

Abstract
Recent advances in single-particle cryogenic-electron microscopy have facilitated an exponential growth in the number of membrane protein structures determined to close to atomic resolution. Nevertheless, despite improvements in microscope hardware, cryo-EM software and sample preparation techniques, challenges remain for structural analysis of small-sized membrane proteins (i.e. < 150 kilodalton). Here we discuss recent examples of structures of macromolecules from this category determined by cryo-EM. We analyze the underlying difficulties, the enabling technologies such as the use of antibody fragments to gain size and provide fiducials for particle alignment, and the unresolved issues like dislocation of complexes at the air-water interface. Finally, we briefly highlight the biological relevance of some of these success stories, and our predictions for the future.
Keywords: structural biology, membrane proteins, single-particle cryogenic-electron microscopy, antibody phage display
Introduction
Integral membrane proteins play an essential role in many diverse biological processes, from regulating membrane potential, signaling between cells, transporting nutrients across the cell membrane, and building and modifying the membrane itself. In humans, many of these proteins are also of major pharmacological relevance. For example, members of the G-protein coupled receptor (GPCR) family are currently the target for approximately 27% of all therapeutic drugs on the market [1].
Structural biology has had a tremendous impact on our understanding of how membrane proteins function at a molecular level. Initially with X-ray crystallography at center stage, starting from the landmark work on the photoreaction center of Rhodopseudomonas viridis [2], to the first ion channel [3], to an exponentially-growing number of structures that followed. Contributors to this success have been progressive improvements in our knowledge on how to prepare membrane protein samples for structure determination, as well as the use of structural genomics approaches to identify well-expressed, detergent-stable orthologs for these studies [4]. However, the very limited success rate at the crystallization step remains a bottleneck for X-ray crystallographic studies of membrane proteins.
Therefore, when hardware and software improvements in single-particle cryogenic electron microscopy (cryo-EM) [5, 6] allowed the structural determination of the first TRP channel [7], it was a eureka moment for membrane protein structural biology, not least because the so often fatal crystallization hurdle had been successfully bypassed.
Subsequent development of faster and better software and more stable microscopes have made it possible to solve high resolution single-particle cryo-EM structures of proteins as small as 52 kDa streptavidin [8, 9], 64 kDa human hemoglobin [10], 64 kDa human methemoglobin and 82 kDa horse liver alcohol dehydrogenase [11].
Despite recent advances, determining a structure of a small-sized (i.e. < 150 kilodalton) membrane protein remains challenging. For membrane proteins in general, the problems most commonly encountered are low expression, heterogeneity and stability of the sample as well as dynamics within the proteins. The smaller proteins present additional challenges as the resulting particles have low contrast and low signal-to-noise, especially so when imaged in the presence of detergent micelles. Additionally, the lack of features outside of the membrane, which is often the case for small membrane proteins, makes alignment of the particles challenging. Nevertheless, there have been steady improvements in recent times, and the limitation in the size of membrane proteins tractable by cryo-EM has progressively been challenged (Figure 1 & Table 1).
Figure 1. Progress in the size limitation of membrane protein structures determined by cryo-EM.
