

Abstract
Several different self-assembling peptide systems that form nanofibers have been investigated as vaccine platforms, but design principles for adjusting the character of the immune responses they raise have yet to be well articulated. Here we compared the immune responses raised by two structurally dissimilar peptide nanofibers, one a β-sheet fibrillar system (Q11), and one an α-helical nanofiber system (Coil29), hypothesizing that integrated T-cell epitopes within the latter would promote T follicular helper (Tfh) cell engagement and lead to improved antibody titers and quality. Despite significantly different internal structures, nanofibers of the two peptides exhibited surprisingly similar nanoscale morphologies, and both were capable of raising strong antibody responses to conjugated peptide epitopes in mice without adjuvant. Both were minimally inflammatory, but as hypothesized Coil29 nanofibers elicited antibody responses with higher titers and avidities against a conjugated model epitope (OVA323–339) and a candidate peptide epitope for vaccination against S. aureus. Subsequent investigation indicated that Coil29 nanofibers possessed internal CD4+ T cell epitopes: whereas Q11 nanofibers required co-assembly of additional CD4+ T cell epitopes to be immunogenic, Coil29 nanofibers did not. Coil29 nanofibers also raised stronger germinal center B cell responses and follicular helper T cell (Tfh) responses relative to Q11 nanofibers, likely facilitating the improvement of the antibody response. These findings illustrate design strategies for improving humoral responses raised by self-assembled peptide nanofibers.
Graphical Abstract
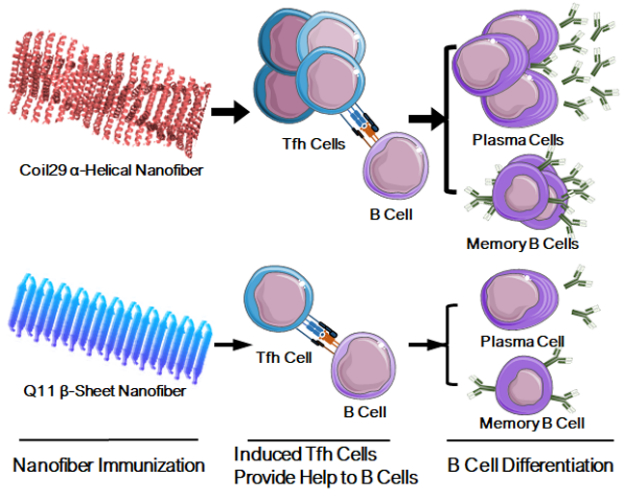
Coil29 nanofibers with integral T cell epitopes generated strong humoral responses and B cell memory by eliciting additional Tfh cells.
Introduction
Recent progresses in personalized vaccines,1, 2 synthetic adjuvant design,3–6 structural-based vaccine design,7, 8 and delivery strategies9, 10 have all greatly improved the ability to generate antigen-specific cellular and humoral immune responses to combat infectious diseases and cancer. However, for many pathogens it remains challenging to raise high-affinity protective antibodies with vaccines, including HIV and malaria.11 High-quality protective antibodies are generated by B cells that have undergone somatic hypermutation and affinity maturation in germinal centers,12, 13 where they receive help from T follicular helper (Tfh) cells through cognate interactions.14–16 Tfh cells play crucial roles in humoral immunity, including selecting B cells for germinal center formation,17 directing B cells affinity maturation,18, 19 and driving the development of memory B cells 20. Much current research is focused on enhancing immunity with adjuvants that act primarily on innate immune cells or by improving antigen trafficking to relevant lymphoid tissues,9, 10, 21–23 but the therapeutic potential of promoting Tfh generation specifically is less investigated, with limited technological platform available.10, 22−24
Adjuvants have been utilized extensively to promote humoral responses.25 Many adjuvants, such as CpG and monophosphoryl lipid A (MPLA), are agonists for various toll-like receptors.24, 26 Other adjuvants based on emulsions or particulates, such as MF59 or alum primarily rely on NLRP3 inflammasome activation to stimulate innate immunity.27–29 Although such adjuvants are commonly quite useful for enhancing immune responses to vaccines, they do not always generate high levels of Tfh cell responses,30, 31 and the strong inflammatory responses they induce may be difficult to balance with immunogenicity, challenging to implement clinically,32 or even counterproductive with respect to vaccine efficacy.33 Recent emerging platforms, including self-assembling protein nanoparticles,34, 35 peptide antigen-conjugated synthetic particles,36, 37 and self-assembling peptide nanofibers38–41 have been found to induce strong cellular and humoral responses without explicitly requiring such inflammatory agonists. Such biomaterial-based and nanomaterial-based platforms provide an alternative strategy to elicit strong humoral responses while minimizing potentially detrimental inflammation.
We have previously reported self-adjuvanting self-assembled peptide nanofiber vaccines based on two considerably different modes of self-assembly. Nanofibers of the first type have been based on β-sheet fibrillizing peptides,38–40, 42 primarily the peptide Q11 (QQKFQFQFEQQ). Nanofibers of the second type are comprised of peptides forming nearly entirely α-helical structures, namely the peptide Coil29 (QARILEADAEILRAYARILEAHAEILRAD).41 In this system, the constituent peptides form α-helical ladder-like structures in which each layer consists of four peptides with their C-termini pointing inward towards the fiber axis and the N-termini projecting radially from the fiber axis.43 The core Coil29 peptide was originally designed by Egelman, Conticello, and co-workers,43 and in subsequent work we found that it was still capable of self-assembling into helical nanofibers even when its N-terminus was appended with various peptide epitopes, including the universal T-cell epitope PADRE, a tumor-specific B-cell epitope from the epidermal growth factor receptor class III variant, or the model CD8+ T-cell epitope SIINFEKL.41 Both β-sheet and α-helical nanofibers have been previously found to raise strong humoral and cellular immune responses without supplemental adjuvants, indicating that neither secondary structure per se is essential for the adjuvanting effects of self-assembled peptide nanofibers.
Given the considerable differences in primary, secondary, and supramolecular structures between these two platforms, we hypothesized that they would elicit different immune phenotypes, and in the present study we sought to elucidate these differences, hypothesizing that potential endogenous T cell epitopes within the Coil29 sequence could provide platform-specific T cell help for humoral responses and improve Tfh cell differentiation relative to other peptide nanofibers. We tested this hypothesis by comparing immune responses raised by Coil29 and Q11, first using the model T- and B-cell epitope OVA323–339 (ISQAVHAAHAEINEAGR), and subsequently using a B-cell epitope of interest for vaccination against S. aureus, E214.44 We comprehensively evaluated a wide range of immune responses, including the strength and quality of antibody responses, any inflammatory responses associated with the immunizations, dendritic cell activation, and Tfh cell generation. Our findings shed light on the features of peptide self-assemblies influencing the character of the immune responses they raise.
