

Abstract
Given the scale and rapid spread of the coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2, or 2019-nCoV), there is an urgent need to identify therapeutics that are effective against COVID-19 before vaccines are available. Since the current rate of SARS-CoV-2 knowledge acquisition via traditional research methods is not sufficient to match the rapid spread of the virus, novel strategies of drug discovery for SARS-CoV-2 infection are required. Structure-based virtual screening for example relies primarily on docking scores and does not take the importance of key residues into consideration, which may lead to a significantly higher incidence rate of false-positive results. Our novel in silico approach, which overcomes these limitations, can be utilized to quickly evaluate FDA-approved drugs for repurposing and combination, as well as designing new chemical agents with therapeutic potential for COVID-19. As a result, anti-HIV or antiviral drugs (lopinavir, tenofovir disoproxil, fosamprenavir and ganciclovir), antiflu drugs (peramivir and zanamivir) and an anti-HCV drug (sofosbuvir) are predicted to bind to 3CLPro in SARS-CoV-2 with therapeutic potential for COVID-19 infection by our new protocol. In addition, we also propose three antidiabetic drugs (acarbose, glyburide and tolazamide) for the potential treatment of COVID-19. Finally, we apply our new virus chemogenomics knowledgebase platform with the integrated machine-learning computing algorithms to identify the potential drug combinations (e.g. remdesivir+chloroquine), which are congruent with ongoing clinical trials. In addition, another 10 compounds from CAS COVID-19 antiviral candidate compounds dataset are also suggested by Molecular Complex Characterizing System with potential treatment for COVID-19. Our work provides a novel strategy for the repurposing and combinations of drugs in the market and for prediction of chemical candidates with anti-COVID-19 potential.
Keywords: MCCS, COVID-19, residue energy contribution, drug repurposing, drug combination
Introduction
The outbreak of the coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [1–6], quickly spread over 213 countries/regions, infecting >21 930 883 individuals globally with more than 774 997 deaths worldwide as reported of 17 August 2020. New and effective anti-COVID-19 drugs are urgently needed, whereas a new drug discovery takes >8 years and costs >$2.6 billion with an approval rate of less than 12% [7]. A few medications including lopinavir-ritonavir, remdesivir and hydroxychloroquine/chloroquine have been used in clinics, but there are still not enough medical data confirming these drugs work and are safe to cure COVID-19. Although lopinavir/ritonavir shows benefit in some secondary endpoints, no benefit is observed with treatment beyond standard care for severe COVID-19 patients [8], and future trials will be carried out to verify this result. Moreover, a report from France’s drug safety agency disclosed that hydroxychloroquine can have serious cardiovascular side effects [9]. Gilead Sciences tracked the responses to remdesivir intervention therapy for 53 patients with COVID-19 and observed a 13% death rate [10]. However, study groups were spread out across several countries with small numbers of patients, and it is hard to draw definitive conclusions from these data. Since most of the drugs or drug combinations currently used in clinical trials are chosen empirically, there is a critical need to leverage innovative technology with available medical resources to rapidly repurpose FDA-approved drugs and select drug combinations for anti-COVID-19 before vaccines are available.
SARS-CoV-2, Middle East Respiratory Syndrome Coronavirus (MERS-CoV) and Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) are members of the coronavirus (CoV) family [11]. These CoVs can all transmit from human-to-human [12–14]. CoV infection can be as mild as the common cold or as deadly as the SARS infection. Based on the confirmed cases, the symptoms of SARS-CoV-2 include fever, malaise, dry cough, shortness of breath, respiratory distress and more. Although there are no approved medications or vaccines for COVID-19, new information regarding SARS-CoV-2 is being revealed daily. Recently, the SARS-CoV-2 has been sequenced and several important proteins have been resolved [15–19], thereby facilitating exploration and discovery of effective treatments. The rationale for repurposing and discovery of drugs for treatment of SARS-CoV-2 is based on the premise that: (i) the entry of SARS-CoV-2 into human host cells relies on the binding of spike protein (S-protein) [19] to the angiotensin-converting enzyme 2 (ACE2) [17, 20], and (ii) the replication of nCoV requires the viral 3C-like cysteine protease (3CLPro) [18] or RNA-dependent RNA polymerase (RdRp) [15], with both 3CLPro and RdRp highly conserved within the coronavirus family. These proteins and/or enzymes are potential targets since they are involved in the mechanism of COVID-19 infection, and drugs or chemical agents targeting them may provide therapeutic potential for the treatment of COVID-19.
In recent years, computational techniques have become more common in drug design because of advantages such as time-saving, high efficiency and high sensitivity. Many in silico tools or approaches have been used to design and discover the potential drug candidates for COVID-19. For example, Jin et al. [18] recently resolved the 3D complex of 3CLpro-N3. Based on this crystal structure, they applied Glide algorithm to carry out a virtual screening against their in-house database and identified cinanserin (IC50: 125 μM) as a potential inhibitor for 3CLpro of SARS-CoV-2 [18]. Similarly, Wang [21] applied virtual docking screening using Glide and molecular dynamics simulation with MM-GBSA to predict the potential drug candidates for 3CLpro. Recently, Liu et al. [22] developed and applied their computational protocol SCAR (steric-classes alleviating receptors) to discover potential covalent drugs for SARS-CoV-2. In addition, several computational tools or databases have been developed to meet the urgent need of drug discovery for COVID-19, including a network-based approach for drug repurposing [23], MolAICal [24] (a de novo approach, https://molaical.github.io/quickstart.html), COVID-19 Docking Server (https://ncov.schanglab.org.cn/) [25] and more. Among these methods, structure-based virtual docking screening is a popular approach for the drug discovery of COVID-19. However, structure-based virtual docking screening relies primarily on docking scores and does not take the contributions of key residues into consideration of selecting the binding pose, which may lead to a significantly higher incidence rate of false-positive results.
We have elaborated on a scoring function-based computing protocol Molecular Complex Characterizing System (MCCS) [26] to aid in rational drug design. Briefly, MCCS first calculates the energy contribution of each residue involved in the binding, which helps in determining the key residues and their energy contributions. Then, a (protein) sequence-based vector embedded with individual residue energy contribution is constructed to represent the recognition pattern or feature between a receptor and its ligand. MCCS takes the contribution and importance of key residue into consideration for selecting a more accurate binding pose during the process. Finally, the reliable energy contribution vectors are used to select potential candidates, which may improve the accuracy of virtual screening.
In the present work, we first determine the residue energy contributions in the binding pocket of 3CLPro, the main viral protease required for SARS-CoV or SARS-CoV-2 replication, toward binding an inhibitor by applying our recently developed MCCS [26] to the 3CLPro-inhibitor co-crystal structure. Then, several 3CLPro-ligand complexes are used to create energy contribution vector(s) to characterize the recognition pattern of 3CLPro in SARS-CoV/SARS-CoV-2 and virtual screening is carried out subsequently against the DrugBank database and the CAS COVID-19 antiviral candidate compounds dataset. Anti-HIV/antiviral drugs (lopinavir, tenofovir disoproxil, fosamprenavir and ganciclovir), antiflu drugs (peramivir and zanamivir) and an anti-HCV drug (sofosbuvir) are predicted to bind to 3CLPro in SARS-CoV-2 with therapeutic potential for COVID-19 infection. Moreover, 10 additional antiviral chemicals from the CAS dataset are also predicted as promising agents for COVID-19. Finally, we also utilize our novel virus chemogenomics knowledgebase platform [27] to identify potential combinations of medications for COVID-19.
Materials and Methods
Scoring function
The scoring function in nature is not limited to the conformational search and optimization process in docking, but also it is mainly used to evaluate an entire protein-ligand complex, or more specifically, a ligand conformation inside a receptor binding site. By revisiting the atom level and aggregating the energy per residue instead of per protein, we serve the distinguishing characteristics of each residue in the process of protein-ligand binding, which also further enables the feature modeling of a protein in terms of binding.
