

Abstract
Since the discovery of CHD1L in 2008 it has emerged as an oncogene implicated in the pathology and poor prognosis of a variety of cancers, including gastrointestinal cancers. However, a mechanistic understanding of CHD1L as a driver of colorectal cancer (CRC) has been limited. Until now, there have been no reported inhibitors of CHD1L, also limiting its development as a molecular target. We sought to characterize the clinicopathological link between CHD1L and CRC, determine the mechanism(s) by which CHD1L drives malignant CRC, and discover the first inhibitors with potential for novel treatments for CRC. The clinicopathologic characteristics associated with CHD1L expression were evaluated using microarray data from 585 CRC patients. Further analysis of microarray data indicated that CHD1L may function through the Wnt/TCF pathway. Thus, we conducted knockdown and overexpression studies with CHD1L to determine its role in Wnt/TCF-driven epithelial-mesenchymal transition (EMT). We performed high-throughput screening (HTS) to identify the first CHD1L inhibitors. The mechanism of action, antitumor efficacy, and drug-like properties of lead CHD1L inhibitors were determined using biochemical assays, cell-models, tumor organoids, patient derived tumor organoids, and in vivo pharmacokinetics (PK) and pharmacodynamics (PD). Lead CHD1L inhibitors display potent in vitro antitumor activity by reversing TCF-driven EMT. The best lead CHD1L inhibitor possesses drug-like properties in PK/PD mouse models. This work validates CHD1L as a druggable target and establishes a novel therapeutic strategy for the treatment of CRC.
Introduction
The integrity of the genome is maintained by conformational changes to chromatin structure that regulate accessibility to DNA for gene expression and replication. Chromatin structure is maintained by post-translational modifications of histones and rearrangement of nucleosomes.(1-3) ATP-dependent chromatin remodelers are enzymes that alter chromatin by changing histone composition, and evicting or translocating nucleosomes along DNA. Their activity plays a critical role in cellular function by regulating gene expression and the accessibility of DNA for replication, transcription, and DNA repair.(4,5) Dysregulation of chromatin remodeling is associated with human disease, particularly cancer.(6,7) In the last decade, the chromatin remodeler known as CHD1L (chromodomain helicase/ATPase DNA binding protein 1-like), also known as ALC1 (amplified in liver cancer 1), has emerged as an oncogene implicated in the pathology of prominent human cancers.(8,9) CHD1L is also involved in multi-drug resistance, ranging from upregulation of drug resistance efflux pumps (e.g. ABCB1)(10) to PARP1 mediated DNA repair(11,12), and anti-apoptotic activity.(13,14) Moreover, amplification or overexpression of CHD1L are correlated with poor prognoses for patients, including low overall survival (OS) and metastatic disease.(15-17) Therefore, the multifunctional oncogenic mechanisms of CHD1L make it an attractive therapeutic target in cancer.
While the cancer driving mechanisms of CHD1L have been studied in liver(13,14), breast(18), and lung(10) cancer, little is known about the pathological mechanisms associated with CHD1L in colorectal cancer (CRC). A majority of CRC patients possess mutations in the Wnt signaling pathway, leading to aberrant T-Cell Factor/Lymphoid Enhancer Factor-transcription, denoted henceforth as TCF-transcription or TCF-complex.(19,20) The TCF-complex is orchestrated by TCF4 (a.k.a. TCFL2), which is activated through interactions with an array of coactivators such as β-catenin, PARP1, and CREB Binding protein (CBP).(21) Recently, TCF4 was shown to be a specific driver of both early metastasis from adenomas (i.e. polyps) and from late stage metastatic CRC (mCRC).16,17 Moreover, we and others have shown that TCF-transcription functions as a master regulator of epithelial-mesenchymal transition (EMT)(22-24), a process that can transform relatively benign epithelial tumor cells into mesenchymal cells with increased cancer stem cell (CSC) stemness and other malignant properties that drive mCRC.(25) Currently, no drug has been clinically approved that target the Wnt/TCF pathway.(26) To this end, we have evaluated the clinicopathological characteristics of CHD1L in CRC, which led us to hypothesize that CHD1L is a druggable target involved in TCF-transcription.
In this study, we propose a mechanism for CHD1L-mediated TCF-transcription. Utilizing high-throughput screening (HTS), we have identified the first small molecule inhibitors of CHD1L. We show that lead inhibitors are able to prevent TCF-transcription, reverse EMT, and other malignant properties in a variety of cell models including tumor organoids and patient derived tumor organoids (PDTOs). The top lead CHD1L inhibitor displays drug-like pharmacological properties, including in vivo pharmacokinetic (PK) and pharmacodynamic (PD) profiles, important for translational development towards the treatment of CRC and other cancers.
Materials and Methods
Clinicopathological characterization of CHD1L.
Transcriptome expression data of 585 CRC patients from the CIT cohort (GEO: GSE39582) were used for in silico validation (GSE39582).(27) Gene expression analyses were performed by the Affymetrix Human Genome U133 Plus 2.0 Array. RMA was used for data preprocessing and COMBAT for batch correction. Signal intensity was log2 normalized. The CHD1L cutoff for CRC risk stratification based on disease specific survival was determined by the receiver operating characteristic (ROC) curve. Cutoff for CHD1L expression was set to 6.45. Differences in OS were estimated by the Kaplan-Meier method and compared using the log-rank test. For the comparison of categorical variables we used the Fisher’s exact test. The Mann-Whitney U test was used for 2 groups of continuous variables and in case of more than two groups data has been analyzed by the Kruskal-Wallis test. For all 2-sided P-values, the unadjusted significance level of 0.05 was applied.
The CHD1L cutoff and clinicopathologic characteristics have been evaluated by multiple cox regression analysis. Only variables that were significant in univariate analyses had been integrated in the cox regression model using the Wald forward algorithm for significance determination. All variables including more than 2 groups had been categorized and the stepwise entry criterion for covariates was P<0.05 and the removal criterion was P>0.1. Statistical analysis had been performed using SPSS (IBM, Armonk, NY), GraphPad Prism (San Diego, CA), JMP (SAS, Cary, NC), and R Studio.
University of Colorado Cancer Center (UCCC) Patient sample RNA-seq analysis.
RNA-seq data were from CRC patient tumor xenograft explants were obtained from the UCCC GI tumor tissue bank, and analyzed as previously described.(28) Briefly, gene expression was Log2 normalized and measured by FPKM (Fragments Per Kilobase of transcript per Million mapped reads). The Wnt signaling pathway defined by the Kyoto Encyclopedia of Genes and Genomes (KEGG) was used as the gene set in this study. Samples with expression of CHD1L <1 FPKM were considered low expression and were removed from this study. Genes with significant Spearman correlations (P<0.05) were displayed as heatmap using matrix2png (gene-wise Z-normalized).
Cell lines.
Cell lines were purchased directly from ATCC and used as indicated. Engineered cell lines previously reported were STR profiled for authenticity. All cell lines were tested for bacterial and mycoplasma contamination before use. Deidentified patient sample cells were obtained from the UCCC GI tissue bank, which are maintained, cataloged and annotated by the tissue bank.
CHD1L overexpression and shRNA knockdown.
Full length CHD1L was synthesized in a pGEX-6P-1 plasmid (GenScript, Piscataway, NJ). The CHD1L sequence flanked by EcoRI and NotI was digested out and ligated to a lentiviral backbone to create pCDH1-CMV-CHD1L-EF1-puro plasmid for overexpression of CHD1L in human CRC cells. Mission shRNA (scrambled) and TRCN0000013469 and TRCN0000013470 (sh69 and sh70) specific for CHD1L were purchased from Sigma-Aldrich (St. Louis, MO). Virus was produced in HEK293T cells using TransIT®−293 reagent (Mirus, Madison, WI), and plasmids pHRdelta8.9 and pVSV-G. CRC cells were transduced with overexpression or shRNA knockdown virus and selected with 2 μg/ml puromycin for 7 days.
Western Blots.
CRC cell lines and homogenized tumor tissue samples from mice were resuspended in RIPA lysis buffer (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2 EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3 VO4, 0.1 mM PMSF). Protein concentration was determined using the Pierce BCA protein assay kit (ThermoFisher, Waltham, MA). Forty micrograms of sample were run on 10% Bis-Tris gels. Following electrophoresis, the proteins were transferred to a nitrocellulose membrane. The membranes were blocked at room temperature with 5% non-fat milk in TBS/tween 20 (TBST) for 1 hour at room temperature. Membranes were washed three times with TBST. Blots were incubated with the appropriate primary antibody in 5% nonfat milk in TBST overnight at 4 °C. Membranes were washed three times with TBST and then incubated with appropriate secondary antibody for one hour. Membranes were washed again with TBST three times. Blots were exposed using SuperSignal West Pico PLUS Chemiluminescent Substrate (ThermoFisher, Waltham, MA) and imaged using a Bio-Rad ChemiDoc imaging system (Hercules, CA).
TOPflash TCF-transcriptional reporter assay.
