

Abstract
Drug screening in oncology, especially for triple-negative breast cancer (TNBC), has high demand but remains unsatisfactory. Currently available models are either nonrepresentative of the complex tumor microenvironment or only suitable for low throughput screening, resulting in a low-yield success for drug development. To tackle these issues, we developed the L-TumorChip system, a three-layered microfluidic tumor-on-a-chip platform integrating tumor microvasculature and tumor-stromal microenvironment with high throughput screening capability. Its layered and modular design is readily scalable through simple integration of multiple units. We validated L-TumorChip with a TNBC model. Our L-TumorChip system emulated certain tumor-stroma complexities and tumor-endothelium interactions, including TNBC invasion through the leaky microvasculature and angiogenesis. Additionally, with this L-TumorChip, we investigated the influence of different stromal cells, including normal fibroblasts, mesenchymal stem cells and cancer-associated fibroblasts (CAF), on cancer cell growth as well as the stromal effects on drug responses to doxorubicin treatment. The presence of CAF delayed drug pharmacokinetics, while apoptotic responses indicated by caspase-3 activities was higher in coculture with normal fibroblasts. Collectively, our L-TumorChip system represents a translational high-throughput screening toolkit that enables drug screening with a scenario closer to the in vivo conditions. This potential use may therefore facilitate development of new cancer drugs.
Keywords: Tumor-on-a-chip, microfluidics, drug screening, tumor microenvironment
Graphical Abstract
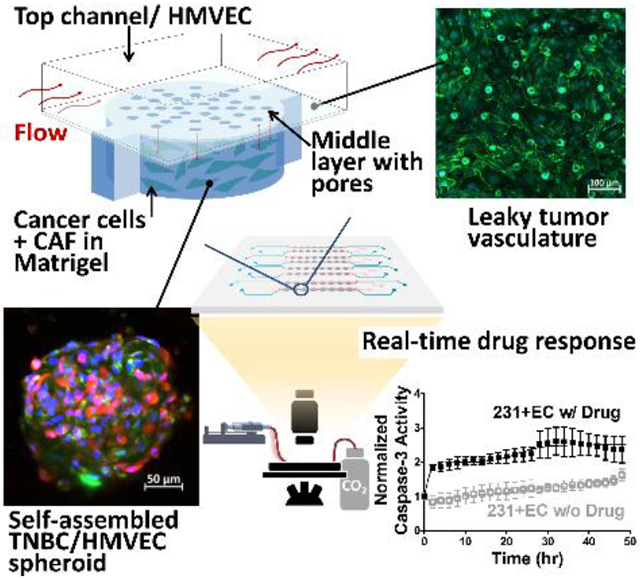
A microfluidic tumor-on-a-chip with integration of tumor-microvasculature and tumorous-stromal microenvironment offers a more representative model to study the cell interactions within tumor microenvironment and between tumor and endothelium barrier. This platform enables more precise high throughput drug screening and drug pharmacokinetic studies.
Oncology is a field with significant demand of drug development due to the large patient population and disease complexity. Despite a significant number of preclinical projects and clinical trials in the pipeline, oncology drug development remains unsatisfactory with few exceptions. For example, according to the reports analyzing clinical developments of over 21,000 compounds, the success rate for oncology drug development from phase 1 clinical trials to FDA-granted approval is less than 4%.[1] The low success rate for compounds already in clinical trials illuminates the shortcomings in current pre-screening models. Especially triple-negative breast cancer (TNBC), the most intractable cancer type in women, one of the earliest immunotherapy drugs for use against TNBC was approved by FDA only as recently as in 2019, demonstrating the difficulty of oncology drug development.[2] This can be attributed to the gap between the experimental models and in vivo physiological and pathological realities. Compound screening in a more physiologically faithful manner is therefore an unmet need for improving the efficiency of the drug development pipeline. The current gold standard uses patient-derived xenograft mouse models, which only offer a murine physiological microenvironment to transplanted human cells. The main bottlenecks in mouse models that hinder their use in new drug discovery are throughput and batch-to-batch variations. Consequently, current strategies of drug discovery and development still mainly rely on conventional monolayer culture (2D culture) for pre-screening. However, there is strong evidence that cancer cells in 2D culture behave differently in proliferation,[3] gene expressions,[4] and drug responses[5] from those in natural tissue architecture. There is therefore a need for novel approaches better replicating the in vivo microenvironment for a more faithful and clinical relevance.