Below are the five core terms used by the scoring function in Vina [28] and idock [29]:
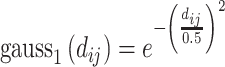 |
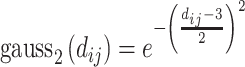 |
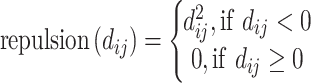 |
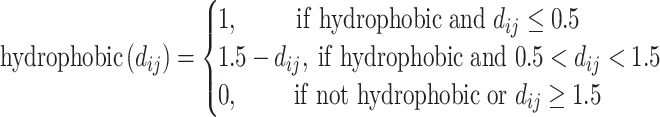 |
 |
In a typical docking process, a computed atom pair consists of one atom from the protein and the other from the small molecule. Essentially the energy terms are related to three variables: the distance between, and the van der Waals radii of the two interacting atoms. By introducing a new variable 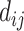 to represent the surface distance between the interacting atoms, the functions can be reduced to unary functions of
to represent the surface distance between the interacting atoms, the functions can be reduced to unary functions of 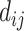 . In this model, atom interactions are divided into three kinds: regular interaction, hydrophobic interaction or hydrogen bond interaction. The
. In this model, atom interactions are divided into three kinds: regular interaction, hydrophobic interaction or hydrogen bond interaction. The 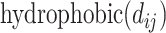 and the
and the 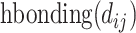 functions evaluate to a nonzero value only if the atom pair is of the hydrophobic interaction or the hydrogen bond, respectively.
functions evaluate to a nonzero value only if the atom pair is of the hydrophobic interaction or the hydrogen bond, respectively.
The weighted sum of the five terms forms the total score, where the coefficients are also given in the original Vina literature:
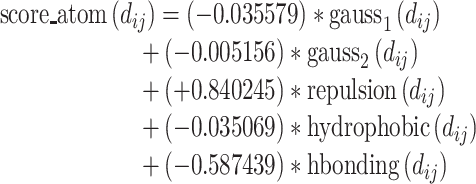 |
The residue energy contribution is calculated as
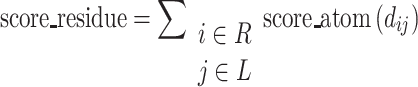 |
where 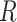 is the set of atoms in the residue being considered, and
is the set of atoms in the residue being considered, and 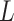 is the set of atoms in the ligand whose coordinates are either computed from Monte Carlo based docking or determined by X-ray crystallography or cryo-EM. Since the hydrophobic and hydrogen bond interactions are both one term more than a regular interaction, the additional terms are the key to making a residue prominent.
is the set of atoms in the ligand whose coordinates are either computed from Monte Carlo based docking or determined by X-ray crystallography or cryo-EM. Since the hydrophobic and hydrogen bond interactions are both one term more than a regular interaction, the additional terms are the key to making a residue prominent.
Similarity and clustering
To quantify the similarity between two vectors, a real-valued similarity function is used in statistics and related fields. Such similarity measures include cosine similarity, the Pearson correlation coefficient (the PCC), Euclidean distance, etc. Among all the various similarity measures, cosine similarity and the PCC are commonly used for real-valued vectors, which are suitable in our scenario.
Given two vectors of residue energy contribution, 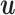 and
and 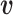 , the cosine similarity,
, the cosine similarity, 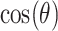 , is represented using a dot product and vector length as
, is represented using a dot product and vector length as
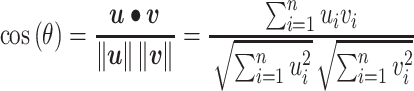 |
where 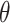 is the angle between the two vectors and the subscript
is the angle between the two vectors and the subscript 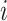 refers to the residue numbering under the selected scheme. The function gives a positive value (up to one) for similar vectors and either zero or a negative value for distinct vectors.
refers to the residue numbering under the selected scheme. The function gives a positive value (up to one) for similar vectors and either zero or a negative value for distinct vectors.
The PCC is defined in the same way except it subtracts the mean from every vector element:
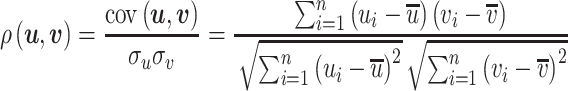 |
With any similarity measure, protein clustering can be carried out in such a way that proteins in the same group are more alike, in terms of binding mode, to each other than to that of other groups. In the visualization, a heatmap of an 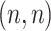 -sized similarity matrix is commonly used to show similarities among a set of
-sized similarity matrix is commonly used to show similarities among a set of 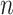 vectors, with a dendrogram for demonstration of the clustering. The grids of the heatmap use a color scale to display a color mapped from its numeric value which represents the similarity between two vectors. The clustering can be carried out directly with the
vectors, with a dendrogram for demonstration of the clustering. The grids of the heatmap use a color scale to display a color mapped from its numeric value which represents the similarity between two vectors. The clustering can be carried out directly with the 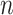 vectors or with the similarity rows or columns of the
vectors or with the similarity rows or columns of the 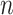 vectors.
vectors.
DrugBank dataset and CAS COVID-19 antiviral candidate compounds dataset
The DrugBank dataset (https://www.drugbank.ca/releases/latest) with 1814 FDA-approved drugs was downloaded and filtered to eliminate metals or mixtures of isotopes. Moreover, CAS COVID-19 antiviral candidate compounds dataset with nearly 50 000 chemical substances was download via https://www.cas.org/covid-19-antiviral-compounds-dataset. VEGA [30] is applied to prepare the small molecules by adding the polar hydrogens, Vina force field and Gasteiger charges. To determine whether the tertiary (3°) amide of the small molecule should be protonated, PROPKA (version 3.1) [31] was utilized to predict the corresponding pKa values. With such values, our program-MCCS [26] is able to donorize the nitrogen atoms whose computed pKa value is greater than or equal to the given pH (7.4 by default). Finally, the torsions (branches) were also defined by VEGA, and the file format was transformed into PDBQT. The PDBQT files of protein and ligand and the pKa file of the ligand form the input of our method.
Virtual screening by MCCS
Starting with the code base of the current stable version 2.2.3 of idock [29] that adopts the exact scoring function of AutoDock Vina [28], we developed an even more efficient variant integrating the ability to calculate the residue contributions of the binding energy, named jdock [26]. MCCS is open source under Apache License 2.0 and is freely available on GitHub at https://github.com/stcmz/jdock/ and https://github.com/stcmz/mccsx. MCCS was then applied to carry out the virtual screening using the crystal structure of 3CLPro in SARS-CoV-2 against the DrugBank dataset and the CAS COVID-19 antiviral candidate compounds dataset.
Virus-associated disease-specific chemogenomics knowledgebase (Virus-CKB)
On the basis of our established domain specific chemogenomics databases [32–35] and our novel computational techniques [32, 36–39], we constructed and reported a novel virus-associated disease-specific chemogenomics knowledgebase (Virus-CKB, https://www.cbligand.org/g/virus-ckb) [27] and applied our computational systems pharmacology-target mapping (CSP-Target Mapping) to rapidly identify the FDA-approved drugs for repurposing into new indications by fast progress into clinical trials to meet the urgent demand due to the COVID-19 outbreak. Virus-CKB, a one-stop computing platform describes the chemical molecules, genes, proteins and signaling pathways involved in the regulation of virus-associated diseases. To date, the Virus-CKB archived 65 antiviral drugs in the market, 107 virus-related proteins or enzymes with 189 available 3D crystal or cryo-EM structures [40–42], and ~2609 chemical agents reported for these target proteins and enzymes. In addition, the Virus-CKB is implemented with our developed machine-learning computational algorithms and computing tools for the prediction of the important protein targets, and the output data analysis and visualization, including HTDocking [32, 36–38], TargetHunter [32, 36–39], BBB predictor [32, 37, 38], NGL viewer [43], Spider Plot [32, 36, 37], etc.
