

Abstract
The spike (S) glycoprotein of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is responsible for the binding to the permissive cells. The receptor-binding domain (RBD) of SARS-CoV-2 S protein directly interacts with the human angiotensin-converting enzyme 2 (ACE2) on the host cell membrane. In this study, we used computational saturation mutagenesis approaches, including structure-based energy calculations and sequence-based pathogenicity predictions, to quantify the systemic effects of missense mutations on SARS-CoV-2 S protein structure and function. A total of 18 354 mutations in S protein were analyzed, and we discovered that most of these mutations could destabilize the entire S protein and its RBD. Specifically, residues G431 and S514 in SARS-CoV-2 RBD are important for S protein stability. We analyzed 384 experimentally verified S missense variations and revealed that the dominant pandemic form, D614G, can stabilize the entire S protein. Moreover, many mutations in N-linked glycosylation sites can increase the stability of the S protein. In addition, we investigated 3705 mutations in SARS-CoV-2 RBD and 11 324 mutations in human ACE2 and found that SARS-CoV-2 neighbor residues G496 and F497 and ACE2 residues D355 and Y41 are critical for the RBD–ACE2 interaction. The findings comprehensively provide potential target sites in the development of drugs and vaccines against COVID-19.
Keywords: missense mutation, computational saturation mutagenesis, SARS-CoV-2 S stability, RBD–ACE2 interaction, COVID-19
Introduction
Coronaviruses are a large family of enveloped RNA viruses [1]. The paradigm shift in the mode of transmission of three pathogenic coronaviruses, including severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV), has resulted in recent outbreaks [2, 3]. The most recent outbreak of Coronavirus Disease 2019 (COVID-19) is caused by a novel coronavirus, SARS-CoV-2. The virus is closely related to the SARS-like CoV, RaTG13 virus, which is found in bats [4].
The spike (S) glycoprotein is critical in terms of the virulence of pathogenic coronaviruses. The homotrimeric protein is responsible for mediating virus entry of SARS-CoV-2 via the angiotensin-converting enzyme 2 (ACE2) receptor on the host cell membrane [5, 6]. Thus, the analysis of SARS-CoV-2 S protein is a high research priority for vaccine design against COVID-19. The S protein consists of an S1 and an S2 subunit [5]. The receptor-binding domain (RBD) of S1 includes a core and a receptor-binding motif (RBM) that specifically recognizes ACE2. RBD is the key determinant of cross-species and human-to-human transmissibility [7]. The interactions between RBD and ACE2 are critical for the host range and cross-species infections of SARS-CoV-2. Recent studies showed that SARS-Cov-2 S protein has a higher affinity to ACE2 compared with those of SARS-CoV [5] and bat coronavirus S [8, 9].
Structure analyses revealed the atomic details of SARS-Cov-2 S proteins and the binding interface between RBD and ACE2. A prefusion ectodomain trimer in the open and closed conformational states of the SARS-COV-2 S was determined using the cryogenic electron microscopy (cryo-EM) study [5]. SARS-CoV-2 S protein can recognize the ACE2 to initiate the viral entry in the open state. Crystallographic study showed that SARS-CoV-2 S RBD can bind hACE2 via its RBM [10]. The RBD residues of SARS-CoV-2 S1 could establish more atomic bonds with the hACE2 when compared with the RBD–ACE2 complex of SARS-CoV. These interface variations are thought to alter the affinity of the RBD for the ACE2 receptor.
The genome of RNA virus can easily generate mutations as virus spreads. Coronaviruses have high mutation rates compared with other RNA viruses [11]. The constant emergence of new mutations in SARS-CoV-2 is the major challenge for the ongoing development of broad neutralizing antibodies. The bioinformatics methods can readily quantify the effects of deleterious mutations on protein function and structure [12–14], and they have been applied to SARS-CoV-2 research [8]. However, a comprehensive effect map of SARS-CoV-2 S mutations still lacks for identifying the target sites for vaccine design. Computational saturation mutagenesis provides a fast methodology to investigate all possible mutations and identify the potential functional sites [15]. The infection of SARS-CoV-2 in permissive cells is largely depending on the interaction of the viral S protein and ACE2. The RBD–ACE2 binding affinity and the S stability are hence important to be investigated. In this study, we applied computational saturation mutagenesis to investigate 18 354 SARS-CoV-2 S missense mutations, 3705 RBD mutations and 11 324 hACE2 mutations in experimentally determined protein structures. We identified the potential target sites critical for the designs of antiviral drugs and vaccines against COVID-19.
Methods
Structure preparation
All structures are collected from Protein Data Bank (PDB) [16]. We collected the structure of the SARS-CoV-2 S-2P (P986 and P987 substitutions) trimer in the open state (PDB ID: 6VYB). The P986K and P987V mutations were introduced to S-2P trimer to generate wide type S trimer using FoldX [17]. We calculated the effects of mutations on protein stabilities of wide type S trimer and monomer (A chain). The RBD–ACE2 complex (6LZG) was used to investigate the binding affinity and protein stability of SARS-CoV-2 RBD and hACE2. The SARS-CoV-2 S structure in closed state (6VXX), SARS-CoV S (6ACG) and RBD–ACE2 complex (2AJF) and MERS-COV S protein (5W9J) were collected for the comparison studies. PyMOL (http://www.pymol.org/) was used to generate protein structural images and perform structure alignments.
Mutation collection
To determine the effects of all mutations on protein structures, we applied saturation mutagenesis to mutate all residues in the corresponding structures to all other 19 amino acid types. Viral variations were collected from 2019nCoVR [18], which integrates the variation information based on the SARS-CoV-2 strains worldwide. As of 30 April 2020, we collected 385 viral missense variations in 325 residue positions of SARS-CoV-2 S protein.
Energy calculation
The effects of mutations on protein stability of S and binding affinity of RBD with hACE2 were estimated by the folding energy change (∆∆G) and the binding energy change (∆∆∆G) between the mutant structure (MUT) and wild-type (WT) structure, respectively. FoldX [17] was used for energy calculations. The performance of the FoldX compares favorably with other random-based approaches for protein engineering research including therapeutic antibody design [19]. Particularly, FoldX is widely used for computational saturation mutagenesis in biomedical studies [15, 20]. All protein structures were repaired using the ‘RepairPdb’ command. The ‘BuildModel’ command was used for stability analysis. The folding energy change was calculated using:
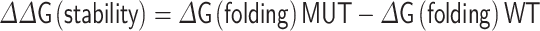 |
A negative ∆∆G value suggests that the mutation can stabilize the protein and a positive value indicates that it makes the protein unstable. The structure-based tools DUET [21] and CUPSAT [22] were applied to check the reliability of FoldX for protein stability predictions. The ‘AnalyseComplex’ command was carried out for interaction analysis. The binding energy change was computed by:
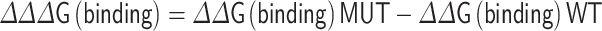 |
A negative ∆∆∆G value suggests that the mutation strengthens the binding affinity, whereas a positive value indicates that the mutation weakens the RBD–ACE2 interaction.
Sequence analysis
The amino acid sequences of S protein of SARS-CoV-2 (Entry: P0DTC2), SARS-CoV (P59594) and MERS-CoV (K9N5Q8) and human ACE2 (Q9BYF1) were downloaded from UniProt [23]. The pairwise and multiple sequence alignments were carried out using EMBOSS Water and Clustal Omega [24]. The protein sequences were submitted to SNAP [25] to predict the effects of mutations on protein function for SARS-CoV-2 S and hACE2. The prediction scores range from −100 (neutral) to 100 (effect). SNAP outperform classical mutation pathogenicity tools and generate predictions for all possible mutations in a protein [26]. The predictions from SNAP and FoldX are corrected and can be used for mutation analysis on protein function and stability [27]. PolyPhen-2 [28] and SIFT [29] were used to compare with SNAP for predicting mutation pathogenicity effects.
R package (https://www.r-project.org/) was used to generate graphs and perform analysis of variance test and t-test in statistical comparisons of energy changes and SNAP scores.
