genes more highly expressed in the invasive compared to the intraductal regions of tumors generated from IGO transplant of Slow-progressor organoids (Fig. 5E) were quite similar to the classes of genes more highly expressed in Fast- compared to Slow-progressor tumors (Fig. 3B). For example, as was observed in the comparison of Fast- and Slow-progressors, genes from the Squamous and Basal-like PDAC and “Janes KRAS gene signatures” (42) were enriched in invasive areas relative to the intraductal areas of IGO transplants of Slow-progressor organoids (Fig. 5F; Supplementary Fig S6F), while the Progenitor and Classical PDAC signature were enriched in the intraductal regions (Fig. 5F; Supplementary Fig S6F). These results support a role for increased RAS signaling in promoting a more invasive and basal-like phenotype in the IGO model.
KRAS hyperactivation promotes a basal-like phenotype
To further determine whether differences in RAS signaling could underlie the different phenotypes of Fast- and Slow-progressors, we analyzed KRAS copy number (CN) in both groups using FACETS (Fraction and Allele-Specific Copy Number Estimates from Tumor Sequencing). KRAS CN was significantly higher in Fast-progressor compared to Slow-progressor organoids (Fig. 6A). In line with these results, expression of DUSP6, a target gene of KRAS signaling, correlated positively with KRAS CN (Fig. 6B). We next sought to determine whether increased KRAS signaling is sufficient to promote an invasive phenotype. To this end, we chose two Slow-progressor organoids harboring high CN of the classical subtype marker GATA6 and low CN of KRAS (KRASG12D) (hF23 and hF27, Supplementary Fig. S7A). We transfected these organoids with a cDNA encoding an estrogen receptor fused to oncogenic KRASG12D, that functions in a 4-hydroxytamoxifen (4-OHT)-inducible manner. Hyperactivation of oncogenic ER-KRASG12D in vitro by 4-OHT treatment resulted in increased levels of p-ERK and expression of DUSP6 (Supplementary Fig. S7B and S7C). Additionally, hyperactivation of KRASG12D resulted in a change in hF23 and hF27 organoid morphology, characterized by the development of multiple cell layers resembling the morphology of Fast-progressor organoids cultured in vitro (Supplementary Fig. S3C; Fig. 6C and 6D). We observed no changes in proliferation rates upon hyperactivation of oncogenic ER-KRASG12D in vitro by 4-OHT treatment (Supplementary Fig. S7D), which reflected the lack of differences in the proliferation rates in Fast- and Slow- progressor organoids (Supplementary Fig. S3A and S3B).
Figure 6. KRAS hyperactivation promotes a basal-like phenotype.
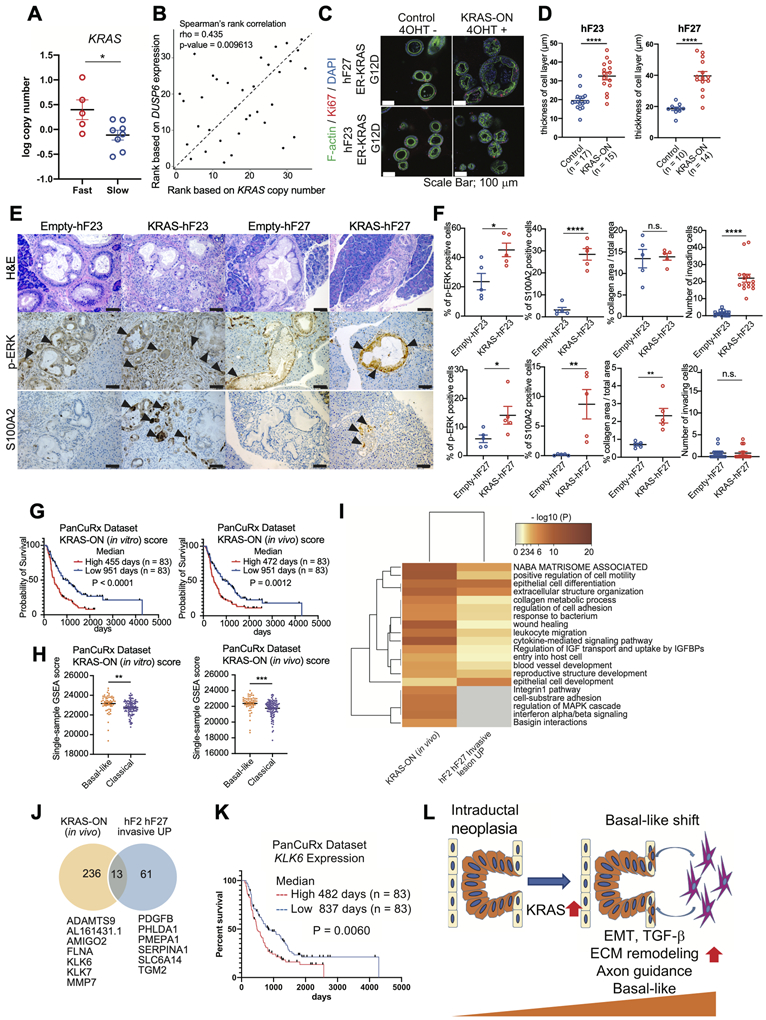
A) FACETS log KRAS copy number in Fast- (n=5, red) and Slow- (n=8, blue) progressor organoids. P value, unpaired Student’s t test.
B) Spearman’s rank correlation of FACETS KRAS copy number with normalized read counts of DUSP6 in organoids cultured in vitro (n = 35). Rho value and two-tailed P value are shown.
C) Representative IF staining for Ki67 (red) and F-actin (green) with DAPI counterstain of the indicated ER-KRASG12D-expressing organoids treated with 0.1 μM 4-OHT or untreated for 48 hours. Scale Bars, 100 μm.
D) Quantification of the thickness of cell layers of indicated ER-KRASG12D-expressing organoids treated with 0.1 μM 4-OHT or untreated for 48 hours as described in Figure 5C. Numbers of organoids analyzed were indicated. Average diameters of 3 locations in each organoid are shown. Data are means SEMs. P value, unpaired Student’s t test.
E) Representative H&E images (top), and IHC staining for p-ERK (middle panels) and S100A2 (bottom panels) in pancreata of mice transplanted with ER-KRASG12D-expressing organoids, 4 weeks after tamoxifen induction. Scale bars, 100 μm.
F) Quantification of percentage of p-ERK positive cells (left panels), S100A2 positive cells (second left panels), percent of collagen area relative to total tumor area (second right panels), and number of invading cells (right panels) of IGO transplant with ER-KRASG12D-expressing organoids as described in (E). Data are means SEMs (n = 5). P value, unpaired Student’s t test.
G) Kaplan-Meier curve analysis showing overall survival based on data deposited in the PanCuRx dataset for patients with high (red) and low (blue) KRAS (in vitro) score (left) and KRAS (in vivo) score (right). Median survival of 2 groups and log rank (Mantel-Cox) test P value are shown.
H) Single-sample GSEA (ssGSEA) scores of KRAS (in vitro) (left) and KRAS (in vivo) (right) in PanCuRx patients whose tumors were classified as classical (purple) and basal-like (orange) subtype.
I) Heatmap showing the Gene Ontology overlap between genes significantly up-regulated in KRAS-IGOs relative to Empty-IGOs (KRAS-ON (in vivo)) and genes significantly up-regulated in invasive relative to intraductal lesions of 2 IGO-derived tumors (hF2 and hF27) from the LCM experiment (hF2 hF27 Invasive UP).
J) Venn diagram showing the overlap between genes significantly up-regulated in KRAS-IGOs relative to Empty-IGOs (yellow) and genes significantly up-regulated in invasive relative to intraductal lesions of 2 IGO-derived tumors (hF2 and hF27) from the LCM experiment (blue). Numbers of genes and names of 13 shared genes between these data sets are indicated.
K) Kaplan-Meier curve of the percent overall survival for PanCuRx patients with high (red) or low (blue) expression of KLK6. Median survival of 2 groups and log rank (Mantel-Cox) test P value are shown.
L) Model explaining the KRAS-driven shift from intraductal neoplasia to a basal-like phenotype and a list of gene pathways associated with the basal-like shift.
Next, RNA-seq analysis was used to compare gene expression in organoids where the ER-KRASG12D was hyperactivated by 4-OHT treatment (KRAS-ON) compared to untreated ER-KRASG12D organoids (Control). DEG analysis indicated that 446 and 291 genes were significantly up- and down-regulated respectively in KRAS-ON compared to Control organoids (designated “KRAS-ON (in vitro)” and “Control (in vitro)” signatures). Similar to the pathways found up-regulated in invasive regions of IGO tumors, GSEA revealed the enrichment of EMT and TNFɑ signaling via NFKB pathways as well as the Squamous and Basal-like PDAC signature in KRAS-ON relative to Control organoids, while E2F signaling, Interferon-ɑ response pathways and the Progenitor PDAC signature were enriched in Control organoids (Supplementary Fig. S7E and S7F). Accordingly, S100A2 and GATA6 protein levels were increased in KRAS-ON and Control organoids, respectively (Supplementary Fig. S7G).
