

Abstract
Emerging from the origins of supramolecular chemistry and the development of selective chemical receptors that rely on lock-and-key binding, activity-based sensing (ABS)–which utilizes molecular reactivity rather than molecular recognition for analyte detection–has rapidly grown into a distinct field to investigate the production and regulation of chemical species that mediate biological signaling and stress pathways, particularly metal ions and small molecules. Chemical reactions exploit the diverse chemical reactivity of biological species to enable the development of selective and sensitive synthetic methods to decipher their contributions within complex living environments. The broad utility of this reaction-driven approach facilitates application to imaging platforms ranging from fluorescence, luminescence, photoacoustic, magnetic resonance, and positron emission tomography modalities. ABS methods are also being expanded to other fields, such as drug and materials discovery.
Keywords: activity-based probes, bioconjugation chemistry, bio-orthogonal reactions, molecular imaging, sensors
Graphical Abstract
1. Introduction
The development and application of new sensors to visualize dynamic chemical species with high selectivity and sensitivity is a powerful approach to help decipher the contributions of small molecules and metals in living systems at a molecular level.[1] In this context, one prominent and broad class of chemosensors are for reactive small-molecule species in the oxygen,[2-5] nitrogen,[4, 6] carbon,[7, 8] and sulfur[9-11] families, which are actively produced and metabolized in a diverse array of native biological processes and play dichotomous signal and/or stress roles in both healthy and diseased states.[12-14] Another important class of chemical species in biology are metals, which play key roles as structural and metabolic cofactors within proteins and nucleic acids, redox sources in respiration and metabolism, and in generating transient ion fluxes mediated by channels, transporters, and allosteric enzymes that contribute to cell homeostasis and physiology.[15, 16]
In this Review, we provide an overview of the field of activity-based sensing (ABS), starting with a historical perspective on the development of ABS originating from supramolecular chemistry and binding-based methods for sensing and imaging in biological environments. We then lay out the principles of ABS, emphasizing reaction development with a view towards selectivity and biocompatible probe design, summarizing the major types of chemical reactions that have been employed to selectively detect analytes in biological environments. We follow with a discussion on select applications of ABS across various imaging modalities that enable the use of these reagents in native biological settings. We then conclude by discussing current and future opportunities for ABS methods applied to new avenues of research, including functionalized materials, therapeutics, bioconjugation, and chemoproteomics. Owing to space limitations and the rapid growth of the field, our intent is not to provide an all-inclusive cataloged list of ABS methods, but rather to provide a foundation and motivation for further development by highlighting key conceptual and practical advances that have pushed the field forward. We restrict our discussion to synthetic small-molecule ABS indicators, noting that proteins, nucleic acids, and other synthetic and biological polymers are also viable platforms for this approach. We refer readers to other leading reviews that summarize various aspects of activity-based sensors.[4, 17-23]
2. Historical Origins of Activity-Based Sensing
2.1. From Supramolecular Chemistry to Binding-Based Sensing (BBS)
The origins of ABS stem from the development of supramolecular chemistry, starting with the lock-and-key recognition complexes pioneered by Pedersen (crowns), Lehn (cryptands), Cram (spherands), and their co-workers in the 1960s and 1970s (Figure 1). This work was recognized by the 1987 Nobel Prize in Chemistry and launched the field of molecular recognition, where receptors to bind specific classes of metals, other ions, and small molecules were created by modifying the structure, intermolecular interactions, and stereoelectronic properties to impart binding selectivity. In 1967 Pedersen reported cyclic polyethers, more commonly referred to as crown ethers, as the first synthetic molecules capable of forming stable complexes with cationic metals and oxygen atoms through ion–dipole interactions.[24, 25] Realization of the initial crown ether complex and further studies relied on optical sensing with ultraviolet/visible (UV/Vis) spectroscopy to characterize the binding of aryl phenols that undergo a sharpening of their characteristic absorption band at 275 nm and development of a second band at 281 nm upon complexation with the ion. Pedersen expanded the size and coplanarity of the ligand interior and modulated the number of coordinating aliphatic and aromatic oxygen atoms to enact selective binding of ions, including Li, Na, K, Cs, Ag, Ca, Cd, Hg, and Pb metal ions in various oxidation states, as well as charged nitrogen species.
Figure 1.

Historical origins of activity-based sensing (ABS). Key advances in the development of the field of ABS stemming from the evolution of fundamental supramolecular host–guest chemistry of crown ethers, cryptands, and spherands to BBS of calcium and other metal ions. Likewise, the advent of chemodosimeters led to the concept of ABS, where the development of synthetic methods can be leveraged in a retrosynthetic manner to cage and uncage fluorescent dyes and other molecular imaging agents for the detection of biological analytes by using them for selective protection-deprotection chemistry.
Expanding upon the work of Pedersen, in a seminal 1969 report, Lehn, Dietrich, and Sauvage developed three-dimensional cavities, termed cryptands, for the recognition of a variety of analytes.[26] Macrocyclic topology increases the stability and retention of the substrate, leading to enhanced binding and selectivity.[27] Cryptands increased the selectivity of the ligand inclusion complexes by modulating the cavity size to create reporters for cations,[28] anions,[29] and small molecules.[30] In the next decade, Cram et al. advanced the field further through the development of spherands, enhanced by the design of a preorganized ligand complex to achieve higher selectivity.[31] The rigidity of complexation ligands supported by a covalent network allows unprecedented control for creating spherical coordination sites.[32, 33] Artz and Cram expanded the field to create derivatives such as hemispherands,[34] and Gutsche et al. pioneered the development of calixeranes,[35, 36] which can recognize and bind a variety of chemical species. Modern methods of supramolecular chemistry continue to expand and create new molecules to develop advanced materials,[37] improve upon recognition capabilities,[38] and apply these techniques to biological and bio-inspired sensing.[39] Indeed, the 2016 Nobel Prize shared by Sauvage,[40] Stoddart,[41] and Feringa[42] for the design and synthesis of molecular machines is a prime example of new function derived from supramolecular chemistry, namely the discovery of the mechanical bond as a fundamentally new type of chemical linkage.
In the late 1970s and continuing throughout the 1980s, Tsien carved out a new path for synthetic lock-and-key methods by realizing their potential in biology, as illustrated by his pioneering work on the development of selective chelators,[43] and later fluorescent sensors[44] and photocaged compounds,[45, 46] for the detection and regulation of calcium, a central signaling metal in living systems. We denote these types of probes as operating through a binding-based sensing (BBS) mechanism.[23] Specifically, Tsien developed 1,2-bis(o-aminophenoxy)ethane-N,N,-N′,N′-tetraacetic acid (BAPTA), an expanded version of the canonical metal chelator ethyl-enediaminetetraacetic acid (EDTA) but with an extended diether backbone to make a tetracarboxylate binding site that would favor formation of 8-coordinate CaII complexes.[47] Moreover, because BAPTA was generally adaptable to incorporation onto fluorescent scaffolds, Tsien and co-workers were able to create metal-sensing platforms that respond to metal binding by either a turn-on increase in the fluorescence[48, 49] or a ratiometric shift in the excitation/emission wavelengths[44] before and after metal binding. Further optimizations of the chelator enabled selective imaging of CaII with 4-fold, 12-fold, and 40-fold selectivity for MgII, MnII, and ZnII respectively. In particular, the ratiometric calcium sensor Fura-2 enabled the study of calcium in systems previously incompatible with sensing platforms, and this work launched the field of BBS for a wide range of biological analytes, including alkali, alkaline-earth, other physiologically relevant cations,[50] and transition-metal ions,[18] as well as a host of applications in molecular sensors and molecular logic gates.[51] More recent directions in synthetic methods for BBS have focused on fluorescent probes for copper,[18, 52-56] zinc,[57, 58] and other essential metal nutrients.[23, 59, 60]
2.2. From Chemodosimeters to Activity-Based Sensing (ABS)
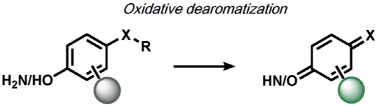
As the application of supramolecular molecular recognition principles led to the development of selective binding-based calcium sensors for widespread biological use, work in the 1990s by Czarnik on chemodosimeters in abiotic contexts led to the development of activity-based sensors for use in living systems. (Figure 1).[61, 62] Chemodosimeters utilize tailored chemical reactivity, rather than binding or recognition-based approaches, to selectively measure classes of organic or inorganic analytes in a dose-dependent manner. In a seminal 1992 report, Chae and Czarnik used the transformation of thioamide-functionalized anthracene for the detection of thiophilic heavy metals.[63] Hydrolysis of the fluorescencequenching thioamide to the fluorescent amide induced by thiophilic metals such as HgII and AgI results in a dose-dependent, highly selective turn-on fluorescence response. Another key advance was realized by Nagano and co-workers in the late 1990s by creating dosimeters for measuring specific classes of biological analytes, which we term biodosimeters. This work culminated in the design and synthesis of probes for reactive nitrogen species (RNS) that exploit the oxidative reactivity of nitric oxide (NO) in the presence of dioxygen to form triazenes from vicinal diamines, an expansion on work carried out by Isom and co-workers.[64, 65] Diaminofluorescein (DAF)[65] and diaminorhodamine (DAR)[66] probes bearing a diaminophenyl group on the bottom ring of a xanthone core elicit a turn-on fluorescence response through a diamine-to-triazene transformation in the presence of NO and oxygen. Stimulation of rat muscle cells through cytokine activation and inhibition revealed fluorescence changes in a RNS-dependent manner with high sensitivity; the use of DAF, DAR, and congeners have unveiled new NO biology.[67-69]
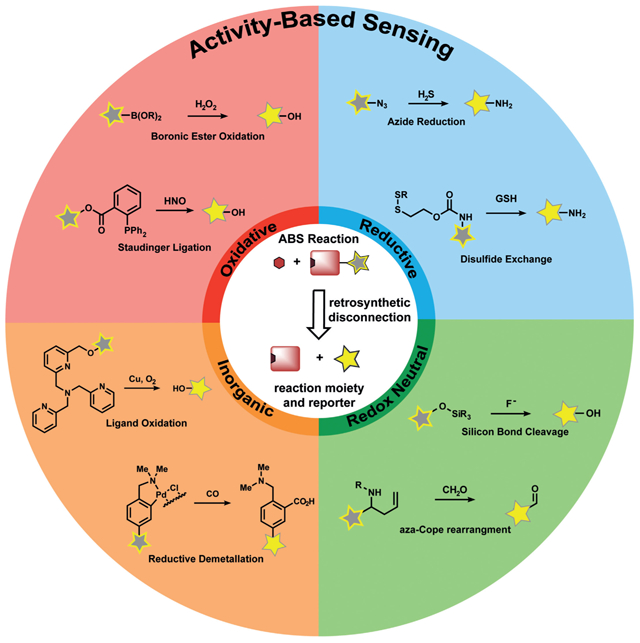
Against this backdrop, our group initiated a program in the early 2000s to create new types of sensor platforms by uncaging a fluorophore through a chemospecific reaction, which we now term activity-based sensing (Figure 2a). ABS reagents can be designed by retrosynthetic disconnection of a reporter modality into an analyte-responsive moiety and an analyte-specific chemotype (Figure 2b). A first-generation example of this strategy is Peroxyfluor-1 (PF1), where the hydrogen peroxide mediated oxidation of boronates to phenols generates fluorescein as the product from a caged diboronate precursor.[70] Indeed, the chemoselective oxidation of PF1 by hydrogen peroxide (H2O2), but not metabolites from other reactive oxygen species (ROS), can proceed in aqueous solution and in living cells, thereby providing the means to report on this important oxygen metabolite with molecular-level specificity in a variety of biological settings. Most importantly, the boronate reaction can be used with other scaffolds, thus providing a general design strategy for the detection of H2O2 and, by extension, other biological analytes, by an ABS method. Figure 1 depicts a timeline linking key advances in supramolecular host–guest chemistry to binding-based sensors and chemodosimeters to ABS sensors. In the following sections, design principles and selected examples to highlight progress and prospects in the ABS field are provided.
Figure 2.

a) General scheme for analyte detection using ABS. The analyte binds to a reactive moiety covalently or noncovalently, thereby Initiating a subsequent reaction to modulate sensing with an imaging modality. b) ABS probes may be designed by linking a reporter motif to a moiety which is sensitive to the chemical reactivity of the analyte. c) A workflow approach to the design of ABS probes.
3. Design Principles for Activity-Based Sensing
The design of effective probes for ABS must meet several criteria; most importantly a chemoselective reaction trigger must be devised that can operate under biologically compatible conditions (Figure 2). Drawing from concepts in supramolecular chemistry,[71] BBS,[48] chemodosimetry,[63] and bio-orthogonal chemistry,[72] ABS offers a unique strategy for analyte detection owing to its focus on molecular reactivity, rather than molecular recognition, to achieve specificity since only the desired analyte can facilitate the targeted chemical transformation. It is this dynamic nature of ABS reactions that offers opportunities to apply retrosynthetic analysis, physical organic chemistry, and other tools and tactics of synthesis in this methods development problem for chemical biology (Figure 2).
A key feature of ABS is to separate the analyte-sensing module from the imaging modality to impart selectivity through chemical reactivity. In a typical ABS probe, a specific biological analyte is reversibly bound to a reactive modality on an imaging platform, which triggers a subsequent irreversible chemical reaction if the analyte reactivity is matched to the reactive modality. This reaction cleaves, adds, and/or modulates the imaging platform in some way to elicit a measurable signal change. In this way, analyte sensing can then be widely applied to a variety of imaging modalities and therapeutic platforms, since a chemically responsive group, rather than a relatively limited chemical modulation of a specific sensing platform, is being exploited to sense an analyte.
The ABS concept is broad by definition and can potentially encompass any biocompatible chemical reaction. A host of organic, organometallic, and inorganic transformations that have traditionally been used in chemical synthesis can be repurposed and/or modified to sense analytes which are present in biological systems, and new reactions can also be developed for ABS purposes. The following sections provide select examples of synthetic reactions which have been employed for ABS, and highlight applications of these synthetic methods to imaging, materials, therapeutics, and proteomics. We will focus on ABS probes for diffusible small-molecule and metal analytes, noting that such approaches for interrogating enzyme and nucleic acid activity can be applied to expand the ABS repertoire.[73] Recent examples include work from Dickinson and co-workers to monitor the depalmitoylation activity of acylprotein thioesterases,[74] a study by Sessler, Shabat, Kim, and co-workers to accelerate cleavage of a biomimetic electron reservoir for detection of NADPH: quinone oxidoreductase,[75] and a report by the Urano and Kamiya groups on prostate-specific membrane antigen (PSMA) carboxypeptidase.[76]
4. Reaction Classes Employed for Activity-Based Sensing
The foundation for the ABS approach is the development of synthetic methods. In the following sections, organic ABS probes are categorized with respect to their change of oxidation state (defined with respect to the probe) as oxidative, reductive, or redox-neutral. ABS probes that employ inorganic/organometallic reactivity are presented together in a fourth section. The present section will describe chemical reactions developed to create ABS reagents, with a brief mention of the imaging modalities utilized to demonstrate the utility of the synthetic method. Selectivity among structurally similar analytes is generally obtained by leveraging slight differences in chemical reactivity, physical organic chemistry principles, and experimentation. Readers are directed towards the reviews and papers on specific analytes for further details. More in-depth discussions on imaging and other applications will be described in subsequent sections.
