

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is accountable for the cause of coronavirus disease (COVID-19) that causes a major threat to humanity. As the spread of the virus is probably getting out of control on every day, the epidemic is now crossing the most dreadful phase. Idiopathic pulmonary fibrosis (IPF) is a risk factor for COVID-19 as patients with long-term lung injuries are more likely to suffer in the severity of the infection. Transcriptomic analyses of SARS-CoV-2 infection and IPF patients in lung epithelium cell datasets were selected to identify the synergistic effect of SARS-CoV-2 to IPF patients. Common genes were identified to find shared pathways and drug targets for IPF patients with COVID-19 infections. Using several enterprising Bioinformatics tools, protein–protein interactions (PPIs) network was designed. Hub genes and essential modules were detected based on the PPIs network. TF-genes and miRNA interaction with common differentially expressed genes and the activity of TFs are also identified. Functional analysis was performed using gene ontology terms and Kyoto Encyclopedia of Genes and Genomes pathway and found some shared associations that may cause the increased mortality of IPF patients for the SARS-CoV-2 infections. Drug molecules for the IPF were also suggested for the SARS-CoV-2 infections.
Keywords: SARS-CoV-2, idiopathic pulmonary fibrosis, differentially expressed genes, gene ontology, protein–protein interactions, hub gene, drug molecule
Introduction
The current world is going through a rough patch for the outbreak of coronavirus disease (COVID-19). Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the virus responsible for COVID-19. SARS-CoV-2 is a virus belonging to the Coronaviridae family [1]. Spike glycoproteins of coronavirus raise the entry of the virus into cells and ACE2 of the virus binds with human ACE2 [2]. Among the most serious risk factors of COVID-19, idiopathic pulmonary fibrosis (IPF) is considered to be the most vital one [3]. As viral infections can immensely enhance IPF risks so patients recovering from COVID-19 can face numerous complications because of having IPF [4, 5].
In late 2019, the COVID-19 case was first discovered in a city named Wuhan which is situated in China and at the end of 2019 World Health Organization (WHO) declared COVID-19 as a serious epidemic of 21st century [6]. The clustering result of the affected people in the early days of the pandemic was linked to the Wuhan seafood market and with the contact of wild animals [7]. In 2020, COVID-19 spread all over the world. As of 3 March 2020, the virus has already spread in most of the provinces in China including 80 151 numbers of confirmed cases and 10 566 numbers of confirmed cases in another 72 countries of the world [8]. Until 6 June 2020, the numbers of confirmed cases all over the world were 6 663 304 including 392 802 death (https://covid19.who.int/).Accordingto Worldometer, United States, Brazil, Russia, Spain, UK are among the top five countries where SARS-CoV-2 has spread the most. The first confirmed case in the United States was in January 2020 and the female patient visited China a few days before she had pneumonia and hospitalized and finally got herself SARS-CoV-2 positive [9]. From January 2020 to June 2020 United States had to witness a lethal face of COVID-19. Until 7 June 2020 according to WHO (https://covid19.who.int/), 1 886 794 cases were confirmed as COVID-19 positive including 109 038 deaths. A current study shows that Brazil has the highest transmission rate among all the countries of the world which makes Brazil a hotspot for COVID-19 [10, 11]. The first patient of COVID-19 was identified on 25 February 2020 who came back from Italy where the epidemic was ever so serious than other countries [12].
IPF is a chronic lung disease that causes serious decay of lung functionality [13]. Breathing complexity and dry cough are the primary symptoms of IPF [13]. A pathological study on IPF suggests that continuous lung injury might be one of the reasons of IPF [14]. IPF results in lung failure and respiratory complexities and the survival time range is between 3 and 5 years starting from diagnosis time [15]. The current study exhibits that SARS-CoV-2 contains S protein that has higher interaction for ACE2 and IPF patients contain a significant level of ACE2 that proved IPF as a risk factor for COVID-19 [16, 17]. These researches raise concerns about a number of interconnection between IPF and COVID-19.
In the field of biomedical research, high throughput methodologies are becoming significant and microarray data analysis is one of the most prominent techniques of high throughput methodologies that are used for analyzing gene expression in large-scale [18]. Microarray study simultaneously assists genetic researchers to study in terms of genetic expression [19]. Previous research demonstrates high throughput sequencing analysis for SARS-CoV which shows the prominent result in the assessment of data quality and gene expression [20]. Microarray data analysis for SARS-CoV-2 and risk factor IPF is not presented yet.
This study attempts to find biological pathways and the relationship between COVID-19 and IPF. Two datasets were selected for analysis of the research. GSE147507 was selected for SARS-CoV-2 infection in humans and GSE35145 was selected for IPF gene expression analysis. Both the datasets were collected from the Gene Expression Omnibus (GEO) database. The initial work was to identify differentially expressed genes (DEGs) for GSE147507 and GSE35145 and then find common DEGs for COVID-19 and IPF. The common DEGs are the prime data for the entire study. Based on the common genes, further analysis was accomplished including gene set enrichment analysis and pathway analysis to have an understanding of biological processes of genome-based expression studies. Identification of hub genes from common DEGs is the most essential work as finding drug molecules mostly depends on hub genes. To achieve this protein–protein interactions (PPIs) network is designed to gather hub genes from the PPIs network. The workflow of the present research is displayed in Figure 1.
Figure 1.
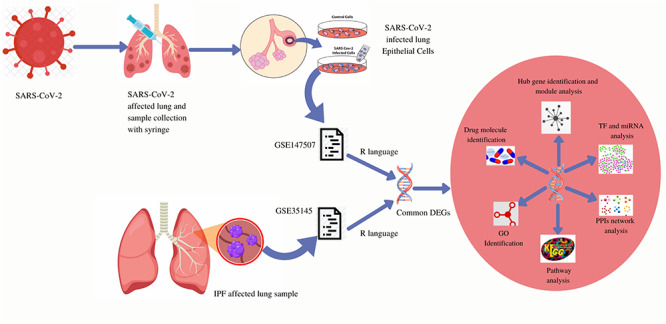
Methodical workflow for the current investigation. Two type of samples (control cells, SARS-CoV-2 infected cells) were collected from SARS-CoV-2 infected lung epithelial cells and both are included in the GSE147507 dataset. GSE147507 dataset contains a sample of SARS-CoV-2 infected lung epithelial cells and the GSE35145 dataset contains IPF affected lung samples. Common DEGs were identified from both the datasets using the R programming language. From the common DEGs, GO identification, KEGG pathway, PPIs network, TF and miRNA analysis, hub gene identification and module analysis was designed and based on those analysis drug molecule identification was performed.
Methodology
Collection of the dataset
Dataset (GSE147507) illustrates infections of SARS-CoV-2 in transcriptional responses and dataset (GSE35145) represents interchange of gene expression in IPF and both datasets were compiled from GEO database [21]. GEO database was introduced for gene expression analysis using high throughput methodology under the National Center for Biotechnology Information platform [22]. Illumina NextSeq 500 platform was used for the GSE147507 dataset for extracted RNA sequence analysis and the GPL10558 (Illumina HumanHT-12 V4.0 expression bead chip) platform is used for GSE35145 dataset. GSE147507 dataset was contributed by Blanco-Melo D et al. [23]. Dataset for IPF (GSE35145) was presented by Yan Y Sanders et al. [24]. COVID-19 dataset (GSE147507) provides samples including SARS-CoV-2 infection in lung epithelium and lung alveolar cells of humans. The IPF dataset (GSE35145) contains eight samples including genetic expression alteration in IPF cells and normal tissue of the lung which is more suitable datasets for this study. GSE35145 dataset is a subset of the GSE35147 dataset and it indicates DNA methylation profile. For our analysis we have selected GSE35145 dataset because of microarray-based analysis for IPF samples.