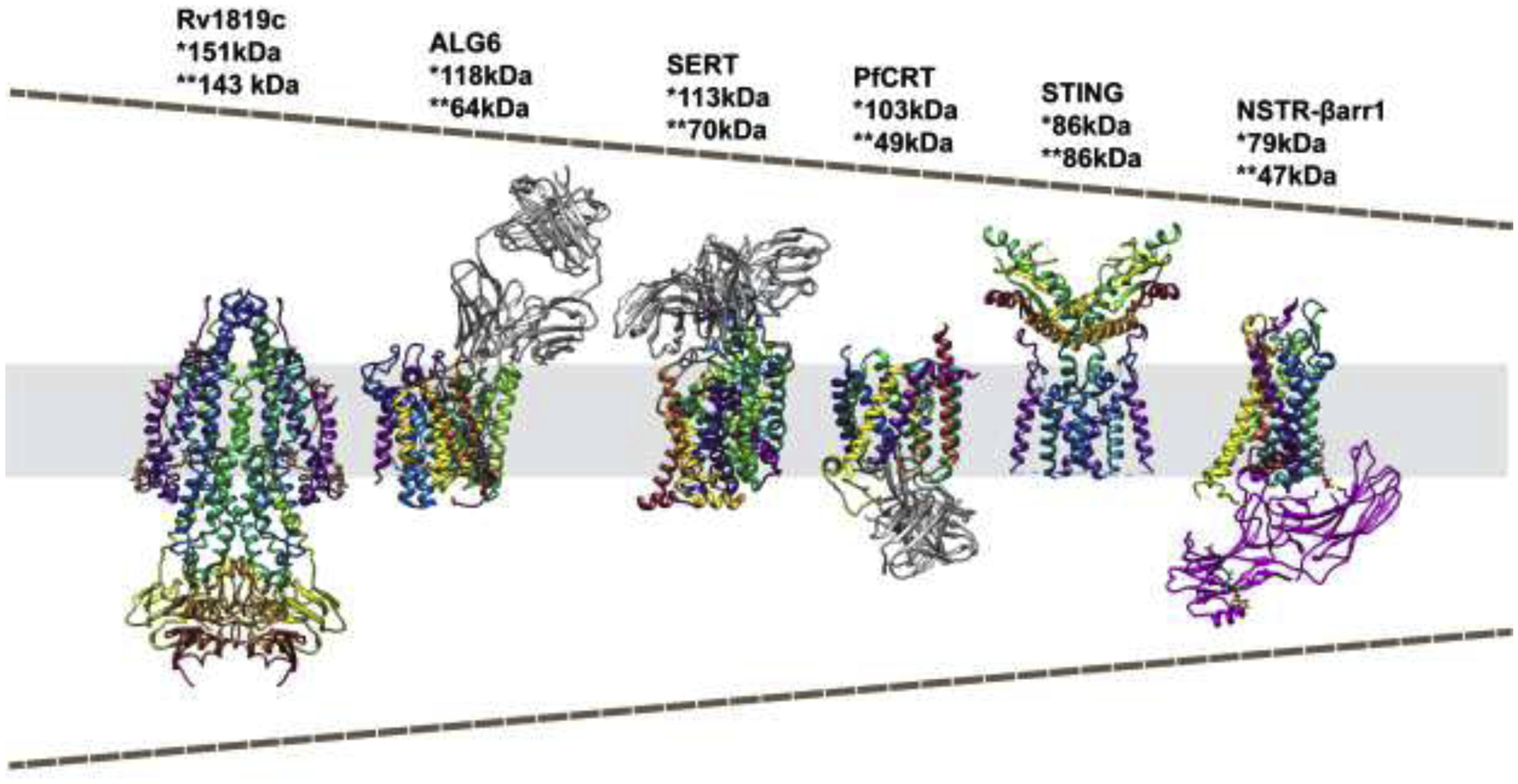
The figure demonstrates a selection of structures of small membrane proteins (colored in rainbow) recently determined by cryo-EM in order of the particle mass that was imaged; ABC exporter (Rv1819c) [75], dolichyl pyrophosphate Man9GlcNAc2 α −1,3-glucosyltransferase (ALG6) [35••], serotonin transporter (SERT) [46••], Plasmodium falciparum chloroquine resistance transporter (PfCRT) [32••], simulator of interferon genes (STING) [73], neurotensin receptor and arrestin 1 complex (NSTR-βarr1) [67••]. Fabs are colored in dark grey, and β-arrestin 1 is colored in magenta. *Molecular weight of the structure that was deposited in the Protein Database. **Molecular weight of membrane protein.
Table 1.
List of small membrane protein structures determined by cryo-EM.
| Protein | ||||||||
|---|---|---|---|---|---|---|---|---|
| Hedgedog receptor Patched 1 (Ptch1) | Homo sapiens | 3.5 Å | 6OEU | 2018 | Digitonin | 163 kDa | 160.5kDa | [76] |
| Hedgedog receptor Patched 1 (Ptch1) | Homo sapiens | 3.9 Å | 6DMB | 2018 | Digitonin | 153 kDa | Ptch1 160.5kDa | [77] |
| ABC exporter Rv1819c | Mycobacterium tuberculosis | 3.5 Å | 6TQF | 2020 | DDM | 151 kDa | Rv1819c 143kDa | [75] |
| ABC exporter TrmAB | Thermus thermophilus | 2.8 Å | 6RAH | 2019 | Nanodiscs | 151 kDa | TrmAB 133kDa | [33] |
| Otopetrin (Otop3) | Xenopus tropicalis | 3.9 Å | 6O84 | 2019 | DDM, CHS | 147 kDa | Otop3 154kDa | [78] |
| Hedgedog receptor Patched 1 (Ptch1) | Mus musculus | 3.6 Å | 6MG8 | 2018 | Amphipol | 146 kDa | Ptch1 159kDa | [79] |
| ABCG2 | Homo sapiens | 3.1 Å | 6HBU | 2018 | Nanodiscs | 146 kDa | ABCG2 144kDa | [80] |
| Urea channel of Helicobacter pylori (HpUreI) | Helicobacter pylori | 2.7 Å | 6NSJ | 2019 | Amphipol | 146 kDa | HpUreI 130kDa | [22] |
| Glutamate transporter homologue GltTK | Thermococcus kodakarensis | 3.2Å | 6XWR | 2020 | Nanodiscs | 137 kDa | GltTK 135kDa | [30] |
| Otopetrin (Otop1) | Danio rerio | 3.0 Å | 6NF4 | 2019 | Nanodiscs | 135 kDa | Otop1 131kDa | [31] |
| L-type amino acid transporter (LAT1–4F2hc complex) | Homo sapiens | 3.3 Å | 6IRS | 2019 | Digitonin | 131 kDa | LAT1 55kDa; 4F2hc 68kDa | [81] |
| GPR52-mini-Gs | Homo sapiens | 3.3 Å | 6LI3 | 2020 | LMNG, CHS | 130 kDa | GPR52 41kDa; mini-Gαs;Gβ1;Gγ2;Nb35 | [63] |
| Otopetrin (Otop3) | Gallus gallus | 3.3 Å | 6NF6 | 2019 | Nanodiscs | 129 kDa | Otop3 135kDa | [31] |
| Dolichyl pyrophosphate Man9GlcNAc2 α - 1,3- Glucosyltransferase (ALG6) | Saccharomyces cerevisiae | 3.0 Å | 6SNI | 2020 | Nanodiscs | 118 kDa | ALG6 64kDa; 6AG9-Fab | [35] |
| Cytochrome bd-I oxidase | Escherichia coli | 2.7 Å | 6RKO | 2019 | Nanodiscs | 114 kDa | Cytochrome bd-I Oxidase 108kDa | [34] |
| Serotonin transporter (SERT) | Homo sapiens | 4.1 Å | 6DZY | 2019 | DDM, CHS | 113 kDa | SERT 70kDa; 15B8 Fab-8B6 scFv | [46] |
| Plasmodium falciparum Chloroquine resistance transporter (PfCRT) 7G8 isoform | Plasmodium falciparum | 3.2 Å | 6UKJ | 2019 | Nanodiscs | 103 kDa | PfCRT 49kDa; CDC-Fab | [32] |
| Stimulator of interferon genes (STING) | Homo sapiens | 4.1 Å | 6NT5 | 2019 | DDM, CHS | 86kDa | STING alone | [73] |
| Neurotensin Receptor and arrestin 1 complex (NSTR1-βarr1) | Homo sapiens | 4.2 Å | 6UP7 | 2020 | LMNG, CHS | 79kDa | NSTR1 47kDa; βarr1 47kDa; | [67] |
Here, we address the typical issues one might encounter in determining the structure of small membrane proteins using cryo-EM. We will present examples of recently solved structures and discuss some of the approaches used to succeed, as well as summarize what insights we have gained from these accomplishments. To the best of our knowledge, we have analyzed all if not most membrane protein structures determined by cryo-EM in the last two years, after having placed an arbitrary cutoff at ~150 kDa or smaller, and determined at 4.5 Å resolution or better.