TOPflash (Millipore, Billerica, MA) was used to evaluate TCF transcriptional activity in CRC cells. A total of 20,000 cells per well were plated into 96-well white plates and transfected with TransLT1 reagent (Mirus, Madison, WI). Cells were incubated with transfection mix for 24 h. Next, cells were washed with phosphate-buffered solution (PBS) and a 1:1 ratio of PBS: One-Glo luciferase reagent Promega (Madison, WI) was added and the luminescence was detected within 10 min. A duplicate experiment was conducted to measure cell viability using CellTiter-Glo (Promega, Madison, WI), which was used to normalize TOPflash luminescence to obtain the fold change in TCF activity. Experiments were replicated 2x (n = 3 for each experiment). A duplicate plate was generated to measure the total protein content with BCA assay at the end-point. One-Glo signal (TCF activity) was normalized to the protein amount first before the data of CHD1L knockdown or overexpression groups were normalized to the scr or EV controls, respectively.
Co-ImmunoPrecipitation (Co-IP).
Nuclear cell lysates were generated from untreated SW620 cells. For the input control, 100 μL of 1 mg/mL nuclear extract was saved and used as the input. IP was conducted with Dynabeads protein-A IP Kit (ThermoScientific, Waltham, MA). Briefly, 300 μg of lysate incubated with 2 μg of the anti-TCF4 and anti-CHD1L IP antibody, anti-rabbit IgG and anti-mouse IgG were used as nonspecific binding controls and were rotated at 4 °C for 2 h. After preincubation, 50 μL of beads were transferred to the preincubated antibody/lysate mixture followed by overnight incubation at 4 °C. The flow through was collected and the beads were washed 3x with PBST. Proteins were eluted with 20 μL of 50 mM glycine (pH = 2.8) at 70 °C for 10 min.
Chromatin Immunoprecipitation (ChIP).
Using detailed methods previously described(23), cells were cross-linked with 1.42% formaldehyde for 15 min and quenching with 125 mM glycine for 5 min. Cells were lysed with Szak’s RIPA buffer and sonicated. The IP steps were conducted at 4 °C as follows: 50 μL of protein A/G agarose beads were prewashed with cold Szak’s RIPA buffer and incubated with 1 mg of lysate for 2 h. 0.3 mg/mL of salmon sperm DNA was added and incubated for 2 h. 100 μL of the lysate was set aside as the input control. 2 μg of anti-CHD1L was added to the remainder and incubated overnight. Beads were washed and the supernatant was aspirated to 100 μL followed by the addition of 200 μL of 1.5x-Talianidis elution buffer. To elute immunocomplexes and reverse crosslink, 12 μL of 5M NaCl was added and the mixture was incubated at 65 °C for 16 h. The supernatant was mixed with 20 μg of proteinase K and incubated for 30 min at 37 °C. DNA was extracted with phenol/chloroform and precipitated with ethanol. The IP product was amplified with PowerUp SYBR Green Master Mix (Applied Biosystems, Austin, TX) using known published primers.23
Clonogenic Assay.
Colony formation was assessed after CHD1L knockdown in SW620 cells or overexpression in DLD1 cells as previously described using the well-established crystal violet or the area methods.(23,24) Cells were plated at 1,000 cells/well in six-well plates and medium was changed 2x per week over a 10-day time course. To assess CHD1L inhibitors for their ability to suppress CSC stemness, HCT116 or CHD1L overexpressing DLD1 cell lines were pre-treated in monolayer cultures for 24 h with vehicle control (0.5% DMSO) or CHD1L inhibitors at the concentrations indicated. Pretreated viable cells were plated at 1,000 cells/well in 6-well plates or 200 cells/well in a 24-well plates. For the crystal violet method(23), colonies were analyzed as previously reported. For the area method(24), colonies were analyzed using the Incucyte S3 2018A software with the following parameters modified from default: (1) for HCT116 cells segmentation adjustment = 0.6; Min area (μm2) = 3x104; Max area (μm2) = 1.6x106; Max eccentricity = 0.9; (2) for DLD1CHD1L OE cells segmentation adjustment = 1; Min area (μm2) = 1x104; Max area was not constrained; Max eccentricity = 0.95. Experiments were replicated 2x (n = 2 for each experiment).
Tumor organoid Culture.
As previously described(23,24), cell lines were cultured as tumor organoids using phenol red free RPMI-1640 containing 5% FBS and by seeding 5,000 cells/well into un-coated 96-well U-bottom Ultra Low Attachment Microplates (PerkinElmer, Hopkinton, MA) followed by centrifugation for 15 min at 1,000 rpm to promote cells aggregation. A final concentration of 2% Matrigel was added and tumor organoids were allowed to self-assemble over 72 h under incubation (5% CO2, 37 °C, humidity) before treatment, and maintained under standard cell culture conditions during treatment time courses.
VimPro-GFP and EcadPro-RFP reporter 3D high-content imaging assays.
Stable VimPro-GFP or EcadPro-RFP SW620 reporter cells were generated using pCDH-VimPro-GFP-EF1-puro virus or pCDH-EcadPro-mCherry-EF1-puro virus as previously reported.(23,24) The stable fluorescently labeled reporter cells were used to generate tumor organoids as described herein. Tumor organoids were treated with CHD1L inhibitors at 10 μM for an additional 72 h. Following treatment, tumor organoids were stained with 16 μM of Hoechst 33342 for 1 h (nuclei stain). Images were taken with a 5x air objective. Z-stacks were set at 26.5 μm apart for a total of 15 optical slices. Imaging and high-content analysis were performed using an Opera Phenix and Harmony software (PerkinElmer, Hopkinton, MA). Nuclei were identified within each layer and cells were found with either GFP or mCherry channel. The fluorescence intensities of each channel were calculated and thresholds were set based on the background intensities. Percentages of GFP or mCherry RFP positive cells were calculated and normalized to the DMSO treated group.
Tumor organoid cytotoxicity.
SW620 tumor organoids were cultured as described herein. CellTox™ Green cytotoxicity assay solution was prepared per manufacturer’s protocol (Promega, Madison, WI). Briefly, tumor organoids were treated for 72 h with CellTox™ Green reagent (0.5X) and various doses of CHD1L inhibitors over a range of 0-to-100 μM. Organoids were imaged using the Opera Phenix with excitation at 488 nm and emission at 500-550 nm. Mean intensity of the whole well was utilized for calculating cytotoxicity with Lysis Buffer (Promega, Madison, WI) as the 100% cytotoxicity control and 0.5% DMSO as the 0% cytotoxicity control. Intensity values were normalized to these controls using GraphPad Prism (San Diego, CA).
Invasion assays.
HCT116 cells were plated at 60,000 cells/well into an ImageLock 96-well plate (Sartorius, France) and allowed to attach overnight. A wound was created in all wells using the WoundMaker then washed 2x with PBS. The plate was brought to 4 °C using a Corning XT Cool Core to avoid polymerization of Matrigel during the preparation of the invasion conditions (Corning, Corning, NY). Wells were coated with 50 μL of 50% Matrigel in RPMI-1640 media. Plates were centrifuged at 150 rpm at 4 °C for 3 min, using a swing bucket rotor to ensure even Matrigel coating with no air bubbles. Afterwards, plates were placed on a CoolSink XT prewarmed inside a cell culture incubator (5% CO2, 37 °C, humidity) for 10 min to evenly polymerize the Matrigel, followed by the addition of CHD1L inhibitors dissolved in 50 μL of RPMI-1640 media containing 5% FBS. Finally, the plate was placed in an Incucyte S3 live cell imager (Sartorius, France) for 48 h. The wound was imaged every h using the phase contrast channel and 10x objective in wide mode.
Cloning and purification of recombinant CHD1L.
Cat-CHD1L (residues 16-61) and fl-CHD1L (residues 16-879) constructs were a generous gift from Helena Berglund at the Karolinska Institute, Department of Medical Biochemistry and Biophysics. Proteins were expressed in Rosetta 2 (DE3) pLysS cells (Promega, Madison, WI) in Terrific Broth (ThermoFischer, Waltham, MA). Cultures were induced with 0.2 mM IPTG at OD600 = 2.0 at 18 °C for 16 h. Cells were harvested and resuspended in buffer-A, containing 20 mM HEPES, pH 7.5, 500 mM NaCl, 50 mM KCl, 20 mM imidazole, 10 mM MgCl2, 1 mM TCEP, 10% glycerol and 500 μM PMSF. Cells were lysed by sonication and cellular debris was removed by centrifugation. The supernatant was loaded onto a NiNTA resin column (Qiagen, Hilden, Germany). Protein bound to the column was washed with 1x with buffer-A, 1x with buffer-A containing 10 mM ATP, and an additional time with buffer-A. Proteins were eluted using buffer-B (buffer-A with 500 mM imidazole) with a gradient from 20 to 500 mM imidazole. Following affinity purification, Cat-CHD1L was dialyzed overnight into 50 mM Tris, pH 7.5, 200 mM NaCl, and 1 mM DTT. Similarly, fl-CHD1L was dialyzed overnight into 20 mM MES, pH 6.0, 300 mM NaCl, 10% glycerol, and 1 mM DTT. Protein was then purified by ion-exchange chromatography, cat-CHD1L was bound to a Q-sepharose column (GE Healthcare, Chicago, IL) and fl-CHD1L was bound to a S-sepharose column (GE Healthcare, Chicago, IL), proteins were eluted using a NaCl gradient of 0.2 – 1M for cat-CHD1L and 0.3 −1M fl-CHD1L. Pure fractions were pooled, concentrated, and further purified by size-exclusion chromatography using an Superdex 200 column (GE Healthcare, Chicago, IL) in 20 mM HEPES, pH 7.5, 100 mM NaCl, 1 mM TCEP, 10% Glycerol. Protein purifications were conducted using an ACTA Start FPLC (GE Healthcare, Chicago, IL)
CHD1L ATPase assay.
