

Abstract
Genetic Code Expansion (GCE) technologies incorporate non-canonical amino acids (ncAAs) into proteins at amber stop codons. To avoid unwanted truncated protein and improve ncAA-protein yields, genomically recoded strains of E. coli lacking Release Factor 1 (RF1) are becoming increasingly popular expression hosts for GCE applications. In the absence of RF1, however, endogenous near-cognate amber suppressing tRNAs can lead to contaminating protein forms with natural amino acids in place of the ncAA. Here, we show that a 2nd-generation amino-acyl tRNA synthetase (aaRS)/tRNACUA pair for site-specific incorporation of 3-nitro-tyrosine could not outcompete near-cognate suppression in an RF1-deficient expression host and therefore could not produce homogenously nitrated protein. To resolve this, we used Rosetta to target positions in the nitroTyr aaRS active site for improved substrate binding, and then constructed of a small library of variants to subject to standard selection protocols. The top selected variant had an ~2-fold greater efficiency, and remarkably this relatively small improvement enabled homogeneous incorporation of nitroTyr in an RF1-deficient expression host and thus eliminates truncation issues associated with typical RF1-containing expression hosts. Structural and biochemical data suggest the aaRS efficiency improvement is based on higher affinity substrate binding. Taken together, the modest improvement in aaRS efficiency provides a large practical impact and expands our ability to study the role protein nitration plays in disease development through producing homogenous, truncation-free nitroTyr-containing protein. This work establishes Rosetta-guided design and incremental aaRS improvement as a viable and accessible path to improve GCE systems challenged by truncation and/or near-cognate suppression issues.
Keywords: nitro-tyrosine, genetic code expansion, tRNA synthetase, near-cognate suppression, Release Factor 1, recoded organisms, Rosetta
Graphical Abstract
INTRODUCTION
Recently developed E. coli strains with “reduced” genetic codes open exciting options for Genetic Code Expansion (GCE) and other types of synthetic biology engineering.[1–6] One example is E. coli strains engineered to no longer rely on the amber codon (UAG) as a specific translational stop signal. To recode these organisms, chromosomal TAG codons were mutated to an alternate stop codon, and then the gene encoding Release Factor 1 (RF1, the E. coli protein responsible for terminating translation at amber codons) was deleted. In GCE applications, amber codons can then be placed in genes of interest to guide the translational incorporation of non-canonical amino acids (ncAAs) into proteins via expression of (i) an orthogonal amino-acyl tRNA synthetase (aaRS) engineered to recognize a specific ncAA, and (ii) its cognate tRNACUA harboring an anti-codon able to base pair with UAG codons. The lack of competition between RF1 and the UAG-suppressing GCE machinery is considered beneficial in that ncAA incorporation efficiency is enhanced, and the buildup of prematurely truncated peptide during ncAA-protein synthesis is mitigated or eliminated.[1–3, 7, 8] The lack of truncated peptide is particularly advantageous for GCE applications involving expression of oligomeric proteins, where truncated and full-length protein would otherwise co-purify,[9] or proteins that require purification and solubility enhancing tags at the N-terminus (e.g. GST, SUMO, MBP) for efficient expression.[10] Significant quantities of truncated peptide alongside ncAA-protein production complicates efforts to delineate the specific effects an ncAA-protein might have on in vivo cellular processes.
These recoded E. coli strains also bring new challenges related to GCE translational fidelity. The lack of RF1 in recoded E. coli results in ribosome stalling, mis-translation (e.g. frameshift), or misincorporation of natural amino acids at encoded UAG sites.[11, 12] Mis-incorporation of natural amino acids at UAG codons can occur via (i) near-cognate suppression by endogenous tRNAs whose anticodons are able to pair with UAG using non-Watson-Crick (i.e. wobble) interactions, or (ii) natural UAG codon suppressors formed by spontaneous chromosomal mutations.[11, 13–15] Thus, in these RF1-deficient strains, even though GCE-derived amber suppressing tRNACUA’s no longer must outcompete RF1, they now must outcompete endogenous near-cognate suppressors in order to produce homogeneously modified protein containing the ncAA (Fig. 1).
Figure 1.
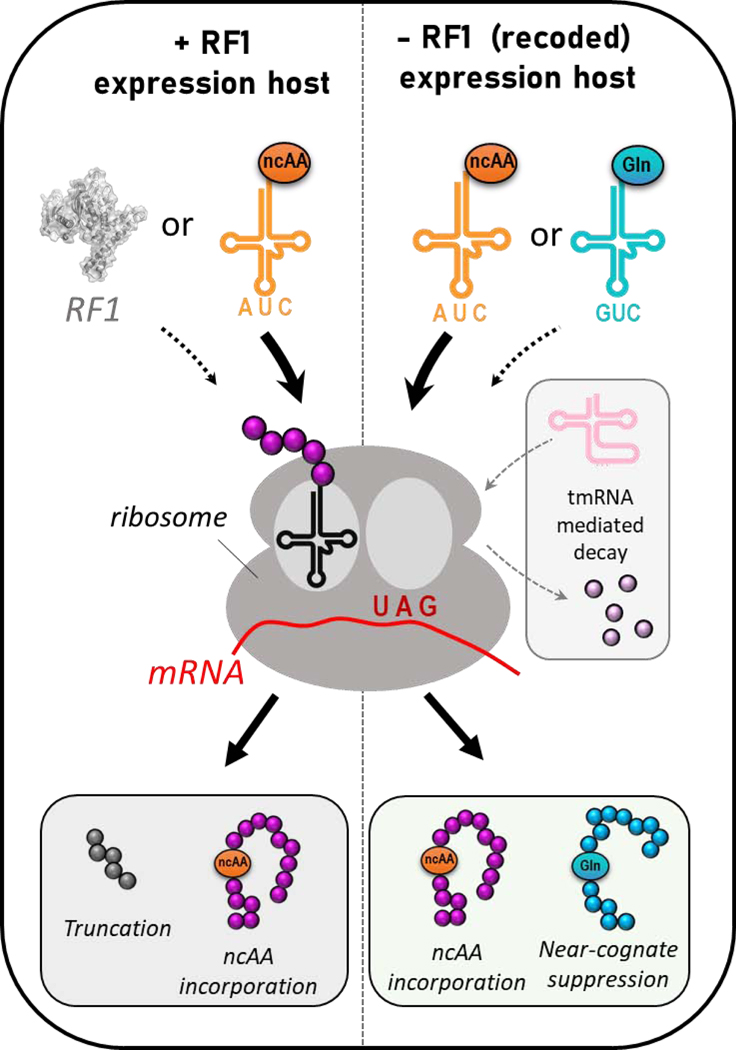
Translational outcomes for non-canonical amino acid (ncAA) incorporation into proteins at UAG amber stop codons in RF1-containing expression hosts (left) and RF1-deficient expression hosts (right). In +RF1 hosts, either ncAA-protein or truncated peptide is typically produced. In −RF1 hosts, UAG codons can be suppressed by either the GCE-derived amber suppressor tRNACUA for accurate ncAA insertion or endogenous near-cognate suppressing tRNAs which can insert e.g. Gln at UAG codons. Transfer-messenger RNA (tmRNA) can also resolve stalled ribosomes and initiate peptide decay.
Favorable competition of GCE amber suppressing systems is facilitated by sufficient buildup of intracellular ncAA-aminoacylated tRNACUA[16, 17] as well as the use of strains with minimal UAG suppressor activity.[1, 14] Previous work has shown that GCE derived aaRS/tRNACUA systems have significantly lower catalytic activity than their natural counterparts,[18] leading to low levels of ncAA-tRNACUA and insufficient competition with near-cognate suppressors. Some relief to this has been demonstrated by increasing aaRS and tRNA expression levels[15, 19], removing near-cognate suppressing tRNAs (for cell-free expression)[20] or increasing concentrations of ncAA[21]. While these strategies can partially compensate for the poor catalytic activity of GCE derived aaRS/tRNACUA systems, we hypothesize that a more effective and general solution would be to improve the catalytic activity of the encoded GCE system. This optimization of GCE catalytic efficiency would reduce the abundance of GCE components moving toward the goal of stable cells containing genomically encoded GCE systems with the added benefit of reducing cellular stress. Here, we show that it is in fact true that the misincorporation of natural amino acids in an RF1-deficient system can be overcome by a readily achievable improvement in the catalytic activity of the encoded aaRS/tRNACUA pair.
Previously, our laboratory evolved an aaRS/tRNACUA pair for the incorporation of 3-nitrotyrosine (nitroTyr), a post-translational modification found in increased abundance on proteins during oxidative stress and disease.[22] This 1st-generation aaRS suffered from low efficiency, which prompted efforts to generate a more efficient 2nd-generation system via improved selection strategies and crystallographic analysis of the aaRS/nitroTyr complex.[23] Despite the ~10-fold enhancement in nitroTyr-protein this yielded, we showed that the kinetic properties of this nitroTyr aaRS/tRNA pair were still at least 3 orders of magnitude worse than its natural counterpart.[18] Here, we show that even this improved 2nd-generation nitroTyr GCE system does not fully outcompete near-cognate suppression and therefore cannot produce homogenously nitrated protein in an RF1-deficient expression host. The homogeneous encoding of nitroTyr is necessary to understand the role of this posttranslational modification (PTM) in biology. Extensive effort to optimize selections from our current library of 108 aaRS variants did not yield discovery of a further improved variant, and intuitive alterations of the aaRS active site, based on its crystal structure, did not yield variants with improved activity.[23] We describe here the use of computationally-guided library design to evolve a 3rd-generation nitroTyr aaRS that is incrementally (~2-fold) improved and that this improvement is sufficient to overcome the fidelity challenges and homogeneously incorporate nitroTyr into proteins in a recoded, RF1-deficient expression host. Crystallographic analysis of this 3rd-generation nitroTyr aaRS reveals the structural basis for its improved utility. We propose that similar computationally-assisted library design and directed evolution strategies are a simple, generally applicable avenue to improve the efficiency of aaRS/tRNACUA systems and expand their compatibility with other recoded expression hosts.