Results
Effects of mutations on SARS-CoV-2 protein stability
Since S protein is critical for SARS-CoV-2 infection, we want to anatomize its amino acids that affect its stability. We used the cryo-EM structure of the SARS-CoV-2 S in the open state (PDB ID: 6VYB) as the full-length S structure. To determine the effects of the systematic mutations on SARS-CoV-2 S stability, we generated 18 354 mutations by mutating all 966 residues in this structure to all other 19 amino acid types and computed the folding energy changes (ΔΔG) introduced by these mutations in monomer (Supplementary Table 1) and trimer (Supplementary Table 2). Of 18 354 mutations, 31.3% mutations have strong effects (ΔΔG > 2.5 kcal/mol) and 30.4% mutations have moderate effects (0.5 < ΔΔG ≤ 2.5 kcal/mol) on monomer destabilization (Figure 1A). The heatmap in Figure 2A also suggests that the most mutations can decrease SARS-CoV-2 full-length S monomer stability. In contrast, only 0.2% mutations have strong effects (ΔΔG < −2.5 kcal/mol) and 10.4% mutations have moderate effects (−2.5 ≤ ΔΔG < −0.5 kcal/mol) on S monomer stabilizing. The stability effects of mutations on S trimer are highly correlated and share similar patterns and common top values with those on monomer (Supplementary Figure 1). We also calculated the ΔΔG based on the cryo-EM structure of the SARS-CoV-2 S in the closed state (6VXX) and found that the results are highly correlated with those in the open state (Supplementary Figure 2A). As the line chart shown in Figure 2A, the mean value of ΔΔG in monomer stability at each SARS-CoV-2 full-length S residue position range from 29.41 kcal/mol in G431 to −1.54 kcal/mol in S514. We listed the amino acid positions that strongly stabilize or destabilize S protein in the Table 1 according to the mean values of ΔΔG of mutations in each position. The mutations at glycine residues G431, G648 and G35 residues have maximum destabilizing effects on the full-length S protein. In contrast, the mutations at serine residues S514, S735 and S50 residues have induced highly stabilizing effects on the overall protein. Notably, both G431 with maximum ΔΔG and S514 with minimum ΔΔG are in the RBD region. Among all mutations destabilizing full-length S protein, G431W has introduced the largest positive ΔΔG for trimer (189.93 kcal/mol) and monomer (59.92 kcal/mol).
Figure 1.
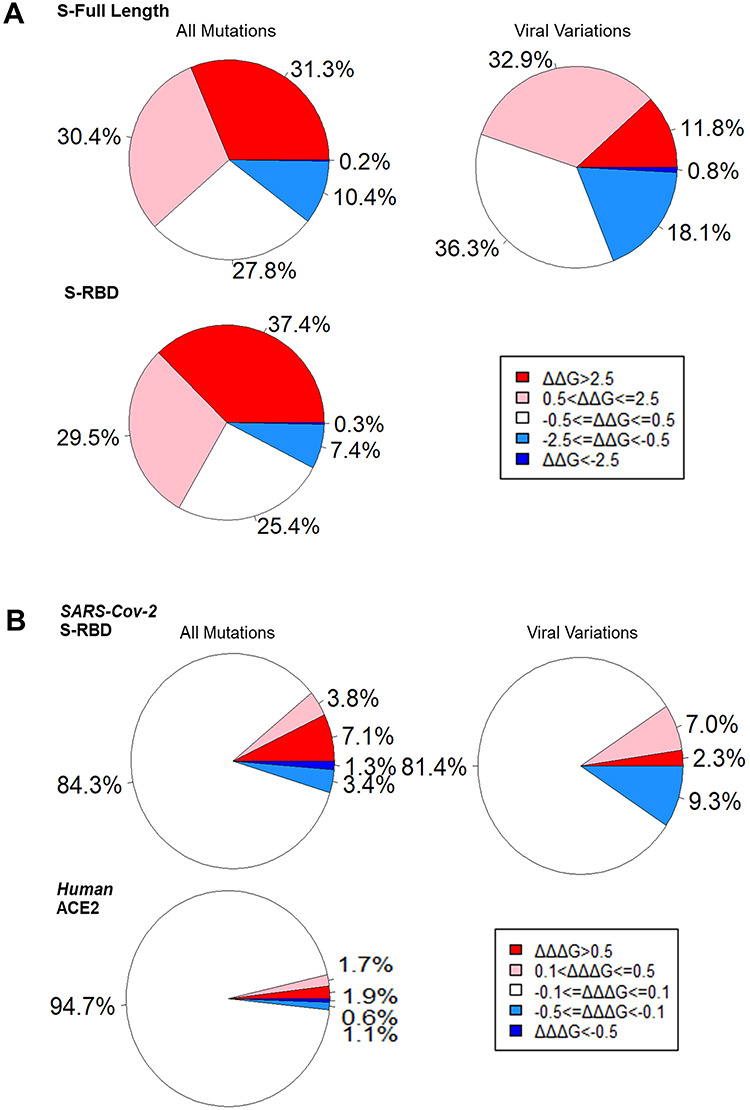
The contribution of mutation effects on protein stability and binding affinity. (A) The stability effects of all mutations and viral variations on SARS-Cov-2 full-length S monomer and all mutations on RBD. (B) The binding affinity effects of SARS-Cov-2 RBD all mutations and viral variations and all human ACE2 mutations.
Figure 2.

Effects of residues and mutations in SARS-Cov-2 (A) full-length S monomer and (B) RBD on protein stability. Line charts summarize the folding energy changes for ΔΔG mean of residues (bar) and ΔΔG of substitutions to alanine (circle). Heatmaps show the ΔΔG of all mutations and the mutations in key residues. Maximum (yellow) and minimum (magenta) ΔΔG values are labeled for each residue position. The ΔΔG values of viral variations are shown in green boxes. The key residues G431 and K514 are marked in the line charts and heatmaps.
Table 1.
Effects of key residues and mutations on SARS-Cov-2 S stability
| Residue | ΔΔG (Mean) | Mutation with maximum ΔΔG | Mutation with minimum ΔΔG | ||
|---|---|---|---|---|---|
| Full-length S monomer | |||||
| G431 | 29.41 | G431W | 59.923 | G431A | 8.588 |
| G648 | 24.798 | G648W | 43.326 | G648A | 6.700 |
| G35 | 21.392 | G35Y | 49.843 | G35A | 5.919 |
| G526 | 18.364 | G526Y | 35.971 | G526A | 4.396 |
| D614 | 0.386 | D614P | 4.419 | D614G | −0.784 |
| S514 | −1.54 | S514P | 2.649 | S514F | −3.304 |
| S735 | −1.267 | S735P | 3.710 | S735M | −2.929 |
| S50 | −1.123 | S50G | −0.073 | S50L | −2.614 |
| V976 | −0.562 | V976P | 2.523 | V976D | −4.014 |
| T385 | −0.431 | T385H | 0.192 | T385E | −1.125 |
| RBD | |||||
| G431 | 26.266 | G431W | 55.323 | G431A | 6.642 |
| G526 | 16.502 | G526H | 27.218 | G526C | 9.39 |
| P507 | 14.592 | P507W | 49.799 | P507A | 2.400 |
| S438 | 6.065 | S438H | 21.956 | S438C | −0.671 |
| V483 | 0.008 | V483P | 3.162 | V483R | −0.779 |
| S514 | −1.658 | S514P | 0.755 | S514F | −3.390 |
| T385 | −0.967 | T385W | −0.102 | T385P | −1.733 |
| N394 | −0.616 | N394P | 2.574 | N394W | −2.033 |
| A397 | 1.563 | A393W | 17.545 | A397L | −3.582 |
| V341 | 2.804 | V341F | 7.216 | V341I | −1.256 |
Note: The residues with the largest or smallest ΔΔG means and mutations with the largest or smallest ΔΔG values in all predictions are shown as bold fonts. The dominant pandemic form D614G is underlined.