Results
Coil29 nanofibers elicit strong humoral responses
We initially studied whether the α-helical Coil29 nanofiber system raised antibody responses of differing quality and strength compared to the β-sheet fibrillized Q11 system and found key differences between the two. Using microwave-assisted solid phase peptide synthesis, we synthesized Coil29, Q11, and both peptides bearing the MHC class II-restricted ovalbumin peptide (OVA323–339) at their N-termini (Table S1 lists all peptide sequences). Coil29 nanofibers bearing OVA were created by co-assembling the OVA-Coil29 and Coil29 peptides in a 1:2 ratio in PBS using methods previously described.41 We prepared Q11 β-sheet nanofibers bearing OVA epitopes in the same molar ratio by co-assembling OVA-Q11 with unmodified Q11 peptide. The 1:2 ratio was selected for optimal OVA-Coil29 nanofiber formation, as we observed irregularities in nanofiber formation at higher loading of OVA-Coil29 within the nanofibers. (Figure S1) Nanofibers from each peptide system exhibited remarkably similar morphologies under transmission electron microscopy (Figure 1A) despite their markedly different internal structures, Coil29 nanofibers being almost entirely α-helical and Q11 nanofibers being rich in β-sheet structure, consistent with previous reports38, 41 (circular dichroism shown in Figure S2). Solutions of the two different types of nanofibers also exhibited similar viscosity (Figure S3) suggesting similar length and entanglement of the fibers, as the entanglement between nanofibers is likely the main contributor to the mechanical properties of these two systems.45 Both nanofibers were able to deliver the OVA epitope such that it was efficiently acquired and presented by BMDCs (Figure S4). Mice were immunized with each of the pOVA-carrying nanofiber constructs, and as a frame of reference we also immunized mice with OVA peptide (pOVA) adjuvanted by either Alum or Sigma adjuvant system (SAS). Alum is known to promote Th2-biased responses, while SAS, comprised of Monophosphoryl Lipid A (MLPA), trehalose dicorynomycolate, and squalene, generates more balanced humoral responses and has been shown to be effective for subunit peptide vaccines.42 C57BL/6 mice were immunized subcutaneously and subsequently boosted 4 weeks later, and anti-pOVA serum antibodies were measured in two-week intervals by IgG ELISA.
Figure 1. Coil29 nanofibers promoted humoral responses with high IgG antibody titers, superior antibody quality, and more antigen-specific memory B cells relative to Q11 nanofibers.

Groups of C57BL/6 mice (n=5) were immunized s.c. either with 120 μg of OVA peptides (0.067 mmol) delivered with Alum salt or SAS adjuvant, with pOVA-Coil29, or pOVA-Q11 nanofibers carrying equivalent molar amounts of the OVA peptide, followed by booster injections with half of the primary dose on week 4. All values shown are mean ± SEM. (A) Both pOVA-Coil29 and pOVA-Q11 self-assemblies exhibited similar nanofiber morphologies, with diameters of 10.9±0.5 nm and 10.2±0.2 nm respectively by TEM (B) Total serum anti-OVA IgG titers measured by ELISA (* p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 compared to pOVA/Alum by two-way ANOVA with Dunnett post-hoc test). (C) Representative surface plasma resonance binding curves of polyclonal sera from the four immunization groups. The window indicates the region where kd was calculated. (D) Serum antibody avidity scores (the ratio of binding responses to the dissociation rate, RU/kd) on week 4(prior to boosting), week 5 (after boosting), week 24 (end point) (** p<0.01, **** p<0.0001 by two-way ANOVA with Tukey post-hoc test). (E) Frequency of OVA-specific memory B cells among memory B cells from immunized mice 24 weeks after primary immunizations (gating, left; quantification, right; p-value calculated using one-way ANOVA with Dunnett post-hoc test, comparing to pOVA-Q11 group).
The Coil29 platform was superior in its ability to elicit antibody responses, showing the greatest response of all materials tested after primary immunization, after boosting, and throughout the 24-week duration of the experiment (Figure 1B). Q11 also raised durable antibody responses, but they were of a lower magnitude. Alum was a poor adjuvant for pOVA, consistent with previous findings,39 and SAS raised titers similar to Coil29, although only after boosting. To assess the quality of antibodies elicited by different platforms, we utilized surface plasmon resonance (SPR) using methods adapted from Lynch et al.46 Briefly, in this method the antibody avidity score is defined as the ratio of binding responses to the dissociation rate (RU/kd, s−1). The early kinetic binding response (relative unit, RU) primarily reflects changes in antibody concentration, whereas the early dissociation constant (kd) indicates antibody binding affinity to the peptide antigens, independent of antibody concentration, which can be calculated from the slope of the curve (see methods for complete details). By taking both antibody concentration and binding affinities into consideration, the avidity scores indicate overall antibody quality (Figure 1C, D, and Figure S5). Utilizing this scoring system, we found that at peak titers (week 5, immediately after the boost), Coil29 elicited higher-quality antibodies than Alum (p<0.0001), SAS (p < 0.01) or Q11 (p < 0.0001) (Figure 1D). The Coil29 group continued to have the highest average scores at week 24, but by this timepoint the differences were not statistically significant.
We next examined antigen-specific B cell memory 24 weeks after immunization, finding that unadjuvanted Coil29 nanofibers also raised significantly more pOVA antigen-specific memory B cells (CD138-lgD-CD38+CD95+B220+pOVA+) compared to Q11 (Figure 1E). These levels were also on average greater than Alum or SAS, but not to a statistically different degree. Overall, these data suggested that Coil29 nanofibers, without adjuvant, elicited antibody responses with higher titers, higher quality, and better memory B-cell responses than Q11.
Coil29 contains integral T-cell epitopes
Having observed the superior ability of Coil29 nanofibers to raise antibody responses, we hypothesized that potential novel internal T-cell epitopes in the Coil29 sequence may be responsible, so we next investigated T-cell responses to the different platforms. Because our search of the immune epitope database (www.iedb.org) did not yield any known T cell epitope for Coil29 peptide, we decided to utilize ELISPOT assays to determine T cell responses against the Coil29 peptides. C57BL/6 mice were immunized subcutaneously with OVA-Coil29, OVA-Q11, or adjuvanted peptides and boosted once 14 days later. On day 21, spleens were harvested and splenocytes were stimulated with soluble pOVA peptide, Coil29, or Q11 and evaluated using ELISPOT. Platform-specific cellular responses (i.e. against the core nanofiber-forming Coil29 or Q11 sequences) were found only in mice immunized with Coil29-based nanofibers, and not against Q11 (Figure 2A, B). This implied that only the Coil29 sequence contained competent T-cell epitopes. With respect to responses against the OVA epitope, Coil29 and Q11 nanofibers elicited similar levels of pOVA-specific IFNγ-producing and IL-4-producing cellular responses, suggesting a similar ability of the two nanofibers to generate T-cell responses to attached T-cell epitopes. SAS adjuvant elicited higher and more variable IFNγ responses, and Alum generated slightly lower IFNγ responses.
Figure. 2. Multiple T-cell epitopes were detected in Coil29 peptide sequences.

C57BL/6 mice were immunized s.c. with equivalent molar amounts of pOVA epitope peptides delivered by either pOVA/Alum (green), pOVA/SAS emulsion (purple), pOVA-Coil29 nanofibers (red), or pOVA-Q11 nanofibers (blue) on day 0 (0.2 ×10−3 mol peptide) and day 14 (0.1 ×10−3 mol peptide) (n=4 per group). Splenocytes were harvested on day 21 for ELISPOT. All values shown are mean ± SEM. (A, B) Both nanofibers elicited similar levels of OVA-specific cellular responses secreting IFNγ (A) and IL4 (B), but only mice immunized with pOVA-Coil29 nanofibers generated significant responses against the relevant nanofiber-forming portions of the peptides. A series of overlapping 15-mer peptides spanning portions of the Coil29 sequence (C) were used to stimulate splenocytes from mice immunized with pOVA-Coil29 or pOVA-Q11 ELISPOT, identifying multiple T cell epitopes toward the C-terminus of Coil29 (D), * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 by two-way ANOVA multiple comparison with Sidak post-hoc test.
To search for CD4+ T-cell epitopes within the Coil29 sequence, we created a library of five different overlapping 15-mer Coil29 fragment peptides, Coil29a-e (Figure 2C). In an IFNγ ELISPOT assay, splenocytes from mice immunized with either pOVA-Coil29 or pOVA-Q11 nanofibers were stimulated with one of these fragment peptides, the full-length Coil29, or pOVA. Significant cellular responses were detected in Coil29-experienced splenocytes when stimulated with Coil29, Coil29c, Coil29d, or Coil29e, and statistically insignificant responses were generated by Coil29a or Coil29b (Figure 2D). Again, comparable numbers of pOVA-specific spot-forming cells were detected for mice immunized with the full OVA-Coil29 and OVA-Q11 peptides. These findings confirmed the presence of multiple T cell epitopes in the Coil29 sequence, with strongest activity near the C-terminus.
