

Background.
Portopulmonary hypertension (POPH), pulmonary arterial hypertension (PAH) that develops in the setting of portal hypertension, affects 5%–6% of patients with liver disease and is associated with significant morbidity and mortality. A mean pulmonary arterial pressure (mPAP) threshold of 35 mm Hg is used to stratify perioperative risk and liver transplant eligibility in treated POPH patients but does not take into account the specific factors that contribute to the pressure elevation.
Methods.
In this case series, we describe the characteristics and posttransplant outcomes of patients with treated POPH and an mPAP ≥35 mm Hg and pulmonary vascular resistance (PVR) <250 dynes-s-cm−5 at or just before liver transplantation (LT). We also describe the effect of PAH therapy on pulmonary hemodynamics in patients with POPH.
Results.
Sixteen patients were included. All patients were on PAH therapy at the time of LT. PAH therapy resulted in a decrease of mPAP (median 18.4%; interquartile range [IQR] 8.9%–27.0%) with a reduction in PVR (median 50.5%; IQR, 45.4%–70.7%), and an increase in both cardiac output (CO) (median 28.1%; IQR 5.7%–63.8%) and PAWP (median 50.0%; IQR 16.7%–108.3%) before LT. One year posttransplant survival was 69% (11/16); however, only 1 death was attributed to POPH. At 1-year posttransplant, 63.6% (7/11) of patients were weaned off all PAH therapy with clinical and echocardiographic resolution of POPH.
Conclusions.
In treated POPH patients with an mPAP ≥35 mm Hg and PVR < 250 dynes-s-cm−5 before LT, 1-year posttransplant survival was 69% and the majority of patients were able to discontinue PAH therapy.
INTRODUCTION
Portopulmonary hypertension (POPH) is characterized by an elevated mean pulmonary arterial pressure (mPAP) and pulmonary vascular resistance (PVR) in the setting of portal hypertension. POPH affects 5%–6% of patients with liver disease and is associated with significant morbidity and mortality.1 POPH is treated with pulmonary arterial hypertension (PAH) targeted therapy and can sometimes resolve with liver transplantation (LT), but poorly controlled POPH can also preclude LT due to increased perioperative risk.1
The diagnosis and severity of POPH has implications for perioperative risk assessment.1 Twenty years ago, a retrospective analysis of 43 patients with POPH reported 50% perioperative cardiopulmonary mortality in patients with an mPAP of 35−50 mm Hg at the time of LT.2 Patients with an mPAP ≥50 mm Hg had 100% perioperative mortality. Notably, the majority of patients included in this study were not treated with PAH-targeted therapy and were transplanted in an era in which PAH treatment options were limited.2 A subsequent multicenter study of 36 POPH patients described favorable outcomes with no posttransplant deaths in the subgroup of POPH patients with an mPAP < 35 mm Hg and PVR < 250 dynes-s-cm−5.3 These studies provided the foundation for the POPH model for end-stage liver disease (MELD) exception criteria. According to these criteria, posttreatment mPAP must be <35 mm Hg to qualify for POPH MELD exception points.4 Notably, the criteria have not been significantly modified since 2006 and were developed based upon transplant experiences before 2001. Patients with a mPAP ≥35 mm Hg are not eligible for a standardized POPH MELD exception and may lose previously accrued exception points if mPAP has increased above that threshold on subsequent right heart catheterizations. Importantly, the mPAP threshold is absolute and does not take into account the cause of the pressure elevation, which may be due to causes other than POPH, such as volume overload or a hyperdynamic cardiac output.
Recent studies suggest that the mPAP threshold of 35 mm Hg may not be optimal for assessing perioperative risk in patients with POPH.5,6 Specifically, mPAP may be elevated due to an increased PVR secondary to obstruction to pulmonary arterial blood flow, an increased pulmonary artery wedge pressure (PAWP) due to volume overload, an increased cardiac output (CO) related to the hyperdynamic state of cirrhosis, or not uncommonly, combinations of all 3 factors. Appropriate treatment varies and depends on the underlying cause as assessed by right heart catheterization. For example, a patient with treated POPH and a persistently elevated mPAP and PVR would warrant additional PAH-targeted therapy while a persistently elevated mPAP due to a hyperdynamic CO with a normal PVR would not.
We hypothesized that patients with treated POPH and an mPAP ≥35 mm Hg at the time of LT with a normal PVR (defined here as <250 dynes-s-cm−5) and preserved right ventricular function could have successful LT with minimal perioperative cardiopulmonary mortality. We performed a case series study to describe the overall posttransplant outcomes of patients with POPH and an mPAP ≥35 mm Hg and PVR < 250 dynes-s-cm−5 at the time of LT.