To determine whether the expression of genes associated with KRAS hyperactivation promotes basal-like features in our IGO model in vivo, we transplanted Empty vector (Empty), and ER-KRASG12D (KRAS)- expressing hF23 and hF27 intraductally and induced KRAS activation in vivo by tamoxifen treatment for 4 weeks (Supplementary Fig. S8A). Increased p-ERK and S100A2 positive cells were observed in IGO tumors upon KRAS activation (Fig. 6E and 6F). There were no differences in tumor size and Ki67 staining between Empty IGO tumors and KRAS IGO tumors (Supplementary Fig. S8B-D). Notably, however, we did observe an increased number of invading cells in KRAS-hF23 IGO tumors and high-density level of collagen deposition in KRAS-hF27 IGO tumors compared to Empty IGO tumors (Fig. 6E and 6F; Supplementary Fig. S8C). This demonstrates that increases in oncogenic KRAS is sufficient to promote the invasive phenotype and desmoplastic reaction in vivo.
Next, RNA-seq analysis was used to compare gene expression in KRAS IGO tumors compared to Empty IGO tumors. DEG analysis indicated that 249 and 116 genes were significantly up- and down-regulated in KRAS IGO tumors compared to Empty IGO tumors (designated “KRAS-ON (in vivo)” and “Control (in vivo)” signatures). EMT and TNFɑ signaling via NFKB pathways were enriched in KRAS IGO tumors relative to Empty IGO tumors, while E2F and G2M signaling pathways were enriched in Empty IGO tumors (Supplementary Fig. S8E). Additionally, the Squamous and Basal-like PDAC signatures were enriched in KRAS IGO tumors (Supplementary Fig. S8F). In contrast, the Progenitor PDAC signature was enriched in IGO tumors (Supplementary Fig. S8F).
To identify common gene signatures induced by KRAS hyperactivation between in vitro and in vivo, we performed gene ontology analysis using overlapping genes between KRAS-ON (in vitro) and KRAS-ON (in vivo) (Supplementary Fig. S8G). Wound healing and cell migration pathways were upregulated both in KRAS-ON (in vitro) and KRAS-ON (in vitro) (Supplementary Fig. S8H). The cell junction organization pathway was upregulated only in KRAS-ON (in vitro) reflecting the morphological change induced in vitro by KRAS hyperactivation (Fig 6C; Supplementary Fig. S8H). Interferon signaling and chemotaxis pathways were upregulated only in KRAS-ON (in vivo), suggesting crosstalk exists between cancer and stromal cells in vivo (Supplementary Fig. S8H). To determine whether KRAS-ON signatures are relevant to human PDAC, we calculated KRAS-ON (in vitro) and KRAS-ON (in vivo) scores of PDAC patients in the PanCuRx dataset using single sample GSEA (ssGSEA). Patients with high KRAS-ON (in vitro) and KRAS-ON (in vivo) scores both exhibited poorer prognosis (Fig. 6G). Moreover, patients with tumors classified as basal-like subtype exhibited high KRAS-ON scores, suggesting that the KRAS-ON signatures correlates with a basal-like subtype (Fig. 6H).
To understand how KRAS hyperactivation contributes to the invasion and basal-like features in our model, we compared genes up-regulated in invasive compared to intraductal areas of Slow-progressor tumors from our LCM expression data (Fig. 4F and G) with genes defining our KRAS-ON (in vivo) signature. Between the two gene sets, we identified 13 overlapping genes (Fig. 6I and 6J). Among them, AMIGO2 functions in axon guidance (43), and PMEPA1, ADAMTS9, MMP7, SERPINA1, TGM2, KLK6, and KLK7 function in EMT and ECM remodeling (44-50). For 4 out of these 13 genes (AMIGO2, KLK6, PMEPA1, and TGM2), high expression was significantly correlated with poorer prognosis in patients from the PanCuRx dataset (Fig. 6K; Supplementary Fig. S8I). Additionally, among those 4 genes, AMIGO2, KLK6, and TGM2 are known to be associated with the squamous phenotype (4,17). Taken together, these results demonstrate that increased KRAS signaling induces EMT, axon guidance, and ECM remodeling pathways, and together these pathways may promote a basal-like subtype in PDAC. Importantly, these cellular and molecular features of advanced PDAC can be recapitulated and systematically evaluated in an intraductal xenotransplantation model (Fig. 6L).
Discussion
Personalized models of PDAC have the potential to provide insight into the heterogeneous biology of the disease and to facilitate the identification of novel biomarkers and more effective therapies. To date, studies of the early stages of human PDAC development and the progression of this disease have relied largely on findings from surgically resected specimens. However, these specimens cannot be perturbed for functional studies, and such specimens do not fully recapitulate the more advanced stages of PDAC, which generally are not resectable. Our previous studies have shown that our organoid culture platform can be exploited for genomic and functional studies in order to predict chemosensitivity and to inform personalized treatment (24). Additionally, we previously demonstrated that a xenograft model in which organoids are injected into the pancreatic interstitial space (the OGO model) forms tumors that recapitulate the histologic heterogeneity of the tissue from which the organoids were derived (25). Here, we present a new xenograft model involving intraductal injection of dissociated organoids for the study of tumorigenesis and progression of human PDAC. This IGO xenograft model leads to initial engraftment of organoids as bona fide intraepithelial neoplasms in the host pancreatic duct system and to the development of intraductal and invasive lesions resembling human disease. Our IGO model offers distinct advantages over the prior OGO model because we can clearly identify demarcation of Fast- and Slow-progressors as determined by a bimodal distribution of survival. The Fast-progressors are hyperproliferative, invasive, and can activate cancer associated fibroblasts when compared to Slow-progressors, and transcriptional and molecular analyses show that the Fast- and Slow-progressor phenotypes recapitulate the basal-like/squamous and classical/progenitor subtypes of human PDAC, respectively (4,6,7). Furthermore, the distribution of molecular subtypes in the IGO model reflects the relative distribution of subtypes of human PDAC tumors described previously (19). Therefore, the IGO model may be a new tool to explore PDAC subtypes with correlation to human specimens for the purpose of discovering fundamental insights and developing translational applications.
The origin of PDAC is still debated, but several studies suggest that a subset of preneoplastic cells migrate from the intraductal lesions throughout the duct system, resulting in multiple lesions characterized by spatial and genetic divergences (51,52). PanIN lesions are well-known precursor lesions to PDAC, and high-grade PanIN commonly harbors signature mutations in KRAS and CDKN2A, and less commonly in TP53 or SMAD4, that are prevalent in invasive PDAC (28). Invasive PDAC has been reported to spread into and within the duct system, which is known as cancerization of the pancreatic ducts (29). Such lesions recapitulate the histopathologic features of high-grade PanIN lesions but harbor genetic alterations more characteristic of late-stage, invasive PDAC. In an alternate model of PDAC progression, Notta and colleagues demonstrate that a subset of PDAC tumors may not progress through a linear series of histologic stages associated with the gradual accumulation of driver mutations (53). Instead, within this subset, catastrophic mitotic events occur with the development of invasive cancer. Using our IGO model, we analyzed how cancer cells with pre-existing driver mutations behave within the non-transformed murine ductal system in the normal pancreata, where cancer cells arise originally, and found that injected cancer cells engraft at the peripheral ducts before expanding intraductally or invading into tissues. The propensity for cancer cells to invade into tissues is driven by cancer cell-intrinsic features, as IGO tumors derived from some organoids quickly invaded tissues, while tumors derived from other organoids were more likely to remain in the duct. We found that a subset of proliferating cancer cells invades through the basement membrane, replacing acinar cells, and inducing an abundant stromal reaction. Alternatively, neoplastic cells that expand intraductally migrate throughout the duct, histologically resembling cancerization of the ducts (51). These results support two possible behaviors of intraductal expansion of tumor cells.