Coil29 nanofibers promoted germinal center B cells
To generate high quality antibodies, B cells need to be activated during antigen acquisition and must receive sufficient T cell help to proliferate and differentiate into germinal center B cells. Germinal center B cells will then undergo somatic hypermutation and affinity maturation, with the help of T cells, to eventually mature into antibody-producing plasma cells and memory B cells.13 Considering the improved antibody responses observed for Coil29 nanofibers, we hypothesized that with their internal T cell epitopes they could promote the differentiation of germinal center B (GCB) cells by providing additional T cell help. To test this hypothesis, we measured germinal center B cells (GL7+Fas+ B cells) in the draining lymph nodes 3 weeks after primary immunizations and found that Coil29 nanofibers stimulated the highest proportion of GCB cells among all B cells, an average percentile of 5.0% (Figure 3A). SAS adjuvant generated 3.2% GCB cells, and both Q11 nanofibers and Alum generated 1.3% GCB. While Coil29 generated higher titers and higher proportions of germinal center B cells, they did not generate the highest proportion of activated B cells. Rather, both adjuvanted formulations generated the highest proportion of activated B cells (MHCII+CD86+), with Coil29 and Q11 generating relatively fewer activated B cells (Figure 3B). We also did not find measurable differences in plasma cell generation in the four groups (Figure S6). Taken together with the titer data, these results indicated that Coil29 nanofibers’ improved antibody titers were associated with improved germinal center B cell formation, but not necessarily B cell activation or plasma cell formation.
Figure 3. Coil29 nanofibers promoted germinal center B cell differentiation relative to Q11 nanofibers.

C57BL/6 mice (n=3) were immunized s.c. with pOVA-Coil29 or pOVA-Q11 nanofibers containing the same molar amount (67 μmol) of OVA epitopes. Control groups received the same amount of peptide delivered with Alum or SAS adjuvant. Draining lymph nodes were harvested from immunized mice at indicated time points to count germinal center B cells (A; DAPI-CD3-F4/80-GL7+Fas+B220+) and activated B cells (B; DAPI-CD3-F4/80-MHCII+CD86+B220+) via flow cytometry. Representative flow plots (left) and frequency among B cells (right) are shown. All values reported are mean ± SEM. * p<0.05, ** p<0.01, *** p<0.001 by two-way ANOVA with Dunnett post-hoc test, as compared to Alum (A), and Q11 (B).
Coil29 nanofibers are minimally inflammatory
It has been found previously that although Q11 nanofibers are immunogenic, they generate almost no inflammatory responses, in stark contrast with most other adjuvants.39 We hypothesized that Coil29 nanofibers would be similarly non-inflammatory, because both nanofiber platforms lack known pathogen-associated molecular pattern (PAMP) or damage-associated molecular pattern (DAMP) molecules and exhibit similar physical properties after assembly. To investigate this, we measured the concentration of inflammatory chemokines (MCP-1/CCL2) and cytokines (G-CSF, TNF, IL-5, IL-6, IL-1β), along with the numbers of recruited immune cells to the intraperitoneal (i.p.) space after i.p. immunization (Figure 4). Twelve hours after injections, SAS adjuvant induced significant levels of MCP-1 (~ 990 pg/mL) and G-CSF (~ 580 pg/mL) (Figure 4A). Alum adjuvant stimulated the highest production of other examined cytokines (TNF, IL-1β, IL-5, and IL-6). In stark contrast, both Coil29 and Q11 elicited minimal cytokine responses, indistinguishable from PBS injections, with the one exception being MCP-1 (~ 96 pg/mL), which was induced by Coil29 in 3-fold higher amount than Q11 nanofibers. With respect to cellular infiltration after i.p. immunization (Figure 4B), neither pOVA-Coil29 nor pOVA-Q11 nanofibers recruited significant levels of inflammatory cells in comparison with the two adjuvants. However, we noted that pOVA-Coil29 nanofibers recruited more neutrophils (3-fold higher) and eosinophils (5-fold higher) than Q11. This is consistent with the higher concentration of MCP-1 chemokine observed in Coil29 group, as MCP-1 is one of the key chemokines to regulate migration of monocytes,47 but the reason why Coil29 nanofibers induced higher level of MCP-1 secretion remains to be explored. Despite these small differences, overall we interpreted the minimal inflammatory response exhibited by Coil29 nanofibers as an indication that they do not stimulate humoral responses via highly inflammatory pathways, similarly to Q11 and other peptide nanofibers.
Figure 4. Both Coil29 and Q11 nanofibers induced low levels of inflammatory cytokines and inflammatory cell influx, relative to both Alum and SAS adjuvanted groups.

Cytokine responses and cellular responses in the lavage fluid were analyzed for C57BL/6 mice intraperitoneally immunized with either nanofibers or peptide/adjuvant formulations. All values reported are mean ± SEM. (A) Cytokine secretion levels were determined using cytokine ELISA 12 hours after i.p. immunization. Abbreviation: MCP-1 (monocyte chemotactic protein-1), G-CSF (granulocyte colony-stimulating factor), TNF (tumor necrosis factor), IL (interleukin). (B) Immune cell populations in lavage fluid were analyzed 1 hour and 12 hours after immunization using flow cytometry. For both (A) and (B): * p<0.05, ** p<0.01, *** p<0.001 **** p<0.0001 by two-way ANOVA with Tukey post-hoc test.
Coil29 nanofibers are avidly acquired by dendritic cells in vivo
Dendritic cell (DC) uptake and activation are crucial for the generation of antigen-specific humoral immunity, so we evaluated the ability of DCs to internalize antigen delivered by Coil29 using TAMRA-conjugated nanofibers and flow cytometry after i.p. injections. In peritoneal lavage fluid collected from mice 12 hours after immunization, over 40% of DCs were found to internalize Coil29 nanofibers, greater than Q11 (25%, Figure 5). For adjuvanted groups and Q11, between 20% and 29% of dendritic cells were found to be TAMRA-positive. Moreover, mean fluorescence intensity (MFI) of TAMRA in DCs for Coil29 groups was about 4-fold higher than the other three groups (Figure 5B). The ability of nanofibers to activate DCs was assessed by measuring the upregulation of CD80 and CD86 (Figure 5C). We previously showed that for Q11 nanofibers, only DCs that acquired the materials were activated.39 Here, Coil29 nanofibers also exhibited the same behavior: DCs that internalized TAMRA-pOVA-Coil29 nanofibers exhibited increased CD80 and CD86, whereas DCs that did not acquire TAMRA-labeled nanofibers did not become activated (Figure 5C). Moreover, the MFI values for both CD86 and CD80 were about 2–3-fold higher for Coil29 compared to Q11, potentially indicating a greater activation of the DCs.
Figure 5. Coil29 nanofibers were acquired by DCs and activated DCs to a greater degree than Q11 or adjuvanted formulations.