MATERIAL AND METHODS
We performed a multicenter case series study of patients with POPH at University of Wisconsin and Mayo Clinic Rochester, Arizona, and Florida. Patients were identified from review of available clinical POPH databases at each institution. POPH was defined by an elevated mPAP >25 mm Hg with an elevated PVR >3 WU in accordance with current guidelines.1 We included patients with an elevated PAWP at baseline if the transpulmonary gradient was >12 mm Hg.7 Alternative causes of PH were evaluated and excluded as part of their comprehensive PH evaluation. Patients who underwent LT between 2000 and 2019 were included. Baseline hemodynamic data refer to the initial diagnostic right heart catheterization. Pretransplant hemodynamic data refer to the right heart catheterization hemodynamic data most immediately before LT. Clinical, laboratory, echocardiographic, and hemodynamic data and outcomes were collected from review of the medical record. Right ventricular function was visually estimated on echocardiogram as per each institution’s protocol. The percent changes in hemodynamics (mPAP, PVR, CO, and PAWP) were calculated as follows: ([pretransplant value-initial diagnostic value]/initial diagnostic value × 100) for patients who had repeated right heart catheterizations before and after PAH therapy. The case series was exempt from Institutional Review Board approval. Data are summarized as median (interquartile range [IQR]) or number (percent).
RESULTS
Sixteen patients with an elevated mPAP ≥35 mm Hg at or just before LT were included. Patient characteristics are summarized in Table 1. Individual hemodynamic data are detailed in Table 2. All patients underwent deceased donor LT. Patients were treated with various PAH-targeted therapies at the time of LT, including phosphodiesterase 5 inhibitors, endothelin receptor antagonists, prostacyclin analogs, and inhaled nitric oxide. Before LT, PAH therapy resulted in a mild decrease of mPAP (median 18.4%; IQR 8.9%–27.0%) with a reduction in PVR (median 50.5%; IQR 45.4%–70.7%) and an increase in both CO (median 28.1%; IQR 5.7%–63.8%) and PAWP (median 50.0%; IQR 16.7%–108.3%) (Figure 1). All patients had normal, borderline, or mildly reduced right ventricular function on echocardiogram before LT (Table 2). Additionally, the majority of patients were treated with a loop diuretic (93.8%, 15/16) and a potassium-sparing diuretic (93.8%, 15/16) before LT. Five out of 16 (31.3%) patients underwent perioperative hemodialysis or ultrafiltration within 6 months of LT.
TABLE 1.
Patient characteristics
| Characteristic | |
|---|---|
| Female | 11, 68.8% |
| Age at pulmonary hypertension diagnosis, y | 50 (41–57) |
| Age at LT, y | 51 (45–58) |
| Liver disease pathogenesis | |
| Alcohol | 7, 43.8% |
| Hepatitis C | 7, 43.8% |
| Nonalcoholic steatohepatitis | 1, 6.3% |
| Cryptogenic cirrhosis | 2, 12.5% |
| Autoimmune hepatitis | 1, 6.3% |
| Primary sclerosing cholangitis | 1, 6.3% |
| Other | 2, 12.5% |
| MELD-Na score at LT | 18 (11–31) |
| Portopulmonary hypertension MELD exception | 7, 43.8% |
| Initial pulmonary hemodynamics | |
| Mean pulmonary artery pressure, mm Hg | 48 (44–54) |
| Pulmonary artery wedge pressure, mm Hg (n = 15) | 12 (8–17) |
| Transpulmonary gradient, mm Hg (n = 15) | 36 (30–39) |
| Right atrial pressure, mm Hg (n = 15) | 10 (5–18) |
| Cardiac output, L/min (n = 15) | 5.8 (5.4–7.4) |
| Cardiac index, L/min/m2 (n = 15) | 3.0 (2.8–3.6) |
| Pulmonary vascular resistance, dynes-s-cm−5 (n = 15) | 455 (344–592) |
| Pretransplant pulmonary hemodynamics | |
| Mean pulmonary artery pressure, mm Hg | 38 (35–42) |
| Pulmonary artery wedge pressure, mm Hg (n = 15) | 20 (14–22) |
| Transpulmonary gradient, mm Hg (n = 15) | 20 (18–23) |
| Right atrial pressure, mm Hg (n = 15) | 13 (10–14) |
| Cardiac output, L/min | 8.5 (7.3–9.8) |
| Cardiac index, L/min/m2 (n = 14) | 4.4 (4.1–5.2) |
| Pulmonary vascular resistance, dynes-s-cm−5 (n = 15) | 191 (172–216) |
| Time between pretransplant right heart catheterization and liver transplant, d | 40 (11–72) |
| PAH therapy at LT | |
| Phosphodiesterase 5 inhibitor | 10, 62.5% |
| Endothelin receptor antagonist | 3, 18.8% |
| Parenteral prostacyclin therapy | 8, 50% |
| Inhaled nitric oxide | 1, 6.3% |
| Outcomes | |
| Acute cardiopulmonary related mortality within 30 d of LT | 1, 6.3% |
| 1-y survival | 11, 68.8% |
| Off PAH therapy at 1 y | 7/11, 63.6% |
Data reported as number, % or median (range). Individual patients may have had >1 liver disease etiology and may have been treated with >1 pulmonary arterial hypertension targeted therapy.