Next, we wonder whether S1 RBD stability could be affected by amino acid substitutions. In doing so, we used the crystal structure of RBD–ACE2 complex (6LZG) and analyzed ΔΔG values of 3705 mutations generated from 195 residues of its RBD chain (Supplementary Table 3). Consistent to the analysis results of the full-length S protein, there are more mutations causing destabilization than stabilization of RBD (Figure 1A). We showed that the effects of mutations on RBD stability are highly correlated with those on full-length S protein (Supplementary Figure 2B). This finding suggests that the energy calculations based on the crystal structure of RBD–ACE2 complex are consistent with those based on the cryo-EM structure of full-length S structure. The ΔΔG mean at each SARS-CoV-2 S residue position range from 26.266 kcal/mol in G431 to −1.658 kcal/mol in S514 (Figure 2B). Notably, G431 and S514 are also the top residues altering the full-length S stability. Interestingly, G431A has the maximum ΔΔG value among all substitutions to alanine in both full-length S and RBD, and S514A has the minimum ΔΔG value among all substitutions to alanine in RBD. These results indicate that the side chains of G431 and S514 have important contributions to the RBD stability (Figure 2). In all RBD mutations, G431W introduced the highest positive ΔΔG at 55.323 kcal/mol, and all mutations in G431 could highly reduce the protein stabilities of RBD and full-length S (Figure 2).
Effects of mutations on RBD–ACE2 binding affinity
Missense mutations in RBD region could change the key interaction site and affect the binding affinity of RBD and ACE2. We calculated the binding energy changes (ΔΔΔG) of total RBD 3705 mutations in the RBD–ACE2 complex (Supplementary Table 4). Of 3705 mutations, 263 (7.1%) mutations can decrease (ΔΔΔG > 0.5 kcal/mol) and only 48 mutations (1.3%) can increase (ΔΔΔG < −0.5 kcal/mol) the binding affinity of RBD–ACE2 complex (Figure 1B). In total, 3.8% mutations have small effects on destabilizing (0.1 < ΔΔΔG ≤ 0.5 kcal/mol), whereas 3.4% mutations have small effects on stabilizing (−0.5 ≤ ΔΔΔG < −0.1 kcal/mol) RBD–ACE2 binding affinity. Most of the mutations (84.3%) have no effects (−0.1 ≤ ΔΔΔG < 0.1 kcal/mol) on RBD–ACE2 interaction. G496 has the maximum ΔΔΔG mean in RBD–ACE2 binding affinity at 4.694 kcal/mol (Figure 3). G496W and G496Y induced the largest binding energy change at 17.418 kcal/mol in all RBD mutations (Table 2). Interestingly, G496s neighbor residues F497 has minimum ΔΔΔG mean at −0.476 kcal/mol. These two residues are in a 10-residue sequence Y495GFQPTNGVG504. Many mutations and residues in this motif have significant effects on RBD–ACE2 binding affinity. For example, G504 and Y495 have the second and third smallest ΔΔΔG mean values at −0.245 kcal/mol and −0.209 kcal/mol, respectively. The largest negative binding free energy change is introduced by N501E at −2.490 kcal/mol. In contrast, G502 has the second largest positive ΔΔΔG mean at 2.922 kcal/mol and G502P (ΔΔΔG = 11.767 kcal/mol) has the highest destabilizing effect on RBD–ACE2 complex.
Figure 3.
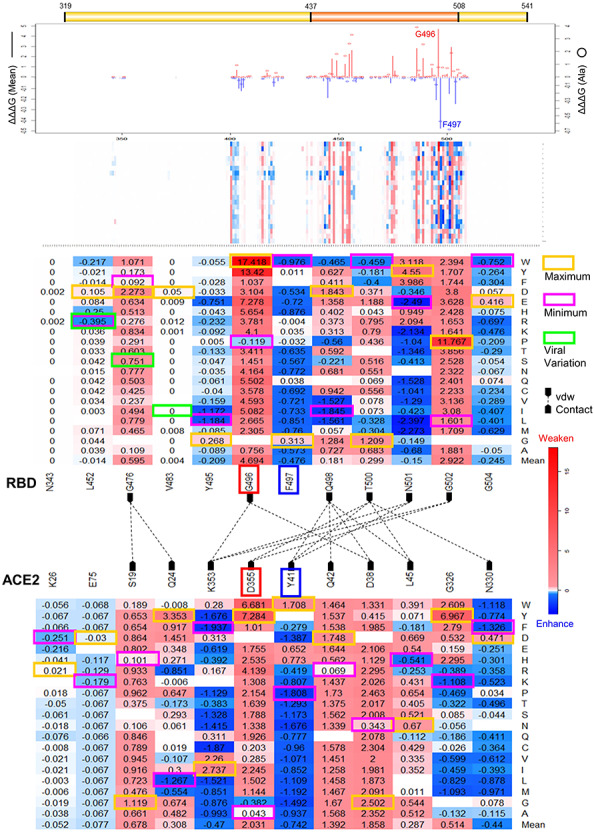
Effects of SARS-Cov-2 RBD residues and mutations on RBD–ACE2 interaction. Line chart summarizes the binding energy changes for ΔΔΔG mean of residues (bar) and ΔΔΔG of substitutions to alanine (circle) in SARS-Cov-2 RBD residues. Heatmaps show the ΔΔΔG of all RBD mutations and the mutations in key residues. The RBD and ACE2 that have vdw contacts are marked and connected with dashed lines. Maximum (yellow) and minimum (magenta) ΔΔΔG values are labeled for each residue position. The ΔΔΔG values of viral variations are shown in green boxes. The key residues G496 and F497 are marked in the line chart and heatmaps.
Table 2.
Effects of key residues and mutations on RBD–ACE2 interaction
| Residue | ΔΔΔG (mean) | Mutation with maximum ΔΔΔG | Mutation with minimum ΔΔΔG | ||
|---|---|---|---|---|---|
| SARS-Cov-2 S RBD | |||||
| G496 | 4.694 | G496W | 17.418 | G496P | −0.119 |
| G502 | 2.922 | G502P | 11.767 | G502L | 1.601 |
| G476 | 0.595 | G476D | 2.273 | G476F | 0.092 |
| T500 | 0.299 | T500G | 1.209 | T500W | −0.459 |
| Q498 | 0.181 | Q498D | 1.843 | Q498I | −1.845 |
| V483 | 0.004 | V483D | 0.050 | 0.000 | |
| F497 | −0.476 | F497G | 0.313 | F497W | −0.976 |
| G504 | −0.245 | G504E | 0.416 | G504W | −0.752 |
| Y495 | −0.209 | Y495G | 0.268 | Y495L | −1.184 |
| N501 | −0.150 | N501Y | 4.550 | N501E | −2.490 |
| L452 | −0.014 | L452D | 0.667 | L452R | −0.395 |
| N343 | 0.000 | 0.002 | 0.000 | ||
| Human ACE2 | |||||
| D355 | 2.031 | D355Y | 7.284 | D355G | −0.382 |
| D38 | 1.858 | D38G | 2.502 | D38N | 0.343 |
| Q42 | 1.392 | Q42D | 1.748 | Q42R | 0.069 |
| S19 | 0.678 | S19G | 1.119 | S19H | 0.101 |
| G326 | 0.514 | G326Y | 6.967 | G326K | −1.108 |
| Q24 | 0.308 | Q24Y | 3.353 | Q24L | −1.267 |
| L45 | 0.287 | L45N | 0.670 | L45H | −0.541 |
| Y41 | −0.742 | Y41W | 1.708 | Y41P | −1.808 |
| K353 | −0.47 | K353I | 2.737 | K353F | −1.937 |
| N330 | −0.44 | N330D | 0.471 | N330F | −1.326 |
| E75 | −0.077 | E75D | −0.03 | E75K | −0.179 |
| K26 | −0.052 | K26R | 0.021 | K26D | −0.251 |
Note: The residues with the largest or smallest ΔΔΔG means and mutations with the largest or smallest ΔΔΔG values in all predictions are shown as bold fonts.
