

Abstract
In this work, we developed a novel “click”-ready pH-cleavable phosphoramidate linker for controlled-release of monomethyl auristantin E (MMAE) in antibody- and small molecule-drug conjugates application. This water-soluble linker was found to have tremendous stability at physiological pHs while rapidly releasing its payload at acidic pH. The linker can also be tailored to release payloads of diverse functional groups, broadening its applications.
Keywords: Cleavable linker, Monomethyl auristatin E, MMAE, Phosphoramidate, Payload click-ready
Since the introduction of Antibody-Drug Conjugates (ADCs) nearly three decades ago, cleavable linker technology has been a proven requirement for the design of safe and efficacious ADCs [1–4]. Traditionally, the mechanism of action of an ADC requires antibody-antigen binding on the target-cell surface, internalization, and lysosomal processing for controlled release of the cytotoxic payload [5]. For an ADC to be considered selective and potent, the cleavable linker must possess essential properties such as robust stability in circulation to prevent premature release of cytotoxic payloads, high water solubility to aid bioconjugation, and efficient release of payloads at the site of action. Furthermore, intracellular endocytic trafficking of the ADC plays a pivotal role in the functionality of these linkers, as they provide unique acidic pH, hydrolytic enzymes, and endogenous molecules for the triggered release of the payloads.