Identification of DEGs and common gene identification between COVID-19 and IPF
Identification of DEGs for GSE147507 and GSE35145 datasets is the primary task of the research. To identify DEGs for GSE147507, the limma package of R programming language is implemented. Data that are produced from microarray analysis is retrieved through DESeq2 [25] and limma package [26]. Cut-off criteria was obtained for GSE147507 using adjusted P-value < 0.05 and log2-fold change (absolute) > 1.0. DEGs for the GSE35145 dataset were analyzed through GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/) web tool which also uses limma package for identifying DEGs. Benjamini-Hochberg was applied for both the datasets for controlling of false discovery rate (FDR) [27]. The common gene identification between DEGs of GSE147507 and GSE35145 datasets was obtained using the R programming language.
Gene ontology and pathway finding in terms of Gene set enrichment analysis
Gene set enrichment analysis undertakes gene sets that have general biological functions and chromosomal locations [28]. For gene product annotation gene ontology (GO) term is used which is organized in three categories including biological process, molecular function and cellular component [29]. The principal reason for identifying GO terms is because of the understanding of molecular activity, cellular role and the location in a cell where the genes execute their functions. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway is usually used in the understanding of metabolic pathways and contains significant use over gene annotation [30]. In the purpose of significant pathway analysis WikiPathways [31], Reactome [32] and BioCarta databases were also used alongside the KEGG pathway. GO terms and all the pathways were obtained through web-based platform Enrichr (https://amp.pharm.mssm.edu/Enrichr/) for the common genes that were identified in the previous step. For experimented genome-wide genes Enrichr provides gene set enrichment analysis in web platforms [33].
Analysis of PPIs network
The activity of PPIs is considered to be the prime target of cellular biology study and works as a precondition for system biology [34]. Proteins perform their operation inside a cell with the interaction of another protein and information that is produced from a PPIs network raises perception about the function of the protein [35]. Common DEGs are inserted in Search Tool for the Retrieval of Interacting Genes (STRING) (https://string-db.org/) for generating PPI network. STRING delivers experimental and predicted interaction-based information and the interaction produced through the web tool is defined with 3D structures, accessory information and confidence score [36]. The confidence score was also used for the current PPIs network with a medium confidence score of 0.400. The confidence score was set using the STRING platform that is considered to be a medium confidence score. For a superior visual representation of the network and the purpose of identifying hub genes, the obtained PPIs are analyzed through Cytoscape (https://cytoscape.org/). Cytoscape software acts as the most powerful one when it comes to integration with larger databases of genetic interactions, protein–protein and protein-DNA interactions [37].
Identification of hub genes and module analysis
PPIs network contains a number of nodes and edges and represents their interactions and among the nodes that have the most interaction is considered to be a hub gene. PPIs network analysis for the current research is attained through Cytoscape. Hub genes for the corresponding PPIs network are pointed out by using cytoHubba (http://apps.cytoscape.org/apps/cytohubba) which is also a plugin of Cytoscape software. The interface of cytoHubba is user-friendly which makes it the most prominent among all the hub detection plugins of Cytoscape and it contains 11 methods for topological analysis [38]. The hub genes for the current research are revealed by using the degree topological algorithm. The reason behind choosing degree algorithm rather than any other algorithm is that degree algorithm indicates the number of interactions for each gene in the PPIs network and it also assists the research by suggesting mostly dense modules in the PPIs network. Hub genes create concentrated areas that can be detected as an essential module from the PPIs network. Molecular Complex Detection (MCODE) (http://apps.cytoscape.org/apps/mcode) plugin of Cytoscape software is used to detect the most profound modules from the PPIs network. Highly interconnected portions are identified through MCODE clustering that assists the research in effective drug designing. For representing molecular complexes in the PPIs network MCODE is used by detecting the densely connected areas [39].
TF-gene interactions
TF-gene gene interaction with the identified common DEGs evaluates the outcome of TF on functional pathways and expression levels of the genes [40]. NetworkAnalyst (https://www.networkanalyst.ca/) platform is used to identify TF-gene interaction with identified common genes. NetworkAnalyst is a comprehensive web platform for performing gene expression for numerous species and also enables them to perform meta-analysis [41]. The network produced for the TF-gene interaction network is obtained from the ENCODE (https://www.encodeproject.org/) database which is included in the NetworkAnalyst platform.
TF-miRNA coregulatory network
Interactions for TF-miRNA coregulatory were collected from the RegNetwork repository [42] which assists to detect miRNAs and regulatory TFs that regulate DEGs of interest at the post-transcriptional and transcriptional level. TF-miRNA coregulatory network was visualized using NetworkAnalyst. NetworkAnalyst assists researchers in the easiest way to navigate complex datasets to identify biological features and functions which leads to effective biological hypothesis [43].
Identification of candidate drugs
Drug molecule identification is the key component of the ongoing research. Based on the common DEGs for COVID-19 and IPF diseases drug molecule is designed using the Drug Signatures database (DSigDB), which consists of 22 527 gene sets. The access of the DSigDB database is acquired through Enrichr (https://amp.pharm.mssm.edu/Enrichr/) platform. Enrichr is mostly used as an enrichment analysis platform that represents numerous visualization details on collective functions for the genes that are provided as input [44].
Results
Identification of DEGs and common gene identification between COVID-19 and IPF
GSE147507 dataset is used for the identification purpose of DEGs for COVID-19. One hundred and eight DEGs were obtained including 93 upregulated and 15 downregulated genes. For IPF dataset GSE35145 is used and a total of 359 DEGs were identified and among them 159 genes were upregulated and 200 genes were downregulated. Collected 108 genes for COVID-19 and 359 genes for IPF were compared using R programming language and identified 11 (SAA2, MMP9, SAA1, S100A8, ICAM1, PI3, SOD2, C8orf4, SERPINA3, S100A12, S100A9) common DEGs. The comparing of common DEGs between two datasets is visualized through a Venn diagram in Figure 2. The results of the Venn diagram exhibit that the common DEGs are 2.4% among total 467 DEGs.
Figure 2.
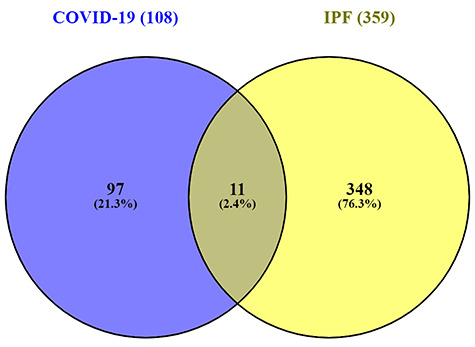
Common differentially expressed genes representation through a Venn diagram. Eleven genes were found common from the 108 differentially expressed genes of SARS-CoV-2 infection and 359 differentially expressed genes of IPF patients. The common differentially expressed genes were 2.4% among total 467 differentially expressed genes.
