

Abstract
Basic neuroscience research employs antibodies as key reagents to label, capture, and modulate the function of proteins of interest. Antibodies are immunoglobulin proteins. Recombinant antibodies are immunoglobulin proteins whose nucleic acid coding regions, or fragments thereof, have been cloned into expression plasmids that allow for unlimited production. Recombinant antibodies offer many advantages over conventional antibodies including their unambiguous identification and digital archiving via DNA sequencing, reliable expression, ease and reliable distribution as DNA sequences and as plasmids, and the opportunity for numerous forms of engineering to enhance their utility. Recombinant antibodies exist in many different forms, each of which offers potential advantages and disadvantages for neuroscience research applications. I provide an overview of recombinant antibodies and their development. Examples of their emerging use as valuable reagents in basic neuroscience research are also discussed. Many of these examples employ recombinant antibodies in innovative experimental approaches that cannot be pursued with conventional antibodies.
Keywords: Immunocytochemistry, immunohistochemistry, intrabody, brain, neuron
ANTIBODIES IN NEUROSCIENCE RESEARCH
Antibodies are a mainstay of basic neuroscience research. Antibodies have diverse uses in labeling, capturing, and modulating the function of target proteins (Greenfield, 2014; Harlow & Lane, 1988). Antibodies are immunoglobulin proteins, the most common being immunoglobulin G or IgG, which are heterodimers formed by two heavy (H) and two light (L) chains (Fig. 1). The wide use of antibodies in research is based on a number of exceptional characteristics of IgG proteins. Useful antibodies exhibit high-affinity and selective binding to their intended target. Procedures for their development are well-established. Antibodies are also unusually stable proteins resistant to misfolding and degradation, which facilitates their production, purification, distribution and storage. Their stability facilitates their use under many different experimental conditions, many of which would cause loss of activity of other proteins. Moreover, due to their long use in neuroscience research, many researchers are familiar with basic considerations about antibodies themselves and antibody-based research applications. As illustrative examples of their widespread use, when looking at two common applications employing antibodies, there are ≈100,000 PubMed citations for “brain immunohistochemistry”, and >40,000 for “brain western blot”. However, in spite of their extensive long-term use, there remain numerous areas in which the use of antibodies in neuroscience research is still evolving. One of these is the application of recombinant antibodies to basic neuroscience research, oftentimes in innovative ways not possible with conventional antibodies.
Fig. 1.

Comparison of typical mammalian IgG and camelid heavy-chain only IgG and their derivatives. Left. A typical mammalian IgG molecule is a heterotetramer comprising two heavy and two light chains. Light chains comprise one variable (VL, light orange) and one constant domain (CL, light green). Heavy chains comprise one variable domain (VH, dark orange) and three constant domains (CH1–3, dark green). The primary region of covalent disulfide bond linkage of the two identical H + L chain heterodimers is shown by a purple bar. The functional antigen binding unit is formed by noncovalent association of the VL and VH domains. Typical mammalian H + L chain IgGs can be miniaturized to various forms including Fabs and ScFvs. The schematic of the VH domain shows the arrangement and typical sizes of the constituent FR and CDR segments. Approximate sizes of FR and CDR segments were derived from the IMTG database http://www.imgt.org/. Right. Camelid H chain-only IgGs lack light chains and exist as a homodimer of two identical H chains. The primary region of covalent disulfide bond linkage of the two identical H chain monomers is shown by a purple bar. The functional antigen binding unit is formed by a single VHH domain. This VHH domain can function autonomously as a nAb. The schematic of the VHH domain shows the arrangement and typical sizes of the constitutive FR and CDR segments, note elongated CDR3 relative to typical mammalian VH domain. Approximate sizes of FR and CDR segments were derived from Mitchell and Colwell, 2018.
Recombinant antibodies are immunoglobulin proteins whose nucleic acid coding regions, or fragments thereof, have been cloned into expression plasmids (Fig. 2) that when introduced into mammalian cells or E. coli allow for unlimited production of the recombinant antibody. This is in contrast to ‘conventional monoclonal antibodies’, which are endogenously encoded in the genome of B lymphocytes in animals and in hybridoma cells in culture. Recombinant monoclonal or mAbs were developed in the 1980s for therapeutic purposes. This procedure first involved making conventional mAbs in mice which then required extensive engineering before being suitable for introduction into patients. Therapeutic mAbs now represent a major sector of the modern pharmaceutical industry (Buss, Henderson, McFarlane, Shenton, & de Haan, 2012; Chiu & Gilliland, 2016), and all are recombinant. In contrast, recombinant mAbs and other forms of recombinant antibodies have been slow to infiltrate research. Therapeutic mAbs need to be humanized to avoid eliciting an immune response in patient. As humanization was not needed for research antibodies, most research antibody developers did not invest the time, expense and effort to convert conventional antibodies into recombinant form. As such it remains that the vast majority of antibodies used in biomedical research are conventional. This is supported by data from the publicly accessible commercial antibody database on the CiteAb website https://www.citeab.com/. CiteAb lists ≈4.9M antibodies from 205 suppliers, of which ≈4.7M are identified as conventional, and only ≈0.2M as recombinant.
Fig. 2.
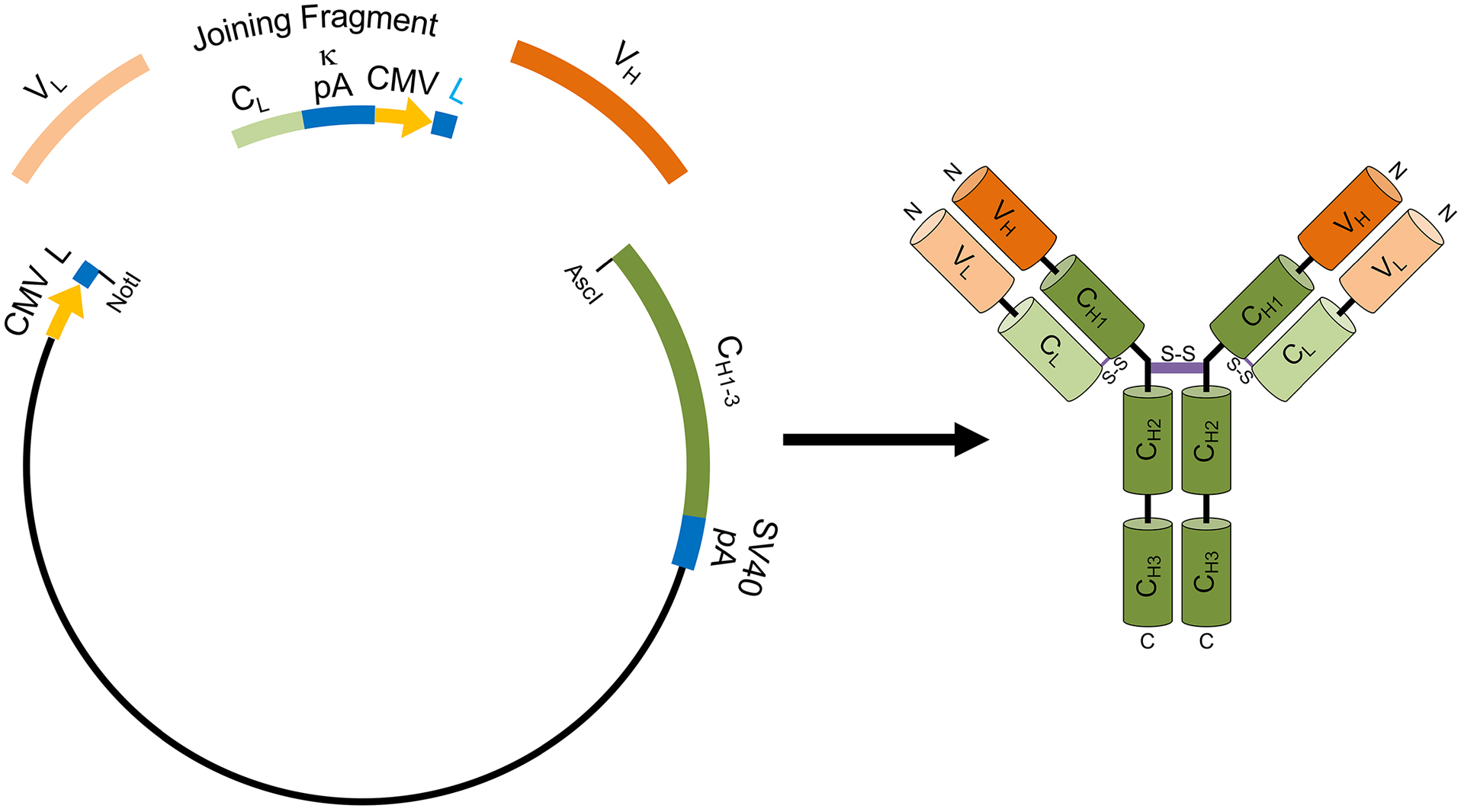
Example of an R-mAb expression plasmid. Schematic to the left shows the elements of the R-mAb expression plasmid as developed by Gavin Wright and colleagues (Crosnier et al., 2010) that we have used for our R-mAb expression (Andrews et al., 2019). This plasmid allows for coexpression of H + L chains as driven by two CMV promoters (yellow). In transfected mammalian cells the H + L chains coassemble to generate an intact IgG (right). Hybridoma-derived VL (light orange) and VH (dark orange) region PCR products or synthetic gene fragments are fused to a joining fragment comprising a κ light chain constant domain (CL) and the κ light chain polyA tail sequences (κ pA), a CMV promoter for heavy chain expression, and an ER signal/leader sequence (L) for translocation of the heavy chain across the ER membrane. PCR-mediated fusion of these three elements is followed by their insertion into the p1316 plasmid that contains an upstream CMV promoter for light chain expression, and an ER signal/leader sequence (L) for translocation of the light chain across the ER membrane. Downstream of the insert are the heavy chain constant regions (CH1–3) that are either γ1 or γ2a depending on the plasmid variant used, followed by the SV40 polyA tail (SV40 pA). Such plasmids can also be generated using Gibson assembly of the four elements (VL, joining fragment, VH and plasmid backbone).
However, the use of recombinant antibodies in biomedical research, including neuroscience, is growing. This is in part due to advances in recombinant DNA techniques that reduce the investment required to develop recombinant antibodies, and also the recognition that there are substantial advantages of recombinant research antibodies. These include 1) unambiguous identification of recombinant antibodies as produced in expression systems via DNA sequencing of the expression plasmid; 2) a permanent digital archive of DNA sequence versus storage as protein or cryopreserved hybridoma cells; 3) more reliable and less variable expression; 4) easier distribution as DNA sequences and as plasmids; and 5) the opportunity for subsequent engineering. These outcomes are entirely consistent with best practices in research transparency, rigor and reproducibility, including ensuring permanent access to critical research reagents, as noted in prior publications (A. Bradbury & A. Pluckthun, 2015; A. R. Bradbury & A. Pluckthun, 2015).
As underscored by examples provided below, one of the major advantages of recombinant antibodies for research use is that they are amenable to different forms of engineering (Fig. 3). There are numerous modifications to allow for easier detection, by changing the specificity of secondary antibody binding, or by adding epitope tags or other short sequences (e.g., Halo or sortase tags) for direct site-specific labeling of the antibody with fluorescent organic molecules, biotin, gold particles, DNA bar codes, etc., or by adding the sequence of a fluorescent protein (Fig. 3). It is also possible to join recombinant antibodies with protein partners such as fluorescent protein partners for detection, as well as partners that are directed to specific target proteins or subcellular sites through their antibody binding partner (Fig. 3). These include partner proteins and/or sequences that lead to enhanced degradation of the target protein such as ubiquitin ligases and degrons, and enzymes to modify the local environment of the cell at the region of antibody accumulation. It is also possible to engineer mutations that increase target binding affinity and/or target selectivity, and/or enhance the folding and/or stability of the antibody (Fig. 3). As such, subsequent engineering of recombinant antibodies allows the same progenitor recombinant antibody to be used to study and manipulate the function of neurons and non-neuronal cells.
Fig. 3.