All reactions were carried out using low volume non-binding surface 384-well plates (Corning Inc., Corning NY). 100 nM cat-CHD1L or fl-CHD1L and 200 nM c-Myc DNA or mononucelosome (Active Motif, Carlsbad, CA) were added to a buffer containing 50 mM Tris pH 7.5, 50 mM NaCl, 1 mM DTT, 5% glycerol, and the reaction was initiated by the addition of 10 μM ATP (New England Biolabs, Ipswich, MA) to a total volume of 10 μL and incubated at 37 °C for 1 h. ATPase activity was measured by adding 500 nM of Phosphate Sensor (Life Technologies, Carlsbad, CA) measuring excitation 430 and emission 450 immediately on an Envision plate reader (PerkinElmer, Hopkinton, MA). An inorganic phosphate standard curve was used to convert the fluorescence to [Pi], and enzyme kinetics were determined using GraphPad Prism (San Diego, CA).
HTS drug discovery for inhibitors of CHD1L.
Assay composition was the same as described above using cat-CHD1L, except that the reaction mixture volume was modified to accommodate addition of drug or DMSO. Using a Janus liquid handler (PerkinElmer, Hopkinton, MA), compounds dissolved in 100% DMSO were mixed with 50 mM Tris pH 7.5, 50 mM NaCl, 1 mM DTT, 5% glycerol buffer to 200 μM in 10% DMSO. Next, 1 μL of each compound was added to the enzyme mixture to give a final concentration of 20 μM. The negative control used was 1% DMSO and 10 mM EDTA was used as a positive control. Reactions were initiated with the addition of 10 μM ATP and incubated at 37 °C for 1 h. ATPase activity was measured by fluorescence by adding 500 nM Phosphate Sensor. Cat-CHD1L was screened against a 20,000-compound diversity set from Life Chemicals (Woodbridge, CT) and a Kinase inhibitor library from Selleck Chemicals (Houston, TX). Both libraries were prescreened before purchase to remove Pan-assay interference compounds (PAINS).
Patient derived tumor organoid (PDTO) culture and viability assay.
CRC patient tumor tissues were obtained from the UCCC GI tissue bank and expanded following established protocols.(29) Briefly, cells were seeded at 5,000 cells per well in 96-well plates and cultured by established methods(30) allowing PDTO formation over 72 h. PDTOs were treated with DMSO (0.5%) or compound 6 with various concentrations for an additional 72 h to obtain a dose response. PDTO cell viability was measured using CellTiter Blue (Promega, Madison, WI). 80 μL of media was aspirated from wells and 80 μL of CellTiter Blue was added and incubated for 4 h and cell viability was measured by fluorescence intensity using excitation 560 excitation and 590 emission.
Evaluation of apoptosis.
SW620 cells were plated at 30,000 cells/well in 96-well plates. Cells were treated with DMSO (negative control), SN-38 (apoptosis positive control), and compound 6 at concentrations indicated for 12 h. Cells were then rinsed 2x with cold PBS, 1x with cold Annexin-V staining buffer (10 mM HEPES, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2,), and then incubated with Annexin-V FITC at 1:100 for 30 min in the dark. Cells were then rinsed 2x with Annexin-V staining buffer and FITC intensity was measured using an Envision plate reader (PerkinElmer, Hopkinton, MA).
Evaluation of DNA damage by γ-H2AX.
DLD1CHD1L-OE cells were seeded into a 96-well PerkinElmer Cell Carrier plate and allowed to adhere overnight. Cells were then treated with the appropriate compound at 10 μM (0.5% DMSO) or with CHD1L inhibitor in combination with SN-38 (1 μM), oxaliplatin (10 μM), and etoposide (10 μM). Cells were treated for 6 h. Media was aspirated and cells were washed with cold PBS. Cells were then fixed with 3% paraformaldehyde for 15 min at room temperature, fixed cells were washed with PBS three times. Cells were blocked for 1 hour at room temperature in 5% BSA, 0.3% Triton X-100 in PBS. Cells were then immunostained with phospho-(S139)-γ-H2AX rabbit mAb using a 1:800 dilution in 1% BSA, 0.3% Triton X-100 in PBS at 4 C overnight. Primary antibody was aspirated and cells were washed with PBS. Cells were incubated for 2 h at room temperature with goat anti-rabbit Alexa Fluor Plus™ 647 fluorescent secondary antibody at a concentration of 5 μg/mL in 1% BSA, 0.3% Triton X-100 in PBS. Cells were then washed with PBS, Hoechst 33342 stain was diluted to a concentration of 1:1000 in PBS, and added to cells for 15 min at room temperature. Cells were then imaged using a 20X water objective on the PerkinElmer Phenix HCS imaging system. Synergy was defined determined using the coefficient of drug interaction (CDI) equation. CDI = (A+B)/(AB), synergy was determined with a CDI < 0.8, additivity was 0.8-1.2, and antagonism was defined by a CDI > 1.2.
Aqueous solubility and CLogP.
Using a recently reported detailed method(24), aqueous solubility was tested for lead compound 6. The PBS UV absorption spectra were compared to a fully saturated solution in 1-propanol and the solubility of compound 6 in 10% DMSO in PBS (pH7.4) was determined using linear regression analysis. The solubility in PBS was conducted in duplicate experiments. The consensus LogP (CLogP) values were obtained using the SwissADME web tools.49
Microsome stability studies.
The microsomal stability of compound 6 was determined using female CD-1 mouse microsomes (M1500) purchased from Sekisui XenoTech (Kansas City, KS), following our recently reported method.(24) Samples were centrifuged at 20,000g for 10 min and the supernatant was transferred to an autosampler vial for LCMS analysis. The following mass transition (m/z, amu) was monitored for compound 6 (molecular weight = 393.5).
In vivo pharmacology.
All animal studies were conducted in accordance with the animal protocol procedures approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Colorado Anschutz Medical Campus (Aurora, CO) and Colorado State University (Fort Collins, CO).
Pharmacokinetics.
Nine-week old female CD-1 mice, purchased from Charles River (Wilmington, MA), were used for PK studies using methods we recently reported.(24) Briefly, the PK studies were designed to cover a range of 0.25-to-24 h with 3 mice/time point for a total of 21 mice/compound 6. Each mouse was dosed with a single i.p. injection of compound 6 at 50 mg/kg prepared in 100% DMSO. Whole blood was harvested at specific time points and the separated plasma frozen at −80 °C for storage or used for LC-MS/MS analysis.
Pharmacodynamics and liver toxicity:
Two million VimPro-GFP SW620 cells suspended in 100 μL of a 1:1 mixture of Matrigel and RPMI 1640 were injected into the flanks of 9-week old female athymic nude mice (Nude-Foxn1nu (069)) (Envigo, Huntingdon, Cambridgeshire, UK). Growth was monitored with caliper measurements 3x per week. At four weeks, mice were randomized into 2 groups and treated with 50 mg/kg of compound 6 in 200 μL of vehicle (10% DMSO, 40% PEG 400, 50% PBS pH=7.4) or with vehicle control. Treatments were administered i.p. QD over five days. Mice were sacrificed 2 h after the final dose on day five of the treatment. Tumors and livers were collected for analysis of compound 6 accumulation measured by LCMS, Western blot analysis measuring effects on EMT and apoptosis, and liver toxicity.
Statistical Analysis.
Data were subjected to unpaired two-tailed Student’s t-test with Welch’s correction statistical analysis or as otherwise stated using Prism (GraphPad, La Jolla, CA). All experiments were replicated 3x (n=3) or as described in the methods.
Results
Clinicopathological characterization of CHD1L in patients with CRC.
CHD1L expression is correlated with poor prognosis in several cancers but limited information about the pathology of CHD1L in CRC is known. As a result, we sought to expand the pathogenic characterization and mechanisms of pathology for CHD1L in CRC patients. The clinicopathological characteristics of 585 patients with CRC were analyzed from the Cartes d’Identite des Tumeurs (CIT) program with respect to CHD1L expression (GEO: GSE39582) and are summarized (Supplementary Table 1).(27)
Follow up information was available for all patients in the CIT cohort over a period of 15 years. For the entire patient cohort, high CHD1L expression is associated with lower OS (P = 0.0167) and median survival (MS) of 8.8 years for high CHD1L patients. Median survival was not reached in the low CHD1L cohort as 72% (115/159) of patients were censored and 26% (42/159) were deceased (Figure 1A). We evaluated patient data using the TNM staging system. As Stage I and IV patients have a high likely hood of survival or death, respectively, we evaluated survival of Stage II and III CRC patients. High CHD1L expression was associated with a lower OS (P = 0.0191) and MS of 11 years for Stage II and III CRC, again median survival was not reached in the low CHD1L cohort (Figure 1B). Survival was also analyzed with respect to CHD1L expression for each stage of CRC. Stage II patients showed a significant difference in survival (P = 00319) with a M.S. of 11 years, no significant difference was observed for Stage I, III or IV patients (Supplementary Figure 1). Welch’s one-way ANOVA analysis of CHD1L expression indicates a significant increase in CHD1L levels from cancer stages I through IV (Figure 1C). We evaluated patients with Stage I and II versus patients with Stage III and IV CRC and observed a significant increase in CHD1L expression in the Stage III and IV versus early stage cohorts (P = 0.0051) (Supplemental Figure 2A). Analysis of CHD1L expression with respect to lymph node metastasis suggests that CHD1L is overexpressed in patients with increased regional lymph node metastasis (N1 P = 0.0128, N2 P = 0.05 compared to N0) (Figure 1D). Although the trend of CHD1L expression was the same for the N3 cohort, no significance was determined due to the limited number of patient samples available. We found no significant difference in CHD1L expression with respect to tumor size, metastasis, or location (Supplemental Figure 2B-D).