Of the cleavable linkers reported, enzyme-cleavable linkers, such as the cathepsin-B cleavable Valine-Citrulline linker, have found the most widespread use [6]. Glutathione-reducible disulfide linkers [7] and acid-labile hydrazone linkers [8,9] have also been utilized with varying degrees of success. Acid cleavable ADC linkers were initially promising but the stringent stability requirements imparted by highly potent payloads remains a challenge [8]. Although the disulfide and dipeptide technologies are well established, there have been issues with solubility, plasma stability, and a lack of versatility in releasing payloads with diverse functional groups.
We recently reported on a phosphoramidate scaffold that can be tuned for the controlled and pH-triggered release of amine-containing payloads [10–12]. With this scaffold, controlled release was found to be dependent upon the proximity of a neighboring acidic group (e.g. carboxylic acid or pyridinium) to the phosphorus center (Fig. 1). Herein, we report the development of a click-ready pH-triggered phosphoramidate linker capable of releasing amine-containing payloads. Likely, this linker can be adapted to carry payloads through alcohol, phenol, and aniline functional groups. For this study, we selected a homoserine-type scaffold that would possess the appropriate structural requirements - 4 atoms between the CO2H moiety and the phosphoramidate center - for the necessary stability at physiological pH, while rapidly releasing a 2° amine payload under acidic conditions. This selection criteria was based on our earlier studies in which we found that a longer spacer between the phosphorus atom and the CO2H moiety led to an insufficiently slow release of cargo under acidic conditions, while a shorter spacer provided inadequate stability at physiological pH11. To test this hypothesis, we utilized monomethyl auristatin E (MMAE, a commonly used synthetic antineoplastic agent [13]), a truncated synthetic MMAE surrogate (MMAE Model), and N-methylphenethylamine as representative payloads.
Fig. 1.
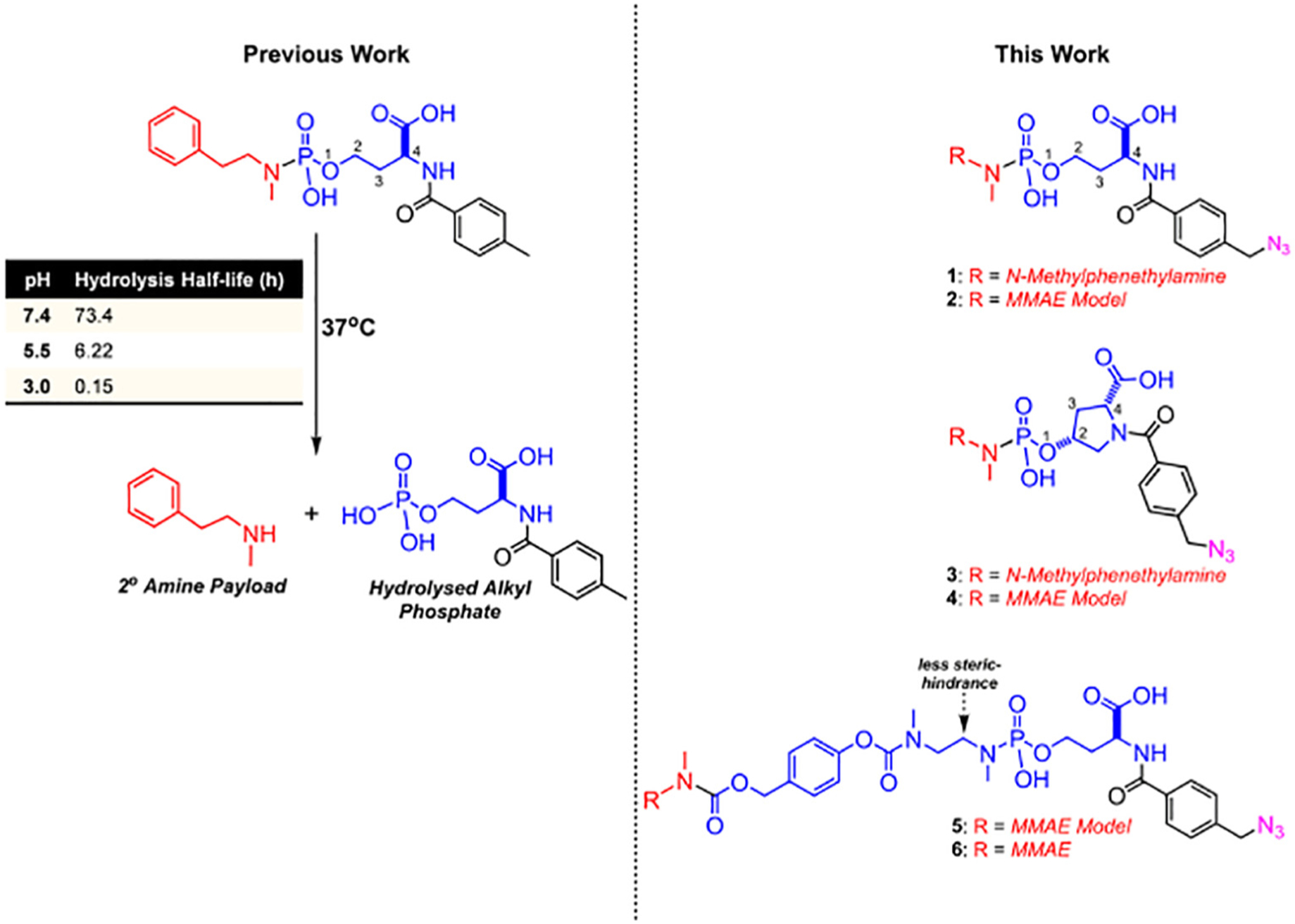
Phosphoramidate-based pH-triggered scaffold for releasing amine-based payloads.
The phosphoramidate payload-linker modules (1–4) were prepared by the reaction of silyl-protected H-phosphonates intermediates (7, 8) with the corresponding amines (MMAE Model and N-methylphenethylamine) in an Atherton-Todd P—N coupling reaction [14], resulting in the O-silyl protected phosphoramidates (9–12). Global O-silyl deprotection with CsF afforded the final phosphoramidate modules (1–4) in moderate yields (Scheme 1). Linker modules 5 and 6 were synthesized via an alternative pathway (Scheme 2). The silyl-protected phosphoramidate 14 was prepared similarly to 9–12, followed by a multistep sequence (Section V., ESI) to couple either the MMAE surrogate or MMAE to the linker scaffold, followed by a final O-silyl deprotection to yield 5 and 6.