The human ACE2 mutations located in RBD–ACE2 interface could affect ACE2 interaction with SARS-Cov-2 RBD. We then scrutinized the effects of total 11 324 mutations in ACE2 chain of crystal structure of RBD–ACE2 complex (Supplementary Table 5). Of 11 324 mutations, 210 (1.9%) mutations can decrease the binding affinity and 192 (1.7%) mutations have small effects on destabilizing RBD–ACE2 complex (Figure 1B). In contrast, only small portions, 63 mutations (0.6%) with ΔΔΔG < −0.5 kcal/mol and 130 mutations (1.1%) with −0.5 ≤ ΔΔΔG < −0.1 kcal/mol, can increase the binding affinity. As shown in Table 2, the mutations at residues D355, D38 and Q42 induce strong destabilizing effects on RBD–ACE2 binding affinity. Residue D355 (ΔΔΔG mean = 2.031 kcal/mol) and mutation D355Y (ΔΔΔG = 7.284 kcal/mol) in this position have maximum effects on destabilizing the RBD–ACE2 complex. However, the mutations at residues Y41, K353 and N330 can increase the RBD–ACE2 binding affinity. Residue Y41 (ΔΔΔG mean = −0.742 kcal/mol) and mutation D355Y (ΔΔΔG = −1.808 kcal/mol) can increase the RBD–ACE2 interaction. K353F has the smallest ΔΔΔG at −1.937 kcal/mol.
Effects of viral variations on S stability and RBD–ACE2 interaction
We also searched and listed the viral mutations that occurred naturally in the strains of SRS-CoV-2. As of 30 April 2020, 384 experimentally identified viral missense variants have occurred in 325 positions (25.5%) of total 1273 amino acids of SARS-Cov-2 full-length S protein. We examined stability effects of 237 viral missense variations that can be mapped to the SARS-Cov-2 full-length S. As shown in the Figure 1A, 32.6% viral mutations have strong effects and 11.8% mutations have moderate destabilizing effects on S protein. Remarkably, there are more viral mutations (18.1%) that have moderate stabilizing effects compared with those of all computationally predicted mutations (10.4%) in the full-length S protein. In addition, more mutations with no effects were observed in viral variations (36.3%) compared with those of all mutations (27.8%). Of 384 viral variations, 47 mutations occur in 40 positions of RBD region and 40 mutations can be mapped to the crystal structure of RBD. As shown in Figure 1B, we observed less viral mutations (2.3%) can reduce the RBD–ACE2 binding affinity on RBD, compared with those of all mutations (7.1%). We did not find viral mutations with ΔΔΔG < −0.5 kcal/mol. However, there are more viral mutations (9.3%) that have small effects (−0.1 < ΔΔG ≤ −0.5 kcal/mol) on stabilizing RBD–ACE2 complex, compared with those of all mutations (3.4%).
We investigated the effects of common viral variations and the mutations with significant effects (Table 3). The most common variation, D614G in 5703 virus strains, has stabilizing effects on the S protein (ΔΔG = −0.784 kcal/mol). Similarly, D936Y (ΔΔG = −0.304 kcal/mol) in 37 strains and H49Y (ΔΔG = −1.902 kcal/mol) in 25 strains can make S protein more stable. However, G1124V (ΔΔG = 3.595 kcal/mol) in 24 strains and S943I (ΔΔG = 0.299 kcal/mol) in 26 strains reduce the stability of S protein. Mutations of S50L (ΔΔG = −2.614 kcal/mol), T724I (ΔΔG = −2.590 kcal/mol) and T240I (ΔΔG = −2.476 kcal mol) have strong stabilizing effects on SARS-Cov-2 full-length S protein. In RBD region, the largest folding energy change takes place in S438F (ΔΔG = 18.399 kcal/mol) for S stability. Consistently, S438F has the maximum ΔΔG at 10.980 kcal/mol for RBD stability. In contrast, V341I has the minimum folding free energy change at −1.256 kcal/mol and can increase RBD stability. V483A in 24 strains have small effects (ΔΔG = −0.196 kcal/mol) and V367F in 13 strains has moderate stabilizing effects (ΔΔG = −0.597 kcal/mol) on RBD. However, G476S in eight strains, A520S in seven strains and Q414E in six strains can destabilize the RBD region. We further investigated the effects of viral mutations on RBD–ACE2 binding affinity. The ΔΔΔG of common variants V483A and V367F are close to 0, meaning that these mutations have no effects on binding affinity. We observed that G476S can decrease RBD–ACE2 binding affinity (ΔΔΔG = 0.751 kcal/mol), but L452R has small stabilizing effect (ΔΔΔG = −0.395 kcal/mol) on RBD–ACE2 complex.
Table 3.
Effects of viral variations on SARS-Cov-2 S stability and RBD–ACE2 interaction
| Virus number with variation | Viral variation | ΔΔG (mean) | SNAP score | Mutation with maximum ΔΔG | Mutation with minimum ΔΔG | ||
|---|---|---|---|---|---|---|---|
| Full-length S monomer stability | |||||||
| 5703 | D614G | −0.784 | −52 | D614P | 4.419 | D614G* | −0.784 |
| 37 | D936Y | −0.304 | 62 | D936P | 2.127 | D936F | −0.393 |
| 26 | S943I | 0.299 | −55 | S943V | 0.582 | S943K | −0.435 |
| 25 | H49Y | −1.902 | −40 | H49P | 3.498 | H49W | −3.068 |
| 24 | G1124V | 3.595 | 0 | G1124P | 4.969 | G1124M | 1.507 |
| 1 | V341I | −1.005 | −82 | V341P | 3.573 | V341I | 1.005 |
| 7 | S50L | −2.614 | −69 | S50G | −0.073 | S50L | −2.614 |
| 1 | T724I | −2.590 | −68 | T724G | 1.797 | T724M | −3.422 |
| 5 | N439K | −0.197 | −61 | N439F | 1.919 | N439I | −1.628 |
| 2 | S438F | 18.399 | −9 | S438Y | 22.163 | S438V | −2.182 |
| RBD stability | |||||||
| 24 | V483A | −0.196 | −69 | V483P | 3.162 | V483K | −0.851 |
| 13 | V367F | −0.597 | −47 | V367P | 0.937 | V367Q | −0.829 |
| 8 | G476S | 2.122 | −20 | G376P | 6.207 | G476S | 2.122 |
| 7 | A520S | 0.904 | −90 | A520P | 6.477 | A520G | −0.882 |
| 6 | Q414E | 0.914 | −90 | Q414P | 3.914 | Q414K | −0.197 |
| 1 | V341I | −1.256 | −82 | V341F | 7.216 | V341I | −1.256 |
| 1 | L452R | 0.021 | −53 | L452P | 4.085 | L452R | 0.021 |
| 1 | V503F | −0.251 | −34 | V503H | 0.843 | V503P | −1.241 |
| 5 | N439K | −0.910 | −61 | N439P | 1.566 | N439L | −1.081 |
| 2 | S438F | 10.980 | −9 | S438H | 21.958 | S438C | −0.671 |
| RBD–ACE2 binding affinity | ΔΔΔG (Mean) | Maximum ΔΔΔG | Minimum ΔΔΔG | ||||
| 24 | V483A | 0.000 | −69 | V483D | 0.05 | 0 | |
| 13 | V367F | 0.000 | −47 | 0 | 0 | ||
| 8 | G476S | 0.751 | −20 | G476D | 2.273 | G476F | 0.092 |
| 6 | Q414E | 0.000 | −90 | Q414W | 0.002 | 0 | |
| 7 | A520S | 0.000 | −90 | 0 | 0 | ||
| 1 | V341I | 0.000 | −82 | V341R | 0.002 | 0 | |
| 1 | L452R | −0.395 | −53 | L452D | 0.105 | L452R | −0.395 |
| 1 | V503F | −0.264 | −34 | V503D | 0.705 | V503W | −0.487 |
| 5 | N439K | 0.178 | −61 | N439E | 0.363 | N439R | −0.183 |
| 2 | S438F | 0.002 | −9 | S438K | 0.003 | 0 | |
Note: The mutations with maximum or minimum ΔΔG/ΔΔΔG values in the same positions of viral variations are shown. The most common viral variations and variations with the largest or smallest ΔΔG/ΔΔΔG are bold. The viral variations that have minimum ΔΔG/ΔΔΔG values in their positions are underlined.