LT, liver transplantation; MELD-Na, model for end-stage liver disease sodium; PAH, pulmonary arterial hypertension.
TABLE 2.
Individual hemodynamic data and outcomes
| Initial portopulmonary hypertension diagnosis | Preliver transplantation | Posttransplant outcomes | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mPAP,mm Hg | PAWP,mm Hg | CO,L/min | PVR, dynes-s-cm−5 | RV function | PAH therapy | mPAP,mm Hg | PAWP,mm Hg | CO,L/min | PVR,dynes-s-cm−5 | RV function | PAH therapy at 1 y | 30-d survival | 1-y survival | |
| 1 | 36 | 6 | 7.4 | 326 | Mild dysfunction | Sildenafil | 37 | 14 | 10.2 | 181 | Borderlinedysfunction | N/A | No | No |
| 2 | 38 | 7 | 5.4 | 462 | Mild dysfunction | Ambrisentan and Tadalafil | 35 | 17 | 6.7 | 216 | Normal | N/A | No | No |
| 3 | 47 | 15 | 7.4 | 344 | Normal | Sildenafil | 43 | 22 | 8.0 | 210 | Normal | N/A | Yes | No |
| 4 | 48 | 8 | 5.4 | 592 | Moderate dysfunction | Epoprostenol | 37 | 14 | 8.2 | 226 | Normal | Tadalafil and ambrisentan | Yes | Yes |
| 5 | 45 | 16 | 5.3 | 438 | Normal | Inhaled nitric oxide | 35 | 5.6 | Mild dysfunction | None | Yes | Yes | ||
| 6 | 60 | Not assessed | Epoprostenol | 43 | 16 | 11.0 | 196 | Borderlinedysfunction | None | Yes | Yes | |||
| 7 | 60 | 23 | 6.4 | 464 | Normal | Remodulin | 35 | 15 | 6.5 | 248 | Normal | Tadalafil | Yes | Yes |
| 8 | 69 | 18 | 5.9 | 691 | Mild dysfunction | Epoprostenol and Ambrisentan | 41 | 21 | 16.7 | 96 | Normal | None | Yes | Yes |
| 9 | 48 | 10 | 8.5 | 360 | Mild dysfunction | Epoprostenol and sildenafil | 41 | 20 | 9.3 | 180 | Milddysfunction | Epoprostenol, sildenafil and ambrisentan | Yes | Yes |
| 10 | 48 | 7 | 4.8 | 683 | Normal | Remodulin and sildenafil | 46 | 28 | 7.2 | 201 | Milddysfunction | Remodulin and sildenafil | Yes | Yes |
| 11 | 42 | 12 | 8.8 | 272 | Mild dysfunction | Sildenafil | 35 | 14 | 8.8 | 191 | Normal | None | Yes | Yes |
| 12 | 51 | 15 | 3.1 | 929 | Markeddysfunction | Epoprostenol and sildenafil | 35 | 23 | 7.4 | 130 | Milddysfunction | N/A | No | No |
| 13 | 45 | 12 | 5.8 | 455 | Mild dysfunction | Sildenafil | 39 | 12 | 9.5 | 227 | Normal | None | Yes | Yes |
| 14 | 56 | 17 | 8.7 | 360 | Normal | Ambrisentan | 43 | 23 | 9.0 | 177 | Normal | None | Yes | Yes |
| 15 | 40 | 17 | 5.8 | 317 | Normal | Sildenafil | 36 | 20 | 7.4 | 172 | Normal | None | Yes | Yes |
| 16 | 50 | 12 | 5.5 | 553 | Mild dysfunction | Epoprostenol and sildenafil | 40 | 20 | 10.0 | 161 | Normal | N/A | Yes | No |
CO measurements rounded to the nearest tenth.