Examples of generic forms of recombinant antibody engineering. Cartoon of various ways that recombinant antibodies (IgG, ScFVs, nAbs) can be engineered to enhance their utility. The top box shows a cartoon of various approaches to impact detection of recombinant antibodies. These include alternate heavy chain constant regions (CH1–3, in pink) to confer selective recognition by species- and or IgG subclass-specific secondary antibodies on R-mAbs, epitope and/or site-specific labeling tags (light blue) on R-mAbs, ScFVs and nAbs, and GFP (bright green) or other fluorescent proteins on ScFV and nAbs (primarily on those used as intrabodies). The middle box shows a cartoon of an ScFV and a nAb (primarily on those used as intrabodies) fused to a protein (dark blue) that confers functionality (protein degradation, enzymatic activity, etc.) to influence target protein, subcellular compartment and/or cellular function. The bottom box shows a cartoon demonstrating mutations (red dots) generated within the VH and VL regions of R-mAbs or ScFVs, or within the VHH region of nAbs. As these regions comprise the antigen (target) binding domains, such mutations can impact the efficacy and specificity of antigen (target) binding.
OVERVIEW OF ANTIBODY STRUCTURE AS RELATED TO DEVELOPING RECOMBINANT ANTIBODIES
Intact IgG antibodies from almost all mammalian species comprise two identical heavy (H) chain and two identical light (L) chain polypeptides in a protein of ≈150 kD (Fig. 1, left). The intact IgG antibody is a dimer of H+L heterodimers, with each H+L dimer having the capacity to bind to antigen, such that intact antibodies have two antigen binding sites, one on each arm (Fig. 1, left). This enhances antibody binding beyond the inherent affinity of each antigen binding site through avidity effects. The variable domains of the heavy and light chains (VH and VL) are located at the N-terminus of each subunit and form the antigen binding region, determining target specificity (Fig. 1, left). Within the variable domains the three complementarity determining regions or CDRs are interspersed between four highly conserved framework regions or FRs (Fig. 1, left, illustrated for VH). The CDRs form the binding interface and determine antibody specificity. The CDRs exhibit the most divergent sequences when comparing the sequences of different antibodies. The FRs provide the stable scaffold upon which the CDRs are displayed. Recombinant antibodies can be roughly classified as intact antibodies and antibody fragments, such as Fabs and ScFVs (Fig. 1, left) which can retain the specificity of the source antibody. In addition to typical mammalian H + L chain IgGs, camelid mammals produce unique H chain only IgGs (Fig. 1, right). In this case the VH domain alone (in this case termed a VHH domain to distinguish it as being from a unique heavy chain only IgG) forms the entire antigen binding region. As such the VHH domain can function autonomously as a nanobody or nAb (Fig. 1, right). The overall arrangement of the FRs and CDRs within the VHH domain is similar to that of the VH domain (Fig. 1, right). However, the VHH CDR3 region is elongated to provide additional structural diversity (Mitchell & Colwell, 2018). There also exist antibody mimetic proteins that are not derived from immunoglobulins but that can function in an analogous manner in terms of their high affinity and selective binding to specific targets. Each of these forms has inherent advantages as renewable recombinant affinity reagents as introduced above.
PRIMARY FORMS OF RECOMBINANT ANTIBODIES
Intact R-mAbs
The H and L chains within each identical half of the intact IgG antibody have extensive non-covalent interactions within both their variable and constant domains and are also covalently attached to one another via disulfide bonds. The two H+L heterodimers associate with one another in the intact IgG via their respective H chains, which are also non-covalently and disulfide bonded to one another (Fig. 1, left). Disulfide bonds form and are retained in oxidizing environments, such as the lumen of the endoplasmic reticulum (ER) and the extracellular environment. Both the H and L chains have N-terminal leader sequences that direct their cotranslational translocation across the ER membrane, after which the leader sequences are cleaved. The chains fold and assemble into the heterotetramer in the ER lumen, after which the intact IgG molecule traffics through the secretory pathway and is secreted into the extracellular fluid.
In all cases recombinant antibodies are monoclonal, no matter whether their form is an intact IgG or a smaller fragment. Here I will use R-mAb as an abbreviation for the intact IgG form of recombinant monoclonal antibody. Much effort has been put towards optimizing the characteristics of recombinant expression plasmids and the host cell expression background to provide robust and efficient expression of intact R-mAbs. To efficiently form IgG molecules, it is important that the H and L chains are co-expressed. To facilitate this, one can use either a plasmid with a single promotor driving expression of both H and L chain coding region cDNA inserts with an intervening IRES sequence or a dual promoter plasmid in which each chain has its own promotor. We have used a dual promotor plasmid (Fig. 2) developed by Gavin Wright and colleagues (Crosnier, Staudt, & Wright, 2010) for our R-mAb expression (Andrews et al., 2019).
One advantage of R-mAbs is that they are the recombinant antibody form most similar to conventional antibodies. As such researchers will be familiar with their properties as related to their use, storage and detection with secondary antibodies. Moreover, if derived from a conventional mAb their efficacy and specificity should be highly similar if not virtually identical. However, conversion into recombinant form creates the opportunity for further engineering to enhance their characteristics (Fig. 3). This can include replacing the endogenous heavy chain constant region to confer detection with alternate secondary antibodies, and creating R-mAbs distinct from their progenitor mAb in their secondary antibody binding but not their target binding efficacy or specificity. This provides additional flexibility for pairing such R-mAbs with other Abs in simultaneous multiplex labeling experiments. We have done this for a collection of mouse mAbs, allowing for their use in simultaneous multiplex labeling with subclass-specific secondary antibodies (Manning, Bundros, & Trimmer, 2012) in combinations not possible with the progenitor conventional mAbs (Andrews et al., 2019). Rabbits do not have IgG subclasses (Knight, Burnett, & McNicholas, 1985), such that such engineering is required to distinguish one rabbit R-mAb from another. Other forms of engineering to enhance detection without impacting the R-mAbs target binding efficacy or specificity include the addition of an epitope tag or other short sequences to facilitate direct site-specific labeling (Fig. 3).
Recombinant Antibody Fragments and Mimetics
Recombinant antibody fragments can have advantages over intact IgG antibodies, due to their smaller size and also that some forms comprise single polypeptide chains. Numerous forms of antibody fragments with coassembled H and L chains can be produced recombinantly, including monovalent Fab fragments (Fig. 1, left). Single chain antibody fragments can be produced by fusing VH and VL regions into a single polypeptide as with ScFVs (Fig. 1, left), or by generating miniaturized versions of H chain-only camelid antibodies as with nAbs (Fig. 1, right). While recombinant Fab fragments have been extensively employed as therapeutics and for clinical diagnostic purposes, I will focus on ScFVs and nAbs, which are gaining increased use as tools for basic neuroscience research.
Single chain variable fragments (ScFvs)
One widely used form of recombinant antibody fragment is a monomeric single chain variable fragment or ScFV, generated by fusing a VH and VL region to one another via a flexible linker domain (Fig. 1, left). ScFVs have been widely used as therapeutics and as probes for diagnostic imaging since their development in the 1980s (Bird et al., 1988; Huston et al., 1988). The small size of ScFvs facilitates their penetration into tissue, which enhances their efficacy in these applications. ScFvs have also been used widely as research reagents, as both immunolabels and as intrabodies. The latter term was originally coined in reference to intracellularly-expressed ScFvs (S. Y. Chen, Bagley, & Marasco, 1994). The concept of ScFVs is to replicate in a single polypeptide the coassembly of the VH and VL regions with one another in a manner similar to that which occurs within an intact IgG (Fig. 1, left). This is complicated by the fact that in an IgG molecule the similar sized VH and VL regions are aligned in parallel, with their respective free N-termini aligned at one end and their C-termini aligned at the other (Fig. 1, left). Since it is impossible to have two free N-termini in a single polypeptide creates problems that the ScFV design attempts to circumvent by attaching the C-terminus of the leading V region to the N-terminus of the lagging V region via a flexible linker segment of sufficient length that the respective V regions can align in parallel (Fig. 1, left). While this results in one of the N-termini being free as it is in the intact IgG, the other is fused to the flexible linker segment. This can negatively impact the correct folding of the V regions, ScFV expression and stability, as well as the efficacy and specificity of target binding.
There are numerous other potential challenges with the successful conversion of intact R-mAbs into ScFVs (Fig. 4), and the generation of high quality ScFV libraries for de novo development of ScFVs (Fig. 5) that have been well-documented in the extensive literature on the subject, and that contribute to the variable success rate in converting any IgG into an ScFV. However, there are also numerous strategies for circumventing these problems [e.g., (Chiu, Goulet, Teplyakov, & Gilliland, 2019; Schaefer, Honegger, & Pluckthun, 2010; Tiller & Tessier, 2015) and many others]. This begins with the design of the flexible linker in terms of its overall length and amino acid sequence, and the orientation of the VH and VL regions relative to one another (i.e., VH-linker-VL versus VL-linker-VH). Once an ScFV is obtained, whether from conversion from an existing R-mAb or from de novo screening, it can be further optimized by altering specific amino acids within the VH and VL regions to facilitate expression, folding, and stability (Fig. 3). Each variant can then be tested for its efficacy and specificity for target binding, and for its structure and biophysical properties. While this is justified and has been performed for ScFVs of high value for use as therapeutics or in clinical diagnostic imaging, it is not typically pursued for ScFvs research use. Fortunately, extensive research in this area has led to some basic design guidelines that can often yield a functional ScFV without further engineering. We have primarily used a generic approach employing a VH-linker-VL orientation and a flexible (GGGGS)4 linker and have used expression in mammalian cells, regardless of the sequence of the VH and VL regions. While our experience is limited, to date we have had an ≈60% (16/27) first pass success rate using this approach to convert mAbs into ScFVs without any further engineering.
Fig. 4.

Flow chart of generic pipelines for developing R-mAbs and ScFVs from existing mAbs. Top two rows show a schematic overview of the major steps involved in generation of R-mAbs from mAb-producing hybridomas. The top row shows a PCR-based cloning based approach whereby VH and VL region fragments are amplified using degenerate primer sets, cloned into an expression plasmid such as that shown in Fig. 2, the R-mAbs expressed and functionally characterized relative to the corresponding conventional mAb. Positive clones are then sequenced and archived. The middle row shows a high-throughput sequencing approach based approach whereby VH and VL region fragments are amplified using degenerate primer sets including bar codes and the amplicons from many different hybridomas pooled and sequenced. The primary VH and VL region fragments are synthesized and cloned into an expression plasmid such as that shown in Fig. 2, the R-mAbs expressed and functionally characterized relative to the corresponding conventional mAb. The bottom row shows a schematic of typical approach used to develop ScFVs from existing R-mAbs. One of the approaches above is used to obtain R-mAb VH and VL region sequences. These are used to design a synthetic gene fragment that has these sequences fused with an intervening flexible linker sequence. This gene fragment is then cloned into a mammalian or bacterial expression plasmid with suitable promoter and leader sequences and used for expression of secreted ScFVs, or expression plasmids without a leader sequence for expression as intrabodies.
Fig. 5.

Flow chart of generic pipelines for de novo ScFV, R-mAb and nAb development. The top row shows a PCR-based cloning based approach to develop ScFVs. The VH and VL region fragments are amplified from mammalian (non-camelid) B cells using degenerate primer sets, and are used to generate a high complexity cDNA library of ScFVs by fusing the VH and VL region fragments with an intervening flexible linker, which are cloned into an expression plasmid, and expressed ScFVs that bind to the target protein isolated by phage display or another appropriate selection method. Selected ScFVs are further characterized and sequenced. The middle row shows a schematic of one approach used to develop R-mAbs from ScFVs obtained from such a de novo approach. Selected ScFV-derived VH and VL region fragments are cloned into an expression plasmid such as that shown in Fig. 2, and the encoded R-mAbs expressed and functionally characterized. The bottom row shows a PCR-based cloning based approach to develop nAbs. VHH region fragments are amplified from camelid B cells using degenerate primer sets, and are used to generate a high complexity cDNA library of nAbs which are cloned into an expression plasmid, and expressed nAbs that bind to the target protein isolated by phage display or another appropriate selection method. Selected nAbs are further characterized and sequenced.
Camelid VHH domain fragments or Nanobodies (nAbs)
Camelids are unique among mammals in having IgG antibodies consisting of only a heavy chain (Fig. 1, right) (Muyldermans, 2013). The variable or VHH domain from these H chain only antibodies functions as a single chain antibody fragment, termed a “nanobody” (nAb), the smallest functional antibody fragment. Camelid VHH variable domains have a longer CDR3 (Fig. 1, right) and structural variation in their CDR1 and CDR2 loops compared to other mammalian VH domains. These modifications are thought to contribute to the structural diversity of camelid antibodies compared to the conventional mammalian H plus L chain antibodies (Mitchell & Colwell, 2018). Nanobodies have numerous advantages, beginning with their small size of 15 kD, which is ≈1/10 the size of a conventional IgG antibody. That they are a single chain makes their expression in E. coli and in mammalian cells simpler as it does not require coassembly of disulfide-linked H and L chains. They are also exceptionally stable (Muyldermans, 2013). Uses of nAbs in basic neuroscience research are detailed below.