RESULTS
Choice of RF1 deficient expression host.
RF1 deficient strains of E. coli differ primarily by the strain from which they were derived and the number of chromosomal TAG codons converted to TAA stop codons. Here, we use the B-95(DE3) ΔAΔfabR strain (referred to hereafter as B-95) engineered by Sakamoto and colleagues, which is a partially recoded strain having 95 of 273 chromosomal TAG stop codons altered to TAA.[2] B-95 is derived from BL21(DE3), a traditional protein overexpression strain compatible with the widely used T7 expression system. It displays minimal endogenous amber suppressor capacity and grows at nearly the same rate as BL21(DE3) in a wide range of temperatures and media, including minimal and auto-induction media.[2, 10] Compatibility with auto-induction media is advantageous for GCE protein expression compared to manual based induction methods.[24]
Mis-incorporation of natural amino acids during ncAA-protein production using B-95 cells.
Our previously evolved 2nd-generation nitroTyr aaRS, referred to previously[23] and hereafter as the “5B” aaRS, was sufficient for homogeneous incorporation of one and two nitroTyr moieties into super-folder green fluorescent protein (sfGFP) when expressed in RF1-containing expression hosts (Figs. 2C and 2E, Supplemental Table S1). These observations confirm this “5B” aaRS/tRNA pair is orthogonal in RF1-containing expression hosts in that it neither accepts canonical amino acids nor is its cognate amber suppressor tRNACUA charged by endogenous aaRSs. However, when these sfGFP variants were expressed in B-95 cells, the majority of the detected protein was non-nitrated (in the case of sfGFP-150TAG) or incompletely (in the case of 134/150TAG-sfGFP) nitrated (Fig. 2D and 2F). The masses of the non-nitrated species were consistent, within the error of the mass spectrometer (+/− 2 Da), with incorporation of a canonical amino acid – either Gln, Lys and/or Glu – at the programmed UAG sites (Supplemental Table S1). These canonical amino acids have masses within 1 Da of each other and are a signature of near-cognate suppression since all have been identified to be incorporated at UAG sites in RF1 deficient E. coli.[15] Indeed, this same non-nitrated species was produced by B-95 cells expressing sfGFP-150TAG in the absence of GCE machinery (Fig. 2B), providing further support that it arises from near-cognate suppression and not from infidelity of the added GCE aaRS/tRNA pair. Though Tyr and Trp have also been shown to be incorporated at UAG codons by near-cognate suppression,[6] these were not observed.
Figure 2.
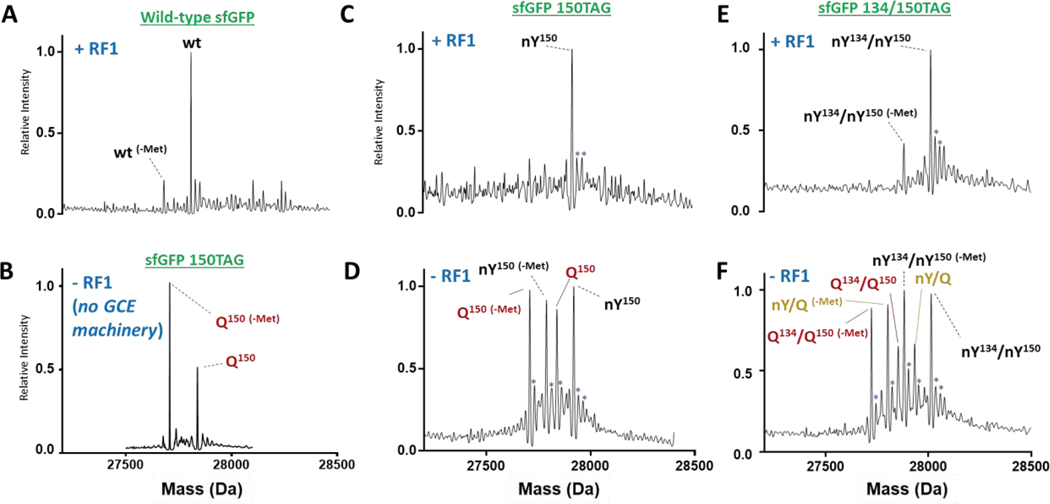
Whole protein mass spectrometry of purified, nitrated sfGFP expressed using the 2nd-generation 5B aaRS in a +RF1 and a −RF1 expression host shows mis-incorporation of natural amino acids in the - RF1 system. (A) Wild-type sfGFP, (B) sfGFP-150TAG protein expressed in RF1-deficient (B-95) cells without any GCE machinery contains peaks consistent with insertion of Gln, Glu or Lys at site 150 (although we cannot distinguish between Q, E and K, for clarity we denote these peaks as simply Q), (C) sfGFP-150-nitroTyr (nY150) expressed in +RF1 strain, (D) 150-nitroTyr (nY150) expressed in −RF1 strain (E) sfGFP-134/150-nitroTyr (nY134/nY150) expressed in +RF1 strain and (F) sfGFP-134/150-nitroTyr (nY134/nY150) expressed in −RF1 strain, which contains peaks consistent with one (nY/Q) or two (Q134/Q150) Gln, Glu or Lys residues inserted. -Met: loss of N-terminal methionine. Measured and expected mass are omitted for clarity and listed in Supplemental Table S1. Purple asterisks are peaks with masses consistent with Na+/K+ adducts.
To assess whether or not the mis-incorporation of natural amino acids in the B-95 strain is unique to this nitroTyr “5B” GCE system, we evaluated the incorporation fidelity for four evolved aaRSs with different specificities: 4-azido-phenylalanine (AzF)[25], p-benzoyl-phenylalanine (Bpa)[26], Tetrazine2.0 (Tet2.0)[27], and trans-cyclo-octene-lysine (TCO-K)[28]. This set was chosen because they are widely used in the field and represent two common GCE platforms, namely the Tyr-based Methanocaldococcus jannaschii (AzF, Tet2.0 and Bpa) and pyrrolysine-based Methanosarcina barkeri (TCO-K) systems. Whereas all of these systems enabled homogeneous incorporation of their respective ncAAs into sfGFP in an RF1-containing expression host, only those for AzF and Bpa provided homogeneous ncAA incorporation into sfGFP in B-95 while those for Tet2.0 and TCO-K produced sfGFP adducts with masses consistent with mis-incorporation of Gln, Lys and/or Glu at the UAG codons (Supplemental Fig. S1). These results collectively exclude the possibility that mis-incorporation is inherent to the B-95 expression strain, is a consequence of our expression methods, or is due to some incompatibility of the GCE machinery platforms with B-95 cells. The AzF and Bpa synthetases are known to be particularly robust, and are widely used, consistent with the hypothesis that misincorporation is not a problem if the evolved aaRS/tRNACUA pair works well enough.
Computational screening for improved 3-nitroTyr aaRSs.
Based on the above, we reasoned that a more efficient nitroTyr aaRS would provide increased ncAA-tRNACUA levels in the cell and mitigate misincorporation of natural amino acids by out-competing endogenous near-cognate suppressing tRNAs more effectively. Having carried out extensive prior efforts to find improved aaRSs from our current 108 member library,[22, 23] alongside thorough kinetic analyses of the 1st and 2nd generation nitroTyr aaRSs,[18] we employed computational methods to explore variants outside the previously sampled library space for improved active sites. Rosetta software has been used successfully to design small molecule binders[29–31] suggesting it could also be effective for designing aaRSs with improved ncAA binding properties. In fact, Rosetta has been used previously to generate a large library (1039) of potential aaRS sequences that enabled the discovery of one improved variant selective for a photocaged tyrosine derivative.[32] In order to find computational metrics that can more reliably predict the best aaRS for a given ncAA, we first ran a benchmark analysis on seven different Methanocaldococcus jannaschii ncAA/aaRS crystal structures (PDB IDs: 1J1U, 1ZH0, 1ZH6, 2AG6, 2HGZ, 2PXH, 4NDA). We made all possible combinations of seven aaRSs and four ncAAs, and evaluated their interface quality using a range of Rosetta-based metrics (Supplemental Dataset). These results indicated that high shape complementarity and low ligand interface energy together were the most reliable metrics by which the cognate ncAA substrate was predicted for each aaRS (Supplemental Fig. S2).
We therefore used these two metrics to assess variants of the nitroTyr “5B” aaRS for improved substrate binding. Rosetta GreedyOpt algorithm was used to mutate, in silico, ten residues within 8 Å of the nitroTyr ligand in the nitroTyr “5B” active site to all 20 amino acids (Fig. 3A, Table S2). Comparing the shape complementarity and ligand interface energy trends across these 200 variants (Fig. 3B, Fig. S3, Supplemental Dataset) led us to select two, C70A and S158C, for follow up characterization based on the following criteria: (i) one metric was improved and other either improved or marginally altered, (ii) the mutations were not likely to break critical hydrogen bonding interactions, particularly with water molecules, and (iii) the variants had not been previously experimentally evaluated. In vivo evaluation of these two single site variants, as well as the double site variant (C70A/S158C), revealed they all possess similar levels of activity as the original nitroTyr “5B” aaRS when nitroTyr was supplemented to the media at 1.0 mM nitroTyr, and lower activity with 0.1 mM nitroTyr (Fig. 3C). Interestingly, the chosen metrics accurately predicted the modestly better performance of C70A over S158C at 0.1 mM nitroTyr. Based on a standard curve of purified sfGFP, the reported fluorescence of “5B”, C70A, S158C and C70A/S158C cultures correspond to yields of ~110, 90, 75 and 100 mg nitroTyr-sfGFP per liter culture at 1 mM nitroTyr.