Scheme 1.
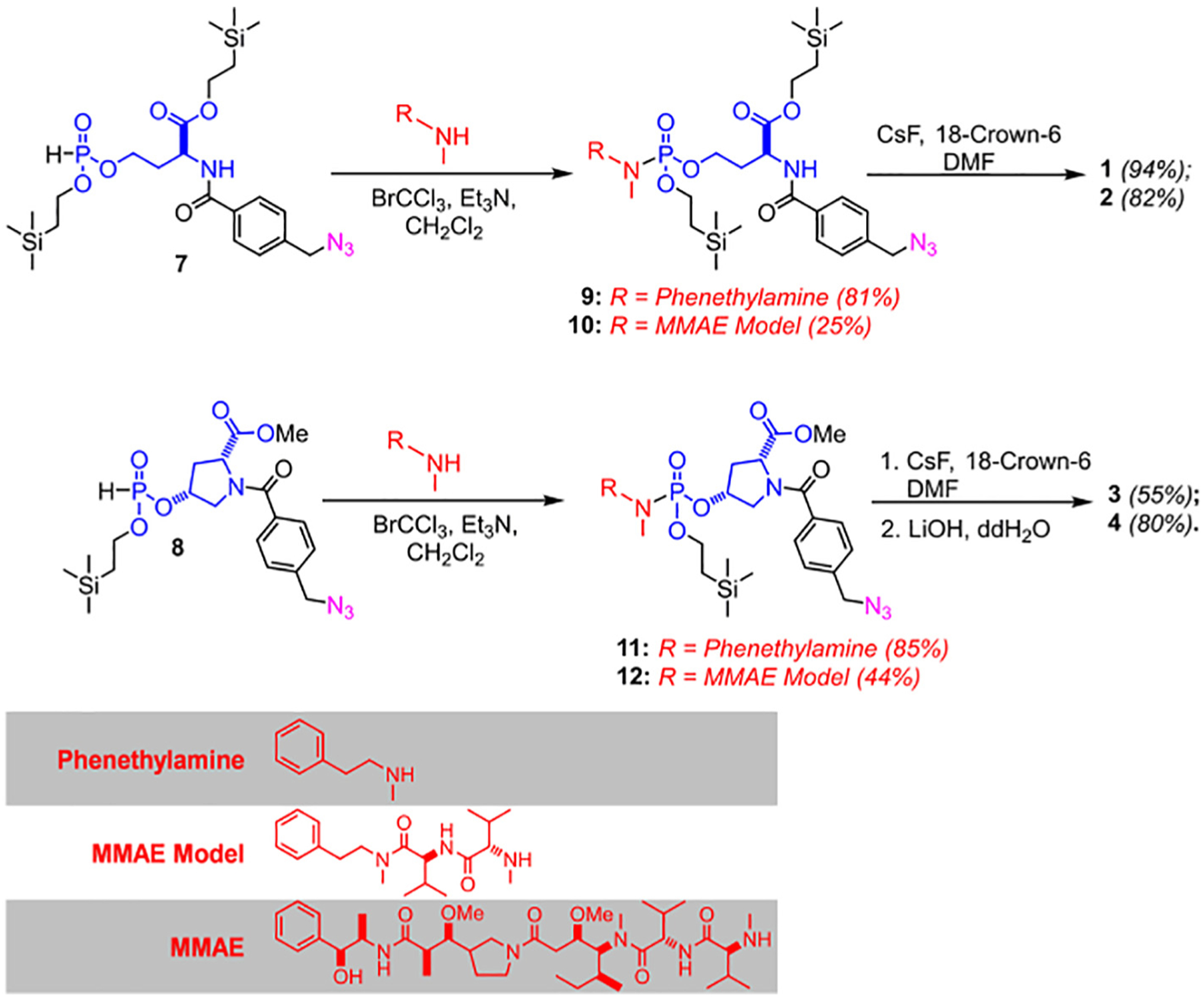
Synthesis of cleavable phosphoramidate linkers, 1–4.
Scheme 2.
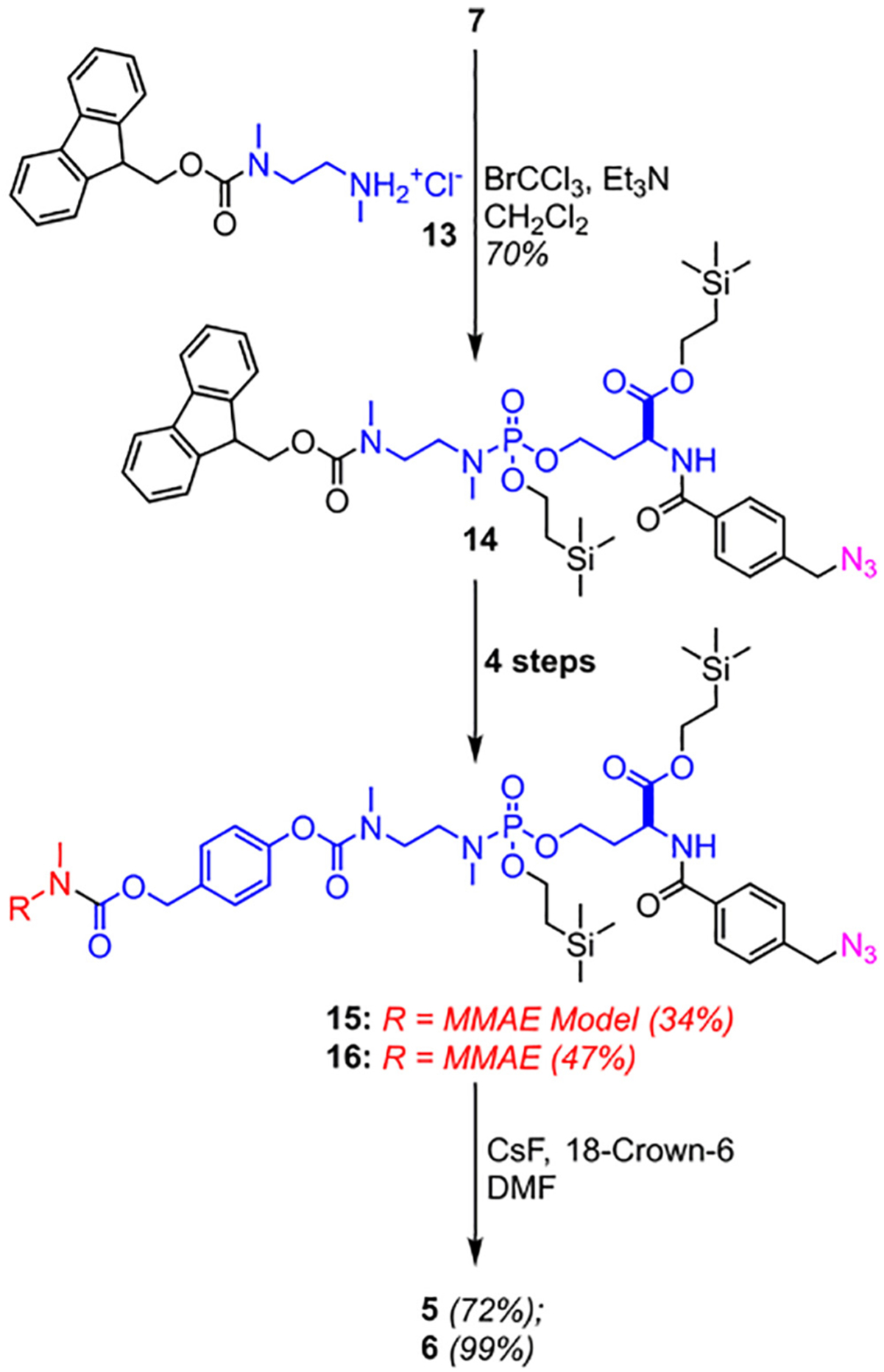
Alternative scheme for synthesis of cleavable phosphoramidate linkers, 5–6.
Once prepared, the hydrolytic P—N bond degradation of linker modules 1–4 was monitored by 31P NMR in various buffers: pH 4.5 (lysosomal pH), 5.0, 5.5, 6.0 (endosomal pH), and 7.4 (physiological) over 8 h at 37 °C. The NMR array data was compiled using MNova 12.0 software and the peak area for each compound was normalized to a constant concentration of external standard (triphenylphosphine oxide, TPPO) (Fig. 3a).
Fig. 3.
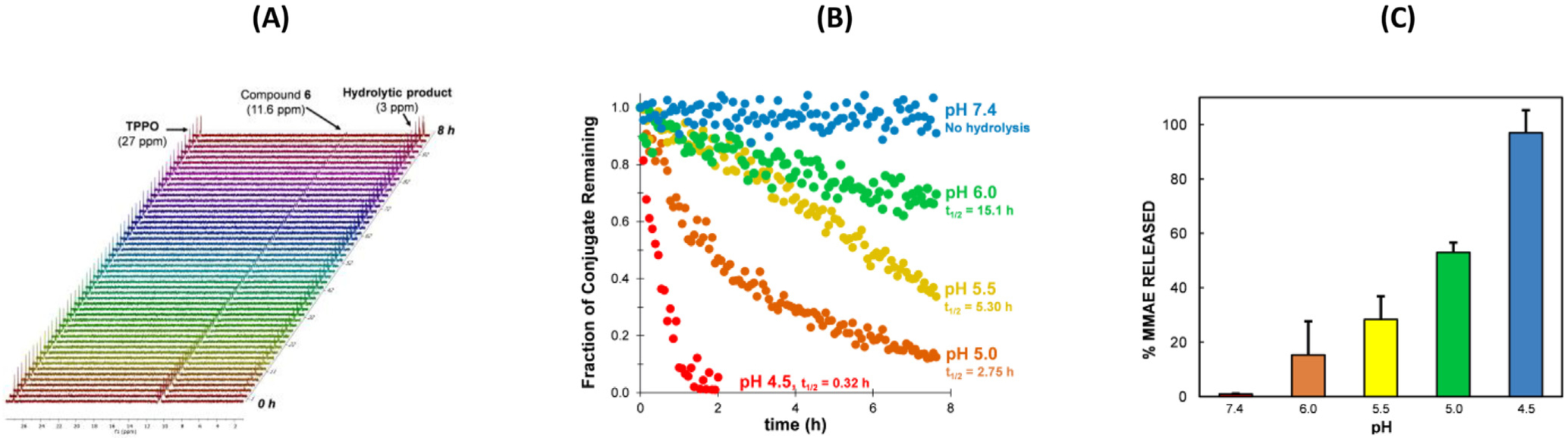
(a) Raw-stacked 31P NMR data for the hydrolysis of 6 (11.6 ppm) at pH 5.0 and formation of its hydrolytic product (3.0 ppm), referenced to the external standard (40 mM TPPO, 27 ppm). (b) Compiled and fitted pH-dependent decay data for the area of 6 at pH 7.4 (blue), pH 6.0 (green), pH 5.5 (yellow), pH 5.0 (orange) and pH 4.5 (red) normalized to a 40 mM TPPO standard. For pH 4.5, hydrolysis complete after 2 h. Half-lives calculated based on linearized first-order kinetics model (p < 0.0001 for all pHs except pH 7.4). (c) HPLC studies showing amount of MMAE released after 8 h, using 1 as an internal standard. Studies was carried out in 100 mM buffers (in triplicate) and quenched with 1 N NaOH after 8 h.