Groups of C57BL/6 mice (n=3) were immunized i.p. with either PBS, TAMRA-labeled OVA peptides formulated with Alum or SAS adjuvants, TAMRA-labeled pOVA-Coil29, or pOVA-Q11 nanofibers. DCs (DAPI-F4/80-CD11c+MHCII+) in lavage fluid were counted 12 hours after immunization using flow cytometry. All values shown are mean ± SEM. (A) Representative flow panels indicating the percentage of TAMRA+ DCs among DCs (left), and combined results (right). (B) Mean fluorescence intensity of TAMRA in dendritic cells. (* P<0.05, by one-way ANOVA with Tukey post-hoc test) (C) Representative fluorescence histograms (left) and combined results (right) of CD86 and CD80 in antigen+ (TAMRA-pOVA+) dendritic cells 12 hours after immunization, compared to the DC population that did not acquire antigen (TAMRA-pOVA-). Asterisks indicate statistical significance as determined by one-way ANOVA with Tukey post-hoc test (* p<0.05).
Coil29 nanofibers generated Coil29-specific Tfh cells
To examine the extent to which additional T cell epitopes contributed to the improved humoral responses elicited by the Coil29 platform, we first compared the T follicular helper (Tfh) cell frequency in vaccine-draining lymph nodes between pOVA-Coil29 and pOVA-Q11 nanofibers. Lymphocytes were harvested 7 days after primary immunization and stained for Tfh cell markers (CD4+CD44+PD-1+CXCR5+). Higher frequencies of Tfh cells among antigen-experienced CD4+ T cells (CD4+CD44+) were identified in the lymph nodes of Coil29-immunized mice compared to Q11-immunized mice (p = 0.0286). To further investigate the presence of Tfh cells specific to Coil29, we utilized the activation induced marker (AIM) assay pioneered by the Crotty group48, 49 and performed in vitro stimulation with either pOVA peptide or Coil29 peptide on lymphocytes harvested from draining lymph nodes 10 days after immunization with nanofibers (Figure 6B). Tfh cell activation markers (CD25 and OX40) were measured 30 h after incubation with peptide antigens. Stimulation with pOVA peptides resulted in comparable levels of Tfh cell activation between the two groups. However, incubation with Coil29 peptide led to significant upregulation of activation markers in Tfh cells isolated from the Coil29 group, but not from the Q11 group (p=0.011). Because the frequency of pOVA-specific Tfh cells were similar between the two nanofiber platforms, the higher levels of total Tfh cells in the Coil29 group was likely caused by the additional Coil29-specific Tfh cells. Considering that Tfh cells are essential in humoral immunity as they govern hyper somatic mutation for antibody affinity maturation, the increase of Coil29-specific Tfh cells was consistent with our previous finding that Coil29 nanofibers generated higher-quality antibody responses.
Figure 6. Coil29 nanofibers induced greater numbers of follicular T helper (Tfh) cells than Q11 nanofibers.

(A) Draining lymph nodes were harvested 7 days after immunizations with either pOVA-Coil29 or pOVA-Q11 nanofibers. Tfh (CXCR5+PD-1+) were counted among CD44+CD4+ T cells. Representative flow cytometry plots (left) and Tfh cell frequency (right) are shown (n = 4, p-value calculated with unpaired t-test). (B) Coil29 nanofibers induced platform-specific Tfh cell responses. Lymphocytes from draining lymph nodes of immunized mice were split into three groups (unstimulated, pOVA-stimulated, Coil29 stimulated). CD25+OX40+ cells were counted by flow cytometry 18 hours after stimulation treatments. Representative flow plots (left) and antigen-specific Tfh cell frequency (right) are shown. P value was calculated with multiple t test using Holm-Sidak method for multiple comparison correction.
Coil29 nanofibers promoted humoral responses and strong B cell memory against a vaccine-relevant epitope from S. aureus
We finally examined the capacity of Coil29 nanofibers to evoke humoral responses against a candidate epitope for vaccination against methicillin-resistant S. aureus. This peptide, E214 (Ac-KFEGTEDAVETIIQAIEA-amide), is a B cell epitope from the S. aureus enolase protein. Conjugated on a carrier protein, it can raise antibodies with protective capacities against methicillin-resistant S. aureus when delivered with adjuvants.50 Previously, Q11 nanofibers exhibited an ability to raise antibodies against E214 epitopes, but only when E214 peptides were also co-assembled with a CD4+ T-cell epitope peptide, PADRE (H2N-aKXVAAWTLKAa-amide, where “X” is cyclohexylalanine and “a” is D -alanine).44 Consistent with our previous findings, PADRE T-cell help was again found to be essential for Q11 nanofibers to generate detectable IgG titers against E214 peptide, reiterating that humoral responses against E214 peptide are T-dependent and that Q11 nanofibers alone cannot provide the T-cell help required (Figure 7A). Conversely, Coil29 nanofibers conjugated to E214 alone stimulated strong anti-E214 IgG antibody production over 30 weeks, and the addition of PADRE within the co-assembly yielded only a slight increase in IgG titers that was not statistically significant (Figure 7A). The splenocytes from all immunized mice were collected for the analysis of class-switched E214-specific memory B cells (CD138-lgD-CD38+CD95+B220+E214+) (Figure 7B), and it was found that E214/PADRE Coil29 nanofibers generated the highest frequency of antigen-specific memory B cells (average 4.1%). Q11 bearing E214 raised considerably lower memory B cell responses, whereas Q11 nanofibers bearing E214 and PADRE performed more similarly to Coil29 bearing E214 and SAS-adjuvanted formulations. Alum-adjuvanted E214 raised poor memory B cell responses. Taken together, Coil29 nanofibers alone can provide T-cell help required for T-dependent humoral responses, and with additional help from incorporated T cell epitopes, antibody responses can be improved further.
Figure 7. Coil29 nanofibers induced strong long-term E214-specific IgG humoral responses and promoted higher antigen-specific memory B cell frequency when coassembled with additional (0.01 mmol) PADRE T cell epitopes.

Groups of C57BL/6 mice (n=5) were immunized s.c. either with 134 μg of E214 peptides (0.067 mmol) delivered with Alum or SAS adjuvant, with E214-Coil29 (E214-C), or E214-Q11 (E214-Q) nanofibers carrying equivalent amounts of E214 peptides, with or without co-assembled PADRE-Coil29 or PADRE-Q11 (PAD-C or PAD-Q, respectively) followed by booster injections on week 4. All values shown are mean ± SEM. (A) Total serum anti-E214 IgG titers measured by ELISA (* p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 compared to E214/Alum group by two-way ANOVA with Dunnett post-hoc test). (B) Frequency of E214-specific memory B cells among memory B cells from immunized mice 30 weeks after primary immunizations. *p<0.05 compared to E214-Q group by ordinary one-way ANOVA with Dunnett post-hoc test.
Discussion
Biomaterials have been investigated extensively to promote strong protective immune responses by prolonging antigen duration,9, 10 codelivering adjuvants,51–53 facilitating antigen trafficking,54, 55 or recruiting specific immune cells.56–58 Much attention has focused on innate immune responses, for example facilitating activation and antigen presentation by APCs, but the B cell affinity maturation process is not always improved by the adjuvants delivered,59 and sometimes it is even impaired.33 This highlights the complexity of humoral immune responses and also reveals the unpredictability associated with enhancing humoral responses using adjuvants alone.
We took advantage of the inherent T cell epitopes within the α-helical Coil29 nanofiber system that we identified through ELISPOT assays and focused on the quality of humoral responses elicited by Coil29 nanofibers. We found that Coil29 nanofibers rapidly generated substantial antibody titers after primary immunizations (103 on week 3), while Q11 nanofibers elicited comparatively lower titers. Although the MPLA/squalene-based SAS adjuvant raised OVA-specific antibodies at comparable titers, SPR experiments revealed that Coil29-generated antibodies had higher avidity scores, suggesting that SAS adjuvant amplified the magnitude of responses without significantly improving the affinity maturation process. In addition, Coil29 nanofiber immunizations led to higher levels of OVA-specific memory B cells in splenocytes relative to Q11 nanofibers.