Figure 3.
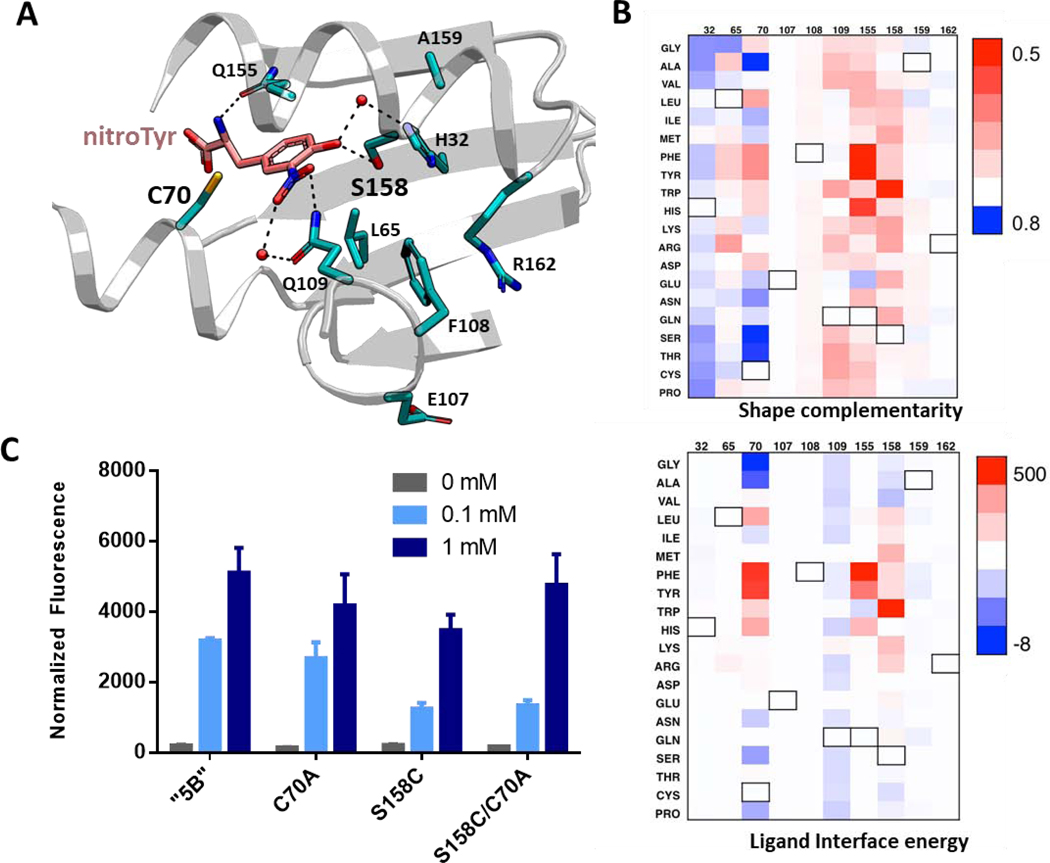
Analysis of nitroTyr binding on the active site of the “5B” aaRS modified by single residue alterations. (A) Crystal structure of the “5B” nitroTyr aaRS active site highlighting the ten residues analyzed for mutational analysis by Rosetta (PDB 4nda). (B) Analysis of shape complementarity (top) and ligand binding interface energy (bottom) for nitroTyr to each modified residue (numbers are listed on top of each panel). Black outlined boxes indicate wild-type amino acids. Value for each amino acid position is subtracted from the original value to show the absolute effect of each mutation. In both plots, blue means improved over original value. Values are provided in the Supplemental Dataset. (C) Variants of “5B” aaRS C70A, S158C and C70A/S158C predicted to have better activity were tested for their ability to express sfGFP-150TAG in vivo in RF1-containing BL21 cells by measuring in-cell fluorescence normalized to culture density. Cultures were supplemented with 0, 0.1 and 1 mM nitroTyr as indicated. Error bars represent the standard deviation of three independent cultures.
Crystal structures of the C70A, S158C and C70A/S158C variants were solved (Supplemental Table 3). Consistent with their comparable in vivo activity to the parent “5B” nitroTyr aaRS, all three variants bind the nitroTyr substrate in a similar manner as the nitroTyr “5B” aaRS (Supplemental Fig. 4). Hydrogen bonds across all structures are preserved and the nitro groups are rotated out of the plane of the phenolic ring. The extent of the rotation was slightly less for the C70A and C70A/S158C enzymes (16° and 18°, respectively, compared to 22° for “5B”) and much greater for the S158C variant (34°). Rosetta was unable to capture this nitro-group rotation, even when the nitroTyr ligand was allowed to move (data not shown). Yet, scoring analyses of shape complementarity and ligand interface energy for the experimentally derived crystal structure conformations were slightly improved, but not notably different than those predicted (Supplemental Fig. S5). These data suggest the current scoring is not very sensitive to the effects of these slight modification in ligand conformation on binding. It is important to note that Rosetta’s interface metrics are only reflective of binding affinity and cannot predict catalytic improvements, thus it is possible that these variants still bind to nitroTyr better than the partner.
Saturation mutagenesis at sites 70 and 158: parallel in vivo and in silico selections.
Since the computational work suggested positions 70 and 158 as targets for optimization (Fig. 3B), and the previously screened library only contained three amino acid types at site 158 (Ser, Val, Gly) while varying C70 to all 20 amino acids (Supporting Table S2), we decided to make and screen a “5B” nitroTyr aaRS library covering all possible combinations of amino acids at both sites (20 × 20 = 400 unique protein variants, of which 20 × 17 = 340 were not present in the original library). In parallel, we evaluated the interface metrics for all combinations using Rosetta in both flexible and fixed backbone format, and calculated ligand interface energy and shape complementarity for each.
The top sixteen performing hits from the in vivo selections were sequenced, revealing seven unique variants (Fig. 4A). The highest performing (“A7”) was the most frequently sequenced and contained two alterations: C70T and S158H. Three of the other seven unique variants were theoretically present in the original library (1-H8, 2-D5 and 2-E5) but were not retrieved in prior selection processes, perhaps due to incomplete coverage during its original assembly. The in silico Rosetta calculations showed a better separation between identified variants and those that failed when flexible backbone was used compared to fixed backbone (Supplemental Figure S6), consistent with prior structure knowledge of the flexible nature of the nitroTyr aaRS active site.[23] The results of this analysis show the “A7”, as well as other better performing variants, scored computationally slightly worse than the parent “5B” (Fig. 4B, Supplemental Fig. S5, Supplemental Dataset), suggesting that computational analysis loses its predictability when finer comparisons are drawn. However, His at position 158 was predicted in two of the top five ligand interface energy scoring variants (Supplemental Table S4), and Thr at position 70 was the third best scoring for shape complementarity in the initial Rosetta GreedyOpt prediction (Fig. 3B, Supplemental Fig. S3). Indeed, this single site C70T variant was among the top experimentally identified hits (1-H8, Fig. 4A). The fact that the top in vivo selection hits (except for 1-A12: C70L/S158T) were among top 20% best computational hits provides compelling evidence that Rosetta is an effective tool to narrow down the mutational space needed for library design and experimental selection.
Figure 4.