CO, cardiac output; mPAP, mean pulmonary arterial pressure; PAH, pulmonary arterial hypertension; PAWP, pulmonary artery wedge pressure; PVR, pulmonary vascular resistance; RV, right ventricular.
FIGURE 1.
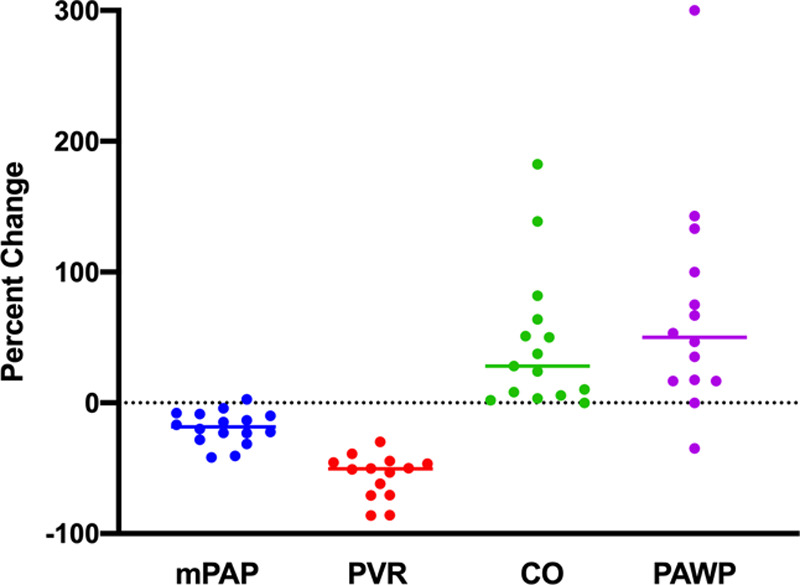
Percent change in pulmonary hemodynamics after treatment with pulmonary arterial hypertension targeted therapy. Percent changes in mPAP, PVR, CO, and PAWP from baseline after treatment with pulmonary arterial hypertension targeted therapy are shown. PAH therapy resulted in a mild reduction in mPAP with a moderate reduction in PVR and an increase in both CO and PAWP. Lines represent the median percent change. CO, cardiac output; mPAP, mean pulmonary arterial pressure; PAH, pulmonary arterial hypertension; PAWP, pulmonary artery wedge pressure; PVR, pulmonary vascular resistance.
One-year posttransplant survival was 69% (11/16). Of the 5 patients that died within the first year following LT, only 2 patients died related to cardiopulmonary causes. In the first case, which occurred 15 years ago before the availability of many current PAH therapies, the patient died 2 months following LT. This patient was treated with high-dose sildenafil monotherapy, and the death was attributed to POPH and right heart failure. In the second case, which occurred over 10 years ago, the patient had a cardiopulmonary arrest at the time of LT. The cause of death was unknown but was not felt to be directly related to POPH, given improved hemodynamics and preserved right ventricular function on echocardiogram before LT. There were no deaths attributed to cardiopulmonary causes within the last decade in the more modern PAH treatment era. Other patients died from hemorrhage (n = 1) and infection (n = 2). Although POPH may have contributed to impaired cardiopulmonary reserve in these patients, POPH was not considered to be the primary cause of death. At 1-year post-LT, 7/11 (63.6%) patients were weaned off all PAH therapy with clinical and echocardiographic resolution of POPH. Among patients who discontinued PAH therapy, no patients required restarting of PAH therapy, and there was no clinical or echocardiographic evidence of POPH recurrence over time.
Three-year and 5-year survival rates were both 53.8% (7/13). Beyond 1-year posttransplant, 1 patient died 1.8 years posttransplant of pneumonia and sepsis. Three patients were transplanted within the last 3 years, so they did not have follow-up data at 3 and 5 years.