We interpret these results as indicating that platform-specific T cell responses elicited by OVA-Coil29 nanofibers potentially took part in the antibody generation process by providing T cell help to OVA-specific B cells. To this end, we demonstrated that higher frequencies of germinal center B cells were induced by Coil29 nanofibers, while similar frequencies of GC B cells were found in Q11 nanofibers and Alum-adjuvanted groups. This hypothesis was further confirmed upon finding improved Tfh cell frequencies in Coil29-immunized mice and by identifying the presence of Coil29-specific Tfh cells. As the OVA epitopes and Coil29 peptides are covalently linked via a short peptide linker (SGSG), it is reasonable to assume that some B cells can present Coil29 epitopes within their MHC class-II molecules once they have internalized the entire OVA-Coil29 peptide, making the cognate interaction between B cells and Coil29-specific T cells possible. Interestingly, neither nanofiber system was as potent as adjuvanted groups for activating B cells. This may be due to the minimally inflammatory nature of nanofibers compared to adjuvanted groups.
Tfh cells are crucial in T-dependent humoral immunity because they maintain germinal centers and influence the selection of high-affinity B cells, ultimately leading to long-term serological memory and B cell memory.60, 61 Carrier proteins such as tetanus toxoid and keyhole limpet hemocyanin have been extensively utilized to improve the immunogenicity of subunit vaccines, in part owing to their ability to generate T cell help.62, 63 However, carrier protein-conjugated subunit vaccines are generally administered with adjuvants to generate immune responses, and the control over antigen density on the protein is limited. Conversely, the present epitope-bearing nanofiber platform can self-assemble in a well-defined fashion and thus yield improved control over epitope dosing. Moreover, Coil29 nanofibers not only exhibited strong self-adjuvanting humoral immunogenicity similar to Q11 nanofibers, but they also significantly improved the generation of memory B cells and the quality of antibodies. Although the exact mechanism is unclear, the additional platform-specific T cell responses stimulated by Coil29 peptides, especially the Coil29-specific Tfh cells, potentially contributed to B cell maturation during germinal center formation, subsequently driving memory B cell differentiation for the two epitopes tested in the current study. This finding raised important considerations for biomaterials vaccine delivery platforms, especially for inducing strong humoral immunity. It is worth noting that these findings may not apply to all B cell epitopes, however, as Coil29 nanofibers were previously shown to only generate low levels of IgG against a tumor-specific growth factor receptor variant III (PEPvIII) without additional T cell epitopes,41 which may have arisen from low immunogenicity of PEPvIII B cell epitopes. Given the T cell immunogenicity of the Coil29 sequence, future experiments could be carried out to determine the effectiveness of Coil29 as a T cell epitope in direct comparison with other T cell epitopes such as PADRE.
Another interesting observation is that when compared to adjuvanted groups, both Q11 and Coil29 nanofibers induced low levels of inflammatory cytokine secretion and inflammatory cell influx. This minimally inflammatory character may be a general feature of peptide self-assemblies, making them useful in applications where inflammation must be minimized, for example in active immunotherapies against autologous targets.64
There are other important differences in the immunogenicity of these two nanofiber platforms revealed by the present study. For example, Coil29 nanofibers stimulated slightly higher levels of macrophage recruitment, but this did not necessarily result in significant increases in the inflammatory cytokines IL-1β, IL-6, or TNF, confirming the minimally inflammatory nature of Coil29 nanofibers. Also, Coil29 nanofibers were more avidly internalized by dendritic cells than Q11 nanofibers after i.p. administration. The improvement in dendritic cell uptake and activation by Coil29 could possibly be caused by the modestly increased inflammatory reactions induced by Coil29, but this remains to be fully investigated. Another potential reason for the differences in DC internalization may be that the α-helical secondary structure of Coil29 nanofibers engage DC surface receptors more effectively than Q11, as some cytokines share similar helical structures.65 It is also possible that the potentially more stable β-sheet Q11 nanofibers can withstand shearing during injections better than Coil29 nanofibers, and any resulting Coil29 nanofiber fragments could be more accessible to DCs. However, future studies are needed to pinpoint the underlying immunologic mechanisms causing the observed differences between these two nanofiber systems.
The present study focused on comparing the immunogenicity of two different nanofiber systems in physiological conditions. Both the thermal stability and long-term shelf life of the nanofiber solutions will need to be determined in the future, although we previously demonstrated that Q11 nanofibers maintain their immunogenicity after extensive exposure to heat.66 The impact of changes in ionic strength or serum proteins would also be interesting to explore, as it is possible that the two nanofiber systems may be variably stable in vivo.
Overall, α-helical peptide nanofibers constructed using the Coil29 platform share considerable similarities with structurally dissimilar β-sheet nanofibers constructed using the Q11 platform. Both raise antibody titers without additional adjuvants, and both were found to be non-inflammatory. However, the Coil29 platform was found to raise antibody responses of a higher titer and quality, which was associated with better germinal center B cell formation, better acquisition by and activation of DCs, and better Tfh cell responses. In vaccine applications where maximization of antibody quality and titer is placed at a premium, these considerations may favor selection of Coil29. Countervailing factors to consider include that the greater length of the Coil29 sequence may be associated with greater costs of synthesis and purification. Moreover, Q11 nanofibers have been previously found to be stable for extended durations in temperature regimes outside the standard “cold chain” range,66 and it is not yet known the extent to which Coil29 nanofibers may be similarly stable.
Conclusion
This work indicated that peptide nanofibers constructed with vastly different internal peptide structures share the ability to raise antibody responses in the absence of adjuvant. Relative to Q11 nanofibers, however, Coil29 nanofibers raised antibody responses of higher titers and quality, enhanced GC formation, and improved B cell memory. ELISPOT assays revealed the presence of T cell epitopes within the helical self-assembling structure of Coil29, and the significant role of such platform-specific T cell responses in driving humoral immunity was suggested by the observation of Coil29-specific Tfh cells. Despite improved immunogenicity of Coil29 nanofibers relative to Q11 nanofibers, they remained minimally inflammatory, even though dendritic cells avidly internalized Coil29-based nanofibers and were effectively activated by them. Although self-adjuvanting properties are shared by dissimilar peptide nanofibers, the present study suggests a strategy to augment humoral responses by providing additional T cell help within the self-assembling domains of fibrillar peptide materials. Such an approach may be broadly applicable for improving the efficacy of subunit peptide vaccines.
Materials and methods
Peptide and vaccine preparation
All peptides were synthesized using Fmoc solid-phase chemistry on a CEM Liberty Blue microwave synthesizer and purified with HPLC and MALDI-MS. Biotinylated peptides were prepared on-resin by reacting Biotin-ONp (Novabiochem) in DMF overnight with amine-terminated peptides in a 3-fold excess before cleavage. TAMRA-labeled peptides were synthesized on-resin by reacting 5(6)-TAMRA (Anaspec Inc.) in a 3-fold excess in the presence of N,N’-Diisopropylcarbodiimide (DIC) and 6-Chloro-1-hydroxybenzotriazole (HOBt-Cl).
To prepare peptide fiber solutions for immunizations, lyophilized peptides were weighed separately and mixed in predetermined molar ratios of 1:2 (epitope-bearing peptides: unmodified fiber-forming peptides). Mixed Coil29 peptides were then dissolved in 10 mM sterilized acetate buffer (pH 4) at a total peptide concentration of 8 mM and incubated overnight at 4 °C. The peptide solutions were subsequently diluted to 2 mM in 1×PBS, and incubated for 3 h at room temperature before immunizations. Epitope-bearing Q11 nanofibers were prepared by dissolving pre-mixed peptide powders in sterile water at 8 mM. After overnight incubation at 4 °C, the peptide solutions were diluted and incubated similarly before injections. To prepare alum peptide formulations, OVA or E214 peptides were dissolved at 1.34 mM in 2×PBS, and an equal volume of alum salt (Thermo Fisher, 77161) was mixed with the peptide solutions and vigorously vortexed for 45 min to form emulsions and yield the final peptide epitope concentration of 0.67 mM, equivalent to the epitope concentration in nanofiber formulations. Sigma Adjuvant System (Sigma, S6322) emulsions were prepared following the manufacturer’s protocol. Briefly, antigen peptides were dissolved at 1.34 mM in 1×PBS, and equal volumes of SAS and peptide solutions were mixed and warmed to 37 °C in a water bath and vortexed before immunization.
