

Minimal residual disease (MRD) monitoring in acute myeloid leukemia (AML) plays an important role in outcome prediction, risk-stratification and therapy adjustment as well as in the early detection of impending relapse.1 The most common genetic markers utilizable for MRD monitoring in AML are fusion genes (FG). Either FG themselves or their transcripts (fusion transcripts, FT) can be monitored via quantitative PCR (qPCR). The uniform copy number per leukemic cell makes FG an ideal target for unambiguously interpretable assessment of MRD levels. However, the use of FG-based MRD monitoring has been limited by the laboriousness of PCR-based genomic fusion sequence identification2–4; thus, only the FT-based approach has been widely used so far.1,5 Although highly feasible, it has several flaws. The number of FT copies per leukemic cell is unknown and possibly inconstant. Moreover, the diagnostic FT expression levels vary significantly among patients (>2 logs),5,6 and in the patients with lower expression levels, the sensitivity of MRD monitoring is thus reduced.
Various methods utilizing the next generation sequencing (NGS) technology have become widely available during the last few years, offering alternative tools for the identification of genomic fusions.7,8 In this study, we investigated the feasibility of genomic breakpoint identification via targeted NGS, the performance of patient-specific assays for genomic breakpoint quantification and the benefit of FG as targets for MRD monitoring in AML patients with PML-RARA/CBFB-MYH11/RUNX1-RUNX1T1 fusions.
We performed targeted sequencing utilizing hybridization to a custom-designed probe set for target enrichment (Supplementary Fig. 1). With a median sequencing output of 504 k reads mapped to target region, we successfully identified genomic fusion sequences in all 23 studied patients. In 20/23 patients we found > 1 fusion sequence (Supplementary Results, Supplementary Table 1), that is, > 1 possible MRD target. One fusion sequence per patient was selected to design a qPCR assay for MRD monitoring. All 23 assays were successfully optimized reaching sufficiently deep quantitative range and sensitivity (10−4–10−5) without non-specific, off-target amplification. We used these in-house established assays to quantify FG and standard qPCR assays5 to quantify FT, and assessed MRD levels in 265 follow-up (FU) samples (Supplementary Table 2, Fig. 1A, Supplementary Fig. 2–6).
Figure 1.
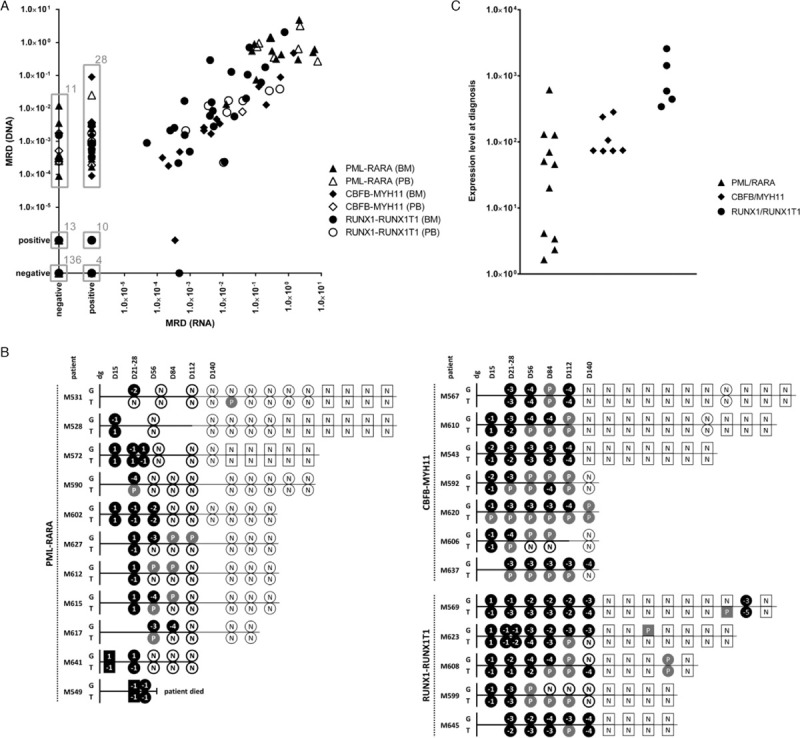
Results of minimal residual disease monitoring and diagnostic levels of fusion transcripts. (A) Comparison of minimal residual disease (MRD) levels in 265 follow-up samples of patients harboring the PML-RARA (triangle), CBFB-MYH11 (diamond) and RUNX1-RUNX1T1 (circle) fusions measured by the FG-based approach (DNA) versus by the FT-approach (RNA). Grey boxes surround specific clusters of samples whose counts are indicated by the numbers at top right corners. (B) Schematic representation of MRD levels in individual patients harboring the PML-RARA, CBFB-MYH11 and RUNX1-RUNX1T1 fusions during their treatment courses as assessed by the FG-based approach (G) versus by the FT-based approach (T). Bone marrow (BM) samples are shown as circles, peripheral blood (PB) samples as squares. If paired BM and PB samples were analyzed at the particular time-point, only BM is shown. MRD levels ≥0.5 are coded as “1”, < 0.5 – ≥0.05 as “−1”, < 0.05 – ≥0.005 as “−2”, < 0.005 – ≥0.0005 as “−3”, < 0.0005 – ≥0.00005 as “−4”, < 0.00005 as “−5”. All samples with