Sample preparation for structure determination of membrane proteins by cryo-EM
Expression and purification still remain a major bottleneck for structure determination of membrane proteins, irrespective of their molecular weight. The expression levels obtained for membrane proteins, eukaryotic ones in particular, are typically low, making them extremely challenging. However, this issue has in part been alleviated by technological advances, and the substantially decreased sample requirements for cryo-EM (microgram) as compared to X-ray crystallography (milligram). The need for less sample has even made determining structures of membrane proteins from natural resources more feasible [12–15], although this appears thus far to have been limited to larger proteins and multi-component macromolecular complexes. Indeed, for smaller membrane proteins, expression in a heterologous system remains the more efficient method of producing sufficient sample for structure determination. Apropos, the rather expensive, yet powerful, approaches such as transient transfection of mammalian cells grown in suspension [16], and transduction of mammalian cells with recombinant baculovirus produced in insect cells [17], are steadily becoming routine for membrane protein production. Structural genomic expansion approaches coupled to efficient screening platforms to biochemically and biophysically characterize targets, have greatly improved the efficiency of identifying membrane proteins suitable for structural studies [4, 18].
In the studies of membrane proteins, detergents have typically been used to extract the protein from the native membrane. However, the presence of detergents often introduces complications in cryo-EM analysis as they reduce contrast in images and can form spurious micellar structures - an issue especially problematic for small membrane proteins because the size of such micelles can be as large as the protein itself [19, 20]. Several approaches are available to stabilize membrane proteins without the presence of detergents. These include amphipols [21, 22], bicelles [23], saposins [24], styrene maleic anhydride (SMA) [25], nanodiscs [26], peptidiscs [27], and more recently proteoliposomes [28, 29].
Of these, the most commonly employed for determining structures of membrane proteins are nanodiscs [30–31, 32••, 33••, 34, 35••]. Nanodiscs consist of two copies of an amphipathic membrane scaffold protein (MSP), derived from apolipoprotein, which embed the target protein within a reconstituted phospholipid bilayer with the MSP wrapped around [26]. In nanodiscs, the protein is in a more native like environment, hence a typically more stabilizing one compared to detergents, and nanodiscs have a more distinct shape than detergent micelles, which helps align the particles. Another example creating widespread interest in the field is styrene maleic anhydride (SMA), an amphipathic polymer, that has been adapted to facilitate the extraction of proteins with lipids bound directly from the native membranes. This reagent was successfully utilized to determine the structure of the multidrug exporter AcrB [36] demonstrating an interaction between transmembrane helices and highly ordered native lipid molecules.
Single-particle cryo-EM structures of small membrane proteins
Small membrane proteins (<150 kDa) are difficult to study by cryo-EM because of the additional complication of the low signal-to-noise ratio of the resulting particles. This impedes particle alignment, thereby making it arduous if not impossible to obtain high resolution information. Also, small particles require very thin ice, as the ideal ice thickness is a monolayer of particles fully embedded in the ice [37, 38•]. However, too thin ice will cause the particles to aggregate, resulting in a very narrow range of ice thickness for optimal data collection, which can be technically challenging to achieve.
Besides the size of the protein per se, another major hurdle for cryo-EM analysis of small membrane proteins is the lack of fiducials. When the entire membrane protein is embedded in the phospholipid bilayer, or in a detergent micelle, without the presence of significant extra-membrane domains, it can often be arduous to align the individual particles during data processing, and thereby difficult to determine a structure at high resolution.
Several different strategies have been applied to overcome the size limitation in cryo-EM, usually requiring a combination of “tricks” in both sample and grid preparation, data collection and processing. From a sample perspective, these include forming a complex with a specific antibody binding to the target protein [39] either directly or, as recently reported, mediated via a genetically-engineered fusion protein BRIL (an engineered variant of apocytochrome b562) for which well-characterized high affinity antibodies are available [40]. A similar approach has involved the development of specific scaffolds for cryo-EM such as green fluorescent protein (GFP), maltose binding protein (MBP), and a modular adaptor protein Designed Ankyrin Repeat Proteins (DARPin) [41, 42].
In particular, Antigen binding-fragments (Fabs) have been widely used in membrane protein structure determination by X-ray crystallography, to promote the formation of stable crystal contacts and increase the probability of diffraction at a higher resolution [43, 44]. Recently, this technique has been transferred to cryo-EM analysis, where it has been successfully implemented to compensate for the size of the particle and lack of fiducials, yielding several structures [32••, 35••, 45, 46••]. One such example is Plasmodium falciparum chloroquine resistance transporter (PfCRT), a 49 kDa integral transmembrane protein localized in the digestive vacuole of the pathogenic parasite Plasmodium falciparum that causes malaria (Figure 1 & Table 1) [32••]. PfCRT harbors mutations that promote efflux of 4-aminoquinoline derivatives chloroquine and piperaquine, a former and current antimalarial drug, respectively, in turn resulting in drug resistance. The authors screened a phage display library [47] to identify Fabs that bind specifically and with high affinity to PfCRT. The use of a Fab fragment was instrumental, as it increased particle mass and size, while aiding in particle alignment. This, combined with a small pixel size (0.52Å) used during data collection - which improved signal to noise for the individual particles [48], thus improving the signal available for particle alignment - together with various features implemented during data processing, such as signal subtraction of a flexible region of the Fab and the nanodisc using Relion [49, 50] as well as non-uniform refinement in cryoSPARC [51•], yielded a structure of PfCRT at 3.2Å resolution. This work revealed the overall architecture of the protein, identifying a putative drug binding site in the structure [32••].