Figure 1: Clinicopathological characteristics associated with CHD1L expression.

(A) OS of CHD1L expression in CRC patients, P = 0.0167. (B) OS of CHD1L expression in Stage II/III patient cohort, P = 0.0191. (C) Differential expression of CHD1L in CRC by stage, P = 0.028. (D) Differential expression of CHD1L among N staging in CRC patients, P = 0.013 (N1), 0.05 (N2) relative to N0. (E) Differential expression of CHD1L among CRC molecular subtypes, P < 0.001. (F) Expression of CHD1L between CRC patients with normal and deficient DNA mismatch repair, P < 0.001.
Evaluation of CHD1L in CRC molecular subtypes.
The association of CHD1L expression with six molecular subtypes of CRC(27): C1 (immune system down, n = 116), C2 (deficient mismatch repair, n = 104), C3 (KRAS mutant, n = 75), C4 (CSC, n = 59), C5 (activated WNT pathway, n = 152), and C6 (chromosomal instability normal, n = 60) was investigated (Figure 1E). There is a significant difference of CHD1L expression among the six molecular subtypes (P < 0.001). CHD1L expression was high in C5, C4, and C3, and low in C2 and C6. The C2 subtype is associated with a decrease in the WNT signaling pathway and deficient for mismatch repair. The C4 and C6 subtypes are associated with poorer relapse free survival compared to other subtypes. The C4 subtype is associated with increased CSC stemness and the C5 subtype is associated with activated WNT signaling and deregulated EMT pathways. The lower CHD1L expression in the C2 (deficient mismatch repair) subtype is consistent with its known function in DNA damage response.(31) Additionally, CHD1L expression was lower in patients with deficient mismatch repair than patients without (P < 0.001) (Figure 1F). CHD1L expression was also higher in patients with KRAS mutations (P = 0.049). The expression of CHD1L in the C3, C4, and C5 molecular subtypes prompted us to further investigate the function of CHD1L expression in EMT, CSC stemness, and the WNT/TCF pathway.
CHD1L expression correlates with Wnt/TCF associated genes.
Utilizing a smaller cohort of CRC patients (n = 26) from the UCCC GI tumor tissue bank, a similar trend was observed as with the larger CIT cohort where CHD1L expression significantly correlated with late stage and metastatic CRC compared to early stage and primary CRC (Supplementary Figure 3). When analyzing CHD1L expression with genes involved in the KEGG WNT pathway, using Spearman correlation we observed a significant positive correlation with 65 of 125 genes. Among these were well-established genes involved in TCF-transcription such as topoisomerase IIα (TOP2A) (r = 0.65, P = 0.004)(23,24), and TCF4 (r = 0.61, P = 0.0012) (Supplementary Figure 4). We also observed a significant positive correlation between known CSC markers CD44 (r = 0.43, P = 0.038), LGR5 (r = 0.55, P = 0.0075) and CHD1L. When comparing the CIT cohort to the UCCC cohort a significant correlation was observed for TOP2A (r = 0.1275, P = 0.0020) and TCF4 (r = 0.1050, P = 0.011). Consistent with this result, we have shown that TOP2A is a required component of the TCF-complex, promoting EMT in CRC.(23,24) Hence, CHD1L may be involved in TCF-transcription and EMT in CRC patients.
CHD1L mediates TCF-transcription in CRC.
Based on the correlation of CHD1L with TCF-complex members, we hypothesized that CHD1L has a mechanistic role in TCF-transcription. To test this, we utilized SW620 and DLD1 cell lines, which have high and low endogenous CHD1L expression, respectively (Supplementary Figure 5). We used shRNA to knockdown CHD1L in SW620 cells (SW620CHD1L-KD) and overexpressed CHD1L in DLD1 cells (DLD1CHD1L-OE). Using the TOPflash luciferase reporter (32) transfected into SW620CHD1L-KD or DLD1CHD1L-OE we determined that overexpression of CHD1L produced a significant increase in TCF-transcription (P < 0.0001) (Figure 2A). Conversely, SW620CHD1L-KD cells displayed a significant decrease in TCF-transcription (P = 0.0006). These results establish CHD1L as a potential factor directly involved in TCF-transcription.
Figure 2: CHD1L mediates TCF-transcription in CRC.
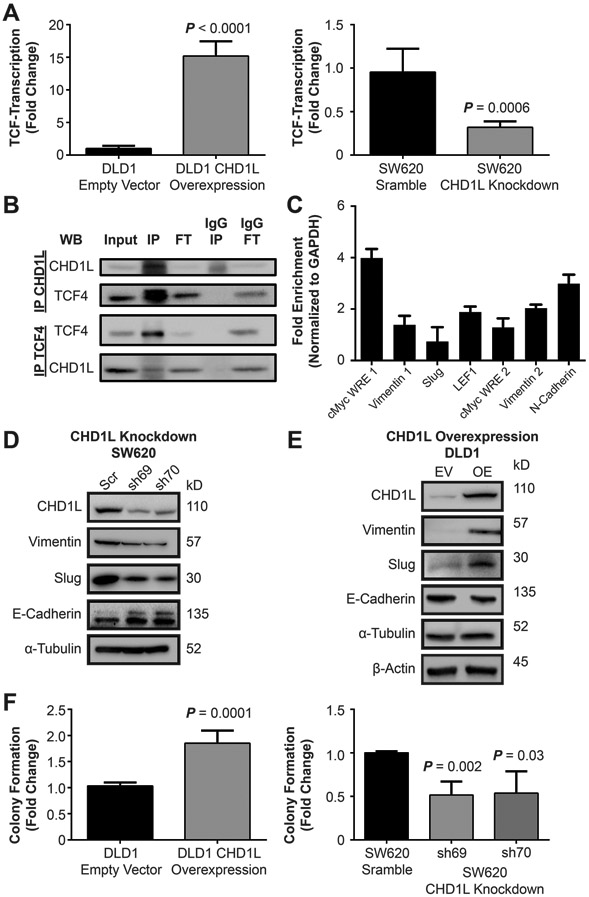
(A) Overexpression of CHD1L in DLD1 cells shows an increase in TCF-transcription using the TOPflash TCF-transcription reporter assay (P < 0.0001), knockdown of CHD1L in SW620 cells using shRNA decreases TCF-transcription (P = 0.0006). Mean value of fold change from three independent experiments ± s.d. are shown. (B) Co-immunoprecipitation of CHD1L with TCF4 from SW620 cells, IP (immunoprecipitation), FT (flow-through). (C) Chromatin immunoprecipitation of CHD1L with WNT response element DNA promoter sites in SW620 cells. (D) Evaluation attenuated gene expression of EMT associated genes with SW620CHD1L-KD cells by Western blot. (E) Evaluation of induction of EMT gene expression in DLD1CHD1L-OE cells by Western blot. (F) Evaluation of CSC colony formation in DLD1CHD1L-OE, (P = 0.0001) and SW620CHD1L-KD (P = 0.002, 0.03). Mean values of fold change are from three independent experiments ± s.d. Representative colony images are shown in the supplemental information.
CHD1L directly interacts with the TCF-transcription complex.
Activation of TCF-transcription is a dynamic process that involves the shedding of co-repressor proteins, binding of co-activator proteins, and remodeling of the chromatin landscape.(1,21) We performed co-immunoprecipitation (Co-IP) studies with TCF4, demonstrating that CHD1L directly binds to the TCF-complex (Figure 2B). CHD1L has been well characterized as a binding partner with PARP1 in DNA damage response.(11,31) PARP1 is also a component of the TCF-complex binding to TCF4 and β-catenin.(33) Our results demonstrate that CHD1L binds to the TCF-complex, which is likely through interactions between TCF4 and PARP1.
To further characterize CHD1L as a component of the TCF-complex, we performed chromatin immunoprecipitation (ChIP) of CHD1L to TCF-complex WNT response elements (WREs) in SW620 cells (Figure 2C). CHD1L was enriched at c-Myc, vimentin, slug, LEF1, and N-cadherin WREs, further supporting that CHD1L is functioning directly with the TCF-complex. Taken together, the data implicate CHD1L as a critical component of the TCF-transcription.
CHD1L mediated TCF-transcription promotes EMT and CSC stemness in CRC.