Antibody mimetics
Antibody mimetics are genetically-encoded molecules whose structure is distinct from antibodies but that exhibit binding efficacies and specificities comparable to antibodies. They are typically comprised of a stable backbone scaffold upon which is built a flexible binding surface. Display libraries are generated from relatively invariant backbone sequences combined with a variety of flexible binding surface sequences. This high complexity library is then subjected to multiple rounds of phage, ribosome, mRNA or other display or selection technology against the target of choice, followed by further characterization as is done for antibodies. While antibody mimetics have not achieved as widespread overall use in neuroscience research as recombinant antibodies, there are notable exceptions.
Monobodies, which are based on a backbone of stable fibronectin repeats (Koide, Bailey, Huang, & Koide, 1998), have been developed against a set of neuronal proteins for use as intrabodies (Gross et al., 2013). These antibody mimetics, termed FingRs (for Fibronectin intrabodies generated with mRNA display) were also engineered with an innovative transcriptional control system to limit FingR expression levels to those which saturate target protein binding, with unbound FingRs acting in a transcriptional feedback loop to inhibit further expression and prevent accumulation of unbound intrabody (Gross et al., 2013). Highly specific FingRs were developed against the excitatory synapse scaffolding protein PSD-95, and the inhibitory synapse scaffolding protein Gephyrin, allowing for selective targeting of fluorescent proteins to these sites in cells expressing these genetically-encoded antibody mimetics. FingRs have been introduced through transfection, in utero electroporation or viral vectors to label synapses in living cells (Bensussen et al., 2020; Kannan, Gross, Arnold, & Higley, 2016; Kwon et al., 2019; Sinnen et al., 2017; Walker et al., 2017). The ability to target fluorescent proteins or other cargo to specific sites in neurons through the expression of these FingRs opens many possibilities for reporting and/or manipulating synaptic structure and function.
DARPins (Designed Ankyrin Repeat Proteins) are another form of antibody mimetics in which a stable backbone, in this case ankyrin repeats, are used as a scaffold to display a variety of flexible binding surfaces (Jost & Pluckthun, 2014; Plückthun, 2015). Anti-GluA4 AMPA receptor DARPins were isolated by ribosome display against a recombinant GluA4 extracellular domain fragment (Hartmann et al., 2019). One of these DARPins was used to coat the surface of AAV particles engineered to be deficient in heparan-sulfate proteoglycan-mediated attachment. When injected into mouse brain, the AAV particles selectively infected parvalbumin-positive interneurons (Hartmann et al., 2019), which are known to express high levels of GluA4 (Geiger et al., 1995).
DEVELOPING RECOMBINANT MABS OR R-MABS
As detailed below, the initial generation steps in developing R-mAbs can involve conversion of existing conventional mAbs into recombinant form (Fig. 4), or de novo generation (Fig. 5). In the latter case antibodies that bind to the target are identified and/or isolated from collections of candidates. The key is that as recombinant reagents, the nucleic acid sequence that encodes the antibody can be archived, amplified and propagated, allowing for production of additional molecular-defined antibody whenever needed, either during screening steps or when subsequently used in basic neuroscience research.
Conversion of existing mAbs into recombinant antibodies
There exist substantial collections of conventional mAbs in academic and commercial collections. Each of these are archived as hybridoma cells cryopreserved in liquid nitrogen. There is substantial cost and effort invested in maintaining these collections, although this represents a fraction of the overall value of the archived hybridomas that serve as a renewable source of mAbs. While this well-established system is for the most part reliable, a number of reasons justify the additional investment to convert existing mAbs into recombinant forms. Cryopreservation requires the successful recovery of viable thawed cells into culture, which is not absolutely guaranteed. Moreover, hybridoma cells can acquire mutations that impact mAb expression and/or the characteristics of the mAb itself. While creating R-mAbs requires an additional investment above and beyond initial mAb development, it is often justified given the substantial cost, time and effort that typically goes into the initial development and characterization of mAbs, and the value of ensuring reliable access to particular high-quality mAbs so that researcher can confirm and extend the research performed with these mAbs. Creating R-mAbs from mAbs (Fig. 4) begins with extraction of RNA from the hybridoma cells, followed by its conversion into cDNA, which is used as a template for PCR amplification of the antibody chains. Sets of degenerate PCR primers have been developed that allow for effective and selective amplification of VH and VL sequences from virtually any hybridoma. (Note: as described above, antibody specificity is determined by the VH and VL regions that form the antigen binding region). These fragments can be inserted (i.e., cloned) into plasmids (Fig. 2) already containing leader and CH and CL region sequences to reconstitute an intact IgG.
In our efforts to convert mouse mAbs into recombinant form, we have employed a plasmid developed by Gavin Wright and colleagues (Crosnier et al., 2010) that contains the mouse IgG H and L chain leader and CH1–3 and CL region coding sequences (Fig. 2). By employing plasmids such as this, one only needs to insert the PCR-amplified VH and VL region fragments in frame into their respective locations in the plasmid to reconstitute intact H and L chain coding sequences. The H and L chain sequences are each preceded by a CMV promoter to drive strong expression of these transcripts in mammalian cells. While we have used this particular expression system for cloning and expression of our mouse mAbs, there are numerous expression plasmids from both commercial, non-profit and academic sources into which hybridoma-derived H and L chain sequences can be inserted and expressed. In general, these plasmids are designed for expression of R-mAbs in mammalian cells, due to the requirement for disulfide bond formation for expression of R-mAbs as intact heterotetrameric IgG molecules. Another method for converting conventional mAbs into recombinant form is to directly sequence the VH and VL regions of individual mAbs from hybridoma derived cDNA (Fig. 4). These ≈400 bp regions can then be synthesized as double stranded gene fragments and used to reconstruct an intact IgG by insertion into plasmids containing CH1–3 and CL regions, as described above for cloning of PCR fragments into such plasmids.
The premise of conventional mAb technology is that clonal hybridomas are the result of the fusion of a single myeloma cell with a single splenocyte, which due to allelic exclusion express a single immunoglobulin H and L chain. One issue when converting conventional mAbs into recombinant form is the presence of additional H and L chain transcripts in certain clonal hybridoma cell lines. Commonly used myeloma cell lines such as SP2/0, NSO and P3-X63-Ag8.653 harbor a nonproductive L chain mRNA transcript containing a premature stop codon just after the VL domain (Carroll, Mendel, & Levy, 1988). This can confound cloning and sequencing efforts as this aberrant L chain transcript can be present at levels substantially higher than the bona fide productive L chain transcript (Juste, Muzard, & Billiald, 2006). This can be removed by restriction digest of the PCR product, although bioinformatics analyses suggest that up to 2% of productive L chains may also be lost in such a digest (Juste et al., 2006). The aberrant L chain transcript can also be removed bioinformatically in approaches employing high throughput sequencing. However, it remains at the core of mAb technology, and as the basis of reconstituting the appropriate pairing of H and L chains in R-mAb expression constructs, that each hybridoma should express only a single productive H and L chain polypeptide and generate a single type of heterotetrameric IgG molecule. A recent study (Bradbury et al., 2018) examined the prevalence of additional productive (i.e., non-aberrant) antibody chains in 185 monoclonal hybridomas from different laboratories. They found that a substantial number (≈30%) expressed more than a single productive H and/or L chain transcript, such that the monoclonal hybridomas were not generating mAbs per se, but a more complex collection of heterotetrameric IgGs comprising a variety of H and L chains pairings. It is not known whether the small sample used in this study is representative of the large number of mAbs in current use. However, it makes the conversion of mAbs into R-mAbs more complicated for determining which H and L chain pairing generates the desired mAb. However, it is yet another compelling reason for converting mAbs to R-mAbs, as in these cases it will “clean up” the antibodies produced by these hybridomas and yield a true “monoclonal” collection of IgG antibodies with the same H and L chain pairing.
Development of ScFVs from R-mAb sequences
The generation of ScFVs from existing hybridoma cells (Fig. 4) is a long-established approach (Toleikis, Broders, & Dubel, 2004). It has the potential of capturing the characteristics of well-characterized, valuable and widely-used mAbs in a miniaturized single-chain format. Advances in gene synthesis and cloning allow researchers to synthesize the entire ScFV coding sequence (≈750 bp with VH, linker and VL sequences) for insertion into their own bacterial or mammalian expression plasmid (Gibson et al., 2009). There are also numerous commercial suppliers who will provide a complete expression-validated plasmid from provided VH and VL sequences. As detailed above, ScFV design can vary in terms of whether the format is VH-linker-VL or VL-linker-VH, as well as the overall length and sequence of the linker. Other differences include expression system-specific leader sequence at the N-terminus, and the addition of various sequences at the C-terminus to enhance the experimental utility of the ScFV (Fig. 3). These can include 6X His or other tags for purification, epitope tags for detection, tags for site-specific labeling or other forms of detection, sequences to promote cell penetration, or any fusion partners which the ScFV can deliver to specific targets, subcellular sites or cells.
De novo generation of recombinant antibodies and antibody fragments by library screening
Another route to develop recombinant antibodies is by direct screening of high complexity cDNA libraries encoding the various forms of recombinant antibodies (Fig. 5). Screening is most commonly accomplished by display technology, which offers the advantage of not only identifying but also capturing the positive clones. This allows for enrichment of these genetically encoded reagents in a form that can be propagated for further characterization and use. The display technology was initially developed using bacteriophage as the vehicle to display the binder (Smith, 1985). Since then other related modalities have been developed including ribosome display and yeast or mammalian cell display. ScFvs, nAbs and Fabs can be isolated directly by this approach, whereas full-length recombinant mAbs are typically first isolated as ScFvs or Fabs (Fig. 5), and then converted into an intact IgG by recombinant engineering to reconstitute the intact H and L chains (Reader, Workman, Maddison, & Gough, 2019).
Phage display was described in detail in a previous contribution to this series (Bradbury, 2010), as such only a brief overview is provided here. Phage display involves expressing a library of the recombinant antibody of interest (ScFV, nAb, etc.) or antibody mimetics on the pili of filamentous phage. This is accomplished by generating a cDNA library in a phagemid (a plasmid cloning vector). Such phagemids can be propagated in plasmid form in E. coli. However, phagemids contain elements that when present in E. coli infected with helper phage, which on their own are inefficient at viral packaging, the plasmid is replicated as single stranded DNA whose incorporation into newly synthesized phage results in efficiently packaging of the reconstituted phage. Importantly, the reconstituted phage expresses any protein encoded by the phagemid, with expression typically controlled by an inducible promotor. An important innovation involved fusing the sequence of encoded protein in frame with the pili PIII protein coding sequence. The encoded PIII fusion protein is expressed or displayed on the surface of the phage. When such phage are exposed to an immobilized target, typically a purified protein, cell or other target moiety, phage exhibiting high-affinity binding are captured. After removing unbound or loosely bound phage by washing steps, bound phage are eluted and used to reinfect fresh bacterial cultures and subsequently amplified. After one or more rounds of selection and amplification, phage are isolated in monoclonal form and individual clones that exhibit binding to the target identified by ELISA or another form of binding assay. Phagemid DNA is then sequenced to identify unique clones. The DNA can also be introduced into a different strain of E. coli bacteria, one common form being TOP10F’, whose phenotype allows the recombinant antibody to be produced in soluble form, and without being fused to the PIII protein, as phagemids used in display typically have an amber stop codon at the junction between the recombinant antibody and PIII sequences. The soluble recombinant antibodies present in conditioned bacterial culture medium, in periplasmic extracts of disrupted bacteria, or as purified preparations can then be used as immunolabels in ELISAs or in other assays to verify anti-target binding, and subsequently in other experiments in which antibodies may be used.