Several static 3D culture approaches, such as spheroid culture or scaffold/hydrogel embedding culture, have been developed to partially recapitulate physiological microenvironment configurations.[6] These static culturing methods cannot reproduce dynamic nutrient transport availed to cells in vivo. To address this deficiency, the tumor-on-a-chip concept emerged to better emulate physical in vivo conditions using human cell sources through (1) flow-induced mechanical stimulation and precise control of nutrient/waste transport, (2) structuring physiologically relevant architecture, and (3) stromal-cancer epithelium interactions. For example, integration of microfluidic chip with spheroid culture without reconstructed microvasculature or stromal components was reported to validate the effects of flow and tumor spheroid size on drug treatment results.[7] Nevertheless, microvasculature in tumor microenvironment plays an important role in regulating drug delivery, cancer cell survival and tumor progression. We and many other groups have reported a wide variety of microfluidic designs to replicate tumor microvasculature to study the endothelial barrier for drug delivery applications.[8] These models enable the study of juxtacrine and paracrine signals involved in the development of chemotherapeutic drug resistance.[9] In addition to the endothelial cells, cancer associated fibroblast (CAF), the most abundant stromal cell in solid tumor, is another key component in tumorigenesis as they can remodel the extracellular matrix and modulate the anti-tumor immune response through interactions with the cancer cells.[10] As such, CAF has also been incorporated into the recent tumor-on-a-chip designs, and these studies demonstrate the pronounced effects of CAF in conjunction with chemotherapeutic drugs on cancer cell migration and survival.[11] Although these systems help to gain insights on the complex tumor hallmarks, their throughput remains a major obstacle that limits their clinical translation. Most of the designs are constructed with all channels in a planar configuration thereby limiting scalable throughput per chip. Some designs vertically stack their final structure, with a filter membrane or hydrogel layer to separate tumor and vasculature zones, but such designs are prone to cross-talk or leakage through these separation layers when scaled up as multi-units on a single chip.
To resolve the aforementioned shortcomings, in this study, we develop the L-TumorChip system to deliver four features (i.e. cancer-stromal interaction, flow, microvasculature and throughput). L-TumorChip is a three-layered polydimethylsiloxane (PDMS) microfluidic array with vertically stacked spatial reconstruction of microvessels and tumor compartments, supported by a controllable continuous perfusion of media. The tumor compartment comprises cancer cells and stromal cells, providing a physiologically relevant tumor microenvironment in a readily scalable fashion for high-throughput drug-screening (Figures 1A). The regenerated endothelial layer was characterized by junctional proteins and permeability measurement. Breast cancer cell invasion and initial angiogenesis are notably present. Furthermore, the influence of stromal cells on cancer cell growth and on real-time drug response were investigated. Altogether, we demonstrated the capability of this L-TumorChip to study tumor-stroma interaction and drug kinetics.
Figure 1.

Design of the three-layer microfluidic device (L-TumorChip) used in this study. (A) Schematic illustation of the microfluidic cell array design. Cancer and stromal cells were encapsulated in the Matrigel and seeded into the bottom layer, taking residence in the bottom chambers and the interconnecting channels between chambers (B) The cross-section view of one unit in the device with simulated flow results by COMSOL. Arrows indicate the flow direction. (C) Variations between chambers when seeded with same numbers of RFP-expressing MDA-MB-231 cells. Fluorescent intensity from each chamber was normalized to the mean of intensity among chambers. Results are reprented as average ± standard deviation (S.D.; n = 16).
L-TumorChip system was built on the foundation of our previous work,[8e] we redesigned the system to better depict the heterogeneous tumor microenvironment. The top channel (990 × 150 μm; width × height), lined with a confluent layer of human microvascular endothelial cells (HMVEC), and the bottom cylindrical chamber (490 μm in radius, 200 μm in height) with Matrigel-encapsulated cancer cells, were separated by a 25μm-thick membrane with a cluster of pores (20 μm in diameter). The device design was geometrically optimized for administering physiological flow parameters by computational fluid dynamics simulation with the prescribed boundary flow conditions described in our previous works.[8e, 12] The inlet velocity boundary condition of the top channel was set to 100 μm/s as set in our experimental conditions, which was in the range of velocity in human capillary, and the hydraulic permeability of 2×10-14 m2 of a 4 mg/mL Matrigel was used for the bottom chamber. As the simulated flow pattern shown in Figure 1B, the patterned pores in the middle PDMS membrane enables controlled cell communication and nutrient/waste exchange between the bottom chamber and the top channel. In addition, this vertical stacking design for each unit allowed the system to be scaled up by simply arraying the units planarly with appropriate interconnections. To ascertain the L-TumorChip’s utility in robust drug screening and biological applications, we first confirmed the cell seeding uniformity between units arrayed in sequence and in parallel across the platform. We seeded our L-TumorChip chambers with the same density of red fluorescent protein (RFP)-expressing breast cancer cells (MDA-MB-231) and separately confirmed the correlation between RFP signal intensity and cell numbers (Figure S1, Supporting Information). From the RFP signal measurement, we detected a minimal cell density variation between units (coefficient of variation <15%, Figure 1C), demonstrating repeatability and feasibility of the arrayed units as replicates.