Results and Discussion
Workflow of MCCS protocol
We here described the general procedure of MCCS protocol as below: (1) the first step of MCCS protocol is to prepare the input files (PDBQTs) of both receptor and ligand, including the ligand protonation with PROPKA [31], residues reparation of the receptor by Chimera [44] and adding force fields and charges to receptor and ligand by VEGA [30]; (2) the second step is to calculate the residue energy contribution by our revised docking algorithm named ‘jdock’, which shared the same scoring function with AutoDock Vina [28] or idock [29]; (3) sequentially, a part of or a full-length protein sequence-based vector embedded with individual residue energy contribution is constructed to represent the binding recognition feature between a receptor and its ligand, name the energy recognition vector; (4) finally, the energy contribution vectors are explored for extensive uses in recognition-pattern generation, protein similarity comparison and clustering, and virtual screening. All these components have been integrated into MCCS with ready-to-use computer scripts developed in-house.
Residue energy contribution
After obtaining the input files, MCCS is used to calculate the energy contribution of each residue, which can help us to understand the role of binding residues for the recognition of ligand(s). With the aggregation accomplished, a list of scores is fetched. Table 1 is the example of outputs of the 3CLPro in the SARS-CoV complexed SID-24808289 (N-(benzo [1,2,3] triazol-1-yl)-N-(benzyl)acetamido)phenyl)carboxamides, inhibitor) (PDB: 4MDS, Resolution: 1.598 Å) [45].
Table 1.
Top 10 residue energy contributions in the complex of 3CLPro with SID-24808289 (PDB: 4MDS) (kcal/mol)
| Residue | Total energy contribution | Sum of steric componentsa | Sum of hydrophobic componentsb | Sum of hydrogen-bonding componentsc |
|---|---|---|---|---|
| Glu166 | −1.9893 | −1.0839 | −0.2428 | −0.6626 |
| Met165 | −1.0111 | −1.0088 | −0.0023 | 0 |
| Met49 | −0.8614 | −0.8246 | −0.0368 | 0 |
| His41 | −0.8522 | −0.8308 | −0.0214 | 0 |
| Asn142 | −0.8434 | −0.7487 | −0.0947 | 0 |
| His163 | −0.7228 | 0.0374 | 0 | −0.7601 |
| Gln189 | −0.5892 | −0.4925 | −0.0967 | 0 |
| Phe140 | −0.4199 | −0.3856 | −0.0342 | 0 |
| Leu167 | −0.4046 | −0.4046 | 0 | 0 |
| His164 | −0.3943 | −0.3943 | 0 | 0 |
aThe sum of steric components is the total of the weighted sum of the first three terms of the scoring function for a residue.
bThe sum of hydrophobic components is the sum of all weighted  terms for a residue.
terms for a residue.
cThe sum of hydrogen-bonding components is the sum of all weighted 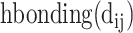 terms for a residue; the three sums amount to the energy contribution.
terms for a residue; the three sums amount to the energy contribution.
The detailed interactions between 3CLPro in SARS-CoV and SID-24808289 were shown in Figure 1. The total binding energy of the crystallized small molecule-SID-24808289 in 3CLpro was −11.7914 kcal/mol, which was calculated from the sum of intra-ligand free energy (−0.0088 kcal/mol) and inter-ligand free energy (−11.7826 kcal/mol). Here, the inter-ligand free energy is the total energy of interacted atom pairs between the small molecule (SID-24808289) and the receptor (3CLPro), which can be computed and further divided into the energy contribution of each residue. Moreover, each residue energy contribution can be further decomposed into (i) the ‘sum of steric components’ that included gauss1, gauss2 and repulsion, (ii) the ‘sum of hydrophobic components’ and (iii) the ‘sum of hydrogen-bonding components.’ Taking Glu166 in 3CLPro (PDB: 4MDS) as an example (Table 1), the total energy contribution of Glu166 to the complex of SID-24808289-3CLPro was −1.9893 kcal/mol, which can be decomposed into (i) −1.0839 kcal/mol from the ‘sum of steric components,’ (ii) −0.2428 kcal/mol from the ‘sum of hydrophobic components’ and (iii) −0.6626 kcal/mol from the ‘sum of hydrogen-bonding components.’ Particularly, we noted that Glu166 and His163 contributed significantly to the recognition of ligand SID-24808289 through hydrogen-bonding interaction (Table 1), as shown in Figure 1.
Figure 1.
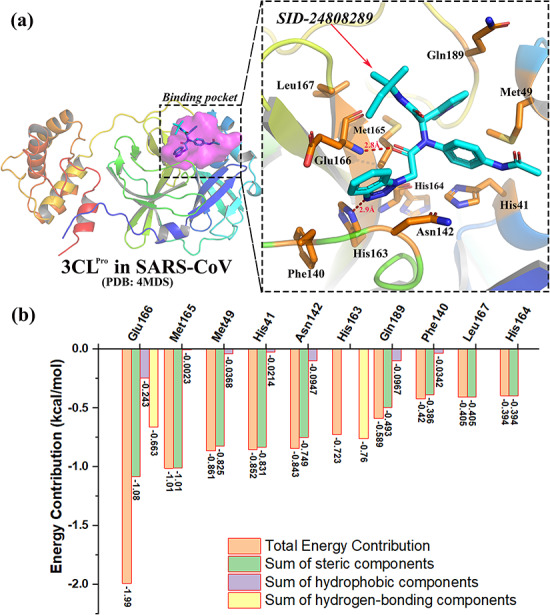
Detailed interactions and top 10 residue energy contributions of 3CLPro-SID-24808289 (PDB: 4MDS, SARS-CoV). (A) The detailed interactions between 3CLPro in SARS-CoV and SID-24808289. The binding pocket in 3CLPro is highlighted in purple surface on the left side. Top 10 residues with higher energy contributions are highlighted in stick on the right side. (B) The energy contribution of top 10 key residues involved in the binding pockets of 3CLPro.
Energy contribution vectors and recognition pattern in the ligand-binding pocket of 3CLPro in SARS-CoV
Then, a protein sequence-based vector embedded with individual residue energy contribution was constructed to represent the recognition feature between a receptor and its ligand, and the most important recognition feature was named as the recognition pattern.
Using our protocol, six representative co-crystal structures of 3CLPro in SARS-CoV complexed with different ligands were selected to calculate the residue energy contribution, including 3SN8 [46], 1WOF [47], 2AMD [47], 2OP9 [48], 3V3M [49] and 4MDS [45]. For each individual complex, the total energy contribution of 73 residues within 8 Å around the co-crystal ligand was used to construct a vector named energy contribution vector (Figure 2).
Figure 2.