We previously reported the presence of molecular subtypes (basal-like and classical) in organoids cultured in vitro. In this study, we uncovered discordance between the molecular subtypes of some organoids when transplanted as IGO tumors compared to those observed in vitro. We found increased expression of squamous and basal-like markers in the invasive areas of Fast-progressor tumors despite low or undetectable expression in most of the corresponding in vitro organoid cultures (4,17,23). Based on the concordance of molecular subtypes observed in vitro and in tumors derived from IGO, we were able to further classify organoids as having a fixed- or hybrid-subtype. The hybrid-subtype exhibited heterogeneity of molecular subtypes within a single tumor consistent with recent single-cell analyses and evolutionary analyses (18,19,39). Our findings suggest that molecular subtypes are regulated by both genetic alterations and dynamic epigenetic modulations consistent with the existence of different trajectories of PDAC progression observed in humans (52,53). In further support of the dynamic nature of PDAC subtypes, single clones of Fast-progressor organoids, labeled by the Autobow system, could form both intraductal and invasive lesions in the IGO model. This finding supports the notion that cell plasticity and not clonality, determines the heterogeneity in histological and molecular phenotypes observed in the hybrid-subtype. By comparing transcriptional signatures and molecular characteristics of the intraductal and invasive lesions, we have identified a set of genes and pathways that determine invasiveness. We hypothesize that these new genes may represent therapeutic targets that when antagonized will either prevent conversion of classical cells to basal-like or cause a reversion of basal-like to classical cells. Future research will better delineate the roles of these invasiveness genes and will test whether targeting such pathways is an effective therapeutic strategy.
The changes in molecular subtypes that we have observed following intraductal transplantation may be attributed to the influence of the microenvironment on the PDAC cells. In support of this hypothesis, while TGF-β signaling is suppressed in traditional organoid culture medium, in tumors derived from IGOs, TGF-β signaling is more highly activated in cancer cells that have invaded through the duct and are embedded in a dense stromal environment compared to those localized within an intraductal environment. EMT and proliferation signaling have been found to be invasive features of cancer cells induced by the stromal environment (22), and these pathways were enriched in invasive lesions compared to intraductal lesions. Further analysis of the crosstalk between stromal cells and cancer cells in the IGO model will help to determine the extent of stromal influence on the development of PDAC heterogeneity (26,54).
Increased allelic frequency and gene dosage of KRAS, a gene mutated in most PDAC tumors, have been shown to alter cellular metabolism, drug sensitivity, and histopathological/clinical outcome (9,19). We identified a RAS/EMT pathway as up-regulated in our bulk analysis of Fast- compared to Slow-progressors derived from IGO. These pathways were also up-regulated in the microdissected invasive areas compared to intraductal areas of Slow-progressor tumors. Consistent with these data, we find that Fast-progressors carry higher KRAS copy number compared to Slow-progressors, and 4/5 of the Fast-progressors harbor the stronger G12V allele whereas 6/8 of Slow-progressors harbor the weaker G12D and G12R alleles (Supplementary Table S2) (55). Moreover, increased mutant KRAS dosage is sufficient to induce basal-like/squamous features in Slow-progressor organoids. Similar to previous reports correlating gene dosage with clinical outcome, we find that expression of a “KRAS-ON” gene signature, which we identified using our KRAS overexpression model, correlates expression of a basal-like/squamous gene signatures and poor prognosis in PDAC patients (41). However, increased KRAS signaling is not required for the basal-like/squamous subtypes since 2/3 of our IGO tumors derived from wild-type KRAS organoids are Fast-progressors (Supplementary Table S2), and prior work has shown that TP63 and GLI2 can dictate a basal-like state in PDAC cells (16,17). Furthermore, KRAS-addicted cells have been previously characterized as more classical and epithelial in monolayer cell cultures (6,56), and KRAS ablation was recently reported to induce a basal-like phenotype in surviving cells in vivo (16). Further experiments are warranted in our IGO system to examine how KRAS addiction controls the molecular subtype and invasive properties of PDAC cells. We identified a set of 13 genes that are up-regulated in the invasive areas in Slow-progressors derived from IGO and upon increasing KRAS dosage in Slow-progressor organoids. This gene set comprises secreted and membrane-localized proteins, which are involved in regulating cell shape, axon guidance, and ECM remodeling—pathways associated with poor patient survival in genomic studies (57). Additionally, we found that a subset of 4 genes (AMIGO2, KLK6, PMEPA1, and TGM2) correlate with poor prognosis in locally advanced PDAC patients. Among them, KLK6, a secreted protein with trypsin-like serine peptidase function, has been implicated in the invasion of ovarian cancer, melanoma, and KRAS-mutant colon cancer cells, and is associated with poor prognosis in multiple human cancers (49,58-61). KLK6 has demonstrated enzymatic activity against fibrinogen and collagens I and IV and may represent a targetable vulnerability for treatment of PDAC (62). Future studies will be aimed at investigating the functional role of KLK6 and other KRAS-ON/Invasive signature genes in subtype switching and PDAC progression.
The IGO model provides, for the first time, an opportunity to follow the trajectory of human pancreatic cancer cells as they proliferate and migrate throughout the pancreatic ductal system to develop extensive intraductal lesions and invasive carcinoma. The histological and transcriptional differences found in Slow- and Fast-progressors may be attributed to differences in the progression of tumors secondary to either the evolution of the tumor microenvironment or the acquisition of early catastrophic genetic events or the gradual accumulation of driver mutations. As such, the IGO model can also serve as a platform for identifying biomarkers for early detection and potential candidates for therapeutic intervention.
Methods
Animal Experiments
All animal experiments were performed according to procedures approved by the IACUC at Cold Spring Harbor Laboratory. NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were bred in-house or purchased from Jackson Laboratories. Mice were enrolled in experimental studies between 3 and 10 weeks of age. For transplant experiments, a single-cell suspension of human patient-derived organoids was generated as previously described (25). For intraductal grafting of organoid (IGO) transplants, 50,000 to 100,000 cells, suspended in 25 μl of PBS, were infused into the pancreatic duct using the modified retrograde infusion technique described previously (30). Mice were anesthetized with 2.5–3% (weight/volume) isoflurane by inhalation. The duodenum was rotated and stretched to expose the biliary-pancreatic duct and its junction with the duodenum, which appears pale. A microclamp (S&T Vascular Clamps 00396-01, F.S.T) was placed on the common bile duct above the branch-point of the pancreatic duct, to prevent infusion into the liver. A small hole opposite to the sphincter of Oddi, 1–2 mm away from the ampulla of Vater, was made with 27 gauge needle, and a 27 gauge blunt-ended Hamilton syringe (80508, HAMILTON) was passed through the small hole that has been created by the needle in the duodenal wall and then into the sphincter of Oddi. The tip of the catheter was positioned at the origin of the branch connecting the pancreatic duct to the common bile duct. If the anatomy was unclear, the tip of the catheter was placed just distal to the sphincter of Oddi. Another microclamp was placed at the sphincter of Oddi to prevent backflow or leakage during the infusion. Cells were injected by manually depressing the plunger into the syringe for 2-3 minutes. The incision point in the duodenum was closed by suturing (6-0 silk, CP Medical Sutures) or using VETCLOSE surgical glue (Henry Schein CA1999). The pancreas and other organs were carefully placed back into the abdomen before suturing the peritoneal cavity and skin incision (separately). Macroscopic fluorescent images were taken using Leica LMD7000 system. For orthotopically grafted of organoid (OGO) transplants, 50,000 cells, suspended in 50 μl of Matrigel (Matrigel, BD) diluted 1:1 with cold PBS, were injected into the tail region of the pancreas using insulin syringes (30 Gauge) as described previously (25). Mice are sacrificed at humane endpoints. For hF23 and hF27, mice were sacrificed when their IGO counterparts reached humane endpoints because the formation of palpable masses was uncommon following OGO transplant. Engraftment was confirmed histologically. 1 mg/kg of body weight (BW) of tamoxifen was dosed by oral gavage for consecutive 5 days for 4 weeks.
Isolation of Cancer Cells from Xenograft Tumors
The murine pancreas was isolated, minced, and digested for 20 minutes at 37°C in digestion buffer (DMEM, 10% FBS, 1% penicillin, 1% streptomycin, 2.5 mg/ml Collagenase D (Sigma-Aldrich), 0.5 mg/ml Liberase DL (Sigma-Aldrich), 0.2 mg/ml DNase I (Sigma-Aldrich)) with triturating every 5 minutes. After washing with flow buffer (PBS, 2% FBS), cell suspensions were collected and run through 100 μm cell strainers. Red red blood cells were lysed using ACK lysis buffer (Gibco). Cancer cells were enriched with human EpCam-coated beads (130-061-101, Miltenyi Biotec) according to the manufacturer’s instruction. Samples were collected in TRIzol reagent for RNA extraction.