After confirming uniform seeding density across units, we characterized and evaluated the endothelial component of the L-TumorChip system. Endothelium plays a key role in controlling the interactions and transport of solutes, proteins and cells between blood vessels and tissues. To mimic the transport phenomena of capillaries in tumor, our L-TumorChip system was composed of a monolayer of microvascular endothelial cells (Figure 2A). To characterize the vasculature structure of this endothelial monolayer in L-TumorChip, HMVECs were first stained with two endothelial junction proteins, CD31 (also known as PECAM-1; Figures 2B and 2C) and VE-cadherin (Figures 2D and 2E). The presented organization of CD31 and VE-cadherin reflect the endothelial junctional integrity. [13] Abundant expressions of these two junction proteins were found in the staining results and displayed cell boundaries that were not tightly adjacent, indicating the leaky properties of endothelial monolayer in L-TumorChip. This mimics the tumor microvasculature, known to be relatively immature and leakier due to the activated proangiogenic pathways in the tumor microenvironment overwhelming the antiangiogenic factor production.[14] We next measured the permeability of regenerated endothelium in L-TumorChip to investigate whether it reflected this visually noticeable leaky characteristic from junction protein staining. The permeability was determined by detecting the fluorescent solutes (10 and 70 kDa dextrans) from the side view of one unit (Figure 1B and Figure S2, Supporting Information). Prior to the permeability measurement, calibration and the sampling depth verification were carried out under the same instrument settings (Figure S3, Supporting Information). Characterizations of endothelial monolayer via cell-cell junction protein staining and the permeability measurement were conducted at day 2 post-seeding. Compared with normal vascular permeability (in 10-7 cm/s magnitude for both dextrans),[15] a relatively higher permeability (4.00×10-5 and 1.82×10-5 cm/s for 10 and 70 kDa dextrans, respectively) of the endothelium layer was measured (Figure 2F), consistent with the staining results. The relatively high permeability of HMVECs in L-TumorChip could be attributed to their interactions with highly metastatic MDA-MB-231 cells and the presence of vascular permeability factor, vascular endothelial growth factor (VEGF),[16] in the commercial HMVEC culture medium (Lonza EGM™-2). The MDA-MB-231 cell, originally derived from a metastatic site of the breast adenocarcinoma patient, is known to be aggressive and highly invasive, spontaneously metastasizing to lung,[17] brain,[18] and bone[19] after intravenous, intracardiac injection or orthotropic implantation in mice. The increased tumor vascular permeability enables the invasion of dissembled tumor cells from primary site.[9d, 20] Cancer cells altered endothelial permeability through the secretion of growth factors (such as VEGF),[16, 21] cytokines[22] and chemokines.[23] To exclude the influence of VEGF in the commercial medium of HMVEC from the observation, another endothelial cell cultured in the media without VEGF was chosen to verify the integrity of regenerated endothelial layer in L-TumorChip. When using the normal human brain endothelial cell line, hCMEC/D3, the endothelium permeability was 2.9×10-6 and 3.7×10-7 cm/s for sodium fluorescein and 70kDa dextran, respectively, and these results were consistent with the measurements of the intact blood-brain-barrier (Figure S4, Supporting Information) from other studies[24], suggesting the simulated representation of leaky tumor vasculature with the aforementioned higher permeability values when tight junctions are notably missing.
Figure 2.

Characteristics of HMVEC monolayer in L-TumorChip at day 2 post-seeding. (A) Representative phase-contrast image of the confluent HMVEC endothelium monolayer. Representative CD-31 immunostained images with (B) 10× and (C) 20× magnifications under the confocal microscope. Images are a maximum intensity projection of the full z-stack images captured from bottom to top layers. Representative images of the endothelium stainted with VE-cadherin antibody with (D) 10× and (E) 20× magnifications under the confocal microscope, focusing on slices above and proximal to the middle layer. The slices obtained from this subset of layers is shown as a maximum intensity projection. (F) Permeability of HMVEC barrier in L-TumorChip under the interaction with MDA-MB-231 cancer cells. Cancer cells were tagged with GFP in (B) and (C) or RFP in (D) and (E). Hoechst dye was used to stain nuclei. Results are represented as average ± S.D. (n = 4). Significance was determined using 2-way ANOVA and Tukey’s Multiple Comparison Test (**, p < 0.01).
Leaky vasculature in tumor may facilitate the cancer invasion,[20, 21b] and our L-TumorChip provides a high-throughput way to study the relationship between endothelial hyperpermeability and tumor metastasis. To study the cell migration in L-TumorChip, we again chose the RFP-expressing MDA-MB-231 and labeled the HMVEC with the PKH67 dye, a long-term green fluorescent cell tracker. After a 7-day culture in L-TumorChip, we observed cancer cells crossing the endothelium to the top channel (Figures 3A and 3B), while HMVECs migrated from the top channel to the tumor cell mass (bottom chamber) and began forming self-assembled spheroids with cancer cells in the bottom chamber (Figures 3A, 3C and 3D). The migration dynamics of GFP-expressing MDA-MB-231 was observed using a wide-field fluorescence microscope. The cancer cells invasively disrupts the endothelial layer as they gradually migrated to the top channel, highlighting their capacity to directly alter endothelial permeability, while concurrently forming more compact spheroid structures within their original seeded chambers (Figure S5, Supporting Information). These results demonstrate that the L-TumorChip recapitulate marked physiological behaviors of cancer cell invasion and initiation of angiogenesis. Interestingly, the presence of endothelial cells in the device correlates with the formation of cancer spheroids. More cell aggregates were observed when coculturing with HMVECs in L-TumorChip (Figure S6, Supporting Information). When cultured without the support of endothelial cells, cancer cell aggregates tended to lose their coherent structures over the time (Figure S6, Supporting Information). The results reveal that the endothelium in L-TumorChip not only functions as a barrier but also participates in tumor progression.
Figure 3.

Cell migration patterns of MDA-MB-231 and HMVEC at day 7 post-seeding. (A) Representative 3D-reconstructed image of one chamber in L-TumorChip obtained from Z-axis sliced confocal images (B) Representative image taken with focal plane at top channel Arrows indicate the migrated RFP-expressing breast cancer cells having invaded the green fluorescent endothelial barrier at the top channel. White dashed oulines indicate the outline of channel in the observing plane and gray round dots depict the outline of bottom chamber in an out-of-view plane. (C) Representative image taken with focal plane at bottom chamber. The white dashed oulines here indicate the bottom chamber in the observing plane and gray round dots depict the outline of the channel in an out-of-view plane. (D) Representative 3D-reconstructed image of the self-assemble MDA-MB-231/HMVEC spheriod. RFP-expressing MDA-MB-231 and PKH67-stained green HMVEC cells were initially seeded separately in the bottom chamber and the top channel, respectively.