GO and pathway finding in terms of gene set enrichment analysis
Enrichr web tool was used for the analysis of gene set enrichment analysis. The current study analyzes GO terms and KEGG pathway for 11(SAA2, MMP9, SAA1, S100A8, ICAM1, PI3, SOD2, C8orf4, SERPINA3, S100A12, S100A9) common DEGs. The three most eminent GO terms include biological process, molecular functions and cellular component. The ongoing study illustrates the top 10 GO terms for each of the subsections (biological process, molecular functions and cellular component), which is presented in Table 1. The data in Table 1 justify that the common DEGs are highly enhanced in neutrophil chemotaxis and granulocyte chemotaxis for the biological process subsection. Molecular function subsection data indicate a transition metal ion binding factor splendidly involved in the common DEGs. Cellular component study exhibits significant involvement of cytoplasmic vesicle lumen factors in common DEGs. KEGG, WikiPathways, Reactome and BioCarta pathway analysis is produced in Table 2. The information attained from Table 2 shows the IL-17 signaling pathway and TNF signaling pathway interaction with the most number of genes according to the KEGG pathway database. A collection of GO terms and pathways according to the combined score is depicted in Figure 3(A and B). A combined score is performed by the Enrichr web tool, which depends on the log of the P-value and z-score. Figures 3(A and B) represents GO terms and pathway analysis results from various pathway databases, respectively.
Table 1.
GO terms, GO pathways and their corresponding P-values and genes for common differentially expressed genes
| Category | GO ID | GO pathways | P-values | Genes |
|---|---|---|---|---|
| GO biological process | GO:0070486 | Leukocyte aggregation | 0.000005766 | S100A9, S100A8 |
| GO:0050832 | Defense response to fungus | 8.380e-8 | S100A12, S100A9, S100A8 | |
| GO:0030593 | Neutrophil chemotaxis | 1.430e-8 | SAA1, S100A12, S100A9, S100A8 | |
| GO:0071621 | Granulocyte chemotaxis | 1.792e-8 | SAA1, S100A12, S100A9, S100A8 | |
| GO:1990266 | Neutrophil migration | 2.069e-8 | SAA1, S100A12, S100A9, S100A8 | |
| GO:1900122 | Positive regulation of receptor binding | 0.003296 | MMP9 | |
| GO:0002693 | Positive regulation of cellular extravasation | 0.003296 | ICAM1 | |
| GO:0032364 | Oxygen homeostasis | 0.003296 | SOD2 | |
| GO:0003085 | Negative regulation of systemic arterial blood pressure | 0.003296 | SOD2 | |
| GO:0051549 | Positive regulation of keratinocyte migration | 0.003296 | MMP9 | |
| GO Molecular Function | GO:0050786 | RAGE receptor binding | 1.038e-8 | S100A12, S100A9, S100A8 |
| GO:0035325 | Toll-like receptor binding | 0.000009879 | S100A9, S100A8 | |
| GO:0046914 | Transition metal ion binding | 0.000001290 | S100A12, SOD2, MMP9, S100A9, S100A8 | |
| GO:0008270 | Zinc ion binding | 0.00001547 | S100A12, MMP9, S100A9, S100A8 | |
| GO:0004866 | Endopeptidase inhibitor activity | 0.001625 | SERPINA3, PI3 | |
| GO:0030145 | Manganese ion binding | 0.01909 | SOD2 | |
| GO:0030414 | Peptidase inhibitor activity | 0.01963 | PI3 | |
| GO:0061135 | Endopeptidase regulator activity | 0.01963 | PI3 | |
| GO:0005507 | Copper ion binding | 0.02233 | S100A12 | |
| GO:0046872 | Metal ion binding | 0.00006865 | S100A12, SOD2, S100A9, S100A8 | |
| GO Cellular Component | GO:0060205 | Cytoplasmic vesicle lumen | 4.624e-9 | SERPINA3, SAA1, S100A12, S100A9, S100A8 |
| GO:0071682 | Endocytic vesicle lumen | 0.009858 | SAA1 | |
| GO:0034774 | Secretory granule lumen | 0.00001872 | SERPINA3, S100A12, S100A9, S100A8 | |
| GO:0005881 | Cytoplasmic microtubule | 0.02071 | SAA1 | |
| GO:1904724 | Tertiary granule lumen | 0.02984 | MMP9 | |
| GO:0031093 | Platelet alpha granule lumen | 0.03625 | SERPINA3 | |
| GO:0045111 | Intermediate filament cytoskeleton | 0.03837 | S100A8 | |
| GO:0005856 | Cytoskeleton | 0.002467 | S100A12, S100A9, S100A8 | |
| GO:0031091 | Platelet alpha granule | 0.04841 | SERPINA3 | |
| GO:0035578 | Azurophil granule lumen | 0.04841 | SERPINA3 |
Table 2.
Top pathways from KEGG, WikiPathways, Reactome and BioCarta databases and their corresponding P-values and genes for common differentially expressed genes
| Databases | Pathways | P-value | Genes |
|---|---|---|---|
| KEGG | IL-17 signaling pathway | 0.00001563 | MMP9, S100A9, S100A8 |
| TNF signaling pathway | 0.001596 | MMP9, ICAM1 | |
| Leukocyte transendothelial migration | 0.001654 | MMP9, ICAM1 | |
| African trypanosomiasis | 0.02017 | ICAM1 | |
| Bladder cancer | 0.02233 | MMP9 | |
| Fluid shear stress and atherosclerosis | 0.002531 | MMP9, ICAM1 | |
| Malaria | 0.02663 | ICAM1 | |
| Viral myocarditis | 0.03198 | ICAM1 | |
| Staphylococcus aureus infection | 0.03678 | ICAM1 | |
| Peroxisome | 0.04473 | SOD2 | |
| WikiPathways | Vitamin B12 Metabolism WP1533 | 3.630e-11 | SERPINA3, SAA1, SAA2, SOD2, ICAM1 |
| IL1 and megakaryocytes in obesity WP2865 | 2.489e-7 | MMP9, S100A9, ICAM1 | |
| Folate Metabolism WP176 | 1.525e-10 | SERPINA3, SAA1, SAA2, SOD2, ICAM1 | |
| Selenium Micronutrient Network WP15 | 5.914e-10 | SERPINA3, SAA1, SAA2, SOD2, ICAM1 | |
| Mammary gland development pathway - Involution (Stage 4 of 4) WP2815 | 0.005488 | MMP9 | |
| Photodynamic therapy-induced NF-kB survival signaling WP3617 | 0.0001620 | MMP9, ICAM1 | |
| Osteopontin Signaling WP1434 | 0.007129 | MMP9 | |
| Platelet-mediated interactions with vascular and circulating cells WP4462 | 0.009313 | ICAM1 | |
| Cells and Molecules involved in local acute inflammatory response WP4493 | 0.009313 | ICAM1 | |
| Extracellular vesicles in the crosstalk of cardiac cells WP4300 | 0.01040 | MMP9 | |
| Reactome | DEx/H-box helicases activate type I IFN and inflammatory cytokines production Homo sapiens R-HSA-3134963 | 0.00002138 | SAA1, S100A12 |
| Advanced glycosylation endproduct receptor signaling Homo sapiens R-HSA-879415 | 0.00002138 | SAA1, S100A12 | |
| Scavenging by Class B Receptors Homo sapiens R-HSA-3000471 | 0.002747 | SAA1 | |
| RIP-mediated NFkB activation via ZBP1 Homo sapiens R-HSA-1810476 | 0.00005742 | SAA1, S100A12 | |
| TRAF6 mediated NF-kB activation Homo sapiens R-HSA-933542 | 0.00007540 | SAA1, S100A12 | |
| ZBP1(DAI) mediated induction of type I IFNs Homo sapiens R-HSA-1606322 | 0.00008873 | SAA1, S100A12 | |
| TAK1 activates NFkB by phosphorylation and activation of IKKs complex Homo sapiens R-HSA-445989 | 0.00008873 | SAA1, S100A12 | |
| Formyl peptide receptors bind formyl peptides and many other ligands Homo sapiens R-HSA-444473 | 0.004392 | SAA1 | |
| Cytosolic sensors of pathogen-associated DNA Homo sapiens R-HSA-1834949 | 0.0005787 | SAA1, S100A12 | |
| TRAF6 Mediated Induction of proinflammatory cytokines Homo sapiens R-HSA-168180 | 0.0006883 | SAA1, S100A12 | |
| BioCarta | Inhibition of Matrix Metalloproteinases Homo sapiens h reckPathway | 0.004392 | MMP9 |
| Cardiac Protection Against ROS Homo sapiens h flumazenilPathway | 0.006035 | SOD2 | |
| Erythropoietin mediated neuroprotection through NF-kB Homo sapiens h eponfkbPathway | 0.007129 | SOD2 | |
| The IGF-1 Receptor and Longevity Homo sapiens h longevity pathway | 0.008767 | SOD2 |
Figure 3.