quantifiably positive MRD levels are shown as black symbols, samples with non-quantifiably positive and negative MRD levels are shown as grey symbols with “P” code and white symbols with “N” code, respectively. Bold versus thin symbol borders and time-lines represent intensive versus maintenance treatment phases. Time course in days (D) is shown at the top of scheme. dg = diagnosis; positive – non-quantifiably positive. (C) Fusion transcript expression levels (number of fusion transcript copies per 1000 copies of GUS) at diagnosis in patients with PML-RARA (n = 11), CBFB-MYH11 (n = 7) and RUNX1-RUNX1T1 (n = 5).
MRD was negative and positive by both the FG-based and FT-based approaches in 136 and 100 samples, respectively, positive only by the FG-based approach in 24 and only by the FT-based approach in 5 samples. Out of 100 “double-positive” samples, in 61 the MRD levels differed by ≤1 log (n = 51) or were non-quantifiably positive (NQP) by both methods (n = 10). In 32 “double-positive” samples, MRD was higher by > 1 log using the FG-based compared to the FT-based approach (n = 4), or was quantifiably positive only by the FG-based while NQP by the FT-based approach (n = 28). Vice-versa, in 7 “double-positive” samples MRD was higher by > 1 log using the FT-based compared to the FG-based approach (n = 6) or NQP only by the FG-based approach (n = 1).
We analyzed how discrepancy in MRD data translates into the evaluation of response to therapy (Supplementary Table 2, Fig. 1B). In 8/18 patients monitored up to molecular remission (MR), the negativity of both FG and FT was achieved at the same time-point. Of the remaining 10 patients, MRD negativity was reached at an earlier time-point by the FG-based in 2 patients and by the FT-based approach in 8 patients. Besides better assessment of MR, the FG-based approach also improved our insight into the dynamics of MRD clearance as it provided quantitative data on MRD levels in 39 samples of 16 patients that were NQ-positive or negative by the FT-based approach. Notably, if early treatment response would be classified according to MRD levels at the end of induction treatment into categories “negative”/“positive <1E−03 (ie, <0.1%)”/“positive ≥1E−03 (ie, ≥0.1%)”, the classification would be skewed depending on the approach in 8/22 patients. Five patients were only low/NQ-positive by the FT- while ≥1E−03 by FG-quantification, 1 patient was positive ≥1E−03 by FT- and <1E−03 by FG-, and 2 patients positive by FG- were negative by FT-based approach.
Conversion from negativity to positivity was detected simultaneously by both methods in 1 patient, only by the FG-based approach in 1 patient and only by the FT-based approach in 2 patients.
Altogether, the FG-based assays were more sensitive for MRD detection compared to the FT-based approach. We have analyzed in more detail the differences in target detection sensitivities in the samples with discrepant results of MRD monitoring. The FT levels in diagnostic samples varied over > 3 logs among patients (Fig. 1C, Supplementary Table 2). Importantly, low diagnostic FT expression dramatically limits the sensitivity of FT-detection in FU samples (Supplementary Table 2). We thus expected that the inferior sensitivity of FT-detection compared to FG-detection in the patients with low diagnostic FT levels could significantly contribute to the MRD discrepancy. Surprisingly, in a non-negligible subset of samples, the discrepancies in MRD levels were not convincingly attributable to the differences in sensitivities (Table 1).
