

Supplemental Digital Content is available in the text.
Keywords: hypertension, pulmonary; macitentan; prognosis; rare diseases; survival
Abstract
Background:
Conducting randomized controlled trials to investigate survival in a rare disease like pulmonary arterial hypertension has considerable ethical and logistical constraints. In many studies, such as the Study with an Endothelin Receptor Antagonist in Pulmonary Arterial Hypertension to Improve Clinical Outcome (SERAPHIN) randomized controlled trial, evaluating survival is further complicated by bias introduced by allowing active therapy among placebo-treated patients who clinically deteriorate.
Methods and Results:
SERAPHIN enrolled and followed patients in the same time frame as the US Registry to Evaluate Early And Long-term PAH Disease Management, providing an opportunity to compare observed survival for SERAPHIN patients with predicted survival had they received real-world treatment as in the Registry to Evaluate Early And Long-term PAH Disease Management. From the Registry to Evaluate Early And Long-term PAH Disease Management (N=3515), 734 patients who met SERAPHIN eligibility criteria were selected and their data used to build a prediction model for time to death up to 3 years based on 10 baseline prognostic variables. The model was used to predict a survival curve for each of the 742 SERAPHIN patients via their baseline variables. The average of these predicted survival curves was compared with observed survival of the placebo (n=250) and macitentan 10 mg (n=242) groups using a log-rank test and Cox proportional hazard model. Observed mortality risk for patients randomized to placebo, 62% of whom were taking background pulmonary arterial hypertension therapy, tended to be lower than that predicted for all SERAPHIN patients (16% lower; P=0.259). The observed placebo survival curve closely approximated the predicted survival curve for the first 15 months. Beyond that time, observed risk of mortality decreased compared with predicted mortality, potentially reflecting the impact of crossover of patients in the placebo group to active therapy. Over 3 years, risk of mortality observed with macitentan 10 mg was 35% lower than predicted mortality (P=0.010).
Conclusions:
These analyses show that, in a rare disease, real-world observational data can complement randomized controlled trial data to overcome some challenges associated with assessing survival in the setting of a randomized controlled trial.
Clinical Trial Registration:
https://www.clinicaltrials.gov. Unique identifiers: NCT00660179 and NCT00370214.
WHAT IS KNOWN
Demonstrating survival benefits of new therapies for rare diseases such as pulmonary arterial hypertension is challenging, as recruiting a sufficient number of patients to detect differences in survival may not be feasible, particularly as potential new medicines should be evaluated against or in addition to available therapies.
In long-term randomized clinical trials, evaluating survival is further complicated by the provision of rescue therapy to patients in the comparator arm who experience clinical deterioration.
WHAT THE STUDY ADDS
This study illustrates that real-world observational data can complement clinical trial data to explore survival in rare diseases.
A tailored model, built using data from the Registry to Evaluate Early And Long-term PAH Disease Management (REVEAL) registry, was used to predict survival of patients with pulmonary arterial hypertension from the randomized controlled Study with an Endothelin Receptor Antagonist in Pulmonary Arterial Hypertension to Improve Clinical Outcome (SERAPHIN) had they received real-world treatment, and this predicted survival was compared with the observed survival of patients in SERAPHIN receiving macitentan 10 mg as well as those randomized to placebo.
The observed mortality risk in the macitentan-treated group was 35% lower than the predicted mortality risk for the overall SERAPHIN population (P=0.010) and may be more representative of the real treatment effect had there not been any crossover of placebo patients to active therapy.
An estimated 350 million people worldwide have a rare disease.1 As these diseases are often life-threatening,2 there is a substantial unmet need for therapies with a proven survival benefit. However, demonstrating a treatment effect on survival in a rare disease represents a substantial challenge. One of the biggest hurdles is the rarity of these diseases per se because recruiting sufficient numbers of patients to demonstrate a survival benefit may not be feasible.3 When therapies that lead to improvements are available, potential new medicines should be evaluated against or in addition to the available medicine,4 and this may require even larger sample sizes to provide sufficient statistical power.5 Furthermore, in diseases where patients worsen before death, there is an ethical obligation to provide rescue therapy to deteriorating patients. Contamination of the control group by rescue therapy may bias survival estimates in the control group, leading to underestimation of the true treatment effect. This scenario may further increase the challenge of demonstrating a survival benefit,6,7 and, even in cases where a survival benefit is demonstrated statistically, the true effect size of the drug may not be established.
Pulmonary arterial hypertension (PAH) is a progressive, rare disease with a poor prognosis.8 Multiple PAH therapies have been approved, mostly on the basis of short-term improvements in exercise capacity.9,10 Recently, event-driven randomized controlled trials (RCTs) have investigated long-term outcomes using composite morbidity-mortality end points.11–14 In the SERAPHIN trial, PAH patients were treated with the endothelin receptor antagonist (ERA) macitentan or placebo for up to 3.6 years; the majority also received background PAH therapy (64% of patients).15 In this study, macitentan 10 mg significantly reduced the risk of the primary composite end point of morbidity and mortality by 45% versus placebo (P<0.001) up to the end of treatment. The study also reported a 23% decrease in the risk of all-cause mortality in an intention-to-treat analysis with macitentan 10 mg versus placebo (P=0.25) up to the end of the study.11 The latter observation was made in the context of many placebo patients crossing-over to receive active therapy, which can lead to an underestimation of the treatment effect.6,11 While long-term, event-driven studies mark a major advancement in the PAH field, they were neither designed nor powered to detect a statistically significant survival benefit. A survival benefit has been reported with intravenous epoprostenol in patients with severe PAH16; however, that study was conducted >20 years ago, when no other therapies were available. In the current treatment era, it is estimated that over 4000 patients would be needed to show a 20% difference in survival in PAH. Therefore, a study that is sufficiently powered to assess mortality is not feasible in this rare disease.
Given the limitations associated with assessing mortality in RCTs, we explored the use of real-world data in conjunction with RCT data to provide further insights into the effect of macitentan treatment on survival. The large US Registry to Evaluate Early And Long-term PAH Disease Management (REVEAL) in PAH provides a real-world data set that was collected during a similar period as SERAPHIN. An earlier analysis of the REVEAL data resulted in the identification of a number of baseline prognostic variables and the development of a prognostic equation and simplified mortality risk calculator.17,18 The objective of the current exploratory analysis was to develop a survival prediction model, based on baseline prognostic variables from a cohort of patients enrolled in REVEAL who met the SERAPHIN eligibility criteria, and thus tailored to the SERAPHIN population. This model was then used to predict the survival of the SERAPHIN population had they been treated in the real world, that is, with no access to macitentan. The predicted survival of the SERAPHIN patients was then compared with the survival observed in the study to provide an estimate of the treatment effect of macitentan.