Tumor microenvironment is a highly heterogeneous mixture composed of endothelial cells, fibroblasts, infiltrated immune cells, epithelial cells, adipocytes and mesenchymal stem cells (MSCs) embedded in the extracellular matrix. Among these, cancer-associated fibroblast (CAF) is the most abundant stromal cell type in tumor microenvironment.[25] CAFs modulate cancer progression through direct contacts with the cancer cells, paracrine signaling and extracellular matrix remodeling. To replicate the fibroblast-mediated complex tumor microenvironment in L-TumorChip, normal fibroblasts or CAFs were introduced in the bottom chambers and co-cultured with the MDA-MB-231 cells. For CAF generation, we followed the previously reported methods to differentiate MSCs into CAFs by culturing MSCs in the MDA-MB-231 cell-conditioned media for 30 days.[26] Following the same reported characterization methods, we confirmed that CAFs were successfully derived from MSCs noted by their elevated expression of myofibroblast biomarkers, including α-smooth muscle actin (α-SMA), fibroblast activation protein (FAP) and vimentin (Figure 4A). We observed similar patterns from the flow cytometry analyses, the FAP expression in the derived CAF was upregulated by 1.7 times compared with the untreated MSC (Figure 4B). In contrast, normal fibroblast and MSC were reported to have low FAP expression level.[27] The stromal cells (normal fibroblasts, CAFs or MSCs) were mixed with the RFP-expressing MDA-MB-231 cells in a 1:1 ratio and seeded in Matrigel in the bottom chamber. At 24h post-seeding, HMVECs were seeded in the top channel. The proliferation of cancer cells was interpreted by measuring the RFP signal in the entire chamber captured using the z-slicing fluorescence imaging microscope.
Figure 4.

Characterizations of MSC-derived CAF and its influence on cancer cell growth when cocultured in L-TumorChip. (A) Representative images of differentiated cells stained with the CAF-specific biomarkers (CD31 used as a negative control here). (B) Flow cytometry analysis on FAP/α-SMA expressions in MSC and the derived CAF. Results are represented as average ± S.D. (n = 3). Significance was determined using Student t-test (*, p < 0.05). (C) The proliferation and (D) doubling time of GFP-expressing MDA-MB-231 under different tumor microenvironment conditions in the device. Results are represented as average ± S.D. (n = 4). Significance was determined using one-way ANOVA and Tukey’s Multiple Comparison Test (*, p < 0.05; ***, p < 0.001).
As shown in Figures 4C and 4D, cancer cell grew slightly slower without statistical significance in the presence of HMVEC compared with cancer cell monoculture, agreeing with the in vivo observations from the study that co-injected human microvascular endothelial cells suppressed tumor growth compared with MDA-MB-231 injection without any endothelial cells, while lymphatic endothelial cells promoted tumor growth.[28] However, some studies reported contradictory findings related to effects of endothelial cells on cancer cell growth. For instance, colorectal cancer cells pre-treated with liver parenchymal endothelial cell-conditioned medium prior to the administration resulted in increased tumor growth in vivo.[9b] Blood flow has been known to affect endothelial cell functions.[29] Thus the difference may be partly attributable to collecting conditioned medium from static culture. The inconsistent observations of endothelial effects on tumor cell growth in literature may be caused by different experimental setting including different types of endothelial cells, dynamic/static culture conditions and 2D/3D culture.
For all coculture conditions of cancer cells with endothelial cells shown in Figure 4C and 4D, normal fibroblasts reduced cancer cell growth, indicated by a longer doubling time while both CAF and MSC promoted cancer cell proliferation when compared against cancer cells cultured alone with endothelial cells. CAF has been reported to have both anti- and pro-tumor activities, depending on the stage of tumor progression. At the initial stage, fibroblasts of a tumor tend to suppress the tumor growth as it emulates a “wound” state, but tumors being “wounds that do not heal”, [30] the constant actions of CAF on tumor sites can gradually become predominantly pro-tumorigenic.[31] This anti-/pro-tumor transition may be attributed to one of the suggested origins of CAFs being resident fibroblasts in the tumor microenvironment, as our results with normal fibroblast suppressing cancer cell growth and CAFs instead promoting such growth simulates this transitional behavior. Similarly, studies have also demonstrated that MSCs have both promoting and suppressive influence on tumor progression, depending on its origins, cancer types and the amount of MSCs.[27, 32] CAFs count in a range of 30-70% of all the cell types in the typical tumor microenvironment,[25] where only 0.01-1.1% account for MSC.[33] Therefore, our experimental setting (50%) reflects the average in vivo scenario for CAF but may overemphasize the MSC effect. In this study, we have demonstrated the effects of stromal cells on cancer cell growth. Interactions between endothelium and stromal cells may also influence on the growth of cancer cells, which warrants further studies by comparing the tumor-stromal coculture with and without the presence of an endothelial layer.