Mutation pathogenicity of SARS-Cov-2 S and ACE2
We analyzed the mutation pathogenicity of all mutations of S and ACE2 proteins based their protein sequences. We generated SNAP [25] predictions for 15 295 mutations in human ACE2 and 24 187 mutations in SARS-Cov-2 full-length S including 4237 RBD mutations. Of 24 187 SARS-Cov-2 S mutations, 12, 678 (52.42%) mutations have damaging effects on S function and 11 509 (47.58%) mutations are predicted as neutral variations (Figure 4A). In RBD region, the distribution of damaging mutations is decreased to 47.01%. Interestingly, most of viral variations (75%) have neutral effects on S function and only 25% are predicted as deleterious mutations. In human ACE2 protein, 9628 (62.9%) mutations are predicted as deleterious variations and 5667 (37.05%) mutations have neutral effects on protein function. The heatmap of hACE2 also suggests that most of the mutations have damaging effects on ACE2 function (Figure 4B). Protein destabilization is a common mechanism by which mutations cause human diseases [30]. As shown in Figure 4C, we observed the SNAP scores of mutations with strong effects (ΔΔG > 2.5 or ΔΔG < −2.5 kcal/mol) on the stabilities of full-length S and RBD are higher than the mutations with moderate effects (0.5 < ΔΔG ≤ 2.5 or −2.5 ≤ ΔΔG < −0.5 kcal/mol). The mutations with no effects (−0.5 ≤ ΔΔG ≤ 0.5 kcal/mol) have the lowest average SNAP scores. The differences are statistically distinguishable among these groups in SARS-Cov-2 S and RBD (Both P-value <2e−16). The correlations between mutation damaging effects and FoldX folding energy changes were found to be strong for the mutations with significant of ΔΔG values [27]. Consistent with this finding, our results suggested that the ΔΔG can be used to distinguish the damaging and neutral mutations in SARS-Cov-2 S and RBD. To avoid any biasness in the predictions, we selected 20 key mutations and compare their FoldX and SNAP results with those from other stability and pathogenicity tools. All these predictors return highly consensus effects for most of the key mutations (Supplementary Table 6). Lastly, we used combined analyses of protein stability, binding affinity and mutation pathogenicity to identify 113 functional important mutations including 21 beneficial mutations that can strength RBD–ACE2 interaction, stabilize RBD and have neutral effects on S function and 92 harmful mutations that weaken binding affinity, dramatically destabilize RBD and cause deleterious functional effects (Supplementary Table 7). These target mutations can be used for further experimental design of antiviral drugs and development of the neutralizing antibodies against COVID-19 [31].
Figure 4.
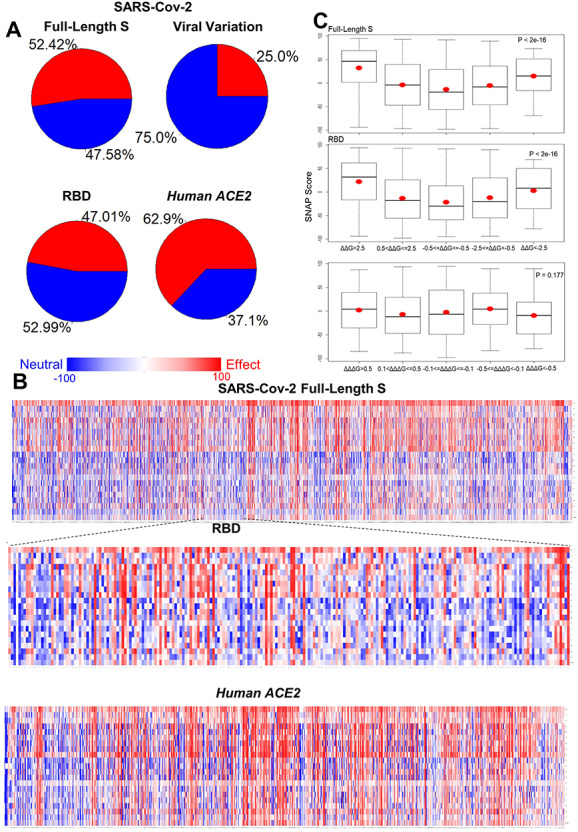
Mutation pathogenicity analysis. (A) Pie charts summarize the contribution of neutral (blue) and damaging (red) all mutations and viral variations in SARS-Cov-2 full-length S and those mutations in RBD and human ACE2. (B) Heatmaps of mutation pathogenicity for SARS-Cov-2 full-length S, its RBD region and human ACE2. (C) Boxplots for the SNAP scores of mutation groups with different folding (ΔΔG) and binding (ΔΔΔG) energy changes. The mean values are shown as red dots.
Discussion
S protein stability is critical in producing therapeutic antibodies against current pathogenic coronaviruses [32]. A prefusion-stabilized MERS-CoV S 2P protein was rationally designed by introducing two consecutive proline mutations V1060P and L1061P [33]. We calculated the folding energy changes and showed that V1060P and L1061P have the minimum ΔΔG values at −2.225 and −0.821 kcal/mol in their positions, respectively. These results are consistent with the findings in experimental structure analysis [33]. Even though the 2P positions are not conserved among MERS-Cov, SARS-Cov and SARS-Cov-2 S amino acid sequences, K986 and V987 of SARS-Cov-2 S and K968 and V969 of SARS-Cov S at equivalent positions are in the linker between two helices and share similar structures (Supplementary Figure 3). SARS-Cov-2 mutations K986P and V987P have the minimum ΔΔG values at −0.839 and −1.999 kcal/mol, respectively. Most mutations in V987 (ΔΔG mean = −0.334 kcal/mol) can stabilize the SARS-Cov-2 S protein. The experimental study showed that MERS-Cov 2P protein can stay in the prefusion conformation and retain high binding affinity of RBD to its receptor and various neutralizing antibodies [33]. Thus, we investigated the SARS-Cov-2 residues and mutations that have significant effects on protein stability and RBD–ACE2 binding affinity.
We wonder whether any other residues and mutations can induce higher stabilizing effects on SARS-Cov-2 S protein. Various mutations in S514, S735 and S50 can highly increase the stability of SARS-Cov-2 full-length S, and many mutations in S514, T385 and N394 have strong stabilizing effects on RBD (Table 1 and Figure 2). S514 has the minimum ΔΔG mean value on both full-length S and RBD and is in a β-strand of RBD (Figure 5A). S514F induce the strongest stabilizing effect on both full-length and RBD region. A serine to phenylalanine substitution can make this residue buried inside the protein core and increase the protein stability. Destabilizing mutations can be damaging for protein function (Figure 4C) and residues with high destabilizing effects could be important active sites of S proteins. We observed that all mutations at glycine residues G431, G648, G35 and G526 residues can destabilize SARS-Cov-2 full-length S and RBD region (Figure 2). Glycine is the smallest amino acid; mutations to any other large residues will result in unfavorable conformation changes and make protein unstable. Among all destabilizing mutations, G431W introduced the highest folding energy change on full-length S trimer, monomer and RBD, indicating this mutation can strongly reduce the protein stability. Interestingly, the G431 and S514 are very close structurally. The distance between α-carbons of G431 and S514 is only 4.4 Å (Figure 5A). This finding indicates the interaction between two key residues is critical for SARS-Cov-2 S and RBD stability. Notably, G431 is in the upstream region of RBM and S514 is closed to the end of RBM. These two residues may have an impact on RBM stability. G431 and S514 are conserved in SARS-Cov S sequence (Figure 6A). The folding energy calculations showed that all mutations in SARS-Cov residue G418, which at the equivalent position of G431 in SARS-Cov-2, can also reduce the S stability (Figure 6B). Similarly, most of the mutations in S500 of SARS-Cov have stabilizing effects like those in its conserved position S514 in SARS-Cov-2. The structure alignment of SARS-Cov-2 S and SARS-Cov S suggests similar structures between two S proteins. The distance from G418 to S500 in SARS-Cov is increased to 5.3 Å, which still makes interactions between two residues. These results indicate that the mechanisms of these spatial residues affecting the stabilities of S proteins are similar in SARS-Cov and SARS-Cov-2.
Figure 5.