A variation of phage display is ribosome display (Hanes & Pluckthun, 1997), in which the cDNA inserts encoding the recombinant antibodies are generated with a spacer region substituting for the stop codons naturally present at the end of the protein coding sequence. When mRNAs transcribed from these cDNAs are translated in vitro, the recombinant antibody remains attached to the translational complex (the recombinant antibody polypeptide, the ribosome and the recombinant antibody-encoding mRNA). The translational complex library is then subjected to selection against immobilized target in a manner analogous to phage display. The bound and eluted translational complexes are then reverse transcribed, and an enriched cDNA sublibrary generated, with the potential for multiple rounds of selection to enrich for high-affinity binders. Selected cDNAs are then cloned into plasmids for subsequent expression and validation. Notably, the use of phage and ribosome display are not confined to selection of single chain recombinant antibodies (ScFvs and nAbs) and single chain non-antibody binders (monobodies and DARPins). In particular, Fab fragments (Fig. 1), which contain the variable regions of the H and L chains that define antibody specificity, can also be screened by both techniques. One advantage of ribosome display is that it circumvents the need for high efficiency bacterial transformation of a phagemid library as is necessary for effective phage display.
Display libraries are often generated from cDNA isolated from B cells obtained from an animal immunized with one or more target proteins. Such libraries would typically be used only for those projects. However, it is also common to use naïve libraries, in which B cells from an nonimmunized animal is the source of cDNA, or completely synthetic libraries (Almagro, Pedraza-Escalona, Arrieta, & Perez-Tapia, 2019). The same naïve or synthetic libraries are then used for any target protein. This approach is much faster and requires much lower quantities of target protein than approaches employing animal immunization, which entails repeated immunization of animals with the immunizing protein over a period of weeks or even months. Many commercial and academic organizations have put considerable efforts into developing high-quality libraries with substantial complexity as to the number of unique recombinant antibodies they contain. As such, screening such libraries is likely to yield a sufficient number of candidates for subsequent validation. However, in many cases the use of naïve or synthetic libraries provides candidates that in themselves exhibit do not sufficiently high affinity binding to the target. These initial candidates are typically subjected to in vitro mutagenesis and selection for higher affinity antibodies, a process similar to that which occurs in the adaptive immune system in response to immunization.
CHARACTERIZATION AND VALIDATION OF RECOMBINANT ANTIBODIES FOR NEUROSCIENCE RESEARCH APPLICATIONS
Regardless of the development approach used, the goal is to identify recombinant antibodies that bind with high affinity to a purified protein or cell line expressing a protein target of interest. It is critical that the screening process used yields a population of candidates that can then be screened for their ability to bind to the target protein in a native neurobiological system. We have used this approach extensively in developing conventional mAbs to use for immunoblotting and immunohistochemistry in brain neurons (Gong, Murray, & Trimmer, 2016) and we (Andrews et al., 2019; J. X. Dong et al., 2019) and others (see examples below) have employed similar approaches in recombinant antibody development. Validation or R-mAbs (and other forms of recombinant antibodies) follows the same principles as for other antibodies [e.g., (Taussig, Fonseca, & Trimmer, 2018; Uhlen et al., 2016), etc.]. However, antibody validation for neuroscience research is impacted by certain unique considerations. There are not many cell line models suitable for screening or subsequent validation of antibodies for subsequent use in brain, as such validation assays need to be performed in brain tissue or in primary cultures of brain cells. This limits the use of knockdown/KO approaches, especially those employing powerful CRISPR/Cas9 approaches (Zotova, Zotov, Filatov, & Mazurov, 2016), that have gained widespread use in antibody validation for use in other cell types. However, one can advantageously employ the neuroanatomical complexity of the brain when screening and validating antibodies, whereby knowledge of the specific subcellular compartment (axon, dendrite, synapse, node of Ranvier, etc.), cell type and circuit in which a target protein is (and is not) likely to be expressed can be effectively used to evaluate immunolabeling in brain sections and in primary cultures of brain cells (Gong et al., 2016; Rhodes & Trimmer, 2006).
Many of the efforts aimed at developing recombinant antibodies for neuroscience research initially employ a set of high throughput proxy assays in heterologous cell lines as an intermediate step between isolation and ELISA validation and experiments in brain neurons. These proxy assays are aimed at recapitulating many of the experimental conditions that are typical for the subsequent use of these antibodies in brain samples. However, instead of evaluating large numbers of antibody candidates in samples of brain neurons within or derived from animals, cultured cell lines expressing the target protein are used. This is a useful filter for the ELISA-positive antibodies that will not recognize their intended target in samples prepared for commonly used applications such as immunocytochemistry (ICC)/immunohistochemistry (IHC), and Western/immunoblotting, as well as the unique sample preparation conditions associated with immunoelectron microscopy, array tomography, expansion microscopy, volumetric imaging of cleared tissue, etc. These proxy assays can also entail expressing the recombinant antibody inside of transfected cells and assaying for its ability to function as an intrabody in being capable of recognizing its target in the intracellular environment. In either case, the experiment is typically performed in cells that have been transiently transfected to express the target protein. This experimental approach provides a sample with a mosaic of cells with and without target protein expression, which can be easily distinguished by an independent marker (fluorescence of a fused fluorescent protein, antibody labeling of an epitope tag, etc.). This provides a powerful assay system for examining target-specific binding in a complex mammalian cell background. For evaluating recombinant antibodies for immunolabeling, one simply looks for immunolabeling present in expressing cells but not in nonexpressing cells (Bekele-Arcuri et al., 1996; Gong et al., 2016; Rhodes & Trimmer, 2006). For assaying function as intrabodies, the recombinant antibody is expressed with and without target protein coexpression. One then assays the cells for whether the localization of the intrabody is altered in coexpressing cells, indicative of target protein binding (J. X. Dong et al., 2019). This assay is facilitated by the intrabody and target having distinct subcellular localizations such that binding leads to interaction-stabilized change in the localization of one protein or the other. These assays were frequently employed in the development of recombinant antibodies used in the examples of neuroscience research applications below. While there are many caveats associated with using heterologous cells to evaluate antibodies to be used in brain neurons, these assays are advantageous to filtering out ELISA-positive antibodies that do not function under these application-specific experimental conditions. This makes such assays valuable steps in antibody screening, especially when evaluating a substantial number of candidates.
The candidates that emerge from these proxy assays are then evaluated in samples of cultured brain neurons, brain tissue or in intact brain in vivo. Depending on the nature of their intended use, these candidates can be evaluated for use as immunolabels for ICC, IHC, immunoblotting, or as intrabodies when expressed in brain neurons or for any other needs as dictated by the nature of the research. Below I provide examples of different strategies that researchers have taken to develop and validate recombinant antibodies for basic neuroscience research. Each typically begins with a set of somewhat generic antibody generation and screening/evaluation procedures that one would pursue regardless of the intended neuroscience or non-neuroscience research application. However, there are also examples in which even the earliest screening steps are focused on a distinct neuroscience-specific use.
Recombinant Antibodies as Immunolabels
Once recombinant candidates have been isolated by display technology, or through recombinant cloning from existing antibodies, recombinant antibodies to be used as immunolabels are typically expressed from either mammalian cell lines or in E. coli, although other expression systems are also used (cell free/in vitro systems, yeast, Sf9 insect cells, etc.). Mammalian expression is most commonly used for intact R-mAbs, to utilize the inherent components of these cells that promote efficient folding, assembly and disulfide bond attachment of the H and L chains to one another. Mammalian expression is also commonly used for ScFVs. Nanobodies are typically produced in bacteria. In each case expression-specific leader sequences need to be employed to direct the expressed protein to an extracellular compartment from which the antibody can be harvested. Other modifications including codon optimization can also be employed.
One item to note is that like conventional antibodies, recombinant antibodies can be present in different preparations/formulations whose non-antibody components can impact experimental results. In general, expression in mammalian cells generates “conditioned media” or in mAb vernacular “tissue culture supernatants” or “TC supes” that can contain substantial amounts of antibody. Highly engineered and proprietary mammalian expression systems used in industry can generate TC supes containing up to 5 mg/mL recombinant antibody (Farid, Baron, Stamatis, Nie, & Coffman, 2020). However, concentrations of 50 μg/mL of antibody are more typical of research lab production levels. Such TC supes can be used in most applications without further purification, as the working concentrations of antibodies is typically on the order of 1–10 μg/mL. Importantly, the other components of the TC supe preparation (mammalian tissue culture medium) do not interfere with most types of immunolabeling. ScFV expression in mammalian cells follows the same considerations. Research laboratory expression of nAbs in E. coli can generate comparable concentrations of tens of mgs/L of bacterial culture (Jovcevska & Muyldermans, 2020) with the additional benefit that as nAbs are ≈1/10 the size of IgG, their molar concentration at an equivalent level of protein is 10X higher. One consideration for bacterial expression is that due to the bacterial cell wall, the recombinant nAb is distributed between the periplasmic space and the bacterial culture medium. Consistent with previously published protocols [e.g., (Ghassabeh, Saerens, & Muyldermans, 2010)] we have found that in spite of this, the level of nAb in bacterial culture supernatants is sufficient for most screening purposes (J. X. Dong et al., 2019), saving the time and effort required to produce periplasmic extracts. However, most nAb development protocols employ periplasmic extracts or even purified nAbs in early screening steps [e.g., (Pardon et al., 2014)]. In each case, the recombinant antibodies present in the conditioned culture medium can be purified using a simple one-step chromatographic purification, employing beads with attached antibody binding proteins such as protein A for intact IgG, or other attached affinity tags (i.e., nickel-coated beads for binding to 6XHis tags).
Recombinant Antibodies as Intrabodies
A distinct use of recombinant antibodies is as intrabodies, whereby genetically-encoded antibodies are expressed in the cytoplasm of living cells. The expressed intrabodies then bind to their targets within the cell. Intrabodies can be developed to be state-specific, binding to their target when it is in a particular conformational or (posttranslational modification state). Intrabodies can be used to label target proteins in living cells by tagging with fluorescent molecules. They can also be used to deliver reporters for intracellular Ca2+, membrane voltage, as well as functional cargo to these sites to modulate the function of the target protein and/or and the specific compartments in which it resides (Fig. 3). For the most part, use of recombinant antibodies as intrabodies is restricted to small single-chain antibodies such as ScFVs and nAbs, as the machinery in ER lumen required for the assembly of H and L chains into the intact heterotetrameric IgG antibody molecule, and the stabilization of this complex via disulfide bonds, is inefficient/absent in the cytoplasm. Other non-antibody forms of binders (monobodies, DARPins, etc.) can also be used as intrabodies.
EXAMPLES OF THE USE OF RECOMBINANT ANTIBODIES IN BASIC NEUROSCIENCE RESEARCH
ScFV use in basic neuroscience research:
ScFvs have been extensively used in brain for clinically directed purposes, including as biological therapeutics, and to target probes for non-invasive diagnostic imaging modalities such as PET. The use of ScFVs in brain for these purposes has been covered in recent review articles [e.g., (Bates & Power, 2019; Belanger et al., 2019; Messer & Butler, 2020), etc.]. These uses of ScFVs take advantage of their small size. This in itself can confer better tissue penetration/bioavailability including crossing the blood-brain barrier (BBB). ScFVs against brain targets are also amenable to conversion into bispecific antibodies, in which the two arms of the reconstituted IgG molecule are engineered to have distinct binding specificities (Riethmuller, 2012). One arm targets a BBB receptor that allows for more efficient uptake into the brain by receptor-mediated transcytosis, while the other arm carries the functionality against the brain therapeutic target (Watts & Dennis, 2013). ScFVs have been used for a variety of purposes in basic neuroscience research, and include those derived from previously generated mAbs (Fig. 4), and those developed de novo from ScFV libraries (Fig. 5). The development of examples of such ScFVs, and their use in a variety of basic neuroscience research applications, are summarized chronologically below.
ScFV-mediated inhibition of neurotrophin receptor function.
The anti-TrkA mAb MNAC13 was developed using conventional hybridoma technology from mice immunized with a stable cell line expressing human TrkA (Cattaneo et al., 1999). MNAC13 is a potent antagonist for NGF-mediated activation of TrkA receptors [e.g., (Pesavento, Margotti, Righi, Cattaneo, & Domenici, 2000), etc.]. Parallel with the original development and characterization of MNAC13, VH and VL regions were amplified from hybridoma-derived cDNA and used to generate a VL-VH ScFV mini-library which was subjected to phage display against the recombinant extracellular domain of TrkA. One of the selected ScFvs inhibited NGF-mediated differentiation of PC12 cells, demonstrating that this ScFV recapitulated the biological activity of the progenitor MNAC13 mAb (Cattaneo et al., 1999).
ScFV-mediated cell-type-specific gene transfer.