DISCUSSION
In summary, in this case series of patients with treated POPH, patients with an elevated mPAP ≥ 35 mm Hg and PVR < 250 dynes-s-cm−5 at or just before LT had a 1-year posttransplant survival of 69%, and more than half were able to discontinue PAH therapy at 1-year post-LT. Prior studies have described 1-year posttransplant survival rates of 63%–92% in POPH.8-14 The 1-year posttransplant survival of 69% in patients with POPH and an elevated mPAP in our cohort is roughly similar to these prior estimates and higher than the 50% survival reported in older studies for patients with predominantly untreated POPH and an elevated mPAP ≥ 35 mm Hg.2 Long-term posttransplant survival rates are also overall similar to our recently reported survival rates for POPH patients at Mayo Clinic (n = 50) (1-y survival 72%, 3-y survival 63%, 5-y survival 60%).15 Additionally, in contrast to older studies, acute perioperative cardiopulmonary mortality was low, and no patients died of POPH or right heart failure in the last decade. Our previously reported experience in patients without known POPH and an mPAP ≥ 35 mm Hg at the time of transplant has also been favorable with 96% (24/25) survival at 1-year posttransplant.5 There was notably no overlap in patients between these 2 studies. Our findings suggest that survival for patients with POPH and an mPAP ≥ 35 mm Hg has improved in the modern PAH treatment era and that other factors, such as PVR and right ventricular function, are important in assessing perioperative risk and LT eligibility. Importantly, we also describe the hemodynamic effect of PAH therapy in POPH patients. PAH therapy was associated with a decrease in mPAP and PVR as expected but also was associated with an increase in both CO and PAWP. The resultant increase in CO and PAWP following initiation of PAH therapy tends to offset the degree of improvement in mPAP and can lead to persistent elevations in mPAP despite normalization of PVR. The majority of patients were treated with loop diuretics and potassium-sparing diuretics to manage volume overload, and several required dialysis or ultrafiltration perioperatively for management of volume overload. These findings demonstrate that PAH therapy has complex hemodynamic effects in patients with POPH and that persistent mPAP elevation related to an elevated CO or PAWP should not necessarily preclude LT.
At our institutions, our practice is to use a combination of factors to assess perioperative risk rather than reliance on the mPAP threshold in isolation. This includes factors such as PVR and right ventricular function. If patients with treated POPH have an elevated mPAP ≥ 35 mm Hg with a PVR < 240–250 dynes-s-cm−5 (varies by institution) and preserved right ventricular function without other contraindications to LT, then we typically proceed with LT if feasible. Although not included in the current MELD exception criteria, right ventricular function is likely an important factor in predicting prognosis and posttransplant outcomes but has been difficult to study due to varied methods of assessment/measurement over time.
According to current MELD exception criteria, however, patients in this cohort would not have been eligible for a standardized POPH MELD exception due to the mPAP elevation ≥35 mm Hg. Additionally, these patients may have been denied LT at some centers that consider an mPAP ≥35 mm Hg to be an absolute contraindication to LT. Without LT, POPH is a lifelong and progressive disease that leads to right heart failure and death.16 Thus, the observed improvement of POPH with discontinuation of PAH therapy in over half of the patients would not have been possible without LT.
We recognize several limitations to our study. These include the retrospective nature of the study, the small sample size, missing data, and the heterogeneous treatment regimens. There was also some heterogeneity in the timing of when hemodynamic data was obtained. In the majority of patients, right heart catheterization was performed in the outpatient setting and reflected the closest data measured before transplant surgery. In 1 patient who was diagnosed at the time of liver transplantation, however, hemodynamic data were obtained at the time of liver transplantation. This small case series, however, does represent the largest description of posttransplant outcomes of patients with treated POPH and an elevated mPAP ≥ 35 mm Hg in the more modern PAH treatment era.
In this study, we illustrate the effect of PAH therapy on mPAP, PVR, CO, and PAWP and describe outcomes of LT in patients with treated POPH and an elevated mPAP ≥ 35 mm Hg at the time of LT. Overall, posttransplant survival in patients with an elevated mPAP and normal PVR and preserved right ventricular function was similar to posttransplant survival estimates in other studies of patients with POPH. Acute perioperative cardiopulmonary mortality was low, and most patients were able to discontinue PAH therapy. Our findings suggest that patients with treated POPH with an elevated mPAP ≥ 35 mm Hg and a PVR < 250 dynes-s-cm−5 with preserved right ventricular function can survive LT and potentially be cured of their POPH. Our findings also suggest that current MELD exception guidelines that rely only on an absolute mPAP threshold of 35 mm Hg without consideration of PVR or right ventricular function could be modified.
Footnotes
Published online 10 November, 2020.
This work was supported by the Mayo Clinic Department of Medicine Catalyst Award for Advancing in Academics.
The authors declare no conflicts of interest.