Another important feature of L-TumorChip is its capability of monitoring drug response in real time, which allows to study drug kinetics in a high throughput manner with tumor microenvironment and continuous perfusion. To demonstrate this feasibility, we chose a commonly-used, FDA-approved breast cancer drug, doxorubicin, to evaluate the drug response of MDA-MB-231 under different stromal conditions. Following a similar fluorogenic caspase-3 assay reported in the 2D culture,[8e] we were able to measure the dynamics of drug responses by monitoring the fluorescence intensity induced by intracellular caspase-3 activity. Doxorubicin and fluorogenic dye-linked caspase-3 enzyme substrate were mixed in medium and introduced into the channel lined with regenerated endothelium at the day 2 post-seeding with a continuous flow for another 48 h. To ensure proper endothelial layer integrity prior to any drug treatment, the permeability was characterized on the same day as drug treatment would begin (2 day post-seeding, Figure 2). Li et al. demonstrated negligibly low adsorption of calcein and 5(6)-carboxytetramethylrhodamine (TMR) on the surface of PDMS, wherein doxorubicin hold physicochemical characteristics intermediate of them but closer to TMR.[34] In a static condition, the adsorbed TMR signals on PDMS was 14% more than that on glass, but lower than polystyrene. In dynamic studies in microchannels dimensionally comparable to our L-TumorChip channels, the adsorption was negligible (<0.002%).
The caspase-3 activity in each bottom chamber with a unique tumor microenvironment condition was monitored for 48 h. As shown in Figures 5A, doxorubicin treatment induced cell apoptosis as higher caspase-3 activities were observed by comparison between treated and non-treated groups. We also observed that the endothelium hampered the apoptotic effects of doxorubicin as noted by comparing the caspase activity between the groups cultured with and without HMVEC. The delayed and attenuated caspase-3 activity observed in the presence of HMVEC monolayer can be fitted to a sequential bi-logistic model:
| (1) |
where S is the activity of caspase-3 interpreted from the normalized fluorescent intensity while ki, ai and bi are the carrying capacity, the activity rate and the inflection timepoint, respectively.
Figure 5.

Drug response of MDA-MB-231 to doxorubicin under different conditions in L-TumorChip. (A) Drug response kinetics of MDA-MB-231 cell in the presence of HMVEC treated with or without doxorubicin and the effect of presence of HMVEC endothelium barrier in the device. Caspase-3 activity in the bottom chamber was normalized to time 0 fluorescence intensity value in each condition. The drug was given at day 2 post-seeding. Results are represented as average ± S.D. (n = 3). Significance between treated and non-treated groups (both cultured with HMVEC) at the endpoint (48h) was determined using Student t-test (*, p < 0.05) (B) Caspase-3 activity of MDA-MB-231 at 48h post-treatment. Drug response kinetics of MDA-MB-231 under different tumor microenvironment conditions. Results are represented as average ± S.D. (n = 3).
Particularly, the extended second inflection timepoint (b2,231alone=18.44, b2,231+EC=22.56) and lower total capacity (k1+k2,231alone=2.78, k1+k2,231+EC=2.50) indicated the influence of endothelium to the drug treatment (Tables S1, Supporting Information). However, the actual mechanism of two-steps response was unclear, warranting further studies. Constructing tumor microenvironment with normal fibroblast and MDA-MB-231 enhanced cell apoptosis as observed in the shortened b2 (b2,231/NF+EC=22.12) and higher k1+k2 (k1+k2,231/NF+EC=3.36) when compared with the group co-cultured with CAFs or MSCs under doxorubicin treatment (Figure 5B and Table S1, Supporting Information). In fact, there was a reverse correlation between the proliferation rate (Figure 4D) and caspase-3 activity in drug-free condition (Figure S7A, Supporting Information). Lower proliferation capacity in coculturing with normal fibroblasts corresponded to the higher apoptotic response. Furthermore, the caspase-3 activity of normal fibroblast coculture at 48 hours of doxorubicin treatment retained the drug-free caspase activity that remained unaffected by the drug itself (Figure S7C, Supporting Information). However, the drug-induced caspase activity of the cocultures other than that with normal fibroblasts showed a significantly more pronounced effect compared to their respective drug-free baseline levels (Figure S7D and E, Supporting Information).
Co-culture with CAF or MSC delayed the drug response, leading to lower activity rate (a2, 231+EC=0.33, a2, 231/CAF+EC=0.28, a2, 231/MSC+EC=0.14) and extended second inflection timepoint (b2, 231+EC=22.56, b2, 231/CAF+EC=33.02, b2, 231/MSC+EC=41.72). Notably, the observed apoptotic response came from the cells in the bottom chamber, and the total number of seeded cells, with or without stromal cells, in each bottom chamber remained the same across all conditions. The higher caspase-3 activity in coculturing with normal fibroblast indicated the increased drug resistance in coculturing with CAF/MSC was not due to the increased signaling distance between cancer cells themselves in the coculture conditions, which only contained half the amount of cancer cells comparing with MDA-MB-231 alone (with endothelium). Evidences have shown that both CAF and MSC can not only boost cancer cells’ proliferation but also reduce apoptosis.[35] Moreover, they can exclude chemotherapeutic drug accumulation in cancer cells.[36] These possible phenomena can explain the delayed caspase-3 activities that we observed in our device. The major difference between the CAF and MSC groups occurred at 40 h, becoming significant after 48 h of the measurement (p < 0.05) with higher caspase activity in MSC co-cultures. In addition, the second activity rate (a2) of the group co-cultured with MSC was twice of that cocultured with CAF (a2, 231/CAF+EC=0.28, a2, 231/MSC+EC=0.14). The lower caspase-3 activity in CAF-carrying tumor microenvironment might also be attributed to the enhanced production of extracellular matrix by CAF,[10b, 37] thereby enhancing the physical resistance against drug transport. We did not expect much cancer cell migration during drug treatment since only minimal migration on both days 3 and 5 post-seeding was visually observable through the confinement of the cancer cells within the bottom chamber perimeter (Figure S5, Supporting Information).. In addition, due to the proliferation-inhibitory nature of doxorubicin, the migration of endothelial cells was not expected. The migration of MBA-MD-231 during drug treatment in a longer experimental time frame might be an interesting point for further investigations.