Table 1.
Sensitivity of Target Detection in Samples with MRD Detectable by One Method Only.
| Patient ID | AML Subtype | Sample Type | Days Post DG | MRD FT | FT Detection Sensitivity | MRD FG | FG Detection Sensitivity |
|---|---|---|---|---|---|---|---|
| M531 | PML-RARA | BM | 27 | NEG | 2,96E−03 | 1,22E−02 | 1,00E−04 |
| M627 | PML-RARA | BM | 56 | NEG | 6,94E−02 | 3,62E−03 | 1,00E−05 |
| M620 | CBFB-MYH11 | PB | 104 | NEG | 9,17E−04 | 1,76E−03 | 1,00E−05 |
| M623 | RUNX1-RUNX1T1 | BM | 168 | NEG | 6,49E−05 | 1,53E−03 | 1,00E−05 |
| M567 | CBFB-MYH11 | PB | 57 | NEG | 9,81E−04 | 5,20E−04 | 1,00E−05 |
| M590 | PML-RARA | PB | 26 | NEG | 1,31E−03 | 3,70E−04 | 1,00E−05 |
| M627 | PML-RARA | PB | 80 | NEG | 9,55E−04 | 3,20E−04 | 1,00E−05 |
| M637 | CBFB-MYH11 | BM | 208 | NEG | 1,63E−04 | 3,00E−04 | 1,00E−05 |
| M615 | PML-RARA | PB | 54 | NEG | 2,34E−02 | 2,60E−04 | 1,00E−05 |
| M606 | CBFB-MYH11 | PB | 28 | NEG | 6,95E−04 | 2,40E−04 | 1,00E−05 |
| M617 | PML-RARA | BM | 85 | NEG | 1,08E−01 | 9,00E−05 | 1,00E−05 |
| M567 | CBFB-MYH11 | PB | 26 | NEG | 6,18E−04 | NQP | 1,00E−05 |
| M592 | CBFB-MYH11 | PB | 90 | NEG | 3,22E−04 | NQP | 1,00E−05 |
| M599 | RUNX1-RUNX1T1 | PB | 59 | NEG | 4,93E−05 | NQP | 1,00E−05 |
| M606 | CBFB-MYH11 | BM | 61 | NEG | 8,43E−04 | NQP | 1,00E−04 |
| M606 | CBFB-MYH11 | BM | 103 | NEG | 3,66E−04 | NQP | 1,00E−04 |
| M612 | PML-RARA | BM | 55 | NEG | 2,45E−03 | NQP | 1,00E−04 |
| M612 | PML-RARA | PB | 55 | NEG | 4,45E−03 | NQP | 1,00E−04 |
| M612 | PML-RARA | BM | 81 | NEG | 5,28E−04 | NQP | 1,00E−04 |
| M615 | PML-RARA | BM | 81 | NEG | 8,09E−03 | NQP | 1,00E−05 |
| M623 | RUNX1-RUNX1T1 | PB | 168 | NEG | 1,26E−04 | NQP | 1,00E−05 |
| M623 | RUNX1-RUNX1T1 | PB | 381 | NEG | 1,76E−05 | NQP | 1,00E−05 |
| M627 | PML-RARA | BM | 80 | NEG | 1,71E−03 | NQP | 1,00E−05 |
| M627 | PML-RARA | BM | 112 | NEG | 2,72E−04 | NQP | 1,00E−05 |
| M608 | RUNX1-RUNX1T1 | BM | 207 | 4,68E-04 | 4,58E−04 | NEG | 1,00E−04 |
| M531 | PML-RARA | BM | 254 | NQP | 1,77E−04 | NEG | 1,00E−04 |
| M569 | RUNX1-RUNX1T1 | PB | 574 | NQP | 2,14E−05 | NEG | 1,00E−05 |
| M599 | RUNX1-RUNX1T1 | BM | 101 | NQP | 1,01E−05 | NEG | 1,00E−05 |
| M599 | RUNX1-RUNX1T1 | BM | 143 | NQP | 1,24E−04 | NEG | 1,00E−05 |
In samples with MRD negative by FT-based approach, but quantifiably positive by FG-approach at the levels above the sensitivity of FT-detection, the FT-detection sensitivity is highlighted in blue. Red color highlights the sensitivities of FG detection deeper compared to those of FT detection in samples with MRD negative by FG- but positive by FT-based approach.