TEM analysis
Nanofiber solutions were prepared in the same way as for immunizations, with further 10-fold dilution to 0.2 mM with 1×PBS. Five microliters of sample solutions were deposited onto 400 mesh carbon copper grids (EMS400-Cu). The sample grids were then rinsed with DI water droplets three times after 1 min incubation at room temperature and were subsequently stained with 1% uranyl acetate for 30 s before washing and blotting away the excess liquid. Samples were imaged using a FEI Tecnai F30 TEM and analyzed with FEI TEM Imaging and Analysis (TIA) software. The diameters of the self-assembled nanofibers were calculated from 20 measurements made within three different images for both Coil29 and Q11 nanofibers.
Circular dichroism (CD) analysis
A Bioz PiStar-180 CD spectrometer was used with a 0.1 cm path length quartz cell. Nanofiber solutions were prepared in the same way as for immunizations, with further dilution to 0.1 mM with 1×PBS. Circular dichroism spectra were collected between 190 nm and 260 nm with a scanning speed of 0.5 nm/s and a bandwidth of 0.5 nm. Each spectrum shown was the average of three scans with the solvent background subtracted.
Mice and immunizations
Female C57Bl/6 mice (7–9 weeks) were purchased from Harlan laboratory and housed at the animal facility of Duke University. All animal procedures were performed in accordance with the guidelines for care and use of laboratory animals of Duke University and approved by the Institutional Animal Care and Use Committee (IACUC) of Duke University. Duke University’s Animal Care and Use Program is fully accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care, International (AAALAC). Mice were assigned in groups of five for each treatment. Anesthetized mice were immunized subcutaneously each with 100 μL of indicated solutions (50 μL at each shoulder) and boosted with half-doses (2×25μL) at indicated time points. Sera were collected from the submandibular vein to analyze for epitope-specific IgG titers via ELISA. Intraperitoneal injections were administered by injecting 100 μL of peptide solutions into the peritoneal cavity. At indicated timepoints, mice were sacrificed, and 1 mL of intraperitoneal lavage fluid was collected from each mouse for subsequent assays.
T-Cell ELISPOT
ELISPOTs were performed as previously described.41 Briefly, splenocytes were harvested from immunized mice one week after final immunizations. After treatment with ACK lysis buffer (Thermo Fisher, A1049201) and gradient centrifugations with Lympholyte M (Cedarlane CL5031), 0.25 million splenocytes were plated in each well of a 96-well ELISPOT plate (Millipore, MSIPS4510). The cells were then stimulated with indicated peptides at 5×10−3 mM of peptide epitopes, PBS as negative controls, or ConA (Sigma, C5275) as positive control. IFNγ ELISPOT pairs (BD, 551881), IL4 ELISPOT pairs (BD, 551878), streptavidin-alkaline phosphatase (Mabtech, 3310–10) and Sigmafast BCIP/NBT substrate (Sigma, B5655) were used to detect and develop antigen-specific spots. Plates were enumerated by ZellNet Consulting using a Zeiss KS ELISPOT system.
ELISA for serum antibodies
Sera were analyzed by ELISA for epitope-specific IgG as previous described.41 Briefly, high-binding ELISA plates (Costar, 3915) were coated with 5 μg/mL streptavidin (Sigma, 85878) overnight at 4 °C and followed by incubation with 10 μg/mL of biotinylated peptide antigen in PBS for 1 hour at room temperature. IgG detection HRP antibodies (Jackson Immunoresearch) were diluted 5000 fold and detected using SigmaFast PNPP substrate solution (Thermo Fisher, 37621).
Surface plasmon resonance (SPR)
Antigen-binding affinities of serum antibodies were analyzed using a GE Biacore T200. Channel 1 and 2 of a GE Biacore Sensor Chip (CM5) were both activated using 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC) and N-hydroxysuccinimide (NHS), and coated with streptavidin at 5 μL/min to reach an equal level of the total relative unit (RU) of about 1500. Biotinylated OVA peptides (25μg/mL) were only immobilized in channel 2 with a flow rate of 5 μL/min for 30 seconds with the RU increased by 140. The instrument was then equilibrated using PBS with 1% BSA and 0.05% Tween 20 (PBST) as running buffer at a flow rate of 5 μL/min for 1 h. For each sample analysis, PBST running buffer was first flowed through both channel 1 and 2 at a rate of 25 μL/min for 2 min, followed by sera solution (10 fold dilution in running buffer) for 2 min, and finally running buffer for another 5 min, before regeneration with 50 mM NaOH for 30 s twice. To remove non-specific background, binding signals from channel 1 (streptavidin coating only) were subtracted from channel 2, followed by substraction of signals for naïve mouse sera. Stability points were taken 30 s after the injection, and the early dissociation factor (kd) was calculated as the slope of the relative response units (RU) between 70 s and 30 s after injection. Avidity is defined as the ratio of the stability response to the early dissociation rate.46
Antibodies and flow cytometry
Flow cytometry was performed using a BD Canto II instrument or a BD Fortessa instrument. FlowJo software (FlowJo 10.6.1) was used for data analysis. Specific staining procedures and clone numbers are provided in the Supporting Information.
Cytokine level analysis
To determine the chemokine and cytokine levels after i.p. immunizations, lavage fluid was collected from mice 12 hours after immunization, and the supernatants were analyzed using cytokine ELISA kits following manufacturer’s protocols (TNF, Thermo Fisher: 88–7324; IL-5 Thermo Fisher: 88–7054-88, IL-6 Thermo Fisher: 88–7064; IL-1beta, Thermo Fisher: 88–7013; G-CSF Thermo Fisher: EMCSF3; MCP-1, Thermo Fisher: BMS6005).
Statistical analysis
Statistical analysis was performed using GraphPad Prism 6 (GraphPad software, Inc.). Data are presented as mean ± SEM. Comparisons between two groups were performed using two tailed unpaired Student’s t-test. Comparisons of multiple groups at a single time point were performed with one-way analysis of variance (ANOVA), and comparisons of multiple groups over time were performed by using two-way ANOVA tests. Dunnett post-hoc test was used when means are compared to a control group. Tukey post-hoc tests were utilized when all possible pairs of means were compared. Multiple t-tests were used when two groups were compared under different independent stimulations, using the Holm-Sidak method for multiple comparison correction. P-values are presented in the figures as: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Supplementary Material
Acknowledgements
This research was supported by the National Institutes of Health (NIBIB 5R01EB009701), the Duke Cancer Institute, and the Biomedical Engineering Department of Duke University. The MALDI was performed on an instrument supported by the North Carolina Biotechnology Center, grant 2017-IDG-1018. Circular dichroism was performed at the University of North Carolina at Chapel Hill using instruments supported by NIH grant P30CA016086.
Footnotes
Competing Interests
JHC and YW are inventors on a patent application associated with the Coil29 technology described.
Supporting Information
Flow cytometry staining protocols, in vitro DOBW assay procedure, a table of all peptide sequences, circular dichroism analyses, viscosity data, DOBW T cell activation data, raw SPR data, and plasma cell measurement data are shown in the Supporting Information.