Plasmid Construction and Transfection
For generation of pBOBI-PGK-puro-EF1-LS (luciferase-2A-mStrawberry), the pBOBI-EF1-LS plasmid was a gift from Scott K Lyons (63), and the puromycin resistance cassette was clonded into pBOBI-EF1-LS. p-lenti-PGK-ER-KRASG12V was a gift from Daniel Haber (Addgene plasmid #35635; http://n2t.net/addgene:35635; RRID:Addgene_35635). To generate the p-lenti-PGK-ER-KRASG12D construct, ER-KRASG12D was PCR amplified and empty linker was annealed to this construct. Then, the annealed construct was subcloned into p-lenti-PGK-puro and confirmed using Sanger sequencing. pCAG-Autobow was a gift from Joshua Sanes (Addgene plasmid # 45182; http://n2t.net/addgene:45182; RRID:Addgene_45182) For generation of the Autobow vector (pPB-CAG-Autobow-neo), Autobow cDNA from the pCAG-Autobow vector was subcloned into the pPB-CAG-EBNXN (Sanger Institute) vector, and a neomycin resistance cassette was added. Organoids were transfected with pCMV-hyPBase (Sanger Institute) and pPB-CAG-Autobow-neo using Xtremegene9 (Roche), and antibiotic selection with Geneticin (1mg/ml) was initiated 3-5 days post-infection.
Organoid Culture, Lentivirus Production, Lentiviral Infection, and 4-OHT Treatment
Establishment and DNA sequencing of human organoids used in this study were previously reported (24). Human organoids were cultured as described previously (24). Briefly, cells were resuspended in 100% Growth-Factor-Reduced Matrigel (Corning) and plated as domes overlaid with Human Complete Feeding Medium (hCPLT): advanced DMEM/F12, 10 mM HEPES, 1X Glutamax, 500nM A83–01, 50 ng/mL hEGF, 100ng/mL mNoggin, 100 ng/mL hFGF10, 0.01 mM hGastrin I, 1.25mM N-acetylcysteine, 10mM Nicotinamide, 1X B27 supplement, 10% (vol/vol) R-spondin1 conditioned medium, 50% (vol/vol) Afamin/Wnt3A conditioned media (36,64). No other authentication of organoids was performed except for DNA sequencing. HEK293T cells were cultured in DMEM with 10% FBS and 1% Penicillin/streptomycin. All organoid and cell lines were routinely tested for Mycoplasma with MycoAlert Mycoplasma Detection Kit (LT07-318, Lonza) at Cold Spring Harbor Laboratory.
Lentivirus was produced in HEK293T cells by transfecting lentiviral constructs with helper plasmids (pMD2.G (Addgene #12259) and psPAX2 (Addgene # 12260)) using Xtremegene9 (Roche) per manufacterer’s instructions. For HEK293T cells in 10 cm dish, 1.25 μg of plasmid DNA, 1.25 μg of pMD2.G, 2.5 μg of psPAX2, and 15 μl of Xtremegene9 were mixed. Lentivirus-containing supernatant was collected at 24, 48, and 72 hours post-transfection, filtered, and pooled together. Lenti-X concentrator (Clontech) was used to concentrate the lentivirus per manufacturer’s instructions. The viral pellet was resuspended in hCPLT containing 8μg/ml polybrene. For organoid infections, single-cell suspensions of organoids were generated (24), and were spinoculated with virus at 600 RCF for 30 minutes at room temperature (100,000 – 250,000 cells). Cells were then incubated at 37°C for 4 to 16 hours, resuspended in Matrigel, and plated under standard organoid culture conditions. Selection was initiated 3-5 days post-infection. Organoids were treated with Geneticin (1mg/ml, Life technologies) or puromycin (2-3μg/mL, Sigma-Aldrich) for selection. To induce Kras hyperactivation, ER-KRASG12D organoids were treated with 4-hydroxytamoxifen (4-OHT) (0.1μM, Sigma-Aldrich) for 48 hours, and untreated organoids were used as a control.
Proliferation Assays
For proliferation assays, organoids were dissociated as previously described (24), and 400 single cells were plated per well in black clear-bottom 384-well plates in 30μl of 10% Matrigel in hCPLT media. Proliferation was measured every 24 hours for 5-6 days using CellTiter-Glo (G7573, Promega) according to manufacturer’s instructions.
Histopathology, Immunohistochemistry and Immunofluorescence
Formalin-fixed, paraffin-embedded (FFPE) blocks were sectioned and de-paraffinized. Antigen retrieval was performed in Citrate buffer, pH 6.0 for 6 minutes in a pressure cooker unless otherwise specified, then rinsed in tap water for 10 minutes. For immunohistochemical (IHC) staining, endogenous peroxidase was quenched in 3% Hydrogen Peroxide in H2O for 15 minutes at room temperature. The slides were then rinsed in 1x TBS + 0.1% Tween-20 (TBST). Slides were blocked in 5% Horse serum for one hour at room temperature and incubated in primary antibody at 4°C overnight. Slides were washed three times in TBST, then incubated in either horseradish peroxidase-conjugated or alkaline phosphatase-conjugated secondary antibodies for 30 minutes (for IHC), or fluorescent secondary antibodies for one hour at room temperature (for immunofluorescence staining). For IHC, slides were washed twice in TBST and once in H2O. ImmPACT DAB (Vector) or ImmPRESS-AP (Vector) was added for 1 to 5 minutes followed by an H2O wash, and slides were incubated in Hematoxylin counterstain for 20 seconds. Slides were then dehydrated, and a coverslip was added using Cytoseal 60 adhesive. Bright field images were obtained with an Axio Imager.A2 (ZEISS) (Improvision, Lexington, MA). For IF, slides were washed three times in PBS. Slides were counterstained with DAPI (D8417, Sigma-Aldrich), mounted in Prolong Gold Antifade Reagent (P10144, Invitrogen), and imaged on a Leica TCS SP8 laser scanning confocal (Boulder Grove Il), controlled by the LAS AF 3.3.10134 software. This confocal was mounted on a DMI 6000 CS inverted microscope, equipped with 4 laser lines and 2 PMTs.
Primary antibodies used for IHC were: p-ERK (4370, Cell Signaling), TP63 (39692, Cell Signaling), S100A2 (ab109484, abcam), GATA6 (AF1700-SP, R&D), AGR2 (13062, Cell Signaling), and αSMA (M0851, Dako). Primary antibodies used for IF were: human mitochondria (MAB1273, Millipore), CK19 (TROMA III, DSHB), Ki67 (RM-9106, Thermo), VIM (5741, Cell Signaling), GFP (ab13970, abcam), YFP (EVN-AB606-C200, Axxora), RFP (EMU107, Kerefast), and F-actin (A12379, Invitrogen). Secondary antibodies used for IF were anti-mouse Alexa Fluor 488 (A21202, Thermo Fisher Scientific), anti-rabbit Alexa Fluor 647 (A21244), anti-rat Alexa Fluor 568 (A11077), anti-chicken Alexa Fluor 488 (A11039), anti-rabbit Alexa Fluor 546 (A11035), and anti-guinea pig Alexa Fluor 647(A21450).
For sequential IHC, DAB (SK-4105, Vector Laboratories) and Red Alkaline Phosphatase substrate (SK-5105, Vector Laboratories) were used for different primary antibodies. Hematoxylin was used as nuclear counterstain. Hematoxylin and Eosin (H&E), Masson’s trichrome and Alcian Blue staining were performed according to standard protocols.
Whole Mount Immunofluorescence of Organoid Cultures
Organoids were plated in small (25μL) Matrigel domes in an 8 well chamber slide (Ibidi #80826) and allowed to grow to 80% confluence. Organoids were fixed with 4% Paraformaldehyde (PFA) at room temperature for 20 minutes. After washing with PBS/Glycine buffer (0.75% Glycine in PBS, pH7.4) three times for 10 minutes each and washing with IF buffer (0.1% BSA, 0.05% NaN3, 0.2% Triton-X, 0.05% Tween-20 in PBS, pH7.4) three times for 10 minutes each, samples were incubated in blocking buffer (10% goat serum in IF buffer) at room temperature for 1 hour. Samples were incubated with primary antibodies diluted in blocking buffer at 4°C overnight, followed by washing with IF buffer three times at room temperature for 10 minutes each. Samples were incubated with secondary antibodies diluted in blocking buffer at room temperature for 1 hour. Slides were DAPI counterstained and mounted in Prolong Gold antifade reagent (P10144, Invitrogen). Organoids were imaged on a Leica TCS SP8 laser scanning confocal as described above.
Three-Dimensional IF Staining
A modified FLASH method (65) was used. The entire pancreas was divided into 3-4 pieces less than 4-5 mm in a diameter and fixed in 4% Paraformaldehyde (PFA) overnight at 4 °C. The PFA-fixed tissues were then washed two times with PBS-T (0.4% Triton X-100 (Sigma-Aldrich) in PBS) at room temperature for 1 hour each, and incubated in 200 mM boric acid (Sigma-Aldrich) with 4% SDS (Sigma-Aldrich) pH 7.0 overnight at 54 °C. For immunostaining, tissues were blocked in FLASH blocking buffer (1% bovine serum albumin (Sigma-Aldrich), 5% DMSO (Sigma- Aldrich), 10% fetal bovine serum (Gibco), 0.02% sodium azide (Sigma-Aldrich), 0.2% Triton X-100 in PBS) for 1 h, before incubation in primary antibodies for 48 hours at room temperature. Secondary antibodies were incubated for 48 hours at room temperature. Cleared tissues were then prepared using a tissue clearing kit (ab243298, Abcam) according to manufacturer’s instructions and imaged using a Z-stack scan on a Leica TCS SP8 laser scanning confocal (Boulder Grove Il), controlled by the LAS AF 3.3.10134 software.