In summary, we demonstrated a high throughput screening device with an in vivo-inspired tumor microenvironment and perfused flow. This L-TumorChip system mimicked the leaky endothelium barrier and partly the complex tumor microenvironment, which subsequently alters the drug response. Physiological cell behaviors (cancer invasion and endothelial angiogenesis) were observed in the L-TumorChip as well. More importantly, it allowed us to monitor the dynamic drug response in real time. The function of a present endothelial barrier in drug response and the drug resistance mediated by CAF were observed in the device, respectively. The results provide a new outlook on the design of a platform for screening oncology compounds in a high throughput manner with mimicked tumor microenvironment. Taken together, a physiologically relevant tumor microenvironment, composed of cancer cells, CAFs, MSCs and endothelial monolayer, has been reconstructed in the microfluidic high throughput screening device, showing the potential of this platform for clinically translatable high-throughput drug screening and drug kinetics studies.
Supplementary Material
Acknowledgements
The authors would like to acknowledge the instrumental support from the nanofabrication facility at CUNY-Advanced Science Research Center and the Flow Cytometry Core Facility at Columbia Center for Translational Immunology. This work is supported by Pershing Square Sohn Cancer Research Alliance (S.W.) and NIH (K.W.L. and B.M.F., UG3/UH3TR002151).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library.
Contributor Information
Chun-Wei Chi, Department of Biomedical Engineering, CUNY- The City College of New York, New York NY, 10031, USA.
Yeh-Hsing Lao, Department of Biomedical Engineering, Columbia University, New York, NY 10027, USA.
A.H. Rezwanuddin Ahmed, Department of Biomedical Engineering, CUNY- The City College of New York, New York NY, 10031, USA.
Elizabeth C. Benoy, Department of Biomedical Engineering, CUNY- The City College of New York, New York NY, 10031, USA
Chenghai Li, Department of Biomedical Engineering, CUNY- The City College of New York, New York NY, 10031, USA.
Zeynep Dereli-Korkut, Department of Biomedical Engineering, CUNY- The City College of New York, New York NY, 10031, USA.
Bingmei M. Fu, Department of Biomedical Engineering, CUNY- The City College of New York, New York NY, 10031, USA
Kam W. Leong, Department of Biomedical Engineering, Columbia University, New York, NY 10027, USA
Sihong Wang, Department of Biomedical Engineering, CUNY- The City College of New York, New York NY, 10031, USA.
References
- [1].Wong CH, Siah KW, Lo AW, Biostatistics 2019, 20, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Leo CP, Leo C, Szucs TD, Nat Rev Drug Discov 2020, 19, 11; [DOI] [PubMed] [Google Scholar]; b) Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Dieras V, Hegg R, Im SA, Shaw Wright G, Henschel V, Molinero L, Chui SY, Funke R, Husain A, Winer EP, Loi S, Emens LA, I. M. T. Investigators, N Engl J Med 2018, 379, 2108. [DOI] [PubMed] [Google Scholar]
- [3].a) Cavo M, Fato M, Penuela L, Beltrame F, Raiteri R, Scaglione S, Sci Rep 2016, 6, 35367; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chitcholtan K, Asselin E, Parent S, Sykes PH, Evans JJ, Exp Cell Res 2013, 319, 75. [DOI] [PubMed] [Google Scholar]
- [4].a) Kapalczynska M, Kolenda T, Przybyla W, Zajaczkowska M, Teresiak A, Filas V, Ibbs M, Blizniak R, Luczewski L, Lamperska K, Arch Med Sci 2018, 14, 910; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lee JM, Mhawech-Fauceglia P, Lee N, Parsanian LC, Lin YG, Gayther SA, Lawrenson K, Lab Invest 2013, 93, 528; [DOI] [PubMed] [Google Scholar]; c) Zschenker O, Streichert T, Hehlgans S, Cordes N, Plos One 2012, 7, e34279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Melissaridou S, Wiechec E, Magan M, Jain MV, Chung MK, Farnebo L, Roberg K, Cancer Cell Int 2019, 19, 16; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Souza AG, Silva IBB, Campos-Fernandez E, Barcelos LS, Souza JB, Marangoni K, Goulart LR, Alonso-Goulart V, Curr Pharm Des 2018, 24, 1689; [DOI] [PubMed] [Google Scholar]; c) Weaver VM, Lelievre S, Lakins JN, Chrenek MA, Jones JC, Giancotti F, Werb Z, Bissell MJ, Cancer Cell 2002, 2, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Langhans SA, Front Pharmacol 2018, 9, 6; [DOI] [PMC free article] [PubMed] [Google Scholar]; Lv D, Hu Z, Lu L, Lu H, Xu X, Oncol Lett 2017, 14, 6999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ran R, Wang HF, Hou F, Liu Y, Hui Y, Petrovsky N, Zhang F, Zhao CX, Adv Healthc Mater 2019, 8, e1900015. [DOI] [PubMed] [Google Scholar]