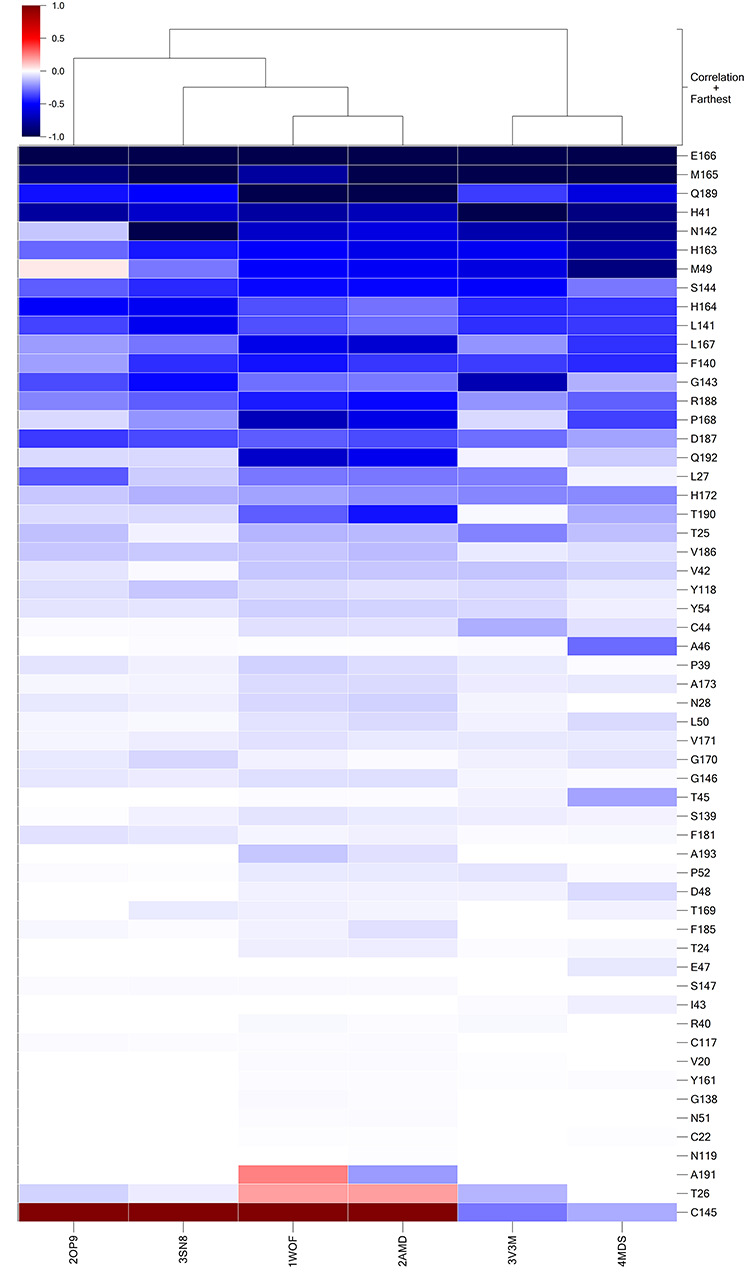
Energy contribution vector (using total energy contribution) in the ligand-binding pocket of 3CLPro in SARS-CoV. The dark blue color in the energy contribution vectors represented that the energy contribution of residue was negative value (≤ −1 kcal/mol), while the dark red color represented that the energy contribution of residue was positive value (≥1 kcal/mol). The residues in the energy contribution vectors were sorted by the average residue energy contribution ascendingly.
As shown in Figure 2, the residue rows were sorted ascendingly according to their average contribution and the protein columns represent energy contribution vectors of each individual complex. The figure consists of 73 residues within 8 Å around the co-crystal ligand. First, our results showed that Glu166 (Figure 2) has the greatest energy contribution to the recognition of ligands with an average energy contribution of −1.5442 kcal/mol. With the insight of the residue energy contribution obtained from the analyses of these six complexes, we found that one of the key contributions of Glu166 was hydrogen-bonding interaction (Figure S1a), which was supported by the detailed interaction shown in Figure 3. Additionally, we found that residues including Gly143, Ser144, His163 and Gln189 also contributed to the recognition of ligands through hydrogen-bonding (Figure S1a). On the other hand, His41, Met49, Met165 and Leu167 contributed to the binding of ligands via hydrophobic interactions, as shown in Figure 3 and Figure S1b. Interestingly, our results showed that Cys145 contributed to the binding by a covalent bond (red color in Figure 2, positive value) and hydrophobic interactions (blue color in Figure 2, negative value).
Figure 3.
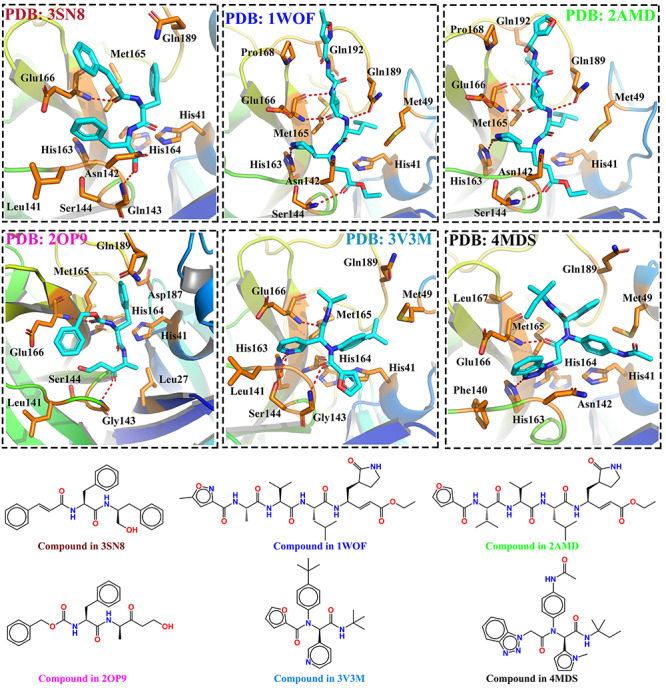
The detailed interactions between SARS-CoV and ligands. The hydrogen-bonding was highlighted in red. The key residues were highlighted in sticks. The structures of small molecules were shown in the bottom.
The energy contribution vectors of proteins could be easily compared and used to cluster similar ligands or proteins. The dendrogram on the top of the grids demonstrated the clustering of similar columns using Pearson’s distance. As shown in Figure 2, we found that ligands with large similarity (or binding interactions) were clustered together, indicating our approach is reasonable for ligands classification or clustering. For example, we observed that the score vector of 1WOF was similar to that of 2AMD and these two proteins were clustered together, supported by the correlation between similarities in their ligands and interactions (Figure 3).
Our program generated nine different energy contribution vectors in total, including (1) Gauss, (2) Gauss1, (3) Gauss2, (4) hydrogen-bonding, (5) hydrophobic, (6) non-steric (hydrogen-bonding+hydrophobic), (7) repulsion, (8) steric (Gauss1 + Gauss2 + repulsion) and (9) total energy contribution, as shown in Figure S2. By analyzing these vectors of 3CLPro in SARS-CoV, we found that the non-steric recognition vector may represent the most important feature (named as recognition pattern) of 3CLPro in SARS-CoV as shown in Figure S3, which included Glu166 (hydrogen bond), Met165 (hydrophobic interaction), Met49 (hydrophobic interaction), etc. They were involved in the ligand-binding pocket in all co-crystal structures with significant contributions to the binding of small molecules. The recognition pattern of 3CLPro in SARS-CoV has taken the contribution of key residues into consideration that will benefit the accuracy of virtual screening, as the traditional docking algorithms are based on docking scores and neglects the importance of key residues.
Repurposing FDA-approved drugs for COVID-19 by recognition pattern-based computing
Using the same method, we first generated the non-steric recognition pattern (including both hydrogen-bonding and hydrophobic components) of 3CLPro in SARS-CoV-2 based on its co-crystal structure (PDB:6 LU7, inhibitor N3) [18]. Then the structure of 3CLPro in SARS-CoV-2 was used to perform a virtual screening against the prepared DrugBank library with 1814 FDA-approved drugs. Finally, the recognition patterns of each drug were used to compare with that of N3 (inhibitor in co-crystal structure), and the pattern similarity between drugs and N3 was used to select the potential hits.