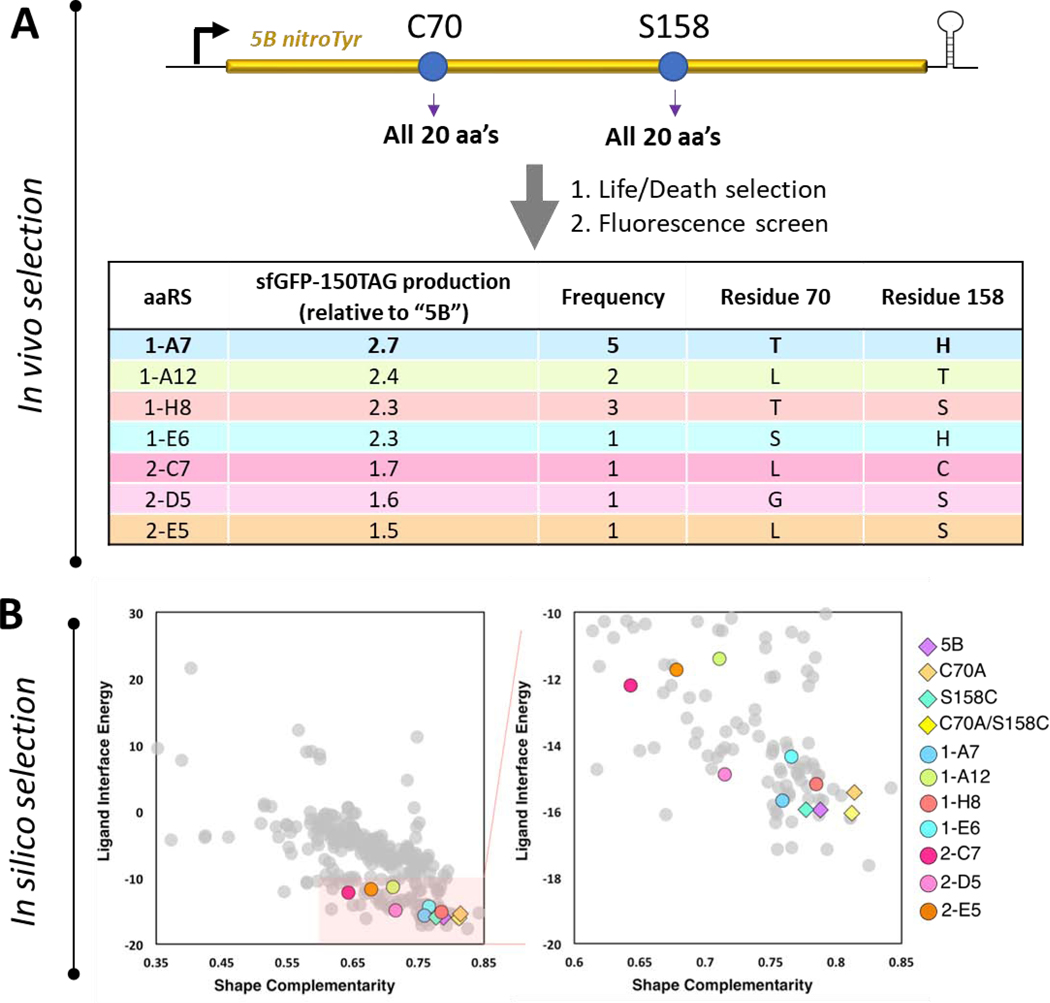
Evaluation of a complete nitroTyr aaRS library at sites 70 and 158 of the “5B” nitroTyr aaRS. (A) The pool of 400 unique protein variants at sites 70 and 158 were selected for their ability to incorporate nitroTyr and no canonical amino acids and surviving members were evaluated for their ability to incorporated nitroTyr into sfGFP-150TAG (see Materials and Methods). Sequences for the top 16 performing hits are indicated, revealing seven unique variants. (B) Rosetta-predicted shape complementarity and ligand interface energy of all 400 variants (left panel), and expanded view of 80 variants with shape complementarity > 0.6 and ligand interface energy < −10 (right panel). The seven new experimental hits (colored circles) and the four previously characterized aaRSs (colored diamonds) were all scored by Rosetta as relatively favorably by these parameters. The top five variants in each category (ligand interface energy and shape complementarity) are listed in Supplemental Table S4.
Efficiency and fidelity of the 3rd-generation nitroTyr “A7”aaRS.
After this initial screening, the nitroTyr “A7” aaRS was selected for further characterization in a more standard pET/pDule GCE expression system in an RF1-containing BL21 cell line (Supplemental Figure S7). These data show that compared to the 2nd-generation “5B” aaRS, ~1.5-fold and ~2.5-fold more singly and doubly nitrated sfGFP was produced when media was supplemented with 1.0 mM nitroTyr and 0.1 mM nitroTyr, respectively (Fig. 5A). The measured fluorescence from the cultures with 1 mM nitroTyr correspond ~160 and 94 mg sfGFP per liter culture for the “A7” and “5B” aaRSs, respectively. The concentration of nitroTyr needed to reach half-maximal sfGFP production (aka the “UP50”)[18] was 23 ± 4 μM for the “A7” aaRS, compared to 120 ± 20 μM for “5B” variant (Fig. 5B), confirming the ability of the “A7” aaRS to function more effectively at lower nitroTyr concentrations. Previous work has shown that lower in vivo UP50 values are indicative of a lower aaRS KM.[18] Interestingly, our results further show that the “5B” aaRS does not incorporate nitroTyr efficiently when expressions are performed in a “complex” media (i.e. containing tryptone and yeast extract) as opposed to “defined” media (see Materials and Methods), whereas the “A7” aaRS is able to efficiently incorporate nitroTyr in both fully-defined and complex media (Fig. 5C). The reason for the poor expression efficiency of the “5B” aaRS in complex media is not understood but could be explained by reduced cellular uptake of nitroTyr during growth on complex media, consistent the “A7” variant having a lower UP50 than “5B”.
Figure 5.
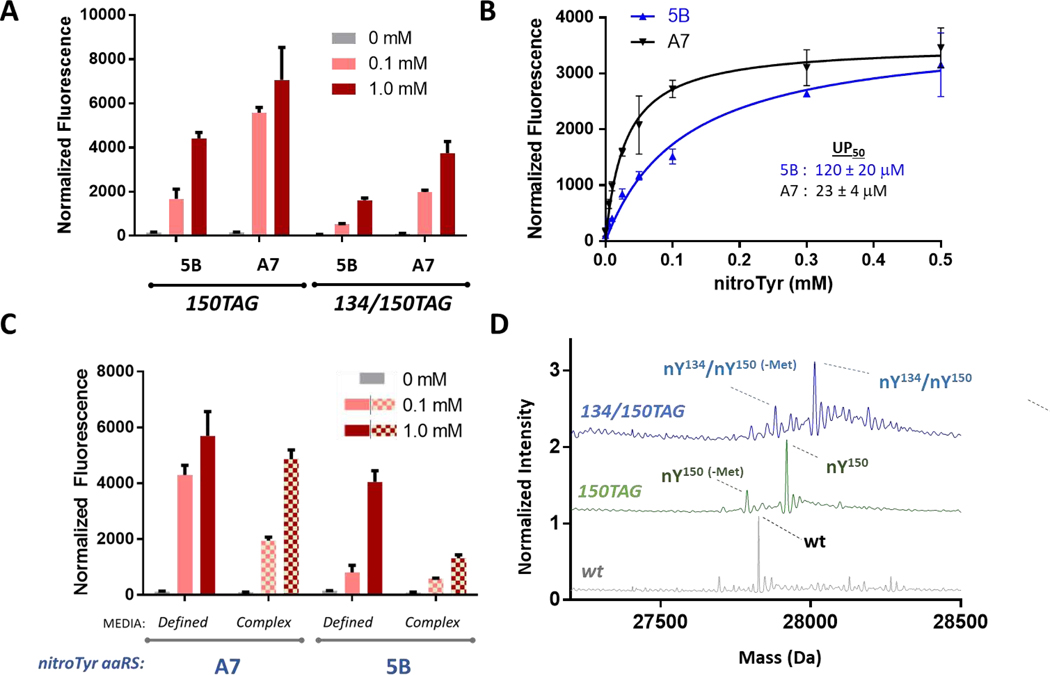
Functional characterization of the “A7” nitroTyr aaRS. (A) In-cell fluorescence normalized to culture density of cells expressing sfGFP-150TAG and sfGFP-134/150TAG using either the “5B” or “A7” aaRS in the presence of either 0, 0.1 and 1 mM nitroTyr in the media. All expressions were performed in RF1-containing, BL21 cells (see Materials and Methods for details). (B) Normalized fluorescence of RF1-containing BL21 cells expressing sfGFP-150TAG measured as a function of nitroTyr supplemented to the media allows for evaluation of the UP50, the concentration at which half maximal fluorescence is achieved. Curves are fitted to a standard Michaelis-Menton like model. (C) Comparison of sfGFP-134/150TAG expression in defined vs. complex (ZY-based) auto-induction media using the “A7” and “5B” nitroTyr aaRSs with RF-1 containing BL21 cells (see Materials and Methods). The dual TAG reporter of sfGFP was used to emphasize efficacy of the “A7” aaRS in complex media compared to the “5B”. (D) Whole protein mass spectra of sfGFP (gray, expected/observed: 27827/27826 Da), sfGFP-150TAG (green, expected/observed: 27921/27919 Da) and sfGFP 134/150TAG (blue, expected/observed: 28013/28012 Da) expressed using the “A7” nitroTyr aaRS in RF1-deficient B-95 cells shows homogenous incorporation of nitroTyr and no detectable natural amino acids. For panels A-C, error bars represent standard deviations from three independent cultures.
The “A7” aaRS enables homogenous, truncation-free incorporation of nitroTyr using the B-95 strain.
We next incorporated nitroTyr into one and two sites of sfGFP with the “A7” aaRS in RF1 deficient B-95 cells and analyzed them by whole protein mass spectrometry. Strikingly, unlike the sfGFP protein produced in B-95 cells using the “5B” aaRS, the proteins made with “A7” aaRS were homogenous populations of protein having masses consistent with the targeted number of nitroTyr residues (Fig. 5D). These results demonstrate the improved functionality of the new “A7” aaRS is enough to overcome the near-cognate suppression of UAG codons in RF1-deficient expression hosts.
In contrast to RF1-containing cells, when using RF1-deficient cells the yields of total sfGFP-150TAG protein are comparable between the “A7” and the “5B” nitroTyr aaRS systems (Supplemental Fig. S8). This results from an increase in total protein production in RF1-deficient cells with the “5B” system, but this increase is not due to enhanced nitroTyr-sfGFP expression but largely increased near-cognate suppression (Figs. 2D and 2E). These results throw caution to the notion that RF1-deficient cells are effective expression hosts for enhancing total ncAA-protein yields, since the observed increase in protein yields may just be the consequence of increased near-cognate suppression rather than an actual enhancement in ncAA-protein production.[33]
Nevertheless, to confirm truncated protein was not formed in the B-95 expression host during nitroTyr-protein production, we expressed a N-terminally His6-tagged SUMO-sfGFP reporter protein in which a TAG site was placed in the flexible linker between SUMO and sfGFP (Fig. 6A). In this construct truncated peptide would be the highly soluble and His6-tagged SUMO domain and would be expected to co-purify with full-length SUMO-sfGFP. Indeed, purification of these SUMO-sfGFP proteins from RF1-containing cells confirmed that truncated SUMO protein co-purified with full-length SUMO-sfGFP. However, when expressed in an RF1-deficient host no truncated protein was co-purified (Fig. 6B). These results confirm, along with the fidelity assessments above, that the “A7” nitroTyr aaRS is a significant improvement over the “5B” nitroTyr aaRS in that it makes possible the production of homogenously nitrated protein without buildup of truncated peptide.