Methods
The data sharing policy of the Sponsor is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for access to the study data can be submitted through Yale Open Data Access Project site at http://yoda.yale.edu.19
Study Population
SERAPHIN and REVEAL were both conducted in accordance with the amended Declaration of Helsinki and the protocols reviewed by local institutional review boards (Tables I and II in the Data Supplement) with written informed consent obtained from all patients.
SERAPHIN was a global, double-blind, randomized, placebo-controlled event-driven, phase 3 study (NCT00660179).11 Patients were enrolled between May 2008 and December 2009 and were randomized (1:1:1) to placebo, macitentan 3 mg, or macitentan 10 mg. Concomitant treatment with a stable dose of phosphodiesterase type 5 inhibitors, oral/inhaled prostanoids, calcium channel blockers, or l-arginine was allowed. Double-blind treatment continued until patients experienced a primary end point event or until 285 events had accrued. Patients were followed until withdrawal from the study or until the end of follow-up (March 2012). Patients who experienced a nonfatal primary end point event and terminated double-blind treatment were eligible to receive another PAH therapy including open-label macitentan 10 mg.
REVEAL was a multicenter, observational, US-based registry designed to provide information about patient characteristics, and disease course and management from 3515 consecutively enrolled patients with newly or previously diagnosed PAH (NCT00370214).8 The registry design and inclusion criteria have been described previously.20 In brief, patients aged ≥3 months with PAH confirmed by right heart catheterization were enrolled at 55 US centers from March 2006 to December 2009. Patients were followed until December 2012 or until time of death or withdrawal from the study. Of note, patients enrolled in an RCT were not eligible for enrollment in REVEAL.
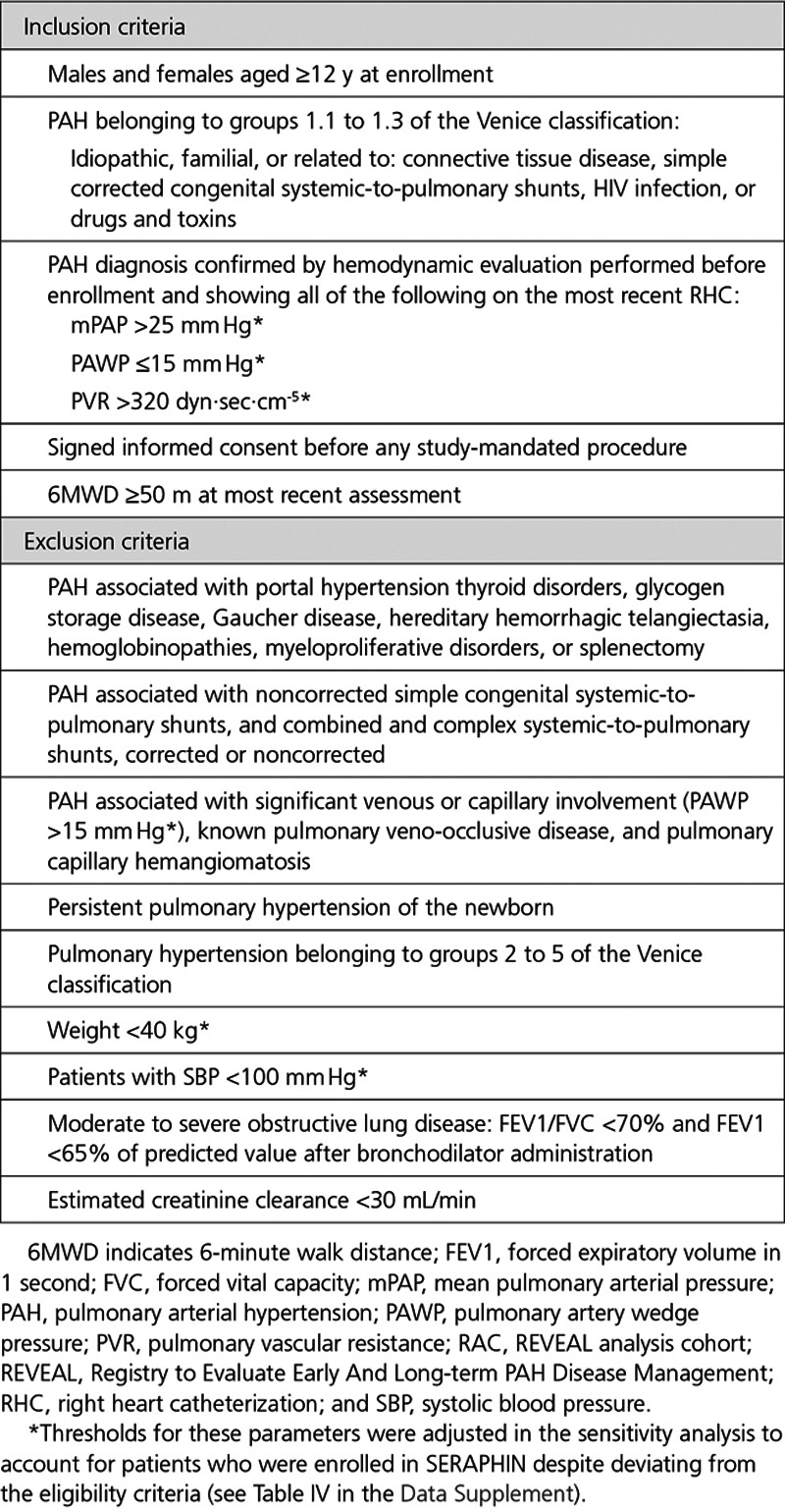
Selection of the REVEAL Analysis Cohort
As REVEAL enrolled a broader population of PAH patients than SERAPHIN in terms of disease classification, severity, and age, the first step was to select a cohort of REVEAL patients (the REVEAL analysis cohort [RAC]) who could have been enrolled in SERAPHIN to align the real-world data set to the RCT population (Figure 1A). The final RAC selection variables were based on SERAPHIN inclusion and exclusion criteria (Table 1). A subset of the SERAPHIN eligibility criteria were not applied for the following reasons: (1) the relevant variables were not available in REVEAL, (2) the criteria related to the safety profile of macitentan and were not considered relevant to the survival analysis, (3) the criteria were subjective, (4) the criteria related to the use of background PAH therapies (a conservative approach to ensure that patients treated with an ERA were not excluded), or (5) the criteria could be controlled via the prediction model in the subsequent step. An overview of the entire SERAPHIN eligibility criteria and reasons for exclusion are provided in Table III in the Data Supplement. Patients with a missing value for one or more selection variables were excluded from the RAC. A sensitivity RAC was defined to account for patients enrolled in SERAPHIN who deviated from some of the eligibility criteria (n=59; 8.0%). For this analysis, the relevant selection criteria were adjusted (Table IV in the Data Supplement), resulting in the inclusion of additional REVEAL patients (Sensitivity RAC-A). A second sensitivity analysis cohort was defined using the cohort of patients included in the RAC who did not receive an ERA at baseline (Sensitivity RAC-B).
Figure 1.