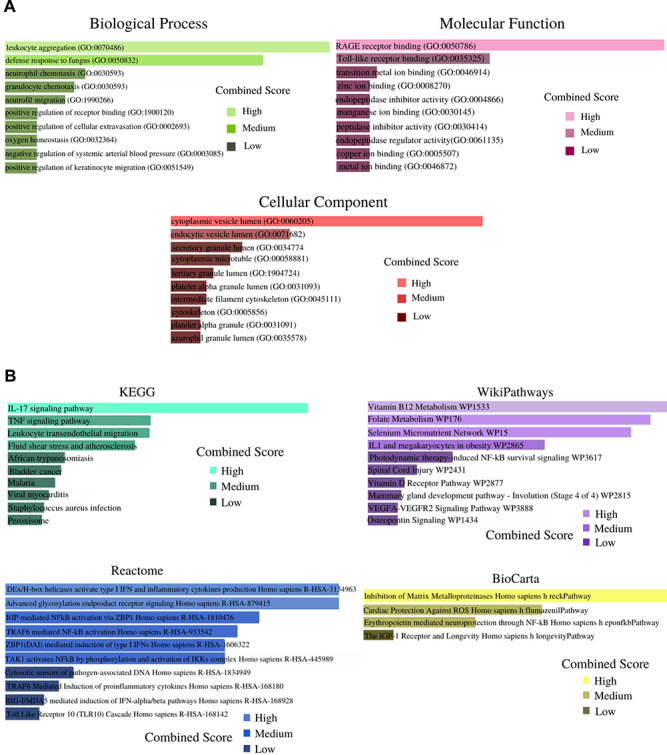
(A) Biological process, molecular function and cellular component related GO terms identification result according to combined score. The higher the enrichment score, the higher number of genes are involved in a certain ontology. (B) Pathway analysis result identification through KEGG, WikiPathways, Reactome and BioCarta. The results of the pathway terms were identified through the combined score.
PPIs network to identify hub genes and module analysis
The common DEGs were provided as an input in STRING and the file produced from the analysis is reintroduced into Cytoscape software for visual representation. The PPIs network is created for further analysis of this study including hub gene detection for identifying drug molecules for COVID-19 and IPF. Eventually the results of the PPIs network connect for suggesting drug compounds that establish the PPIs analysis as a center point of this research. The PPIs network contains 60 nodes and 403 edges, which is picturized in Figure 4.
Figure 4.
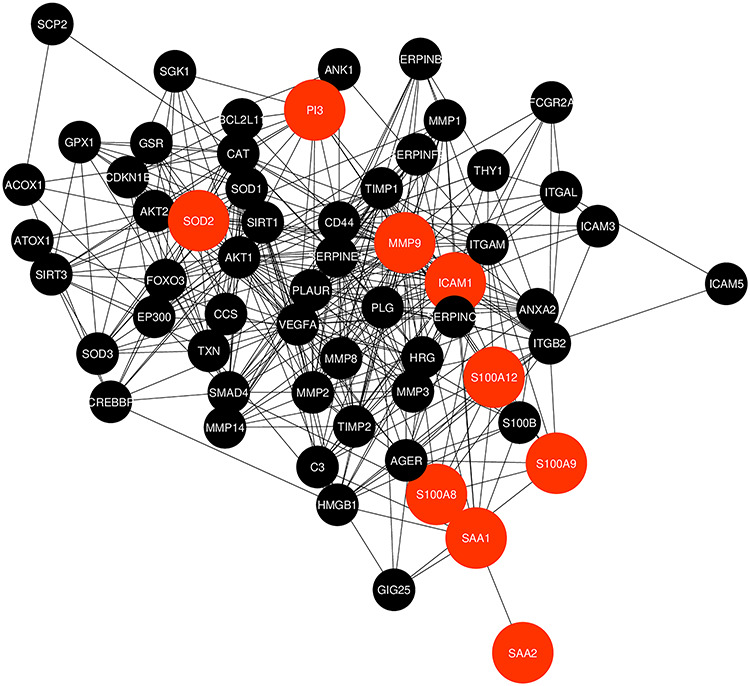
Protein–protein interactions (PPIs) network for identified common differentially expressed genes that are shared by two diseases (COVID-19 and IPF). Nodes in orange color indicate common differentially expressed genes and edges specify the interconnection in the middle of two genes. The analyzed network holds 60 nodes and 403 edges.
Identification of hub genes and module analysis for suggesting therapeutic solutions
To trace the hub genes from the PPIs network which is highlighted in Figure 3, cytohubba is used which is a plugin of Cytoscape software. The hub genes were sorted by their degree value, which indicates the number of interactions of the genes in the PPIs network. Top five identified hub genes are VEGFA, AKT1, MMP9, ICAM1 and CD44. Hub protein interactions with other protein in the PPIs network are demonstrated in Figure 5. The network consists of 53 nodes and 378 edges. Highly dense modules are designed from the PPIs network using MCODE which is also a plugin of Cytoscape software. MMP9 and ICAM1 are the two genes which are highlighted in the module network as these two genes are also the common DEGs between the two datasets. Module analysis is shown in Figure 6. The module analysis network contains 16 nodes and 106 edges. Topological analysis for the hub genes (VEGFA, AKT1, MMP9, ICAM1 and CD44) is identified using cytohubba. The topological analysis result is presented in Table 3.
Figure 5.