Previously, we have characterized TCF-transcription as a master regulator of EMT in CRC.(23) In addition, CHD1L localizes at WREs of EMT effector genes (Figure 2C). Therefore, we evaluated whether knockdown or overexpression of CHD1L modulates EMT by measuring biomarker expression in SW620CHD1L-KD and DLD1CHD1L-OE cells. Knockdown of CHD1L induced reversion of EMT, decreasing vimentin and slug while increasing E-cadherin expression (Figure 2D). Conversely, EMT was induced in DLD1CHD1L-OE cells, evidenced by a decrease in E-cadherin and an increase in vimentin and slug expression (Figure 2E). These results indicate that CHD1L is an EMT effector gene involved in promoting the mesenchymal phenotype in CRC. A hallmark of EMT is an increase in CSC stemness. To characterize the impact of CHD1L expression on stemness, we performed clonogenic colony formation assays (Figure 2F).(34) CSC stemness increased with DLD1CHD1L-OE (P = 0.0001) and decreased in SW620CHD1L-KD (P = 0.002) cells measured by colony formation.
HTS Identifies the first small molecule inhibitors of CHD1L.
After establishing CHD1L as a driver of TCF-mediated EMT, we sought to identify small molecule inhibitors of CHD1L and assess their therapeutic potential. Our drug discovery goal was to target CHD1L DNA translocation or interactions with DNA, which are dependent on its catalytic domain ATPase activity ((Supplemental Figure 6 and 7).(35,36) CHD1L belongs to the SNF2 (sucrose non-fermenter 2) ATPase superfamily of chromatin remodelers that contains a two-lobe ATPase domain. CHD1L also has a macro domain that is unique relative to other chromatin remodelers, which promotes an autoinhibited state through interactions between the macro and the ATPase domains (Supplemental Figure 7A) .(37,38) However, the macro domain binds to PARP1, the major activator of CHD1L, alleviating auto-inhibition.(37,38) Using the methodology by Lehmann et. al.(37) we purified full-length CHD1L (fl-CHD1L) and the catalytic ATPase domain (cat-CHD1L) (Supplemental Figure 7B). The cat-CHD1L provides for a more robust ATPase assay compared to fl-CHD1L, which is consistent with the report from Lehman et al.(37) Therefore, to discover direct inhibitors of CHD1L ATPase we developed a HTS assay in the context of TCF-transcription, including: cat-CHD1L, c-Myc WRE DNA, ATP, and phosphate-binding protein that fluoresces upon binding inorganic phosphate (Pi). We validated this assay and performed pilot screening against clinically relevant kinase inhibitors (Supplementary Figure 6). Moreover, the pilot screen found no hits, demonstrating that CHD1L is not a likely target for kinase inhibitors. Thus, we expect hits identified will have limited off target binding of kinases. Once validated, we conducted primary HTS using 20,000 compounds from the Life Chemicals Diversity Set, which were screened at 20 μM in 1% DMSO with 10 mM EDTA as a positive control (Supplemental Figure 7B). The screen provided robust statistics with an average Z’-factor value of 0.57 ± 0.06 over 64 plates (Supplemental Figure 7C). The average compound activity was 92.3% ± 17.8. As a result, we set the hit limit to be 3 standard deviations from the mean at 39% ATPase activity. This stringent hit limit identified 64 hits, of which 53 hits were confirmed against recombinant CHD1L ATPase activity (Supplemental Figure 7D).
A subset of seven confirmed hits (compounds 1-7) represent a range of pharmacophores with greater than 50% inhibition against cat-CHD1L ATPase. After hit confirmation, compounds were purchased as new batches in solid form from Life Chemicals for lead validation studies (Supplemental Table 2). Compounds 1-7 were subjected to dose response studies against cat-CHD1L ATPase, which validated these hits as potent CHD1L inhibitors with activity between 900 nM to 5 μM (Figure 3A). There are striking similarities in structure between the hits, with hit 1-3 and 6-7 being close derivatives, further validating the HTS results. Next, we tested compound 1-7 in HCT116, SW620, and DLD1CHD1L-OE cells for their ability to inhibit TCF-transcription using the TOPflash reporter system (Figure 3B). Compounds 1-3 were eliminated displaying no significant activity in cells. Compound 4 was also deprioritized due to modest activity with no dose dependent inhibition of TCF-activity. However, compounds 5-7 demonstrate superior dose dependent activity against TCF-transcription in all three CRC cell lines, advancing as lead CHD1L inhibitors. Notably, we observed decreased inhibition of TCF-transcription for 5-7 at the low 2 μM dose in DLD1CHD1L-OE cells, which is evidence of cellular CHD1L target engagement.
Figure 3: Validation of CHD1L inhibitors identified from HTS.
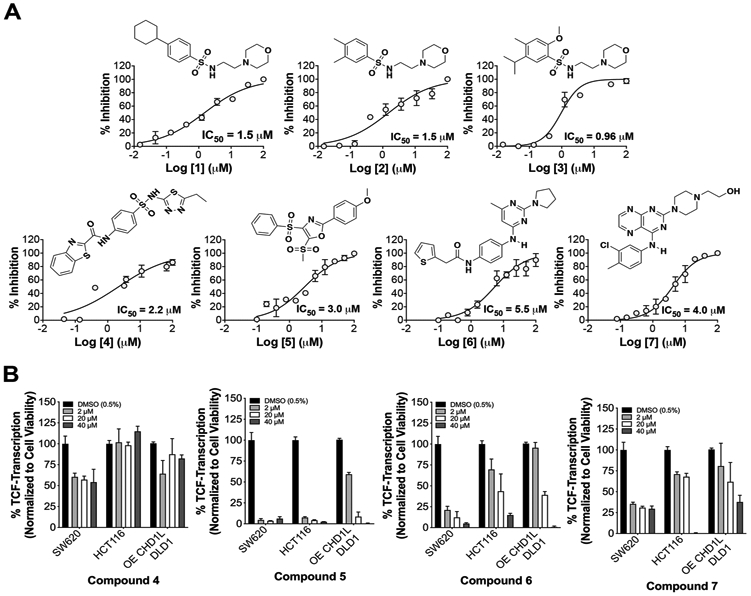
(A) cat-CHD1L ATPase IC50 dose responses with hits 1-7. Mean IC50 values are calculated from three independent experiments and representative graphs are shown. (B) SW620, HCT116, and DLD1CHD1L-OE cells with TOPflash reporter were used to measure inhibition of TCF-transcription using 3 doses over 24 h.
CHD1L inhibitors reverse EMT and malignant properties in CRC.
After validating hits 5-7 against CHD1L mediated TCF- transcription we next evaluated their ability to reverse EMT and other malignant properties in CRC. E-cadherin and vimentin are putative biomarkers for the epithelial and mesenchymal phenotypes, respectively.(39) Loss of E-cadherin and gain of vimentin are also clinical biomarkers of poor prognosis.(40-44) Accordingly, we developed and validated lentiviral promoter driven reporters for E-cadherin (pCDH1-EcadPro-RFP) and vimentin (pCDH1-VimPro-GFP), which faithfully reports E-cadherin and vimentin protein expression.(23,24) SW620 cells transduced with either EcadPro-RFP or VimPro-GFP were cultured as tumor organoids for 72 h, reaching a diameter of 600 μm. Tumor organoids were treated with compounds 5-7 for an additional 72 h to determine the effective concentration 50 percent (EC50) for modulating promoter activity. Changes in promoter expression was quantified using a 3D confocal image based high-content analysis algorithm (Figure 4A-C).(23,24) We observed that 5-7 effectively downregulated vimentin promoter activity with EC50 values of 15.6 ± 1.7 μM (5), 4.7 ± 1.5 μM (6), and 12.8 ± 1.3 μM (7). Conversely, E-cadherin promoter activity was upregulated with EC50 values of 11.9 ± 0.3 μM (5), 11.4 ± 0.3 μM (6), and 28 ± 0.003 μM (7). These results are consistent with our hypothesis that small molecule inhibitors of CHD1L reverse TCF-driven EMT in CRC. To confirm that CHD1L inhibitors reverse EMT we looked at the protein expression of two additional putative biomarkers of EMT, slug (mesenchymal) and zona occludens-1 (ZO-1, epithelial); changes in slug and ZO-1 are considered major criteria for EMT.(45) SW620 tumor organoids treated with CHD1L inhibitors downregulate slug and upregulate ZO-1, further indicating a reversion of EMT (Figure 4D).
Figure 4: CHD1L inhibitors reverse EMT and the malignant phenotype in CRC.

Dose responses for CHD1L inhibitors that modulate EMT measured by high-content imaging of (A) downregulation of VimPro-GFP reporter and (B) Upregulation of EcadPro-RFP reporter. Data curves were fit using Log [CHD1L inhibitor] vs. variable slope (4 parameters) in GraphPad Prism. Mean EC50 values ± SEM are calculated from three independent experiments (C) Representative images exhibiting reversion of EMT by compound 6 in SW620 tumor organoids measured by EMT reporter assays. (D) Western blot analysis showing protein expression changes of additional EMT biomarkers slug and ZO1. (E) CSC stemness measured by clonogenic colony formation after pretreatment with CHD1L inhibitors in DLD1CHD1L-OE and HCT116 cells. (F) Inhibition of invasive potential of HCT116 cells after treatment of CHD1L inhibitors. Welch’s t-test statistical analysis was used to determine significance, where * = P ≤ 0.05, ** = P ≤ 0.01, *** = P ≤ 0.001, **** = P ≤ 0.0001.