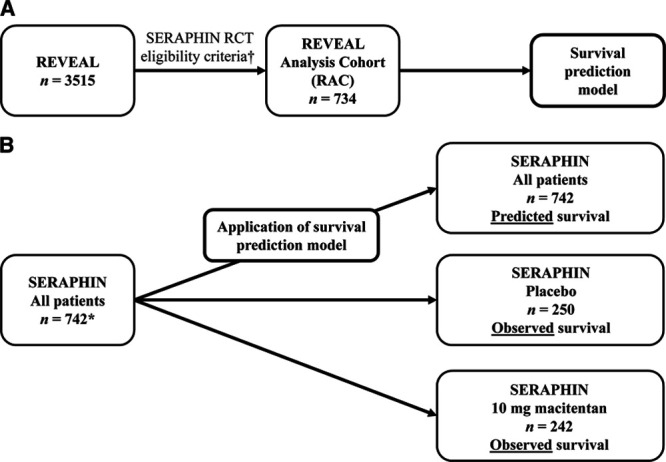
Data Integration Strategy. A, Development and (B) application of the survival prediction model. *In the SERAPHIN study, patients were randomized to placebo (n=250), macitentan 3 mg (n=250), and macitentan 10 mg (n=242). †The exact eligibility criteria used to select the REVEAL analysis cohort (RAC) are detailed in Table III in the Data Supplement. RCT indicates randomized controlled trial; REVEAL, Registry to Evaluate Early And Long-term PAH Disease Management; and SERAPHIN, Study with an Endothelin Receptor Antagonist in Pulmonary Arterial Hypertension to Improve Clinical Outcome.
Table 1.
RAC Selection Variables
Statistical Methods
In these exploratory analyses, time to all-cause death in SERAPHIN and REVEAL was defined as the time between the baseline date (date of randomization [SERAPHIN] or enrollment [REVEAL]) and the date of death due to any cause; patients who had not died within 3 years of baseline were censored at the last known contact date or at 3 years postbaseline, whichever occurred first. A cutoff for censoring at 3 years was chosen as fewer than 10% of SERAPHIN patients were still at risk by this timepoint.
To adjust for the remaining differences between the RAC and SERAPHIN populations, a Cox regression prediction model was developed using the RAC that related time to all-cause death to the patients’ baseline variables (Figure 1A). The first step in the development of the survival prediction model was the identification of candidate prognostic variables. Initially, the 26 variables that were evaluated during the development of the REVEAL model-estimated risk calculator were considered.17,18 Of these, 15 variables were retained, as they were available in SERAPHIN. An additional 3 variables that were available in SERAPHIN and REVEAL and have evidence supporting their prognostic relevance were also included. The resulting 18 prognostic variables (sex, age, race, PAH classification, time since diagnosis, weight, body mass index, World Health Organization functional class, 6-minute walk distance, Borg dyspnea score, pulmonary vascular resistance, mean right atrial pressure, mean pulmonary arterial pressure, pulmonary arterial wedge pressure, cardiac index, systolic blood pressure, heart rate, and mixed venous oxygen saturation) were evaluated using a stepwise regression procedure. For continuous covariates, discretization was applied based on previously determined thresholds17 and both the continuous and discrete variables were tested in the model. A RAC patient had to have a full set of covariates to be included in the final model. Stepwise regression using an α of 0.05 for prognostic variable selection was used to obtain the most parsimonious model. Due to missing data for some variables excluded from the model during the stepwise approach, the final model could include variables with a P value marginally >0.05 and these variables were retained in the model. A cross-validated C statistic was calculated to assess whether the model fit the data adequately. A survival curve for each SERAPHIN patient based on their baseline characteristics was then estimated using the final survival prediction model (Figure 1B). The average temporal profile of the individual survival curves obtained from all 742 patients enrolled in SERAPHIN was then compared with the observed survival curve of the placebo (n=250) and macitentan 10 mg (n=242) groups by sampling a large number of survival times from this average survival curve, and estimating the hazard ratio estimate and log-rank test. The variance estimate of the hazard ratio estimate was based on boot-strapping of the REVEAL data. Hazard ratios were obtained to estimate the size of the differences between groups. Sensitivity analyses were performed using Sensitivity RAC-A and RAC-B. In addition, a sensitivity analysis was performed on the RAC whereby baseline PAH therapy (phosphodiesterase type 5 inhibitors only, prostanoid only, prostanoid and phosphodiesterase type 5 inhibitors in combination, others or no therapy) was also included as a covariate in the model.
Role of the Funding Source
The study was sponsored by Actelion Pharmaceuticals Ltd, Allschwil, Switzerland. The Sponsor participated in the conception and design of the analysis and interpretation of the data, drafting and critical revision of the report, and approved submission of the manuscript. The initial draft of the manuscript was prepared by Adam Torbicki, Raymond Benza, Gérald Simonneau, Richard Channick, Lee-Jen Wei, and Brian Hennessy, with professional medical writing support funded by the sponsor, before review by all authors.
Results
Survival Prediction Model for the RAC
Of the 3515 patients enrolled in REVEAL, 734 (20.9%) met the main SERAPHIN eligibility criteria. These patients were included in the RAC (Figure 1A; Table 2). Of the 2781 (79.1%) patients excluded from the RAC, 1466 (41.7%) were excluded as they did not meet at least one SERAPHIN eligibility criteria and 1315 (37.4%) were excluded as they had one or more missing values for the eligibility criteria. Excluded patients had a worse survival prognosis compared to those included in the RAC (Figure I in the Data Supplement). This was not unexpected given that the RAC selection variables excluded patients with baseline characteristics likely to be associated with an increased mortality risk, including comorbid lung conditions, renal insufficiency, and portopulmonary hypertension (Table III in the Data Supplement). The baseline characteristics for the patients included and excluded from the RAC are shown in Table V in the Data Supplement.
Table 2.