Figure 6.

Evidence that RF1-deficient B-95 expression hosts mitigate truncated peptide side product. (A) N-terminally His6 tagged construct of SUMO-sfGFP with and without a TAG codon in the linker between the SUMO and sfGFP domains. The N-terminal location for the His6 purification tag ensures any truncated SUMO protein will co-purify will full-length SUMO-sfGFP protein. (B) SDS-PAGE gel showing the affinity-purified proteins under each expression condition with positions expected for full-length and truncated proteins indicated. When a TAG construct is expressed in an RF1-containing expression host, ~1/3 of the produced protein is truncated. In contrast, no truncated protein is observed for expression using the B-95 RF1-deficient expression host. Molecular weight markers are indicated. The gel was stained with Coomassie Blue.
We next assessed if the “A7” nitroTyr aaRS could produce biologically relevant homogenously nitrated forms of proteins in an RF1-deficient expression. For this we expressed 14-3-3 and calmodulin with nitroTyr at sites Y130 (β-isoform numbering) and Y99, respectively. Both proteins are key regulators of signaling eukaryotic signaling systems that have been identified as nitrated at these tyrosine residues in vivo under physiologically relevant conditions.[34–36] Using the “A7” nitroTyr aaRS, we expressed and purified nitrated 14-3-3 and calmodulin in both RF1-containing and RF1-deficient hosts with yields of ~75 and 50 mg per liter culture, respectively. Mass spectrometry analysis revealed homogenous nitroTyr incorporation in both proteins when expressed in B-95 cells (Supplemental Figure S9).
Structural basis for the improved “A7” aaRS efficiency.
We next solved the structure of the “A7” aaRS bound to nitroTyr at 1.95 Å resolution (Fig. 7, Supplemental Fig. S10). In the “A7” aaRS structure, the T70-hydroxyl participates in two hydrogen bonds not seen in the “5B” structure: one to the NO2 group of nitroTyr and one to a water molecule that bridges the side chains of T70 and Q109 (Fig. 7). Also, His158 in the “A7” aaRS donates a more linear hydrogen bond to the phenolic oxygen of nitroTyr (158-NδH…O angle of 159° compared to the 158-Ser-OγH…O angle of 134° in the “5B” structure). His158 is held in position by a double hydrogen bond from the guanidinium group of R162 which adopts a different conformation than it does in the “5B” aaRS structure (Fig. 7B). Lastly, the nitro group of the nitroTyr is nearly in plane with the phenolic ring (6° rotation) compared to 22° for the “5B” aaRS (Fig. 7 and Supplemental Figure S10B). Rosetta scoring analysis for ligand interface energy for the experimentally derived “A7” crystal structure conformations was notably better than originally predicted by Rosetta based on minimizations starting with the “5B” aaRS structure (Supplemental Fig. S5). Collectively, the active site architecture of the “A7” aaRS is consistent with a more stable enzyme-substrate complex that serves to lower the UP50 and, in practice, provide for a more efficient GCE system than that of the “5B” nitroTyr aaRS.
Figure 7.
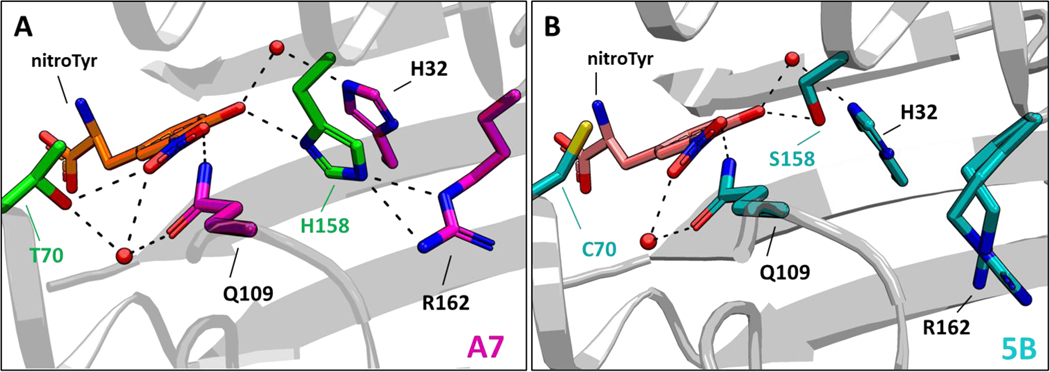
Structural basis of nitroTyr recognition by the “A7” nitroTyr aaRS. (A) Crystal structure of the active site of the “A7” nitroTyr aaRS showing T70 and H158. In the “A7” active site, two additional hydrogen bonds from T70 and a bifurcated hydrogen bond from H158 to the guanidinium group of R162 are shown. (B) Active site configuration of the 2nd-generation “5B” nitroTyr aaR (PDB 4nda). R162 is shown in two equally populated conformations.
DISCUSSION
Rewriting the genetic code and reassigning the function of codons is a key step in engineering novel protein chemistries. As more synonymous codons are condensed,[4] it will be increasingly important to understand how to ensure that the engineered orthogonal translational machineries that introduce ncAAs at these unassigned codons will outcompete the mechanisms by which natural translation systems resolve them.[16, 17] We have shown here that some GCE aaRS/tRNA pairs, like the widely used benzoyl-phenylalanine and para-azidophenylalanine GCE systems, are efficient enough to outcompete near-cognate suppression systems, while other systems like the 2nd-generation nitroTyr, Tet2.0 and TCO-K systems, are not able to do so (Supplemental Fig. S1). This means that the ability to out compete near-cognate suppression is system dependent and likely a function of overall efficiency. As the number of new GCE systems continue to grow rapidly, the ability to favorably balance ncAA incorporation over near-cognate suppression in recoded organisms should be a key measure of the quality of a GCE system. Use of low efficiency GCE systems, or ncAAs with poor cellular uptake, in RF1-deficient cells should be approached with caution, as failure to out-compete near-cognate suppression can lead to production of unintended target protein containing natural amino acids. Here, we show that by optimization of the catalytic pocket of the aaRS active site of a low efficiency system, while keeping all other GCE components the same, endogenous near-cognate suppressors can be outcompeted to make homogenous nitroTyr-containing protein. This test case study illustrates that even a modest ~1.5–2.5-fold improvement in aaRS efficiency (Fig. 5A), can be enough to shift from the near-cognate suppression product accounting for a fairly large fraction of the protein produced (Fig. 2D, 2F) to it being below the limits of detection (Fig. 5D).
The advantages of using RF1-deficient cells lines has been demonstrated before, which include higher yields of ncAA-protein particularly when incorporating multiple ncAA’s and the lack of truncated protein.[3, 8, 10, 19] For in vivo applications, buildup of truncated protein alongside full-length ncAA-protein makes it challenging to deconvolute the specific contribution each has toward a particular cellular response. For ncAA-protein overexpression applications, RF1-deficient cell lines permit the use of N-terminal solubility/folding-enhancing fusion chaperones (e.g. SUMO, GST, MBP) and purification tags commonly used to enhance heterologous protein expression without the hassles of co-purifying truncated protein. Lastly, they also solve issues associated with expression of oligomeric proteins where truncated protein can co-purify with full-length protomers. Indeed, this very issue prevented successful synthesis of homogenous, full-length, dimeric peroxiredoxin-2 with nitroTyr at a biologically relevant site near its C-terminus (Y192).[9] Such examples highlight the advantages of this new 3rd-generation nitroTyr GCE system for expanding our ability to study biologically relevant nitrated proteins for uncovering the effects nitroTyr, a common protein PTM found in diseased and oxidatively stressed cells, has on protein function.[36–38] As a side note, we speculate that an independent avenue via which the efficiency of nitroTyr incorporation could be even further enhanced is by taking advantage of a recently identified biosynthetic pathway for nitroTyr in order to circumvent possible cell import challenges.[39]
The strategies outlined here to improve the nitroTyr aaRS activity should be readily transferable to other GCE systems that could benefit from similar improvements in efficiency. A key component to our successful approach was combining computationally-guided library design with directed evolution. Previous work has shown the utility of Rosetta to narrow down synthetase library sizes to those practical for experimental screening.[32] In that case, 1036 aaRS variants were evaluated for improved substrate binding, from which 108 were selected for tractable library generation and selection, producing one experimentally selected variant with improved activity. While this result is impressive for beginning a library from scratch, one validated hit from 108 sequences is a low computational success rate. Additionally sobering is that the one experimentally derived hit differed at 10 of 17 positions from the consensus sequence of the top scoring design.