Similarly, a Fab identified from screening a phage display library was used to determine the structure of a 64 kDa glycosyltransferase, ALG6, that is involved in endoplasmic reticulum (ER)-luminal N-glycan synthesis. ALG6 transfers glucose moieties from dolichylpyrophosphate onto a pre-assembled mannose glycan in the lipid-linked oligosaccharide biosynthesis. The Fab was bound to the ER-luminal side of the protein without interfering with the catalytic activity, while revealing a fold distinct from other previously reported GT-C fold enzyme structures [35••]. A Fab was also used to elucidate the mechanism of neurotransmitter transport and the action of ibogaine, a non-competitive inhibitor, in the 70 kDa monomeric serotonin transporter (SERT) cryo-EM structure [46••]. SERT is a therapeutic target for anxiety disorders and depression, which regulates neurotransmitter homeostasis by transporting serotonin from the postsynaptic clef into the presynaptic neurons in both sodium- and chloride-dependent manner. The authors illustrate the structural rearrangements between outward-open, occluded and inward-open conformations in the presence of ibogaine, providing superb molecular insights for a potential structure-based drug discovery approach [46••]. While using Fabs for solving high resolution structures of small membrane proteins has proven instrumental in some cases, one potential pitfall is the possibility of stabilizing a non-physiological state, although relatively unlikely, this is something that should always be kept in mind and, where possible, verified.
Despite the typical high-affinity of a Fab-antigen complex, a major issue with this method is that the protein-Fab complex tends to dissociate upon vitrification during grid preparation. When protein particles are vitrified into a thin layer of ice for cryo-EM, these are often adsorbed to the air-water interface introducing several complications including protein denaturation, complex dissociation and preferred orientation [52]. There are several strategies to prevent air-water interface issues, including a rapid-plunge freezing method [52, 53], crosslinking the complex to avoid complex dissociation [54] and/or adding detergent prior to freezing to alter the air-water interface and eliminate particle absorption [55]. Lastly, several different carbon support films have been developed such as hydrolyzed graphene [9, 56–58]. Although these support films can help distance the particles from the air-water interface, one suffers a loss in contrast which can be difficult to overcome especially when working with small membrane proteins.
Small membrane protein dynamics
Membrane proteins are generally dynamic molecules, a property which is essential to their function. Classic examples, which belong to the small membrane protein category, are transporters that need to adopt different states in order to move substrates across the membrane, and GPCRs that translate the effect of a ligand binding on the extracellular side of the protein to a conformational change intracellularly in order to elicit second messenger responses. The dynamic nature of these membrane proteins makes them ever more challenging for cryo-EM analysis due to the presence in the micrographs of particles with proteins in a range of different conformations. The upside is that these different states can be biologically relevant, and cryo-EM offers a unique way of studying different states observed in a single sample preparation by separating out the states during processing. This can of course be challenging, if not impossible, if the states are not discrete, but more a continuum. In one example, cryo-EM was used to illustrate eight different conformational states of 133 kDa heterodimeric ATP-binding cassette (ABC) exporter TmrAB [33••, 59]. The authors performed multiple rounds of 3D classifications to obtain distinct conformations of the ABC exporter to delineate the fundamental steps of the substrate translocation and ATP-hydrolysis cycle [33••]. This impressive work is just one example of how cryo-EM can be used to study the dynamics in a given sample, from the same preparation.
Frequently, it is difficult to distinguish between states within a sample and different techniques need to be applied to bias towards one state in order to obtain maps of an interpretable resolution. One example is the complexes between GPCRs and their downstream effectors [60–62, 63•, 64–66, 67••]. G protein coupled receptors comprise the largest family of transmembrane receptors, they bind a diverse variety of ligands and are one of the most important known drug targets. They signal through their cognate G protein and to some extent β-arrestin [61]. Both signaling complexes have been studied by cryo-EM. GPCRs are intrinsically dynamic proteins [68], which has proven a challenge for structural studies, the dynamic nature and the inherent flexibility of the interaction between GPCRs and their cognate G protein makes them tremendously challenging for structural determination. To overcome these challenges, independent groups have used Fabs, single chain variable fragments (svFc) as well as nanobodies in combination with an engineered G protein to stabilize the complex between GPCRs and G proteins and determine their structures using cryo-EM [60–62, 63•, 64–66, 67••, 69, 70]. One example is the structure of GPR52 in complex with a truncated version of Gs (mini-Gs), Gβ,γ and a nanobody (Nb35). Nb35 binds to the G protein, stabilizing the interaction between the subunits [71], making it possible to solve the complex to a global resolution of 3.3 Å [63•]. Recently, the structure of Neurotensin receptor 1 in complex with β-arrestin 1 was determined (Figure 1 & Table 1) [67••]. The authors observed complex dissociation while freezing, and therefore opted for cross-linking with sulfosuccinimidyl 6-(4,4′-azipentanamido)hexanoate (sulfo-LC-SDA) before freezing. Phosphorylation of the receptor is crucial for complex formation, and the receptor was therefore phosphorylated in-vitro by GRK5 prior to complex formation. The structure revealed a number of important phosphorylation sites within the receptor and a conformational change compared to the inactive state within the receptor upon binding to β-arrestin 1 [67••].