Baseline Characteristics of the Patients in the RAC
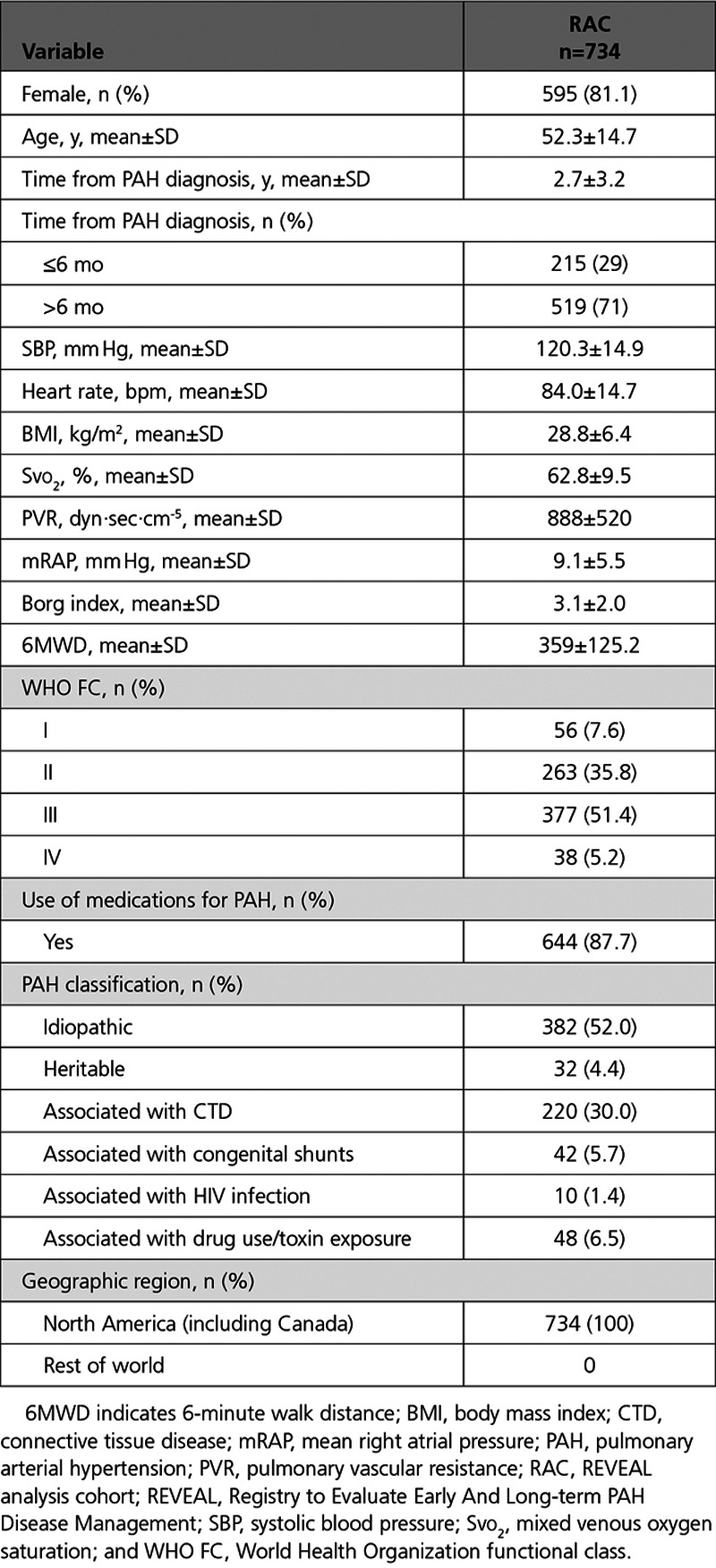
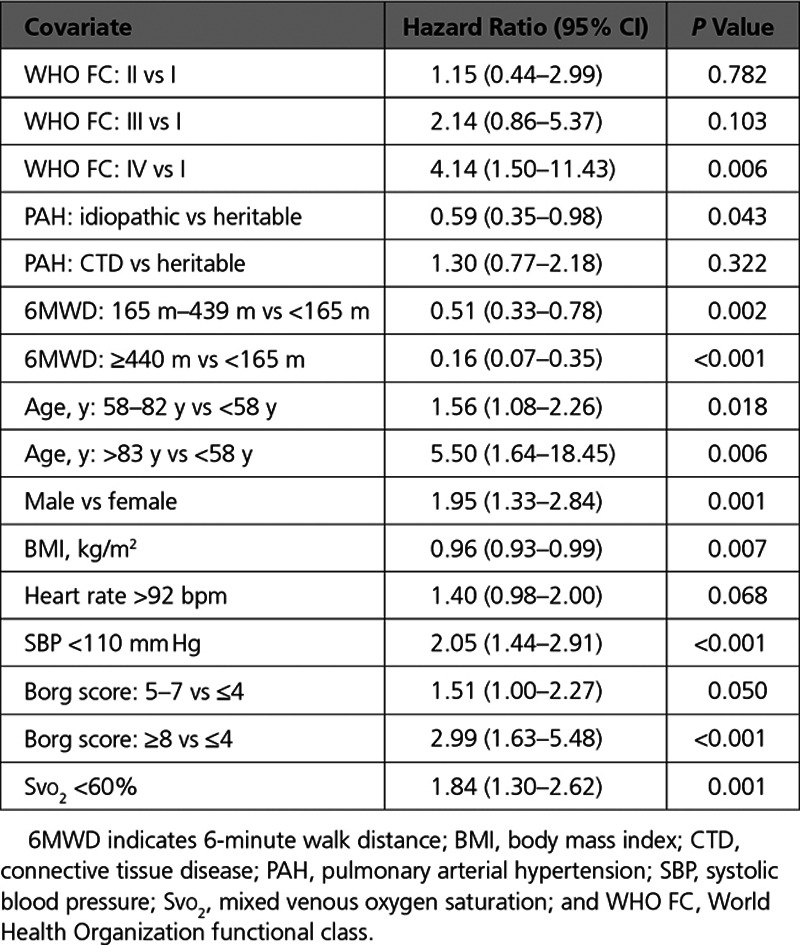
A survival prediction model was built using the baseline data from the RAC (Table 2). After the stepwise regression was performed to evaluate the 18 prognostic variables, 10 of these variables were retained in the survival prediction model (Table 3). The variables included were: World Health Organization functional class, PAH classification, 6-minute walk distance, heart rate, systemic blood pressure, sex, age, body mass index, Borg dyspnea score, and mixed venous oxygen saturation. The cross-validated C statistic, which quantifies the goodness of fit of the final model, was 76%, indicating that the model is a reasonable fit.
Table 3.
Cox Proportional Hazards Prediction Model Parameters
Survival of SERAPHIN Patients up to 3 Years: Predicted and Observed
For each of the 742 patients enrolled in SERAPHIN, a survival probability was estimated by entering their baseline characteristics into the prediction model (Figure 1B). The baseline characteristics of the SERAPHIN population are outlined in Table 4. There were no significant between-group differences at baseline.11
Table 4.