4.1. Oxidative Organic Reactions
The first set of ABS methods utilized oxidative organic reactions that were well-matched to sense the contributions of reactive oxygen species (ROS) and reactive nitrogen species (RNS) in the regulation of physiology and pathology (Tables 1 and 2). The diverse chemistry, transient nature, and propensity of such species to dynamically interconvert between ROS/RNS families highlight the need for new methods to decipher their individual contributions to living systems. In this context, a formative example of ABS was the development of H2O2 probes by our group through the H2O2-mediated transformation of arylboronates into phenols,[70] a reaction that was first reported in the 1950s (Table 1).[77] Subsequent work has shown that peroxynitrite (ONOO−) can also perform this oxidation on certain fluorescent scaffolds,[78, 79] but has a much shorter half-life (t1/2(H2O2) = 1 ms[80] and t1/2(ONOO−) = 0.8 ns[81]) and exists at lower concentrations than H2O2 ([H2O2] = 100 nm and [ONOO−] = 10 nm[82]) during normal physiology. Control experiments using an inhibitor or siRNA/CRISPRi for nitric oxide synthase is a simple way to distinguish H2O2 from ONOO− contributions.[83, 84] By caging various phenols and amines with an arylboronate ABS trigger, the functionalization of a diverse range of sensing platforms enables the sensitive and selective detection of H2O2 in biological environments by a variety of imaging modalities.[2, 5, 85] The wide applicability of the arylboronate trigger has spawned application to fluorescent scaffolds with higher sensitivity,[83, 86] ratiometric calibration,[87-89] various excitation/emission colors,[90-93] subcellular H2O2 sensing,[94-97] lanthanide luminescence,[98] bioluminescence,[99, 100] positron emission tomography,[101] and histochemical platforms.[84] Our group also employed the H2O2-dependent oxidation of α-keto acids to benzoic acid for hyperpolarized 13C NMR imaging of peroxide,[102] and Nagano and co-workers developed a benzil trigger that undergoes a Baeyer–Villiger-type reaction to form benzoic anhydride, which undergoes a subsequent hydrolysis to a benzoic acid upon reaction with H2O2.[103]
Table 1:
Selected ABS methods using oxidative organic reactions (A).
| Primary Reaction | Associated Secondary Reactions |
Analytes | Associated Imaging Modalities |
Seminal Refs./Reviews |
|---|---|---|---|---|
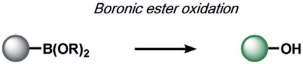 |
vinylogous elimination | H2O2 | fluorescence, chemiluminescence, bioluminescence, immunofluorescence, PET, histochemical | 70, 78, 79 |
| ONOO− | ||||
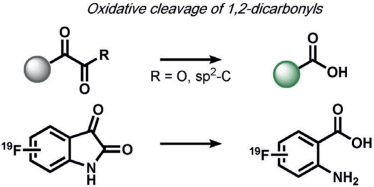 |
hydrolysis, oxidative benzylic acid rearrangment, Baeyer-Villiger rearrangment | H2O2 | fluorescence, NMR | 102, 103 |
| ONOO− | 122 | |||
 |
hydrolysis, vinylogous elimination | HOCl | fluorescence | 104, 105 |
| NO, Hg2+ | 119 | |||
 |
– | HOCl | fluorescence | 106 |
 |
hydrolysis | HOCl | fluorescence | 109, 110 |
 |
– | HOCl | fluorescence | 111, 112 |
 |
– | HOCl | photoacoustic imaging | 113 |
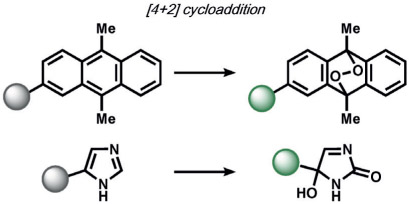 |
Kornblum-DeLamare fragmentation | 1O2 | fluorescence | 114, 115 |
Table 2:
Selected ABS methods using oxidative organic reactions (B).[a]
| Primary Reaction | Associated Secondary Reactions |
Analytes | Associated Imaging Modalities |
Seminal Refs./Reviews |
|---|---|---|---|---|
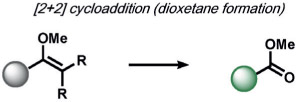 |
(formal) retro [2+2] cycloaddition | 1O2 | chemiluminescence | 116 |
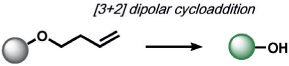 |
Criegee fragmentation, b-elimination | O3 | fluorescence | 117 |
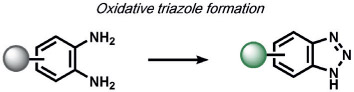 |
dehydration | NO+O2 | fluorescence | 65 |
 |
dehydration, electrophilic aromatic substitution | NO+O2 | fluorescence | 118 |
 |
5-exo-tet cyclization, hydrolysis | ONOO− | fluorescence | 120, 121 |
 |
5-exo-trig cyclization, trans-esterification | HNO | fluorescence | 123 |
 |
dehydration, 5-exo-trig cyclization | HNO | fluorescence, chemiluminescence | 124 |
 |
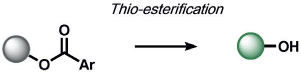
5-exo-trig cyclization, thio-esterification | RS-(S)n-SR, H2S2, or S8 | fluorescence | 128 |
| HSnR (n>1); RSH, S8 | 129 |
EDG = electron-donating group.
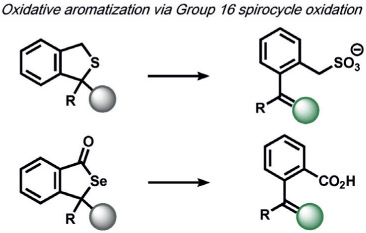
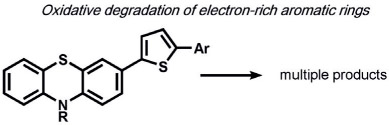
ABS approaches can be tailored to achieve specificity for other ROS. For example, for the detection of hypochlorous acid (HOCl), Nagano and co-workers have leveraged HOCl-dependent spirolactam thiol oxidation to create HySOx and related fluorescent probes by modulating the open and closed forms of fluorescent rhodamines (Table 1).[104, 105] Liu and Wu reported related work on selenium oxidation,[106] which led to a number of fluorescent probes based on selenium and tellurium oxidation.[107, 108] The oxidation of p-methoxyphenol and related aryl ethers by HOCl was exploited by the Nagano[109] and Yang[110] groups to create benzoquinone-responsive colorimetric and fluorescent indicators. Deoximation reactions were utilized by Tan and co-workers for the ratiometric fluorescence imaging of HOCl,[111] while Yang and co-workers used a related reaction for cleavage of diamino-maleonitrile.[112] The Pu group exploited the HOCl-mediated oxidation of phenothiazine to induce nanoparticle degradation for the generation of ratiometric acoustogenic sensors.[113] Nagano and co-workers utilized the cycloaddition of singlet oxygen (1O2) and 9,10-dimethylanthracene to modulate the fluorescent on/off states of fluorescein (Table 1)[114] The Tang group reported a related oxidative 1,4-cycloaddition of histidine and 1O2 (Table 2).[115] Shabat and co-workers described an innovative enol-ether oxidation to create a chemiluminescent dioxetane product.[116] The Koide group has reported a unique ABS method for the detection of ozone (O3) through the reaction of O3 with olefins to form molozonide and a resulting aldehyde, which then undergoes a β-elimination to yield a fluorescent compound.[117]
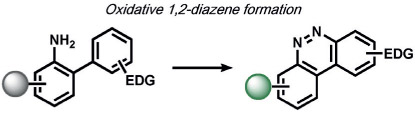
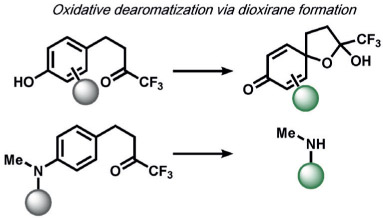
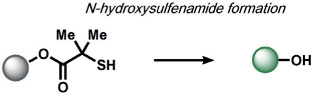
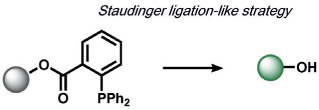
In the area of RNS detection, building on the vicinal diamine motifs for aerobic NO oxidation,[65] the Shear and Anslyn groups reported an N-nitrosoaniline oxidation that undergoes a reaction cascade after exposure to NO to yield a fluorescent diazene ring system. This is a clever example of utilizing a reactive analyte for the in situ synthesis of a fluorophore in living systems (Table 2).[118] Ma and co-workers also reported a selenolactone ring-opening oxidation upon formation of a Se─NO bond, which they used to create fluorescent probes for the detection of NO.[119] The Yang group developed a versatile ABS method for the detection of ONOO− by the oxidation of anisole-activated ketones to dioxiranes.[120] Second-generation trifluoroketone triggers undergo ONOO−-induced N-dearylation reactions with improved selectivity and sensitivity.[121] Lippert and co-workers developed a general isatin-based oxidative decarbonylation reaction to selectively detect ONOO− using 19F MRI (Table 1).[122] Finally, several ABS methods have been reported for the detection of nitroxyl (HNO; Table 2). Chan and co-workers developed a thiol sulfenamide ABS trigger that is converted in the presence of HNO into an N-hydroxysulfenamide, which undergoes an intramolecular cyclization to yield the free reporter molecule.[123] The reaction between HNO and phosphines, based on the Staudinger ligation reaction pioneered by Bertozzi and Saxon for bio-orthogonal chemistry[124] and expanded by the Hackenberger group and others,[125, 126] has also been exploited for the development of fluorescent and chemiluminescent ABS by the Lin[6] and Lippert[127] groups, respectively.
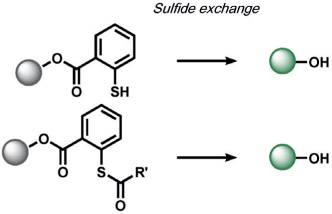
Xian and co-workers developed a selective sulfane-sulfur (composed of persulfide, polysulfide, and elemental sulfur) induced benzodithiolone formation from a mercaptobenzoic acid in live cells (Table 2).[128] The same group detected hydrogen polysulfides (H2Sn) by utilizing 2-fluoro-5-nitrobenzoic esters as the trigger, which undergoes a similar benzodithiolone formation.[129]
4.2. Reductive Organic Reactions
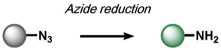
Reductive reactions are underexplored compared to their oxidative counterparts, but still constitute an important type of reactivity that can be leveraged for the selective detection of biological analytes (Table 3). The H2S-mediated reduction of azides to amines has emerged as a useful reaction for the development of ABS probes and benefits from the privileged nature of organic azides as bio-orthogonal groups in chemical biology[10] as well as the facile installation of azides onto amine scaffolds.[10, 130] Our group[131] and Wang and co-workers[132] published the first reports of azide-based fluorescent probes for H2S reduction. We reported Sulfidefluors 1 and 2 (SF1/SF2),[131] where H2S transforms azide-derivatized rhodamines into the parent rhodamine dyes with high specificity over other biological thiols as a result of the smaller size and higher acidity of H2S (pKa = 7) compared with common thiols such as cysteine/glutathione (pKa = 8–9). In parallel, Wang and co-workers reported a dansyl azide analogue for the detection of H2S,[132] with a 2,6-dansyl azide structural isomer showing improved solubility and sensitivity.[133] We expanded the SF family to bisazido derivatives to lower the background signal, and added ester trapping groups to increase cell permeability and retention;[134] similar derivatives were reported by Sun and co-workers.[135] Indeed, H2S-mediated azide reduction has been applied to a diverse array of fluorophore scaffolds[9, 10] including coumarin,[136-138] fluorescein,[134] rhodol,[139, 140] BODIPY,[141, 142] and 1,8-naphthalimide[143] dye classes, which are commonly utilized for cellular imaging, as well as 2-(2-aminophenyl)benzothiazole,[144] dicyanomethylenedihydrofuran,[145] and resorufamine[146] platforms. In addition, NIR and two-photon H2S imaging can be achieved using dicyanomethylene-4H-chromene[147, 148] and tetraphenylethene,[149] with ratiometric behavior displayed in some cases. Chemi-[150] and bioluminescent[151] azide probes have also been reported, which circumvent potential issues of off-target photoactivation of azides with a high light intensity. In addition to the reduction of azides, Montoya and Pluth developed the H2S-mediated reduction of nitro groups to amines as a complementary ABS method to create H2S sensors.[143] Owing to the potential sensitivity of the nitro functional group towards endogenous reductases in cells, appropriate controls must be employed to confirm that H2S production is responsible for the signal changes.
Table 3:
Selected ABS methods using reductive organic reactions.
| Primary Reaction | Associated Secondary Reactions |
Analyte | Associated Imaging Modalities |
Seminal Refs./Reviews |
|---|---|---|---|---|
 |
vinylogous elimination, 5-exo-trig cyclization, esterification | H2S | fluorescence, chemiluminescence, bioluminescence | 134 |
| vinylogous elimination | H2S | fluorescence | 143 | |
 |
5-exo-trig cyclization | glutathione | fluorescence | 153 |
 |
− | selenocysteine | fluorescence | 154 |
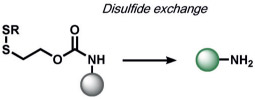
Glutathione (GSH) is a major redox buffer in cells and can reduce disulfide bonds.[152] Exploiting this native chemical reactivity, Pires and Chmielewski created an innovative disulfide trigger to yield nucleophilic sulfhydryl groups.[153] Juxtaposition of the sulfhydryl groups with carbamate bonds causes intramolecular breakdown to yield free anilines, which the authors applied to create fluorescent rhodamine products. These probes can detect changes in GSH levels in cells.
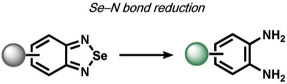
A final example from Tang and co-workers for the detection of the rare amino acid selenocysteine utilized a benzeneselenadiazol reduction to cleave a pair of Se─N bonds to yield a vicinal diamine, which they applied to modulate the fluorescence of a hemicyanine dye for detection in living cells.[154]
4.3. Redox-Neutral Organic Reactions
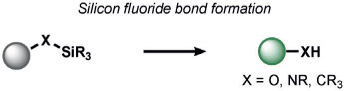
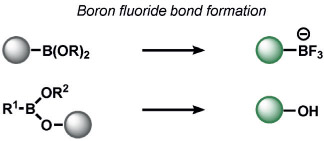
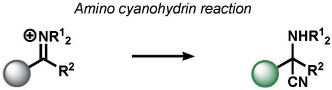
Redox-neutral transformations constitute a major fraction of ABS methods for a broad range of analytes (Tables 4-7). Most of these methods exploit nucleophile-electrophile reactions to elicit a signal change. A leading example of a redox-neutral organic reaction is the detection of fluoride through the fluoride-mediated deprotection of silyl alcohols (Table 4). Yoon and co-workers have reviewed this work extensively;[155] most probes rely on the selective cleavage of Si─X (X = C, N, O) bonds by fluoride to modulate fluorescent scaffolds.[156, 157] Fluoride detection with these methods is compatible with living organisms and selective over other nucleophiles. Other common nucleophilic anions such as cyanide, derived from hydrogen cyanide (HCN), can be sensed with the fluorescent spiropyran probes developed by Shiraishi et al. In this case, the spirocarbon atom serves as an electrophile for CN─ in an amino-cyanohydrin reaction.[158] Although the probe is selective to a number of biological nucleophiles, it was not evaluated in living systems. Sessler and co-workers reported 1-methylenepyyrolidinium electrophiles for HCN. These had similar selectivity profiles and were capable of probing changes in HCN concentrations in the mitochondria of living cells.[159]
Table 4:
Selected ABS methods using redox-neutral organic reactions (A).[a]
| Primary Reaction | Associated Secondary Reactions |
Analytes | Associated Imaging Modalities |
Seminal Refs./Reviews |
|---|---|---|---|---|
 |
vinylogous elimination | F− | fluorescence | 155 |
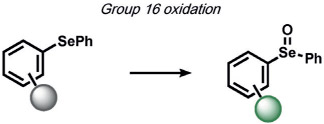
 |
vinylogous elimination | F− | fluorescence | 156, 157 |
 |
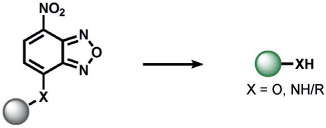
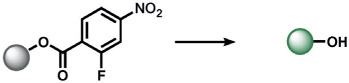
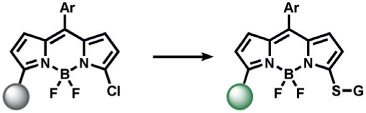
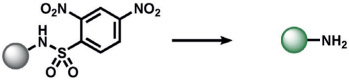
spiro-aminal opening | CN− | fluorescence | 158 |
 |
hemithioacetal formation, 5-exo-trig cyclization, thioesterification | H2S | fluorescence | 161, 162 |
 |
5-exo-trig cyclization, 1,4-conjugate addition | H2S | fluorescence | 163 |
EWG = electron-withdrawing group.