To improve the utility of Rosetta in guiding aaRS redesign, we sought to find metrics that were more informative than the Rosetta score for capturing the quality of substrate binding. The Rosetta score reflects the overall energy of the system and so need not reflect differences in substrate binding energies if they are offset by energy changes elsewhere in the structure. Our results led us to two metrics (ligand interface energy and shape complementarity) that provided much better guidance for evaluating ncAA-aaRS interactions. It is important to note that these metrics are only considered to be informative within a similar system (i.e. for comparing binding to one ncAA), and it is not meaningful to compare the absolute scores of binding aaRS/ncAA pairs where both aaRS and ncAA are different.[40] We used these metrics to create a focused library (n=400) that was evaluated, in parallel, both experimentally and computationally. This approach for evaluating ncAA-aaRS interactions – using a protocol that allowed the protein backbone to move – was powerfully validated by its generation of at least seven improved aaRSs of which the best was the “A7” aaRS (Fig. 4A). Even with this success, we have no doubt that there is still room for improvements in the scoring function and, for instance, the ability to include water, especially since the active sites of the nitroTyr aaRSs all contain key interactions with water molecules. Nevertheless, this study highlights the utility of using Rosetta to generate small, focused libraries (<103) which can be synthesized and screened with minimal time and resource investment.
The crystal structure and UP50 analyses both support the conclusion that the improvements and enhanced utility seen in the “A7” nitroTyr aaRS compared to the “5B” aaRS is based on it having a more stable enzyme/substrate complex. The “A7” mutations C70T and S158H provide a series of additional stabilizing interactions within the active site, and the nitroTyr substrate adopts a more energetically relaxed conformation which could lead to a lower enzyme KM thereby explaining the lower UP50 compared to the 5B variant. Whether a lower KM is the primary kinetic basis for its improved activity awaits to be confirmed with a full in vitro kinetics study, but worth noting is that our computational strategy to identify an improved nitroTyr aaRS was founded on predictive methods meant not for improving catalytic rates but rather optimizing enzyme/substrate interactions. In closing, we emphasize that even though we already had in hand an improved 2nd-generation nitroTyr aaRS obtained via advances in selection protocols and detailed structural analyses, it was still not sufficient to overcome near-cognate suppression in RF1-deficient expression systems, and better nitroTyr aaRSs were still possible. This means that such a computationally guided approach is well worth trying for any aaRS/tRNACUA pair for which truncation and/or near-cognate suppression lead to inhomogeneous products.
MATERIALS and METHODS
Strains.
The RF1-deficient E.coli strain B-95(DE3) ΔAΔfabR strain was provided by RIKEN BRC through the National BioResource Project of the MEXT/AMED, Japan.[2] The RF1-containing strains DH10b and BL21-ai were purchased from ThermoFisher.
Non-canonical amino acids.
3-nitro-tyrosine was purchased from Alfa Aesar (Cat. no. A11018), para-azido-phenylalanine was purchased from Bachem (Cat. no. 4096192), and TCO-K (axial isomer of trans-Cyclooct-2-ene-L-Lysine) was purchased from SiChem (Cat. no. SC-8008). Stock solutions were prepared fresh for each experiment by suspending the amino acids in water and solubilizing with 1–2 molar equivalents of NaOH. 4-(6-methyl-s-tetrazin-3-yl)phenylalanine (Tet-v2.0) was synthesized as previously described.[27, 41] Stock solutions were prepared in DMF.
Assessment of pAzF, pBpa, Tet-v2.0 and TCO-K incorporation fidelity in the B-95 expression host.
Chemically competent BL21-ai or B-95(DE3) ΔAΔfabR (referred to B-95) were co-transformed with pET28-sfGFP-150TAG (containing a C-terminal His6 affinity purification tag, Addgene #85493) and the appropriate GCE machinery plasmid. For para-azido-phenylalanine (pAzF), para-benzoyl-phenylalanine (pBpa) and 4-(6-methyl-s-tetrazin-3-yl)phenylalanine (Tet-v2.0) incorporation, the GCE machinery plasmids were pDule2-pCNF (Addgene 85495), pDule2-Bpa, and pDule2-Tet-v2.0 (Addgene 85497), respectively. All pDule2 plasmids contain a p15a origin of replication and constitutively express the indicated amber suppressing Methanocaldococcus jannaschii (Mj) amino-acyl tRNA synthetase (aaRS)/tRNACUA pair (Supplemental Figure S7). For TCO-K, the pEVOL-AF machinery plasmid conferring chloramphenicol resistance was used,[28] which expresses two copies of the Methanosarcina barkeri pyrrolysine-based Y306A/Y384F aaRS (one copy under the control of a constitutive promoter and the other under control of an arabinose inducible promoter), as well its cognate amber suppressing tRNACUA. After transformation, cells were recovered in SOC media for 1 h at 37°C, plated on LB/agar containing 50 μg/ml kanamycin and either 100 μg/ml spectinomycin (for pDule2 machinery plasmids) or 25 μg/ml chloramphenicol (for pEVOL-AF), and grown overnight at 37°C. The next day, several colonies were scrapped and used to inoculate defined non-inducing media (Supplemental Table S5) supplemented with the same antibiotic combinations. After overnight growth at 37 °C, these cultures were used to inoculate 50 ml of defined auto-inducing media (Supplemental Table S5) with the appropriate antibiotics. Non-canonical amino acids were added to the media at 1 mM final concentration at the time of inoculation. Cultures were grown in 250 ml baffled flasks for ~24 h at 37°C, at which time the cells were harvested by centrifugation and stored at −80°C until protein purification.
sfGFP purification.
Cell pellets containing sfGFP were thawed, resuspended in Lysis Buffer (50 mM Tris pH 7.5, 500 mM NaCl and 5 mM imidazole), and lysed by microfluidization. Soluble cell lysate was obtained by centrifugation at 20,000 × g for 30 min, to which TALON metal affinity resin was added. His6-tagged protein was allowed to bind to the TALON resin for 30–60 min with gentle rocking. Resin was collected and extensively washed with Lysis Buffer, and then protein was eluted with Lysis Buffer supplemented with 300 mM imidazole. Purified sfGFP was buffer exchanged into 25 mM Tris, 150 mM NaCl using PD-10 columns (GE Healthcare), flash frozen in liquid nitrogen and stored at −80°C.
Whole protein mass spectrometry.
Purified protein was exchanged into LC-MS grade water or 50 mM triethyl-ammonium bicarbonate with PD-10 columns, diluted to 50 μM and analyzed with the Waters Synapt G2 Mass Spectrometer at the Mass Spectrometry Facility at Oregon State University. The deconvoluted masses were obtained by using Waters MassLynx MaxEnt1 software.
Benchmark analysis:
7 ncAA/aaRS pairs were used for initial benchmark analysis (PDB IDs: 1J1U, 1ZH0, 1ZH6, 2AG6, 2HGZ, 2PXH, 4NDA). Each of the ligands (Tyrosine:TYR, beta-2-naphthyl-Alanine:NAL, 4-acetyl-Phenylalanine:4AF, 4-boromo-Phenylalanine:4BF, para-benzoyl-Phenylalanine:PBF, 3-(2,2’-bipyridine-5-yl)-L-Alanine:BP5, 3-nitro-Tyrosine:NIY) was parameterized using ligand_to_params_file.py public python script in Rosetta. Different combinations of ncAAs and aaRSs were then prepared by manually aligning the ncAA backbone to exactly match the parent backbone and phenolic ring placement observed in 1J1U:Tyr. Each combination was then relaxed with coordinate constraints using Rosetta relax app (Supplemental Methods). Some of the combinations resulted in huge clashes between the protein and the ncAA and thus were removed from further analysis. In addition, shape complementarity could not be calculated for BP5 and PBF due to undefined atom types; these ncAAs were also removed from analysis.
After preparation of the files, each was scored and the binding interaction between the ncAA and the protein was analyzed with different Rosetta metrics. The “scoring script” is shown in the Supplemental Methods. The same scoring was used for the obtained crystal structures.
GreedyOpt algorithm.
Greedy optimization was performed using GreedyOpt mover in Rosetta running the “Greedyopt algorithm” in Supplemental Methods.
In silico saturation mutagenesis at sites 70 and 158 of the “5B” nitroTyr aaRS.
The 20 × 20 combination of mutations at C70 and S158 site were generated using “In silico library generation script” shown in Supplemental Methods. Each mutation then went through both a fixed and flexible backbone scoring analysis. The score function used for these analyses was beta_genpot.wts due to its superior performance on ligand-protein interactions. The script “In silico flexible backbone analysis of site 70 and 158 library” in Supporting Methods shows the run for flexible backbone. To run with fixed_backbone, the movemap name in FastRelax mover was simply changed to fixed_bb. Average of 25 structures were generated and used for analysis.
In vivo saturation mutagenesis at sites 70 and 158 of the “5B” nitroTyr aaRS.
The pBK plasmid was used for nitroTyr aaRS library selection work, as previously described (Supplemental Fig. S7).[23] To mutagenize sites 70 and 158 of the nitroTyr “5B” aaRS to all combinations of amino acids, primers containing the degenerate NNK codon (N = A/T/C/G and K = G/T) at residues 70 and 158 were designed and synthesized by Integrated DNA Technologies. A DNA fragment encoding the entire aaRS gene and containing 322 = 1024 unique genetic variants (202 = 400 unique full-length protein variants) was created by standard PCR overlap extension protocols and was then inserted into NdeI/PstI digested pBK vector using SLiCE.[42] Following assembly, the library was electroporated into DH10b cells. Transformed cells were recovered for 1 h in SOC, and 50 μL were set aside for plating onto LB/agar plates containing 50 μg/ml kanamycin to determine library assembly efficiency. The remaining SOC recovery culture was added to 100 mL of liquid 2xYT media containing 50 μg/ml kanamycin and grown overnight at 37°C. The next day, ~ 109 cells were passaged into another 100 mL of 2xYT with kanamycin and grown to an OD600 ~1–2. Cells were then harvested by centrifugation and library plasmid DNA was extracted. The resulting pBK-70+158:5B-nitroTyr library plasmid DNA was sequenced to confirm degenerate NNK codons for residues 70 and 158. A total of 40,000 transformants were obtained ensuring complete coverage of the library.