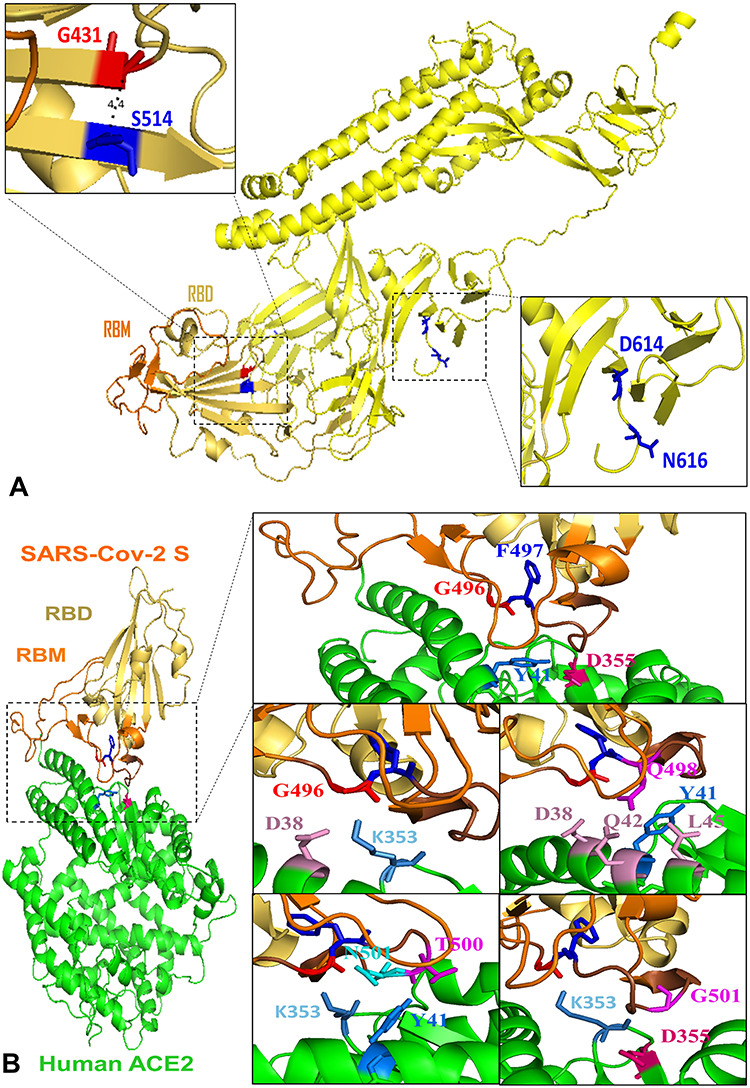
Structural representation of key residues. (A) Key residues altering protein stability in SARS-Cov-2 full-length S (yellow). G431 and K514 are spatial residues (4.4 Å) located in RBD (yellow orange) and closed to RBM (orange). Viral common variation D614G located in D614 is closed to N-linked glycosylation site N616. (B) Key residues altering the binding affinity between SARS-Cov-2 S (RBD: yellow orange; RBM: orange) and human ACE2 yellow. G496 (red) and F497 (blue) are located in the interacting motif Y495GFQPTNGVG504 (brown) and form an interaction network with ACE2 residues D355 (hot pink) and Y41 (marine). RBD residue G496 has vdw contacts with ACE2 residues D38 (pink) and K353 (sky blue). Q498 has contacts with residues D38, Q42, L45 (pink) as well as Y41 (marine). RBD residues T500 (magenta) and N501 (cyan) interact with ACE2 residues K353 (sky blue) and Y41 (marine). RBD residue G502 (magenta) has vdw contacts with D355 (hot pink) and K353 (sky blue) in ACE2.
Figure 6.
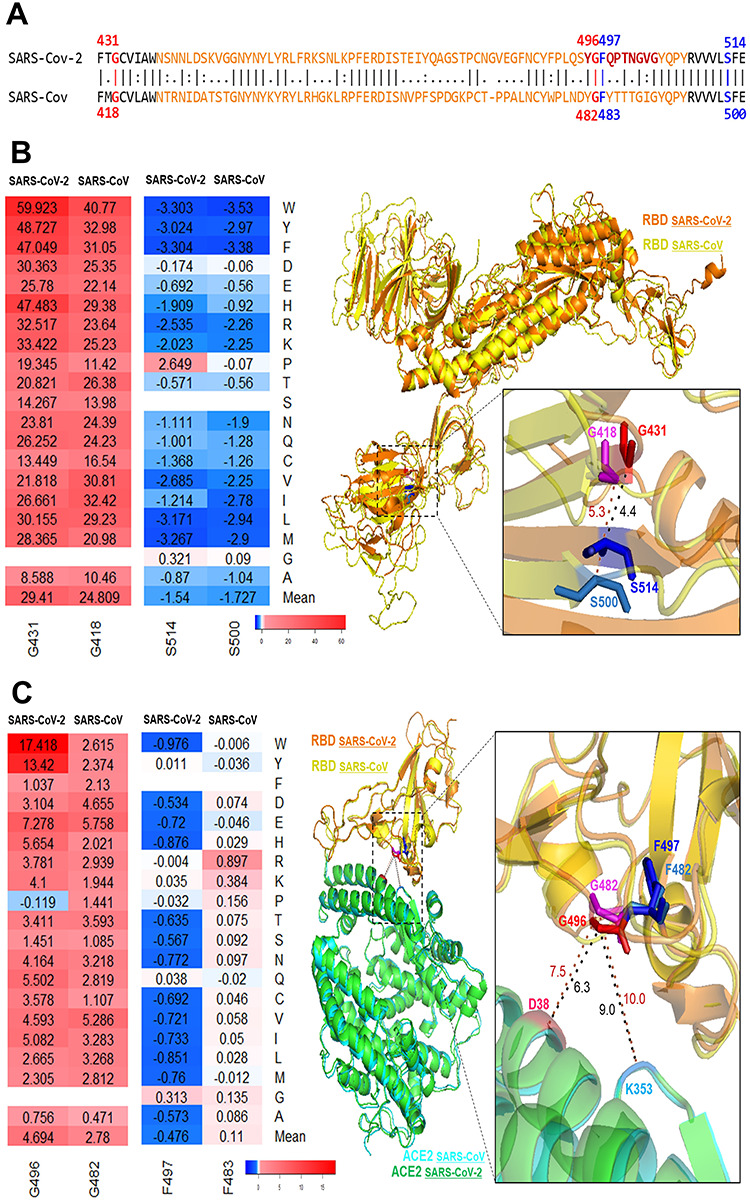
Comparison between SARS-CoV-2 and SARS-CoV. (A) Sequence alignment for the SARS-CoV-2 and SARS-CoV. G431 and S514 are closed to RBM (orange). G496 and F497 are located in the interacting motif Y495GFQPTNGVG504 (brown). These four residues are conserved in SARS-CoV. (B) Superimposition of the SARS-CoV-2 S (orange) and SARS-CoV S (yellow). Heatmap and structural representation of SARS-CoV-2 residues G431 (red) and S514 (blue) and SARS-CoV residues G418 (magenta) and S500 (marine). (C) Superimposition of the SARS-CoV-2 RBD (orange) with its ACE2 (green) and SARS-CoV RBD (yellow) with its RBD. Heatmap and structural representation of SARS-CoV-2 residues G496 (red) and F497 (blue) and SARS-CoV residues G482 (magenta) and F482 (marine). The distances to ACE2 residues D38 (hot pink) and K353 (sky blue) are measured.
Glycosylation, a posttranslational modification, plays important roles in viral pathobiology and host immune responses [34]. Glycosylation sites could shield specific epitopes in the S protein from antibody recognition and facilitate immune evasion. SARS-CoV-2 S includes 22 N-linked glycosylation sequons and these oligosaccharides are critical for S protein folding [5]. The oligomannose-type and complex-type glycans were observed in these N-linked glycosylation sites [35]. We investigated the mutations stability effects in 15 sites that could be mapped to full-length S structure (Figure 7). N122 with oligomannose-type glycan introduces the minimum ΔΔG mean at −0.452 kcal/mol. Most of the mutations in the N-linked glycosylation sites with complex-type glycans, N616 (ΔΔG mean = −0.433 kcal/mol) and N1134 (ΔΔG mean = −0.398 kcal/mol), can highly increase the S protein stability. There have been four observed viral mutations (N74K, N149H, N603K and N1194S) to N-linked glycosylation sites. We can calculate the ΔΔG of N603K at −0.5521 kcal/mol, indicating this mutation could increase full-length S stability. We also investigated two validated O-linked glycosylation sites T323 and S325. Most of the mutations in both sites can increase the stability of SARS-CoV-2 full-length S (Figure 7). One viral mutation T323I was identified on this site and this mutation can increase the protein stability (ΔΔG = −0.827 kcal/mol). Remarkably, both viral mutations N603K and T323I are not the mutations introducing the strongest stabilizing effects. Of 22 N-linked glycosylation sites, only N343 with complex-type glycan can be mapped to the RBD–ACE2 complex structure. The mutations in N343 have no effects (ΔΔΔG = 0 or 0.002 kcal/mol) on binding affinity (Figure 3). Interestingly, a recent study showed that glycosylation deletion including N343 can reduce infectivity and N343Q cause 20-fold reduction in viral infectivity [36]. Many mutations in N343 (ΔΔG mean = 0.227 kcal/mol) including N343Q (ΔΔG = 0.552 kcal/mol) induce the destabilizing effects on S protein stability that are unfavorable on virus function.