Histologic Quantification
Slides were scanned using an Aperio CS2 scanner (Leica). Tumor and non-tumor areas on H&E stained slides were manually annotated and were measured using ImageJ software. The number of Ki67-positive and total nuclei of tumor cells in intraductal area and the tumor cells adjacent to murine duct cells in Fig. 1I and J were counted manually. The number of Ki67-positive and total nuclei in organoids in Supplementary Fig. S3C and S7D and in invasive and intraductal area of IGO and OGO tumors in Supplementary Fig. S1I and J were counted automatically using ImageJ software in at least three representative high-powered fields. To quantify AGR2, S100A2 and p-ERK IHC, the percentage of strong positive pixels were counted relative to the entire section using ImageJ software.
Protein Isolation and Immunoblot Analysis
Organoids were harvested in Cell Recovery Solution (Corning), incubated for 15 minutes on ice, pelleted and washed twice in ice-cold PBS. Resulting cell pellets were lysed in 1% Triton X-100, 50mM Tris-HCl pH 7.5, 150mM NaCl, 5mM EDTA with 1X protease (11836170001, Roche) and phosphatase inhibitor cocktails (4906845001, Roche) on ice for 30 minutes. The lysate was clarified at 16,000 RCF for 10 minutes at 4°C, and aliquoted into new tubes for storage at −80°C.
Protein lysates were separated on 4-12% Bis-Tris NuPAGE gels (Life technologies) run with MOPS Running Buffer (Life technologies), transferred to PVDF membrane (Millipore), and blocked in 5% BSA (Sigma-Aldrich) in TBST for one hour. The membranes were then incubated in primary antibody overnight with rocking at 4°C followed by incubation in appropriate secondary antibodies conjugated to horse radish peroxidase for one hour and detection by ECL (GE Healthcare).
Primary antibodies used were: TP63 (39692, Cell Signaling), S100A2 (ab109484, abcam), GATA6 (AF1700-SP, R&D), HSP90α (07-2174, EMD Millipore), SMAD4 (sc-7966, Santa Cruz), p-ERK1/2 (4370, Cell Signaling), and ERK1/2 (4695, Cell Signaling).
Genomic DNA Isolation from organoid
Organoids were harvested in Cell Recovery Solution (Corning), incubated for 15 minutes on ice, pelleted and washed twice in ice-cold PBS. Genomic DNA from organoids was collected using DNeasy Blood & Tissue Kit (Qiagen) and was sonicated using a Covaris focus acoustics system. Libraries were prepared using the NEBNext® Ultra™ II DNA Library Prep kit (NEB). Libraries were evaluated using BioAnalyzer (Agilent 2100) and sequenced using Illumina NextSeq platform (Cold Spring Harbor Laboratory Genome Center, Woodbury).
Genomic DNA Isolation from Formalin-Fixed, Paraffin-Embedded (FFPE) sections of IGO tumors
Formalin-fixed, paraffin-embedded (FFPE) blocks were transferred to PEN membrane glass slides. Adjacent sections were stained with H&E, S100A2, and GATA6 for pathology review. Laser capture microdissection was performed on a LMD7000 system (Leica) to isolate intraductal lesions with GATA6 staining and invasive lesions with S100A2 staining, DNA was extracted using Arcturus® PicoPure® DNA Extraction Kit (Thermo), and was sonicated using a Covaris focus acoustics system. Libraries were prepared using the NEBNext® Ultra™ II DNA Library Prep kit (NEB). Libraries were evaluated using BioAnalyzer (Agilent 2100) and sequenced using Illumina NextSeq platform (Cold Spring Harbor Laboratory Genome Center, Woodbury).
KRAS Copy Number Analysis Based on Organoid Exome Sequencing
In addition, we made use of copy number analysis performed using whole-exome sequencing data from organoids as previously described (24). Whole exome sequencing data for organoids were processed by the New York Genome Center somatic preprocessing pipeline. Copy-number variants (CNV) were detected using FACETS v0.5.2 (66) for exome, resulting in segmented profiles where the copy number is approximated by a piecewise-constant function of the genomic position.
Copy Number Analysis of bulk DNA from organoids and FFPE sections
The copy number analysis was performed as described previously (67). Briefly, the genome was divided into either 5000 or 20,000 bins. After the normalization, the normalized bincount vectors were segmented using Bioconductor DNAcopy package version 1.50.5 as described previously (67). DNAcopy parameters used were alpha = 0.02, nperm = 1000, undo.SD = 0.5, and min.width = 3. Copy number heatmaps were made using the heatmap function in R using the segmented bin values. The distance function used was “manhattan”, and the hierarchical clustering agglomeration method used was “ward.D2”.
RNA Extraction and Quantitative RT-PCR
RNA was extracted from samples in TRIzol Reagent (15596-018, Thermo Fisher Scientific) using the PureLink RNA Mini Kit (12183018A, Thermo Fisher Scientific) per the manufacturer’s instructions. cDNA was synthesized using 1μg of total RNA and TaqMan Reverse Transcription Reagents (N808-0234, Applied Biosystems). RT-qPCR analysis was performed with gene-specific Taqman primers (Supplementary Table S4) (ThermoFisher Scientific) and Taqman Universal Master Mix II , no UNG (ThermoFisher Scientific) on a QuantStudio 6-Flex Real time-PCR instrument (Applied Biosystems). Relative gene expression quantification was performed using the ΔΔCt method with the QuantStudio Real-Time PCR software v1.1 (Applied Biosystems). Expression levels were normalized to HPRT or ACTB.
Tumor RNA-Sequencing Experiments
For RNA-sequencing (RNA-seq) comparison of OGO to IGO-derived tumors, OGO- and IGO transplants of hM1A, hF2, hF3, hT3, hF23, hF24, hT30, hF32, hF43, hT85, hT93, hT102, hM17D and hM19B were performed. Transplant-derived tumors were harvested at endpoint. For RNA-seq comparison of Fast- vs. Slow-IGO tumors, 6 IGO transplants of Fast-progressors (hM1A, hF3, hT3, hF24, hF43 and hM19B) and 9 IGO transplants of Slow-progressors (hF2, hF23, hF27, hT30, hF32, hT85, hT93, hT102, and hM17D) were performed, and transplant-derived tumors were harvested at endpoint. RNA from bulk tumors was extracted using the PureLink RNA Mini Kit with TRIzol Reagent as described above.
RNA-seq libraries were constructed using a TruSeq Stranded Total RNA Kit with RiboZero Human/Mouse/Rat (RS-122-2202, Illumina) with 0.2–1 μg RNA per sample, per manufacter’s instructions. Libraries were evaluated using BioAnalyzer (Agilent 2100) and then sequenced using Illumina NextSeq platform (Cold Spring Harbor Laboratory Genome Center, Woodbury).
RNA-Seq of Laser Capture Microdissected IGO Tumors
Formalin-fixed, paraffin-embedded (FFPE) blocks were transferred to PEN membrane glass slides. Adjacent sections were stained with H&E and S100A2 for pathology review. Laser capture microdissection was performed on a LMD7000 system (Leica) to isolate regions containing cancer cells within the duct (intraductal, “INTRA”) and outside the duct (invasive, “INVASIVE”) from IGO transplants of hM1A clonal organoids, cancer cells within the duct (intraductal) and outside the duct (invasive) from IGO transplants of hF2 and hF27, and cancer cells from IGO transplants of Empty vector (Empty) and ER-KRASG12D (ER-KRAS)- expressing hF23 and hF27. RNA was extracted using NucleoSpin® totalRNA FFPE XS (Clontech), and libraries were prepared using the SMARTer® Stranded Total RNA-Seq Kit v2 - Pico Input Mammalian (Takara). Libraries were evaluated using BioAnalyzer (Agilent 2100) and sequenced using Illumina NextSeq platform (Cold Spring Harbor Laboratory Genome Center, Woodbury).