BM = bone marrow, DG = diagnosis, FG = fusion gene, FT = fusion transcript, MRD = minimal residual disease, PB = peripheral blood.
Although in all 24 samples positive only by the FG-based approach the sensitivity of FG-detection was deeper compared to the calculated FT-detection sensitivity, the MRD level in 40% (4/10) of the quantifiably positive samples was above the sensitivity of FT-detection. Strikingly, also the sensitivity of FG-detection was equal or deeper compared to FT-detection in all 5 samples positive only by the FT-based approach.
Various PCR-based methods used to obtain genomic fusion sequences in the past decades generally suffered from low amplification efficiency and laboriousness, they were time-consuming and frequently required a large amount of high molecular weight DNA.2–4 In the first part of our study we showed that these laborious methods can be replaced by targeted sequencing which enables an efficient identification of genomic fusion sequences within 1 week from diagnosis and requires only 50 ng of diagnostic DNA.
In laboratories where NGS-based methods are well established within routine diagnostics (eg, panel/whole-exome/whole-transcriptome sequencing, NGS-based screening of immunoreceptor gene-rearrangements in acute lymphoblastic leukemia), pooling of the FG targeted sequencing with other NGS-based experiments is fully feasible. This significantly reduces total expenses; in an optimal setting, the final costs of the genomic fusion identification could be even lower compared to the PCR-based approach.
In the subsequent part of our study we showed, that, similarly to the identification of fusion sequences, the optimization of patient-specific qPCR assays was straightforward, facilitated by the sequence uniqueness and thus the lack of non-specific amplification.
When comparing the MRD levels measured by the two approaches, we have encountered both possible types of discrepancy; in a quarter of samples (68/265), MRD levels assessed by the FG-based approach were either significantly higher or lower compared to those assessed by the FT-based approach.
The first type of discrepancy (>80% discordant samples), demonstrates that MRD levels can be frequently underestimated by the FT-based approach. Primarily, this can be a result of different sensitivity; indeed, in a majority of samples, the FT approach was less sensitive than the FG-based assay. However, in some samples, the calculated FT detection sensitivity should have been sufficient to yield the same result as the FG-based approach. This data demonstrates that, unlike in the FG approach, the expression level – and thus the target to cell ratio - is inconstant, which we consider a major pitfall of the FT-based MRD monitoring. The change in expression can go in both directions, as shown by the samples with the second type of discrepancy, where the FT-based MRD levels were significantly higher compared to FG quantification. This situation was less common and, notably, we detected such samples at the earliest treatment time-points in several patients.
Both overestimation and underestimation of MRD levels caused by expression changes and variable levels of FT per cell are certainly undesirable as they skew the evaluation of response to therapy. The only exception could be an earlier detection of molecular relapse, possibly caused in some cases by high FT levels in relapse-driving cells, resulting in an increased sensitivity of its detection. However, our data on this subject is very limited.
Multiple studies have demonstrated that levels of MRD at certain time-points during therapy are highly predictive of patients’ outcome.1 In the AML subtypes presented in our report, those studies mainly utilized data from FT-based MRD monitoring9–15; the predictive value of MRD levels assessed by the FG-based approach has not been evaluated yet. Our study included a limited number of patients, none of which relapsed so far. Thus, we could not compare the prognostic significance of MRD assessed by the two approaches.
In summary, our study shows that both the identification of genomic fusion sequences and the FG-based MRD monitoring are highly feasible in PML-RARA-/CBFB-MYH11-/RUNX1-RUNX1T1-positive AML. Quantification of FG, a stable target with a constant level per cell, enables precise assessment of the proportion of positive cells and represent a technically superior tool for the evaluation of therapy response than the so far widely used FT-based monitoring. We believe that our data provides rationale for additional studies addressing the question whether such an improvement of evaluation of response to therapy could translate into an improvement of risk prediction and therapy tailoring – and, finally, of patients’ outcome.
Sources of Funding
This study was supported by grants from the Czech Health Research Council (NU20-07-00322 and NV19-07-00329) and Charles University (UNCE 204012), by the project 1000 braves (Foundation Nation for Children) and by the project (Ministry of Health, Czech Republic) for conceptual development of research organization 00064203 (University Hospital Motol, Prague, Czech Republic). Research infrastructure was supported by the Ministry of Education, Youth and Sports (NPU I no. LO1604 and LM2015091).