Table 7:
Selected ABS methods using redox-neutral organic reactions (D).
| Primary Reaction | Associated Secondary Reactions |
Analytes | Associated Imaging Modalities |
Seminal Refs./Reviews |
|---|---|---|---|---|
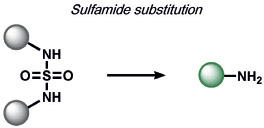 |
− | glutathione (G-SH) | fluorescence | 180 |
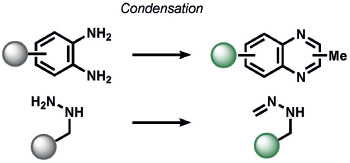 |
aromatization | fluorescence | 185 | |
| − | CH2O | 192 | ||
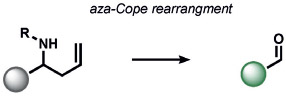 |
b-elimination, condensation, hydrolysis | CH2O | fluorescence, chemiluminescence, PET | 186, 187 |
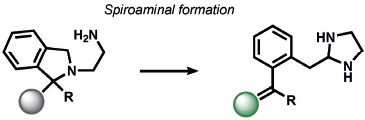 |
condensation, aromatization | CH2O | fluorescence | 193 |
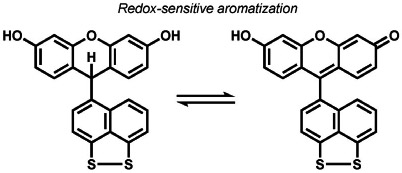 |
proton transfer, reversible disulfide cleavage | e− | fluorescence, magnetic resonance imaging | 194, 195, 208 |
Interestingly, many redox-neutral ABS methods have been developed to detect redox-active analytes, most commonly by exploiting analyte nucleophilicity. An early non-oxidative H2O2 ABS method was developed by Miyamoto and co-workers based on a nucleophilic aromatic substitution (SNAr) mechanism, where H2O2 reacts with a pentafluorobenzenesulfonyl trigger to unmask fluorophores (Table 5).[160]
Table 5:
Selected ABS methods using redox-neutral organic reactions (B).
| Primary Reaction | Associated Secondary Reactions |
Analytes | Associated Imaging Modalities |
Seminal Refs./Reviews |
|---|---|---|---|---|
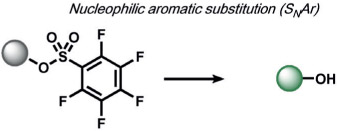 |
hemi-sulfite formation, 5-endo-dig cyclization, 5-exo-trig cyclization, vinylogous elimination | H2O2 | fluorescence | 160 |
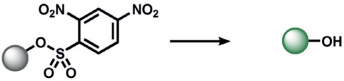 |
H2S | 164, 165 | ||
 |
H2S | 167 | ||
 |
H2Sn (n > 1) | 172 | ||
 |
glutathione (G-SH) | 175 | ||
 |
selenocysteine | 184 | ||
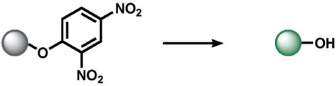 |
selenocysteine | 184 |
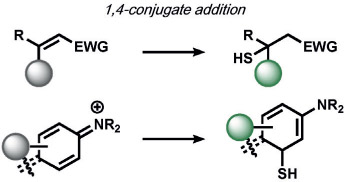
In parallel, several types of redox-neutral reactions have been applied to sense nucleophilic reducing molecules such as H2S. Zhao, He, and co-workers utilized an innovative tandem 1,4-conjugate addition of H2S to an aldehyde to form a hemithioacetal.[161] Selectivity is achieved by the juxtaposition of an α,β-unsaturated acrylate methyl ester and aldehyde to form a stable thioacetal product, a sequence which does not occur for other reactive sulfur species (RSS). Another creative ABS approach by Xian and co-workers to determine H2S exploits the dual reactivity of H2S to cleave disulfide[162] and ester[163] electrophiles, thereby allowing triggers based on thioester reactions to be incorporated onto fluorescent scaffolds for high selectivity. Likewise, He and co-workers explored the SNAr reaction of 2,4-dinitrophenyl (DNP) triggers to sense H2S, although DNP can have potential cross-reactivity with other RSS and related nucleophiles (Table 5).[164, 165] In a similar manner, SNAr reactions of nitrobenzofurans,[166] 7-nitrobenz-2-oxa-1,3-diazole,[167] phenanthroimidazole,[168] cyanine,[169] and nitro triggers[170, 171] have been explored.
The redox-neutral reactions developed by the Xian group for the detection of H2Sn utilized a 2-fluoro-5-nitrobenozic ester trigger (Table 6).[172] Selectivity for H2Sn was achieved by introducing bis-electrophilic groups (the fluorobenzoic ester and a fluorescent scaffold) that will only cleave if two nucleophilic residues are present in the biological analyte. The group also discovered that the high nucleophilicity of H2Sn enabled selective ring-opening reactions of aziridines over other nucleophilic analytes, which they utilized to manipulate twisted intramolecular charge-transfer properties to create fluorescent probes.[173] Chang and co-workers developed an ABS trigger for sulfite based on the intramolecular cyclization of levulinate and subsequent uncaging of the phenol, although the reaction was not tested in cells.[174]
Table 6:
Selected ABS methods using redox-neutral organic reactions (C).
| Primary Reaction | Associated Secondary Reactions |
Analytes | Associated Imaging Modalities |
Seminal Refs./Reviews |
|---|---|---|---|---|
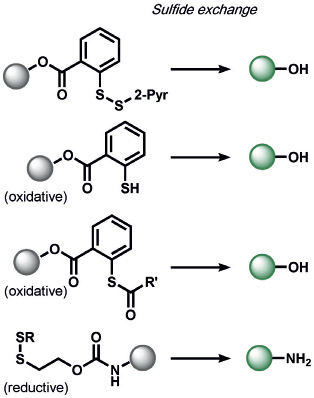 |
5-exo-trig cyclization, thio-esterification | H2S | fluorescence | 162 |
| RS-(S)n-SR, H2S2, or S8 | 128 | |||
| HSnR (n>1); RSH, S8 | 128 | |||
| glutathione | 153 | |||
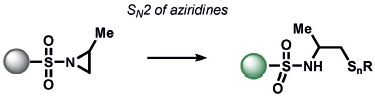 |
− | HSnR (n > 1) | fluorescence | 173 |
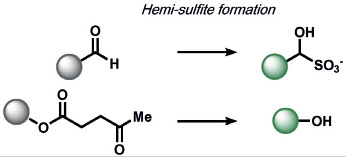 |
5-exo-trig cyclization, trans-esterification | HSO3−, SO32− | fluorescence | 174 |
Redox-neutral ABS methods for GSH were developed by Yang and co-workers through thiol-halogen nucleophilic aromatic substitution on fluorescent BODIPY scaffolds (Table 5).[175] The probe is compatible with living cells and is selective for cysteine and homocysteine thiol nucleophiles, which undergo an intramolecular S→N rearrangement to produce products with distinct photophysical properties. Wang and co-workers reported a reversible Michael addition reaction for the detection of GSH by modulating the α,β-substituents of an unsaturated acrylate;[176] this strategy has since been expanded to coumarins[177, 178] and silicon rhodamines.[179] Zhang et al. utilized GSH cleavage of a sulfamide functional group to create a ratiometric GSH sensor,[180] expanding on work by the Urano,[181] Kim,[182] and Yin[183] groups. Fang and co-workers utilized a nucleophilic aromatic substitution of 2,4-dinitrobenzenesulfonyl chloride to detect selenocysteine, thereby achieving a > 100-fold selectivity over thiols by adjusting the electron density of the probe.[184]
Recent ABS methods have also targeted the detection of electrophilic species in biological systems, such as electrophilic reactive carbon species (RCS; Table 7). For example, Spiegel and co-workers utilized a methyl-modified diaminobenzene-BODIPY probe for imaging methylglyoxal (MGO).[185] Despite the common use of vicinal phenylenediamine groups in NO sensing, the unique formation of quinoxalines through the reactivity of MGO was sufficient to achieve selective detection in live cells.
Our group[186] and Chan’s group[187] concomitantly reported the first ABS probes for formaldehyde (FA) based on the aza-Cope rearrangement (Table 7). Formaldehyde Probe 1 (FAP-1) from our studies[186] contains an appended aza-Cope trigger to favor a closed lactone form of a silicon rhodamine that would rearrange to an open form upon aza-Cope reaction and aqueous hydrolysis. This reaction offers a prime example of ABS detection, as many potential aldehydes can bind the homoallyl trigger in a reversible manner, but only FA can achieve ABS through the aza-Cope reaction. The broad utility of such ABS reactivity was demonstrated with a second-generation trigger involving geminal dimethyl groups to increase the rate of the aza-Cope rearrangement through the Thorpe–Ingold effect; this produced FA probes with ratiometric readouts[188] and an expanded color palette.[189] Moreover, this aza-Cope cage has enabled ABS of FA by positron emission tomography (PET)[190] and chemiluminescence[191] in live animals. Parallel efforts by Lin and co-workers led to the development of a notable suite of formimine[192] and aminal[193] ABS probes to sense FA in living cells and animals by electronic modulation of fluorescent scaffolds.[7]
A final class of redox-neutral transformations are electron-transfer processes that involve reversible cycling between oxidation and reduction steps (Table 7). Our group reported Redoxfluor-1 (RF1), which has an embedded dithiol/disulfide switch coupled with aromatization to monitor reversible oxidation-reduction cycles in cells.[194] Miller et al. have developed molecular wire constructs as a general approach to optical voltage sensing with small molecules, where changes in the membrane voltage alters the photo-induced electron transfer within a donor–acceptor pair that inserts into cellular membranes. Voltagefluors 2.1.Cl and 2.4.Cl (VF2.1.Cl/VF2.4.Cl) were the first-generation ABS sensors for voltage imaging in living cells by this design strategy.[195] From this starting point, a broad family of voltage sensors have been created with various excitation/emission profiles.[196-202] This approach has enabled the tuning of molecular wires from styryl to fluorene,[203] targeting of cells by bio-orthogonal labeling,[204] esterase cleavage,[205] or light activation,[206] as well as quantitative voltage measurements by fluorescence lifetime imaging microscopy (FLIM).[207] The New group has exploited flavins as bioderived reversible redox switches, which can monitor dynamic fluxes of oxidation and reduction events in cells.[208] Innovative work using redox-sensitive small molecules coupled to sensor platforms led to nicotinimidium-coupled MRI contrast agents,[209] as well as naphthalimide,[210] subcellular-targetable,[211] and ratiometric[212] fluorescent scaffold-coupled flavin redox sensors.
4.4. Inorganic and Organometallic Reactions
Inorganic and organometallic methods for ABS offer unique opportunities to incorporate a broader and more complex range of reactions by expanding the number of chemical elements employed. Such metal-mediated ABS reactions can be classified into two main types: the utilization of metals with biocompatible sensor moieties (Tables 8 and 9) or direct ABS sensing of the biological metals themselves (Tables 10 and 11). These methods combine BBS approaches, often inspired by supramolecular chemistry and bioinorganic chemistry, to create versatile tools to exploit redox chemistry, which is inherently rich in inorganic and organometallic systems. We refer the reader to a more detailed review on the use of inorganic chemistry principles for ABS[23] and provide a selected subset of examples here.
Table 8:
Selected examples of metal-mediated ABS platforms for the detection of small molecules (A).[a]
| Reaction | Analyte | Associated Imaging Modalities |
Seminal Refs./Reviews |
|---|---|---|---|
 |
NO | fluorescence | 210 |
 |
219 | ||
 |
H2S | fluorescence | 217 |
| CN− | 218 | ||
 |
NO | fluorescence | 213 |
Por = porphyrin.
Table 9:
Selected examples of metal-mediated ABS platforms for the detection of small molecules (B).[a]
| Reaction | Analyte | Associated Imaging Modalities |
Seminal Refs./Reviews |
|---|---|---|---|
 |
CO | fluorescence | 223 |
 |
H2O2 | fluorescence | 220 |
| Zn2+, OH− | 246 | ||
 |
CH2=CH2 | fluorescence | 230 |
Mes = mesityl.
Table 10:
Selected examples of ABS methods for the detection of metal Ions (A).
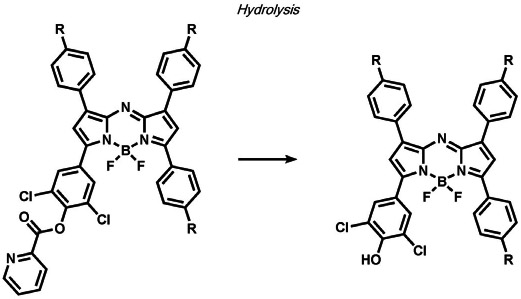
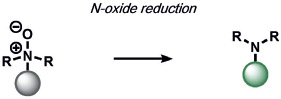
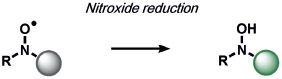
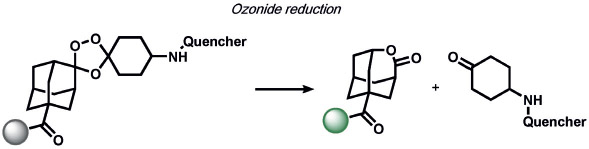
Table 11:
Selected examples of ABS methods for the detection of metal Ions (B).
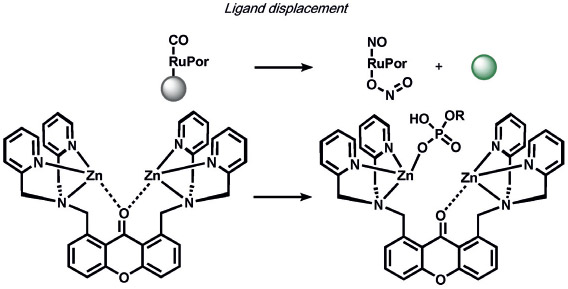
A variety of inorganic and organometallic reactions have been employed to detect various biological analytes, but the most common approach is to utilize a metal bound to a fluorophore as a fluorescent quencher, which can be released upon reaction with a small-molecule analyte (Table 8). Lippard and co-workers pioneered an early example for the direct and sensitive detection of NO by using CuII-dependent metal displacement with NO-induced fluorophore nitrosylation;[210] this was later expanded to piperazine spacer ligands for ratiometric fluorescence detection in living cells[213] as well as ruthenium-porphyrin-coordinated fluorophores.[214] Copper-based displacement methods have also been employed for sensing HNO, as reviewed previously.[215] In addition, Chang and co-workers utilized copper displacement from fluorescein functionalized with a DPA (DPA = dipicolylamine) ligand for H2S sensing;[216] later work by the Nagano group utilized more stable Cu-cyclen adducts[217] for the precipitation of copper sulfide. HCN was detected by a similar CuII decomplexation in living cells by Kim and co-workers.[218] The Hamachi group showed that phosphate anions could be detected through ratiometric fluorescence sensing through coordination to a zinc binding site.[219] Metal redox processes have also been exploited for ABS of small-molecule analytes. For example, Kodera and co-workers utilized the H2O2-mediated uncaging of resorufin fluorescent scaffolds of iron complexes.[220] Magnetic resonance imaging (MRI) probes from the Que group utilized H2O2-mediated cobalt oxidation, with detection shown through magnetic anisotropy changes in the 19F signal,[221] while Au-Yeung and co-workers have developed copper-fluorophore ABS probes for superoxide.[222]
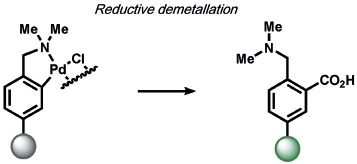
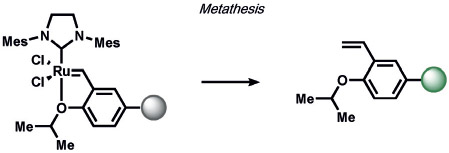
On the organometallic front, our group developed Carbon Monoxide Probe 1 (COP-1), which utilizes a palladium-mediated carbonylation reaction to selectively detect carbon monoxide (CO) by release of a Pd quencher from a fluorophore with concomitant C─C bond formation (Table 9).[223] As many diatomic compounds of similar size exist in biological systems (e.g. O2, NO), this ABS approach has proved general for expansion to benzimidazole,[224] Nile Red,[225-227] carbazole-coumarin,[228] and Raman spectroscopy nanosensors.[229] Finally, Michel and co-workers have reported an innovative approach for the selective and sensitive fluorescence detection of ethylene in plant and mammalian cells by olefin metathesis using a Hoveyda–Grubbs catalyst[230] which promises to be a useful way to detect this essential signaling molecule.