References
- 1.Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, Chen C, Olive O, Carter TA, Li S, Lieb DJ, Eisenhaure T, Gjini E, Stevens J, Lane WJ, Javeri I, Nellaiappan K, Salazar AM, Daley H, Seaman M, Buchbinder EI, Yoon CH, Harden M, Lennon N, Gabriel S, Rodig SJ, Barouch DH, Aster JC, Getz G, Wucherpfennig K, Neuberg D, Ritz J, Lander ES, Fritsch EF, Hacohen N and Wu CJ, Nature, 2017, 547, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Löwer M, Bukur V, Tadmor AD, Luxemburger U, Schrörs B, Omokoko T, Vormehr M, Albrecht C, Paruzynski A, Kuhn AN, Buck J, Heesch S, Schreeb KH, Müller F, Ortseifer I, Vogler I, Godehardt E, Attig S, Rae R, Breitkreuz A, Tolliver C, Suchan M, Martic G, Hohberger A, Sorn P, Diekmann J, Ciesla J, Waksmann O, Brück A-K, Witt M, Zillgen M, Rothermel A, Kasemann B, Langer D, Bolte S, Diken M, Kreiter S, Nemecek R, Gebhardt C, Grabbe S, Höller C, Utikal J, Huber C, Loquai C and Türeci Ö, Nature, 2017, 547, 222–226. [DOI] [PubMed] [Google Scholar]
- 3.Xia Y, Wu J, Wei W, Du Y, Wan T, Ma X, An W, Guo A, Miao C, Yue H, Li S, Cao X, Su Z and Ma G, Nat Mater, 2018, 17, 187–194. [DOI] [PubMed] [Google Scholar]
- 4.Luo M, Wang H, Wang Z, Cai H, Lu Z, Li Y, Du M, Huang G, Wang C, Chen X, Porembka MR, Lea J, Frankel AE, Fu Y-XX, Chen ZJ and Gao J, Nat Nanotechnol, 2017, 12, 648–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tom JK, Dotsey EY, Wong HY, Stutts L, Moore T, Davies HD, Felgner PL and Esser-Kahn AP, ACS Cent Sci, 2015, 1, 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu TY-H, Singh M, Miller AT, De Gregorio E, Doro F, D’Oro U, Skibinski DAG, Mbow ML, Bufali S, Herman AE, Cortez A, Li Y, Nayak BP, Tritto E, Filippi CM, Otten GR, Brito LA, Monaci E, Li C, Aprea S, Valentini S, Calabrό S, Laera D, Brunelli B, Caproni E, Malyala P, Panchal RG, Warren TK, Bavari S, O’Hagan DT, Cooke MP and Valiante NM, Sci Transl Med, 2014, 6, 263ra160–263ra160. [DOI] [PubMed] [Google Scholar]
- 7.Liu H, Moynihan KD, Zheng Y, Szeto GL, Li AV, Huang B, Egeren DS, Park C and Irvine DJ, Nature, 2014, 507, 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bradley T, Fera D, Bhiman J, Eslamizar L, Lu X, Anasti K, Zhang R, Sutherland Laura L., Scearce Richard M., Bowman Cindy M., Stolarchuk C, Lloyd Krissey E., Parks R, Eaton A, Foulger A, Nie X, Karim Salim S. A, Barnett S, Kelsoe G, Kepler Thomas B., Alam SM, Montefiori David C., Moody MA, Liao H-X, Morris L, Santra S, Harrison Stephen C. and Haynes Barton F., Cell Rep, 2016, 14, 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pauthner M, Havenar-Daughton C, Sok D, Nkolola JP, Bastidas R, Boopathy AV, Carnathan DG, Chandrashekar A, Cirelli KM, Cottrell CA, Eroshkin AM, Guenaga J, Kaushik K, Kulp DW, Liu J, McCoy LE, Oom AL, Ozorowski G, Post KW, Sharma SK, Steichen JM, de Taeye SW, Tokatlian T, Torrents de la Peña A, Butera ST, LaBranche CC, Montefiori DC, Silvestri G, Wilson IA, Irvine DJ, Sanders RW, Schief WR, Ward AB, Wyatt RT, Barouch DH, Crotty S and Burton DR, Immunity, 2017, 46, 1073–1088.e1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tam HH, Melo MB, Kang M, Pelet JM, Ruda VM, Foley MH, Hu JK, Kumari S, Crampton J, Baldeon AD, Sanders RW, Moore JP, Crotty S, Langer R, Anderson DG, Chakraborty AK and Irvine DJ, Proc Natl Acad Sci U S A, 2016, 113, E6639–E6648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rappuoli R and Aderem A, Nature, 2011, 473, 463–469. [DOI] [PubMed] [Google Scholar]
- 12.Nutt SL, Hodgkin PD, Tarlinton DM and Corcoran LM, Nat Rev Immunol, 2015, 15, 160–171. [DOI] [PubMed] [Google Scholar]
- 13.Victora GD and Nussenzweig MC, Annu Rev Immunol, 2012, 30, 429–457. [DOI] [PubMed] [Google Scholar]
- 14.Silva N and Klein U, Nat Rev Immunol, 2015, 15, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crotty S, Immunity, 2014, 41, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heesters BA, Myers RC and Carroll MC, Nat Rev Immunol, 2014, 14, 495–504. [DOI] [PubMed] [Google Scholar]
- 17.Schwickert TA, Victora GD, Fooksman DR, Kamphorst AO, Mugnier MR, Gitlin AD, Dustin ML and Nussenzweig MC, J Exp Med, 2011, 208, 1243–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gitlin AD, Shulman Z and Nussenzweig MC, Nature, 2014, 509, 637–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ise W, Fujii K, Shiroguchi K, Ito A, Kometani K, Takeda K, Kawakami E, Yamashita K, Suzuki K, Okada T and Kurosaki T, Immunity, 2018, 48, 702–715.e704. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, Shi J, Yan J, Xiao Z, Hou X, Lu P, Hou S, Mao T, Liu W, Ma Y, Zhang L, Yang X and Qi H, Nat Immunol, 2017, 18, 921–930. [DOI] [PubMed] [Google Scholar]
- 21.Jewell CM, López SC and Irvine DJ, Proc Natl Acad Sci U S A, 2011, 108, 15745–15750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moon JJ, Suh H, Li AV, Ockenhouse CF, Yadava A and Irvine DJ, Proceedings of the National Academy of Sciences, 2012, 109, 1080–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cirelli KM, Carnathan DG, Nogal B, Martin JT, Rodriguez OL, Upadhyay AA, Enemuo CA, Gebru EH, Choe Y, Viviano F, Nakao C, Pauthner MG, Reiss S, Cottrell CA, Smith ML, Bastidas R, Gibson W, Wolabaugh AN, Melo MB, Cossette B, Kumar V, Patel NB, Tokatlian T, Menis S, Kulp DW, Burton DR, Murrell B, Schief WR, Bosinger SE, Ward AB, Watson CT, Silvestri G, Irvine DJ and Crotty S, Cell, 2019, 177, 1153–1171.e1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baumjohann D, Preite S, Reboldi A, Ronchi F, Ansel KM, Lanzavecchia A and Sallusto F, Immunity, 2013, 38, 596–605. [DOI] [PubMed] [Google Scholar]
- 25.Reed SG, Orr MT and Fox CB, Nat Med, 2013, 19, 1597–1608. [DOI] [PubMed] [Google Scholar]
- 26.Shirota H, Tross D and Klinman DM, Vaccines, 2015, 3, 390–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Hagan DT, Expert Rev Vaccines, 2007, 6, 699–710. [DOI] [PubMed] [Google Scholar]
- 28.Del Giudice G, Rappuoli R and Didierlaurent AM, Semin Immunol, 2018, 39, 14–21. [DOI] [PubMed] [Google Scholar]