As expected, the rate of hydrolysis of all the linker modules followed first-order kinetics. In addition, compound 1 exhibited a similar hydrolytic pH-dependence to a previously published compound [11] (Fig. 2). In addition, the azido group had little effect on the rate of hydrolysis. Because both compounds 1 and 2 were linked as 2°-amines to the phosphoramidate center, we expected that their hydrolysis rates would be similar. However, compound 2 exhibited unanticipated stability, with hydrolysis being observed only at pH 4.5 (t½ = 42 h). We surmised that the unusual stability of 2 was due to steric congestion imparted by the proximal side-chain of the N-methylvaline residue. To counter this, we designed the cis-D-hydroxyproline based compounds 3 and 4 to constrain the linker structure with respect to the proximity of the carboxylic acid group. Surprisingly, 3 and 4 showed greater hydrolytic stability at all pH values compared to the respective analogs 1 and 2. We believe that this additional unanticipated stability was a result of additional steric hinderance about the phosphorus center imparted by the structural alkyl features around the 2°-alcohol of the cis-D-hydroxyproline residue, thus blocking the backside approach of water to the phosphoramidate center. To circumvent the apparently additive steric constraints presented by both the leaving amine and the alcohol of the phosphoramidate center, 5 was designed to retain the desirable properties of 1, while being insensitive to the structural complexity of the amine-linked payload. Hence, the incorporation of the well-studied self-immolating spacer [15–17] provided a hydrolytic stability profile for 5 that was similar to the simple model compound 1 (Fig. 2).
Fig. 2.
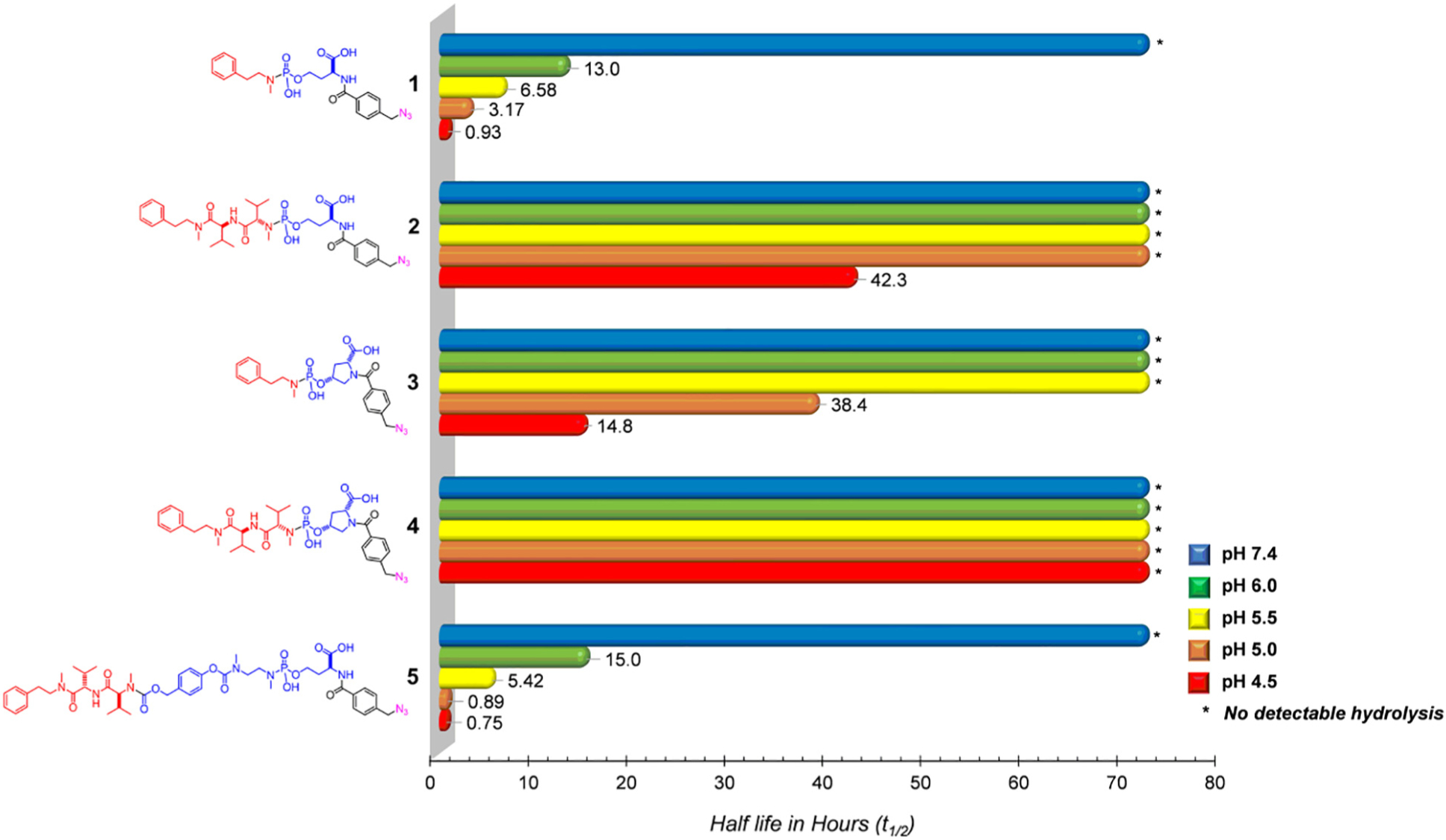
Stability (reported as half-lives) of Model linkers at pH 4.5, 5.0, 5.5, 6.0 and 7.4. Each study was carried out for 8 h.