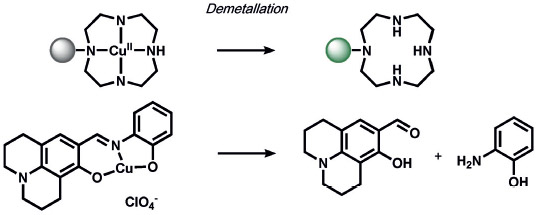
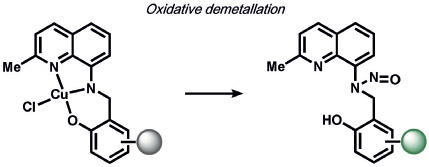
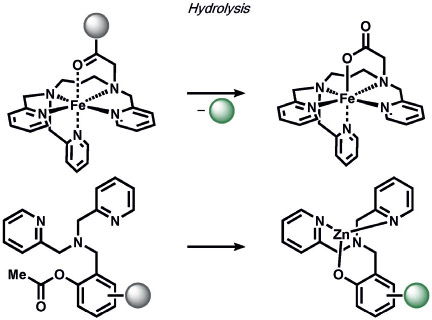
ABS methods can also be employed for metal detection, as illustrated by the diversity of reactions for tracking physiologically relevant metals as well as heavy noble and coinage metals used for technological purposes and that are potential environmental contaminants (Tables 10 and 11). An early reaction of a metal-directed ABS detection of copper was developed by Taki et al.,[231, 232] who used a bio-inspired tris(2-pyridyl)methylamine (TPA) receptor based on synthetic models of bio-inorganic copper sites that can uncage phenols of imaging scaffolds with CuI- and O2-dependent cleavage of a benzyl ether bond (Table 10). This work has inspired other probe scaffolds that rely on biomimetic, metaland O2-dependent cleavage processes for metal detection. For example, we adapted this trigger to develop Copper Caged Luciferin-1 (CCL-1), a probe that enables whole-animal imaging of copper pools in vivo. Our group also devised Cobalt Probe 1 (CP1) through a related oxidative cleavage reaction, using an N3O receptor for live-cell CoII imaging;[233] this was further elaborated for bioluminescence imaging.[234] We also developed a first-generation probe, Iron Probe 1 (IP1), for labile FeII imaging. This probe can visualize changes in labile iron pools with a trispyridine carboxylate receptor for iron activity.[235]
Other ABS methods for iron detection have been developed (Table 10).[236] Hirayama, Nagasawa et al. have developed a versatile N-oxide reduction switch that is selectively reduced to the amine by FeII. Starting from the first-generation probe RhoNox-1,[237] multicolor imaging has been achieved using coumarin,[238] rhodol,[238, 239] and silicon rhodamine[238] derivatives as well as organelle-targeted detection of labile FeII pools.[240] Wang and co-workers developed an FeII-mediated cyclization reaction to modulate the fluorescence of coumarin dyes in living cells.[241] Our group as well as the Renslo group have designed endoperoxide-based probes inspired by antimalarial drugs such as artemisinin. These probes undergo an FeII-dependent cleavage reaction to create a suite of ABS reagents for the detection of labile iron pools, including histochemical,[242] ratiometric,[243] bioluminescent,[244] and PET probes.[245] Lippard and co-workers exploited a dual ABS/BBS approach by combining the Lewis acidity of zinc to hydrolyze an ester group to unveil a free polypyridine receptor with a concomitant fluorescent turn-on response upon subsequent zinc binding.[246] Chan and co-workers utilized the CuII-dependent cleavage of picolinic esters for the photoacoustic detection of copper.[247]
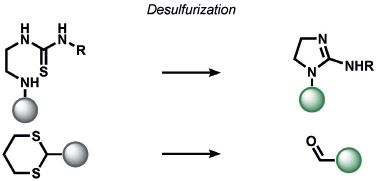
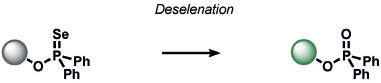
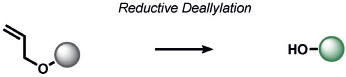
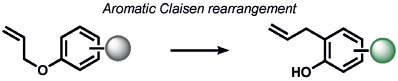
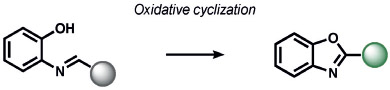
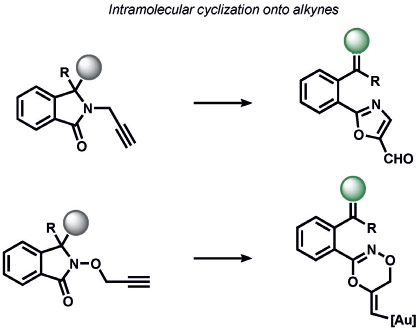
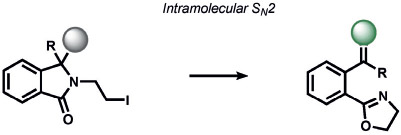
Heavy noble and coinage metals are also amenable to ABS detection (Table 11). For example, mercury-promoted desulfurization of cyanine dyes has been developed by Tian and co-workers, although such probes were not utilized in living systems.[248] Tang et al. utilized a mercury-selective deselenation reaction to uncage fluorescein for detection in living cells.[249] A similar approach for modulating the fluorescein spirocyclization of a selenolactone was utilized by Shin, Yoon, and co-workers.[250] The Koide group has exploited classic Tsuji–Trost allylic oxidative insertion reactions to deprotect allyl-protected fluorescent scaffolds, thereby enabling the detection of palladium and platinum in various oxidation states.[251] Aromatic Claisen rearrangements can also be exploited for the same purpose.[252] Pang and co-workers reported a PdII-mediated benzoxazole cyclization to form a fluorescent product[253] and Mahapatra et al. reported a CdII-mediated thioacetal desulfurization for the detection of these heavy metals, but neither of these triggers was examined in living systems.[254] On the coinage metal side, Ahn and co-workers utilized a vinylgold intermediate to form formyloxazole products to detect AuI and AuIII.[255] In a related study, Tae and co-workers reported the AuIII-promoted cyclization of aryl amides and alkynes to form acylsemicarbazides, a reaction that can operate in cells.[256] Ahn and co-workers also developed a similar method for the detection of silver through a AgI-promoted ring opening of alkyl iodides to form oxazolines.[257]
5. Activity-Based Sensing for Imaging Applications
ABS methods are broadly applicable to a variety of imaging platforms, largely as a result of the separation of the analyte-sensing modality from the sensing platform. In this section, we highlight select examples of ABS methods utilized for more in-depth biological investigations to help decipher signaling and/or stress contributions of a given biological analyte. The following selected examples, spanning many different types of imaging modalities, showcase the power of this chemical method for biological discovery.
5.1. Fluorescence Imaging
The vast majority of ABS methods utilize fluorescence readouts to establish the sensitivity and selectivity of a given chemical reaction trigger toward a biological analyte of interest. Indeed, the availability of fluorescence imaging systems and the ability of dyes to be tuned for excitation/emission color, subcellular to tissue-level localization, and changes in fluorescence intensity, wavelength, and/or lifetime offer a diverse set of chemical tools for biological inquiry. We provide select examples of where fluorescence ABS probes have revealed new biological principles (Figure 3) and refer readers to other leading reviews for fluorescence-based ABS detection of small molecules[21] and metals[18, 258] in living systems.
Figure 3.

Select examples of fluorescence ABS probes for biological discovery. a) Boronate-based H2O2 probes such as Peroxy Green 1 (PG1), Peroxy Yellow 1 Methyl Ester (PY1-ME), and Peroxyfluor-6 Acetoxymethyl Ester (PF6-AM) Identify endogenous, NADPH oxidase derived H2O2 fluxes generated upon stimulation of growth factors,[83] specific aquaporin channels that mediate membrane H2O2 transport,[259] and the physiological importance of H2O2 signaling in neural stem cell proliferation and neurogenesis pathways as well as oncogene-induced cell senescence.[260] b) The azide-based H2S probe Sulfidefluor-7 Acetoxymethyl Ester (SF7-AM) reveals crosstalk between oxidative H2O2 and reductive H2S signals generated with growth factor stimulation.[134] c) The diamine-based MGO probe MBo detects methylglyoxal (MG) fluxes (blue fluorescence) that trigger the central transcription factor YAP to translocate to the nucleus (red fluorescence).[261] d) The aza-Cope formaldehyde (FA) indicator Formaldehyde Probe 1 (FAP-1) provides supporting evidence that specific folate derivatives in the folate cycle are metabolic sources of this one-carbon unit.[262] e) The combined activity/binding-based zinc indicator Diacetyl Zinpyr-1 (DA-ZP1) reveals intracellular attenuation of labile ZnII pools upon sound stimulation[263] (R = (2-aminoethyl)triphenylphosphonium). f) The endoperoxide-based fluorescent probe FRET Iron Probe 1 (FIP-1) identifies dynamic elevations in labile FeII pools for epigenetic regulation upon activation with the second messenger cyclic AMP (cAMP).[264]
In terms of oxidative reactions, our group as well as Tonks and co-workers utilized boronate-based H2O2 probes to reveal several principles of physiological H2O2 signaling. Examples include the discovery of specific aquaporin water channel isoforms that have additional functions as peroxide channels to regulate intracellular ROS levels under stimulatory conditions,[259] H2O2-dependent neural stem cell proliferation and neurodegenesis,[86] and H2O2-induced cell senescence pathways through transient inhibition of the phosphatase PTP1B[260] (Figure 3a). The reductive azide-based H2S ABS probe SF7-AM was used to monitor H2O2-dependent H2S production through stimulation of the vascular endothelial growth factor (VEGF; Figure 3b).[134] Redox-neutral ABS probes such as the MGO fluorescent probe (MBo)[185] from Spiegel and co-workers have been employed to interrogate MGO signaling of the transcriptional coactivator yes-associated protein (YAP) that regulates tumor growth (Figure 3c).[261] Additionally, Patel and co-workers applied the formaldehyde indicator FAP-1[186] to discover that specific intracellular folate metabolites such as tetrahydrofolate, but not others, spontaneously decompose to release FA to regulate one-carbon metabolism in the cell (Figure 3d).[262] A prime example of ABS probes for metal detection was reported by Tzounopoulos, Lippard, and co-workers, who utilized the fluorescent zinc probe DA-ZP1[246] to discover that endogenous zinc inhibits α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) subtype glutamate receptors to tune neurotransmission and modulate the signaling of these receptors (Figure 3e).[263] Additionally, application of iron probe 1 (FIP-1)[243] by Wang and co-workers led to the discovery that cyclic adenosine monophosphate (cAMP) regulates gene transcription by modulating cellular labile FeII pools (Figure 3f).[264]
5.2. Luminescence and Whole-Animal Imaging
As complementary tools to fluorophores that are commonly employed for in vitro, cell, tissue, and zebrafish imaging, bioluminescence and chemiluminescence reagents enable in vivo imaging in living animals. Luminescence imaging also exhibits minimal background fluorescence, since the light emission is generated by decomposition of chemically excited luminescent scaffolds. Recent reviews highlight the growing palette of ABS methods for luminescence imaging.[265-267]
For example, the Shabat and Dvir groups utilized a boronate chemiluminescent probe to evaluate immune responses of mice to autologous, xenogeneic, and allogeneic implants,[268] and joint work between us and the Shabat group showed that in vivo metabolism of folate can produce FA[191] (Figure 4a). Zhang and co-workers utilized their bioluminescent probe BP-PN to monitor in vivo ONOO− levels generated from LPS-induced inflammation stress in tissues (Figure 4b).[269] In a joint study between us and the Stahl group, the CCL-1 reagent was utilized to probe the status of labile CuI pools in a diet-induced mouse model of non-alcoholic fatty liver disease (NAFLD, Figure 4c).[232] By applying our bioluminescent FeII reporter ICL-1, Skaar and co-workers identified significant increases in the iron-dependent luminescent signal in mice infected with A. baumannii (Figure 4d).[244]
Figure 4.

Select examples of luminescent ABS probes for biological discovery. a) Chemiluminescent probes based on Schaap’s dioxetane can sense in vivo H2O2 production triggered by LPS[270] as well as formaldehyde by tetrahydrofolate (THF) consumption.[191] b) The bioluminescent probe BP-PN was used to visualize endogenous ONOO− production triggered by liposaccharide (LPS) inflammation.[269] c) The TPA-based probe Copper Caged Luciferin 1 (CCL-1) revealed an early labile CuI deficiency localized to the liver in a diet-induced model of non-alcoholic fatty liver disease (NAFLD), showing more copper accumulation in the liver in mice fed a normal diet (ND) relative to mice fed a high-fat diet (HFD).[232] d) The endoperoxide probe Iron Caged Luciferin 1 (ICL-1) revealed elevations in labile FeII pools upon bacterial infection, correlating increases in labile FeII with sites of higher pathogen burden.[244]
5.3. Magnetic Resonance Imaging
Magnetic resonance imaging (MRI) is one of the most commonly used medical imaging platforms, and analyte-responsive MRI contrast agents have been extensively reviewed.[271, 272] As the field has primarily focused on BBS approaches for modulating MRI signals, opportunities for developing ABS methods for MRI are plentiful. In this context, a unique feature of ABS probes using this particular imaging modality is the ability to modulate signal intensity based on the chemical shift of a specific single atom on a given chemical scaffold, thereby allowing for a more diverse set of chemical reaction triggers to be created and explored. Additionally, one ABS probe may be utilized for the selective, in parallel, detection of multiple analytes that result in differing chemical shifts of the reporter atom. A prominent example comes from hyperpolarized MRI using a 13C-labeled analogue of the natural metabolite pyruvate, which can be utilized to trace metabolic pathways through conversion of pyruvate into a variety of well-defined downstream products[273-275] (Figure 5a). Recent work from McMahon and Zhou utilized 19F NMR spectroscopy of a labeled furan-malononitrile conjugated to a Michael acceptor that modulates the chemical shift of a fluorine atom upon reaction with biological cysteine, homocysteine, and GSH. (Figure 5b).[276]
Figure 5.

Select examples of magnetic resonance ABS probes for biological discovery. a) [1-13C]pyruvate for the NMR detection of an alteration In the lactate dehydrogenase (LDH) expression in tumors. Observation of increased conversion of [1-13C]pyruvate to [1-13C]lactate in tumors using 3D spectral arrays from differing 13C chemical shifts.[274] b) Furan-malononitrile probe for the NMR detection of biological thiols. Michael addition of thiol to the acrylamide results in differing 19F chemical shifts for cysteine, homocysteine, and glutathione detection.[276] c) MRI contrast agent EgadMe for the NMR detection of gene expression in zebrafish. The NMR signal from Gd3+ is altered upon galactopyranose cleavage in the presence of marker gene coding for β-galactosidase.[277] d) NMR redox detection through differing 13C chemical shifts of dehydroascorbate (DHA) and vitamin C (VitC) reveals an altered redox state in prostate tumors.[278]
Despite advances in ABS methods that operate in the MRI modality, there has been relatively limited use of such reagents in vivo. A classic example showcasing the power of MR detection for in vivo imaging is work by Meade, Fraser, and co-workers on gene expression using a β-galactosidase-responsive MRI contrast agent (Figure 5c).[277] Likewise, Wilson, Keshari, Kurhanewicz, and co-workers have exploited a hyperpolarized 13C-dehydroascorbate probe as a redox sensor for in vivo metabolic imaging of the elevated oxidation status in a transgenic prostate cancer mouse model[278] (Figure 5d). These and other studies provide a starting point for the application of ABS probes for biological MRI studies.
5.4. Photoacoustic Imaging
Photoacoustic imaging is emerging as an exciting new direction in the ABS field. This modality relies on the detection of sound generated from the absorption of light by chromophores that undergo relaxation energy loss in the form of heat and result in thermal expansion of biological tissue.[280, 281] Commonly used ultrasound detectors can be applied for detection, and the small interference of sound waves compared to light waves from biological tissue scattering allows visualization of analyte regulation in living animals. The design criteria for achieving high photoacoustic signal intensities are complementary and inverse to fluorescence/luminescence signals, where the former seeks to promote high yields of nonradiative decay and heat from excited states rather than light emission. As such, one can envision designing a “turn-on” response of the photoacoustic signal by leveraging a “turn-off” response of the fluorescence/luminescence and vice versa. Photoacoustic probes using existing ABS methods have been developed for CuII,[247] NO,[282] hypoxia,[283] H2S,[284] and fluoride.[285] As this area is still in its infancy and imaging instrumentation is less available compared to fluorescent counterparts, in vivo studies with photoacoustic ABS remain rare. A prime example comes from Chan and co-workers, who developed APNO-5, a NO-responsive photoacoustic probe that operates by N-nitrosation of an N-methylaniline reaction-based trigger, and showed endogenous NO generation in vivo with inflammation induced by LPS (Figure 6).[282] This work sets the stage for further application of acoustogenic reporters for cell, tissue, and animal imaging.
Figure 6.

Photoacoustic detection of endogenous NO generated upon liposaccharide (LPS) inflammation in a living mouse by APNO-5.[282]
5.5. Positron Emission Tomography Imaging
Positron emission tomography (PET) is another modality that is widely used in medical imaging and adaptable to ABS methods. ABS methods applied to the PET modality must circumvent inherent challenges in radiochemical synthesis and application, as well as the need to accumulate sufficient amounts of a tracer in a specific location to achieve a measurable signal. However, since radiolabels can also be directly applied to the analyte itself or upstream/downstream metabolites in addition to the ABS trigger or associated moiety, many opportunities are available to expand PET detection by ABS. For example, our group, in collaboration with the Wilson group, has developed Peroxy Caged Fluorothymidine 1 (PC-[18F]-FLT-1), a boronate-based PET probe that cages the clinically employed tracer [18F]-fluorothymidine (18F-FLT). Accumulation of this tracer in proliferating cells can only occur upon H2O2-induced deprotection of the boronate cage (Figure 7a).[101] We expanded PET ABS to in vivo imaging through the development of Formaldehyde Caged Fluorodeoxyglucose 1 (FAC-[18F]-FDG-1), which masks the canonical PET tracer [18F]-fluorodeoxyglucose 1 to enable monitoring FA in cells as well as in a PC3 xenograft model of prostate cancer (Figure 7b).[286] Other ROS-responsive PET tracers have been reported by the groups of Wilson and Valliant.[287, 288] Renslo, Evans, and co-workers created PET probes for tracking labile iron pools in living animals, which revealed systemic changes in labile iron disposition in mice and in vivo imaging of spontaneous tumors arising in a genetically engineered model of prostate cancer driven by loss of the phosphatase and tensin homologue (PTEN; Figure 7c).[245] This area of ABS is ripe for further investigation owing to the widespread use of PET imaging in research and clinical settings.
Figure 7.