- 29.Hutchison S, Benson RA, Gibson VB, Pollock AH, Garside P and Brewer JM, FASEB J, 2012, 26, 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martins KAO, Cooper CL, Stronsky SM, Norris SLW, Kwilas SA, Steffens JT, Benko JG, van Tongeren SA and Bavari S, EBioMedicine, 2016, 3, 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ciabattini A, Pettini E, Fiorino F, Pastore G, Andersen P, Pozzi G and Medaglini D, Front Immunol, 2016, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reed SG, Bertholet S, Coler RN and Friede M, Trends in Immunology, 2009, 30, 23–32. [DOI] [PubMed] [Google Scholar]
- 33.Akkaya M, Akkaya B, Kim AS, Miozzo P, Sohn H, Pena M, Roesler AS, Theall BP, Henke T, Kabat J, Lu JH, Dorward DW, Dahlstrom E, Skinner J, Millers LH and Pierce SK, Nat Immunol, 2018, 19, 255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaba SA, Brando C, Guo Q, Mittelholzer C, Raman S, Tropel D, Aebi U, Burkhard P and Lanar DE, J Immunol, 2009, 183, 7268–7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaba SA, McCoy ME, Doll TAPF, Brando C, Guo Q, Dasgupta D, Yang YK, Mittelholzer C, Spaccapelo R, Crisanti A, Burkhard P and Lanar DE, Plos One, 2012, 7, e48304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fifis T, Mottram P, Bogdanoska V, Hanley J and Plebanski M, Vaccine, 2004, 23, 258–266. [DOI] [PubMed] [Google Scholar]
- 37.Ulery BD, Kumar D, Ramer-Tait AE, Metzger DW, Wannemuehler MJ and Narasimhan B, Plos One, 2011, 6, e17642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rudra JS, Sun T, Bird KC, Daniels MD, Gasiorowski JZ, Chong AS and Collier JH, ACS Nano, 2012, 6, 1557–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen J, Pompano RR, Santiago FW, Maillat L, Sciammas R, Sun T, Han H, Topham DJ, Chong AS and Collier JH, Biomaterials, 2013, 34, 8776–8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chesson CB, Huelsmann EJ, Lacek AT, Kohlhapp FJ, Webb MF, Nabatiyan A, Zloza A and Rudra JS, Vaccine, 2014, 32, 1174–1180. [DOI] [PubMed] [Google Scholar]
- 41.Wu Y, Norberg PK, Reap EA, Congdon KL, Fries CN, Kelly SH, Sampson JH, Conticello VP and Collier JH, ACS Biomater Sci Eng, 2017, 3, 3128–3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rudra JS, Tian YF, Jung JP and Collier JH, Proc Natl Acad Sci U S A, 2010, 107, 622–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Egelman EH, Xu C, DiMaio F, Magnotti E, Modlin C, Yu X, Wright E, Baker D and Conticello VP, Structure, 2015, 23, 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pompano RR, Chen J, Verbus EA, Han H, Fridman A, McNeely T, Collier JH and Chong AS, Adv Healthc Mater, 2014, 3, 1898–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rodriguez LMDL, Hemar Y, Cornish J and Brimble MA, Chem Soc Rev, 2016, 45, 4797–4824. [DOI] [PubMed] [Google Scholar]
- 46.Lynch HE, Stewart SM, Kepler TB, Sempowski GD and Alam SM, J Immunol Methods, 2014, 404, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deshmane SL, Kremlev S, Amini S and Sawaya BE, J Interferon Cytokine Res, 2009, 29, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dan JM, Lindestam Arlehamn CS, Weiskopf D, da Silva Antunes R, Havenar-Daughton C, Reiss SM, Brigger M, Bothwell M, Sette A and Crotty S, J Immunol, 2016, 197, 983–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reiss S, Baxter AE, Cirelli KM, Dan JM, Morou A, Daigneault A, Brassard N, Silvestri G, Routy J-P, Havenar-Daughton C, Crotty S and Kaufmann DE, PLOS ONE, 2017, 12, e0186998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cope L, Fridman A, Joshi A, Mcneely TB, Pak I, Smith S, Patent WO2012065034 A1, 2012.
- 51.Shae D, Becker KW, Christov P, Yun DS, Lytton-Jean AKR, Sevimli S, Ascano M, Kelley M, Johnson DB, Balko JM and Wilson JT, Nat Nanotechnol, 2019, 14, 269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koshy ST, Cheung AS, Gu L, Graveline AR and Mooney DJ, Adv Biosyst, 2017, 1, 1600013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lynn GM, Laga R, Darrah PA, Ishizuka AS, Balaci AJ, Dulcey AE, Pechar M, Pola R, Gerner MY, Yamamoto A, Buechler CR, Quinn KM, Smelkinson MG, Vanek O, Cawood R, Hills T, Vasalatiy O, Kastenmüller K, Francica JR, Stutts L, Tom JK, Ryu K, Esser-Kahn AP, Etrych T, Fisher KD, Seymour LW and Seder RA, Nat Biotech, 2015, 33, 1201–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A and Moon JJ, Nat Mater, 2016, 16, 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jiang H, Wang Q and Sun X, J Control Release, 2017, 267, 47–56. [DOI] [PubMed] [Google Scholar]
- 56.Tokatlian T, Read BJ, Jones CA, Kulp DW, Menis S, Chang JYH, Steichen JM, Kumari S, Allen JD, Dane EL, Liguori A, Sangesland M, Lingwood D, Crispin M, Schief WR and Irvine DJ, Science, 2018, 363, 649–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leleux J, Atalis A and Roy K, J Control Release, 2015, 219, 610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ali OA, Huebsch N, Cao L, Dranoff G and Mooney DJ, Nat Mater, 2009, 8, 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Francica JR, Sheng Z, Zhang Z, Nishimura Y, Shingai M, Ramesh A, Keele BF, Schmidt SD, Flynn BJ, Darko S, Lynch RM, Yamamoto T, Matus-Nicodemos R, Wolinsky D, Program NCS, Barnabas B, Blakesley R, Bouffard G, Brooks S, Coleman H, Dekhtyar M, Gregory M, Guan X, Gupta J, Han J, Ho S.-l., Legaspi R, Maduro Q, Masiello C, Maskeri B, McDowell J, Montemayor C, Mullikin J, Park M, Riebow N, Schandler K, Schmidt B, Sison C, Stantripop M, Thomas J, Thomas P, Vemulapalli M, Young A, Nason M, Valiante NM, Malyala P, De Gregorio E, Barnett SW, Singh M, O’Hagan DT, Koup RA, Mascola JR, Martin MA, Kepler TB, Douek DC, Shapiro L and Seder RA, Nature Communications, 2015, 6, 6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crotty S, Immunity, 2019, 50, 1132–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kurosaki T, Kometani K and Ise W, Nat Rev Immunol, 2014, 15, 149–159. [DOI] [PubMed] [Google Scholar]
- 62.Pollard AJ, Perrett KP and Beverley PC, Nat Rev Immunol, 2009, 9, 213–220. [DOI] [PubMed] [Google Scholar]
- 63.Pichichero ME, Hum Vaccin Immunother, 2013, 9, 2505–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mora-Solano C, Wen Y, Han H, Chen J, Chong AS, Miller ML, Pompano RR and Collier JH, Biomaterials, 2017, 149, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reche PA, Cytokine, 2019, 116, 161–168. [DOI] [PubMed] [Google Scholar]
- 66.Sun T, Han H, Hudalla GA, Wen Y, Pompano RR and Collier JH, Acta Biomater, 2016, 30, 62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.