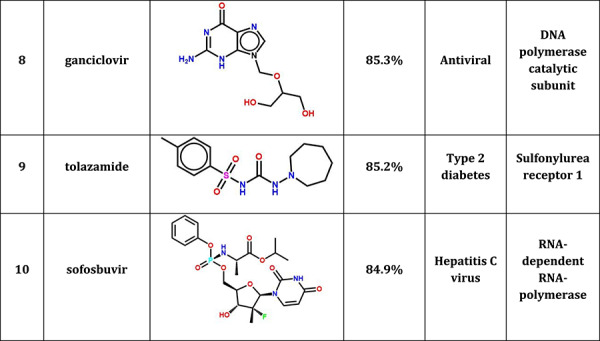
Our results in Table 2 shows 10 FDA-approved drug candidates may bind to 3CLPro with therapeutic potential for COVID-19. Table S1 shows the ranking and pattern similarity of the top 100 drug candidates. For comparison, Table S2 shows the results of 10 drug candidates with the lowest pattern similarity when compared to the N3. From Table 2, we found that four anti-HIV/antiviral drugs (lopinavir, tenofovir disoproxil, fosamprenavir and ganciclovir), two antiflu drugs (peramivir and zanamivir) and one anti-HCV drug (sofosbuvir) were predicted to bind to 3CLPro in SARS-CoV-2, which could be the possible treatments for COVID-19. These drugs shared a high pattern similarity with a reported N3 inhibitor in the co-crystal complex, especially in lopinavir which shared up to 89.7% pattern similarity with N3. The comparison of detailed interactions between lopinavir and N3 in SARS-CoV-2 was shown in Figure 4. Our results showed that these two compounds shared very similar non-steric interactions (hydrogen-bonding and hydrophobic interactions), indicating that lopinavir may bind to 3CLPro in SARS-CoV-2 with therapeutic potential for the treatment of COVID-19, which is consistent with the current clinical trial of lopinavir/ritonavir for COVID-19.
Table 2.
10 FDA-approved drugs with therapeutic potential for COVID-19
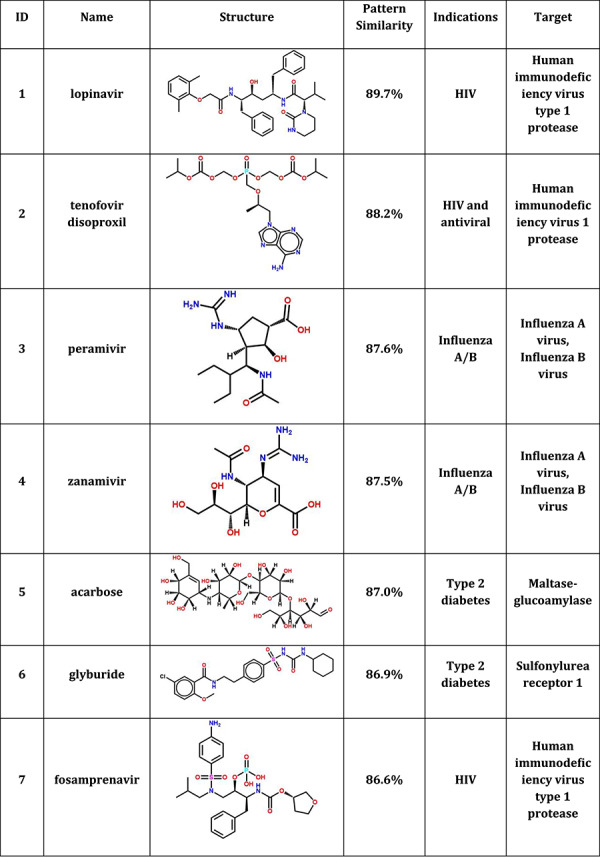
|
|
Figure 4.
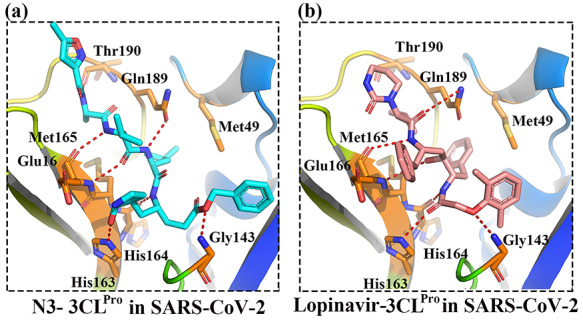
The comparisons between N3 (co-crystal inhibitor) and Lopinavir in SARS-CoV-2. The hydrogen-bonding was highlighted in red. The key residues were highlighted in sticks.
In addition, recent studies [50, 51] showed that type II diabetes patients with COVID-19 develop a more severe condition compared to those without diabetes. From Table 2, we also predicted three antidiabetic drugs (acarbose, glyburide and tolazamide) with the potential to treat COVID-19 coexisting with type II diabetes [52]. Our predictions are consistent with recent work on COVID-19. For example, Wang et al. [53] suggested the roles of 21 drugs included acarbose in inflammatory response prevention in patients with COVID-19 for the first time. In addition, Paniri et al. [54] proposed that glyburide might be a useful drug to combat SARS-CoV-2 by blocking the wide spectrum of molecules related to inflammatory cascade. Certainly, these in silico predictions will require the in vitro target validation experiments in the future.
Repurposing CAS COVID-19 antiviral candidate compounds dataset for COVID-19 by recognition pattern-based computing
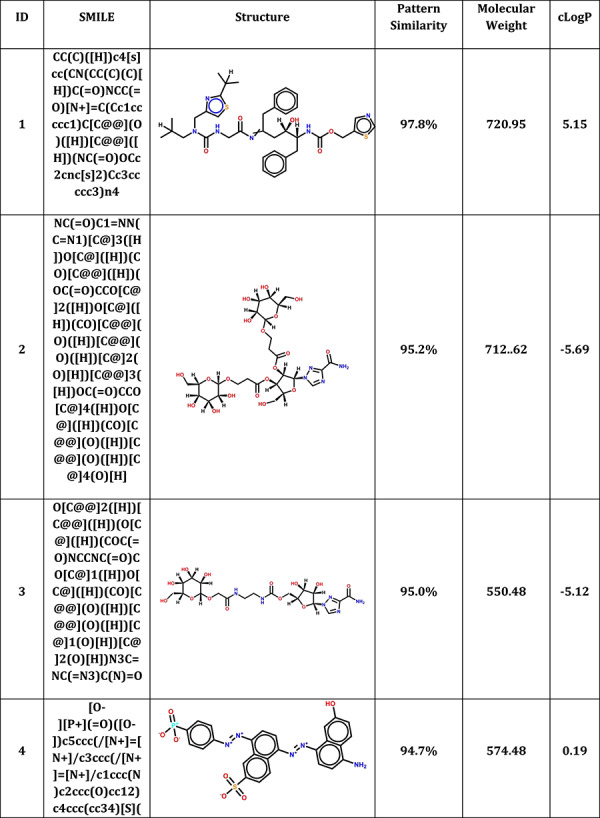
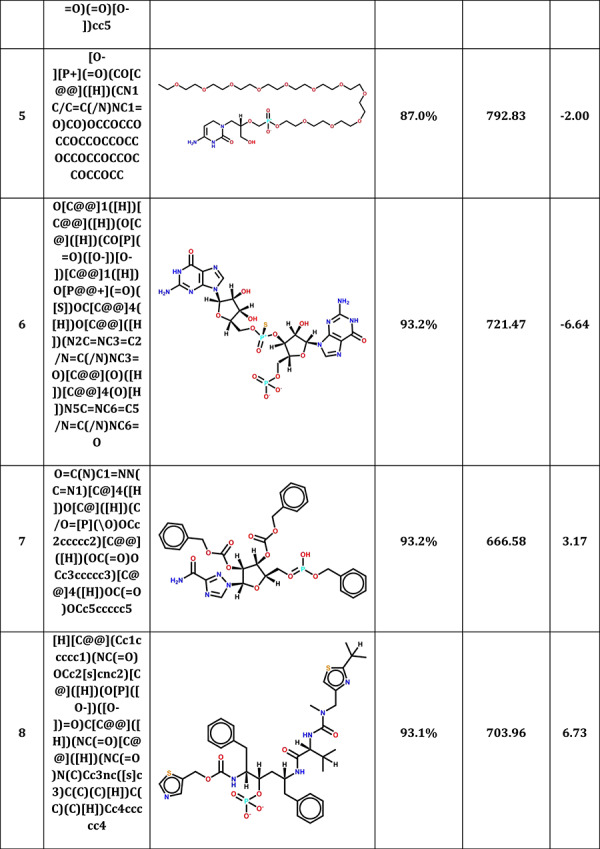
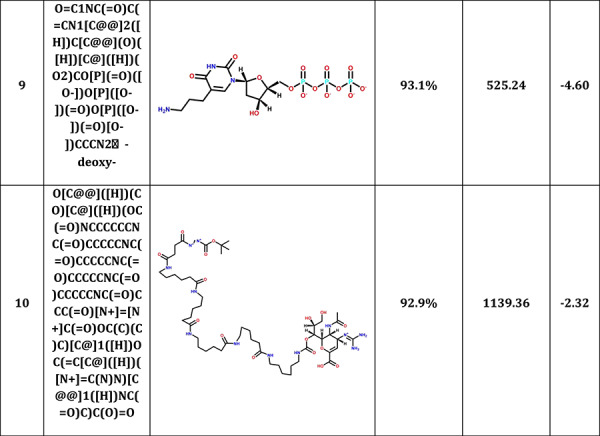
MCCS was also used to predict the potential candidates from CAS COVID-19 antiviral candidate compounds dataset (https://www.cas.org/covid-19-antiviral-compounds-dataset). Among 50 000 compounds, 44 665 small molecules were successfully docked into 3CLPro. Table 3 shows top 10 antiviral candidates may bind to 3CLPro with therapeutic potential for COVID-19. These drugs also shared a high pattern similarity with a reported N3 inhibitor in the co-crystal complex, especially the top compound (2,7,10,12-Tetraazapentadecanoic acid, 4-hydroxy-14-methyl-12-[[2-(1-methylethyl)-4-thiazolyl]methyl]-8,11-dioxo-3,6-bis(phenylmethyl)-, 5-thiazolylmethyl ester, (3S,4S,6S)-) which shared up to 97.8% pattern similarity with N3. The planning experiments will be carried out for further validations.