Representative PET ABS probes. a) Exogenous H2O2 visualized in endothelial cells with PC-FLT.[101] b) FAC-FDG-1 visualization of FA in a living mouse.[289] c) Visualization of iron accumulation in a prostate cancer model by 18F-TRX (SV = seminal vesicle, Mu. = hindlimb muscle, Pr. = whole prostate).[290]
6. Activity-Based Sensing Beyond Imaging
The high molecular selectivity afforded by ABS methods can also be leveraged to make functional small molecules, proteins, polymers, and other materials that respond to endogenous stimuli in living systems. In this section, we highlight progress and opportunities for expanding ABS approaches beyond imaging. Emphasis is placed on therapeutic applications, where selective ABS reactions can enable the development of drugs with greater specificity and/or efficacy, often by releasing an active agent at a localized site of action. To illustrate how a single, robust ABS reaction method can be utilized in a diverse array of potential therapeutic applications, we will focus this discussion on examples using the H2O2-mediated transformation of boronates to phenols as a privileged ABS reaction, which has also been discussed more generally in two recent reviews.[291, 292] We refer readers to reviews by Sessler, Kang, Kim, and co-workers on the development of ABS prodrugs for RSS, ROS, and enzymes that are upregulated in cancer,[293] and by Gu and co-workers on platforms that exploit selective H2O2 reactivity for biomedical applications,[294] as well a recent example from Renslo and co-workers on prodrugs for infectious diseases that exploits the FeII-mediated cleavage of endoperoxides to deliver therapeutics specifically at sites of elevated iron.[290]
In early work on prodrug strategies that exploit ABS methods, Franz and co-workers,[295] along with the Guo group,[296] exploited H2O2-dependent boronate oxidation to develop prochelators that can combat metal-catalyzed oxidative stress and damage by selectively uncaging free chelators only in situations with elevated ROS levels (Figure 8, yellow Prochelators section). Initial work by Franz and co-workers made use of prochelators to block Fenton reactions by quenching both ROS and redox-active iron.[295] Next-generation boronate-based prochelators for iron provided significant cytoprotection against oxidative damage,[297] prevented metal-dependent amyloid aggregation,[298] and captured metals in a peroxide-dependent manner with concomitant fluorescent reporting.[299] More recently, Franz, Thiele, and co-workers reported a H2O2-responsive prochelator that unmasks upon the generation of ROS in activated macrophages, and then complexes copper to elicit fungal killing.[300]
Figure 8.

Selected examples of ABS approaches to prodrugs and related therapeutics through a common privileged reaction motif. The H2O2-mediated transformation of boronates to phenols was applied to various classes of therapeutics including prodrug activation, caged analyte donors, prochelator coordination, protein conjugate activation, and responsive materials.
In addition to chelators, caged enzyme inhibitors can also be developed using ABS approaches (Figure 8, blue Prodrugs section). For example, Major Jourden and Cohen applied the boronate cage to create matrix metalloproteinase (MMP) prodrugs that release MMP inhibitors selectively in the presence of hydrogen peroxide,[301] which has potential to neutralize both damaging ROS and inhibit degradative MMPs in situations such as ischemia-repurfusion injury. Wang and co-workers developed boronate-caged belinostat, an inhibitor of histone deacetylases, which allowed for improved biocompatibility and reduction of tumor volume.[302]
ABS approaches have also been employed with other types of prodrug platforms, such as creating caged analyte donors (Figure 8, purple Caged Small Molecules section). Zhao and Pluth have reported a variety of peroxide-responsive H2S/COS donors using boronate cages. A first-generation reagent, PeroxyTCM, was the first H2O2-triggered H2S donor engineered to release carbonyl sulfide (COS) upon oxidative activation. The COS subsequently generates H2S and provides protection against oxidative damage induced by H2O2 in cells.[303] Likewise, the Matson group utilized caged boronate-caged persulfide donors for related purposes, which led to the sustained release of the persulfide of N-acetylcysteine (NAC) in cell culture models.[304]
In addition to small-molecule prodrug systems, ABS methods can be employed for activity-based drug delivery using macromolecular scaffolds (Figure 8, green Caged Materials section). In one early example, Fréchet and co-workers reported a boronate-capped dextran carrier micro-particle system that releases its payload in response to elevations of H2O2, which they applied for testing vaccine efficacy.[305] Likewise, the Murthy group targeted ROS more generally and developed an orally delivered thioketal nanoparticle loaded with siRNA for detection of TNF-α as an oxidation-sensitive carrier to target inflammation in vivo.[306] As a complement to these drug delivery materials, Zhang and co-workers developed materials-based therapeutics that directly quench ROS to serve as antioxidants and anti-inflammatory therapeutics.[307]
Finally, ABS approaches to biologics have also been demonstrated (Figure 8, orange Caged Proteins section). For example, Raines and co-workers created a H2O2-responsive masked protein conjugate for angiogen, a ribonuclease compromised in amyotrophic lateral sclerosis, that cleaves and activates enzyme activity under oxidative stress but does not contribute to increased cell proliferation.[308]
7. Conclusions and Future Prospects
ABS offers a versatile approach to leverage the power of synthetic methods development to enable biological studies. Chemoselective host–guest complexes that form the basis of supramolecular chemistry have laid the foundations for the development of a diverse array of sensor systems spanning BBS approaches that rely on molecular recognition and lock- and-key binding to ABS methods that exploit the intrinsic differences in chemical reactivity among molecules to distinguish between them in complex biological settings. Early ABS work focused on manipulating chemical structure and intermolecular interactions to create selectivity for binding analytes, particularly reactive metal ions and small molecules, in solution. Further efforts to create more selective recognition moieties that are compatible with living biological environments has greatly expanded the utility of these tools and tactics to address important questions at the interface of chemistry and biology. Indeed, a growing number of ABS methods are available for the highly sensitive and selective detection of a wide range of analytes, and current efforts in the development of ABS probes have expanded from sensing and imaging to creating new therapeutics and bioresponsive materials.
Against this backdrop, many opportunities await further exploration in the ABS field. Fundamental synthetic advances to improve the sensitivity and selectivity for key biological analytes, as well as opening a new space to investigate underexplored analytes, will have a lasting impact in the field. One limitation of the vast majority of ABS probes is that, unlike many of their binding-based counterparts, the reactions employed for detection are often irreversible and consume the analyte of interest. Although low doses of probe are generally employed for minimal perturbation, creating reversible ABS reaction methods through analyte regeneration is a promising area for development to improve the sensitivity and bio-orthogonal nature of ABS probes. Innovative reports on ABS probes with analyte regeneration and/or signal amplification include sensors for heavy metals,[309] fluoride,[310] H2O2,[311] H2S,[312] and FA.[313] We refer readers to recent reviews on self-immolative linkers to improve ABS reactivity beyond stoichiometric reactions[314] and self-immolative polymers to create new ABS-responsive materials.[315]
Beyond reaction development, the versatility of ABS allows expansion from high-resource research in laboratory and clinical settings to low-resource field applications, thereby enabling broader, public use. Phillips and co-workers developed paper-based microfluidic devices for ABS, where oxidation of a boronate by H2O2 cleaves a hydrophobic detection reagent that is deposited into defined regions of the microfluidic conduits prior to assembling the devices to generate hydrophilic by-products, thus allowing the sample to wick through the device and wet a detection region in an analyte concentration-dependent manner.[316] Lippert and co-workers also developed innovative methods for chemiluminescent smartphone imaging of the H2O2-mediated conversion of an oxalate to form a chemiluminescent 1,2-dioxetanedione.[317]
Finally, the outgrowth of ABS approaches from detection and imaging to diagnostics and therapeutics applications has parallels with the advent of activity-based protein profiling[318-322] (ABPP) by Cravatt and other leading scientists in the field of chemical biology. This field has undergone a paradigm shift in how proteins are studied in living systems by focusing not only on a protein’s expression level, but on its chemical activity. At the core of this technology is new bioconjugation chemistry to “sense” amino acids that are hyperreactive within native proteomes through bond-forming ligation reactions, which can identify hotspots for protein function and therapeutic intervention.[323-329] A landmark approach in the field is the development of a chemoproteomic platform for the identification of hyperreactive cysteine residues by isotopic Tandem Orthogonal Proteolysis–Activity-Based Protein Profiling (isoTOP-ABPP).[330] Building upon extensive work in the ABPP field on cysteine,[331, 332] lysine,[333, 334] and serine[335] detection, we and others have been pursuing bioconjugation strategies of other native amino acids, including methionine,[336-338] histidine,[339] tyrosine,[340-342] and tryptophan.[343] For example, our group and the Toste group have co-developed a new ABS method for methionine bioconjugation termed Redox-Activated Chemical Tagging (ReACT), where we exploit the native redox properties of the oxidation of methionine to methionine sulfoxide to develop oxaziridine reagents that perform the isoelectronic nitrene transfer reaction to generate a sulfimide adduct.[336] Gaunt and co-workers developed a complementary approach by probing the nucleophilicity of methionine with a hypervalent iodine reagent.[337] Further synthetic developments are sure to advance the continuum between ABS and ABPP. These foregoing discussions and future prospects highlight ABS as a powerful chemical approach for deciphering biology, with opportunities to impact basic chemistry and biology as well as translation to modern imaging, diagnostics, and drug discovery and development.
Acknowledgements
We thank the NIH (GM 79465, ES28096, ES4705) and the Novartis Institutes for BioMedical Research and the Novartis-Berkeley Center for Proteomics and Chemistry Technologies (NB-CPACT) for research support. K.J.B. thanks the National Science Foundation for a graduate fellowship. S.W.M.C. is supported by the AGBT-Elaine R. Mardis Fellowship in Cancer Genomics from the Damon Runyon Cancer Research Foundation and The Genome Partnership, Inc. (DRG-2395-20). C.J.C. is a CIFAR Senior Fellow.
Biography

Kevin J. Bruemmer received his B.S. in Chemistry and Mathematics from Southern Methodist University in 2014, where he worked with Prof. Alexander Lippert on the development of fluorescence and magnetic resonance probes for reactive nitrogen and sulfur species. He then moved to UC Berkeley to continue work on studying reactive species with Prof. Chris Chang in 2015, where his work as an NSF graduate fellow focuses on developing and applying activity-based sensing methods to study the physiological roles of formaldehyde.

Steven W. M. Crossley received his B.Sc. in Chemistry from the University of British Columbia in 2012. After a gap year, he pursued Ph.D. studies as an NSERC postgraduate scholar at The Scripps Research Institute with Prof Ryan Shenvi to work on first-row transition-metal hydrofunctionalization of alkenes and total synthesis to support interrogation of GABAA receptor neurobiology. In 2018, he joined the group of Prof. Chris Chang, where he is developing amino acid specific reaction-based probes for cancer therapeutics discovery.

Christopher J. Chang is the Class of 1942 Chair Professor of Chemistry and Molecular and Cell Biology at the University of California, Berkeley, and Faculty Scientist at Lawrence Berkeley National Laboratory. He graduated with a B.S. and M.S. from Caltech in 1997 working with Prof. Harry Gray, spent a year as a Fulbright scholar with Dr Jean-Pierre Sauvage, and earned his Ph.D.from MIT in 2002 with Prof. Dan Nocera. After postdoctoral studies with Prof. Steve Lippard at MIT, he joined the Berkeley faculty in 2004. His research focuses on the study of metals and redox-active molecules in biology and energy.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Contributor Information
Kevin J. Bruemmer, Department of Chemistry, University of California, Berkeley Berkeley, CA 94720 (USA).
Steven W. M. Crossley, Department of Chemistry, University of California, Berkeley Berkeley, CA 94720 (USA).
Christopher J. Chang, Department of Chemistry, University of California, Berkeley Berkeley, CA 94720 (USA); Department of Molecular and Cell Biology and Helen Wills Neuroscience Institute, University of California, Berkeley Berkeley, CA 94720 (USA)
References
- [1].Tsien RY, Nat. Rev. Mol. Cell. Biol 2003, September;Suppl:SS16–21 [PubMed] [Google Scholar]
- [2].Dickinson BC, Chang CJ, Nat. Chem. Biol 2011, 7, 504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chen X, Tian X, Shin I, Yoon J, Chem. Soc. Rev 2011, 40, 4783–4804. [DOI] [PubMed] [Google Scholar]
- [4].Yang Y, Zhao Q, Feng W, Li F, Chem. Rev 2013, 113, 192–270. [DOI] [PubMed] [Google Scholar]
- [5].Brewer TF, Garcia FJ, Onak CS, Carroll KS, Chang CJ, Annu. Rev. Biochem 2015, 84, 765–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dong B, Kong X, Lin W, ACS Chem. Biol 2018, 13, 1714–1720. [DOI] [PubMed] [Google Scholar]
- [7].Bruemmer KJ, Brewer TF, Chang CJ, Curr. Opin. Chem. Biol 2017, 39, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tang Y, Ma Y, Yin J, Lin W, Chem. Soc. Rev 2019, 48, 4036–4048. [DOI] [PubMed] [Google Scholar]