Selection of 3rd-generation “A7” nitroTyr aaRS.
Life/death selections were performed on the Pbk-70+158:5B-nitroTyr library to identify members encoding aaRSs specific for nitroTyr exactly as previously described.[23] Briefly, functional aaRS variants of the pBK-70+158:5B-nitroTyr library were identified by first co-transforming the library plasmid DNA with the positive selection plasmid pCG (which expresses a UAG codon-interrupted chloramphenicol resistance cassette and the cognate Mj tRNACUA) into DH10b cells and plating the recovered cells on LB/agar plates containing kanamycin (50 μg/ml), and tetracycline (25 μg/ml), as well as chloramphenicol (20 μg/ml) and 1 mM nitroTyr to ensure that only cells expressing a functional aaRS survive. After overnight growth, cells were scraped, pooled together and their pBK plasmids were isolated. Next, to remove aaRS variants that conferred survival in this first selection step due to their ability to incorporate natural amino acids, the extracted pBK plasmid DNA was then co-transformed into DH10b cells with a negative selection plasmid (pNEG) that encodes a UAG codon-interrupted, arabinose inducible toxic barnase gene. Cells were grown on LB/agar media with kanamycin (50 μg/ml), chloramphenicol (25 μg/ml) and arabinose (0.2%) but lacking nitroTyr. Surviving cells unable to express a functional barnase gene grew, were scraped and pooled, and final pBK library DNA was extracted. Following this single round of positive and negative selection, the resulting pBK library was co-transformed into DH10b cells with the pALS- sfGFP-150TAG reporter plasmid and plated on defined auto-inducing agar plates containing 1 mM nitroTyr, 50 μg/ml kanamycin and 25 μg/ml tetracycline (Supplemental Table S5). The ninety-six most fluorescent colonies were picked to inoculate 0.5 ml of defined non-inducing media with the same antibiotics, and grown overnight in a 96-well block at 37°C. After overnight growth, these non-inducing cultures were used to inoculate two 96-well blocks containing 0.5 ml defined auto-inducing media in each well; one block contained media lacking nitroTyr while the other was supplemented with 1 mM nitroTyr. In-cell sfGFP fluorescence and culture density (OD600) were measured at 24 and 48 hrs of growth at 37°C. The top sixteen performing hits, as measured by highest normalized fluorescence only in the presence of nitroTyr, were selected for sequencing.
Evaluating amber codon suppression efficiency.
DNA fragments encoding nitroTyr “5B” aaRS variants C70A, S158C, C70A/S158C and “A7” were PCR amplified from their respective pBK plasmids and inserted into the pDule2 machinery plasmid for compatible plasmid pairing with the pET28-sfGFP fluorescent reporter plasmid system and expression in BL21 cell lines (Supplemental Fig. S7). All constructs were confirmed by DNA sequencing.
To characterize aaRS suppression efficiency, pDule2 plasmids expressing the associated nitroTyr aaRS variants were co-transformed with either pET28-sfGFP150TAG or pET28-sfGFP150/134TAG into BL21-ai (RF1-containing cells). Defined non-inducing media starter cultures (3 ml) were inoculated from fresh colonies on agar plates and grown overnight at 37 °C. These cultures were used to inoculate 0.5 ml cultures of defined auto-inducing media supplemented with 0 to 1 mM nitroTyr in 96-well blocks. Each expression culture was grown in triplicate, at 37°C with shaking at 300 rpm. After 24 and 48 h of growth/expression, sfGFP expression was measured by in-cell fluorescence and normalized by culture density (OD600). Reported errors are standard deviations from three replicate cultures.
Expression, purification and crystallization of nitroTyr aaRSs.
DNA fragments encoding the C70A, S158C, C70A/158C and the “A7” nitroTyr aaRSs from pBK plasmids were cloned into pET28 with a C-terminal His6 affinity purification tag for overexpression and purification. Proteins were expressed and purified as previous described [23], with the exception that the proteins were expressed at 18°C (instead of 37°C), and the “A7” aaRS was additionally gel-filtered with an S200 16/60 size exclusion column (GE Healthcare) in 20 mM Tris, 50 mM NaCl, 10 mM β-mercaptoethanol, pH 8.5. The purified proteins were concentrated, frozen in liquid nitrogen and stored at −80°C for subsequent crystallization.
Crystals of all four nitroTyr aaRS proteins were grown by the hanging drop vapor diffusion method by first pre-incubating protein with 1 mM nitroTyr and then mixing 2 μl at 16 – 20 mg/mL (in 20 mM Tris, 50 mM NaCl, 10 mM β-mercaptoethanol, pH 8.5) with 2 μl of reservoir solution containing 22– 23% polyethylene glycol (PEG) 300, 5% PEG 8000, 10% glycerol, and 100 mM Tris pH 8.0–8.3. Crystals grew at room temperature to full size in 4 – 6 days, at which time the crystals were incubated in the same reservoir solution containing 100 mM nitroTyr for 5 min to ensure full occupancy of the substrate and then flash frozen in liquid nitrogen.
Data collection and Refinement of nitroTyr aaRSs.
Data were collected using beamlines 5.0.1, 5.0.2 and 5.0.3 at the Advanced Light Source (Lawrence Berkeley National Laboratory) at a temperature of 100 K. Data were processed and scaled in space group P43212 by XDS, and 5% of the data were randomly flagged for use in Rfree. Data collection statistics are reported in Supplemental Table S3.
Structures were determined by molecular substitution using the previously solved structure of the nitroTyr “5B” aaRS (PDB 4nda) and refined with Phenix. Standard criteria were used for modeling water molecules (>1 ρrms intensity in the 2Fo – Fc map, >2.4 Å distance from nearest contact). Translation/libration/screw (TLS) refinement of B-factors for the C70A, S158C/C70A and “A7” aaRS was performed for each structure using 4 groups (residues 1–77, 78–155, 156–232 and 233–311), while TLS for the S158C aaRS was performed using two groups which roughly correspond to the N- and C-terminal domains (residues 1–194 and 195–311). Additional refinement statistics are reported in Supplemental Table S3.
Expression and purification of human 14-3-3β 130nitroTyr and calmodulin 99nitroTyr.
The gene encoding 14-3-3β (1–240) with a TEV cleavable C-terminal His6 affinity purification tag was codon optimized, synthesized by Integrated DNA Technologies (Coralville, IA) and inserted into NcoI/XhoI digested pET28 by PPY-based SLiCE techniques.[42] The gene encoding human calmodulin (1–149) with a C-terminal His6 tag was codon optimized for expression in E. coli synthesized by Integrated DNA Technologies (Coralville, IA) as described previously[36], and inserted into pET28. Codons encoding residues Y130 of 14-3-3β and Y99 of calmodulin were mutated to TAG for nitroTyr incorporation. These pET28 plasmids were co-transformed with the pDule2-nitroTyr “A7” machinery plasmid into BL21-ai and B-95 cells. After overnight growth on LB/agar plates (with kanamycin and spectinomycin) at 37 °C, multiple colonies were used to inoculate defined non-inducing media culture, which were grown at 37°C for 18 h. Defined auto-inducing cultures were then inoculated with the appropriate strains and grown to an OD ~1.5 at 37°C, at which time the temperature was lowered to 25°C. After 20 h of expression, cells were harvested by centrifugation, frozen in liquid nitrogen and stored at −80°C. 14-3-3β and calmodulin proteins were purified by metal affinity and prepared for whole-protein MS analysis exactly as described above for sfGFP. Additionally, 14-3-3β proteins were TEV-cleaved overnight, and the His6 affinity purification tag and TEV protease were removed by subtractive metal affinity purification.
Supplementary Material
HIGHLIGHTS.
Near-cognate amber codon suppression in “recoded” E. coli expression hosts that lack Release Factor 1 creates a general fidelity problem for Genetic Code Expansion (GCE)
Improving the efficiency of an amino-acyl tRNA synthetase/tRNA pair overcame these near-cognate suppression problems
Improvements were made using Rosetta-guided library design and directed evolution strategies
This new 3-nitro-tyrosine GCE system expands our ability to produce site-specifically nitrated proteins in E. coli
ACKNOWLEDGEMENTS
This work was supported in part by the National Institute of Health [1R01GM114653-01 and 5R01GM131168-02 to R.A.M, 3F32GM120791-01S1 to P.H., and 1S10RR025628-01 to the Oregon State University Mass Spectrometry Facility], the Medical Research Foundation at Oregon Health Sciences University [to R.B.C.], and the Collins Medical Trust [to R.B.C.]. We are grateful for the assistance from Jeff Moore for mass spectrometry data acquisition and processing.
Footnotes
Accession Codes
Coordinates and structure factors for the C70A, S158C, C70A/S158C and “A7” (C70T/S158H) nitroTyr aaRSs have been deposited in the Protein Data Bank under accession numbers 6WRN, 6WRQ, 6WRT and 6WRK, respectively.