GluA2 and GluA4 cross-reactive ScFVs were isolated from a custom ScFV library. The library was generated from splenocyte cDNA obtained from mice immunized with full-length recombinant GluA4 reconstituted into liposomes. The GluA4 proteoliposomes were then used in phage display panning of this custom library, yielding 10 unique clones (Jespersen, Kuusinen, Orellana, Keinanen, & Engberg, 2000). None recognized GluA4 on immunoblots, suggesting conformational epitopes. All were able to capture GluA4 in immunoprecipitation reactions performed on lysates from transfected cells, and many also effectively captured GluA2. One of the GluA2/GluA4 cross-reactive ScFVs was subsequently used to direct ScFV-coated lentivirus to specific neuronal cell types (Anliker et al., 2010). A similar approach employing proteoliposomes as immunogens to create an immunized library for proteoliposome-mediated phage display was subsequently used to develop ScFVs against the human M2 muscarinic acetylcholine receptor (Suharni et al., 2014).
ScFV-mediated enhancement of neurite outgrowth.
A set of externally directed anti-CHL1 adhesion molecule ScFvs were selected from a naïve ScFV library by phage display panning against a recombinant CHL1 extracellular domain fragment (L. Dong, Chen, Richter, & Schachner, 2002). A mutagenized library from a single ScFV was used to isolate affinity matured ScFVs. Two of these exhibited immunolabeling of exogenously expressed CHL1 in transfected cells. These also enhanced neurite outgrowth in hippocampal and cerebellar granule neurons in culture. A set of externally directed anti-L1 adhesion molecule ScFvs were subsequently isolated by an analogous approach. Two of these exhibited immunolabeling of exogenously expressed L1 in transfected cells and cerebellar granule neurons. These also enhanced neurite outgrowth in cerebellar granule, motor and dorsal root ganglion neurons in culture, and survival of cerebellar granule neurons in culture. The same approach was used to isolate a distinct set of anti-L1 ScFVs from a different naïve ScFV library. Among these were ScFvs that either blocked or enhanced neuroblastoma cell proliferation, transmigration, and neurite outgrowth, and the L1-mediated signaling pathways involved in these processes (Wang et al., 2012).
ScFV-mediated knockdown of an inhibitory synaptic protein and suppression of synaptic activity.
ScFVs against the inhibitory synapse cytoplasmic scaffolding protein gephyrin were isolated from a naïve ScFV library by a yeast two hybrid screen using a recombinant fragment of gephyrin as bait (Zacchi et al., 2008). When the anti-gephyrin ScFVs were expressed as cytoplasmic intrabodies in heterologous HEK293 cells coexpressing gephyrin, the localization of both proteins was altered such that they were now found colocalized in cytoplasmic aggregates, indicative of ScFV-gephyrin binding. Expression of the anti-gephyrin ScFVs in cultured hippocampal neurons led to loss of endogenous gephyrin expression at inhibitory synapses, and whole cell glycine but not AMPA currents were reduced in these cells (Zacchi et al., 2008). These ScFVs were subsequently used to knockdown gephyrin expression in a subsequent studies that showed that gephyrin plays a role in supporting both phasic and tonic inhibition in cultured rat hippocampal neurons (Marchionni et al., 2009), and that ScFV-mediated suppression of GABAergic synaptic transmission could be rescued by overexpression of neuroligin 2, a gephyrin binding protein (Varley et al., 2011).
ScFV-mediated knockdown of neurotrophin receptor expression.
A set of externally directed anti-P75NTR ScFvs were isolated by phage display panning of a naïve ScFV library against a recombinant P75NTR extracellular domain fragment (Zhang et al., 2012). Three anti-P75NTR ScFVs were selected that exhibited binding to transfected HEK293T cells exogenously expressing P75NTR, and to live rat pheochromocytoma PC12 cells (known to endogenously express P75NTR). These ScFVs were then cloned into a mammalian expression plasmid as fusions with an N-terminal leader sequence to provide translation into the lumen of the endoplasmic reticulum, and appended with a C-terminal KDEL ER retrieval sequence. When expressed in PC12 cells and in mouse motor neuron neuroblastoma NSC19 cells, they reduced surface expression of P75NTR, presumably by binding to the luminally localized extracellular domains of newly synthesized p75NTR thereby preventing its plasma membrane expression. These ScFVs also inhibited NGF-induced neurite outgrowth in PC12 cells (Zhang et al., 2012).
ScFV-based tracking of a specific state of an excitatory synapse scaffolding protein.
The ScFV PF11 was selected by phage display panning against the palmitoylated form of the synaptic scaffolding molecule PSD-95 (Fukata et al., 2013). Although PF11 does not bind to the palmitoylation site itself, it exhibits selective binding to a palmitoylation-dependent conformational state of PSD-95. This ScFV was fused to a human heavy chain Fc region and used as an immunolabel to define sites at which palmitoylated PSD-95 was found in cultured neurons, with the human Fc region conferring detection by secondary antibodies. This ScFV was also used as a GFP-tagged intrabody to track the palmitoylated form of PSD-95 in living neurons, superresolution STED imaging revealed that their localization corresponded to nanodomains within the postsynaptic density (Fukata et al., 2013). This ScFV was subsequently used in a study that demonstrated that only the palmitoylated form of PSD-95 associated with AMPA and NMDA receptors (Jeyifous et al., 2016).
ScFV-guided neurotrophin receptor-mediated uptake and retrograde labeling.
ScFvs against external domains of p75NTR were used to mediate uptake and retrograde transport of expressing phage from sciatic nerve into spinal cord. Anti-p75NTR ScFVs were first selected from a naïve ScFV library by phage display against pure p75NTR recombinant protein. These were pooled to make a mini-library that was screened in vivo for ScFVs that exhibited retrograde transport to sites of ligation in injured sciatic nerve (Tani, Osbourn, Walker, Rush, & Ferguson, 2013). ScFV-coated phage also exhibit retrograde transport from uninjured sciatic nerve into spinal cord.
ScFV-mediated GABAergic synapse labeling.
Anti-GABA-A receptor ScFVs were selected from a naïve ScFV library by phage display against synthetic peptides (Koduvayur et al., 2014). These were used to immunolabel exogenously expressed receptors in Xenopus oocytes. The α1-specific ScFV exhibited a pattern of immunolabeling in retina sections similar to that obtained with an α1-specific commercial mAb.
Higher resolution imaging of the microtubule system in neurons.
Anti-tubulin ScFvs were used for high resolution imaging of tubulin (note that the same paper also used anti-tubulin nAbs) (Mikhaylova et al., 2015). The ScFvs used were developed previously (Nizak et al., 2003) by screening a naïve ScFV library against purified subcellular fractions of microtubule binding proteins from HeLa cells, which led to the isolation of the anti-tubulin ScFvs used in this study.
ScFV-mediated ion channel blockade to protect cells from ischemic cell death.
An anti-ASIC1 ScFV was developed that protects cells from ischemic death (Qiang et al., 2018). Purified recombinant ASIVC1 fragment immobilized on nanodiscs were used for phage display of a naïve ScFV library. One ScFV was an antagonist for ASIC1 channel activity, protected CHO-K1 cell line from acidosis-induced cell death, and displayed activity in vivo in a mouse MCAO ischemia-reperfusion model when the ScFV was introduced by intracerebroventricular injection three hours post-ischemia and the infarct size assayed 24 hours later.
ScFV-guided GABAergic synapse-specific retrograde gene transfer.
ScFVs were derived from hybridoma cells producing the anti-GABA-A receptor β2/β3-specific mAb 62–3G1, which binds to an external domain of receptors containing these subunits. The VH and VL regions were cloned and sequenced commercially and used to generate ScFVs in both orientations (VH-linker-VL and VL-linker-VH). Subsequent analyses demonstrated that only the ScFV in the VH-linker-VL orientation exhibited activity similar to the parent mAb in various in vitro binding assays (Nagayach, Singh, De Blas, & Geller, 2019). This ScFV was tagged with an N-terminal leader sequence for insertion into the lumen of dense core vesicles, a form of synaptic secretory vesicles, and appended with His tag for detection. When expressed in brain neurons, the ScFV was secreted and through its high affinity binding to β2/β3-containing GABA-A receptors yielded retrograde labeling of neurons synaptically connected via GABAergic synapses. Specific gene transfer was then accomplished by coating lentivirus gene transfer vectors with anti-His tag antibodies, which through their binding to the His-tagged ScFV were specifically focused to targets that had been retrogradely labeled with the ScFV (Nagayach, Singh, De Blas, & Geller, 2019).
ScFV-guided glutamatergic synapse-specific retrograde gene transfer.
ScFVs against mGluR5 were used for a similar purpose for glutamatergic synapses. One technical distinction is that the authors purchased a vial of commercial anti-mGluR5 mAb from R&D Systems (RRID: AB_2295161, MAB 45141) and had the VH and VL amino acid sequences determined commercially by mass spectrometry-based proteomics. From the obtained amino acid sequences, they synthesized the corresponding cDNA sequences for these regions, which were used to generate ScFVs. In this case both VH-linker-VL and VL-linker-VH orientations yielded active ScFVs, which were used to direct lentiviral gene transfer vectors to glutamatergic synapses (Nagayach, Singh, & Geller, 2019).
ScFV-based tracking of newly synthesized proteins in neurons.
The previously published sequence of the commonly used anti-HA mAb 12CA5 was used as the starting point to generate an anti-HA ScFV that could function inside cells as an intrabody. The 12CA5 sequence itself yielded ScFVs that failed to fold in the cytoplasm of mammalian cells, so ScFVs were engineered that had the backbone of successful ScFv intrabodies into which key specificity-determining residues from 12CA5 were incorporated to create a 12CA5 ScFV. This was fused to GFP and its efficacy to function was shown by its specific targeting to the HA-tagged neuronal ion channel Kv2.1 when coexpressed in transfected cells. This ScFV was then used to report on the sites and rates of translation of different HA-tagged proteins in cultured neurons (Zhao et al., 2019). The plasmid encoding this and related ScFVs is available from Addgene.
Examples of nanobody use in basic neuroscience research:
Nanobodies have been primarily used as immunolabels, due to the advantage gained by their small size that results in better penetration into tissue and subcellular domains with dense protein architecture (postsynaptic density, axon initial segment, etc.) and the smaller linkage error between target and dye relative to conventional antibodies. In basic neuroscience research this has been pursued most prominently using anti-GFP nAbs, which have been widely available for over a decade (Kirchhofer et al., 2010), to label GFP or GFP-tagged proteins expressed in neurons. Anti-GFP nAbs have also been used as intrabodies in a variety of research pursuits in living neurons. More recently nAbs have been developed against neuronal proteins that recognize endogenous target proteins, which will allow for more widespread application in samples from non-genetically modified organisms.
Anti-GFP nanobodies employed in high resolution immunolabeling
Quantum dots were conjugated to commercially available anti-GFP nAbs to yield higher resolution imaging of a variety of GFP-tagged proteins including extracellularly tagged GluA2 AMPA receptor subunit. The nAb-quantum dot conjugate was also used to track synaptic GluA2-containing receptors exogenously expressed in live cultured hippocampal neurons (Wang et al., 2014). A similar approach was used to track diffusion of synaptic receptors in live cultured hippocampal neurons exogenously expressing GFP-tagged GABA-A and AMPA receptor subunits (Modi, Higgs, Sheehan, Griffin, & Kittler, 2018). This nAb-quantum dot conjugate was also used to track GABA-A receptor dynamics in biolistically transfected organotypic hippocampal slice cultures. In all cases higher resolution imaging was obtained with nAbs than with dye conjugated nAbs.
In addition: 1) live neurons expressing an externally GFP-tagged neurexin-1b were labeled with an Atto594-labeled anti-GFP nAb (Chamma et al., 2016). This allowed for determination of diffusion rates of neurexin-1b and showed that the small, monomeric nAbs performed similarly to a newly developed monomeric streptavidin. 2) Atto647-labeled anti-GFP nAbs were trapped in synaptic vesicles in neurons expressing GFP-tagged VAMP2 (Joensuu et al., 2016). After partial photobleaching to allow for discrimination of single synaptic vesicles, detailed live cell imaging was performed to provide new insights into the dynamics of endocytic pathways in cultured hippocampal neurons. 3) published anti-GFP nAb sequences were used to design nAbs more amenable to site-specific labeling that allowed for their use in a variety of proof-of-concept assays, primarily in cultured heterologous cells but culminating in immunolabeling of retina (Yamagata & Sanes, 2018).