Baseline Characteristics of the Patients in SERAPHIN11
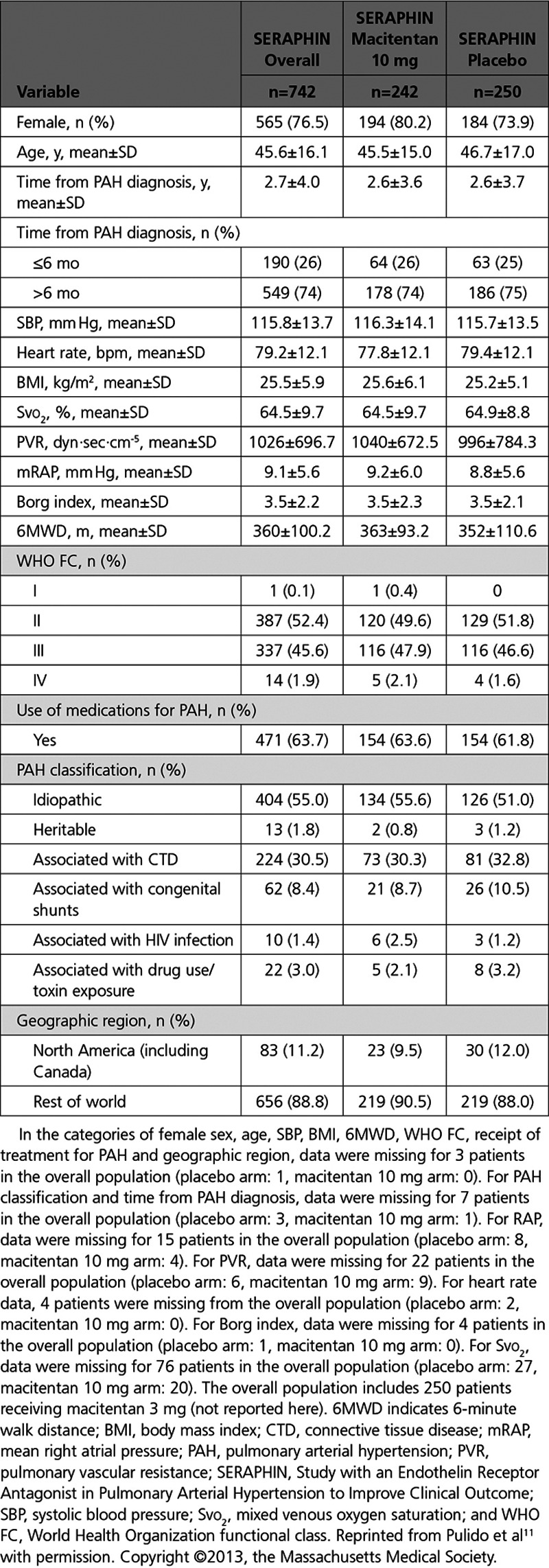
The predicted survival for the overall SERAPHIN population was compared to the observed survival for the SERAPHIN placebo patients (Figure 2A). Over 3 years, although the risk of mortality was numerically lower in the placebo arm compared with the predicted mortality of all SERAPHIN patients had they received real-world treatment (16% lower; hazard ratio, 0.84; 95% CI, 0.62–1.14; P=0.259), this difference was not statistically significant. For the predicted survival curve, the risk of mortality was constant for the 3 years of follow-up, that is, no change in risk was observed over time. Over the first 15 months, a similar constant risk was observed for the placebo patients and their survival curve very closely approximated the predicted curve. However, after this time, the observed risk of mortality in the placebo group appeared to decrease and the 2 curves separated.
Figure 2.
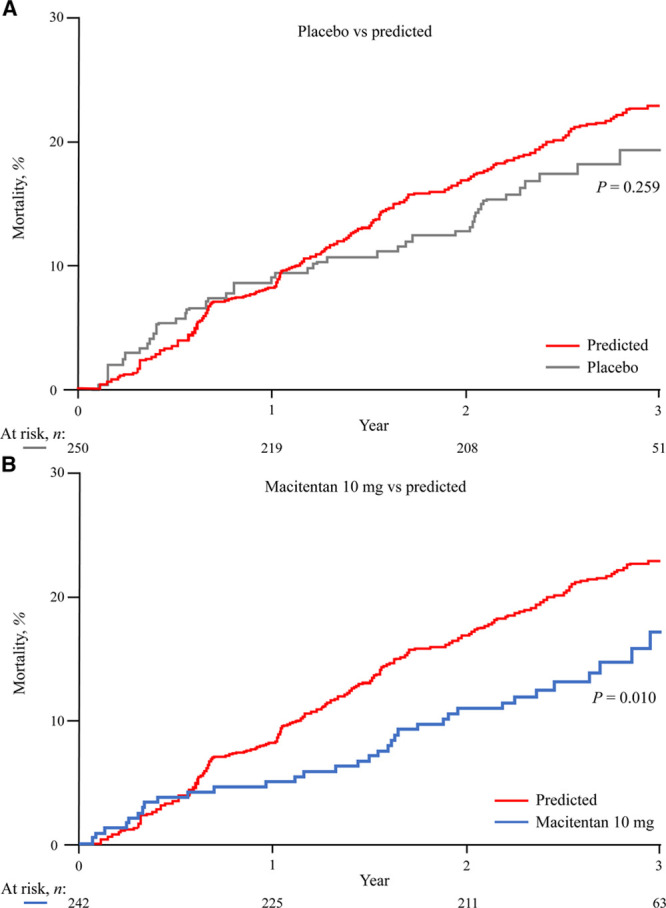
Observed vs predicted mortality in SERAPHIN. Observed mortality for patients receiving (A) placebo or (B) macitentan 10 mg in SERAPHIN compared with predicted mortality for the overall SERAPHIN patient population had they received real-world treatment. n=742 for the predicted curve (the number of patients at risk at each timepoint are not available for the predicted curves as the Kaplan-Meier estimates are based on probability predictions). SERAPHIN indicates Study with an Endothelin Receptor Antagonist in Pulmonary Arterial Hypertension to Improve Clinical Outcome.
The predicted survival for all SERAPHIN patients was also compared to the observed survival for the SERAPHIN patients treated with macitentan 10 mg (Figure 2B). The 2 curves separated within a few months and the observed risk of mortality was consistently lower than that predicted for the overall population. Over 3 years, the risk of mortality observed with macitentan 10 mg was 35% lower than the predicted mortality (hazard ratio, 0.65; 95% CI, 0.46–0.90; P=0.010).
Sensitivity Analyses
For the sensitivity analysis that was conducted on the RAC to include baseline PAH therapy as an additional covariate in the model, baseline PAH therapy was not a significant covariate. When baseline PAH therapy was forced into the model, the results were consistent with the main analysis (macitentan 10 mg versus predicted: hazard ratio, 0.65; 95% CI, 0.46–0.91; P=0.012; Table VI in the Data Supplement).
Sensitivity RAC-A (n=1013) comprised the 734 RAC patients and an additional 279 REVEAL patients who were included after the thresholds for some criteria were adjusted to reflect the enrolled SERAPHIN population, rather than the criteria per se. Sensitivity RAC-B comprised the 391 RAC patients who were not receiving ERA therapy at baseline. For analyses based on both Sensitivity RAC-A and RAC-B, the fit of the sensitivity survival prediction model and the comparisons between predicted and observed survival of SERAPHIN patients were consistent with the findings reported based on the RAC survival prediction model (Table VI in the Data Supplement).