Table 3.
Top 10 CAS antiviral candidate compounds with therapeutic potential for COVID-19
|
|
|
Moreover, we applied our method to explore the details of other reported SARS-CoV-2 inhibitors, including ebselen and tideglusib from the work of Jin et al. [18] (Nature volume 582, pages289–293(2020)). Based on the detailed docking results and residue energy contribution from Figure S4, we found that several key residues in 3CLPro in SARS-CoV-2 contributed to the binding of these two ligands, including Cys145, His163, Leu141, Met49, Phe140, Asn142, His164, His41, Met165, Gln189 and Glu166. Importantly, we found that their inhibitory activities against COVID-19 may mainly attribute to a potential covalent bond formation with the thiol group of the Cys145, as shown in Figure S4a and S4b. Our results are consistent with the finding from Jin’s work [18].
Virus-CKB to accelerate drug combinational therapy for COVID-19 treatment
Recently, COVID-19 patients who met the criteria for hospital discharge or discontinuation of quarantine show positive reverse transcriptase polymerase chain reaction test results even after 5 to 13 days after recovery. This suggests that at least a proportion of recovered patients may still be virus carriers [55]. In addition, co-existing diseases such as diabetes, hypertension and cardiovascular disease are found in about one-third to one-half of reported COVID-19 patients and tend to worsen the patients’ prognosis [56]. These findings indicate that COVID-19 is a complex disease involving simultaneous production of signals from a multitude of transduction pathways. Therefore, a traditional single-target drug, though it may be highly selective and potent, may not be sufficient to effectively treat and cure COVID-19. An alternative strategy is to seek simultaneous modulation at multiple nodes in the network of virus signaling pathways through either a multi-target drug or drug–drug combinations.
On the basis of our established domain specific chemogenomics databases [32, 35] and our novel computational techniques, we constructed a virus-associated disease-specific chemogenomics knowledgebase (Virus-CKB, https://www.cbligand.org/g/virus-ckb) [27] and applied our computational systems pharmacology-target mapping (CSP-Target Mapping) to accelerate drug combinational therapy to meet the urgent demand due to the COVID-19 outbreak.
To first validate our Virus-CKB, we predicted potential FDA-approved drugs that may bind to 3CLpro. As shown in Figure 5, our results showed that eltrombopag (for thrombocytopenia), anidulafungin (for fungal infections), imatinib (for cancer) and proscillaridin (anti-cardiovascular) were predicted to bind to 3CLpro (Node: PR_SARS2) of SARS-CoV-2. Our findings is supported by a recent study, which eltrombopag, anidulafungin, imatinib and proscillaridin exhibited antiviral efficacy (0.1 μM < IC50 < 10 μM) against SARS-CoV-2 [57, 58].
Figure 5.

Spider Plot for data virtualization and analysis for four antiviral drugs. The average docking scores are displayed as connection labels and the protein targets on which the query compound is active are displayed as circular discs.
Then, combinations of drugs with therapeutic potential for the treatment of COVID-19 are identified by our Virus-CKB and systems pharmacology-mapping program. As shown in Figure 6, our Virus-CKB platform reveals that one green node, representing a drug target NA_INFB (Neuraminidase, Influenza B virus), connects to an antiflu drug oseltamivir with green solid lines, which is congruent with the fact that oseltamivir shows high biological activity at its known target NA_INFB. Moreover, bictegravir (anti-HIV drug) and paritaprevir (anti-HCV drug) are also identified through Virus-CKB data analysis and computing, showing that these two drugs bind to a target of PR_SARS2, which is3CLpro of SARS-CoV-2, as highlighted by the yellow color node in Figure 6. Thus, a plausible combinational therapy is made by combining oseltamivir (antiflu medication) with bictegravir (anti-HIV drug) and paritaprevir (anti-HCV drug) as they are targeting NA_INFB and PR_SARS2, respectively. Such combination will create a drug synergy in clinical treatment. As proof-of-evidence, some of the predictions are congruent with ongoing clinical trials registered at ClinicalTrials.gov, such as ASC09F + Oseltamivir and Ritonavir+Oseltamivir (https://clinicaltrials.gov/ct2/results?cond=2019nCoV&term=&cntry=&state=&city=&dist).
Figure 6.
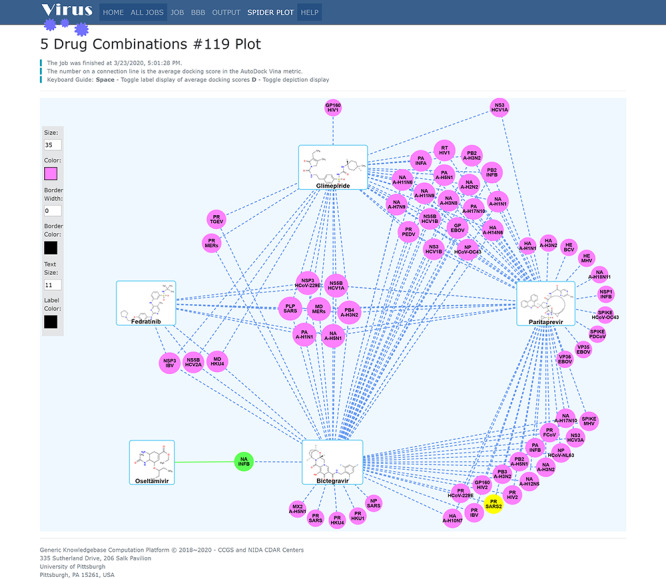
The predicted drug combinations for COVID-19 by CSP-Target Mapping that was implemented in our Virus-CKB. The green circles and solid lines represented the known targets and the interacted drugs, while the purple circles and dash lines represent the predicted targets and interaction, https://www.cbligand.org/g/virus-ckb.