Available nAbs were also engineered with sortase tags for site-specific fluorescent labeling. These were employed in confocal microscopy correlated with electron microscopy on brain sections (Fang et al., 2018). Sortase tag Alexa647-labeled nAbs against GFP were used to label YFP-expressing neurons in brain sections from Thy-1 line H mice. The authors engineered sortase tags onto existing nAbs against CD11b to label microglia, Ly-6C/6G to label endothelial cells, and GFAP to label glial cells, each conjugated to a dye with a distinct emission allowing for four color simultaneous multiplex labeling. Following confocal imaging the sample was subjected to serial electron microscopy to correlate the fluorescence signals with the underlying ultrastructure. The combination of serial EM allowed for 3D volumetric reconstruction of the associate cells and structures. The authors found that the enhanced penetration of the nAbs compared to conventional antibodies allowed for effective immunolabeling of brain sections in the absence of detergent, which resulted in enhanced preservation of ultrastructure.
nAbs against endogenous neuronal proteins
nAbs as immunolabels
nAbs against cytoplasmic domains of VGLUT1 were first isolated from a library prepared from a llama immunized with recombinant rat VGLU1 by phage display panning against reconstituted VGluT1 liposomes (Schenck et al., 2017). In native or FP-fused forms a subset of these nAbs effectively immunolabeled cultured neurons in a pattern indistinguishable from conventional anti-VGluT1 antibodies. Certain nAbs also inhibited VGLUT1-mediated glutamate uptake into reconstituted VGluT1 liposomes and synaptic vesicles.
Nanobodies against a variety of brain proteins were developed as part of a BRAIN Initiative effort (J. X. Dong et al., 2019). Nanobodies were isolated from a cDNA library generated from a llama immunized with recombinant fragments from five different proteins by phage display panning against the individual fragments. Unique nAbs were identified by sequencing, and then tested as immunolabels for ICC against fixed and permeabilized target-expressing transfected heterologous cells. ICC-positive nAbs were assayed for efficacy and specificity in IHC against brain sections, and for ICC against cultured neurons. In the latter case, Alexa647-conjugated anti-Homer1 nAbs were used in super-resolution GSD microscopy of Homer1 at synapses. These yielded higher resolution imaging than unconjugated nAb labeling detected with dye-conjugated anti-HA epitope tag antibody, and much higher resolution than conventional mAb labeling as detected with dye-conjugated secondary antibody. A separate branch of the screening pipeline was used to identify which of the developed nAbs functioned as intrabodies as detailed below.
Nanobodies against SNAP-25 and Syntaxin 1A were used in super-resolution STED microscopy of synapses in cultured neurons (Maidorn, Olichon, Rizzoli, & Opazo, 2019). NAbs were isolated from an immunized alpaca library by phage display panning, with purified recombinant SNAP-25 and Syntaxin 1A proteins used as both the immunogen and panning bait. Unique nAbs were identified by sequencing, and then tested as immunolabels for ICC against fixed and permeabilized target-expressing transfected heterologous cells. One nAb against each target was selected or further evaluation, which included binding affinity against purified recombinant protein, crude epitope mapping versus recombinant protein fragments, and cross-reactivity against paralogs. Immunoblots were performed against samples of recombinant protein, transfected cells and brain samples, and immunoprecipitations revealed the nAbs could also bind their targets when they were assembled in SNARE complexes. The nAbs were then directly conjugated to dye and used to label their respective targets in the neuronal PC12 cell line, and cultured hippocampal neurons. Atto647N-conjugated nAbs provided access to epitopes not possible with conventional mAbs which revealed novel details of the synaptic and extrasynaptic subcellular localization of these proteins.
nAbs as modulators of neuronal cell surface receptor function.
Nanobodies were developed against the extracellular domain of the EphA4 receptor tyrosine kinase (Schoonaert et al., 2017) by immunizing an alpaca with recombinant human EphA4 ligand binding domain, followed by phage display panning against the same protein. Following ELISA confirmation and sequencing, a set of purified nAbs was tested. None recognized denatured EphA4 on Western blots, but a subset immunoprecipitated pure recombinant EphA, with some differences between human and mouse EphA4. Quantitative binding measurements against purified recombinant EphA4 revealed a subset of nAbs with KDs in the 10s of nM range, and all exhibited some cross-reactivity with other Eph receptors, except for two that were selected for further consideration. These two nAbs effectively blocked binding of ephrin ligands to EphA4, inhibited ligand-induced EphA4 activation, and ephrin-B3-mediated growth cone collapse in cortical neuron cultures. These nAbs target and antagonize EphA4 function and represent candidates for manipulating the Ephrin-EphA signaling pathway for future research exploring potential therapeutic utility.
Nanobodies were developed against extracellular domains of the Gi-coupled GPCR mGluR2 (Scholler et al., 2017). Llamas were immunized with HEK293T cells heterologously expressing rat and human mGluR2. Phage display panning was performed against purified recombinant mGuR2 immobilized on nanodiscs, followed by panning against HEK293T cells expressing rat mGluR2. A variety of assays characterizing binding to mGluR2, cross-reactivity to other mGluRs, and state-dependent binding to drug-stabilized active and inactive forms of mGluR2 were performed, the latter identifying both state-independent and active-state-dependent nAbs. Moreover, the different nAbs exhibited preferred binding to either homodimeric or heterodimeric mGluR2-containing mGluRs. Two active state-dependent nAbs were found to be positive allosteric modulators of mGluR2 function, and their pattern of activity to allosterically enhance agonist-induced mGlu2-mdiated modulation of mossy fiber nerve terminals suggested that the agonist acted through mGluR2 homodimers. One of these nAbs acted to potentiate the agonist-induced inhibition of contextual fear memory when infused with agonist into mouse hippocampus but had no impact when introduced alone. These data are consistent with nAbs acting as positive allosteric modulators of the mGluR2 GPCR.
Anti-GFP nAbs as intrabodies:
nAb-mediated manipulation of neuronal gene expression
Anti-GFP nAbs (Kirchhofer et al., 2010) were employed to mediate GFP-triggered transcription (Tang et al., 2013). The authors used anti-GFP nAbs that could tolerate fusion partners and had distinct binding sites on GFP to create a system whereby GFP acted as a dimerizer for the nAbs and their fused cargo. Many transcription factors comprise separable and autonomous DNA binding and activation domains, which when brought together can reconstitute transcription factor activity (the basis of the yeast two hybrid system (Fields & Song, 1989)). After extensive optimization, a nAb-mediated dimerization system was employed to use GFP as a cell-specific regulator of gene expression in electroporated mouse retina samples expressing GFP in specific sets of neurons. This included using GFP to manipulate expression of endogenous genes, as well as expression of exogenously expressed optogenetic actuator channelrhodopsin-2. Proof of concept studies revealed that this system also worked in intact zebrafish embryos. The authors extended these technique to develop destabilized/conditionally unstable nAbs whose stability was enhanced by target binding (Tang et al., 2016). This allowed for selective accumulation of nAb-cargo fusions only in cells that express the target, thereby allowing for a single nAb to mediate the desired function. They showed proof of concept in studies again employing GFP as a target, but also showed one could manipulate HIV-1 infected human T cells by employing an anti-capsid nAb. Combining this technology with the growing collection of publicly available nAbs directed against neuronal targets will allow for selective target-dependent manipulation of cell function.
nAb-mediated neuronal ribosome profiling:
Anti-GFP nAbs were employed in an innovative way to capture actively translated transcripts in brain neurons in a circuit-specific manner (Ekstrand et al., 2014). Transgenic mice were generated that express an anti-GFP nAb (Rothbauer et al., 2006) fused to a ribosomal protein (Rpl10a). The authors then introduced GFP into neurons using a retrogradely transported virus injected into specific brain regions. They then captured ribosomes from neurons projecting to that region by using beads coated with an anti-GFP mAb that binds to an epitope distinct from the nAb to capture ribosomes with nAb-bound GFP. This approach was used to map gene expression in neurons in specific circuits. The authors subsequently used a similar approach to define gene expression in a distinct subset of midbrain neurons that participate in reward circuitry (Pomeranz et al., 2017).
nAb-mediated enhancement of neuronal GFP reporting:
The transgenic mice used for ribosome profiling (Ekstrand et al., 2014) that express an anti-GFP nAb fused to a ribosome protein were employed to substantially enhance the GFP signal used to mark populations of neurons in GFP reporter mice (Y. Chen et al., 2020). Previous studies had shown that the binding of certain anti-GFP nAbs enhanced GFP fluorescence in vitro (Kirchhofer et al., 2010; Kubala, Kovtun, Alexandrov, & Collins, 2010). Another contributor to the enhanced GFP signal is that the GFP is highly concentrated in the cell body through its capture by the ribosome localized nAb which prevents the GFP from diffusing throughout the large area of the axon and dendrites. The GFP may also be stabilized by its inclusion in a complex at the ribosome, which is turned over very slowly in neurons (Retz & Steele, 1980).
nAbs against endogenous neuronal proteins as intrabodies
Nanobodies against endogenous neuronal proteins targeting fluorescent reporters to specific sites in neurons.
As detailed above, nAbs against a variety of targets with restricted subcellular localization in neurons were developed (J. X. Dong et al., 2019). nAb-GFP fusions were assayed for their ability to recognize their target when expressed as intrabodies. This was initially evaluated by expression of nAb-GFP fusions in target-expressing transfected heterologous cells, and then in cultured hippocampal neurons, and colocalization with exogenous and endogenous target proteins, respectively, by ICC (J. X. Dong et al., 2019).
Nanobodies employed to knockdown cell surface expression of endogenous neuronal proteins.
nAbs against the cytoplasmic Cavβ auxiliary subunit of high voltage-activated Ca2+ channels were developed from a phage display library prepared from a llama immunized with recombinant Cavβ1 and Cavβ3 proteins (Morgenstern, Park, Fan, & Colecraft, 2019). One of these anti-Cavβ1/Cavβ3 nAbs was used to generate a tripartite fusion protein with cyan fluorescent protein (CFP) and the PKC C1 domain, the latter of which led to the plasma membrane association of the nAb-CFP-C1 fusion upon phorbol ester treatment of transfected cells. The intrabody function of this nAb fusion was supported by the phorbol ester-induced translocation of coexpressed Cavβ1 in nAb expressing cells. The nAb was then fused to a ubiquitin E3-ligase and was shown to function to selectively knockdown plasma membrane expression and function of Cavβ-containing high voltage-gated Ca2+ channels exogenously expressed in heterologous cells, and endogenously expressed in dorsal route ganglion neurons, and in cardiomyocytes and pancreatic beta cells (Morgenstern et al., 2019).
CONCLUSIONS
These examples underscore the varied and often powerful and innovative uses of recombinant antibodies in basic neuroscience research. The availability and use of these and other recombinant antibodies will only continue to increase. In the future there will be many additional opportunities to make further gains in ongoing research, and in opening new fields of inquiry by applying these powerful, unambiguously identified and renewable research reagents to basic neuroscience research.
SIGNIFICANCE STATEMENT.
Antibodies are fundamental reagents to basic neuroscience research. Recombinant antibodies offer many advantages over conventional antibodies including their unambiguous identification and digital archiving via DNA sequencing, reliable expression, facile and reliable dissemination as DNA sequences and as plasmids, and the opportunity for numerous forms of engineering to enhance their utility. This overview describes various forms of recombinant antibodies and examples of their emerging and innovative use in basic neuroscience research.
Acknowledgements
Our work developing recombinant antibodies for neuroscience research was supported by National Institutes of Health Grants U24NS050606 and R24NS092991, and National Institutes of Health BRAIN Initiative Grants U01NS099714 and U24NS109113 to J. S. Trimmer.
Footnotes
Conflict of Interest
The author has no conflict of interest to declare.