A hallmark of EMT is an increase in CSC stemness and cell invasion. Therefore, we tested the ability of compounds 5-7 to inhibit migration and invasion in HCT116 and DLD1CHD1L-OE cells. All three compounds demonstrate a significant inhibition of CSC stemness (Figure 4E). However, 5 and 6 display more potent dose dependent inhibition. Interestingly, DLD1CHD1L-OE cells form two times more colonies than HCT116 cells, which have moderate CHD1L expression. This observation is consistent with CHD1L’s oncogenic and tumorigenic properties. Next, using HCT116 cells with uniform scratch wounds imbedded in 50% Matrigel, we treated cells with CHD1L inhibitors and monitored invasion over 72 h. Compounds 5-7 exhibited a dose dependent inhibition of invasion (Figure 4F, Supplementary Figure 8), with compound 6 displaying the most potent activity.
Inhibition of CHD1L enhances efficacy of DNA damaging drugs
CHD1L is known to function in PARP1 mediated DNA damage repair, which is a mechanism of drug resistance to DNA damaging chemotherapy.(10,31,38) For example, drug resistance to cisplatin in lung cancer was observed in cells overexpressing CHD1L, and the efficacy of cisplatin was restored after CHD1L knockdown.(10) In addition, knockdown of CHD1L alone does not increase in DNA damage.(31) In order to determine if CHD1L inhibitors could increase the efficacy of DNA damaging drugs against low CHD1L expressing DLD1 cells transduced with empty vector (DLD1CHD1L-EV ) and overexpressing DLD1CHD1L-OE cells, we evaluated compound 6 alone and in combination with SN-38 (the active pharmacophore of prodrug irinotecan), oxaliplatin, and etoposide. To assess DNA damage we measured the phosphorylation of H2AX (γ-H2AX) by immunofluorescence (Supplemental Figure 9), which is used as a biomarker for DNA damaging chemotherapy.(31) Compound 6 alone showed no significant DNA damage when treating cells at 10 μM and measuring γ-H2AX activity, which is consistent with previously reported CHD1L knockdown studies.(31) However, combination treatments in DLD1CHD1L-OE cells with 6 synergized with etoposide (10 μM) and SN-38 (1 μM), significantly increasing DNA damage compared to etoposide and SN-38 alone. In DLD1CHD1L-EV cells only the combination of etoposide and 6 displayed significant synergy. Under the experimental conditions used we observed no synergy with oxaliplatin. Nevertheless, SN-38 (i.e. irinotecan) combination therapy, known as FOLFIRI, is a standard of care chemotherapy in the treatment of CRC. Therefore, the enhanced DNA damage that occurs with 6 in combination with SN-38 supports our hypothesis that CHD1L inhibitors may increase the efficacy of CRC standard of care DNA damaging chemotherapies.
CHD1L inhibitors reverse EMT prior to the induction of cell death.
CHD1L has been reported to confer anti-apoptotic activity by inhibiting activation of caspase-dependent apoptosis.(13,46) Additionally, reversal or inhibition of EMT is known to restore apoptotic activity of cancer cells.(47) To determine if CHD1L inhibitors reverse EMT prior to induction of cell death, we monitored E-cadherin expression by EcadPro-RFP reporter activity, and cytotoxicity was measured using the CellTox™ Green assay. Cells were treated with CHD1L inhibitors for 72 h and imaged every 2 h. We observed a significant increase in E-cadherin expression prior to induction of cytotoxicity for compound 6 relative to DMSO (Figure 5A).
Figure 5: Lead compound 6 induces apoptosis in CRC cell lines and PDTOs.

(A) Time course evaluation of the induction E-cadherin expression using Ecad-ProRFP reporter assay and cytotoxicity using Cell-Tox Green. (B) Western blot analysis of induction of cleaved E-cadherin after treatment of 5 and 6 for 48 hours in CRC organoids. (C) Western blot analysis of induction of caspase dependent apoptosis cadherin after treatment of 6 for 48 hours in CRC organoids. (D) Annexin V-FITC staining analysis of apoptosis after treatment of SN-38 and 6 for 12 hours (E) Cytotoxicity of 6 in PDTO CRC102 using CellTiter-Blue. Mean EC50 values ± s.d. are calculated from six independent experiments and representative graph is shown with inset of a representative PDTO. Welch’s t-test statistical analysis was used to determine significance, where * = P ≤ 0.05, ** = P ≤ 0.01, *** = P ≤ 0.001, **** = P ≤ 0.0001.
We then evaluated if CHD1L inhibitors are able to induce apoptosis in CRC, we performed western blot from SW620 tumor organoids and observed that E-cadherin becomes cleaved after treatment with 5 and 6 (Figure 5B), which is a marker of apoptosis.(48) Next, using the more potent CHD1L inhibitor 6, we measured increases in cleaved PARP1, cleaved caspase 8, and cleaved caspase 3 relative to DMSO control (Figure 5C). These results suggest compound 6 induces extrinsic apoptosis that is consistent with E-cadherin mediated apoptosis through death receptors.(47) To further characterize the apoptotic activity of CHD1L inhibitors, we performed annexin-V staining in SW620 cells over 12 h. Compound 6 induced significant apoptosis compared to DMSO and had similar activity to the positive control SN-38 (Figure 5D).
CHD1L inhibitors are effective against patient-derived tumor organoids (PDTOs)
The use of PDTOs in preclinical drug development has been established as predictive in vitro cell model for clinical efficacy.(49) After establishing the ability of compound 6 to reverse EMT and induce apoptosis using cell line based models, we evaluated the efficacy of 6 in PDTOs produced from patient sample CRC102 obtained from the University of Colorado Cancer Center (UCCC) gastrointestinal (GI) tissue bank (Figure 5E). Consistent with our results in CRC cell lines, compound 6 showed potent cytotoxicity in PDTOs with an EC50 of 11.6 ± 2 μM. This result further validates 6 as a priority lead CHD1L inhibitor.
In vitro and in vivo PK, PD, and liver toxicity of lead CHD1L inhibitor 6.
To assess the drug-like potential and properties of 6 we conducted in silico, in vitro, and in vivo PK studies assessing CLogP, aqueous solubility, stability in mouse liver microsomes, and PK in CD-1 mice (Figure 6 and Supplemental Figure 10). Compound 6 has an excellent balance of lipophilicity (CLogP = 3.2) and aqueous solubility that is relatively stable to liver metabolizing enzymes, and an excellent PK disposition when administered to CD-1 mice. Compound 6 reaches a high plasma drug concentration CMax (~30,000 ng/mL) and AUC (~80,000 ng/mL/h) with a relatively long half-life (T1/2λ) of 3 h after intraperitoneal (i.p.) administration.
Figure 6: In vivo pharmacology and proposed mechanism of action.

(A) Summary of compound 6 in vivo and in vitro pharmacokinetic parameters. The consensus LogP (CLogP) values were obtained using the SwissADME web tools.(50) Compound 6 was administered by i.p. injection to athymic nude mice QD for 5 days to measure (B) Accumulation in SW620 xenograft tumors and (C) Histopathological assessment of liver toxicity. Representative photomicrograph sections (5x magnification) of liver in both vehicle and compound 6 treated animals. The images demonstrate normal hepatic cord and lobule architecture, with no evidence of hepatocyte degeneration, necrosis, hyperplasia, or parenchymal inflammation. (D) Western blots from xenograft tumors for vehicle and 6 were done to measure PD effects on EMT. (E) Densitometry of blot intensity showing mean value and SEM of four tumor xenograft samples. (F) Proposed mechanism of action of CHD1L mediated TCF-transcription where CHD1L is activated through binding TCF-complex members. (1) Once activated, CHD1L is directed to hindered WREs localized on chromatin. (2) Chromatin remodeling and DNA translocation occurs exposing WRE sites. (3) TCF-complex binds to exposed WREs facilitated by CHD1L, promoting EMT genes and other genes associated with mCRC. CHD1L ATPase inhibitors effectively prevent step 1, leading to the reversion of EMT and other malignant properties of CRC. Welch’s t-test statistical analysis was used to determine significance, where * = P ≤ 0.05
Next, we conducted a second acute in vivo experiment using a maximum tolerated dose of 6 (50 mg/kg) administered to athymic nude mice by i.p. QD over five days. The goals of this experiment were to (1) determine if compound 6 causes acute toxicity to livers, (2) accumulates in VimPro-GFP SW620 xenograft tumors, and (3) to determine PD effects. Compound 6 accumulates in SW620 tumors at a concentration of 10,533 ± 5,579 ng/mL (n=4). As expected, when comparing the ratio of compound 6 accumulation in tissue/plasma, we observed 2.7 times more accumulation in liver compared to tumor (Figure 6B). However, there was no apparent liver toxicity resulting from 6 at the dose and schedule administered (Figure 6C and Supplementary Table 2). Overall, there were no significant histological differences between the livers of vehicle or compound 6 treated mice. The primary histological change observed were minimal fibrosis and inflammation of the hepatic capsule in both vehicle and compound 6 treated animals. This change suggests a very low grade, sub-clinical peritonitis, and is consistent with being secondary to i.p. drug administration. We also did not observe and changes in weight loss to animals treated with compound 6 during the course of study.