For structure determination of calcitonin receptor [64], a class B GPCR, in complex with Gs, the use of a Volta phase plate made high resolution structural determination possible. The Volta phase plate provide another strategy to increase the signal proteins, the technology provides in focus phase contrast in a transmission election microscope, leading to higher contrast images which can be especially important if not crucial when solving structures of small membrane proteins [64, 72, 73].
Future perspectives
In the modern era of structural biology, cryo-EM has established itself as a powerful tool to investigate the structures of intricate membrane proteins that has previously been intractable to other techniques including X-ray crystallography. With the continuous development in technology, cryo-EM has proven to be a well-suited approach to determine structures of membrane proteins smaller than 150 kDa. The implementation of Fab technology combined with cryo-EM has been successfully applied to determine several recent structures of small membrane proteins, by increasing the size of the particles while also providing features for particle alignment. Along with the improvements in cryo-EM, novel approaches to stabilize membrane proteins in detergent-free solution have also been advanced. Going forward, the possibility of solving structures of membrane proteins incorporated into liposomes unlocks the prospect of being able to image a membrane protein while applying different solutions on either side of the bilayer [28, 74]. This opens up a host of new experiments which are not possible in the typical isocratic setting of a single-particle or crystallization experiment. These include imaging membrane proteins in a native like membrane environment across a pH or ionic gradients, according to their respective energetic driving forces. So far, this approach has only been successful for large membrane proteins, but its extension to smaller macromolecules is no longer inconceivable. Together, it makes for an exciting time to use cryo-EM to investigate the molecular mechanism at atomic resolution of many previously intractable and progressively smaller biologically and clinically relevant integral membrane proteins, with many more surely to come.
Acknowledgements
We thank Yong Zi Tan for helpful discussions. This work was supported by NIH grants R01 AI147628 and R35 GM132120 (to F.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
Nothing declared.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Hauser AS, Attwood MM, Rask-Andersen M, Schioth HB, and Gloriam DE, Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov, 2017. 16(12): p. 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deisenhofer J, Epp O, Miki K, Huber R, and Michel H, Structure of the protein subunits in the photosynthetic reaction centre of Rhodopseudomonas viridis at 3A resolution. Nature, 1985. 318(6047): p. 618–24. [DOI] [PubMed] [Google Scholar]
- 3.Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, and MacKinnon R, The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science, 1998. 280(5360): p. 69–77. [DOI] [PubMed] [Google Scholar]
- 4.Mancia F and Love J, High throughput platforms for structural genomics of integral membrane proteins. Curr Opin Struct Biol, 2011. 21(4): p. 517–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McMullan G, Faruqi AR, Clare D, and Henderson R, Comparison of optimal performance at 300keV of three direct electron detectors for use in low dose electron microscopy. Ultramicroscopy, 2014. 147: p. 156–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuhlbrandt W, Biochemistry. The resolution revolution. Science, 2014. 343(6178): p. 1443–4. [DOI] [PubMed] [Google Scholar]
- 7.Liao M, Cao E, Julius D, and Cheng Y, Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature, 2013. 504(7478): p. 107–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fan X, Wang J, Zhang X, Yang Z, Zhang JC, Zhao L, Peng HL, Lei J, and Wang HW, Single particle cryo-EM reconstruction of 52 kDa streptavidin at 3.2 Angstrom resolution. Nat Commun, 2019. 10(1): p. 2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han Y, Fan X, Wang H, Zhao F, Tully CG, Kong J, Yao N, and Yan N, High-yield monolayer graphene grids for near-atomic resolution cryoelectron microscopy. Proc Natl Acad Sci U S A, 2020. 117(2): p. 1009–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khoshouei M, Radjainia M, Baumeister W, and Danev R, Cryo-EM structure of haemoglobin at 3.2 A determined with the Volta phase plate. Nat Commun, 2017. 8: p. 16099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herzik MA Jr., Wu M, and Lander GC, High-resolution structure determination of sub-100 kDa complexes using conventional cryo-EM. Nat Commun, 2019. 10(1): p. 1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zalk R, Clarke OB, des Georges A, Grassucci RA, Reiken S, Mancia F, Hendrickson WA, Frank J, and Marks AR, Structure of a mammalian ryanodine receptor. Nature, 2015. 517(7532): p. 44–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yan Z, Bai X, Yan C, Wu J, Li Z, Xie T, Peng W, Yin C, Li X, Scheres SHW, et al. , Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature, 2015. 517(7532): p. 50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan Z, Zhou Q, Wang L, Wu J, Zhao Y, Huang G, Peng W, Shen H, Lei J, and Yan N, Structure of the Nav1.4-beta1 Complex from Electric Eel. Cell, 2017. 170(3): p. 470–482 e11. [DOI] [PubMed] [Google Scholar]