Discussion
Using real-world data to complement trial data is one approach to exploring the impact of therapies on survival. This approach was used in the evaluation of alglucosidase alfa (Myozyme®) for the treatment of the fatal infantile-onset Pompe disease, as no active comparator was available. The assessment of alglucosidase alfa using data from 2 single-arm studies and from a separate cohort of untreated patients resulted in the approval of this medication.21,22 In PAH, the National Institutes of Health prognostic equation was developed from survival data obtained from untreated PAH patients enrolled in the National Institutes of Health registry between 1981 and 1985.23 This equation has been applied to randomized clinical trial data sets to estimate survival in comparison with treated patients.24
In the analyses reported here, we used PAH registry data to develop a survival prediction model, which was applied to SERAPHIN data to estimate the effect of macitentan on survival. Although our approach was conceptually similar to previous studies, the methodology has a number of novel aspects. For example, we selected an appropriate observational data set on which to develop our prediction model. We used data from REVEAL because this registry was conducted contemporaneously to SERAPHIN. Using data from 2 studies conducted during the same time period allows us to minimize the unknown confounders and biases that can be introduced as a result of changes in patient management over time. To further ensure the appropriateness of the observational data set, we selected a subgroup of patients in REVEAL who would have met the eligibility criteria for SERAPHIN. This allowed for sufficient overlap of patient characteristics in the RAC and SERAPHIN populations. The regression model itself then serves to account for any remaining differences between the 2 populations. Such models are frequently used to adjust for imbalances in prognostic factors between groups of patients25 and have been used previously to correct for lack of randomization when comparing observational and trial data.26 In our survival prediction model, 10 baseline variables were retained and were considered sufficient (cross-validated C statistic of 76%, which is indicative of very good prediction) to describe the predicted survival of a given patient.
For each patient enrolled in SERAPHIN, their individual baseline values were entered into the model to predict their survival had they been treated in a real-world setting. The cumulative predicted real-world survival of all patients was compared to the observed survival for patients assigned to placebo and to those assigned to macitentan 10 mg. For patients randomized to placebo, the predicted survival curve closely approximated the observed survival for 15 months of follow-up, after which the 2 curves separated as the mortality risk in the placebo arm decreased. Over 3 years, the mortality risk in the placebo arm was 16% lower than that predicted, although this difference was not statistically significant. For patients randomized to macitentan 10 mg, the observed risk of mortality was 35% lower than the predicted mortality risk. This reduction is greater than the 23% lower mortality risk observed for macitentan 10 mg versus placebo in the SERAPHIN trial.11 By month 18, the majority (85%) of patients randomized to placebo who had experienced a morbidity event were receiving open-label macitentan. This crossover to active therapy may be a contributing factor in the higher mortality risk of the predicted curve versus the observed placebo curve, as well as the higher treatment effect on survival with macitentan 10 mg compared with the predicted curve versus the observed placebo curve.11 While these findings are consistent with our hypothesis about the impact of crossover to active therapy, other factors may also play a role, and are discussed below.
Similar to other analyses using observational data, our study has limitations related to the comparability of the 2 populations being studied. There are differences between the SERAPHIN and REVEAL populations that could impact survival analyses. One consideration is a difference in background PAH therapy use in the RAC (87.7% of patients) versus SERAPHIN (63.7% of patients). We excluded baseline PAH therapy from the main model because therapy use could change after enrollment. These changes are likely to differ between the RAC and the SERAPHIN populations and such changes cannot be accounted for in the model. We did, however, perform 2 sensitivity analyses for PAH therapy: one analysis forced baseline PAH therapy into the model, the second analysis used the Sensitivity RAC-B, that is, excluding patients who were receiving ERA therapy at baseline, to develop the model. The results of both sensitivity analyses were consistent with the main analysis; this is perhaps not surprising because PAH medications lead to changes in modifiable risk factors and many of these are included in our model. In addition to the sensitivity analyses described, major efforts were made to minimize the impact of potential differences between the populations by rigorously selecting REVEAL patients for the RAC. Despite these efforts some differences remain, such as the more structured follow-up schedule in an RCT versus a registry and differences in geographic location, as well as the possibility of unknown confounding factors, and these should be considered. Another potential limitation is the exclusion of NT-proBNP (N-terminal pro brain natriuretic peptide) from our model. NT-proBNP is a known prognostic variable but was not included as these data were not available for almost one-third of patients enrolled in SERAPHIN.
Long-term, event-driven RCTs represent an important step forward in the evaluation of PAH therapies. The analyses presented here provide an additional means of exploring potential survival benefits that complement RCTs and support the use of risk equations, such as the REVEAL prognostic equation,17,18 in the real world and clinical trial environments. In the future, analyzing real-world and single-arm trial data in parallel could allow researchers to enrich study populations and gain insights into the survival benefits offered by new therapies. This approach may be particularly valuable for rare diseases, where recruitment of large numbers of patients is not practical and where patients receive active therapy in response to disease progression. Prospective planning should improve the robustness and validity of such future analyses, by ensuring that all relevant variables are collected from both the registry and trial patients to address some of the limitations related to missing data. A prespecified analysis plan should be agreed and finalized before any data collection to ensure the integrity of the analysis.
Conclusions
Demonstrating a survival benefit of therapies for rare diseases, such as PAH, is difficult. We used a tailored, model-based approach to compare the observed survival of patients treated with macitentan in an RCT (SERAPHIN) with that predicted for all patients in SERAPHIN, based on a large contemporaneous real-world data set (REVEAL). Over 3 years, the risk of mortality observed in the active treatment arm (macitentan 10 mg) was 35% lower than the predicted mortality (P=0.010). This exploratory analysis shows that, notwithstanding its limitations, real-world observational data can complement RCT data to provide a means of exploring survival benefits in rare diseases.
Acknowledgments
All authors contributed to the conception and design of the analyses and the interpretation of the data. Statistical analyses were performed by B. Hennessy, Dr Bacchi, and Dr Wei. All authors reviewed and edited the article for intellectual content and approved the final version for publication. All authors had full access to all the data in the study and take responsibility for its integrity and the data analysis. Medical writing assistance was provided by Stephanie Carter (nspm ltd, Meggen, Switzerland) and funded by Actelion Pharmaceuticals Ltd (Allschwil, Switzerland).
Sources of Funding
This study was supported by Actelion Pharmaceuticals Ltd, Allschwil, Switzerland. The Sponsor participated in the conception and design of the analysis and interpretation of the data, drafting and critical revision of the report, and approved submission of the article.