- [9].Yu F, Han X, Chen L, Chem. Commun 2014, 50, 12234–12249. [DOI] [PubMed] [Google Scholar]
- [10].Lin VS, Chen W, Xian M, Chang CJ, Chem. Soc. Rev 2015, 44, 4596–4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hartle MD, Pluth MD, Chem. Soc. Rev 2016, 45, 6108–6117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Smith BC, Marletta MA, Curr. Opin. Chem. Biol 2012, 16, 498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Filipovic MR, Zivanovic J, Alvarez B, Banerjee R, Chem. Rev 2018, 118, 1253–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Parvez S, Long MJC, Poganik JR, Aye Y, Chem. Rev 2018, 118, 8798–8888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bertini I, Gray HB, Lippard SJ, Valentine JS, Bioinorganic Chemistry, University Science Books, Mill Valley, CA, 1994. [Google Scholar]
- [16].Lippard SJ, Berg JM, Principles of Bioinorganic Chemistry, University Science Books, Mill Valley, CA, 1994. [Google Scholar]
- [17].Chan J, Dodani SC, Chang CJ, Nat. Chem 2012, 4, 973–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Aron AT, Ramos-Torres KM, Cotruvo JA, Chang CJ, Acc. Chem. Res 2015, 48, 2434–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lee MH, Kim JS, Sessler JL, Chem. Soc. Rev 2015, 44, 4185–4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Li J, Yim D, Jang WD, Yoon J, Chem. Soc. Rev 2017, 46, 2437–2458. [DOI] [PubMed] [Google Scholar]
- [21].Jiao X, Li Y, Niu J, Xie X, Wang X, Tang B, Anal. Chem 2018, 90, 533–555. [DOI] [PubMed] [Google Scholar]
- [22].Kolanowski JL, Liu F, New EJ, Chem. Soc. Rev 2018, 47, 195–208. [DOI] [PubMed] [Google Scholar]
- [23].Iovan DA, Jia S, Chang CJ, Inorg. Chem 2019, 58, 13546–13560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pedersen CJ, J. Am. Chem. Soc 1967, 89, 7017–7036. [Google Scholar]
- [25].Pedersen CJ, Angew. Chem. Int. Ed. Engl 1988, 27, 1021–1027; Angew. Chem. 1988, 100, 1053–1059. [Google Scholar]
- [26].Dietrich B, Lehn JM, Sauvage JP, Tetrahedron Lett 1969, 10, 2889–2892. [Google Scholar]
- [27].Lehn JM, Acc. Chem. Res 1978, 11, 49–57. [Google Scholar]
- [28].Albrecht M, Röttele H, Burger P, Chem. Eur. J 1996, 2, 1264–1268. [Google Scholar]
- [29].Kang SO, Llinares JM, Day VW, Bowman-James K, Chem. Soc. Rev 2010, 39, 3980–4003. [DOI] [PubMed] [Google Scholar]
- [30].Zhang M, Yan X, Huang F, Niu Z, Gibson HW, Acc. Chem. Res 2014, 47, 1995–2005. [DOI] [PubMed] [Google Scholar]
- [31].Cram DJ, Angew. Chem. Int. Ed. Engl 1988, 27, 1009–1020; Angew. Chem. 1988, 100, 1041–1052. [Google Scholar]
- [32].Sogah GDY, Cram DJ, J. Am. Chem. Soc 1979, 101, 3035–3042. [Google Scholar]
- [33].Cram DJ, Kaneda T, Helgeson RC, Brown SB, Knobler CB, Maverick E, Trueblood KN, J. Am. Chem. Soc 1985, 107, 3645–3657. [Google Scholar]
- [34].Artz SP, Cram DJ, J. Am. Chem. Soc 1984, 106, 2160–2171. [Google Scholar]
- [35].Gutsche CD, Dhawan B, No KH, Muthukrishnan R, J. Am. Chem. Soc 1981, 103, 3782–3792. [Google Scholar]
- [36].Gutsche CD, Calixarenes, Royal Society Of Chemistry, Cambridge, 2008. [Google Scholar]
- [37].Amabilino DB, Smith DK, Steed JW, Chem. Soc. Rev 2017, 46, 2404–2420. [DOI] [PubMed] [Google Scholar]
- [38].Busschaert N, Caltagirone C, Van Rossom W, Gale PA, Chem. Rev 2015, 115, 8038–8155. [DOI] [PubMed] [Google Scholar]
- [39].You L, Zha D, Anslyn EV, Chem. Rev 2015, 115, 7840–7892. [DOI] [PubMed] [Google Scholar]
- [40].Dietrich-Buchecker CO, Sauvage JP, Kintzinger JP, Tetrahedron Lett. 1983, 24, 5095–5098. [Google Scholar]
- [41].Anelli PL, Spencer N, Stoddart JF, J. Am. Chem. Soc 1991, 113, 5131–5133. [DOI] [PubMed] [Google Scholar]
- [42].Harada N, Koumura N, Zijlstra RWJ, van Delden RA, Feringa BL, Nature 1999, 401, 152–155. [DOI] [PubMed] [Google Scholar]
- [43].Tsien RY, Biochemistry 1980, 19, 2396–2404. [DOI] [PubMed] [Google Scholar]
- [44].Grynkiewicz G, Poenie M, Tsien RY, J. Biol Chem 1985, 260, 3440–3450. [PubMed] [Google Scholar]
- [45].Adams SR, Kao JPY, Grynkiewicz G, Minta A, Tsien RY, J. Am. Chem. Soc 1988, 110, 3212–3220. [Google Scholar]
- [46].Ellis-Davies GCR, Methods Enzymol. 2003, 360, 226–238. [DOI] [PubMed] [Google Scholar]
- [47].Tsien RY, Nature 1981, 290, 527–528. [DOI] [PubMed] [Google Scholar]
- [48].Minta A, Kao JPY, Tsien RY, J. Biol. Chem 1989, 264, 8171–8178. [PubMed] [Google Scholar]
- [49].Kao JPY, Harootunian AT, Tsien RY, J. Biol. Chem 1989, 264, 8179–8184. [PubMed] [Google Scholar]
- [50].Valeur B, Leray I, Coord. Chem. Rev 2000, 205, 3–40. [Google Scholar]
- [51].de Silva AP, Gunaratne HQN, Gunnlaugsson T, Huxley AJM, McCoy CP, Rademacher JT, Rice TE, Chem. Rev 1997, 97, 1515–1566. [DOI] [PubMed] [Google Scholar]
- [52].Que EL, Domaille DW, Chang CJ, Chem. Rev 2008, 108, 4328. [DOI] [PubMed] [Google Scholar]
- [53].Fahrni CJ, Curr. Opin. Chem. Biol 2013, 17, 656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Cotruvo JA Jr., Aron AT, Ramos-Torres KM, Chang CJ, Chem. Soc. Rev 2015, 44, 4400–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ramos-Torres KM, Kolemen S, Chang CJ, Isr. J. Chem 2016, 56, 724–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ackerman CM, Lee S, Chang CJ, Anal. Chem 2017, 89, 22–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Hirano T, Kikuchi K, Nagano T, in Rev. Fluoresc. 2004, Springer US, Boston, MA, 2004, pp. 55–73. [Google Scholar]
- [58].Nolan EM, Lippard SJ, Acc. Chem. Res 2009, 42, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].New EJ, Wimmer VC, Hare DJ, Cell Chem. Biol 2018, 25, 7–18. [DOI] [PubMed] [Google Scholar]
- [60].Wu D, Chen L, Lee W, Ko G, Yin J, Yoon J, Coord. Chem. Rev 2018, 354, 74–97. [Google Scholar]
- [61].Czarnik AW, Acc. Chem. Res 1994, 27, 302–308. [Google Scholar]
- [62].Czarnik AW, Chem. Biol 1995, 2, 423–428. [DOI] [PubMed] [Google Scholar]
- [63].Chae MY, Czarnik AW, J. Am. Chem. Soc 1992, 114, 9704–9705. [Google Scholar]
- [64].Gunasekar PG, Kanthasamy AG, Borowitz JL, Isom GE, J. Neurosci. Methods 1995, 61, 15–21. [DOI] [PubMed] [Google Scholar]
- [65].Kojima H, Nakatsubo N, Kikuchi K, Kawahara S, Kirino Y, Nagoshi H, Hirata Y, Nagano T, Anal. Chem 1998, 70, 2446–2453. [DOI] [PubMed] [Google Scholar]
- [66].Kojima H, Hirotani M, Nakatsubo N, Kikuchi K, Urano Y, Higuchi T, Hirata Y, Nagano T, Anal. Chem 2001, 73, 1967–1973. [DOI] [PubMed] [Google Scholar]
- [67].Nakatsubo N, Kojima H, Kikuchi K, Nagoshi H, Hirata Y, Maeda D, Imai Y, Irimura T, Nagano T, FEBS Lett. 1998, 427, 263–266. [DOI] [PubMed] [Google Scholar]
- [68].Itoh Y, Ma FH, Hoshi H, Oka M, Noda K, Ukai Y, Kojima H, Nagano T, Toda N, Anal. Biochem 2000, 287, 203–209. [DOI] [PubMed] [Google Scholar]
- [69].Kitamura T, Kimura K, Makondo K, Furuya DT, Suzuki M, Yoshida T, Saito M, Diabetologia 2003, 46, 1698–1705. [DOI] [PubMed] [Google Scholar]
- [70].Chang MCY, Pralle A, Isacoff EY, Chang CJ, J. Am. Chem. Soc 2004, 126, 15392–15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Lehn J-M, Angew. Chem. Int. Ed. Engl 1988, 27, 89–112; Angew. Chem. 1988, 100, 91–116. [Google Scholar]
- [72].Sletten EM, Bertozzi CR, Angew. Chem. Int. Ed 2009, 48, 6974–6998; Angew. Chem. 2009, 121, 7108–7133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Liu HW, Chen L, Xu C, Li Z, Zhang H, Zhang XB, Tan W, Chem. Soc. Rev 2018, 47, 7140–7180. [DOI] [PubMed] [Google Scholar]
- [74].Kathayat RS, Elvira PD, Dickinson BC, Nat. Chem. Biol 2017, 13, 150–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Son S, Won M, Green O, Hananya N, Sharma A, Jeon Y, Kwak JH, Sessler JL, Shabat D, Kim JS, Angew. Chem. Int. Ed 2019, 58, 1739–1743; Angew. Chem. 2019, 131, 1753–1757. [DOI] [PubMed] [Google Scholar]
- [76].Kawatani M, Yamamoto K, Yamada D, Kamiya M, Miyakawa J, Miyama Y, Kojima R, Morikawa T, Kume H, Urano Y, J. Am. Chem. Soc 2019, 141, 10409–10416. [DOI] [PubMed] [Google Scholar]
- [77].Kuivila HG, Wiles RA, J. Am. Chem. Soc 1955, 77, 4830–4834. [Google Scholar]
- [78].Sikora A, Zielonka J, Lopez M, Joseph J, Kalyanaraman B, Free Radical Biol Med. 2009, 47, 1401–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Zielonka J, Sikora A, Hardy M, Joseph J, Dranka BP, Kalyanaraman B, Chem. Res. Toxicol 2012, 25, 1793–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Reth M, Nat. Immunol 2002, 3, 1129–1134. [DOI] [PubMed] [Google Scholar]
- [81].Radi R, J. Biol. Chem 2013, 288, 26464–26472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Cheng AJ, Yamada T, Rassier DE, Andersson DC, Westerblad H, Lanner JT, J. Physiol 2016, 594, 5149–5160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Miller EW, Tulyanthan O, Isacoff EY, Chang CJ, Nat. Chem. Biol 2007, 3, 263–267. [DOI] [PubMed] [Google Scholar]
- [84].Yik-Sham Chung C, Timblin GA, Saijo K, Chang CJ, J. Am. Chem. Soc 2018, 140, 6109–6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Lippert AR, Van De Bittner GC, Chang CJ, Acc. Chem. Res 2011, 44, 793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Dickinson BC, Peltier J, Stone D, Schaffer DV, Chang CJ, Nat. Chem. Biol. 2011, 7, 106–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Albers AE, Okreglak VS, Chang CJ, J. Am. Chem. Soc. 2006, 128, 9640–9641. [DOI] [PubMed] [Google Scholar]
- [88].Srikun D, Miller EW, Domaille DW, Chang CJ, J. Am. Chem. Soc 2008, 130, 4596–4597. [DOI] [PubMed] [Google Scholar]
- [89].Chung C, Srikun D, Lim CS, Chang CJ, Cho BR, Chem. Commun 2011, 47, 9618–9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Miller EW, Albers AE, Pralle A, Isacoff EY, Chang CJ, J. Am. Chem. Soc 2005, 127, 16652–16659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Albers AE, Dickinson BC, Miller EW, Chang CJ, Bioorg. Med. Chem. Lett 2008, 18, 5948–5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Dickinson BC, Huynh C, Chang CJ, J. Am. Chem. Soc 2010, 132, 5906–5915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Karton-Lifshin N, Segal E, Omer L, Portnoy M, Satchi-Fainaro R, Shabat D, J. Am. Chem. Soc 2011, 133, 10960–10965. [DOI] [PubMed] [Google Scholar]
- [94].Dickinson BC, Chang CJ, J. Am. Chem. Soc 2008, 130, 9638–9639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Srikun D, Albers AE, Nam CI, Iavarone AT, Chang CJ, J. Am. Chem. Soc 2010, 132, 4455–4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Dickinson BC, Tang Y, Chang Z, Chang CJ, Chem. Biol 2011, 18, 943–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Dickinson BC, Lin VS, Chang CJ, Nat. Protoc 2013, 8, 1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Lippert AR, Gschneidtner T, Chang CJ, Chem. Commun 2010, 46, 7510–7512. [DOI] [PubMed] [Google Scholar]
- [99].Van de Bittner GC, Dubikovskaya EA, Bertozzi CR, Chang CJ, Proc. Natl. Acad. Sci. USA 2010, 107, 21316–21321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Van De Bittner GC, Bertozzi CR, Chang CJ, J. Am. Chem. Soc 2013, 135, 1783–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Carroll V, Michel BW, Blecha J, Vanbrocklin H, Keshari K, Wilson D, Chang CJ, J. Am. Chem. Soc 2014, 136, 14742–14745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Lippert AR, Keshari KR, Kurhanewicz J, Chang CJ, J. Am. Chem. Soc 2011, 133, 3776–3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Abo M, Urano Y, Hanaoka K, Terai T, Komatsu T, Nagano T, J. Am. Chem. Soc 2011, 133, 10629–10637. [DOI] [PubMed] [Google Scholar]
- [104].Kenmoku S, Urano Y, Kojima H, Nagano T, J. Am. Chem. Soc 2007, 129, 7313–7318. [DOI] [PubMed] [Google Scholar]
- [105].Koide Y, Urano Y, Hanaoka K, Terai T, Nagano T, J. Am. Chem. Soc 2011, 133, 5680–5682. [DOI] [PubMed] [Google Scholar]
- [106].Liu SR, Wu SP, Org. Lett 2013, 15, 878–881. [DOI] [PubMed] [Google Scholar]
- [107].Manjare ST, Kim Y, Churchill DG, Acc. Chem. Res 2014, 47, 2985–2998. [DOI] [PubMed] [Google Scholar]
- [108].Mulay SV, Choi M, Jang YJ, Kim Y, Jon S, Churchill DG, Chem. Eur. J 2016, 22, 9642–9648. [DOI] [PubMed] [Google Scholar]
- [109].Setsukinai K. i., Urano Y, Kakinuma K, Majima HJ, Nagano T, J. Biol. Chem 2003, 278, 3170–3175. [DOI] [PubMed] [Google Scholar]
- [110].Zhang W, Guo C, Liu L, Qin J, Yang C, Org. Biomol. Chem 2011, 9, 5560–5563. [DOI] [PubMed] [Google Scholar]
- [111].Lin W, Long L, Chen B, Tan W, Chem. Eur. J 2009, 15, 2305–2309. [DOI] [PubMed] [Google Scholar]
- [112].Yuan L, Lin W, Song J, Yang Y, Chem. Commun 2011, 47, 12691–12693. [DOI] [PubMed] [Google Scholar]
- [113].Yin C, Zhen X, Fan Q, Huang W, Pu K, ACS Nano 2017, 11, 4174–4182. [DOI] [PubMed] [Google Scholar]
- [114].Tanaka K, Miura T, Umezawa N, Urano Y, Kikuchi K, Higuchi T, Nagano T, J. Am. Chem. Soc 2001, 123, 2530–2536. [DOI] [PubMed] [Google Scholar]
- [115].Xu K, Wang L, Qiang M, Wang L, Li P, Tang B, Chem. Commun 2011, 47, 7386–7388. [DOI] [PubMed] [Google Scholar]
- [116].Hananya N, Green O, Blau R, Satchi-Fainaro R, Shabat D, Angew. Chem. Int. Ed 2017, 56, 11793–11796; Angew. Chem. 2017, 129, 11955–11958. [DOI] [PubMed] [Google Scholar]
- [117].Garner AL, St Croix CM, Pitt BR, Leikauf GD, Ando S, Koide K, Nat. Chem 2009, 1, 316–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Yang Y, Seidlits SK, Adams MM, Lynch VM, Schmidt CE, Anslyn EV, Shear JB, J. Am. Chem. Soc 2010, 132, 13114–13116. [DOI] [PubMed] [Google Scholar]
- [119].Sun C, Shi W, Song Y, Chen W, Ma H, Chem. Commun 2011, 47, 8638–8640. [DOI] [PubMed] [Google Scholar]
- [120].Sun ZN, Wang HL, Liu FQ, Chen Y, Tam PKH, Yang D, Org. Lett 2009, 11, 1887–1890. [DOI] [PubMed] [Google Scholar]
- [121].Peng T, Yang D, Org. Lett 2010, 12, 4932–4935. [DOI] [PubMed] [Google Scholar]
- [122].Bruemmer KJ, Merrikhihaghi S, Lollar CT, Morris SNS, Bauer JH, Lippert AR, Chem. Commun 2014, 50, 12311–12314. [DOI] [PubMed] [Google Scholar]
- [123].Pino NW, Davis J, Yu Z, Chan J, J. Am. Chem. Soc 2017, 139, 18476–18479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Saxon E, Bertozzi CR, Science 2000, 287, 2007–2010. [DOI] [PubMed] [Google Scholar]
- [125].Serwa R, Wakening L, Del Signore G, Mühlberg M, Claußnitzer I, Weise C, Gerrits M, Hackenberger CPR, Angew. Chem. Int. Ed 2009, 48, 8234–8239; Angew. Chem. 2009, 121, 8382 – 8387. [DOI] [PubMed] [Google Scholar]
- [126].Majkut P, Böhrsch V, Serwa R, Gerrits M, Hackenberger CPR, in Unnatural Amino Acids, Humana Press, Totowa, New Jersey, 2012, pp. 241–249. [Google Scholar]