Literature Cited
- Almagro JC, Pedraza-Escalona M, Arrieta HI, & Perez-Tapia SM (2019). Phage display libraries for antibody therapeutic discovery and development. Antibodies (Basel), 8(3). doi: 10.3390/antib8030044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews NP, Boeckman JX, Manning CF, Nguyen JT, Bechtold H, Dumitras C, … Trimmer JS (2019). A toolbox of IgG subclass-switched recombinant monoclonal antibodies for enhanced multiplex immunolabeling of brain. eLife, 8, e43322. doi: 10.7554/eLife.43322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anliker B, Abel T, Kneissl S, Hlavaty J, Caputi A, Brynza J, … Buchholz CJ (2010). Specific gene transfer to neurons, endothelial cells and hematopoietic progenitors with lentiviral vectors. Nat Methods, 7(11), 929–935. doi: 10.1038/nmeth.1514 [DOI] [PubMed] [Google Scholar]
- Bates A, & Power CA (2019). David vs. Goliath: the structure, function, and clinical prospects of antibody fragments. Antibodies (Basel), 8(2). doi: 10.3390/antib8020028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekele-Arcuri Z, Matos MF, Manganas L, Strassle BW, Monaghan MM, Rhodes KJ, & Trimmer JS (1996). Generation and characterization of subtype-specific monoclonal antibodies to K+ channel alpha- and beta-subunit polypeptides. Neuropharmacology, 35(7), 851–865. [DOI] [PubMed] [Google Scholar]
- Belanger K, Iqbal U, Tanha J, MacKenzie R, Moreno M, & Stanimirovic D (2019). Single-domain antibodies as therapeutic and imaging agents for the treatment of CNS diseases. Antibodies (Basel), 8(2). doi: 10.3390/antib8020027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensussen S, Shankar S, Ching KH, Zemel D, Ta TL, Mount RA, … Han X (2020). A viral toolbox of genetically encoded fluorescent synaptic tags. iScience, 23(7), 101330. doi: 10.1016/j.isci.2020.101330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird RE, Hardman KD, Jacobson JW, Johnson S, Kaufman BM, Lee SM, … Whitlow M (1988). Single-chain antigen-binding proteins. Science, 242(4877), 423–426. [DOI] [PubMed] [Google Scholar]
- Bradbury A, & Pluckthun A (2015). Reproducibility: Standardize antibodies used in research. Nature, 518(7537), 27–29. doi: 10.1038/518027a [DOI] [PubMed] [Google Scholar]
- Bradbury AR, & Pluckthun A (2015). Getting to reproducible antibodies: the rationale for sequenced recombinant characterized reagents. Protein Eng Des Sel, 28(10), 303–305. doi: 10.1093/protein/gzv051 [DOI] [PubMed] [Google Scholar]
- Bradbury ARM (2010). The use of phage display in neurobiology. Curr Protoc Neurosci, 51, 5.12.11–15.12.27. doi:DOI: 10.1002/0471142301.ns0512s51 [DOI] [PubMed] [Google Scholar]
- Bradbury ARM, Trinklein ND, Thie H, Wilkinson IC, Tandon AK, Anderson S, … Dubel S (2018). When monoclonal antibodies are not monospecific: Hybridomas frequently express additional functional variable regions. MAbs, 10(4), 539–546. doi: 10.1080/19420862.2018.1445456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss NA, Henderson SJ, McFarlane M, Shenton JM, & de Haan L (2012). Monoclonal antibody therapeutics: history and future. Curr Opin Pharmacol, 12(5), 615–622. doi: 10.1016/j.coph.2012.08.001 [DOI] [PubMed] [Google Scholar]
- Carroll WL, Mendel E, & Levy S (1988). Hybridoma fusion cell lines contain an aberrant kappa transcript. Mol Immunol, 25(10), 991–995. [DOI] [PubMed] [Google Scholar]
- Cattaneo A, Capsoni S, Margotti E, Righi M, Kontsekova E, Pavlik P, … Novak M (1999). Functional blockade of tyrosine kinase A in the rat basal forebrain by a novel antagonistic anti-receptor monoclonal antibody. J Neurosci, 19(22), 9687–9697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamma I, Letellier M, Butler C, Tessier B, Lim KH, Gauthereau I, … Thoumine O (2016). Mapping the dynamics and nanoscale organization of synaptic adhesion proteins using monomeric streptavidin. Nat Commun, 7, 10773. doi: 10.1038/ncomms10773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SY, Bagley J, & Marasco WA (1994). Intracellular antibodies as a new class of therapeutic molecules for gene therapy. Hum Gene Ther, 5(5), 595–601. doi: 10.1089/hum.1994.5.5-595 [DOI] [PubMed] [Google Scholar]
- Chen Y, Jang H, Spratt PWE, Kosar S, Taylor DE, Essner RA, … Garrison JL (2020). Soma-targeted imaging of neural circuits by ribosome tethering. Neuron, 107(3), 454–469 e456. doi: 10.1016/j.neuron.2020.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu ML, & Gilliland GL (2016). Engineering antibody therapeutics. Curr Opin Struct Biol, 38, 163–173. doi: 10.1016/j.sbi.2016.07.012 [DOI] [PubMed] [Google Scholar]
- Chiu ML, Goulet DR, Teplyakov A, & Gilliland GL (2019). Antibody structure and function: the basis for engineering therapeutics. Antibodies (Basel), 8(4). doi: 10.3390/antib8040055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosnier C, Staudt N, & Wright GJ (2010). A rapid and scalable method for selecting recombinant mouse monoclonal antibodies. BMC Biol, 8, 76. doi: 10.1186/1741-7007-8-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong JX, Lee Y, Kirmiz M, Palacio S, Dumitras C, Moreno CM, … Trimmer JS (2019). A toolbox of nanobodies developed and validated for use as intrabodies and nanoscale immunolabels in mammalian brain neurons. eLife, 8, e48750. doi: 10.7554/eLife.48750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L, Chen S, Richter M, & Schachner M (2002). Single-chain variable fragment antibodies against the neural adhesion molecule CHL1 (close homolog of L1) enhance neurite outgrowth. J Neurosci Res, 69(4), 437–447. doi: 10.1002/jnr.10250 [DOI] [PubMed] [Google Scholar]
- Ekstrand MI, Nectow AR, Knight ZA, Latcha KN, Pomeranz LE, & Friedman JM (2014). Molecular profiling of neurons based on connectivity. Cell, 157(5), 1230–1242. doi: 10.1016/j.cell.2014.03.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang T, Lu X, Berger D, Gmeiner C, Cho J, Schalek R, … Lichtman J (2018). Nanobody immunostaining for correlated light and electron microscopy with preservation of ultrastructure. Nat Methods, 15(12), 1029–1032. doi: 10.1038/s41592-018-0177-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farid SS, Baron M, Stamatis C, Nie W, & Coffman J (2020). Benchmarking biopharmaceutical process development and manufacturing cost contributions to R&D. MAbs, 12(1), 1754999. doi: 10.1080/19420862.2020.1754999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields S, & Song O (1989). A novel genetic system to detect protein-protein interactions. Nature, 340(6230), 245–246. doi: 10.1038/340245a0 [DOI] [PubMed] [Google Scholar]
- Fukata Y, Dimitrov A, Boncompain G, Vielemeyer O, Perez F, & Fukata M (2013). Local palmitoylation cycles define activity-regulated postsynaptic subdomains. J Cell Biol, 202(1), 145–161. doi: 10.1083/jcb.201302071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger JR, Melcher T, Koh DS, Sakmann B, Seeburg PH, Jonas P, & Monyer H (1995). Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron, 15(1), 193–204. doi: 10.1016/0896-6273(95)90076-4 [DOI] [PubMed] [Google Scholar]
- Ghassabeh GH, Saerens D, & Muyldermans S (2010). Isolation of antigen-specific nanobodies In Kontermann R & Dübel S (Eds.), Antibody Engineering (pp. 251–266). Berlin, Heidelberg: Springer Berlin Heidelberg. [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, & Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods, 6(5), 343–345. doi: 10.1038/nmeth.1318 [DOI] [PubMed] [Google Scholar]
- Gong B, Murray KD, & Trimmer JS (2016). Developing high-quality mouse monoclonal antibodies for neuroscience research - approaches, perspectives and opportunities. N Biotechnol, 33(5 Pt A), 551–564. doi: 10.1016/j.nbt.2015.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield EA (2014). Antibodies A laboratory manual, second edition Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Gross GG, Junge JA, Mora RJ, Kwon HB, Olson CA, Takahashi TT, … Arnold DB (2013). Recombinant probes for visualizing endogenous synaptic proteins in living neurons. Neuron, 78(6), 971–985. doi: 10.1016/j.neuron.2013.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanes J, & Pluckthun A (1997). In vitro selection and evolution of functional proteins by using ribosome display. Proc Natl Acad Sci U S A, 94(10), 4937–4942. doi: 10.1073/pnas.94.10.4937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow E, & Lane D (1988). Antibodies A laboratory manual. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Hartmann J, Thalheimer FB, Hopfner F, Kerzel T, Khodosevich K, Garcia-Gonzalez D, … Buchholz CJ (2019). GluA4-targeted AAV vectors deliver genes selectively to interneurons while relying on the AAV receptor for entry. Mol Ther Methods Clin Dev, 14, 252–260. doi: 10.1016/j.omtm.2019.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huston JS, Levinson D, Mudgett-Hunter M, Tai MS, Novotny J, Margolies MN, … et al. (1988). Protein engineering of antibody binding sites: recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc Natl Acad Sci U S A, 85(16), 5879–5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jespersen LK, Kuusinen A, Orellana A, Keinanen K, & Engberg J (2000). Use of proteoliposomes to generate phage antibodies against native AMPA receptor. Eur J Biochem, 267(5), 1382–1389. doi: 10.1046/j.1432-1327.2000.01137.x [DOI] [PubMed] [Google Scholar]
- Jeyifous O, Lin EI, Chen X, Antinone SE, Mastro R, Drisdel R, … Green WN (2016). Palmitoylation regulates glutamate receptor distributions in postsynaptic densities through control of PSD95 conformation and orientation. Proc Natl Acad Sci U S A, 113(52), E8482–E8491. doi: 10.1073/pnas.1612963113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joensuu M, Padmanabhan P, Durisic N, Bademosi AT, Cooper-Williams E, Morrow IC, … Mezunier FA (2016). Subdiffractional tracking of internalized molecules reveals heterogeneous motion states of synaptic vesicles. J Cell Biol, 215(2), 277–292. doi: 10.1083/jcb.201604001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost C, & Pluckthun A (2014). Engineered proteins with desired specificity: DARPins, other alternative scaffolds and bispecific IgGs. Curr Opin Struct Biol, 27, 102–112. doi: 10.1016/j.sbi.2014.05.011 [DOI] [PubMed] [Google Scholar]
- Jovcevska I, & Muyldermans S (2020). The therapeutic potential of nanobodies. BioDrugs, 34(1), 11–26. doi: 10.1007/s40259-019-00392-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juste M, Muzard J, & Billiald P (2006). Cloning of the antibody kappa light chain V-gene from murine hybridomas by bypassing the aberrant MOPC21-derived transcript. Anal Biochem, 349(1), 159–161. doi: 10.1016/j.ab.2005.10.046 [DOI] [PubMed] [Google Scholar]
- Kannan M, Gross GG, Arnold DB, & Higley MJ (2016). Visual deprivation during the critical period enhances layer 2/3 GABAergic inhibition in mouse V1. J Neurosci, 36(22), 5914–5919. doi: 10.1523/JNEUROSCI.0051-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhofer A, Helma J, Schmidthals K, Frauer C, Cui S, Karcher A, … Rothbauer U (2010). Modulation of protein properties in living cells using nanobodies. Nat Struct Mol Biol, 17(1), 133–138. doi: 10.1038/nsmb.1727 [DOI] [PubMed] [Google Scholar]
- Knight KL, Burnett RC, & McNicholas JM (1985). Organization and polymorphism of rabbit immunoglobulin heavy chain genes. J Immunol, 134(2), 1245–1250. [PubMed] [Google Scholar]
- Koduvayur SP, Gussin HA, Parthasarathy R, Hao Z, Kay BK, & Pepperberg DR (2014). Generation of recombinant antibodies to rat GABAA receptor subunits by affinity selection on synthetic peptides. PLoS One, 9(2), e87964. doi: 10.1371/journal.pone.0087964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide A, Bailey CW, Huang X, & Koide S (1998). The fibronectin type III domain as a scaffold for novel binding proteins. J Mol Biol, 284(4), 1141–1151. doi: 10.1006/jmbi.1998.2238 [DOI] [PubMed] [Google Scholar]