- [8].a) Choi Y, Hyun E, Seo J, Blundell C, Kim HC, Lee E, Lee SH, Moon A, Moon WK, Huh D, Lab Chip 2015, 15, 3350; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bai J, Tu TY, Kim C, Thiery JP, Kamm RD, Oncotarget 2015, 6, 36603; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mi S, Du Z, Xu Y, Wu Z, Qian X, Zhang M, Sun W, Sci Rep 2016, 6, 35544. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Pradhan S, Smith AM, Garson CJ, Hassani I, Seeto WJ, Pant K, Arnold RD, Prabhakarpandian B, Lipke EA, Sci Rep 2018, 8, 3171. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Dereli-Korkut Z, Akaydin HD, Ahmed AHR, Jiang XJ, Wang SH, Anal Chem 2014, 86, 2997. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Carvalho MR, Barata D, Teixeira LM, Giselbrecht S, Reis RL, Oliveira JM, Truckenmuller R, Habibovic P, Sci Adv 2019, 5, eaaw1317. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Mintz RL, Lao YH, Chi CW, He S, Li M, Quek CH, Shao D, Chen B, Han J, Wang S, Leong KW, Bioeng Transl Med 2020, 5, e10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Ghiabi P, Jiang J, Pasquier J, Maleki M, Abu-Kaoud N, Rafii S, Rafii A, Plos One 2014, 9, e112424. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang R, Bhattacharya R, Ye X, Fan F, Boulbes DR, Ellis LM, Mol Cancer Res 2019, 17, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang YH, Dong YY, Wang WM, Xie XY, Wang ZM, Chen RX, Chen J, Gao DM, Cui JF, Ren ZG, J Exp Clin Cancer Res 2013, 32, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wolf MJ, Hoos A, Bauer J, Boettcher S, Knust M, Weber A, Simonavicius N, Schneider C, Lang M, Sturzl M, Croner RS, Konrad A, Manz MG, Moch H, Aguzzi A, van Loo G, Pasparakis M, Prinz M, Borsig L, Heikenwalder M, Cancer Cell 2012, 22, 91. [DOI] [PubMed] [Google Scholar]
- [10].a) Liu T, Han C, Wang S, Fang P, Ma Z, Xu L, Yin R, J Hematol Oncol 2019, 12, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu T, Zhou L, Li D, Andl T, Zhang Y, Front Cell Dev Biol 2019, 7, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten FR, Hingorani SR, Hunter T, Hynes RO, Jain RK, Janowitz T, Jorgensen C, Kimmelman AC, Kolonin MG, Maki RG, Powers RS, Pure E, Ramirez DC, Scherz-Shouval R, Sherman MH, Stewart S, Tlsty TD, Tuveson DA, Watt FM, Weaver V, Weeraratna AT, Werb Z, Nat Rev Cancer 2020, 20, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Nashimoto Y, Okada R, Hanada S, Arima Y, Nishiyama K, Miura T, Yokokawa R, Biomaterials 2020, 229, 119547. [DOI] [PubMed] [Google Scholar]; b) Jeong SY, Lee JH, Shin Y, Chung S, Kuh HJ, Plos One 2016, 11, e0159013. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Truong DD, Kratz A, Park JG, Barrientos ES, Saini H, Nguyen T, Pockaj B, Mouneimne G, LaBaer J, Nikkhah M, Cancer Res 2019, 79, 3139. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Hockemeyer K, Janetopoulos C, Terekhov A, Hofmeister W, Vilgelm A, Costa L, Wikswo JP, Richmond A, Biomicrofluidics 2014, 8, 044105. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Shirure VS, Bi Y, Curtis MB, Lezia A, Goedegebuure MM, Goedegebuure SP, Aft R, Fields RC, George SC, Lab Chip 2018, 18, 3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ahmed AHR, Dereli Korkut Z, Akaydin HD, Wang S, presented at ASME 2013 Summer Bioengineering Conference 2013. [Google Scholar]
- [13].a) Dejana E, Orsenigo F, Lampugnani MG, J Cell Sci 2008, 121, 2115. [DOI] [PubMed] [Google Scholar]; b) Gavard J, Cell Adh Migr 2014, 8, 158; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Privratsky JR, Newman PJ, Cell Tissue Res 2014, 355, 607; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, Jain RK, McDonald DM, Am J Pathol 2000, 156, 1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chauhan VP, Stylianopoulos T, Boucher Y, Jain RK, Annu Rev Chem Biomol 2011, 2, 281. [DOI] [PubMed] [Google Scholar]
- [15].Yuan W, Lv Y, Zeng M, Fu BM, Microvasc Res 2009, 77, 166. [DOI] [PubMed] [Google Scholar]
- [16].Fu BM, Shen S, Microvasc Res 2004, 68, 51. [DOI] [PubMed] [Google Scholar]