Conclusion
To overcome the limitation of structure-based virtual screening, we here applied our new in silico recognition pattern-based approach for repurposing FDA approved drugs, combining potential drug candidates, or designing new chemical candidates. We identified four anti-HIV/antiviral drugs, two antiflu drugs, one anti-HCV drug and three antidiabetic drugs for potential treatments of COVID-19. Moreover, by using our virus chemogenomics knowledgebase and CSP-Target Mapping tools, we also proposed that combinations of different types of drug candidates may be useful for the treatment of SARS-CoV-2 infection. Of course, drugs identified for repurposing and/or in combination using in silico approaches will require in vitro target validation experiments as well as human clinical studies, which are currently underway via collaborations. To achieve this goal, bioassay experiments are under way for anti-2019-nCoV testing to identify the approved drugs or chemical agents with therapeutic potential for COVID-19. Biotesting is performed using: (i) the viral cytopathic effect (CPE) inhibition, (ii) virus yield reduction (VYR) assays and (iii) toxicity assay.
Key Points
MCCS is a scoring-function based method for characterizing the recognition pattern between protein and ligand to aid in the rational drug design.
MCCS distinguishes itself from molecular dynamics (MD) simulation-based energy decomposition in its ability to generate the residue energy contribution and the binding recognition feature with reduced time-consumption and high accuracy.
Recognition pattern-based method in MCCS protocol takes the importance of key residues into consideration, which helps to improve the accuracy of structure-based virtual screening.
The use of MCCS and Virus-CKB can facilitate rapid drug development to meet the urgent demand of the COVID-19 outbreak.
Supplementary Material
Acknowledgement
We thank Professor Zihe Rao and his group from Shanghai Institute for Advanced Immunochemical Studies (SIAIS) for the crystal structure of 3CLPro (PDB ID: 6 LU7).
Zhiwei Feng is currently an assistant professor in the School of Pharmacy, University of Pittsburgh. His research interests include (i) the development of algorithms/tools/apps for drug discovery, (ii) chemogenomic knowledgebase design and novel tool development for drug design of small molecules and modulators and (iii) big-data or clinical data analysis and pharmacometrics and systems pharmacology.
Maozi Chen is currently conducting research work in the School of Pharmacy, University of Pittsburgh. His research interests are algorithm design and software development.
Ying Xue is currently an assistant professor in the School of Pharmacy, University of Pittsburgh. Her research interests include clinical pharmacy, outcomes research and systems pharmacology.
Tianjian Liang is currently a second year’s master student in the School of Pharmacy, University of Pittsburgh. His research interests are antibody drug design and systems pharmacology analysis.
Hui Chen is currently a P3 PharmD student in the School of Pharmacy, University of Pittsburgh. Her research mainly focuses on the computer-aided vaccine design.
Yuehan Zhou is currently a P2 PharmD student in the School of Pharmacy, University of Pittsburgh. His research mainly focuses on the computer-aided drug discovery of COVID-19.
Thomas D. Nolin is currently an associate professor in the School of Pharmacy, University of Pittsburgh. His research focuses on developing an understanding of the impact of kidney disease and renal replacement therapy (RRT) on nonrenal metabolism and transport pathways and corresponding effects on drug exposure and response.
Randall B. Smith is a professor and Senior Associate Dean at the University of Pittsburgh School of Pharmacy. He has extensive experience in research and the management of research for drug development and demonstration of the value of pharmaceutical products. Smith’s current research interests are in the development and application of novel systems of health care delivery and education of health professionals.
Xiang-Qun Xie received his PhD degree in 1993 from the University of Connecticut. He is an Associate Dean of the School of Pharmacy, a professor of Pharmaceutical Sciences and Director/PI of NIH Center of Excellence for Computational Drug Abuse Research and Computational Chemogenomics Screening Center at the University of Pittsburgh. He serves as a Charter Member of the US FDA Science Advisory Board. He is Director of Pharmacometrics & System Pharmacology (PSP) Graduate Program for preclinical and clinical education & research. Xie is known for his pioneering research for the development of renowned ‘Big-Data to Knowledge’ diseases-specific chemogenomics knowledgebases platform implemented with GPU-accelerated machine-/deep-learning computational TargetHunter system pharmacology for translational medicinal chem-biology drug discovery.
Contributor Information
Zhiwei Feng, University of Pittsburgh.
Maozi Chen, University of Pittsburgh.
Ying Xue, University of Pittsburgh.
Tianjian Liang, University of Pittsburgh.
Hui Chen, University of Pittsburgh.
Yuehan Zhou, University of Pittsburgh.
Thomas D Nolin, University of Pittsburgh.
Randall B Smith, University of Pittsburgh.
Xiang-Qun Xie, University of Pittsburgh.
Funding
Authors would like to acknowledge the funding support to Xie laboratory from the National Institutes of Health, National Institute on Drug Abuse (P30 DA035778A1).
Authors’ Contributions
Z.F. and X.-Q.X. designed the project. M.C. wrote the code. Z.F. and M.C. prepared the figures and wrote the manuscript. All authors read, edited and approved the final manuscript.
Conflict of interest
The authors declare no conflict of interest.
Code and Data Availability
MCCS is open source under Apache License 2.0 and is freely available on GitHub at https://github.com/stcmz/jdock/ and https://github.com/stcmz/mccsx. The example data is also available at https://github.com/stcmz/mccs-bib-examples.
REFERENCES
- 1. Wu JT, Leung K, Leung GM. Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: a modelling study. The Lancet 2020;395:689–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. The Lancet 2020;395:565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 2020;382:727–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The Lancet 2020;395:497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhou P, Yang X-L, Wang X-G, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020;579:270–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature 2020;579:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sullivan T. A tough road: cost to develop one new drug is $2.6 billion; approval rate for drugs entering clinical development is less than 12%. Policy & Medicine 2019. https://www.policymed.com/2014/12/a-tough-road-cost-to-develop-one-new-drug-is-2.6-billion-approval-rate-for-drugs-entering-clinical-de.html (23 August 2019, date last accessed). [Google Scholar]
- 8. Cao B, Wang Y, Wen D, et al. A trial of lopinavir-ritonavir in adults hospitalized with severe Covid-19. N Engl J Med 2020;382:1787–99 NEJMoa2001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. French Drug-Surveillance Specialist Highlights Hydroxychloroquine Risks. https://www.wsj.com/articles/french-drug-surveillance-specialist-highlights-hydroxychloroquine-risks-11586629800 (4 May 2020, date last accessed).