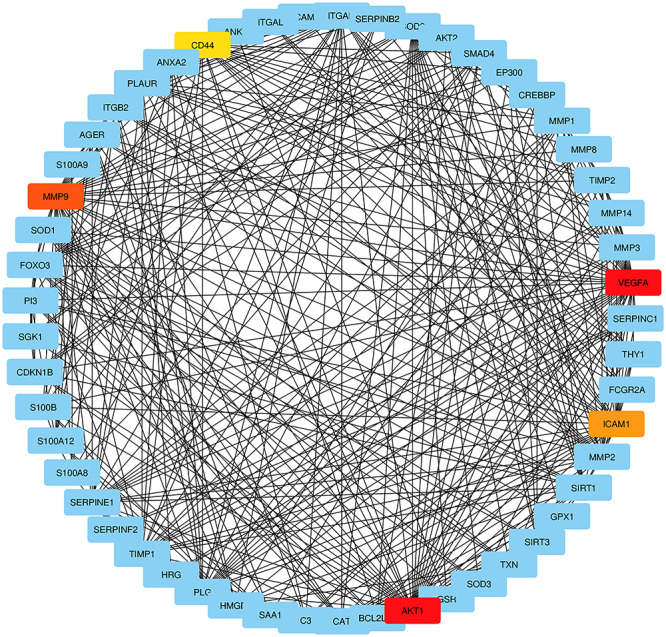
Detection of hub genes from the PPIs network of common differentially expressed genes. The highlighted five genes are VEGFA, AKT1, MMP9, ICAM1 and CD44. These five genes are considered as hub genes according to their degree value. The network has 53 nodes and 378 edges. According to topological analysis, the degree value of VEGFA and AKT1 was 38. The degree value of MMP9, ICAM1 and CD44 were 34, 29 and 27, respectively.
Figure 6.
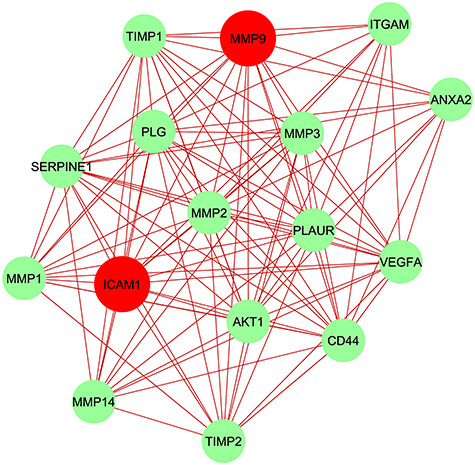
Module analysis network obtained from Figure 4 PPIs network. ICAM1 and MMP9 are highlighted in red color as these two hub nodes are common between GSE147507 and GSE35145. The network represents highly interconnected regions of the PPIs network. The network holds 16 nodes and 106 edges.
Table 3.
Topological result exploration for top five hub genes where the network density is 0.274, network diameter 3 and network radius 2
| Hub gene | Degree | Stress | Closeness centrality | Betweenness centrality | Distance | Eccentricity | Edge betweenness | Transitivity |
|---|---|---|---|---|---|---|---|---|
| VEGFA | 38 | 2248 | 48.1667 | 362.6209 | 1.269230 | 2 | 0.121695 | 0.34424 |
| AKT1 | 38 | 2502 | 48.1667 | 396.249 | 1.269230 | 2 | 0.133888 | 0.35277 |
| MMP9 | 34 | 2126 | 46.5 | 343.5242 | 1.346153 | 2 | 0.095339 | 0.38324 |
| ICAM1 | 29 | 1594 | 44 | 242.5716 | 1.442307 | 2 | 0.064545 | 0.42365 |
| CD44 | 27 | 1060 | 42.3333 | 189.3317 | 1.480769 | 2 | 0.064571 | 0.4359 |
TF-gene interactions
TF-gene interactions were collected using NetworkAnalyst. For the common DEGs (SAA2, MMP9, SAA1, S100A8, ICAM1, PI3, SOD2, C8orf4, SERPINA3, S100A12, S100A9) the TF-genes were identified. TF regulators’ interaction with the common DEGs is visualized in Figure 7. The network contains 142 nodes and 180 edges. The network contains a total of 132 TF-genes. MMP9 is regulated by 22 TF-genes and ICAM1 is regulated by 69 TF-genes. These 132 TF-genes regulate more than one common DEGs of the network, which indicates high interaction of the TF-genes with common DEGs. Figure 7 represents the TF-gene interaction network.
Figure 7.
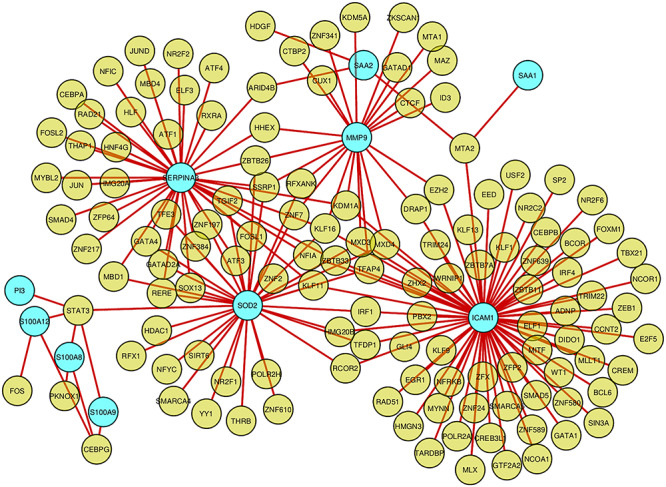
Network for TF-gene interaction with common differentially expressed genes. The highlighted blue color node represents the common genes and other nodes represent TF-genes. The network consists of 142 nodes and 180 edges.
TF-miRNA coregulatory network
TF-miRNA coregulatory network is generated using NetworkAnalyst. The analysis of the TF-miRNA coregulatory network delivers miRNAs and TFs interaction with the common DEGs. This interaction can be the reason for regulating the expression of the DEGs. The network created for TF-miRNA coregulatory network comprises 101 nodes and 131 edges. Thirty-nine miRNAs and 53 TF-genes have interacted with the common DEGs. Figure 8 dispenses TF-miRNA coregulatory network.
Figure 8.
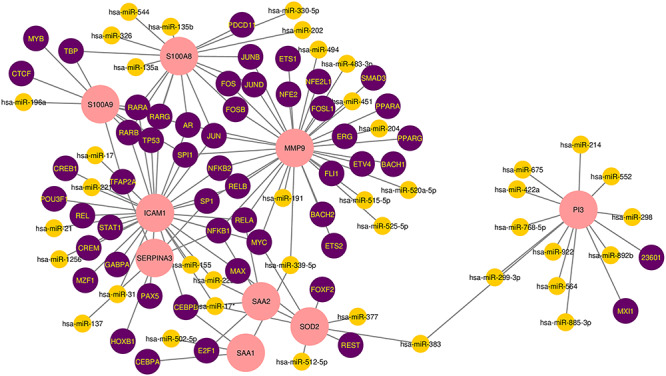
The network presents the TF-miRNA coregulatory network. The network consists of 101 nodes and 131 edges including 53 TF-genes, 39 miRNA and nine differentially expressed genes. The nodes in pink color are the differentially expressed genes, a yellow node represents miRNA and other nodes indicate TF-genes.
Identification of candidate drugs
Enrichr platform is used to identify drug molecules for 11 common DEGs. The data were collected from the DSigDB database. According to P-value and adjusted P-value, the results from the candidate drugs were generated. The analysis depicts that parthenolide CTD 00000087 and MIGLITOL CTD 00002031 are the two drug molecules that most genes are interacted with. As these signature drugs were detected for the common DEGs, these drugs represent common drugs for COVID-19 and IPF. Table 4 points out the candidate drugs from the DSigDB database for common DEGs.