In accordance with accumulation of compound 6 in tumors, we measured PD effects on tumor tissue by Western blot analysis, indicating a significant downregulation of mesenchymal markers vimentin, vimentin reporter (VimPro-GFP), and slug (Figure 6D-E). Although not statistically significant, we also observed upregulation of the epithelial marker ZO-1 and induction of cleaved caspase 3, the putative biomarker of apoptosis. Taken together, we observed PD effects by 6 indicating the reversion of EMT and apoptosis in vivo that were consistent with in vitro cell-based antitumor activity of 6. In summary, compound 6 displays good PK drug-like properties and the ability to alter EMT and induce apoptosis in vivo with no observed liver toxicity.
Discussion
CHD1L is amplified (Chr1q21) and overexpressed in many types of cancer.(8,9) CHD1L overexpression has been characterized as a marker for poor prognosis and metastasis in numerous cancers.(8,9,16,17) While the collective literature demonstrating CHD1L as an oncogene and driver of malignant cancer is compelling, we set out to test the rigor of the prior research and our hypothesis that CHD1L is an oncogene with potential as a molecular target in CRC. We conducted in silico analyses of transcriptome data from a large cohort of 585 CRC patients obtained over 15 years.(27) CHD1L expression correlates with poor survival, with low-CHD1L-patients living significantly longer than high-CHD1L-patients (Figure 1). Using the same cohort, Marisa et al. identified six distinct subtypes for improved clinical stratification of CRC and CHD1L is universally expressed in all subtypes, indicating its potential as a therapeutic target for CRC. CHD1L also correlated with tumor node metastasis, with increased expression moving from N0 (no regional spread) to N3 (distant regional spread). We then analyzed transcriptome data from a UCCC patient cohort and found that CHD1L expression significantly correlated with stage IV and mCRC. Literature reports and our own analyses and data demonstrate that CHD1L is an oncogene promoting malignant CRC and its high expression correlates with poor prognosis and survival of CRC patients.
In this study we have determined a new biological function for CHD1L as a DNA binding factor for the TCF-transcription complex required for promoting TCF-driven EMT and other malignant properties (Figure 2). Using HTS drug discovery we have identified the first known inhibitors of CHD1L (Figure 3), and have characterized several lead compounds that display good pharmacological efficacy in cell-based models of CRC, including PDTOs. CHD1L inhibitors effectively prevent CHD1L-mediated TCF-transcription, leading to the reversion of EMT and other malignant properties, including CSC stemness and invasive potential (Figure 4). Notably, CHD1L inhibitor 6 displays the ability to induce cell death that is consistent with the reversion of EMT and induction of cleaved E-cadherin mediated extrinsic apoptosis through death receptors (Figure 5).(47) Furthermore, compound 6 synergizes with SN-38 displaying potent DNA damage induction compared to SN-38 alone, which is consistent with the inhibition of PARP1/CHD1L mediated DNA repair (Supplemental Figure 9).(31,38) Finally, we have characterized compound 6 as a lead CHD1L inhibitor with drug-like physicochemical properties and favorable in vivo PK/PD disposition with no acute liver toxicity (Figure 6). Therefore, the pharmacophore of 6 has excellent potential for drug development.
Based on the data presented herein, we propose a mechanism of action for CHD1L-mediated TCF-driven EMT involved in CRC tumor progression and metastasis (Figure 6F). We hypothesize that the TCF-complex specifically recruits CHD1L to dynamically regulate metastatic gene expression. Central to this hypothesis, we propose that CHD1L binds to nucleosome hindered WREs when directed by the TCF-complex via protein interactions with TCF4. Importantly, PARP1 is characterized as the major cellular activator of CHD1L through macro domain binding that releases auto inhibition.(37,38) Moreover, PARP1 is a required component of the TCF-complex forming interactions with β-catenin and TCF4.(33) Therefore, it is reasonable to postulate that CHD1L is recruited by the TCF-complex and activated by PARP1 and TCF4. Once activated, CHD1L exposes WREs by nucleosome translocation, facilitating TCF-complex binding to WREs and transcription of malignant genes promoting EMT. CHD1L inhibitors have a unique mechanism of action by inhibiting CHD1L ATPase activity, which prevents exposure of WREs to the TCF-complex, inhibiting transcription of TCF-target genes associated with EMT and mCRC.
In conclusion, since its discovery in 2008, numerous literature reports have characterized the biological role and clinical evaluation of oncogenic CHD1L in various solid tumor types. However, no approaches targeting oncogenic CHD1L are in clinical practice. With this report, we demonstrate that CHD1L is an oncogene and druggable target in CRC. We have identified the first inhibitors of CHD1L that have contributed fundamental knowledge of the mechanism of action of CHD1L in CRC and its potential as a therapeutic target. Future work with CHD1L inhibitors will focus on optimization through medicinal chemistry and conducting in vivo antitumor and anti-metastatic efficacy studies. However, the work herein lays a foundation for the development of CHD1L targeted therapeutics as an effective strategy to treat CRC and other CHD1L driven cancers.
Supplementary Material
Acknowledgements
This research was supported in part by the Department of Defense Peer Reviewed Cancer Research Program (W81XWH-18-1-0142) to D.V.L. The Colorado Cancer Translational Research Accelerator (D.V.L PI and D.L.G co-PI). The Skaggs School of Pharmacy and Pharmaceutical Sciences (SSPPS) ADR grant to D.V.L. The Cancer League of Colorado grant (D.V.L PI and W.A.M. co-PI). S.R. was supported by the Cancer Foundation of Luxembourg. We thank the SSPPS HTS drug discovery and chemical biology core facility, the UCCC shared resource laboratories, particularly the Pharmacology core facility for microsomal and PK studies. The UCCC is a NIH NCI designated cancer center supported by grant number P30CA046934. We are grateful to Helena Berglund at the Karolinska Institute for generously providing the pNIC-CH2 vector.
Abbreviations List
- CHD1L
chromodomain helicase/ATPase DNA binding protein 1-like
- HTS
high-throughput screening
- TCF
transcription
- EMT
epithelial-mesenchymal transition
- PDTO
patient derived tumor organoids
- PK/PD
pharmaco-kinetics/dynamics
Footnotes
Disclosure of Conflict of Interest: The authors declare no conflicts of interest.
References
- 1.Lorch Y, Maier-Davis B, Kornberg RD. Mechanism of chromatin remodeling. Proc Natl Acad Sci U S A 2010;107(8):3458–62 doi 10.1073/pnas.1000398107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kumar R, Li DQ, Muller S, Knapp S. Epigenomic regulation of oncogenesis by chromatin remodeling. Oncogene 2016;35(34):4423–36 doi 10.1038/onc.2015.513. [DOI] [PubMed] [Google Scholar]
- 3.Swygert SG, Peterson CL. Chromatin dynamics: interplay between remodeling enzymes and histone modifications. Biochim Biophys Acta 2014;1839(8):728–36 doi 10.1016/j.bbagrm.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Erdel F, Krug J, Langst G, Rippe K. Targeting chromatin remodelers: signals and search mechanisms. Biochim Biophys Acta 2011;1809(9):497–508 doi 10.1016/j.bbagrm.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 5.Brownlee PM, Meisenberg C, Downs JA. The SWI/SNF chromatin remodelling complex: Its role in maintaining genome stability and preventing tumourigenesis. DNA Repair (Amst) 2015;32:127–33 doi 10.1016/j.dnarep.2015.04.023. [DOI] [PubMed] [Google Scholar]
- 6.Zhang C, Lu J, Zhang P. The Roles of Chromatin Remodeling Proteins in Cancer. Curr Protein Pept Sci 2016;17(5):446–54 doi 10.2174/1389203717666160122120713. [DOI] [PubMed] [Google Scholar]
- 7.Valencia AM, Kadoch C. Chromatin regulatory mechanisms and therapeutic opportunities in cancer. Nat Cell Biol 2019;21(2):152–61 doi 10.1038/s41556-018-0258-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma NF, Hu L, Fung JM, Xie D, Zheng BJ, Chen L, et al. Isolation and characterization of a novel oncogene, amplified in liver cancer 1, within a commonly amplified region at 1q21 in hepatocellular carcinoma. Hepatology 2008;47(2):503–10 doi 10.1002/hep.22072. [DOI] [PubMed] [Google Scholar]