Supplementary Material
Footnotes
The authors declare no conflicts of interest.
Supplemental Digital Content is available for this article.
References
- 1.Schuurhuis GJ, Heuser M, Freeman S, et al. Minimal/measurable residual disease in AML: a consensus document from the European LeukemiaNet MRD Working Party. Blood. 2018;131:1275–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duployez N, Nibourel O, Marceau-Renaut A, et al. Minimal residual disease monitoring in t(8;21) acute myeloid leukemia based on RUNX1-RUNX1T1 fusion quantification on genomic DNA. Am J Hematol. 2014;89:610–615. [DOI] [PubMed] [Google Scholar]
- 3.Kommers IO, Bartley PA, Budgen B, et al. Sensitive monitoring of acute promyelocytic leukemia by PML-RARA DNA Q-PCR. Leuk Lymphoma. 2017;58:1767–1769. [DOI] [PubMed] [Google Scholar]
- 4.Meyer C, Schneider B, Reichel M, et al. Diagnostic tool for the identification of MLL rearrangements including unknown partner genes. Proc Natl Acad Sci U S A. 2005;102:449–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gabert J, Beillard E, van der Velden VH, et al. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia - a Europe Against Cancer program. Leukemia. 2003;17:2318–2357. [DOI] [PubMed] [Google Scholar]
- 6.Beillard E, Pallisgaard N, van der Velden VH, et al. Evaluation of candidate control genes for diagnosis and residual disease detection in leukemic patients using ‘real-time’ quantitative reverse-transcriptase polymerase chain reaction (RQ-PCR) - a Europe against cancer program. Leukemia. 2003;17:2474–2486. [DOI] [PubMed] [Google Scholar]
- 7.Afrin S, Zhang CRC, Meyer C, et al. Targeted next-generation sequencing for detecting MLL gene fusions in leukemia. Mol Cancer Res. 2018;16:279–285. [DOI] [PubMed] [Google Scholar]
- 8.Grossmann V, Kohlmann A, Klein HU, et al. Targeted next-generation sequencing detects point mutations, insertions, deletions and balanced chromosomal rearrangements as well as identifies novel leukemia-specific fusion genes in a single procedure. Leukemia. 2011;25:671–680. [DOI] [PubMed] [Google Scholar]
- 9.Grimwade D, Jovanovic JV, Hills RK, et al. Prospective minimal residual disease monitoring to predict relapse of acute promyelocytic leukemia and to direct pre-emptive arsenic trioxide therapy. J Clin Oncol. 2009;27:3650–3658. [DOI] [PubMed] [Google Scholar]
- 10.Santamaria C, Chillon MC, Fernandez C, et al. Using quantification of the PML-RARalpha transcript to stratify the risk of relapse in patients with acute promyelocytic leukemia. Haematologica. 2007;92:315–322. [DOI] [PubMed] [Google Scholar]
- 11.Testi AM, Pession A, Diverio D, et al. Risk-adapted treatment of acute promyelocytic leukemia: results from the International Consortium for Childhood APL. Blood. 2018;132:405–412. [DOI] [PubMed] [Google Scholar]
- 12.Yin JA, O’Brien MA, Hills RK, et al. Minimal residual disease monitoring by quantitative RT-PCR in core binding factor AML allows risk stratification and predicts relapse: results of the United Kingdom MRC AML-15 trial. Blood. 2012;120:2826–2835. [DOI] [PubMed] [Google Scholar]
- 13.Zhang L, Cao Z, Zou Y, et al. Quantification of PML/RARa transcript after induction predicts outcome in children with acute promyelocytic leukemia. Int J Hematol. 2012;95:500–508. [DOI] [PubMed] [Google Scholar]
- 14.Jourdan E, Boissel N, Chevret S, et al. Prospective evaluation of gene mutations and minimal residual disease in patients with core binding factor acute myeloid leukemia. Blood. 2013;121:2213–2223. [DOI] [PubMed] [Google Scholar]
- 15.Rucker FG, Agrawal M, Corbacioglu A, et al. Measurable residual disease monitoring in acute myeloid leukemia with t(8;21)(q22;q22.1): results from the AML Study Group. Blood. 2019;134:1608–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.