- 15.Ho CM, Beck JR, Lai M, Cui Y, Goldberg DE, Egea PF, and Zhou ZH, Malaria parasite translocon structure and mechanism of effector export. Nature, 2018. 561(7721): p. 70–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jordan M, Kohne C, and Wurm FM, Calcium-phosphate mediated DNA transfer into HEK-293 cells in suspension: control of physicochemical parameters allows transfection in stirred media. Transfection and protein expression in mammalian cells. Cytotechnology, 1998. 26(1): p. 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goehring A, Lee CH, Wang KH, Michel JC, Claxton DP, Baconguis I, Althoff T, Fischer S, Garcia KC, and Gouaux E, Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat Protoc, 2014. 9(11): p. 2574–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawate T and Gouaux E, Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure, 2006. 14(4): p. 673–81. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt-Krey I and Rubinstein JL, Electron cryomicroscopy of membrane proteins: specimen preparation for two-dimensional crystals and single particles. Micron, 2011. 42(2): p. 107–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gewering T, Januliene D, Ries AB, and Moeller A, Know your detergents: A case study on detergent background in negative stain electron microscopy. J Struct Biol, 2018. 203(3): p. 242–246. [DOI] [PubMed] [Google Scholar]
- 21.Tribet C, Audebert R, and Popot JL, Amphipols: polymers that keep membrane proteins soluble in aqueous solutions. Proc Natl Acad Sci U S A, 1996. 93(26): p. 15047–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cui Y, Zhou K, Strugatsky D, Wen Y, Sachs G, Zhou ZH, and Munson K, pH-dependent gating mechanism of the Helicobacter pylori urea channel revealed by cryo-EM. Sci Adv, 2019. 5(3): p. eaav8423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Durr UH, Gildenberg M, and Ramamoorthy A, The magic of bicelles lights up membrane protein structure. Chem Rev, 2012. 112(11): p. 6054–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frauenfeld J, Loving R, Armache JP, Sonnen AF, Guettou F, Moberg P, Zhu L, Jegerschold C, Flayhan A, Briggs JA, et al. , A saposin-lipoprotein nanoparticle system for membrane proteins. Nat Methods, 2016. 13(4): p. 345–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knowles TJ, Finka R, Smith C, Lin YP, Dafforn T, and Overduin M, Membrane proteins solubilized intact in lipid containing nanoparticles bounded by styrene maleic acid copolymer. J Am Chem Soc, 2009. 131(22): p. 7484–5. [DOI] [PubMed] [Google Scholar]
- 26.Bayburt TH and Sligar SG, Membrane protein assembly into Nanodiscs. FEBS Lett, 2010. 584(9): p. 1721–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carlson ML, Young JW, Zhao Z, Fabre L, Jun D, Li J, Li J, Dhupar HS, Wason I, Mills AT, et al. , The Peptidisc, a simple method for stabilizing membrane proteins in detergent-free solution. Elife, 2018. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tonggu L and Wang L, Cryo-EM sample preparation method for extremely low concentration liposomes. Ultramicroscopy, 2020. 208: p. 112849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang L and Sigworth FJ, Structure of the BK potassium channel in a lipid membrane from electron cryomicroscopy. Nature, 2009. 461(7261): p. 292–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arkhipova V, Guskov A, and Slotboom DJ, Structural ensemble of a glutamate transporter homologue in lipid nanodisc environment. Nat Commun, 2020. 11(1): p. 998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saotome K, Teng B, Tsui CCA, Lee WH, Tu YH, Kaplan JP, Sansom MSP, Liman ER, and Ward AB, Structures of the otopetrin proton channels Otop1 and Otop3. Nat Struct Mol Biol, 2019. 26(6): p. 518–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim J, Tan YZ, Wicht KJ, Erramilli SK, Dhingra SK, Okombo J, Vendome J, Hagenah LM, Giacometti SI, Warren AL, et al. , Structure and drug resistance of the Plasmodium falciparum transporter PfCRT. Nature, 2019. 576(7786): p. 315–320. [DOI] [PMC free article] [PubMed] [Google Scholar]; This manuscript describes a strategy of implementing cryo-EM combined with Fab technology to determine a 49 kDa integral transmembrane protein involved in antimalarial drug resistance. The Fab enhanced the size of the molecule and greatly facilitated particle alignment in cryo-EM analysis.
- 33.Hofmann S, Januliene D, Mehdipour AR, Thomas C, Stefan E, Bruchert S, Kuhn BT, Geertsma ER, Hummer G, Tampe R, et al. , Conformation space of a heterodimeric ABC exporter under turnover conditions. Nature, 2019. 571(7766): p. 580–583. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrates a translocation cycle of the heterodimeric ABC exporter, in which the authors performed four rounds of multi-model 3D classification to obtain eight distinct conformations.
- 34.Safarian S, Hahn A, Mills DJ, Radloff M, Eisinger ML, Nikolaev A, Meier-Credo J, Melin F, Miyoshi H, Gennis RB, et al. , Active site rearrangement and structural divergence in prokaryotic respiratory oxidases. Science, 2019. 366(6461): p. 100–104. [DOI] [PubMed] [Google Scholar]
- 35.Bloch JS, Pesciullesi G, Boilevin J, Nosol K, Irobalieva RN, Darbre T, Aebi M, Kossiakoff AA, Reymond JL, and Locher KP, Structure and mechanism of the ER-based glucosyltransferase ALG6. Nature, 2020. 579(7799): p. 443–447. [DOI] [PMC free article] [PubMed] [Google Scholar]; Structure of the 64 kDa glycosyltransferase ALG6 bound to a Fab. The Fab is bound to the ER-luminal side of the protein, revealing a fold distinct from other previously reported GT-C fold enzyme structures.
- 36.Qiu W, Fu Z, Xu GG, Grassucci RA, Zhang Y, Frank J, Hendrickson WA, and Guo Y, Structure and activity of lipid bilayer within a membrane-protein transporter. Proc Natl Acad Sci U S A, 2018. 115(51): p. 12985–12990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Noble AJ, Dandey VP, Wei H, Brasch J, Chase J, Acharya P, Tan YZ, Zhang Z, Kim LY, Scapin G, et al. , Routine single particle CryoEM sample and grid characterization by tomography. Elife, 2018. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cianfrocco MA and Kellogg EH, What Could Go Wrong? A Practical Guide to Single-Particle Cryo-EM: From Biochemistry to Atomic Models. J Chem Inf Model, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]; This review gives a practical guide to cryo-EM. The authors provide a wonderful overview of how to overcome technical issues when initiating a cryo-EM project.