- 9.Cheng W, Su Y, Xu F. CHD1L: a novel oncogene. Molecular cancer 2013;12(1):170 doi 10.1186/1476-4598-12-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y, He LR, Gao Y, Zhou NN, Liu Y, Zhou XK, et al. CHD1L contributes to cisplatin resistance by upregulating the ABCB1-NF-kappaB axis in human non-small-cell lung cancer. Cell Death Dis 2019;10(2):99 doi 10.1038/s41419-019-1371-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pines A, Vrouwe MG, Marteijn JA, Typas D, Luijsterburg MS, Cansoy M, et al. PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J Cell Biol 2012;199(2):235–49 doi 10.1083/jcb.201112132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsuda M, Cho K, Ooka M, Shimizu N, Watanabe R, Yasui A, et al. ALC1/CHD1L, a chromatin-remodeling enzyme, is required for efficient base excision repair. PLoS One 2017;12(11):e0188320 doi 10.1371/journal.pone.0188320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Y, Chen L, Chan TH, Liu M, Kong KL, Qiu JL, et al. SPOCK1 is regulated by CHD1L and blocks apoptosis and promotes HCC cell invasiveness and metastasis in mice. Gastroenterology 2013;144(1):179–91 e4 doi 10.1053/j.gastro.2012.09.042. [DOI] [PubMed] [Google Scholar]
- 14.Chen L, Hu L, Chan TH, Tsao GS, Xie D, Huo KK, et al. Chromodomain helicase/adenosine triphosphatase DNA binding protein 1-like (CHD1l) gene suppresses the nucleus-to-mitochondria translocation of nur77 to sustain hepatocellular carcinoma cell survival. Hepatology 2009;50(1):122–9 doi 10.1002/hep.22933. [DOI] [PubMed] [Google Scholar]
- 15.He LR, Ma NF, Chen JW, Li BK, Guan XY, Liu MZ, et al. Overexpression of CHD1L is positively associated with metastasis of lung adenocarcinoma and predicts patients poor survival. Oncotarget 2015;6(31):31181–90 doi 10.18632/oncotarget.5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hyeon J, Ahn S, Park CK. CHD1L Is a Marker for Poor Prognosis of Hepatocellular Carcinoma after Surgical Resection. Korean J Pathol 2013;47(1):9–15 doi 10.4132/KoreanJPathol.2013.47.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Su Z, Zhao J, Xian G, Geng W, Rong Z, Wu Y, et al. CHD1L is a novel independent prognostic factor for gastric cancer. Clinical & translational oncology : official publication of the Federation of Spanish Oncology Societies and of the National Cancer Institute of Mexico 2014;16(8):702–7 doi 10.1007/s12094-013-1136-8. [DOI] [PubMed] [Google Scholar]
- 18.Wu J, Zong Y, Fei X, Chen X, Huang O, He J, et al. Presence of CHD1L over-expression is associated with aggressive tumor biology and is a novel prognostic biomarker for patient survival in human breast cancer. PLoS One 2014;9(8):e98673 doi 10.1371/journal.pone.0098673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell 1996;87(2):159–70. [DOI] [PubMed] [Google Scholar]
- 20.Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487(7407):330–7 doi 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shitashige M, Hirohashi S, Yamada T. Wnt signaling inside the nucleus. Cancer science 2008;99(4):631–7 doi 10.1111/j.1349-7006.2007.00716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sánchez-Tilló E, de Barrios O, Siles L, Cuatrecasas M, Castells A, Postigo A. β-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc Natl Acad Sci USA 2011;108(48):19204–9 doi 10.1073/pnas.1108977108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou Q, Abraham AD, Li L, Babalmorad A, Bagby S, Arcaroli JJ, et al. Topoisomerase IIα mediates TCF-dependent epithelial-mesenchymal transition in colon cancer. Oncogene 2016;35(38):4990–9 doi 10.1038/onc.2016.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abraham AD, Esquer H, Zhou Q, Tomlinson N, Hamill BD, Abbott JM, et al. Drug Design Targeting T-Cell Factor-Driven Epithelial-Mesenchymal Transition as a Therapeutic Strategy for Colorectal Cancer. J Med Chem 2019;62(22):10182–203 doi 10.1021/acs.jmedchem.9b01065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chaffer CL, San Juan BP, Lim E, Weinberg RA. EMT, cell plasticity and metastasis. Cancer Metast Rev 2016;35(4):645–54 doi 10.1007/s10555-016-9648-7. [DOI] [PubMed] [Google Scholar]
- 26.Lu B, Green BA, Farr JM, Lopes FC, Van Raay TJ. Wnt Drug Discovery: Weaving Through the Screens, Patents and Clinical Trials. Cancers 2016;8(9) doi 10.3390/cancers8090082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marisa L, de Reynies A, Duval A, Selves J, Gaub MP, Vescovo L, et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med 2013;10(5):e1001453 doi 10.1371/journal.pmed.1001453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scott AJ, Song EK, Bagby S, Purkey A, McCarter M, Gajdos C, et al. Evaluation of the efficacy of dasatinib, a Src/Abl inhibitor, in colorectal cancer cell lines and explant mouse model. PLoS One 2017;12(11):e0187173 doi 10.1371/journal.pone.0187173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, et al. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol 2012;9(6):338–50 doi 10.1038/nrclinonc.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernandez-Mateos J, Khan K, et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018;359(6378):920–6 doi 10.1126/science.aao2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahel D, Horejsi Z, Wiechens N, Polo SE, Garcia-Wilson E, Ahel I, et al. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 2009;325(5945):1240–3 doi 10.1126/science.1177321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997;275(5307):1787–90. [DOI] [PubMed] [Google Scholar]
- 33.Idogawa M, Yamada T, Honda K, Sato S, Imai K, Hirohashi S. Poly(ADP-Ribose) Polymerase-1 Is a Component of the Oncogenic T-Cell Factor-4/β-Catenin Complex. Gastroenterology 2005;128(7):1919–36 doi 10.1053/j.gastro.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 34.Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc 2006;1(5):2315–9 doi 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- 35.Ryan DP, Owen-Hughes T. Snf2-family proteins: chromatin remodellers for any occasion. Curr Opin Chem Biol 2011;15(5):649–56 doi 10.1016/j.cbpa.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flaus A, Owen-Hughes T. Mechanisms for ATP-dependent chromatin remodelling: the means to the end. FEBS J 2011;278(19):3579–95 doi 10.1111/j.1742-4658.2011.08281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lehmann LC, Hewitt G, Aibara S, Leitner A, Marklund E, Maslen SL, et al. Mechanistic Insights into Autoinhibition of the Oncogenic Chromatin Remodeler ALC1. Mol Cell 2017;68(5):847–59 e7 doi 10.1016/j.molcel.2017.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gottschalk AJ, Timinszky G, Kong SE, Jin J, Cai Y, Swanson SK, et al. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc Natl Acad Sci U S A 2009;106(33):13770–4 doi 10.1073/pnas.0906920106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McDonald PC, Dedhar S. The role of epithelial-mesenchymal transition in cancer metastasis In: Regad T, Sayers TJ, Rees RC, editors. Principles of stem cell biology and cancer future applications and therapeutics. 1st ed. West Sussex, UK: John Wiley & Sons, Ltd; 2015. p 101–21. [Google Scholar]
- 40.Yun JA, Kim SH, Hong HK, Yun SH, Kim HC, Chun HK, et al. Loss of E-Cadherin Expression Is Associated with a Poor Prognosis in Stage III Colorectal Cancer. Oncology 2014;86(5-6):318–28 doi 10.1159/000360794. [DOI] [PubMed] [Google Scholar]
- 41.Richardson F, Young GD, Sennello R, Wolf J, Argast GM, Mercado P, et al. The evaluation of E-Cadherin and vimentin as biomarkers of clinical outcomes among patients with non-small cell lung cancer treated with erlotinib as second- or third-line therapy. Anticancer Res 2012;32(2):537–52. [PubMed] [Google Scholar]
- 42.Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S, Kurachi K, et al. Delineation of prognostic biomarkers in prostate cancer. Nature 2001;412(6849):822–6 doi 10.1038/35090585. [DOI] [PubMed] [Google Scholar]
- 43.Kashiwagi S, Yashiro M, Takashima T, Nomura S, Noda S, Kawajiri H, et al. Significance of E-cadherin expression in triple-negative breast cancer. Br J Cancer 2010;103(2):249–55 doi 10.1038/sj.bjc.6605735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Toiyama Y, Yasuda H, Saigusa S, Tanaka K, Inoue Y, Goel A, et al. Increased expression of Slug and Vimentin as novel predictive biomarkers for lymph node metastasis and poor prognosis in colorectal cancer. Carcinogenesis 2013;34(11):2548–57 doi 10.1093/carcin/bgt282. [DOI] [PubMed] [Google Scholar]
- 45.Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest 2009;119(6):1429–37 doi 10.1172/JCI36183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun J, Zhang L, Zhao H, Qiu X, Chen W, Wang D, et al. CHD1L Regulates Cell Cycle, Apoptosis, and Migration in Glioma. Cell Mol Neurobiol 2016;36(4):565–76 doi 10.1007/s10571-015-0237-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu M, Marsters S, Ye X, Luis E, Gonzalez L, Ashkenazi A. E-cadherin couples death receptors to the cytoskeleton to regulate apoptosis. Mol Cell 2014;54(6):987–98 doi 10.1016/j.molcel.2014.04.029. [DOI] [PubMed] [Google Scholar]
- 48.Steinhusen U, Weiske J, Badock V, Tauber R, Bommert K, Huber O. Cleavage and shedding of E-cadherin after induction of apoptosis. J Biol Chem 2001;276(7):4972–80 doi 10.1074/jbc.M006102200. [DOI] [PubMed] [Google Scholar]
- 49.Drost J, Clevers H. Organoids in cancer research. Nat Rev Cancer 2018;18(7):407–18 doi 10.1038/s41568-018-0007-6. [DOI] [PubMed] [Google Scholar]
- 50.Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 2017;7(1):1–13 doi 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.