Table 4.
Suggested top drug compounds for the IPF-2 infections
| Name of drugs | P-value | Adjusted P-value | Genes |
|---|---|---|---|
| MIGLITOL CTD 00002031 | 0.00001810 | 0.004285 | S100A12, S100A9 |
| CHEMBL55802 CTD 00003118 | 0.00002876 | 0.005514 | MMP9, ICAM1 |
| Hesperidin CTD 00006087 | 0.00004187 | 0.007024 | MMP9, ICAM1 |
| Cytochalasin D CTD 00007076 | 0.00005197 | 0.007472 | MMP9, ICAM1 |
| Prolinedithiocarbamate CTD 00002658 | 0.00007540 | 0.008928 | MMP9, ICAM1 |
| Parthenolide CTD 00000087 | 0.000002540 | 0.001705 | SAA1, MMP9, ICAM1 |
| FEXOFENADINE HYDROCHLORIDE CTD 00003191 | 0.00008193 | 0.009163 | MMP9, ICAM1 |
| Hydroxytyrosol CTD 00000267 | 0.00008193 | 0.008915 | MMP9, ICAM1 |
| Antimycin A CTD 00005427 | 0.00008873 | 0.009401 | SOD2, ICAM1 |
| Anacardic acid C15:3 CTD 00003117 | 0.00008873 | 0.009160 | MMP9, ICAM1 |
Discussion
IPF is regarded as a risk factor for COVID-19. When the lung tissue of a person gets damaged and that is the time when the functionality of the lung cannot adjust properly to its task. People with lung disease are at higher risk of COVID-19. The study assists to narrate Bioinformatics lessons for meaningful analysis of SARS-CoV-2 affected lung epithelium and lung alveolar samples and IPF affected lung tissue of humans. Methodologies related to Bioinformatics are used for the study to identify 108 and 359 DEGs from GSE147507 and GSE35145, respectively. For establishing relationships and for detecting candidate drugs according to COVID-19 and IPF, common DEGs between GSE147507 and GSE35145 datasets were identified. After identification 11 (SAA2, MMP9, SAA1, S100A8, ICAM1, PI3, SOD2, C8orf4, SERPINA3, S100A12 and S100A9) common DEGs were found. The rest of the research study is continued with the analysis of GO, KEGG pathway analysis, PPIs, TF-gene interactions, TF-miRNA coregulatory network and candidate drug detection.
Identified 11 common DEGs were used for detecting GO terms. GO terms were selected according to the P-values. For biological process leukocyte aggregation, defense response to fungus, neutrophil chemotaxis, granulocyte chemotaxis and neutrophil migration are among the top GO term. In vitro, leukocyte aggregation may cause pseudoleukopenia which is a rare phenomenon [45]. In the cell environment, chemotaxis is basically a reaction that determines neutrophil locomotion direction [46]. GO terms in term of molecular function transition metal ion binding, zinc ion binding, RAGE receptor binding and metal ion binding are considered to be among the top of the list. Zn, which is a transition metal ion binding factor can enhance immunity against viral infections and can stop the replication of SARS-CoV-2 in the human cells [47]. Top GO terms according to the cellular component are cytoplasmic vesicle lumen, secretory granule lumen and cytoskeleton.
The determination of the KEGG pathway is identified for 11 common DEGs. The analysis was achieved form the common DEGs because to find a similar pathway for both COVID-19 and IPF. Top 10 KEGG pathway includes IL-17 signaling pathway, TNF signaling pathway, Leukocyte transendothelial migration, African trypanosomiasis, bladder cancer, fluid shear stress and atherosclerosis, malaria, viral myocarditis, Staphylococcus aureus infection and peroxisome. IL-17 signaling pathway contributes cytokine storm basically in SARS-CoV-2 and also in pulmonary based viral infections [48]. Meanwhile results from WikiPathways show the most interacted gene pathways are Vitamin B12 Metabolism WP1533, Folate Metabolism WP176 and Selenium Micronutrient Network WP15. Results from the Reactome pathway produce DEx/H-box helicases activate type I IFN and inflammatory cytokines production Homo sapiens R-HSA-3134963 pathway.
PPIs network analysis is the most prominent section of the study as hub gene detection, analysis of modules and drug identification thoroughly depends on the PPIs network. Analysis for PPIs was also generated for SAA2, MMP9, SAA1, S100A8, ICAM1, PI3, SOD2, C8orf4, SERPINA3, S100A12 and S100A9 genes, as these genes are common DEGs. According to the PPIs network VEGFA, AKT1, MMP9, ICAM1 and CD44 genes were declared as hub genes because of their high interaction rate or degree value. ICAM1 serum of the median level was higher in IPF patient’s serum samples compared to healthy samples [49]. To focus on the essential regions of the PPIs network, module analyses of the hub genes were achieved. The reason of focusing on highly concentrated area is a more effective drug compound suggestion.
TF-gene interaction was obtained with the common DEGs. TF-genes work as regulators according to genetic expressions which may result in creating cancer cells. From the network, ICAM1 shows a high interaction rate with other TF-genes. The degree value of ICAM1 in the TF-gene interactions network is 69. Among the regulators, STAT3 and KLF16 have significant interaction. The degree value of STAT3 and KLF16 are 5 and 4, respectively in the TF-gene interactions network. The upregulated STAT3 gene is found in lung carcinomas of human and can be a contributing factor for regulation in lung diseases [50].
Regulatory biomolecules act as potential biomarkers in numerous complex diseases. Keeping this part in memory, the activities of miRNAs and TF-genes that are analyzed for the regulation of common DEGs are visualized in the TF-miRNA coregulatory network. Thirty-nine miRNAs and 53 TF-genes are found in the study. Among the most interacted TFs, AR has the higher degree value of 4. Drugs based on androgen modulation can be contributed as treatment factor for SARS-CoV-2 [51]. Proof of changing the miRNA expression in IPF samples is established in various research and family members of miR-200 plays a vital role in the regulation of IPF samples [52]. TF-genes are reactors for the regulation of gene expression and the regulation is completed through binding with targeted genes and miRNAs on the other hand, able to regulate gene expression through mRNA degradation [53].
According to DSigDB database drug molecules were suggested from 11 common DEGs. Among all the candidate drugs, the current study highlights the top 10 significant drugs. MIGLITOL CTD 00002031, CHEMBL55802 CTD 00003118, hesperidin CTD 00006087, cytochalasin D CTD 00007076, prolinedithiocarbamate CTD 00002658, parthenolide CTD 00000087, FEXOFENADINE HYDROCHLORIDE CTD 00003191, hydroxytyrosol CTD 00000267, antimycin A CTD 00005427, anacardic acid C15:3 CTD 00003117 are the peak drug candidates for COVID-19 and IPF. Parthenolide demonstrates role of anti-inflammatory activities against IPF [54] that proves the efficiency of the proposed drugs. The current study uses a number of Bioinformatics methodologies in GSE147507, which indicates SARS-CoV-2 infection in human lung epithelium cell and GSE35145 compare sample between affected and normal IPF tissue of humans. This study hopefully integrates COVID-19 and risk factor IPF treatment. These drugs can be considered for further verification by chemical experiments. As SARS-CoV-2 is a new virus, less research has been done so far. This is the reason for collecting less number of samples for analyzing the results. In future, if more samples are available, the current study would be more effective in the context of the SARS-CoV-2 pandemic.