Analysis of Bulk Tumor, Laser Capture Microdissected Tumor and Organoid RNA-Seq Data
The quality of the RNA-seq reads was quantified using FastQC version 0.11.5 (Babraham Bioinformatics). Next, to classify reads as coming from the mouse stroma or the human cancer cells, we employed the strategy presented by Callari et al. (32). A custom reference genome composed of the GRCm38.p6 mouse genome and GRCh38.p12 human genome was generated, and a combined human and mouse transcriptome annotation composed of GENCODE 28 and M17 (68), respectively, were generated. Reads were aligned against the composite genome using STAR version 2.5.2b (69) by RSEM software version 1.3.0 (70) with following parameters: rsem-calculate-expression –star. Counts per gene were extracted, using the combined human and mouse transcriptome annotation. For the organoid RNA-seq, the alignment was done on the GRCh38.p12 human genome, and counts per gene were extracted from human transcriptome using STAR and RSEM as described above. Differential gene expression analysis was performed using Bioconductor package DESeq2 version 1.24.0 (71), with a pre-filtering step to retain genes with reads in more than one sample. Human and mouse transcriptomes were split to be analyzed separately. In presence of paired data, the DESeq2 design was adapted consistently. Only genes with an adjusted P value<0.05 and an absolute log2 fold change >=1 were retained as significantly differentially expressed.
The Bioconductor package ComplexHeatmap version 2.0.0 (72) was used to generate heatmaps of the DeSeq2-normalized counts shown in Figures 3A, standardized by gene, of the significantly differentially expressed genes. Hierarchical clustering of the genes was based on the complete linkage method using the Spearman correlation coefficient. For the clustering of the samples, Euclidian distance was used.
Gene Set Enrichment Analysis was performed using the GSEA program version 4.0.3 (Broad Institute), with ranked log2 fold change values as input. Hallmark gene sets and C6 oncogenic signatures were downloaded from the Molecular Signatures Database (MSigDB(http://www.broadinstitute.org/gsea/index.jsp)). Squamous-PDAC and Progenitor-PDAC Identity signatures (17), Basal-like, classical, Moffitt-Normal, and Moffitt-Activated signatures (7), Immune-rich and ECM-rich signatures (9), and the JANES-KRAS signature (42) were described previously.
For the heatmap showing transcriptional subtypes of IGO and OGO tumors in Figure 2H, consensus clustering was performed on tumor expression signatures using the top 100 ranked genes from the four malignancy-related NMF components (19) using ConsensusClusterPlus v.1.40.0 (73). Using Pearson correlation distance, clustering was performed with Pearson correlation distance (partitioning around medoids) with 10,000 re-samplings to generate the final consensus.
For the heatmaps showing expression of genes in tumors taken from PanCuRx patients (i.e. Supplementary Fig. S3H), heatmaps of gene expression (log10(FPKM+1)) were generated with the clustermap function in the Python data visualization library Seaborn version 0.9.0 (https://github.com/mwaskom/seaborn/issues/252). Each row shows the gene expression scaled to have zero mean and unit variance. Scaled values were clipped to be within the range [−5, 5] for visualization. Hierarchical clustering was performed using an average-linkage method using Pearson correlation coefficients between each patient or gene signature.
Imputation of Survival Time for Censored Mice
For mice in the survival study that were censored (because the mouse was sacrificed prior to humane endpoint or was still alive at the last observed time), the survival time was imputed by using the median of the last observed survival times for all mice (transplanted with any organoid) that survived as long as or longer than the censored mouse. The survival plot in Fig. 1F was generated with package ggplot2 version 3.1.0 (74).
Classification of Fast and Slow-progressors
Supplementary Figure SM1 displays the workflow of the classification method. For the purpose of determining the Fast- and Slow-progressor classification, when multiple organoid lines from a single patient were used in the study (e.g. hM1A, hM1E, and hM1F; or hM19A, hM19B, hM19C, and hM19D) only a single organoid line per patient (hM1A and hM19B) was retained. For each organoid line, three survival times (corresponding to the number of times available in the group with the smallest number of replicates) were randomly selected. If censoring was present, the imputed survival time was used. For each organoid line, the mean and the standard deviation of the three sampled survival times were used to estimate a normal distribution of survival times, and ten data points (survival times) were randomly selected from the distribution. All selected survival times from all organoid lines were pooled together and used to generate a global distribution of the survival times. The package mixtools version 1.1.0 (75) was then used to detect the two normal curves that best approximate the global distribution of the survival times. The mean (mean survival of 75 days) was associated with the Fast-progressors, while other curve (mean survival of 263 days) was associated with the Slow-progressors. To determine the upper threshold in survival times for the Fast-progressors, we selected the position in the right tail of the first curve where the cumulative normal probability becomes 95% (111 days). To determine the lower threshold in survival times for the Slow-progressors, we selected the position in the left tail of the second curve where the cumulative normal probability becomes 5% (157 days). The area between those two positions is considered unclassified.
Survival analysis of PanCuRx data
KRAS-ON (in vitro) score and KRAS-ON (in vivo) score were obtained using ssGSEA through GenePattern web-tool (https://cloud.genepattern.org/gp/pages/login.jsf). GraphPad Prism software was used for Log-rank (Mantel-Cox) test.
Data Availability
The RNA and DNA sequencing data discussed in this publication have been deposited in NCBI’s Genotypes and Phenotypes database and is accessible through dbGaP study accession number: phs002045.v1.p1
Statistical Analysis
Statistical analysis was performed using GraphPad Prism software using unpaired Student’s t test and Log-rank (Mantel-Cox) test for survival data unless otherwise indicated in figure legend. P values <0.05 were determined to be significant. Asterisks denote P values as follows: *P<0.05, **P<0.01, ***P<0.001.
Supplementary Material
Significance.
Accurate modeling of the molecular subtypes of pancreatic cancer is crucial to facilitate the generation of effective therapies. We report the development of an intraductal organoid transplantation model of pancreatic cancer which models the progressive switching of subtypes, and identify stochastic and Ras-driven mechanisms that determine subtype specification.
Acknowledgements
We thank Dr. Chen-Hua Chuang and Dr. Monte M. Winslow for the instruction of retrograde pancreatic intraductal injection. We acknowledge the Cold Spring Harbor Laboratory Animal, Animal Imaging, Histology, Microscopy, and Next Generation Sequencing Shared Resources, which are supported by the NIH Cancer Center Support Grant P30CA045508. We acknowledge Ela Elyada for the construction of pBOBI-PGK-puro-EF1-LS plasmid. We also acknowledge Vincenzo Corbo for generation of mStrawberry expressing organoids. We thank members of the Tuveson Laboratory for their assistance and advice. DAT is a distinguished scholar of the Lustgarten Foundation and Director of the Lustgarten Foundation-designated Laboratory of Pancreatic Cancer Research. DAT are also supported by the Cold Spring Harbor Laboratory Association, the V Foundation, the Thompson Foundation, and the National Institutes of Health (NIH P30CA45508, P20CA192996, U10CA180944, U01CA224013, U01CA210240, R01CA188134, and R01CA190092). DAT and CRV are supported by the Simons Foundation (552716) and the NIH R01CA229699. YP is supported by NCI R50CA211506. BA is supported by NCI F30CA200240. CRV was supported by Pershing Square Sohn Cancer Research Alliance, the Cold Spring Harbor Laboratory and Northwell Health Affiliation, the National Cancer Institute (NCI) 5P01CA013106-Project 4 and 1RO1CA229699, and the 2016 Pancreatic Cancer Action Network-American Association for Cancer Research (AACR) Career Development Award , supported from the Daniel and Janet Mordecai Foundation, Grant number 16-20-25-VAKO. KM is supported by the Daiichi Sankyo Foundation of Life Science and the Uehara Memorial Foundation, Japan. BT is supported by the German Research Foundation (DFG) (TR 1663/1). DP is supported by the German Research Foundation (DFG) (PL 894/1-1). TDDS was supported by a grant from the State of New York, contract no. C150158. LAB was supported through the NIH (F32CA192904). CIH was supported by NIH F32CA180717. LDW is supported by the Sol Goldman Pancreatic Cancer Research Center. This work was supported by grants to MW from the Simons Foundation, Life Sciences Founders Directed Giving-Research (award numbers 519054), and The Breast Cancer Research Foundation (award number 18-174).The pancreatic cancer patient study was conducted with the support of the Ontario Institute for Cancer Research (PanCuRx Translational Research Initiative) through funding provided by the Government of Ontario. This work was also supported by the Cold Spring Harbor Laboratory and Northwell Health Affiliation (Project Lazarus) as well as the Northwell Health Tissue Donation Program.