- Kubala MH, Kovtun O, Alexandrov K, & Collins BM (2010). Structural and thermodynamic analysis of the GFP:GFP-nanobody complex. Protein Sci, 19(12), 2389–2401. doi: 10.1002/pro.519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon T, Merchan-Perez A, Rial Verde EM, Rodriguez JR, DeFelipe J, & Yuste R (2019). Ultrastructural, molecular and functional mapping of GABAergic synapses on dendritic spines and shafts of neocortical pyramidal neurons. Cereb Cortex, 29(7), 2771–2781. doi: 10.1093/cercor/bhy143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maidorn M, Olichon A, Rizzoli SO, & Opazo F (2019). Nanobodies reveal an extra-synaptic population of SNAP-25 and Syntaxin 1A in hippocampal neurons. MAbs, 11(2), 305–321. doi: 10.1080/19420862.2018.1551675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning CF, Bundros AM, & Trimmer JS (2012). Benefits and pitfalls of secondary antibodies: why choosing the right secondary is of primary importance. PLoS One, 7(6), e38313. doi: 10.1371/journal.pone.0038313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchionni I, Kasap Z, Mozrzymas JW, Sieghart W, Cherubini E, & Zacchi P (2009). New insights on the role of gephyrin in regulating both phasic and tonic GABAergic inhibition in rat hippocampal neurons in culture. Neuroscience, 164(2), 552–562. doi: 10.1016/j.neuroscience.2009.07.063 [DOI] [PubMed] [Google Scholar]
- Messer A, & Butler DC (2020). Optimizing intracellular antibodies (intrabodies/nanobodies) to treat neurodegenerative disorders. Neurobiol Dis, 134, 104619. doi: 10.1016/j.nbd.2019.104619 [DOI] [PubMed] [Google Scholar]
- Mikhaylova M, Cloin BM, Finan K, van den Berg R, Teeuw J, Kijanka MM, … Kapitein LC (2015). Resolving bundled microtubules using anti-tubulin nanobodies. Nat Commun, 6, 7933. doi: 10.1038/ncomms8933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell LS, & Colwell LJ (2018). Comparative analysis of nanobody sequence and structure data. Proteins, 86(7), 697–706. doi: 10.1002/prot.25497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi S, Higgs NF, Sheehan D, Griffin LD, & Kittler JT (2018). Quantum dot conjugated nanobodies for multiplex imaging of protein dynamics at synapses. Nanoscale, 10(21), 10241–10249. doi: 10.1039/c7nr09130c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern TJ, Park J, Fan QR, & Colecraft HM (2019). A potent voltage-gated calcium channel inhibitor engineered from a nanobody targeted to auxiliary Cavbeta subunits. eLife, 8. doi: 10.7554/eLife.49253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muyldermans S (2013). Nanobodies: natural single-domain antibodies. Annu Rev Biochem, 82, 775–797. doi: 10.1146/annurev-biochem-063011-092449 [DOI] [PubMed] [Google Scholar]
- Nagayach A, Singh A, De Blas AL, & Geller AI (2019). Delivery of different genes into pre- and post-synaptic neocortical interneurons connected by GABAergic synapses. PLoS One, 14(5), e0217094. doi: 10.1371/journal.pone.0217094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagayach A, Singh A, & Geller AI (2019). Separate gene transfers into pre- and postsynaptic neocortical neurons connected by mGluR5-containing synapses. J Mol Neurosci, 68(4), 549–564. doi: 10.1007/s12031-019-01317-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nizak C, Martin-Lluesma S, Moutel S, Roux A, Kreis TE, Goud B, & Perez F (2003). Recombinant antibodies against subcellular fractions used to track endogenous Golgi protein dynamics in vivo. Traffic, 4(11), 739–753. doi: 10.1034/j.1600-0854.2003.00132.x [DOI] [PubMed] [Google Scholar]
- Pardon E, Laeremans T, Triest S, Rasmussen SG, Wohlkonig A, Ruf A, … Steyaert J (2014). A general protocol for the generation of nanobodies for structural biology. Nat Protoc, 9(3), 674–693. doi: 10.1038/nprot.2014.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesavento E, Margotti E, Righi M, Cattaneo A, & Domenici L (2000). Blocking the NGF-TrkA interaction rescues the developmental loss of LTP in the rat visual cortex: role of the cholinergic system. Neuron, 25(1), 165–175. doi: 10.1016/s0896-6273(00)80880-6 [DOI] [PubMed] [Google Scholar]
- Plückthun A (2015). Designed ankyrin repeat proteins (DARPins): binding proteins for research, diagnostics, and therapy. Annu Rev Pharmacol Toxicol, 55, 489–511. doi: 10.1146/annurev-pharmtox-010611-134654 [DOI] [PubMed] [Google Scholar]
- Pomeranz LE, Ekstrand MI, Latcha KN, Smith GA, Enquist LW, & Friedman JM (2017). Gene expression profiling with Cre-conditional pseudorabies virus reveals a subset of midbrain neurons that participate in reward circuitry. J Neurosci, 37(15), 4128–4144. doi: 10.1523/JNEUROSCI.3193-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang M, Dong X, Zha Z, Zuo XK, Song XL, Zhao L, … Lerner RA (2018). Selection of an ASIC1a-blocking combinatorial antibody that protects cells from ischemic death. Proc Natl Acad Sci U S A, 115(32), E7469–E7477. doi: 10.1073/pnas.1807233115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reader RH, Workman RG, Maddison BC, & Gough KC (2019). Advances in the production and batch reformatting of phage antibody libraries. Mol Biotechnol, 61(11), 801–815. doi: 10.1007/s12033-019-00207-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retz KC, & Steele WJ (1980). Ribosome turnover in rat brain and liver. Life Sci, 27(25–26), 2601–2604. doi: 10.1016/0024-3205(80)90546-9 [DOI] [PubMed] [Google Scholar]
- Rhodes KJ, & Trimmer JS (2006). Antibodies as valuable neuroscience research tools versus reagents of mass distraction. J Neurosci, 26(31), 8017–8020. doi:26/31/8017 [pii] 10.1523/JNEUROSCI.2728-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riethmuller G (2012). Symmetry breaking: bispecific antibodies, the beginnings, and 50 years on. Cancer Immun, 12, 12. [PMC free article] [PubMed] [Google Scholar]
- Rothbauer U, Zolghadr K, Tillib S, Nowak D, Schermelleh L, Gahl A, … Leonhardt H (2006). Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat Methods, 3(11), 887–889. doi: 10.1038/nmeth953 [DOI] [PubMed] [Google Scholar]
- Schaefer JV, Honegger A, & Pluckthun A (2010). Construction of scFv Fragments from hybridoma or spleen cells by PCR assembly In Kontermann R & Dubel S (Eds.), Antibody Engineering (pp. 21–44). Heidelberg, Germany: Springer Verlag. [Google Scholar]
- Schenck S, Kunz L, Sahlender D, Pardon E, Geertsma ER, Savtchouk I, … Dutzler R (2017). Generation and characterization of anti-VGLUT nanobodies acting as inhibitors of transport. Biochemistry, 56(30), 3962–3971. doi: 10.1021/acs.biochem.7b00436 [DOI] [PubMed] [Google Scholar]
- Scholler P, Nevoltris D, de Bundel D, Bossi S, Moreno-Delgado D, Rovira X, … Pin JP (2017). Allosteric nanobodies uncover a role of hippocampal mGlu2 receptor homodimers in contextual fear consolidation. Nat Commun, 8(1), 1967. doi: 10.1038/s41467-017-01489-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoonaert L, Rue L, Roucourt B, Timmers M, Little S, Chavez-Gutierrez L, … Robberecht W (2017). Identification and characterization of Nanobodies targeting the EphA4 receptor. J Biol Chem, 292(27), 11452–11465. doi: 10.1074/jbc.M116.774141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnen BL, Bowen AB, Forte JS, Hiester BG, Crosby KC, Gibson ES, … Kennedy MJ (2017). Optogenetic control of synaptic composition and function. Neuron, 93(3), 646–660 e645. doi: 10.1016/j.neuron.2016.12.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GP (1985). Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science, 228(4705), 1315–1317. doi: 10.1126/science.4001944 [DOI] [PubMed] [Google Scholar]
- Suharni, Nomura Y, Arakawa T, Hino T, Abe H, Nakada-Nakura Y, … Nomura N (2014). Proteoliposome-based selection of a recombinant antibody fragment against the human M2 muscarinic acetylcholine receptor. Monoclon Antib Immunodiagn Immunother, 33(6), 378–385. doi: 10.1089/mab.2014.0041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang JC, Drokhlyansky E, Etemad B, Rudolph S, Guo B, Wang S, … Cepko CL (2016). Detection and manipulation of live antigen-expressing cells using conditionally stable nanobodies. eLife , 5. doi: 10.7554/eLife.15312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang JC, Szikra T, Kozorovitskiy Y, Teixiera M, Sabatini BL, Roska B, & Cepko CL (2013). A nanobody-based system using fluorescent proteins as scaffolds for cell-specific gene manipulation. Cell, 154(4), 928–939. doi: 10.1016/j.cell.2013.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani H, Osbourn JK, Walker EH, Rush RA, & Ferguson IA (2013). A novel in vivo method for isolating antibodies from a phage display library by neuronal retrograde transport selectively yields antibodies against p75(NTR.). MAbs, 5(3), 471–478. doi: 10.4161/mabs.24112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taussig MJ, Fonseca C, & Trimmer JS (2018). Antibody validation: a view from the mountains. N Biotechnol, 45, 1–8. doi: 10.1016/j.nbt.2018.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiller KE, & Tessier PM (2015). Advances in antibody design. Annu Rev Biomed Eng, 17, 191–216. doi: 10.1146/annurev-bioeng-071114-040733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toleikis L, Broders O, & Dubel S (2004). Cloning single-chain antibody fragments (scFv) from hybridoma cells. Methods Mol Med, 94, 447–458. doi: 10.1385/1-59259-679-7:447 [DOI] [PubMed] [Google Scholar]
- Uhlen M, Bandrowski A, Carr S, Edwards A, Ellenberg J, Lundberg E, … Yamamoto T (2016). A proposal for validation of antibodies. Nat Methods, 13(10), 823–827. doi: 10.1038/nmeth.3995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varley ZK, Pizzarelli R, Antonelli R, Stancheva SH, Kneussel M, Cherubini E, & Zacchi P (2011). Gephyrin regulates GABAergic and glutamatergic synaptic transmission in hippocampal cell cultures. J Biol Chem, 286(23), 20942–20951. doi: 10.1074/jbc.M111.234641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AS, Neves G, Grillo F, Jackson RE, Rigby M, O’Donnell C, … Burrone J (2017). Distance-dependent gradient in NMDAR-driven spine calcium signals along tapering dendrites. Proc Natl Acad Sci U S A, 114(10), E1986–E1995. doi: 10.1073/pnas.1607462114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cai E, Rosenkranz T, Ge P, Teng KW, Lim SJ, … Selvin PR (2014). Small quantum dots conjugated to nanobodies as immunofluorescence probes for nanometric microscopy. Bioconjug Chem, 25(12), 2205–2211. doi: 10.1021/bc5004179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Loers G, Pan HC, Gouveia R, Zhao WJ, Shen YQ, … Schachner M (2012). Antibody fragments directed against different portions of the human neural cell adhesion molecule L1 act as inhibitors or activators of L1 function. PLoS One, 7(12), e52404. doi: 10.1371/journal.pone.0052404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts RJ, & Dennis MS (2013). Bispecific antibodies for delivery into the brain. Curr Opin Chem Biol, 17(3), 393–399. doi: 10.1016/j.cbpa.2013.03.023 [DOI] [PubMed] [Google Scholar]
- Yamagata M, & Sanes JR (2018). Reporter-nanobody fusions (RANbodies) as versatile, small, sensitive immunohistochemical reagents. Proc Natl Acad Sci U S A, 115(9), 2126–2131. doi: 10.1073/pnas.1722491115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacchi P, Dreosti E, Visintin M, Moretto-Zita M, Marchionni I, Cannistraci I, … Cherubini E (2008). Gephyrin selective intrabodies as a new strategy for studying inhibitory receptor clustering. J Mol Neurosci, 34(2), 141–148. doi: 10.1007/s12031-007-9018-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Helmsing S, Zagrebelsky M, Schirrmann T, Marschall AL, Schungel M, … Dubel S (2012). Suppression of p75 neurotrophin receptor surface expression with intrabodies influences Bcl-xL mRNA expression and neurite outgrowth in PC12 cells. PLoS One, 7(1), e30684. doi: 10.1371/journal.pone.0030684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao N, Kamijo K, Fox PD, Oda H, Morisaki T, Sato Y, … Stasevich TJ (2019). A genetically encoded probe for imaging nascent and mature HA-tagged proteins in vivo. Nat Commun, 10(1), 2947. doi: 10.1038/s41467-019-10846-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zotova A, Zotov I, Filatov A, & Mazurov D (2016). Determining antigen specificity of a monoclonal antibody using genome-scale CRISPR-Cas9 knockout library. J Immunol Methods, 439, 8–14. doi: 10.1016/j.jim.2016.09.006 [DOI] [PubMed] [Google Scholar]