- [127].An W, Ryan LS, Reeves AG, Bruemmer KJ, Mouhaffel L, Gerberich JL, Winters A, Mason RP, Lippert AR, Angew. Chem. Int. Ed 2019, 58, 1361–1365; Angew. Chem. 2019, 131, 1375–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Chen W, Liu C, Peng B, Zhao Y, Pacheco A, Xian M, Chem. Sci 2013, 4, 2892–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Chen W, Rosser EW, Matsunaga T, Pacheco A, Akaike T, Xian M, Angew. Chem. Int. Ed 2015, 54, 13961–13965; Angew. Chem. 2015, 127, 14167–14171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Lin VS, Chang CJ, Curr. Opin. Chem. Biol 2012, 16, 595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Lippert AR, New EJ, Chang CJ, J. Am. Chem. Soc 2011, 133, 10078–10080. [DOI] [PubMed] [Google Scholar]
- [132].Peng H, Cheng Y, Dai C, King AL, Predmore BL, Lefer DJ, Wang B, Angew. Chem. Int. Ed 2011, 50, 9672–9675; Angew. Chem. 2011, 123, 9846–9849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Wang K, Peng H, Ni N, Dai C, Wang B, J. Fluoresc 2014, 24, 1–5. [DOI] [PubMed] [Google Scholar]
- [134].Lin VS, Lippert AR, Chang CJ, Proc. Natl. Acad. Sci. USA 2013, 110, 7131–7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Zhang H, Wang P, Chen G, Cheung HY, Sun H, Tetrahedron Lett. 2013, 54, 4826–4829. [Google Scholar]
- [136].Chen B, Li W, Lv C, Zhao M, Jin H, Jin H, Du J, Zhang L, Tang X, Analyst 2013, 138, 946–951. [DOI] [PubMed] [Google Scholar]
- [137].Thorson MK, Majtan T, Kraus JP, Barrios AM, Angew. Chem. Int. Ed 2013, 52, 4641–4644; Angew. Chem. 2013, 125, 4739–4742. [DOI] [PubMed] [Google Scholar]
- [138].Li W, Sun W, Yu X, Du L, Li M, J. Fluoresc 2013, 23, 181–186. [DOI] [PubMed] [Google Scholar]
- [139].Hammers MD, Taormina MJ, Cerda MM, Montoya LA, Seidenkranz DT, Parthasarathy R, Pluth MD, J. Am. Chem. Soc 2015, 137, 10216–10223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Chen W, Pacheco A, Takano Y, Day JJ, Hanaoka K, Xian M, Angew. Chem. Int. Ed 2016, 55, 9993–9996; Angew. Chem. 2016, 128, 10147–10150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Saha T, Kand D, Talukdar P, Org. Biomol. Chem 2013, 11, 8166–8170. [DOI] [PubMed] [Google Scholar]
- [142].Zhao Q, Yin C, Kang J, Wen Y, Huo F, Dye. Pigment 2018, 159, 166–172. [Google Scholar]
- [143].Montoya LA, Pluth MD, Chem. Commun 2012, 48, 4767–4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Zhang J, Guo W, Chem. Commun 2014, 50, 4214–4217. [DOI] [PubMed] [Google Scholar]
- [145].Chen T, Zheng Y, Xu Z, Zhao M, Xu Y, Cui J, Tetrahedron Lett. 2013, 54, 2980–2982. [Google Scholar]
- [146].Chen B, Lv C, Tang X, Anal Bioanal. Chem 2012, 404, 1919–1923. [DOI] [PubMed] [Google Scholar]
- [147].Sun W, Fan J, Hu C, Cao J, Zhang H, Xiong X, Wang J, Cui S, Sun S, Peng X, Chem. Commun 2013, 49, 3890–3892. [DOI] [PubMed] [Google Scholar]
- [148].Zheng Y, Zhao M, Qiao Q, Liu H, Lang H, Xu Z, Dyes Pigm. 2013, 98, 367–371. [Google Scholar]
- [149].Cai Y, Li L, Wang Z, Sun JZ, Qin A, Tang BZ, Chem. Commun 2014, 50, 8892–8895. [DOI] [PubMed] [Google Scholar]
- [150].Cao J, Lopez R, Thacker JM, Moon JY, Jiang C, Morris SNS, Bauer JH, Tao P, Mason RP, Lippert AR, Chem. Sci 2015, 6, 1979–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Ke B, Wu W, Liu W, Liang H, Gong D, Hu X, Li M, Anal. Chem 2016, 88, 592–595. [DOI] [PubMed] [Google Scholar]
- [152].Filomeni G, Rotilio G, Ciriolo MR, Biochem. Pharmacol 2002, 64, 1057–1064. [DOI] [PubMed] [Google Scholar]
- [153].Pires MM, Chmielewski J, Org. Lett 2008, 10, 837–840. [DOI] [PubMed] [Google Scholar]
- [154].Kong F, Hu B, Gao Y, Xu K, Pan X, Huang F, Zheng Q, Chen H, Tang B, Chem. Commun 2015, 51, 3102–3105. [DOI] [PubMed] [Google Scholar]
- [155].Zhou Y, Zhang JF, Yoon J, Chem. Rev 2014, 114, 5511–5571. [DOI] [PubMed] [Google Scholar]
- [156].Kim SY, Hong J, Org. Lett 2007, 9, 3109–3112. [DOI] [PubMed] [Google Scholar]
- [157].Rao MR, Mobin SM, Ravikanth M, Tetrahedron 2010, 66, 1728–1734. [Google Scholar]
- [158].Shiraishi Y, Nakamura M, Hayashi N, Hirai T, Anal. Chem 2016, 88, 6805–6811. [DOI] [PubMed] [Google Scholar]
- [159].Long L, Huang M, Wang N, Wu Y, Wang K, Gong A, Zhang Z, Sessler JL, J. Am. Chem. Soc 2018, 140, 1870–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Maeda H, Fukuyasu Y, Yoshida S, Fukuda M, Saeki K, Matsuno H, Yamauchi Y, Yoshida K, Hirata K, Miyamoto K, Angew. Chem. Int. Ed 2004, 43, 2389–2391; Angew. Chem. 2004, 116, 2443 – 2445. [DOI] [PubMed] [Google Scholar]
- [161].Qian Y, Karpus J, Kabil O, Zhang S, Zhu H, Banerjee R, Zhao J, He C, Nat. Commun 2011, 2, 495–497. [DOI] [PubMed] [Google Scholar]
- [162].Liu C, Pan J, Li S, Zhao Y, Wu LY, Berkman CE, Whorton AR, Xian M, Angew. Chem. Int. Ed 2011, 50, 10327–10329; Angew. Chem. 2011, 123, 10511–10513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Liu C, Peng B, Li S, Park CM, Whorton AR, Xian M, Org. Lett 2012, 14, 2184–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Cao X, Lin W, Zheng K, He L, Chem. Commun 2012, 48, 10529–10531. [DOI] [PubMed] [Google Scholar]
- [165].Maeda H, Matsuno H, Ushida M, Katayama K, Saeki K, Itoh N, Angew. Chem. Int. Ed 2005, 44, 2922–2925; Angew. Chem. 2005, 117, 2982–2985. [DOI] [PubMed] [Google Scholar]
- [166].Wei C, Wei L, Xi Z, Yi L, Tetrahedron Lett. 2013, 54, 6937–6939. [Google Scholar]
- [167].Hammers MD, Pluth MD, Anal. Chem 2014, 86, 7135–7140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Liu Y, Feng G, Org. Biomol. Chem 2014, 12, 438–445. [DOI] [PubMed] [Google Scholar]
- [169].Xiong J, Xia L, Huang Q, Huang J, Gu Y, Wang P, Talanta 2018, 184, 109–114. [DOI] [PubMed] [Google Scholar]
- [170].Pak YL, Li J, Ko KC, Kim G, Lee JY, Yoon J, Anal. Chem 2016, 88, 5476–5481. [DOI] [PubMed] [Google Scholar]
- [171].Gu B, Su W, Huang L, Wu C, Duan X, Li Y, Xu H, Huang Z, Li H, Yao S, Sens. Actuators B 2018, 255, 2347–2355. [Google Scholar]
- [172].Liu C, Chen W, Shi W, Peng B, Zhao Y, Ma H, Xian M, J. Am. Chem. Soc 2014, 136, 7257–7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Chen W, Rosser EW, Zhang D, Shi W, Li Y, Dong W, Ma H, Hu D, Xian M, Org. Lett 2015, 17, 2776–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Choi MG, Hwang J, Eor S, Chang SK, Org. Lett 2010, 12, 5624–5627. [DOI] [PubMed] [Google Scholar]
- [175].Niu L, Guan Y, Chen Y, Wu L, Tung C, Yang Q, J. Am. Chem. Soc 2012, 134, 18928–18931. [DOI] [PubMed] [Google Scholar]
- [176].Jiang X, Yu Y, Chen J, Zhao M, Chen H, Song X, Matzuk AJ, Carroll SL, Tan X, Sizovs A, Cheng N, Wang MC, Wang J, ACS Chem. Biol 2015, 10, 864–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Liu Z, Zhou X, Miao Y, Hu Y, Kwon N, Wu X, Yoon J, Angew. Chem. Int. Ed 2017, 56, 5812–5816; Angew. Chem. 2017, 129, 5906–5910 [DOI] [PubMed] [Google Scholar]
- [178].Jiang X, Chen J, Bajić A, Zhang C, Song X, Carroll SL, Cai ZL, Tang M, Xue M, Cheng N, Schaaf CP, Li F, MacKenzie KR, Ferreon ACM, Xia F, Wang MC, Maletić-Savatić M, Wang J, Nat. Commun 2017, 8, 16087–16163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Umezawa K, Yoshida M, Kamiya M, Yamasoba T, Urano Y, Nat. Chem 2017, 9, 279–286. [DOI] [PubMed] [Google Scholar]
- [180].Zhang H, Wang C, Wang G, Wang K, Jiang K, Biosens. Bioelectron 2016, 78, 344–350. [DOI] [PubMed] [Google Scholar]
- [181].Yoshida M, Kamiya M, Yamasoba T, Urano Y, Bioorg. Med. Chem. Lett 2014, 24, 4363–4366. [DOI] [PubMed] [Google Scholar]
- [182].Kim GJ, Yoon DH, Yun MY, Kwon H, Ha HJ, Kim HJ, Sens. Actuators B 2015, 211, 245–249. [Google Scholar]
- [183].Cao M, Chen H, Chen D, Xu Z, Liu SH, Chen X, Yin J, Chem. Commun 2016, 52, 721–724. [DOI] [PubMed] [Google Scholar]
- [184].Zhang B, Ge C, Yao J, Liu Y, Xie H, Fang J, J. Am. Chem. Soc 2015, 137, 757–769. [DOI] [PubMed] [Google Scholar]
- [185].Wang T, Douglass EF, Fitzgerald KJ, Spiegel DA, J. Am. Chem. Soc 2013, 135, 12429–12433. [DOI] [PubMed] [Google Scholar]
- [186].Brewer TF, Chang CJ, J. Am. Chem. Soc 2015, 137, 10886–10889. [DOI] [PubMed] [Google Scholar]
- [187].Roth A, Li H, Anorma C, Chan J, J. Am. Chem. Soc 2015, 137, 10890–10893. [DOI] [PubMed] [Google Scholar]
- [188].Brewer TF, Burgos-Barragan G, Wit N, Patel KJ, Chang CJ, Chem. Sci 2017, 8, 4073–4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Bruemmer KJ, Walvoord RR, Brewer TF, Burgos-Barragan G, Wit N, Pontel LB, Patel KJ, Chang CJ, J. Am. Chem. Soc 2017, 139, 5338–5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Liu W, Truillet C, Flavell RR, Brewer TF, Evans MJ, Wilson DM, Chang CJ, Chem. Sci 2016, 7, 5503–5507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Bruemmer KJ, Green O, Su TA, Shabat D, Chang CJ, Angew. Chem. Int. Ed 2018, 57, 7508–7512; Angew. Chem. 2018, 130, 7630–7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Tang Y, Kong X, Xu A, Dong B, Lin W, Angew. Chem. Int. Ed 2016, 55, 3356–3359; Angew. Chem. 2016, 128, 3417–3420. [DOI] [PubMed] [Google Scholar]
- [193].He L, Yang X, Ren M, Kong X, Liu Y, Lin W, Chem. Commun 2016, 52, 9582–9585. [DOI] [PubMed] [Google Scholar]
- [194].Miller EW, Bian SX, Chang CJ, J. Am. Chem. Soc 2007, 129, 3458–3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Miller EW, Lin JY, Frady EP, Steinbach PA, Kristan WB, Tsien RY, Proc. Natl Acad. Sci. USA 2012, 109, 2114–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Woodford CR, Frady EP, Smith RS, Morey B, Canzi G, Palida SF, Araneda RC, Kristan WB, Kubiak CP, Miller EW, Tsien RY, J. Am. Chem. Soc 2015, 137, 1817–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Huang YL, Walker AS, Miller EW, J. Am. Chem. Soc 2015, 137, 10767–10776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Deal PE, Kulkarni RU, Al-Abdullatif SH, Miller EW, J. Am. Chem. Soc 2016, 138, 9085–9088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Kulkarni RU, Kramer DJ, Pourmandi N, Karbasi K, Bateup HS, Miller EW, Proc. Natl. Acad. Sci. USA 2017, 114, 2813–2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Kulkarni RU, Vandenberghe M, Thunemann M, James F, Andreassen OA, Djurovic S, Devor A, Miller EW, ACS Cent. Sci 2018, 4, 1371–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Ortiz G, Liu P, Naing SHH, Muller VR, Miller EW, J. Am. Chem. Soc 2019, 141, 6631–6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Franke JM, Raliski BK, Boggess SC, Natesan DV, Koretsky ET, Zhang P, Kulkarni RU, Deal PE, Miller EW, J. Am. Chem. Soc 2019, 141, 12824–12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Boggess SC, Gandhi SS, Siemons BA, Huebsch N, Healy KE, Miller EW, ACS Chem. Biol 2019, 14, 390–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Grenier V, Daws BR, Liu P, Miller EW, J. Am. Chem. Soc 2019, 141, 1349–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Liu P, Grenier V, Hong W, Muller VR, Miller EW, J. Am. Chem. Soc 2017, 139, 17334–17340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Grenier V, Walker AS, Miller EW, J. Am. Chem. Soc 2015, 137, 10894–10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Lazzari-Dean JR, Gest AMM, Miller EW, Elife 2019, 8, e44522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Kaur A, New EJ, Acc. Chem. Res 2019, 52, 623–632. [DOI] [PubMed] [Google Scholar]
- [209].Harris M, Kolanowski JL, O’Neill ES, Henoumont C, Laurent S, Parac-Vogt TN, New EJ, Chem. Commun 2018, 54, 12986–12989. [DOI] [PubMed] [Google Scholar]
- [210].Lim MH, Xu D, Lippard SJ, Nat. Chem. Biol 2006, 2, 375–380. [DOI] [PubMed] [Google Scholar]
- [211].Kaur A, Brigden KWL, Cashman TF, Fraser ST, New EJ, Org. Biomol. Chem 2015, 13, 4343–4349. [DOI] [PubMed] [Google Scholar]
- [212].Kaur A, Haghighatbin MA, Hogan CF, New EJ, Chem. Commun 2015, 51, 10510–10513. [DOI] [PubMed] [Google Scholar]
- [213].Loas A, Lippard SJ, J. Mater. Chem. B 2017, 5, 8929–8933. [DOI] [PubMed] [Google Scholar]
- [214].Lim MH, Lippard SJ, Inorg. Chem 2004, 43, 6366–6370. [DOI] [PubMed] [Google Scholar]
- [215].Rivera-Fuentes P, Lippard SJ, Acc. Chem. Res 2015, 48, 2927–2934. [DOI] [PubMed] [Google Scholar]
- [216].Choi MG, Cha S, Lee H, Jeon HL, Chang SK, Chem. Commun 2009, 7390–7392. [DOI] [PubMed] [Google Scholar]
- [217].Sasakura K, Hanaoka K, Shibuya N, Mikami Y, Kimura Y, Komatsu T, Ueno T, Terai T, Kimura H, Nagano T, J. Am. Chem. Soc 2011, 133, 18003–18005. [DOI] [PubMed] [Google Scholar]
- [218].Jung HS, Han JH, Kim ZH, Kang C, Kim JS, Org. Lett 2011, 13, 5056–5059. [DOI] [PubMed] [Google Scholar]
- [219].Ojida A, Nonaka H, Miyahara Y, Tamaru SI, Sada K, Hamachi I, Angew. Chem. Int. Ed 2006, 45, 5518–5521; Angew. Chem. 2006, 118, 5644 – 5647. [DOI] [PubMed] [Google Scholar]
- [220].Hitomi Y, Takeyasu T, Funabiki T, Kodera M, Anal. Chem 2011, 83, 9213–9216. [DOI] [PubMed] [Google Scholar]
- [221].Yu M, Bouley BS, Xie D, Enriquez JS, Que EL, J. Am. Chem. Soc 2018, 140, 10546–10552. [DOI] [PubMed] [Google Scholar]
- [222].Yu ZH, Chung CY, Tang FK, Brewer TF, Au-Yeung HY, Chem. Commun 2017, 53, 10042–10045. [DOI] [PubMed] [Google Scholar]
- [223].Michel BW, Lippert AR, Chang CJ, J. Am. Chem. Soc 2012, 134, 15668–15671. [DOI] [PubMed] [Google Scholar]
- [224].Sun M, Yu H, Zhang K, Wang S, Hayat T, Alsaedi A, Huang D, ACS Sens. 2018, 3, 285–289. [DOI] [PubMed] [Google Scholar]
- [225].Liu K, Kong X, Ma Y, Lin W, Angew. Chem. Int. Ed 2017, 56, 13489–13492; Angew. Chem. 2017, 129, 13674–13677. [DOI] [PubMed] [Google Scholar]
- [226].Liu K, Kong X, Ma Y, Lin W, Nat. Protoc 2018, 13, 1020–1033. [DOI] [PubMed] [Google Scholar]
- [227].Xu S, Liu H, Yin X, Yuan L, Huan S, Zhang X, Chem. Sci. 2019, 10, 320–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Zheng K, Lin W, Tan L, Chen H, Cui H, Chem. Sci 2014, 5, 3439. [Google Scholar]
- [229].Cao Y, Li D-W, Zhao L, Liu X, Cao X, Long Y, Anal. Chem 2015, 87, 9696–9701. [DOI] [PubMed] [Google Scholar]
- [230].Toussaint SNW, Calkins RT, Lee S, Michel BW, J. Am. Chem. Soc 2018, 140, 13151–13155. [DOI] [PubMed] [Google Scholar]
- [231].Taki M, Iyoshi S, Ojida A, Hamachi I, Yamamoto Y, J. Am. Chem. Soc 2010, 132, 5938–5939. [DOI] [PubMed] [Google Scholar]
- [232].Heffern MC, Park HM, Au-Yeung HY, Van de Bittner GC, Ackerman CM, Stahl A, Chang CJ, Proc. Natl. Acad. Sci. USA 2016, 113, 14219–14224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Au-Yeung HY, New EJ, Chang CJ, Chem. Commun 2012, 48, 5268–5270. [DOI] [PubMed] [Google Scholar]
- [234].Ke B, Ma L, Kang T, He W, Gou X, Gong D, Du L, Li M, Anal. Chem 2018, 90, 4946–4950. [DOI] [PubMed] [Google Scholar]
- [235].Au-Yeung HY, Chan J, Chantarojsiri T, Chang CJ, J. Am. Chem. Soc 2013, 135, 15165–15173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Aron AT, Reeves AG, Chang CJ, Curr. Opin. Chem. Biol 2018, 43, 113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Hirayama T, Okuda K, Nagasawa H, Chem. Sci 2013, 4, 1250. [Google Scholar]