Abbreviations:
- PDAC
pancreatic ductal adenocarcinoma
- EMT
epithelial–mesenchymal transition
- PanIN
Pancreatic Intraepithelial Neoplasia
- OGO
Orthotopically Grafted Organoid
- IGO
Intraductally Grafted Organoid
- ECM
Extracellular Matrix
- GSEA
Gene Set Enrichment Analysis
- ssGSEA
single-sample Gene Set Enrichment Analysis
- HOP
Head of Pancreas
- DEG
Differentially Expressed Gene
- TCGA
The Cancer Genome Atlas
- LCM
Laser Capture Microdissection
- CN
Copy Number
- FACETS
Fraction and Allele-Specific Copy Number Estimates from Tumor Sequencing
- 4-OHT
4-Hydroxytamoxifen
- NSG
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ
- NES
Normalized Enrichment Score
- FDR
False Discovery Rate
- IHC
Immunohistochemical
Footnotes
Declaration of interests: The authors declare no potential conflicts of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69(1):7–34 doi 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Hruban RH, Lillemoe KD. Screening for Pancreatic Cancer Gets a D, But the Student Is Improving. JAMA Surg 2019; 154(9):795–797 doi 10.1001/jamasurg.2019.2832. [DOI] [PubMed] [Google Scholar]
- 3.Aguirre AJ, Nowak JA, Camarda ND, Moffitt RA, Ghazani AA, Hazar-Rethinam M, et al. Real-time Genomic Characterization of Advanced Pancreatic Cancer to Enable Precision Medicine. Cancer Discov 2018;8(9):1096–111 doi 10.1158/2159-8290.CD-18-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531(7592):47–52 doi 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 5.Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017;32(2):185–203.e13 doi 10.1016/j.ccell.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med 2011;17(4):500–3 doi 10.1038/nm.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet 2015;47(10):1168–78 doi 10.1038/ng.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Puleo F, Nicolle R, Blum Y, Cros J, Marisa L, Demetter P, et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 2018;155(6):1999–2013.e3 doi 10.1053/j.gastro.2018.08.033. [DOI] [PubMed] [Google Scholar]
- 9.Maurer C, Holmstrom SR, He J, Laise P, Su T, Ahmed A, et al. Experimental microdissection enables functional harmonisation of pancreatic cancer subtypes. Gut 2019;68(6):1034–43 doi 10.1136/gutjnl-2018-317706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.S NK, Wilson GW, Grant RC, Seto M, O’Kane G, Vajpeyi R, et al. Morphological classification of pancreatic ductal adenocarcinoma that predicts molecular subtypes and correlates with clinical outcome. Gut 2020;69(2):317–28 doi 10.1136/gutjnl-2019-318217. [DOI] [PubMed] [Google Scholar]
- 11.Collisson EA, Bailey P, Chang DK, Biankin AV. Molecular subtypes of pancreatic cancer. Nat Rev Gastroenterol Hepatol 2019;16(4):207–20 doi 10.1038/s41575-019-0109-y. [DOI] [PubMed] [Google Scholar]
- 12.Rashid NU, Peng XL, Jin C, Moffitt RA, Volmar KE, Belt BA, et al. Purity Independent Subtyping of Tumors (PurIST), A Clinically Robust, Single-sample Classifier for Tumor Subtyping in Pancreatic Cancer. Clin Cancer Res 2020;26(1):82–92 doi 10.1158/1078-0432.Ccr-19-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muckenhuber A, Berger AK, Schlitter AM, Steiger K, Konukiewitz B, Trumpp A, et al. Pancreatic Ductal Adenocarcinoma Subtyping Using the Biomarkers Hepatocyte Nuclear Factor-1A and Cytokeratin-81 Correlates with Outcome and Treatment Response. Clin Cancer Res 2018;24(2):351–9 doi 10.1158/1078-0432.Ccr-17-2180. [DOI] [PubMed] [Google Scholar]
- 14.Aung KL, Fischer SE, Denroche RE, Jang GH, Dodd A, Creighton S, et al. Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results from the COMPASS Trial. Clin Cancer Res 2018;24(6):1344–54 doi 10.1158/1078-0432.Ccr-17-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Kane GM, Grunwald BT, Jang GH, Masoomian M, Picardo S, Grant RC, et al. GATA6 Expression Distinguishes Classical and Basal-like Subtypes in Advanced Pancreatic Cancer. Clin Cancer Res 2020. doi 10.1158/1078-0432.Ccr-19-3724. [DOI] [PubMed] [Google Scholar]
- 16.Adams CR, Htwe HH, Marsh T, Wang AL, Montoya ML, Subbaraj L, et al. Transcriptional control of subtype switching ensures adaptation and growth of pancreatic cancer. Elife 2019;8 doi 10.7554/eLife.45313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Somerville TDD, Xu Y, Miyabayashi K, Tiriac H, Cleary CR, Maia-Silva D, et al. TP63-Mediated Enhancer Reprogramming Drives the Squamous Subtype of Pancreatic Ductal Adenocarcinoma. Cell Rep 2018;25(7):1741–55 e7 doi 10.1016/j.celrep.2018.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayashi A, Fan J, Chen R, Ho Y-j, Makohon-Moore AP, Lecomte N, et al. A unifying paradigm for transcriptional heterogeneity and squamous features in pancreatic ductal adenocarcinoma. Nature Cancer 2020;1(1):59–74 doi 10.1038/s43018-019-0010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan-Seng-Yue M, Kim JC, Wilson GW, Ng K, Figueroa EF, O’Kane GM, et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nature Genetics 2020; 52(2):231–240 doi 10.1038/s41588-019-0566-9. [DOI] [PubMed] [Google Scholar]
- 20.Candido JB, Morton JP, Bailey P, Campbell AD, Karim SA, Jamieson T, et al. CSF1R(+) Macrophages Sustain Pancreatic Tumor Growth through T Cell Suppression and Maintenance of Key Gene Programs that Define the Squamous Subtype. Cell Rep 2018;23(5):1448–60 doi 10.1016/j.celrep.2018.03.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016;29(6):832–45 doi 10.1016/j.ccell.2016.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ligorio M, Sil S, Malagon-Lopez J, Nieman LT, Misale S, Di Pilato M, et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 2019;178(1):160–75.e27 doi 10.1016/j.cell.2019.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andricovich J, Perkail S, Kai Y, Casasanta N, Peng W, Tzatsos A. Loss of KDM6A Activates Super-Enhancers to Induce Gender-Specific Squamous-like Pancreatic Cancer and Confers Sensitivity to BET Inhibitors. Cancer Cell 2018;33(3):512–26 e8 doi 10.1016/j.ccell.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tiriac H, Belleau P, Engle DD, Plenker D, Deschenes A, Somerville TDD, et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov 2018;8(9):1112–29 doi 10.1158/2159-8290.CD-18-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015;160(1–2):324–38 doi 10.1016/j.cell.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov 2019;9(2):282–301 doi 10.1158/2159-8290.CD-18-0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Basturk O, Hong SM, Wood LD, Adsay NV, Albores-Saavedra J, Biankin AV, et al. A Revised Classification System and Recommendations From the Baltimore Consensus Meeting for Neoplastic Precursor Lesions in the Pancreas. Am J Surg Pathol 2015;39(12):1730–41 doi 10.1097/pas.0000000000000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hruban RH, Goggins M, Parsons J, Kern SE. Progression model for pancreatic cancer. Clin Cancer Res 2000;6(8):2969–72. [PubMed] [Google Scholar]
- 29.Patra KC, Bardeesy N, Mizukami Y. Diversity of Precursor Lesions For Pancreatic Cancer: The Genetics and Biology of Intraductal Papillary Mucinous Neoplasm. Clin Transl Gastroenterol 2017;8(4):e86 doi 10.1038/ctg.2017.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chiou SH, Winters IP, Wang J, Naranjo S, Dudgeon C, Tamburini FB, et al. Pancreatic cancer modeling using retrograde viral vector delivery and in vivo CRISPR/Cas9-mediated somatic genome editing. Genes Dev 2015;29(14):1576–85 doi 10.1101/gad.264861.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iacobuzio-Donahue CA, Fu B, Yachida S, Luo M, Abe H, Henderson CM, et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J Clin Oncol 2009;27(11):1806–13 doi 10.1200/JCO.2008.17.7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Callari M, Batra AS, Batra RN, Sammut SJ, Greenwood W, Clifford H, et al. Computational approach to discriminate human and mouse sequences in patient-derived tumour xenografts. BMC Genomics 2018;19(1):19 doi 10.1186/s12864-017-4414-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daemen A, Peterson D, Sahu N, McCord R, Du X, Liu B, et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc Natl Acad Sci U S A 2015;112(32):E4410–7 doi 10.1073/pnas.1501605112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Puleo F, Nicolle R, Blum Y, Cros J, Marisa L, Demetter P, et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 2018;155(6):1999–2013 e3 doi 10.1053/j.gastro.2018.08.033. [DOI] [PubMed] [Google Scholar]