Based on the results presented in Fig. 2, we extended the design of 5 to that of the MMAE-linker module 6. Indeed, the stability and pH-dependent hydrolysis of 6 was similar to that of both 1 and 5 (Fig 3b). As expected, the extent of MMAE released from the self-immolative spacer of 6 (after 8 h) was consistent with the extent of the pH-dependent hydrolysis of the P—N bond of 6 (Fig 3c).
Mechanistically, the general acid-catalyzed hydrolysis of the P—N bond of these phosphoramidate linkers has been found to follow pseudo-first order kinetics. The presence [10,11] and pKa [18] of the proximal carboxylic acid moiety plays a key role in modulating the stability and pH-triggered cleavage of these linkers. We recently confirmed that the mechanism for hydrolysis involves the displacement of the phosphoramidate amine by the proximal carboxylate [18]. In the case of compounds 5 and 6, this is followed by the collapse of the self-immolative spacer to ultimately release an amine-linked cargo (Scheme 3).
Scheme 3.
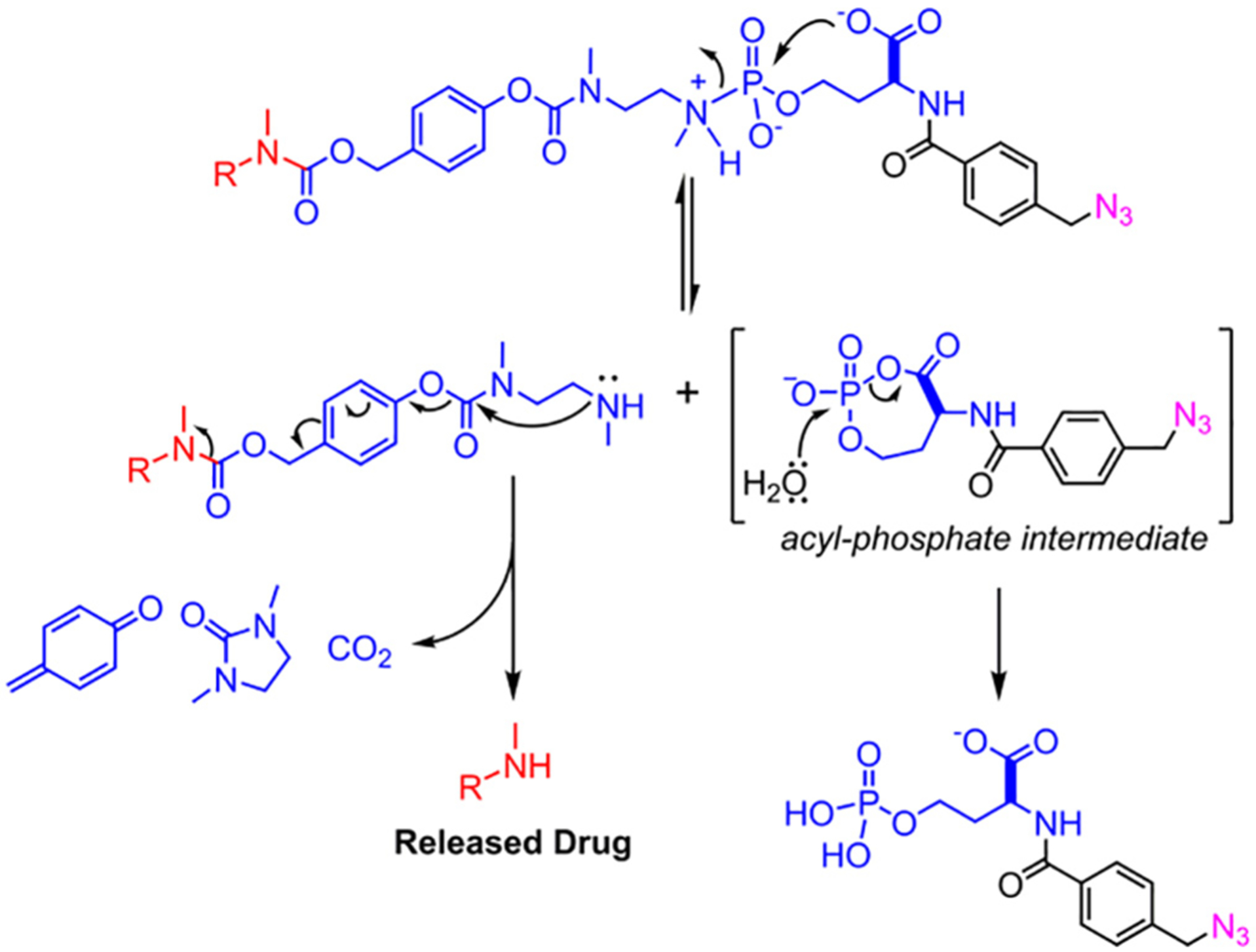
Mechanism of MMAE release from 6.
While the work in this study demonstrates the applicability of phosphoramidate linkers for the controlled release of drug cargo through an amine linkage, we anticipate that these linkers can be adapted more broadly for the controlled release through alcohol-, phenol- (Fig. 4, Site 2), and aniline-type linkages (Fig. 4, Site 1). In addition, we have noted that these phosphoramidate linkers are highly water-soluble because of the ionizable phosphate and carboxylate groups. This property makes the linker an attractive alternative to other cleavable linkers, as it allows for convenient bioconjugation to targeting proteins.
Fig. 4.
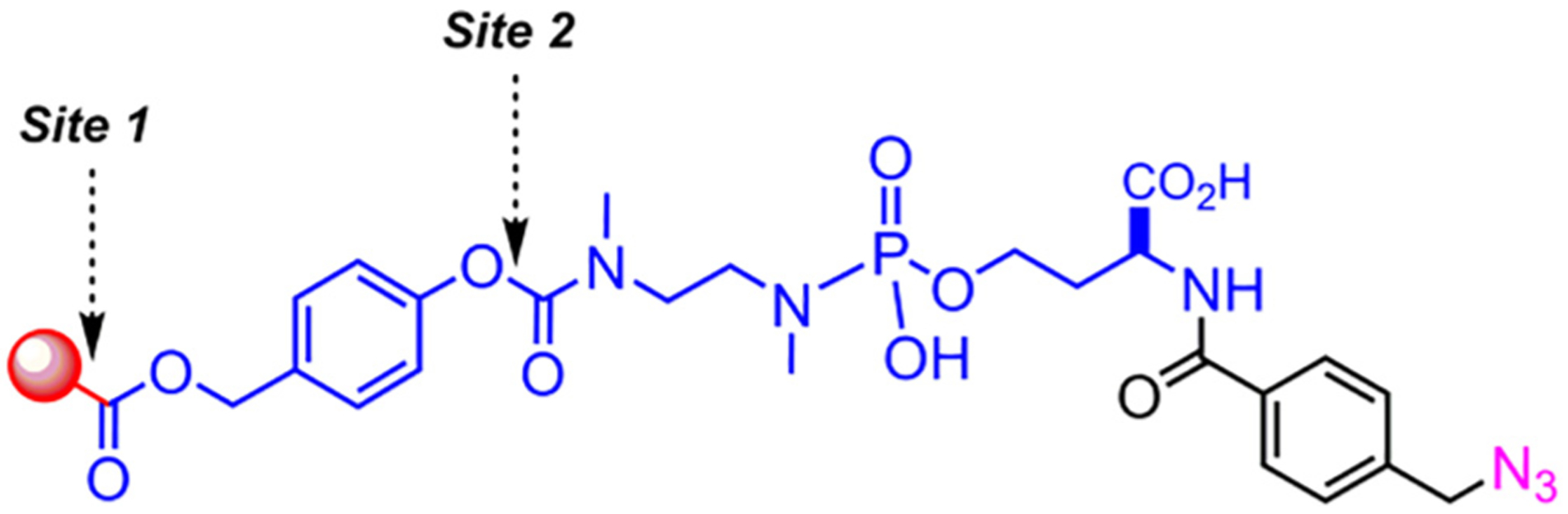
Potential sites for conjugation and release of functionally diverse payloads.