H.M.D., J.R.R., C.J.S., and M.J.K. participated in research design, performance of the research, data analysis, and writing of the article. C.D.B., R.C.-C., C.B.R., T.T., S.L.N., J.K.H., and J.Y.F. participated in critical review of the article.
REFERENCES
- 1.Krowka MJ, Fallon MB, Kawut SM, et al. International liver transplant society practice guidelines: diagnosis and management of hepatopulmonary syndrome and portopulmonary hypertension. Transplantation. 2016; 100:1440–1452 [DOI] [PubMed] [Google Scholar]
- 2.Krowka MJ, Plevak DJ, Findlay JY, et al. Pulmonary hemodynamics and perioperative cardiopulmonary-related mortality in patients with portopulmonary hypertension undergoing liver transplantation. Liver Transpl. 2000; 6:443–450 [DOI] [PubMed] [Google Scholar]
- 3.Krowka MJ, Mandell MS, Ramsay MA, et al. Hepatopulmonary syndrome and portopulmonary hypertension: a report of the multicenter liver transplant database. Liver Transpl. 2004; 10:174–182 [DOI] [PubMed] [Google Scholar]
- 4.Krowka MJ, Fallon MB, Mulligan DC, et al. Model for end-stage liver disease (MELD) exception for portopulmonary hypertension. Liver Transpl. 2006; 12(12 Suppl 3):S114–S116 [DOI] [PubMed] [Google Scholar]
- 5.DeMartino ES, Cartin-Ceba R, Findlay JY, et al. Frequency and outcomes of patients with increased mean pulmonary artery pressure at the time of liver transplantation. Transplantation. 2017; 101:101–106 [DOI] [PubMed] [Google Scholar]
- 6.DuBrock HM, Goldberg DS, Sussman NL, et al. Predictors of waitlist mortality in portopulmonary hypertension. Transplantation. 2017; 101:1609–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krowka MJ, Miller DP, Barst RJ, et al. Portopulmonary hypertension: a report from the US-based REVEAL Registry. Chest. 2012; 141:906–915 [DOI] [PubMed] [Google Scholar]
- 8.Savale L, Guimas M, Ebstein N, et al. Portopulmonary hypertension in the current era of pulmonary hypertension management. J Hepatol. 2020; 73:130–139 [DOI] [PubMed] [Google Scholar]
- 9.Deroo R, Trépo E, Holvoet T, et al. Vasomodulators and liver transplantation for portopulmonary hypertension: evidence from a systematic review and meta-analysis. Hepatology. 2020. doi: 10.1002/hep.31164 [DOI] [PubMed] [Google Scholar]
- 10.Sithamparanathan S, Nair A, Thirugnanasothy L, et al. ; National Pulmonary Hypertension Service Research Collaboration of the United Kingdom and Ireland. Survival in portopulmonary hypertension: outcomes of the United Kingdom National Pulmonary Arterial Hypertension Registry. J Heart Lung Transplant. 2017; 36:770–779 [DOI] [PubMed] [Google Scholar]
- 11.Savale L, Sattler C, Coilly A, et al. Long-term outcome in liver transplantation candidates with portopulmonary hypertension. Hepatology. 2017; 65:1683–1692 [DOI] [PubMed] [Google Scholar]
- 12.Verma S, Hand F, Armstrong MJ, et al. Portopulmonary hypertension: still an appropriate consideration for liver transplantation?. Liver Transpl. 2016; 22:1637–1642 [DOI] [PubMed] [Google Scholar]
- 13.Sadd CJ, Osman F, Li Z, et al. Long-term outcomes and survival in moderate-severe portopulmonary hypertension after liver transplant. Transplantation. 2020. doi: 10.1097/TP.0000000000003248 [DOI] [PubMed] [Google Scholar]
- 14.DuBrock HM, Krowka MJ. The myths and realities of portopulmonary hypertension. Hepatology. 2020; 72:1455–1460 [DOI] [PubMed] [Google Scholar]
- 15.Cartin-Ceba R, Swanson K, Vargas H, et al. Clinical outcomes after liver transplantation in patients with portopulmonary hypertension. Transplantation. 2020. doi: 10.1097/TP.0000000000003490 [DOI] [PubMed] [Google Scholar]
- 16.Swanson KL, Wiesner RH, Nyberg SL, et al. Survival in portopulmonary hypertension: Mayo Clinic experience categorized by treatment subgroups. Am J Transplant. 2008; 8:2445–2453 [DOI] [PubMed] [Google Scholar]