- 39.Wu S, Avila-Sakar A, Kim J, Booth DS, Greenberg CH, Rossi A, Liao M, Li X, Alian A, Griner SL, et al. , Fabs enable single particle cryoEM studies of small proteins. Structure, 2012. 20(4): p. 582–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mukherjee S, Erramilli SK, Ammirati M, Alvarez FJD, Fennell KF, Purdy MD, Skrobek BM, Radziwon K, Coukos J, Kang Y, et al. , Synthetic antibodies against BRIL as universal fiducial marks for single-particle cryoEM structure determination of membrane proteins. Nat Commun, 2020. 11(1): p. 1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Y, Gonen S, Gonen T, and Yeates TO, Near-atomic cryo-EM imaging of a small protein displayed on a designed scaffolding system. Proc Natl Acad Sci U S A, 2018. 115(13): p. 3362–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yeates TO, Agdanowski MP, and Liu Y, Development of imaging scaffolds for cryoelectron microscopy. Curr Opin Struct Biol, 2020. 60: p. 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carpenter EP, Beis K, Cameron AD, and Iwata S, Overcoming the challenges of membrane protein crystallography. Curr Opin Struct Biol, 2008. 18(5): p. 581–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hunte C and Michel H, Crystallisation of membrane proteins mediated by antibody fragments. Curr Opin Struct Biol, 2002. 12(4): p. 503–8. [DOI] [PubMed] [Google Scholar]
- 45.Butterwick JA, Del Marmol J, Kim KH, Kahlson MA, Rogow JA, Walz T, and Ruta V, Cryo-EM structure of the insect olfactory receptor Orco. Nature, 2018. 560(7719): p. 447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coleman JA, Yang D, Zhao Z, Wen PC, Yoshioka C, Tajkhorshid E, and Gouaux E, Serotonin transporter-ibogaine complexes illuminate mechanisms of inhibition and transport. Nature, 2019. 569(7754): p. 141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes three distinct conformational states of a monomeric 70 kDa serotonin transporter (SERT) in the presence of a non-competitive inhibitor determined by cryo-EM. The structures provide greater molecular insights into the mechanism of serotonin transport and the action of inhibition.
- 47.Dominik PK and Kossiakoff AA, Phage display selections for affinity reagents to membrane proteins in nanodiscs. Methods Enzymol, 2015. 557: p. 219–45. [DOI] [PubMed] [Google Scholar]
- 48.McMullan G, Faruqi AR, and Henderson R, Direct Electron Detectors. Methods Enzymol, 2016. 579: p. 1–17. [DOI] [PubMed] [Google Scholar]
- 49.Bai XC, Rajendra E, Yang G, Shi Y, and Scheres SH, Sampling the conformational space of the catalytic subunit of human gamma-secretase. Elife, 2015. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang C, Cantara W, Jeon Y, Musier-Forsyth K, Grigorieff N, and Lyumkis D, Analysis of discrete local variability and structural covariance in macromolecular assemblies using Cryo-EM and focused classification. Ultramicroscopy, 2019. 203: p. 170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Punjani AZ, H.Z.; Fleet DJ, Non-uniform refinement: Adaptive regularization improves single particle cryo-EM reconstruction. bioRxiv, 2020. doi: 10.1101/2019.12.15.877092, [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]; This manuscript introduces a cross-validation approach to derive non-uniform refinement in the cryoSPARC data processing software package. This feature automatically regularizes 3D density maps during iterative refinement to account for spatial variability. This in turn helps to enhance the resolution of 3D maps by removing noise in disordered regions in cryo-EM particle images.
- 52.Noble AJ, Wei H, Dandey VP, Zhang Z, Tan YZ, Potter CS, and Carragher B, Reducing effects of particle adsorption to the air-water interface in cryo-EM. Nat Methods, 2018. 15(10): p. 793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dandey VP, Wei H, Zhang Z, Tan YZ, Acharya P, Eng ET, Rice WJ, Kahn PA, Potter CS, and Carragher B, Spotiton: New features and applications. J Struct Biol, 2018. 202(2): p. 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adamus K, Le SN, Elmlund H, Boudes M, and Elmlund D, AgarFix: Simple and accessible stabilization of challenging single-particle cryo-EM specimens through crosslinking in a matrix of agar. J Struct Biol, 2019. 207(3): p. 327–331. [DOI] [PubMed] [Google Scholar]
- 55.Chen J, Gopalkrishnan S, Chiu C, Chen AY, Campbell EA, Gourse RL, Ross W, and Darst SA, E. coli TraR allosterically regulates transcription initiation by altering RNA polymerase conformation. Elife, 2019. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Palovcak E, Wang F, Zheng SQ, Yu Z, Li S, Betegon M, Bulkley D, Agard DA, and Cheng Y, A simple and robust procedure for preparing graphene-oxide cryo-EM grids. J Struct Biol, 2018. 204(1): p. 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Naydenova K, Peet MJ, and Russo CJ, Multifunctional graphene supports for electron cryomicroscopy. Proc Natl Acad Sci U S A, 2019. 116(24): p. 11718–11724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.D’Imprima E, Floris D, Joppe M, Sanchez R, Grininger M, and Kuhlbrandt W, Protein denaturation at the air-water interface and how to prevent it. Elife, 2019. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zutz A, Hoffmann J, Hellmich UA, Glaubitz C, Ludwig B, Brutschy B, and Tampe R, Asymmetric ATP hydrolysis cycle of the heterodimeric multidrug ABC transport complex TmrAB from Thermus thermophilus. J Biol Chem, 2011. 286(9): p. 7104–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang Y, Sun B, Feng D, Hu H, Chu M, Qu Q, Tarrasch JT, Li S, Sun Kobilka T, Kobilka BK, et al. , Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature, 2017. 546(7657): p. 248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weis WI and Kobilka BK, The Molecular Basis of G Protein-Coupled Receptor Activation. Annu Rev Biochem, 2018. 87: p. 897–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nguyen AH, Thomsen ARB, Cahill TJ 3rd, Huang R, Huang LY, Marcink T, Clarke OB, Heissel S, Masoudi A, Ben-Hail D, et al. , Structure of an endosomal signaling GPCR-G protein-beta-arrestin megacomplex. Nat Struct Mol Biol, 2019. 26(12): p. 1123–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lin X, Li M, Wang N, Wu Y, Luo Z, Guo S, Han GW, Li S, Yue Y, Wei X, et al. , Structural basis of ligand recognition and self-activation of orphan GPR52. Nature, 2020. 579(7797): p. 152–157. [DOI] [PubMed] [Google Scholar]; In this paper the authors present three structures of GPR52. A crystal structure of the ligand free state, and two cryo-EM structures of GPR52 in complex with mini-Gs, Gβ,γ with and without a potential allosteric ligand. For the complex structure a nanobody was added to stabilize the G-protein.