Disclosures
Dr Torbicki has received grants, personal fees, and nonfinancial support from Actelion Pharmaceuticals Ltd, personal fees from Bayer HealthCare, Sanofi, AOP, United Therapeutics, MSD, Janssen, and Arena Pharmaceuticals, and is the Chairperson of the Pulmonary Hypertension Foundation, receiving donations for its statue activities also from the medical industry. Chairperson is an honorary function and receives neither honoraria nor any other personal benefits. Dr Bacchi is an employee of Actelion Pharmaceuticals and holds stock in the parent company Johnson & Johnson. Dr Delcroix has received grants, personal fees, and nonfinancial support from Actelion Pharmaceuticals Ltd during the conduct of the study. Dr Farber has received nonfinancial support, grants, and personal fees from Actelion Pharmaceuticals Ltd, grants and personal fees from Gilead and United Therapeutics, and personal fees from Bayer and Bellerophon. Dr Ghofrani has received nonfinancial support, grants, and personal fees from Actelion Pharmaceuticals Ltd, grants and personal fees from Bayer HealthCare, Novartis Corporation, and Pfizer; personal fees from Gilead Sciences, GlaxoSmithKline, and Merck & Co. B. Hennessy is an employee of Actelion Pharmaceuticals Ltd and holds stock in the parent company Johnson & Johnson. Dr Jansa has received grants, personal fees, and nonfinancial support from Actelion Pharmaceuticals Ltd, and grants and personal fees from United Therapeutics, AOP Orphan, Bayer HealthCare, and GlaxoSmithKline. Dr Mehta has received nonfinancial support, grants, and personal fees from Actelion Pharmaceuticals Ltd, grants and personal fees from Bayer Pharmaceuticals, and grants from Gilead, Ikaria, and United Therapeutics. Dr Perchenet is an employee of Actelion Pharmaceuticals Ltd and holds stock in the parent company Johnson & Johnson. Dr Pulido has received nonfinancial support, grants, and personal fees from Actelion Pharmaceuticals Ltd, grants from United Therapeutics, Bristol-Myers Squibb, and Eli Lilly & Co, and grants and personal fees from Bayer HealthCare and GlaxoSmithKline. Dr Rosenberg is an employee of Actelion Pharmaceuticals and holds stock in the parent company Johnson & Johnson. Dr Rubin has received nonfinancial support and personal fees from Actelion Pharmaceuticals Ltd, Arena Pharmaceuticals, GeNO Pharmaceuticals, SoniVie, Karos Pharmaceuticals, Gilead Sciences, and Pfizer. Dr Sastry has received grants, personal fees, and nonfinancial support from Actelion Pharmaceuticals Ltd, grants from United Therapeutics and grants and personal fees from Cipla. Dr Simonneau has received grants, personal fees, and nonfinancial support from Actelion Pharmaceuticals Ltd, grants, personal fees, and nonfinancial support from Bayer HealthCare; personal fees and nonfinancial support from Eli Lilly, GlaxoSmithKline, Novartis, and Pfizer, and personal fees from Arena Pharmaceuticals. Dr Sitbon has received grants, personal fees, and nonfinancial support from Actelion Pharmaceuticals Ltd, grants and personal fees from Bayer HealthCare, GlaxoSmithKline, and Merck, and personal fees from United Therapeutics and Arena Pharmaceuticals. Dr Souza has received grants and personal fees from Actelion Pharmaceuticals; and personal fees from Bristol-Myers Squibb, Bayer HealthCare, and GlaxoSmithKline. Dr Wei has received nonfinancial support from Actelion Pharmaceuticals Ltd. Dr Channick has received grants, personal fees, and nonfinancial support from Actelion Pharmaceuticals Ltd, grants from United Therapeutics, and personal fees from Bayer. Dr Benza has received nonfinancial support from Actelion Pharmaceuticals Ltd.
Supplementary Material
Footnotes
Drs Channick and Benza contributed equally to this work.
The Data Supplement is available at https://www.ahajournals.org/doi/suppl/10.1161/CIRCOUTCOMES.118.005095.
References
- 1.Walkley SU, Davidson CD, Jacoby J, Marella PD, Ottinger EA, Austin CP, Porter FD, Vite CH, Ory DS. Fostering collaborative research for rare genetic disease: the example of niemann-pick type C disease. Orphanet J Rare Dis. 2016;11:161. doi: 10.1186/s13023-016-0540-x. doi: 10.1186/s13023-016-0540-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Institute of Medicine (US) Committee on Accelerating Rare Diseases Research Orphan Product Development. Rare Diseases and Orphan Products: Accelerating Research and Development. Washington, DC: National Academies Press (US); 2010. [Google Scholar]
- 3.Behera M, Kumar A, Soares HP, Sokol L, Djulbegovic B. Evidence-based medicine for rare diseases: implications for data interpretation and clinical trial design. Cancer Control. 2007;14:160–166. doi: 10.1177/107327480701400209. doi: 10.1177/107327480701400209. [DOI] [PubMed] [Google Scholar]
- 4.Temple R, Ellenberg SS. Placebo-controlled trials and active-control trials in the evaluation of new treatments. Part 1: ethical and scientific issues. Ann Intern Med. 2000;133:455–63. doi: 10.7326/0003-4819-133-6-200009190-00014. doi: 10.7326/0003-4819-133-6-200009190-00014. [DOI] [PubMed] [Google Scholar]
- 5.Holubkov R, Dean JM, Berger J, Anand KJ, Carcillo J, Meert K, Zimmerman J, Newth C, Harrison R, Willson DF, Nicholson C Eunice Kennedy Shriver National Institute of Child Health and Human Development Collaborative Pediatric Critical Care Research Network. Is “rescue” therapy ethical in randomized controlled trials? Pediatr Crit Care Med. 2009;10:431–438. doi: 10.1097/PCC.0b013e318198bd13. doi: 10.1097/PCC.0b013e318198bd13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jönsson L, Sandin R, Ekman M, Ramsberg J, Charbonneau C, Huang X, Jönsson B, Weinstein MC, Drummond M. Analyzing overall survival in randomized controlled trials with crossover and implications for economic evaluation. Value Health. 2014;17:707–713. doi: 10.1016/j.jval.2014.06.006. doi: 10.1016/j.jval.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 7.White IR, Bamias C, Hardy P, Pocock S, Warner J. Randomized clinical trials with added rescue medication: some approaches to their analysis and interpretation. Stat Med. 2001;20:2995–3008. doi: 10.1002/sim.927. doi: 10.1002/sim.927. [DOI] [PubMed] [Google Scholar]