- 10. Grein J, Ohmagari N, Shin D, et al. Compassionate use of remdesivir for patients with severe Covid-19. N Engl J Med 2020;382:2327–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu A, Peng Y, Huang B, et al. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe 2020;27:325–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ralph R, Lew J, Zeng T, et al. 2019-nCoV (Wuhan virus), a novel coronavirus: human-to-human transmission, travel-related cases, and vaccine readiness. J Infect Dev Ctries 2020;14:3–17. [DOI] [PubMed] [Google Scholar]
- 13. Lai CC, Shih TP, Ko WC, et al. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): the epidemic and the challenges. Int J Antimicrob Agents 2020;55:105924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chowell G, Abdirizak F, Lee S, et al. Transmission characteristics of MERS and SARS in the healthcare setting: a comparative study. BMC Med 2015;13:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gao Y, Yan L, Huang Y, et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020;eabb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Littler DR, Gully BS, Colson RN, et al. Crystal structure of the SARS-CoV-2 non-structural protein 9, Nsp9. iScience 2020;23:101258 2020.2003.2028.013920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lan J, Ge J, Yu J, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020;581:215–20. [DOI] [PubMed] [Google Scholar]
- 18. Jin Z, Du X, Xu Y, et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 2020;582:289–93. [DOI] [PubMed] [Google Scholar]
- 19. Wrapp D, Wang N, Corbett KS, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020;367:1260–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hoffmann M, Kleine-Weber H, Krüger N, et al. The novel coronavirus 2019 (2019-nCoV) uses the SARS-coronavirus receptor ACE2 and the cellular protease TMPRSS2 for entry into target cells. bioRxiv 2020.2001.2031.929042. [Google Scholar]
- 21. Wang J. Fast identification of possible drug treatment of coronavirus disease-19 (COVID-19) through computational drug repurposing study. J Chem Inf Model 2020;60:3277–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu S, Zheng Q, Wang Z. Potential covalent drugs targeting the main protease of the SARS-CoV-2 coronavirus. Bioinformatics (Oxford, England) 2020;36:3295–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou Y, Hou Y, Shen J, et al. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov 2020;6:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bai Q, Tan S, Xu T, et al. MolAICal: a soft tool for 3D drug design of protein targets by artificial intelligence and classical algorithm. Brief Bioinform 2020;1–12. https://academic.oup.com/bib/advance-article/doi/10.1093/bib/bbaa161/5890512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kong R, Yang G, Xue R, et al. COVID-19 Docking Server: a meta server for docking small molecules, peptides and antibodies against potential targets of COVID-19. Bioinformatics 2020;1–3. [DOI] [PMC free article] [PubMed]
- 26. Xing C, Zhuang Y, Xu TH, et al. Cryo-EM structure of the human cannabinoid receptor CB2-G(i) signaling complex. Cell 2020;180:645–654.e613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Feng Z, Chen M, Liang T, et al. Virus-CKB: an integrated bioinformatics platform and analysis resource for COVID-19 research. Brief Bioinform 2020;1–14. https://academic.oup.com/bib/advance-article/doi/10.1093/bib/bbaa155/5876604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010;31:455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li H, Leung K-S, Wong M-H. idock: A multithreaded virtual screening tool for flexible ligand docking In: 2012 IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB). San Diego, CA, USA: IEEE, 2012, 77–84. [Google Scholar]
- 30. Pedretti A, Villa L, Vistoli G. VEGA–an open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J Comput Aided Mol Des 2004;18:167–73. [DOI] [PubMed] [Google Scholar]
- 31. Olsson MH, Søndergaard CR, Rostkowski M, et al. PROPKA3: consistent treatment of internal and surface residues in empirical p K a predictions. J Chem Theory Comput 2011;7:525–37. [DOI] [PubMed] [Google Scholar]
- 32. Chen M, Jing Y, Wang L, et al. DAKB-GPCRs: an integrated computational platform for drug abuse related GPCRs. J Chem Inf Model 2019;59:1283–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xu X, Ma S, Feng Z, et al. Chemogenomics knowledgebase and systems pharmacology for hallucinogen target identification-salvinorin A as a case study. J Mol Graph Model 2016;70:284–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang H, Ma S, Feng Z, et al. Cardiovascular disease chemogenomics knowledgebase-guided target identification and drug synergy mechanism study of an herbal formula. Sci Rep 2016;6:33963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang Y, Wang L, Feng Z, et al. StemCellCKB: an integrated stem cell-specific chemogenomics knowledgeBase for target identification and systems-pharmacology research. J Chem Inf Model 2016;56:1995–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen Y, Feng Z, Shen M, et al. Insight into Ginkgo biloba L. extract on the improved spatial learning and memory by chemogenomics knowledgebase, molecular docking, molecular dynamics simulation, and bioassay validations. ACS omega 2020;5:2428–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cheng J, Wang S, Lin W, et al. Computational systems pharmacology-target mapping for fentanyl-laced cocaine overdose. ACS Chem Nerosci 2019;10:3486–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu H, Wang L, Lv M, et al. AlzPlatform: an Alzheimer's disease domain-specific chemogenomics knowledgebase for polypharmacology and target identification research. J Chem Inf Model 2014;54:1050–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang L, Ma C, Wipf P, et al. TargetHunter: an in silico target identification tool for predicting therapeutic potential of small organic molecules based on chemogenomic database. AAPS J 2013;15:395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Burley SK, Berman HM, Bhikadiya C, et al. RCSB Protein Data Bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res 2019;47:D464–d474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang Y, Zhang S, Li F, et al. Therapeutic target database 2020: enriched resource for facilitating research and early development of targeted therapeutics. Nucleic Acids Res 2020;48:D1031–d1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li YH, Yu CY, Li XX, et al. Therapeutic target database update 2018: enriched resource for facilitating bench-to-clinic research of targeted therapeutics. Nucleic Acids Res 2018;46:D1121–d1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rose AS, Bradley AR, Valasatava Y, et al. NGL viewer: web-based molecular graphics for large complexes. Bioinformatics (Oxford, England) 2018;34:3755–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pettersen EF, Goddard TD, Huang CC, et al. UCSF chimera--a visualization system for exploratory research and analysis. J Comput Chem 2004;25:1605–12. [DOI] [PubMed] [Google Scholar]
- 45. Turlington M, Chun A, Tomar S, et al. Discovery of N-(benzo[1,2,3]triazol-1-yl)-N-(benzyl)acetamido(phenyl) carboxamides as severe acute respiratory syndrome coronavirus (SARS-CoV) 3CLpro inhibitors: identification of ML300 and noncovalent nanomolar inhibitors with an induced-fit binding. Bioorg Med Chem Lett 2013;23:6172–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhu L, George S, Schmidt MF, et al. Peptide aldehyde inhibitors challenge the substrate specificity of the SARS-coronavirus main protease. Antiviral Res 2011;92:204–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang H, Xie W, Xue X, et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol 2005;3:e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Goetz D, Choe Y, Hansell E, et al. Substrate specificity profiling and identification of a new class of inhibitor for the major protease of the SARS coronavirus. Biochemistry 2007;46:8744–52. [DOI] [PubMed] [Google Scholar]
- 49. Jacobs J, Grum-Tokars V, Zhou Y, et al. Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J Med Chem 2013;56:534–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xu Z, Wang Z, Wang S, et al. The impact of type 2 diabetes and its management on the prognosis of patients with severe COVID-19. J Diabetes 2020;1–10. https://onlinelibrary.wiley.com/doi/epdf/10.1111/1753-0407.13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Guo W, Li M, Dong Y, et al. Diabetes is a risk factor for the progression and prognosis of COVID-19. Diabetes Metab Res Rev 2020;e3319. doi: 10.1002/dmrr.3319. Epub ahead of print. PMID: 32233013; PMCID: PMC7228407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang D, Hu B, Hu C, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. JAMA 2020;323:1061–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang X, Bin B, Xu Z. et al., Network representation learning-based drug mechanism discovery and anti-inflammatory response against COVID-19. ChemRxiv. Preprint 2020. 10.26434/chemrxiv.12531314.v2. [DOI] [Google Scholar]
- 54. Paniri A, Akhavan-Niaki H. Emerging role of IL-6 and NLRP3 inflammasome as potential therapeutic targets to combat COVID-19: role of lncRNAs in cytokine storm modulation. Life Sci 2020;257:118114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lan L, Xu D, Ye G, et al. Positive RT-PCR test results in patients recovered from COVID-19. JAMA 2020;323:1502–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Guan WJ, Ni ZY, Hu Y, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 2020;382:1708–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jeon S, Ko M, Lee J, et al. Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. Antimicrob Agents Chemother 2020;64:e00819-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Weston S, Coleman CM, Sisk JM, et al. Broad anti-coronaviral activity of FDA approved drugs against SARS-CoV-2 in vitro and SARS-CoV in vivo. J Virol 2020:JVI.01218–20. doi: 10.1128/JVI.01218-20. Epub ahead of print. PMID: 32817221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
MCCS is open source under Apache License 2.0 and is freely available on GitHub at https://github.com/stcmz/jdock/ and https://github.com/stcmz/mccsx. The example data is also available at https://github.com/stcmz/mccs-bib-examples.