Conclusions
In the context of transcriptomic analysis, no other research has been done so far on SARS-CoV-2 and IPF. We have accomplished DEGs analysis between two datasets and filtered the materials through common gene identification and attempted to find infection responses between SARS-CoV-2 and IPF affected lung cells. Analyses regarding SARS-CoV-2 and IPF predict the way of detecting infections for various diseases. The drug targets are suggested logically as they are derived through the identification of hub genes and it possibly plays an active preface for already sanctioned drugs. As SARS-CoV-2 is a recent discovery, there has been little research on its risk factors and infections. Unique research on SARS-CoV-2 will become more and more important with the availability of exceeding datasets.
Key Points
Protein–protein interactions network-based analysis assists to find out only the definite genes related to both SARS-CoV-2 and IPF and the preconditioning step of Systems biology is fulfilled through protein–protein interactions analysis.
Gene set enrichment based analysis predicts Gene ontology terms for both SARS-CoV-2 and IPF affected lung cells and hub gene identification makes the prediction of drug compounds even more effective.
Computer-aided drug suggestion significantly brings out drugs like parthenolide that produces numerous actions including anti-inflammatory based actions. And various pathway-based analyses highlight the usefulness of the biological system for both SARS-CoV-2 and IPF in the context of molecular-based information.
Module analysis focuses on concentrated regions of the protein–protein interactions network that justifies the high involvement of hub nodes and eventually establishes the drug prediction even more logical and efficient.
Acknowledgment
This manuscript has not been published yet and not even under consideration for publication elsewhere. The authors are grateful who have participated in this research work.
Tasnimul Alam Taz is currently pursuing BSc in Software Engineering from the Department of Software Engineering, Daffodil International University. He is working with bioinformatics and data mining. His research interest encircles systems biology, machine learning and artificial intelligence.
Kawsar Ahmed received his BSc and MSc Engineering Degree in Information and Communication Technology (ICT) at Mawlana Bhashani Science and Technology University, Tangail, Bangladesh. He has achieved gold medals for engineering faculty first both in BSc (Engg.) and MSc (Engg.) degree from the university for his academic excellence. Currently, he is serving as an assistant professor in the same department. Before that, he joined as a lecturer in the same department and software engineering department at Daffodil International University. He has more than 200 publications in IEEE, IET, OSA, Elsevier, Springer, ISI and PubMed indexed journals. He has published two books on bioinformatics and photonic sensor design. He is the research coordinator of the Group of Biophotomatiχ. He is also a member of IEEE, SPIE, OSA, professor (assistant), Department of Information and Communication Technology Mawlana Bhashani Science and Technology University, Santosh, Tangail-1902, Bangladesh. He holds the top position at his department as well as a university from 2017 to 2019 and 1st, 2nd and 4th top researcher (Scopus indexed based) in Bangladesh, 2019 to 2017, respectively. His research group received SPIE traveling award and best paper award in IEEE WIECON ECE-2015 Conference. His research interests include sensor design, biophotonics, nanotechnology, data mining and bioinformatics.
Bikash Kumar Paul received his BSc and MSc Engineering degree in 2017, 2019, respectively from the Department of Information and Communication Technology (ICT) at Mawlana Bhashani Science and Technology University, Tangail, Bangladesh. That same year, he joined as a faculty member in the Department of Computer Science and Engineering at Ranada Prasad Shaha University, Narayangong and the department of Software Engineering at Daffodil International University, Dhaka. Currently, he is working as a full-time faculty member in the Department of Information and Communication Technology (ICT) at Mawlana Bhashani Science and Technology University, Tangail, Bangladesh. As an active research member of the Group of Biophotomatiχ, his current research interests are the development of Surface Plasmon Resonance (SPR) based sensors, fiber-optic sensors, biophotonics, low loss terahertz waveguide and bioinformatics. He has authored and coauthored more than 100 international scientific papers and conference presentations. Moreover, he was the 13th and 4th top researcher (Scopus indexed based) in Bangladesh, in the year of 2018 and 2019. In the same years, he has been selected as the ‘Best Academic Research Leader of the Year’ at Daffodil International University. He is also a member of the Institute of Electrical and Electronics Engineers (IEEE), Society of Photographic Instrumentation Engineers (SPIE) and Optical Society of America (OSA) since 2016.
Md Kawsar is currently pursuing BSc in Software Engineering from the Department of Software Engineering, Daffodil International University. He is working with Bioinformatics and Software development. His interests are in theoretical physics and data mining.
Mst. Nargis Aktar is an associate professor at the Department of Information and Communication Technology, Mawlana Bhashani Science and Technology University, Santosh, Tangail-1902, Bangladesh. She has completed her PhD degree from the University of New South Wales, Canberra at the Australian Defence Force Academy. Her research interest includes image processing, optical communication, optical sensor design and bioinformatics.
S M Hasan Mahmud received the BSc degree in Software Engineering from the Shenyang University of Chemical Technology, China, in 2011and the MSc degree in Software Engineering from Hohai University, China, in 2016. He is currently pursuing the PhD degree in Computer Science and Technology with the University of Electronic Science and Technology of China, China. He received a full scholarship from the China Scholarship Council (CSC) for his Master’s and Doctoral studies. He has published several conferences and SCI-indexed journal papers and received the Best Paper Award from the IEEE conference ICCSNT 2016. Since 2013, he is also serving as a faculty member in the Department of Software Engineering, Daffodil International University, Bangladesh. His research interests include machine learning, deep learning, bioinformatics, drug discovery and pattern recognition.
Mohammad Ali Moni is a research fellow and conjoint lecturer at the University of New South Wales, Australia. He received his PhD degree in Bioinformatics from the University of Cambridge. His research interest encompasses artificial intelligence, machine learning, data science and clinical bioinformatics.
Contributor Information
Tasnimul Alam Taz, Department of Software Engineering, Daffodil International University.
Kawsar Ahmed, Information and Communication Technology (ICT) at Mawlana Bhashani Science and Technology University, Tangail, Bangladesh.
Bikash Kumar Paul, Department of Information and Communication Technology (ICT) at Mawlana Bhashani Science and Technology University, Tangail, Bangladesh.
Md Kawsar, Department of Software Engineering, Daffodil International University.
Nargis Aktar, Department of Information and Communication Technology, Mawlana Bhashani Science and Technology University, Santosh, Tangail-1902, Bangladesh.
S M Hasan Mahmud, Shenyang University of Chemical Technology, China.
Mohammad Ali Moni, University of New South Wales, Australia.
Funding
There is no funding for this work.