- [17].a) Fraker LD, Halter SA, Forbes JT, Cancer Res 1984, 44, 5757. [PubMed] [Google Scholar]; b) Price JE, Polyzos A, Zhang RD, Daniels LM, Cancer Res 1990, 50, 717. [PubMed] [Google Scholar]
- [18].Yoneda T, Williams PJ, Hiraga T, Niewolna M, Nishimura R, J Bone Miner Res 2001, 16, 1486. [DOI] [PubMed] [Google Scholar]
- [19].Yoneda T, Sasaki A, Mundy GR, Breast Cancer Res Treat 1994, 32, 73. [DOI] [PubMed] [Google Scholar]
- [20].Garcia-Roman J, Zentella-Dehesa A, Cancer Lett 2013, 335, 259. [DOI] [PubMed] [Google Scholar]
- [21].a) Eum SY, Lee YW, Hennig B, Toborek M, Exp Cell Res 2004, 296, 231. [DOI] [PubMed] [Google Scholar]; b) Lee TH, Avraham HK, Jiang S, Avraham S, J Biol Chem 2003, 278, 5277. [DOI] [PubMed] [Google Scholar]
- [22].a) Galaup A, Cazes A, Le Jan S, Philippe J, Connault E, Le Coz E, Mekid H, Mir LM, Opolon P, Corvol P, Monnot C, Germain S, Proc Natl Acad Sci U S A 2006, 103, 18721; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Padua D, Zhang XH, Wang Q, Nadal C, Gerald WL, Gomis RR, Massague J, Cell 2008, 133, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].a) Dwyer J, Hebda JK, Le Guelte A, Galan-Moya EM, Smith SS, Azzi S, Bidere N, Gavard J, Plos One 2012, 7, e45562; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lee BC, Lee TH, Avraham S, Avraham HK, Mol Cancer Res 2004, 2, 327. [PubMed] [Google Scholar]
- [24].a) Afonso PV, Ozden S, Cumont MC, Seilhean D, Cartier L, Rezaie P, Mason S, Lambert S, Huerre M, Gessain A, Couraud PO, Pique C, Ceccaldi PE, Romero IA, PLoS Pathog 2008, 4, e1000205; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bonakdar M, Graybill PM, Davalos RV, RSC Adv 2017, 7, 42811; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lopez-Ramirez MA, Fischer R, Torres-Badillo CC, Davies HA, Logan K, Pfizenmaier K, Male DK, Sharrack B, Romero IA, J Immunol 2012, 189, 3130. [DOI] [PubMed] [Google Scholar]
- [25].a) Louault K, Bonneaud TL, Seveno C, Gomez-Bougie P, Nguyen F, Gautier F, Bourgeois N, Loussouarn D, Kerdraon O, Barille-Nion S, Jezequel P, Campone M, Amiot M, Juin PP, Souaze F, Oncogene 2019, 38, 3261; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Son GM, Kwon MS, Shin DH, Shin N, Ryu D, Kang CD, Medicine (Baltimore) 2019, 98, e15164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].a) Mishra PJ, Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, Ganesan S, Glod JW, Banerjee D, Cancer Res 2008, 68, 4331; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shangguan L, Ti X, Krause U, Hai B, Zhao Y, Yang Z, Liu F, Stem Cells 2012, 30, 2810. [DOI] [PubMed] [Google Scholar]
- [27].Plava J, Cihova M, Burikova M, Matuskova M, Kucerova L, Miklikova S, Mol Cancer 2019, 18, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lee E, Pandey NB, Popel AS, Sci Rep 2014, 4, 5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang C, Baker BM, Chen CS, Schwartz MA, Arterioscler Thromb Vasc Biol 2013, 33, 2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Dvorak HF, N Engl J Med 1986, 315, 1650. [DOI] [PubMed] [Google Scholar]
- [31].LeBleu VS, Kalluri R, Dis Model Mech 2018, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].a) Lin W, Huang L, Li Y, Fang B, Li G, Chen L, Xu L, Biomed Res Int 2019, 2019, 2820853; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ridge SM, Sullivan FJ, Glynn SA, Mol Cancer 2017, 16, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Brennen WN, Chen S, Denmeade SR, Isaacs JT, Oncotarget 2013, 4, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Li N, Schwartz M, Ionescu-Zanetti C, J Biomol Screen 2009, 14, 194. [DOI] [PubMed] [Google Scholar]
- [35].Borriello L, Nakata R, Sheard MA, Fernandez GE, Sposto R, Malvar J, Blavier L, Shimada H, Asgharzadeh S, Seeger RC, DeClerck YA, Cancer Res 2017, 77, 5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].a) Chen DR, Lu DY, Lin HY, Yeh WL, Biomed Res Int 2014, 2014, 532161; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cheteh EH, Augsten M, Rundqvist H, Bianchi J, Sarne V, Egevad L, Bykov VJ, Ostman A, Wiman KG, Cell Death Dis 2017, 8, e2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].a) Faouzi S, Le Bail B, Neaud V, Boussarie L, Saric J, Bioulac-Sage P, Balabaud C, Rosenbaum J, J Hepatol 1999, 30, 275; [DOI] [PubMed] [Google Scholar]; b) Fiori ME, Di Franco S, Villanova L, Bianca P, Stassi G, De Maria R, Mol Cancer 2019, 18, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.