In conclusion, we have reported on the development of a click-ready pH-triggered, water-soluble phosphoramidate linker that is capable of controlled release of amine-containing therapeutic cargo, such as MMAE, a commonly used cytotoxic payload for ADC applications. Central to the functionality of this phosphorami-date-based linker is a proximal carboxylate that promotes the pH-triggered release of payloads through an amine linkage. This linker system was found to be stable at physiological pH, thus ensuring the minimization of possible off-target effects. It was also found to rapidly release amine-linked cargo under the acidic conditions of the endosomal/lysosomal compartments. Studies to demonstrate the versatility of this linker to release diverse payloads through other functional groups (alcohols, phenols, and anilines) are currently underway. Combined with the click-readiness of this linker provided by the azido group, this phosphoramidate linker system can be employed in antibody drug conjugate (ADC) and small-molecule drug conjugate (SMDC) applications, as well as turn-on and ratiometric dye probes.
Acknowledgements
This work was supported, in part, by the National Institutes of Health (Grant CA223121). The authors extend their gratitude for technical assistance to Dr W. Hiscox (WSU Center for NMR Spectroscopy) and Dr. Gerhard Munske (WSU Laboratory of Biotechnology and Bioanalysis).
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Chau CH, Steeg PS, Figg WD, Lancet 394 (2019) 793–804. [DOI] [PubMed] [Google Scholar]
- [2].Casi G, Neri D, Control J. Release 161 (2012) 422–428. [DOI] [PubMed] [Google Scholar]
- [3].Ducry L, Stump B, Bioconjug. Chem 21 (2010) 5–13. [DOI] [PubMed] [Google Scholar]
- [4].Dal Corso A, Pignataro L, Belvisi L, Gennari C, Chem. Eur. J 25 (2019) 14740–14757. [DOI] [PubMed] [Google Scholar]
- [5].Bargh JD, Isidro-Llobet A, Parker JS, Spring DR, Chem. Soc. Rev 48 (2019) 4361–4374. [DOI] [PubMed] [Google Scholar]
- [6].Beck A, Goetsch L, Dumontet C, Corvaïa N, Nat. Rev. Drug Discov 16 (2017) 315–337. [DOI] [PubMed] [Google Scholar]
- [7].Pillow TH, Sadowsky JD, Zhang D, Yu SF, Del Rosario G, Xu K, He J, Bhakta S, Ohri R, Kozak KR, Ha E, Junutula JR, Flygare JA, Chem. Sci 8 (2016) 366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Doronina SO, Toki BE, Torgov MY, Mendelsohn BA, Cerveny CG, Chace DF, DeBlanc RL, Gearing RP, Bovee TD, Siegall CB, Francisco JA, Wahl AF, Meyer DL, Senter PD, Nat. Biotechnol 21 (2003) 778–784. [DOI] [PubMed] [Google Scholar]
- [9].Hamann PR, Hinman LM, Hollander I, Beyer CF, Lindh D, Holcomb R, Hallett W, Tsou HR, Upeslacis J, Shochat D, Mountain A, Flowers DA, Bernstein I, Bioconjug. Chem 13 (2002) 47–58. [DOI] [PubMed] [Google Scholar]
- [10].Choy CJ, Geruntho JJ, Davis AL, Berkman CE, Bioconjug. Chem 27 (2016) 824–830. [DOI] [PubMed] [Google Scholar]
- [11].Choy CJ, Ley CR, Davis AL, Backer BS, Geruntho JJ, Clowers BH, Berkman CE, Bioconjug. Chem 27 (2016) 2206–2213. [DOI] [PubMed] [Google Scholar]
- [12].Olatunji FP, Kesic BN, Choy CJ, Berkman CE, Bioorganic Med. Chem. Lett 29 (2019) 2571–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Anderl J, Faulstich H, Hechler T, Kulke M, Methods Mol. Biol 1045 (2013) 51–70. [DOI] [PubMed] [Google Scholar]
- [14].Le Corre SS, Berchel M, Couthon-Gourvès H, Haelters JP, Jaffrès PA, Beilstein J Org. Chem 10 (2014) 1166–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].DeWit MA, Gillies ER, J. Am. Chem. Soc 131 (2009) 18327–18334. [DOI] [PubMed] [Google Scholar]
- [16].McBride RA, Gillies ER, Macromolecules 46 (2013) 5157–5166. [Google Scholar]
- [17].Sirianni QEA, Gillies ER, Polymer (Guildf) 202 (2020) 122638. [Google Scholar]
- [18].Backer BS, Choy CJ, Davis AL, Browne ZS, Berkman CE, Tetrahedron Lett. 61 (2020) 151650. [DOI] [PMC free article] [PubMed] [Google Scholar]