- 64.Liang YL, Khoshouei M, Radjainia M, Zhang Y, Glukhova A, Tarrasch J, Thal DM, Furness SGB, Christopoulos G, Coudrat T, et al. , Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature, 2017. 546(7656): p. 118–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Koehl A, Hu H, Maeda S, Zhang Y, Qu Q, Paggi JM, Latorraca NR, Hilger D, Dawson R, Matile H, et al. , Structure of the micro-opioid receptor-Gi protein complex. Nature, 2018. 558(7711): p. 547–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kang Y, Kuybeda O, de Waal PW, Mukherjee S, Van Eps N, Dutka P, Zhou XE, Bartesaghi A, Erramilli S, Morizumi T, et al. , Cryo-EM structure of human rhodopsin bound to an inhibitory G protein. Nature, 2018. 558(7711): p. 553–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang W, Masureel M, Qu Q, Janetzko J, Inoue A, Kato HE, Robertson MJ, Nguyen KC, Glenn JS, Skiniotis G, et al. , Structure of the neurotensin receptor 1 in complex with beta-arrestin 1. Nature, 2020. 579(7798): p. 303–308. [DOI] [PMC free article] [PubMed] [Google Scholar]; This manuscript describes the structure of Neurotensin receptor 1 in complex with β-arrestin. The authors used crosslinking to stabilize the complex and phosphorylation of the receptor to form a stable interaction between the two protein components.
- 68.Nygaard R, Zou Y, Dror RO, Mildorf TJ, Arlow DH, Manglik A, Pan AC, Liu CW, Fung JJ, Bokoch MP, et al. , The dynamic process of beta(2)-adrenergic receptor activation. Cell, 2013. 152(3): p. 532–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Garcia-Nafria J, Lee Y, Bai X, Carpenter B, and Tate CG, Cryo-EM structure of the adenosine A2A receptor coupled to an engineered heterotrimeric G protein. Elife, 2018. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Carpenter B, Nehme R, Warne T, Leslie AG, and Tate CG, Structure of the adenosine A(2A) receptor bound to an engineered G protein. Nature, 2016. 536(7614): p. 104–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. , Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature, 2011. 477(7366): p. 549–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Danev R, Buijsse B, Khoshouei M, Plitzko JM, and Baumeister W, Volta potential phase plate for in-focus phase contrast transmission electron microscopy. Proc Natl Acad Sci U S A, 2014. 111(44): p. 15635–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shang G, Zhang C, Chen ZJ, Bai XC, and Zhang X, Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature, 2019. 567(7748): p. 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pang SS, Bayly-Jones C, Radjainia M, Spicer BA, Law RHP, Hodel AW, Parsons ES, Ekkel SM, Conroy PJ, Ramm G, et al. , The cryo-EM structure of the acid activatable pore-forming immune effector Macrophage-expressed gene 1. Nat Commun, 2019. 10(1): p. 4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rempel S, Gati C, Nijland M, Thangaratnarajah C, Karyolaimos A, de Gier JW, Guskov A, Slotboom DJ, A mycobacterial ABC transporter mediates the uptake of hydrophilic compounds. Nature, 2020. doi: 10.1038/s41586-020-2072-8, [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 76.Qi X, Schmiege P, Coutavas E, Wang J, and Li X, Structures of human Patched and its complex with native palmitoylated sonic hedgehog. Nature, 2018. 560(7716): p. 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gong X, Qian H, Cao P, Zhao X, Zhou Q, Lei J, and Yan N, Structural basis for the recognition of Sonic Hedgehog by human Patched1. Science, 2018. 361(6402). [DOI] [PubMed] [Google Scholar]
- 78.Chen Q, Zeng W, She J, Bai XC, and Jiang Y, Structural and functional characterization of an otopetrin family proton channel. Elife, 2019. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang Y, Bulkley DP, Xin Y, Roberts KJ, Asarnow DE, Sharma A, Myers BR, Cho W, Cheng Y, and Beachy PA, Structural Basis for Cholesterol Transport-like Activity of the Hedgehog Receptor Patched. Cell, 2018. 175(5): p. 1352–1364 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Manolaridis I, Jackson SM, Taylor NMI, Kowal J, Stahlberg H, and Locher KP, Cryo-EM structures of a human ABCG2 mutant trapped in ATP-bound and substrate-bound states. Nature, 2018. 563(7731): p. 426–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yan R, Zhao X, Lei J, and Zhou Q, Structure of the human LAT1–4F2hc heteromeric amino acid transporter complex. Nature, 2019. 568(7750): p. 127–130. [DOI] [PubMed] [Google Scholar]