Figure 7.
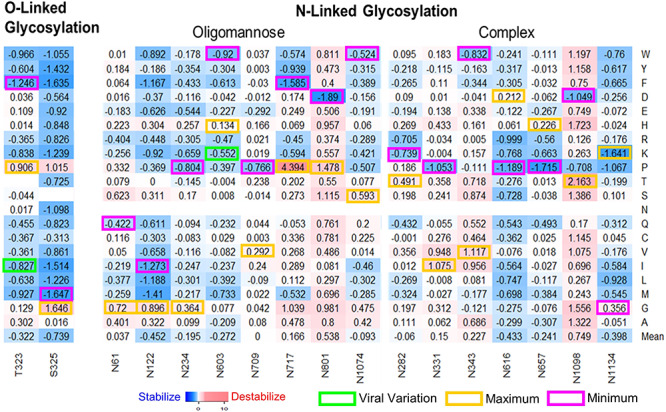
Heatmaps of effects of mutations in glycosylation sites on monomer stability. Maximum (yellow) and minimum (magenta) ΔΔG values are labeled for each N-linked or O-linked glycosylation site. The ΔΔG values of viral variations are shown in green boxes.
We investigated the binding energy changes induced by mutations in 21 RBD residues and 22 ACE2 residues that make van der Waals (vdw) contacts [10]. We observed many mutations in these contact residues could alter RBD–ACE2 binding (Supplementary Figure 4). In the RBD-interacting motif (Y495GFQPTNGVG504), RBD residue G496 with maximum ΔΔΔG mean has contacts with ACE2 residues D38 and K353. F497 with maximum ΔΔΔG mean has no vdw contacts with ACE2 residues. However, its neighbor residue Q498 has vdw contacts with residues D38, Q42, L45 as well as Y41 with minimum ΔΔΔG mean in ACE2. RBD residues T500 and N501 interact with ACE2 residues K353 and Y41, and RBD residue G502 have vdw contacts with D355 and K353 in ACE2. These viral ligand and receptor residues generate a complex network for RBD–ACE2 interaction (Figure 3 and Figure 5B). A recent molecular dynamics study also identified the RBD residues Q498 and N501 and ACE2 residues D355 and K353 are important for RBD–ACE2 hydrogen-bonding network and hydrophobic interactions [37]. SARS-Cov-2 S neighbor residues G496 and F497 introduce the most significant effects on RBD–ACE2 binding. Most of the mutations in G496 can highly reduce the RBD–ACE2 binding affinity. The substitutions from small glycine to other large amino acids will lead to an unfavorable change in protein interface and reduce the binding of complex. In G496s contact residue K353 of ACE2, mutation K353F has the minimum ΔΔΔG at −1.937 kcal/mol. Most mutation in RBD residue F497 can increase the RBD–ACE binding, and mutation F497W has the minimum ΔΔΔG at −0.978 kcal/mol in this position. Both phenylalanine and tryptophan are aromatic amino acids that include delocalized π electrons that interact with other aromatic residues as well as with positively charged residues such as lysine. SARS-Cov RBD residues G482 and F483 are in the equivalent positions of G496 and F497 of SARS-Cov-2 in the sequence and structure alignments (Figure 6A). All mutations in SARS-Cov RBD residue G482 can reduce the RBD–ACE2 binding affinity, but the ΔΔΔG values are relatively small compared with those changes in G496 of SARS-Cov-2. This may be explained by that the distances from SARS-Cov residue G482 to ACE2 residues D38 and K353 are increased to 7.5 and 10.0 Å, compared with the distances from SARS-Cov-2 residue G496 to ACE2 residues D38 and K353 are 6.3 and 9.0 Å (Figure 6C). Interestingly, even though the F483 of SARS-Cov can be superimposed to F497 of SARS-Cov-2, F483 shows the different effects on RBD–ACE2 interaction. Most of the mutations in F483 of SARS-Cov have not effects on binding affinity and F483R can weaken the interaction. However, most of the mutations in F497 of SARS-Cov-2 can enhance the RBD–ACE2 binding affinity. Previous studies suggest that SARS-Cov-2 S protein has a higher RBD–ACE2 binding affinity compared with those of SARS-CoV S [5, 8, 10]. F497 may play important roles for enhancing the RBD–ACE2 interaction for SARS-Cov-2. Our binding energy calculations suggest that these residues are potential binding sites for S protein and its receptor; any alterations in these sites may significantly change the binding affinity of RBD–ACE2 complex.
We found that the most common variant D614G in 5703 strains can induce the stabilizing effects on SARS-Cov-2 full-length S at −0.7838 kcal/mol. Particularly, D614G becomes the dominant pandemic form worldwide. A recent study showed that the patients with D614G strain had higher viral loads and suggested this mutation is important for RBD binding and enhance viral infection and production [38]. D614G has the minimum ΔΔG at −0.7838 kcal/mol among all 19 possible mutations in this position (Figure 2). This suggests that G614 is the most stable form compared to other possible mutations. In addition, D614G is predicted to have neutral effect (SNAP = −52) on protein function, which would benefit the viral S protein function (Table 3). Interestingly, we also found D614 is very closed to N-linked glycosylation site N616 (Figure 5A). Many mutations in N616 can also induce the stabilizing effects (Figure 7). Thus, D614G may enhance the fitness of SARS-Cov-2 through increasing S protein stability and participating in the N-linked glycosylation. The mechanism of D614G infections is still unknown [39], but recent studies showed that D614G can increase infectivity in various cells lines [36, 40] and strengthen the interaction between S1 and S2 domains [40]. The results from these experiments and our analysis suggest that D614G make the S protein more stable, and this change is favorable for virus infection. In addition, common viral mutations D936Y in 37 strains, V483A in 24 strains and V367F in 14 strains can also induce the stabilizing effects on SARS-Cov-2 full-length S or RBD (Table 3). S50L in seven strains and V341I have the minimum ΔΔG among all other possible mutations in their positions (Supplementary Figure 5). Moreover, we observed the percentage of mutations with stabilizing effects is higher in viral mutations (18.9%), compared with the one in all computationally predicted mutations (10.6%) (Figure 1). These results indicate that the mutations with stabilizing effects can make S protein remain sufficiently stable for its function and enhance the resistance of SARS-CoV-2. We noticed that most of the viral mutations are not substitutions with minimum and maximum folding or binding energy changes. As shown in Supplementary Figure 6, the |ΔΔG| mean of residues with viral mutations is lower than those without viral mutations (P < 2.2e−16), and |ΔΔG| of viral mutations is also smaller than other computational predicted mutations (P = 0.00016) in the same positions. The mutations with significant stabilizing or destabilizing effects may interfere with the S protein function (Figure 4C). The selection pressure makes the viral mutations occurred in the residues with small effects on protein stability and maintain SARS-Cov-2 S at its normal functions for transmission.
Conclusions
In conclusion, we applied the computational saturation mutagenesis to investigate 18 354 SARS-CoV-2 S missense mutations and 11 324 human ACE2mutations. We found RBD residue G431 can decrease the S protein stability, but its spatial residue S514 can make the S and RBD more stable. We analyzed 384 viral variations and identified that D614G in 5703 virus strains can stabilize SARS-Cov-2 entire S protein. Moreover, we showed that many mutations in N-linked glycosylation sites can increase the stability of the S protein. In addition, we showed that SARS-CoV-2 neighbor residues G496 and F497 have different effects on RBD–ACE2 binding and ACE2 contact residues D355 and Y41 are critical for this protein–protein interaction. Overall, the analysis is critical for understanding the roles of missense mutations in SARS-CoV-2 S and human ACE2 proteins on the viral pathogenesis of COVID-19.