- 8.Farber HW, Miller DP, Poms AD, Badesch DB, Frost AE, Muros-Le Rouzic E, Romero AJ, Benton WW, Elliott CG, McGoon MD, Benza RL. Five-year outcomes of patients enrolled in the REVEAL Registry. Chest. 2015;148:1043–1054. doi: 10.1378/chest.15-0300. doi: 10.1378/chest.15-0300. [DOI] [PubMed] [Google Scholar]
- 9.Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonssagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT) Eur Respir J. 2015;46:903–975. doi: 10.1183/13993003.01032-2015. doi: 10.1183/13993003.01032-2015. [DOI] [PubMed] [Google Scholar]
- 10.Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M ESC Scientific Document Group. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016;37:67–119. doi: 10.1093/eurheartj/ehv317. doi: 10.1093/eurheartj/ehv317. [DOI] [PubMed] [Google Scholar]
- 11.Pulido T, Adzerikho I, Channick RN, Delcroix M, Galiè N, Ghofrani HA, Jansa P, Jing ZC, Le Brun FO, Mehta S, Mittelholzer CM, Perchenet L, Sastry BK, Sitbon O, Souza R, Torbicki A, Zeng X, Rubin LJ, Simonneau G SERAPHIN Investigators. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369:809–818. doi: 10.1056/NEJMoa1213917. doi: 10.1056/NEJMoa1213917. [DOI] [PubMed] [Google Scholar]
- 12.Galiè N, Barberà JA, Frost AE, Ghofrani HA, Hoeper MM, McLaughlin VV, Peacock AJ, Simonneau G, Vachiery JL, Grünig E, Oudiz RJ, Vonk-Noordegraaf A, White RJ, Blair C, Gillies H, Miller KL, Harris JH, Langley J, Rubin LJ AMBITION Investigators. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med. 2015;373:834–844. doi: 10.1056/NEJMoa1413687. doi: 10.1056/NEJMoa1413687. [DOI] [PubMed] [Google Scholar]
- 13.McLaughlin VV, Channick R, Ghofrani HA, Lemarie JC, Naeije R, Packer M, Souza R, Tapson VF, Tolson J, Al Hiti H, Meyer G, Hoeper MM. Bosentan added to sildenafil therapy in patients with pulmonary arterial hypertension. Eur Respir J. 2015;46:405–413. doi: 10.1183/13993003.02044-2014. doi: 10.1183/13993003.02044-2014. [DOI] [PubMed] [Google Scholar]
- 14.Sitbon O, Channick R, Chin KM, Frey A, Gaine S, Galiè N, Ghofrani HA, Hoeper MM, Lang IM, Preiss R, Rubin LJ, Di Scala L, Tapson V, Adzerikho I, Liu J, Moiseeva O, Zeng X, Simonneau G, McLaughlin VV GRIPHON Investigators. Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med. 2015;373:2522–2533. doi: 10.1056/NEJMoa1503184. doi: 10.1056/NEJMoa1503184. [DOI] [PubMed] [Google Scholar]
- 15.European Medicines Agency. European Public Assessment Report: Opsumit (macitentan). http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002697/WC500160900.pdf. Accessed March 16, 2018.
- 16.Barst RJ, Rubin LJ, Long WA, McGoon MD, Rich S, Badesch DB, Groves BM, Tapson VF, Bourge RC, Brundage BH, Koerner SK, Langleben D, Keller CA, Murali S, Uretsky BF, Clayton LM, Jöbsis MM, Blackburn SD, Shortino D, Crow JW Primary Pulmonary Hypertension Study Group. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med. 1996;334:296–301. doi: 10.1056/NEJM199602013340504. doi: 10.1056/NEJM199602013340504. [DOI] [PubMed] [Google Scholar]
- 17.Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, Frost A, Barst RJ, Badesch DB, Elliott CG, Liou TG, McGoon MD. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation. 2010;122:164–172. doi: 10.1161/CIRCULATIONAHA.109.898122. doi: 10.1161/CIRCULATIONAHA.109.898122. [DOI] [PubMed] [Google Scholar]
- 18.Benza RL, Gomberg-Maitland M, Miller DP, Frost A, Frantz RP, Foreman AJ, Badesch DB, McGoon MD. The REVEAL Registry risk score calculator in patients newly diagnosed with pulmonary arterial hypertension. Chest. 2012;141:354–362. doi: 10.1378/chest.11-0676. doi: 10.1378/chest.11-0676. [DOI] [PubMed] [Google Scholar]
- 19.CORE: Center for Outcomes Research and Evaluation, 2018. The Yoda Project, Yale University. http://yoda.yale.edu/welcome-yoda-project. Accessed May 5, 2019.
- 20.McGoon MD, Krichman A, Farber HW, Barst RJ, Raskob GE, Liou TG, Miller DP, Feldkircher K, Giles S. Design of the REVEAL registry for US patients with pulmonary arterial hypertension. Mayo Clin Proc. 2008;83:923–931. doi: 10.4065/83.8.923. doi: 10.4065/83.8.923. [DOI] [PubMed] [Google Scholar]
- 21.Kishnani PS, Corzo D, Nicolino M, Byrne B, Mandel H, Hwu WL, Leslie N, Levine J, Spencer C, McDonald M, Li J, Dumontier J, Halberthal M, Chien YH, Hopkin R, Vijayaraghavan S, Gruskin D, Bartholomew D, van der Ploeg A, Clancy JP, Parini R, Morin G, Beck M, De la Gastine GS, Jokic M, Thurberg B, Richards S, Bali D, Davison M, Worden MA, Chen YT, Wraith JE. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;68:99–109. doi: 10.1212/01.wnl.0000251268.41188.04. doi: 10.1212/01.wnl.0000251268.41188.04. [DOI] [PubMed] [Google Scholar]
- 22.Nicolino M, Byrne B, Wraith JE, Leslie N, Mandel H, Freyer DR, Arnold GL, Pivnick EK, Ottinger CJ, Robinson PH, Loo JC, Smitka M, Jardine P, Tatò L, Chabrol B, McCandless S, Kimura S, Mehta L, Bali D, Skrinar A, Morgan C, Rangachari L, Corzo D, Kishnani PS. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet Med. 2009;11:210–219. doi: 10.1097/GIM.0b013e31819d0996. doi: 10.1097/GIM.0b013e31819d0996. [DOI] [PubMed] [Google Scholar]
- 23.D’Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, Fishman AP, Goldring RM, Groves BM, Kernis JT, Levy PS, Pietra GG, Reid LM, Reeves JT, Rich S, Vreim CE, Williams GW, Wu M. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115:343–349. doi: 10.7326/0003-4819-115-5-343. [DOI] [PubMed] [Google Scholar]
- 24.McLaughlin VV, Sitbon O, Badesch DB, Barst RJ, Black C, Galiè N, Rainisio M, Simonneau G, Rubin LJ. Survival with first-line bosentan in patients with primary pulmonary hypertension. Eur Respir J. 2005;25:244–249. doi: 10.1183/09031936.05.00054804. doi: 10.1183/09031936.05.00054804. [DOI] [PubMed] [Google Scholar]
- 25.Klein JP, Rizzo JD, Zhang MJ, Keiding N. Statistical methods for the analysis and presentation of the results of bone marrow transplants. Part 2: Regression modeling. Bone Marrow Transplant. 2001;28:1001–1011. doi: 10.1038/sj.bmt.1703271. doi: 10.1038/sj.bmt.1703271. [DOI] [PubMed] [Google Scholar]
- 26.Hlatky MA, Califf RM, Harrell FE, Jr, Lee KL, Mark DB, Pryor DB. Comparison of predictions based on observational data with the results of randomized controlled clinical trials of coronary artery bypass surgery. J Am Coll Cardiol. 1988;11:237–245. doi: 10.1016/0735-1097(88)90086-1. [DOI] [PubMed] [Google Scholar]