Declarations
The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERNCES
- [1].Lajoie MJ, Rovner AJ, Goodman DB, Aerni HR, Haimovich AD, Kuznetsov G, et al. Genomically recoded organisms expand biological functions. Science. 2013;342:357–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mukai T, Hoshi H, Ohtake K, Takahashi M, Yamaguchi A, Hayashi A, et al. Highly reproductive Escherichia coli cells with no specific assignment to the UAG codon. Sci Rep. 2015;5:9699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Johnson DB, Xu J, Shen Z, Takimoto JK, Schultz MD, Schmitz RJ, et al. RF1 knockout allows ribosomal incorporation of unnatural amino acids at multiple sites. Nat Chem Biol. 2011;7:779–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Fredens J, Wang K, de la Torre D, Funke LFH, Robertson WE, Christova Y, et al. Total synthesis of Escherichia coli with a recoded genome. Nature. 2019;569:514–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Heinemann IU, Rovner AJ, Aerni HR, Rogulina S, Cheng L, Olds W, et al. Enhanced phosphoserine insertion during Escherichia coli protein synthesis via partial UAG codon reassignment and release factor 1 deletion. FEBS Lett. 2012;586:3716–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Johnson DB, Wang C, Xu J, Schultz MD, Schmitz RJ, Ecker JR, et al. Release factor one is nonessential in Escherichia coli. ACS Chem Biol. 2012;7:1337–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hong SH, Ntai I, Haimovich AD, Kelleher NL, Isaacs FJ, Jewett MC. Cell-free protein synthesis from a release factor 1 deficient Escherichia coli activates efficient and multiple site-specific nonstandard amino acid incorporation. ACS Synth Biol. 2014;3:398–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zheng Y, Lajoie MJ, Italia JS, Chin MA, Church GM, Chatterjee A. Performance of optimized noncanonical amino acid mutagenesis systems in the absence of release factor 1. Mol Biosyst. 2016;12:1746–9. [DOI] [PubMed] [Google Scholar]
- [9].Randall LM, Dalla Rizza J, Parsonage D, Santos J, Mehl RA, Lowther WT, et al. Unraveling the effects of peroxiredoxin 2 nitration; role of C-terminal tyrosine 193. Free Radic Biol Med. 2019;141:492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhu P, Gafken PR, Mehl RA, Cooley RB. A Highly Versatile Expression System for the Production of Multiply Phosphorylated Proteins. ACS Chem Biol. 2019;14:1564–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ma NJ, Hemez CF, Barber KW, Rinehart J, Isaacs FJ. Organisms with alternative genetic codes resolve unassigned codons via mistranslation and ribosomal rescue. Elife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].George S, Aguirre JD, Spratt DE, Bi Y, Jeffery M, Shaw GS, et al. Generation of phospho-ubiquitin variants by orthogonal translation reveals codon skipping. FEBS Lett. 2016;590:1530–42. [DOI] [PubMed] [Google Scholar]
- [13].Eggertsson G, Soll D. Transfer ribonucleic acid-mediated suppression of termination codons in Escherichia coli. Microbiol Rev. 1988;52:354–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].O’Donoghue P, Prat L, Heinemann IU, Ling J, Odoi K, Liu WR, et al. Near-cognate suppression of amber, opal and quadruplet codons competes with aminoacyl-tRNAPyl for genetic code expansion. FEBS Lett. 2012;586:3931–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Aerni HR, Shifman MA, Rogulina S, O’Donoghue P, Rinehart J. Revealing the amino acid composition of proteins within an expanded genetic code. Nucleic Acids Res. 2015;43:e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mukai T, Lajoie MJ, Englert M, Soll D. Rewriting the Genetic Code. Annu Rev Microbiol. 2017;71:557–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lajoie MJ, Soll D, Church GM. Overcoming Challenges in Engineering the Genetic Code. J Mol Biol. 2016;428:1004–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rauch BJ, Porter JJ, Mehl RA, Perona JJ. Improved Incorporation of Noncanonical Amino Acids by an Engineered tRNA(Tyr) Suppressor. Biochemistry. 2016;55:618–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pirman NL, Barber KW, Aerni HR, Ma NJ, Haimovich AD, Rogulina S, et al. A flexible codon in genomically recoded Escherichia coli permits programmable protein phosphorylation. Nat Commun. 2015;6:8130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gan Q, Fan C. Increasing the fidelity of noncanonical amino acid incorporation in cell-free protein synthesis. Biochim Biophys Acta Gen Subj. 2017;1861:3047–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Steinfeld JB, Aerni HR, Rogulina S, Liu Y, Rinehart J. Expanded cellular amino acid pools containing phosphoserine, phosphothreonine, and phosphotyrosine. ACS Chem Biol. 2014;9:1104–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Neumann H, Hazen JL, Weinstein J, Mehl RA, Chin JW. Genetically encoding protein oxidative damage. J Am Chem Soc. 2008;130:4028–33. [DOI] [PubMed] [Google Scholar]
- [23].Cooley RB, Feldman JL, Driggers CM, Bundy TA, Stokes AL, Karplus PA, et al. Structural basis of improved second-generation 3-nitro-tyrosine tRNA synthetases. Biochemistry. 2014;53:1916–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Muzika M, Muskat NH, Sarid S, Ben-David O, Mehl RA, Arbely E. Chemically-defined lactose-based autoinduction medium for site-specific incorporation of non-canonical amino acids into proteins. RSC Adv. 2018;8:25558–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Miyake-Stoner SJ, Miller AM, Hammill JT, Peeler JC, Hess KR, Mehl RA, et al. Probing protein folding using site-specifically encoded unnatural amino acids as FRET donors with tryptophan. Biochemistry. 2009;48:5953–62. [DOI] [PubMed] [Google Scholar]
- [26].Chin JW, Martin AB, King DS, Wang L, Schultz PG. Addition of a photocrosslinking amino acid to the genetic code of Escherichiacoli. Proc Natl Acad Sci U S A. 2002;99:11020–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Blizzard RJ, Backus DR, Brown W, Bazewicz CG, Li Y, Mehl RA. Ideal Bioorthogonal Reactions Using A Site-Specifically Encoded Tetrazine Amino Acid. J Am Chem Soc. 2015;137:10044–7. [DOI] [PubMed] [Google Scholar]
- [28].Yanagisawa T, Ishii R, Fukunaga R, Kobayashi T, Sakamoto K, Yokoyama S. Multistep engineering of pyrrolysyl-tRNA synthetase to genetically encode N(epsilon)-(o-azidobenzyloxycarbonyl) lysine for site-specific protein modification. Chem Biol. 2008;15:1187–97. [DOI] [PubMed] [Google Scholar]
- [29].Bick MJ, Greisen PJ, Morey KJ, Antunes MS, La D, Sankaran B, et al. Computational design of environmental sensors for the potent opioid fentanyl. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Tinberg CE, Khare SD, Dou J, Doyle L, Nelson JW, Schena A, et al. Computational design of ligand-binding proteins with high affinity and selectivity. Nature. 2013;501:212–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dou J, Vorobieva AA, Sheffler W, Doyle LA, Park H, Bick MJ, et al. De novo design of a fluorescence-activating beta-barrel. Nature. 2018;561:485–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Baumann T, Hauf M, Richter F, Albers S, Moglich A, Ignatova Z, et al. Computational Aminoacyl-tRNA Synthetase Library Design for Photocaged Tyrosine. Int J Mol Sci. 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Fan Z, Song J, Guan T, Lv X, Wei J. Efficient Expression of Glutathione Peroxidase with Chimeric tRNA in Amber-less Escherichia coli. ACS Synth Biol. 2018;7:249–57. [DOI] [PubMed] [Google Scholar]
- [34].Zhao Y, Zhang Y, Sun H, Maroto R, Brasier AR. Selective Affinity Enrichment of Nitrotyrosine-Containing Peptides for Quantitative Analysis in Complex Samples. J Proteome Res. 2017;16:2983–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Nuriel T, Whitehouse J, Ma Y, Mercer EJ, Brown N, Gross SS. ANSID: A Solid-Phase Proteomic Approach for Identification and Relative Quantification of Aromatic Nitration Sites. Front Chem. 2015;3:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Porter JJ, Jang HS, Haque MM, Stuehr DJ, Mehl RA. Tyrosine nitration on calmodulin enhances calcium-dependent association and activation of nitric-oxide synthase. J Biol Chem. 2020;295:2203–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Porter JJ, Jang HS, Van Fossen EM, Nguyen DP, Willi TS, Cooley RB, et al. Genetically Encoded Protein Tyrosine Nitration in Mammalian Cells. ACS Chem Biol. 2019;14:1328–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Porter JJ, Mehl RA. Genetic Code Expansion: A Powerful Tool for Understanding the Physiological Consequences of Oxidative Stress Protein Modifications. Oxid Med Cell Longev. 2018;2018:7607463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Tomita H, Katsuyama Y, Minami H, Ohnishi Y. Identification and characterization of a bacterial cytochrome P450 monooxygenase catalyzing the 3-nitration of tyrosine in rufomycin biosynthesis. J Biol Chem. 2017;292:15859–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ren W, Truong TM, Ai HW. Study of the Binding Energies between Unnatural Amino Acids and Engineered Orthogonal Tyrosyl-tRNA Synthetases. Sci Rep. 2015;5:12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Blizzard RJ, Gibson TE, Mehl RA. Site-Specific Protein Labeling with Tetrazine Amino Acids. Methods Mol Biol. 2018;1728:201–17. [DOI] [PubMed] [Google Scholar]
- [42].Zhang Y, Werling U, Edelmann W. SLiCE: a novel bacterial cell extract-based DNA cloning method. Nucleic Acids Res. 2012;40:e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.