- [238].Hirayama T, Tsuboi H, Niwa M, Miki A, Kadota S, Ikeshita Y, Okuda K, Nagasawa H, Chem. Sci 2017, 8, 4858–4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Niwa M, Hirayama T, Okuda K, Nagasawa H, Org. Biomol. Chem 2014, 12, 6590–6597. [DOI] [PubMed] [Google Scholar]
- [240].Hirayama T, Inden M, Tsuboi H, Niwa M, Uchida Y, Naka Y, Hozumi I, Nagasawa H, Chem. Sci 2019, 10, 1514–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Long L, Wang N, Han Y, Huang M, Yuan X, Cao S, Gong A, Wang K, Analyst 2018, 143, 2555–2562. [DOI] [PubMed] [Google Scholar]
- [242].Spangler B, Morgan CW, Fontaine SD, Vander Wal MN, Chang CJ, Wells JA, Renslo AR, Nat. Chem. Biol 2016, 12, 680–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Aron AT, Loehr MO, Bogena J, Chang CJ, J. Am. Chem. Soc 2016, 138, 14338–14346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Aron AT, Heffern MC, Lonergan ZR, Vander Wal MN, Blank BR, Spangler B, Zhang Y, Park HM, Stahl A, Renslo AR, Skaar EP, Chang CJ, Proc. Natl Acad. Sci. USA 2017, 114, 12669–12674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Muir RK, Zhao N, Wei J, Wang Y, Moroz A, Huang Y, Chen Y, Sriram R, Kurhanewicz J, Ruggero D, Renslo AR, Evans MJ, ACS Cent. Sci 2019, 5, 727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Zastrow ML, Radford RJ, Chyan W, Anderson CT, Zhang DY, Loas A, Tzounopoulos T, Lippard SJ, ACS Sens. 2016, 1, 32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Li H, Zhang P, Smaga LP, Hoffman RA, Chan J, J. Am. Chem. Soc 2015, 137, 15628–15631. [DOI] [PubMed] [Google Scholar]
- [248].Guo Z, Zhu W, Zhu M, Wu X, Tian H, Chem. Eur. J 2010, 16, 14424–14432. [DOI] [PubMed] [Google Scholar]
- [249].Tang B, Ding B, Xu K, Tong L, Chem. Eur. J 2009, 15, 3147–3151. [DOI] [PubMed] [Google Scholar]
- [250].Chen X, Baek K, Kim Y, Kim S, Shin I, Yoon J, Tetrahedron 2010, 66, 4016–4021. [Google Scholar]
- [251].Garner AL, Koide K, Chem. Commun 2009, 86–88. [DOI] [PubMed] [Google Scholar]
- [252].Garner AL, Koide K, J. Am. Chem. Soc 2008, 130, 16472–16473. [DOI] [PubMed] [Google Scholar]
- [253].Chen W, Wright BD, Pang Y, Chem. Commun 2012, 48, 3824–3826. [DOI] [PubMed] [Google Scholar]
- [254].Mahapatra AK, Roy J, Sahoo P, Tetrahedron Lett. 2011, 52, 2965–2968. [Google Scholar]
- [255].Egorova OA, Seo H, Chatterjee A, Ahn KH, Org. Lett 2010, 12, 401–403. [DOI] [PubMed] [Google Scholar]
- [256].Yang Y, Lee S, Tae J, Org. Lett 2009, 11, 5610–5613. [DOI] [PubMed] [Google Scholar]
- [257].Chatterjee A, Santra M, Won N, Kim S, Kim JK, Bin Kim S, Ahn KH, J. Am. Chem. Soc 2009, 131, 2040–2041. [DOI] [PubMed] [Google Scholar]
- [258].Carter KP, Young AM, Palmer AE, Chem. Rev 2014, 114, 4564–4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].Miller EW, Dickinson BC, Chang CJ, Proc. Natl Acad. Sci. USA 2010, 107, 15681–15686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Yang M, Haase AD, Huang F, Coulis G, Rivera KD, Dickinson BC, Chang CJ, Pappin DJ, Neubert TA, Hannon GJ, Boivin B, Tonks NK, Mol. Cell 2014, 55, 782–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [261].Nokin M-J, Durieux F, Peixoto P, Chiavarina B, Peulen O, Blomme A, Turtoi A, Costanza B, Smargiasso N, Baiwir D, Scheijen JL, Schalkwijk CG, Leenders J, De Tullio P, Bianchi E, Thiry M, Uchida K, Spiegel DA, Cochrane JR, Hutton CA, De Pauw E, Delvenne P, Belpomme D, Castronovo V, Bellahcène A, elife 2016, 5, e19375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Burgos-Barragan G, Wit N, Meiser J, Dingler FA, Pietzke M, Mulderrig L, Pontel LB, Rosado IV, Brewer TF, Cordell RL, Monks PS, Chang CJ, Vazquez A, Patel KJ, Nature 2017, 548, 549–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Kalappa BI, Anderson CT, Goldberg JM, Lippard SJ, Tzounopoulos T, Proc. Natl Acad. Sci 2015, 112, 15749–15754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Camarena V, Sant DW, Huff TC, Mustafi S, Muir RK, Aron AT, Chang CJ, Renslo AR, Monje PV, Wang G, eLife 2017, 6, e29750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [265].Lippert AR, ACS Cent. Sci 2017, 3, 269–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [266].Hananya N, Shabat D, Angew. Chem. Int. Ed 2017, 56, 16454–16463; Angew. Chem. 2017, 129, 16674–16683. [DOI] [PubMed] [Google Scholar]
- [267].Green O, Eilon T, Hananya N, Gutkin S, Bauer CR, Shabat D, ACS Cent. Sci 2017, 3, 349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [268].Edri R, Gal I, Noor N, Harel T, Fleischer S, Adadi N, Green O, Shabat D, Heller L, Shapira A, Gat-Viks I, Peer D, Dvir T, Adv. Mater 2019, 31, 1803895. [DOI] [PubMed] [Google Scholar]
- [269].Bin Li J, Chen L, Wang Q, Liu HW, Hu XX, Yuan L, Zhang XB, Anal. Chem 2018, 90, 4167–4173. [DOI] [PubMed] [Google Scholar]
- [270].Green O, Gnaim S, Blau R, Eldar-Boock A, Satchi-Fainaro R, Shabat D, J. Am. Chem. Soc 2017, 139, 13243–13248. [DOI] [PubMed] [Google Scholar]
- [271].Que EL, Chang CJ, Chem. Soc. Rev 2010, 39, 51–60. [DOI] [PubMed] [Google Scholar]
- [272].Xu Z, Liu C, Zhao S, Chen S, Zhao Y, Chem. Rev 2019, 119, 195–230. [DOI] [PubMed] [Google Scholar]
- [273].Golman K, in′t Zandt R, Thaning M, Proc. Natl Acad. Sci. USA 2006, 103, 11270–11275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [274].Nelson SJ, Kurhanewicz J, Vigneron DB, Larson PEZ, Harzstark AL, Ferrone M, van Criekinge M, Chang JW, Bok R, Park I, Reed G, Carvajal L, Small EJ, Munster P, Weinberg VK, Ardenkjaer-Larsen JH, Chen AP, Hurd RE, Odegardstuen L, Robb FJ, Tropp J, Murray JA, Sci. Transl. Med 2013, 5, 198ra108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [275].Gutte H, Hansen AE, Johannesen HH, Clemmensen AE, Ardenkjær-Larsen JH, Nielsen CH, Kjær A, Am. J. Nucl. Med. Mol. Imaging 2015, 5, 548–560. [PMC free article] [PubMed] [Google Scholar]
- [276].Yang S, Zeng Q, Guo Q, Chen S, Liu H, Liu M, McMahon MT, Zhou X, Talanta 2018, 184, 513–519. [DOI] [PubMed] [Google Scholar]
- [277].Louie AY, Hüber MM, Ahrens ET, Rothbächer U, Moats R, Jacobs RE, Fraser SE, Meade TJ, Nat. Biotechnol 2000, 18, 321–325. [DOI] [PubMed] [Google Scholar]
- [278].Keshari KR, Kurhanewicz J, Bok R, Larson PEZ, Vigneron DB, Wilson DM, Proc. Natl. Acad. Sci. USA 2011, 108, 18606–18611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [279].See Ref. [276].
- [280].Knox HJ, Chan J, Acc. Chem. Res 2018, 51, 2897–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [281].Reinhardt CJ, Chan J, Biochemistry 2018, 57, 194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [282].Reinhardt CJ, Zhou EY, Jorgensen MD, Partipilo G, Chan J, J. Am. Chem. Soc 2018, 140, 1011–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [283].Knox HJ, Hedhli J, Kim TW, Khalili K, Dobrucki LW, Chan J, Nat. Commun 2017, 8, 1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [284].Sun L, Wu Y, Chen J, Zhong J, Zeng F, Wu S, Theranostics 2019, 9, 77–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [285].Zeng L, Yuan Y, Jiang C, Mu J, Li F, Wan Y, Xu H, Qu J, Huang P, Lin J, Dyes Pigm. 2019, 165, 408–414. [Google Scholar]
- [286].See Ref. [190].
- [287].Carroll VN, Truillet C, Shen B, Flavell RR, Shao X, Evans MJ, VanBrocklin HF, Scott PJH, Chin FT, Wilson DM, Chem. Commun 2016, 52, 4888–4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [288].Al-Karmi S, Albu SA, Vito A, Janzen N, Czorny S, Banevicius L, Nanao M, Zubieta J, Capretta A, Valliant JF, Chem. Eur. J 2017, 23, 254–258. [DOI] [PubMed] [Google Scholar]
- [289].See Ref. [190].
- [290].Blank BR, Talukder P, Muir RK, Green ER, Skaar EP, Renslo AR, ACS Infect. Dis 2019, 5, 1366–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [291].António JPM, Russo R, Carvalho CP, Cal PMSD, Gois PMP, Chem. Soc. Rev 2019, 48, 3513–3536. [DOI] [PubMed] [Google Scholar]
- [292].Peiró Cadahía J, Previtali V, Troelsen NS, Clausen MH, MedChemComm 2019, 10, 1531–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [293].Lee MH, Sharma A, Chang MJ, Lee J, Son S, Sessler JL, Kang C, Kim JS, Chem. Soc. Rev 2018, 47, 28–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [294].Wang J, Zhang Y, Archibong E, Ligler FS, Gu Z, Adv. Biosyst 2017, 1, 1700084. [DOI] [PubMed] [Google Scholar]
- [295].Charkoudian LK, Pham DM, Franz KJ, J. Am. Chem. Soc 2006, 128, 12424–12425. [DOI] [PubMed] [Google Scholar]
- [296].Wei Y, Guo M, Angew. Chem. Int. Ed 2007, 46, 4722–4725; Angew. Chem. 2007, 119, 4806–4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [297].Jansová H, Macháček M, Wang Q, Hašková P, Jirkovská A, Potůčková E, Kielar F, Franz KJ, Šimůnek T, Free Radical Biol. Med 2014, 74, 210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [298].Dickens MG, Franz KJ, ChemBioChem 2010, 11, 59–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [299].Franks AT, Franz KJ, Chem. Commun 2014, 50, 11317–11320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [300].Festa RA, Helsel ME, Franz KJ, Thiele DJ, Chem. Biol 2014, 21, 977–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [301].Major Jourden JL, Cohen SM, Angew. Chem. Int. Ed 2010, 49, 6795–6797; Angew. Chem. 2010, 122, 6947–6949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [302].Zheng S, Guo S, Zhong Q, Zhang C, Liu J, Yang L, Zhang Q, Wang G, ACS Med. Chem. Lett 2018, 9, 149–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [303].Zhao Y, Pluth MD, Angew. Chem. Int. Ed 2016, 55, 14638–14642; Angew. Chem. 2016, 128, 14858–14862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [304].Powell CR, Dillon KM, Wang Y, Carrazzone RJ, Matson JB, Angew. Chem. Int. Ed 2018, 57, 6324–6328; Angew. Chem. 2018, 130, 6432–6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [305].Broaders KE, Grandhe S, Fréchet JMJ, J. Am. Chem. Soc 2011, 133, 756–758. [DOI] [PubMed] [Google Scholar]
- [306].Wilson DS, Dalmasso G, Wang L, Sitaraman SV, Merlin D, Murthy N, Nat. Mater 2010, 9, 923–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [307].Zhang Q, Zhang F, Chen Y, Dou Y, Tao H, Zhang D, Wang R, Li X, Zhang J, Chem. Mater 2017, 29, 8221–8238. [Google Scholar]
- [308].Hoang TT, Smith TP, Raines RT, Angew. Chem. Int. Ed 2017, 56, 2619–2622; Angew. Chem. 2017, 129, 2663 – 2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [309].Baker MS, Phillips ST, J. Am. Chem. Soc 2011, 133, 5170–5173. [DOI] [PubMed] [Google Scholar]
- [310].Baker MS, Phillips ST, Org. Biomol. Chem 2012, 10, 3595. [DOI] [PubMed] [Google Scholar]
- [311].Brooks AD, Yeung K, Lewis GG, Phillips ST, Anal Methods 2015, 7, 7186–7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [312].Zhao Y, Cerda MM, Pluth MD, Chem. Sci 2019, 10, 1873–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [313].Xu H, Xu H, Ma S, Chen X, Huang L, Chen J, Gao F, Wang R, Lou K, Wang W, J. Am. Chem. Soc 2018, 140, 16408–16412. [DOI] [PubMed] [Google Scholar]
- [314].Yan J, Lee S, Zhang A, Yoon J, Chem. Soc. Rev 2018, 47, 6900–6916. [DOI] [PubMed] [Google Scholar]
- [315].Roth ME, Green O, Gnaim S, Shabat D, Chem. Rev 2016, 116, 1309–1352. [DOI] [PubMed] [Google Scholar]
- [316].Lewis GG, DiTucci MJ, Phillips ST, Angew. Chem. Int. Ed 2012, 51, 12707–12710; Angew. Chem. 2012, 124, 12879–12882. [DOI] [PubMed] [Google Scholar]
- [317].Quimbar ME, Krenek KM, Lippert AR, Methods 2016, 109, 123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [318].Faleiro L, Kobayashi R, Fearnhead H, Lazebnik Y, EMBO J 1997, 16, 2271–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [319].Liu Y, Patricelli MP, Cravatt BF, Proc. Natl. Acad. Sci. USA 1999, 96, 14694–14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [320].Greenbaum D, Medzihradszky KF, Burlingame A, Bogyo M, Chem. Biol 2000, 7, 569–581. [DOI] [PubMed] [Google Scholar]
- [321].Adam GC, Sorensen EJ, Cravatt BF, Mol. Cell. Proteomics 2002, 1, 781–790. [DOI] [PubMed] [Google Scholar]
- [322].Activity-Based Protein Profiling (Eds.: Cravatt BF, Hsu K-L, Weerapana E), Springer International Publishing, Cham, 2019. [Google Scholar]
- [323].Stephanopoulos N, Francis MB, Nat. Chem. Biol 2011, 7, 876–884. [DOI] [PubMed] [Google Scholar]
- [324].Spicer CD, Davis BG, Nat. Commun 2014, 5, 4740. [DOI] [PubMed] [Google Scholar]
- [325].Mizukami S, Hori Y, Kikuchi K, Acc. Chem. Res 2014, 47, 247–256. [DOI] [PubMed] [Google Scholar]
- [326].Shannon DA, Weerapana E, Curr. Opin. Chem. Biol 2015, 24, 18–26. [DOI] [PubMed] [Google Scholar]
- [327].Counihan JL, Ford B, Nomura DK, Curr. Opin. Chem. Biol 2016, 30, 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [328].DeGruyter JN, Malins LR, Baran PS, Biochemistry 2017, 56, 3863–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [329].Hoyt EA, Cal PMSD, Oliveira BL, Bernardes GJL, Nat. Rev. Chem 2019, 3, 147–171. [Google Scholar]
- [330].Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MBD, Bachovchin DA, Mowen K, Baker D, Cravatt BF, Nature 2010, 468, 790–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [331].Chalker JM, Bernardes GJL, Lin YA, Davis BG, Chem. Asian J 2009, 4, 630–640. [DOI] [PubMed] [Google Scholar]
- [332].Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, Gray NS, Chem. Biol 2013, 20, 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [333].Ward CC, Kleinman JI, Nomura DK, ACS Chem. Biol 2017, 12, 1478–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [334].Cuesta A, Taunton J, Annu. Rev. Biochem 2019, 88, 365–381. [DOI] [PubMed] [Google Scholar]
- [335].Simon GM, Cravatt BF, J. Biol. Chem 2010, 285, 11051–11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [336].Lin S, Yang X, Jia S, Weeks AM, Hornsby M, Lee PS, Nichiporuk RV, Iavarone AT, Wells JA, Toste FD, Chang CJ, Science 2017, 355, 597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [337].Taylor MT, Nelson JE, Suero MG, Gaunt MJ, Nature 2018, 562, 563–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [338].Christian AH, Jia S, Cao W, Zhang P, Meza AT, Sigman MS, Chang CJ, Toste FD, J. Am. Chem. Soc 2019, 141, 12657–12662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [339].Jia S, He D, Chang CJ, J. Am. Chem. Soc 2019, 141, 7294–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [340].Ban H, Gavrilyuk J, Barbas CF, J. Am. Chem. Soc 2010, 132, 1523–1525. [DOI] [PubMed] [Google Scholar]
- [341].Seim KL, Obermeyer AC, Francis MB, J. Am. Chem. Soc 2011, 133, 16970–16976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [342].Mortenson DE, Brighty GJ, Plate L, Bare G, Chen W, Li S, Wang H, Cravatt BF, Forli S, Powers ET, Sharpless KB, Wilson IA, Kelly JW, J. Am. Chem. Soc 2018, 140, 200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [343].Antos JM, Francis MB, J. Am. Chem. Soc 2004, 126, 10256–10257. [DOI] [PubMed] [Google Scholar]