Key Points
Structural consequences of 18 354 SARS-CoV-2 spike (S) mutations, 3705 receptor binding domain (RBD) mutations and 11 324 human angiotensin-converting enzyme 2 (ACE2) mutations on protein stability and binding affinity.
Residues G431 and S514 in SARS-CoV-2 RBD are important for S and RBD protein stability.
The dominant pandemic viral variation, D614G, can stabilize the entire S protein.
Various mutations in N-linked glycosylation sites can increase the stability of the S protein.
SARS-CoV-2 S residues G496 and F597 and ACE2 residues D355 and Y41 are critical for RBD–ACE2 interaction.
Supplementary Material
Shaolei Teng is an Assistant Professor of the Department of Biology at the Howard University. He received his PhD from the Clemson University and completed his postdoctoral training at the Cold Spring Harbor Laboratory. His research interests are to develop and apply bioinformatics approaches for analyzing the genetic variations associated with human diseases.
Adebiyi Sobitan is a PhD student in the Department of Biology at the Howard University. He is working on the predictions of protein functions using machine learning approaches.
Raina Rhoades is a PhD student in the Department of Biology at the Howard University. Her work spans structure modeling and next-generation sequencing.
Dongxiao Liu is a PhD student at the Howard University College of Medicine. His research interests include virology and bioinformatics.
Qiyi Tang is a Full Professor at the Howard University College of Medicine. He has more than 20 years of experience in virology, with specific training and expertise in gene regulation, signal transduction, molecular biology and cellular innate defenses.
Contributor Information
Shaolei Teng, Department of Biology at the Howard University, 415 College St. NW, Washington, DC 20059.
Adebiyi Sobitan, Department of Biology at the Howard University, 415 College St. NW, Washington, DC 20059.
Raina Rhoades, Department of Biology at the Howard University, 415 College St. NW, Washington, DC 20059.
Dongxiao Liu, Howard University College of Medicine, 520 W Street NW, Washington, DC 20059.
Qiyi Tang, Howard University College of Medicine, 520 W Street NW, Washington, DC 20059.
Acknowledgements
Authors acknowledge the Howard University Junior Faculty Writing & Creative Works Summer Academy.
Funding
Howard University startup funds [U100193]; National Science Foundation [DBI 2000296, IIS 1924092]; National Institute on Minority Health and Health Disparities of the National Institutes of Health [2U54MD007597].
Disclosure
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author contributions
S.T. and Q.T. are principal investigator on the grant funding and supervised the study. S.T. designed the study and performed the bioinformatics analysis. S.T., A.S., R.R., D.L. and Q.T. analyzed data. S.T. wrote the manuscript, and all authors critically revised the manuscript.
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- 1. Schoeman D, Fielding BC. Coronavirus envelope protein: current knowledge. Virol J 2019;16:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Song Z, Xu Y, Bao L, et al. From SARS to MERS, thrusting coronaviruses into the spotlight. Viruses 2019;11:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ge XY, Li JL, Lou YX, et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013;503:535–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhou P, Lou YX, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020;579:270–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Walls AC, Park YJ, Tortorici MA, et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020;181:281–292.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020;181:271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li F, Li W, Farzan M, et al. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005;309:1864–8. [DOI] [PubMed] [Google Scholar]
- 8. Ortega JT, Serrano ML, Pujol FH, et al. Role of changes in SARS-CoV-2 spike protein in the interaction with the human ACE2 receptor: an in silico analysis. EXCLI J 2020;19:410–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tai W, He L, Zhang X, et al. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Mol Immunol 2020;1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang QQ, Zhang Y, Wu L, et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020;181:894–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Denison MR, Graham RL, Donaldson EF, et al. Coronaviruses: an RNA proofreading machine regulates replication fidelity and diversity. RNA Biol 2011;8:270–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Teng S, Srivastava AK, Schwartz CE, et al. Structural assessment of the effects of amino acid substitutions on protein stability and protein protein interaction. Int J Comput Biol Drug Des 2010;3:334–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Teng S, Madej T, Panchenko A, et al. Modeling effects of human single nucleotide polymorphisms on protein-protein interactions. Biophys J 2009;96:2178–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li L, Jia Z, Peng Y, et al. Forces and disease: electrostatic force differences caused by mutations in kinesin motor domains can distinguish between disease-causing and non-disease-causing mutations. Sci Rep 2017;7:8237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vedithi SC, Rodrigues CHM, Portelli S, et al. Computational saturation mutagenesis to predict structural consequences of systematic mutations in the beta subunit of RNA polymerase in Mycobacterium leprae. Comput Struct Biotechnol J 2020;18:271–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Berman H, Henrick K, Nakamura H, et al. The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res 2007;35:D301–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schymkowitz J, Borg J, Stricher F, et al. The FoldX web server: an online force field. Nucleic Acids Res 2005;33:W382–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhao WM, Song SH, Chen ML, et al. The 2019 novel coronavirus resource. Yi Chuan 2020;42:212–21. [DOI] [PubMed] [Google Scholar]
- 19. Buß O, Rudat J, Ochsenreither K. FoldX as protein engineering tool: better than random based approaches? Comput Struct Biotechnol J 2018;16:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cheng TMK, Goehring L, Jeffery L, et al. A structural systems biology approach for quantifying the systemic consequences of missense mutations in proteins. PLoS Comput Biol 2012;8:e1002738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pires DEV, Ascher DB, Blundell TL. DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res 2014;42:314–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Parthiban V, Gromiha MM, Schomburg D. CUPSAT: prediction of protein stability upon point mutations. Nucleic Acids Res 2006;34:W239–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu CH, Apweiler R, Bairoch A, et al. The universal protein resource (UniProt): an expanding universe of protein information. Nucleic Acids Res 2006;34:D187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Madeira F, Park YM, Lee J, et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res 2019;47:W636–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bromberg Y, Yachdav G, Rost B. SNAP predicts effect of mutations on protein function. Bioinformatics 2008;24:2397–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bromberg Y, Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res 2007;35:3823–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bromberg Y, Rost B. Correlating protein function and stability through the analysis of single amino acid substitutions. BMC Bioinf 2009;10:S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet 2013; Chapter 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vaser R, Adusumalli S, Leng SN, et al. SIFT missense predictions for genomes. Nat Protoc 2016;11:1–9. [DOI] [PubMed] [Google Scholar]
- 30. Wang Z, Moult J. SNPs, protein structure, and disease. Hum Mutat 2001;17:263–70. [DOI] [PubMed] [Google Scholar]
- 31. Renn A, Fu Y, Hu X, et al. Fruitful neutralizing antibody pipeline brings hope to defeat SARS-Cov-2. Trends Pharmacol Sci 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang L, Shi W, Joyce MG, et al. Evaluation of candidate vaccine approaches for MERS-CoV. Nat Commun 2015;6:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pallesen J, Wang N, Corbett KS, et al. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc Natl Acad Sci U S A 2017;114:E7348–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Watanabe Y, Bowden TA, Wilson IA, et al. Exploitation of glycosylation in enveloped virus pathobiology. Biochim Biophys Acta 2019;1863:1480–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Watanabe Y, Allen JD, Wrapp D, et al. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020;369:330–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li Q, Wu J, Nie J, et al. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell 2020;182:1284–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang Y, Liu M, Gao J. Enhanced receptor binding of SARS-CoV-2 through networks of hydrogen-bonding and hydrophobic interactions. Proc Natl Acad Sci USA 2020;117:13967–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Korber B, Fischer WM, Gnanakaran S, et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020;182:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Grubaugh ND, Hanage WP, Rasmussen AL. Making sense of mutation: what D614G means for the COVID-19 pandemic remains unclear. Cell 2020182:794–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang L, Jackson CB, Mou H, et al. The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity. bioRxiv 2020; 2020.06.12.148726. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.