- 35.Takahashi K, Ehata S, Koinuma D, Morishita Y, Soda M, Mano H, et al. Pancreatic tumor microenvironment confers highly malignant properties on pancreatic cancer cells. Oncogene 2018;37(21):2757–72 doi 10.1038/s41388-018-0144-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seino T, Kawasaki S, Shimokawa M, Tamagawa H, Toshimitsu K, Fujii M, et al. Human Pancreatic Tumor Organoids Reveal Loss of Stem Cell Niche Factor Dependence during Disease Progression. Cell Stem Cell 2018;22(3):454–67 e6 doi 10.1016/j.stem.2017.12.009. [DOI] [PubMed] [Google Scholar]
- 37.Palm W, Park Y, Wright K, Pavlova NN, Tuveson DA, Thompson CB. The Utilization of Extracellular Proteins as Nutrients Is Suppressed by mTORC1. Cell 2015;162(2):259–70 doi 10.1016/j.cell.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Connor AA, Denroche RE, Jang GH, Timms L, Kalimuthu SN, Selander I, et al. Association of Distinct Mutational Signatures With Correlates of Increased Immune Activity in Pancreatic Ductal Adenocarcinoma. JAMA Oncol 2017;3(6):774–83 doi 10.1001/jamaoncol.2016.3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Juiz N, Elkaoutari A, Bigonnet M, Gayet O, Roques J, Nicolle R, et al. Basal-like and Classical cells coexistence in pancreatic cancer revealed by single cell analysis. bioRxiv 2020:2020.01.07.897454 doi 10.1101/2020.01.07.897454. [DOI] [PubMed] [Google Scholar]
- 40.Cai D, Cohen KB, Luo T, Lichtman JW, Sanes JR. Improved tools for the Brainbow toolbox. Nat Methods 2013;10(6):540–7. [PubMed] [Google Scholar]
- 41.Mueller S, Engleitner T, Maresch R, Zukowska M, Lange S, Kaltenbacher T, et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature 2018;554(7690):62–8 doi 10.1038/nature25459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018;172(3):578–89 e17 doi 10.1016/j.cell.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 43.Kuja-Panula J, Kiiltomaki M, Yamashiro T, Rouhiainen A, Rauvala H. AMIGO, a transmembrane protein implicated in axon tract development, defines a novel protein family with leucine-rich repeats. J Cell Biol 2003;160(6):963–73 doi 10.1083/jcb.200209074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watanabe Y, Itoh S, Goto T, Ohnishi E, Inamitsu M, Itoh F, et al. TMEPAI, a transmembrane TGF-beta-inducible protein, sequesters Smad proteins from active participation in TGF-beta signaling. Mol Cell 2010;37(1):123–34 doi 10.1016/j.molcel.2009.10.028. [DOI] [PubMed] [Google Scholar]
- 45.Mead TJ, Du Y, Nelson CM, Gueye NA, Drazba J, Dancevic CM, et al. ADAMTS9-Regulated Pericellular Matrix Dynamics Governs Focal Adhesion-Dependent Smooth Muscle Differentiation. Cell Rep 2018;23(2):485–98 doi 10.1016/j.celrep.2018.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fukuda A, Wang SC, Morris JPt, Folias AE, Liou A, Kim GE, et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell 2011;19(4):441–55 doi 10.1016/j.ccr.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramani VC, Kaushal GP, Haun RS. Proteolytic action of kallikrein-related peptidase 7 produces unique active matrix metalloproteinase-9 lacking the C-terminal hemopexin domains. Biochim Biophys Acta 2011;1813(8):1525–31 doi 10.1016/j.bbamcr.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kwon CH, Park HJ, Lee JR, Kim HK, Jeon TY, Jo HJ, et al. Serpin peptidase inhibitor clade A member 1 is a biomarker of poor prognosis in gastric cancer. Br J Cancer 2014;111(10):1993–2002 doi 10.1038/bjc.2014.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ghosh MC, Grass L, Soosaipillai A, Sotiropoulou G, Diamandis EP. Human kallikrein 6 degrades extracellular matrix proteins and may enhance the metastatic potential of tumour cells. Tumour Biol 2004;25(4):193–9 doi 10.1159/000081102. [DOI] [PubMed] [Google Scholar]
- 50.Agnihotri N, Kumar S, Mehta K. Tissue transglutaminase as a central mediator in inflammation-induced progression of breast cancer. Breast Cancer Res 2013;15(1):202 doi 10.1186/bcr3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hutchings D, Waters KM, Weiss MJ, Wolfgang CL, Makary MA, He J, et al. Cancerization of the Pancreatic Ducts: Demonstration of a Common and Under-recognized Process Using Immunolabeling of Paired Duct Lesions and Invasive Pancreatic Ductal Adenocarcinoma for p53 and Smad4 Expression. Am J Surg Pathol 2018;42(11):1556–61 doi 10.1097/PAS.0000000000001148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Makohon-Moore AP, Matsukuma K, Zhang M, Reiter JG, Gerold JM, Jiao Y, et al. Precancerous neoplastic cells can move through the pancreatic ductal system. Nature 2018;561(7722):201–5 doi 10.1038/s41586-018-0481-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Notta F, Chan-Seng-Yue M, Lemire M, Li Y, Wilson GW, Connor AA, et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016;538(7625):378–82 doi 10.1038/nature19823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov 2019;9(8):1102–23 doi 10.1158/2159-8290.CD-19-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith G, Bounds R, Wolf H, Steele RJ, Carey FA, Wolf CR. Activating K-Ras mutations outwith ‘hotspot’ codons in sporadic colorectal tumours - implications for personalised cancer medicine. Br J Cancer 2010;102(4):693–703 doi 10.1038/sj.bjc.6605534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, et al. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 2009;15(6):489–500 doi 10.1016/j.ccr.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012;491(7424):399–405 doi 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Henkhaus RS, Gerner EW, Ignatenko NA. Kallikrein 6 is a mediator of K-RAS-dependent migration of colon carcinoma cells. Biol Chem 2008;389(6):757–64 doi 10.1515/bc.2008.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krenzer S, Peterziel H, Mauch C, Blaber SI, Blaber M, Angel P, et al. Expression and function of the kallikrein-related peptidase 6 in the human melanoma microenvironment. J Invest Dermatol 2011;131(11):2281–8 doi 10.1038/jid.2011.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ogawa K, Utsunomiya T, Mimori K, Tanaka F, Inoue H, Nagahara H, et al. Clinical Significance of Human Kallikrein Gene 6 Messenger RNA Expression in Colorectal Cancer. Clinical Cancer Research 2005;11(8):2889–93 doi 10.1158/1078-0432.Ccr-04-2281. [DOI] [PubMed] [Google Scholar]
- 61.White NM, Mathews M, Yousef GM, Prizada A, Popadiuk C, Dore JJ. KLK6 and KLK13 predict tumor recurrence in epithelial ovarian carcinoma. Br J Cancer 2009;101(7):1107–13 doi 10.1038/sj.bjc.6605280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sananes A, Cohen I, Shahar A, Hockla A, De Vita E, Miller AK, et al. A potent, proteolysis-resistant inhibitor of kallikrein-related peptidase 6 (KLK6) for cancer therapy, developed by combinatorial engineering. J Biol Chem 2018;293(33):12663–80 doi 10.1074/jbc.RA117.000871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rodriguez E, Mannion L, D’Santos P, Griffiths M, Arends MJ, Brindle KM, et al. Versatile and enhanced tumour modelling in mice via somatic cell transduction. J Pathol 2014;232(4):449–57 doi 10.1002/path.4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mihara E, Hirai H, Yamamoto H, Tamura-Kawakami K, Matano M, Kikuchi A, et al. Active and water-soluble form of lipidated Wnt protein is maintained by a serum glycoprotein afamin/alpha-albumin. Elife 2016;5 doi 10.7554/eLife.11621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Messal HA, Alt S, Ferreira RMM, Gribben C, Wang VM, Cotoi CG, et al. Tissue curvature and apicobasal mechanical tension imbalance instruct cancer morphogenesis. Nature 2019;566(7742):126–30 doi 10.1038/s41586-019-0891-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res 2016;44(16):e131 doi 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li S, Kendall J, Park S, Wang Z, Alexander J, Moffitt A, et al. Copolymerization of single-cell nucleic acids into balls of acrylamide gel. Genome Res 2020;30(1):49–61 doi 10.1101/gr.253047.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 2019;47(D1):D766–d73 doi 10.1093/nar/gky955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29(1):15–21 doi 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011;12:323 doi 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016;32(18):2847–9 doi 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 73.Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 2010;26(12):1572–3 doi 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wickham H, SpringerLink (Online service). ggplot2 Elegant Graphics for Data Analysis. Use R!,. p 1 online resource (VIII, 213 p). [Google Scholar]
- 75.Benaglia T, Chauveau D, Hunter DR, Young DS. mixtools: An R Package for Analyzing Mixture Models. 2009 2009;32(6):29 doi 10.18637/jss.v032.i06. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA and DNA sequencing data discussed in this publication have been deposited in NCBI’s Genotypes and Phenotypes database and is accessible through dbGaP study accession number: phs002045.v1.p1