References
- 1. Coronaviridae Study Group of the International Committee on Taxonomy of Viruses.. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol 2020;5(4):536–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Walls AC, Park YJ, Tortorici MA, et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020;181(2):281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. George PM, Wells AU, Jenkins RG. Pulmonary fibrosis and COVID-19: the potential role for antifibrotic therapy. Lancet Respir Med 2020;8(8):807–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sheng G, Chen P, Wei Y, et al. Viral infection increases the risk of idiopathic pulmonary fibrosis: a meta-analysis. Chest 2019;157(5):1175–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sun P, Lu X, Xu C, et al. Understanding of COVID-19 based on current evidence. J Med Virol 2020;92(6):548–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zou L, Ruan F, Huang M, et al. SARS-CoV-2 viral load in upper respiratory specimens of infected patients. N Engl J Med 2020;382(12):1177–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020;181(2):271–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chinazzi M, Davis JT, Ajelli M, et al. The effect of travel restrictions on the spread of the 2019 novel coronavirus (COVID-19) outbreak. Science 2020;368(6489):395–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ghinai I, McPherson TD, Hunter JC, et al. First known person-to-person transmission of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in the USA. Lancet 2020;395(10230):1137–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bhatia S. Short-term forecasts of COVID-19 deaths in multiple countries. Online Rep 2020;04–15. [Google Scholar]
- 11. Lancet T. COVID-19 in Brazil: “So what?”. Lancet 2020;395(10235):1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rodriguez-Morales AJ, Gallego V, Escalera-Antezana JP, et al. COVID-19 in Latin America: the implications of the first confirmed case in Brazil. Travel Med Infect Dis 2020;35:101613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011;183(6):788–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Patti MG, Tedesco P, Golden J, et al. Idiopathic pulmonary fibrosis: how often is it really idiopathic? J Gastrointest Surg 2005;9(8):1053–8. [DOI] [PubMed] [Google Scholar]
- 15. Lindell KO, Olshansky E, Song MK, et al. Impact of a disease-management program on symptom burden and health-related quality of life in patients with idiopathic pulmonary fibrosis and their care partners. Heart Lung 2010;39(4):304–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brake SJ, Barnsley K, Lu W, et al. Smoking upregulates angiotensin-converting enzyme-2 receptor: a potential adhesion site for novel coronavirus SARS-CoV-2 (Covid-19). J Clin Med 2020 2020;9(3):841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sohal SS, Hansbro PM, Shukla SD, et al. Potential mechanisms of microbial pathogens in idiopathic interstitial lung disease. Chest 2017;152(4):899–900. [DOI] [PubMed] [Google Scholar]
- 18. Sturn A, Quackenbush J, Trajanoski Z. Genesis: cluster analysis of microarray data. Bioinformatics 2002;18(1):207–8. [DOI] [PubMed] [Google Scholar]
- 19. Lee MLT, Kuo FC, Whitmore GA, et al. Importance of replication in microarray gene expression studies: statistical methods and evidence from repetitive cDNA hybridizations. Proc Natl Acad Sci 2000;97(18):9834–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Irigoyen N, Firth AE, Jones JD, et al. High-resolution analysis of coronavirus gene expression by RNA sequencing and ribosome profiling. PLoS Pathog 2016;12(2):e1005473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Clough E, Barrett T. The gene expression omnibus database In: Statistical Genomics. New York, NY: Humana Press, 2016, 93–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002;30(1):207–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Blanco-Melo D, Nilsson-Payant B, Liu WC, et al. SARS-CoV-2 launches a unique transcriptional signature from in vitro, ex vivo, and in vivo systems, 2020, BioRxiv.
- 24. Sanders YY, Ambalavanan N, Halloran B, et al. Altered DNA methylation profile in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2012;186(6):525–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smyth GK. Limma: linear models for microarray data In: Gentleman R, Care V, Dudoit S et al. (eds). Bioinformatics and Computational Biology Solutions using R and Bioconductor. New York: Springer, 2005, 397–420. [Google Scholar]
- 27. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B Methodol 1995;57(1):289–300. [Google Scholar]
- 28. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Doms A, Schroeder M. GoPubMed: exploring PubMed with the gene ontology. Nucleic Acids Res 2005;33(suppl_2):W783–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000;28(1):27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Slenter DN, Kutmon M, Hanspers K, et al. WikiPathways: a multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res 2018;46(D1):D661–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fabregat A, Jupe S, Matthews L, et al. The reactome pathway knowledgebase. Nucleic Acids Res 2018;46(D1):D649–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kuleshov MV, Jones MR, Rouillard AD, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016;44(W1):W90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ewing RM, Chu P, Elisma F, et al. Large-scale mapping of human protein–protein interactions by mass spectrometry. Mol Syst Biol 2007;3(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ben-Hur A, Noble WS. Kernel methods for predicting protein–protein interactions. Bioinformatics 2005;21(suppl_1): i38–46. [DOI] [PubMed] [Google Scholar]
- 36. Szklarczyk D, Franceschini A, Kuhn M, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 2010;39(suppl_1):D561–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13(11):2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chin CH, Chen SH, Wu HH, et al. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol 2014;8(S4):S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 2003;4(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ye Z, Wang F, Yan F, et al. Bioinformatic identification of candidate biomarkers and related transcription factors in nasopharyngeal carcinoma. World J Surg Oncol 2019;17(1):60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhou G, Soufan O, Ewald J, et al. NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res 2019;47(W1):W234–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu ZP, Wu C, Miao H, et al. RegNetwork: an integrated database of transcriptional and post-transcriptional regulatory networks in human and mouse. Database 2015;2015:bav095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xia J, Gill EE, Hancock RE. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat Protoc 2015;10(6):823. [DOI] [PubMed] [Google Scholar]
- 44. Chen EY, Tan CM, Kou Y, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 2013;14(1):128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yang D, Guo X, Chen Y, et al. Leukocyte aggregation in vitro as a cause of pseudoleukopenia. Lab Med 2008;39(2):89–91. [Google Scholar]
- 46. Cicchetti G, Allen PG, Glogauer M. Chemotactic signaling pathways in neutrophils: from receptor to actin assembly. Crit Rev Oral Biol Med 2002;13(3):220–8. [DOI] [PubMed] [Google Scholar]
- 47. Rahman MT, Idid SZ. Can Zn be a critical element in COVID-19 treatment? Biol Trace Elem Res 2020;1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wu D, Yang XO. TH17 responses in cytokine storm of COVID-19: an emerging target of JAK2 inhibitor Fedratinib. J Microbiol Immunol Infect 2020;53(3):368–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tsoutsou PG, Gourgoulianis KI, Petinaki E, et al. ICAM-1, ICAM-2 and ICAM-3 in the sera of patients with idiopathic pulmonary fibrosis. Inflammation 2004;28(6):359–64. [DOI] [PubMed] [Google Scholar]
- 50. Qu P, Roberts J, Li Y, et al. Stat3 downstream genes serve as biomarkers in human lung carcinomas and chronic obstructive pulmonary disease. Lung Cancer 2009;63(3):341–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. McCoy J, Wambier CG, Vano-Galvan S, et al. Racial variations in COVID-19 deaths may be due to androgen receptor genetic variants associated with prostate cancer and androgenetic alopecia. Are anti-androgens a potential treatment for COVID-19? J Cosmet Dermatol 2020;19:1542–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang L, Huang W, Zhang L, et al. Molecular pathogenesis involved in human idiopathic pulmonary fibrosis based on an integrated microRNA-mRNA interaction network. Mol Med Rep 2018;18(5):4365–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang HM, Kuang S, Xiong X, et al. Transcription factor and microRNA co-regulatory loops: important regulatory motifs in biological processes and diseases. Brief Bioinform 2015;16(1):45–58. [DOI] [PubMed] [Google Scholar]
- 54. Li XH, Xiao T, Yang JH, et al. Parthenolide attenuated bleomycin-induced pulmonary fibrosis via the NF-κB/snail signaling pathway. Respir Res 2018;19(1):111. [DOI] [PMC free article] [PubMed] [Google Scholar]