

Abstract
For decades, high-resolution structural studies of biological macromolecules with masses of <200 kDa by cryo-EM single-particle analysis were considered infeasible. It was not until several years after the advent of direct detectors that the overlooked potential of cryo-EM for studying small complexes was first realized. Subsequent advances in sample preparation, imaging, and data processing algorithms have improved our ability to visualize small biological targets. In the past two years alone, nearly two hundred high-resolution structures have been determined of small (<200 kDa) macromolecules, the smallest being approximately 39 kDa in molecular weight. Here we summarize some salient lessons and strategies for cryo-EM studies of small biological complexes, and also consider future prospects for routine structure determination.
Introduction
The advent of improved imaging technologies and data processing software in cryo-electron microscopy (cryo-EM) single-particle analysis (SPA) has enabled high-resolution structure determination of biological macromolecules in solution, permitting the direct visualization of complexes that were previously incompatible with other structural techniques due to size, conformational heterogeneity, and/or compositional variability [1,2]. While high-resolution SPA reconstructions of diverse targets are now increasingly routine, the sensitivity of biological complexes to ionizing radiation from the electron beam places a significant constraint on the physical capabilities of cryo-EM. The trade-off between maximizing the signal-to-noise ratio (SNR) of cryo-EM images while minimizing the extent of radiation damage is of non-trivial importance and arguably the crux of future improvements in the field. This compromise has particularly impacted the study of specimens of low molecular mass, which have fewer scattering atoms to contribute to signal, and have consequently largely eluded cryo-EM SPA structure determination due to their poor SNR in vitreous ice. For this reason, it was widely believed for many years that cryo-EM was not a viable technique for structure determination of biological samples with molecular weights below 200 kilodaltons (kDa) [3]. However, in 2016 Merk et al. [4] were the first to break ground in this arena by presenting cryo-EM structures of the ~140 kDa lactase dehydrogenase and the ~93 kDa isocitrate dehydrogenase to 2.8 Å and 3.8 Å resolutions, respectively. The size limits of cryo-EM SPA have since been continuously challenged, and several high-resolution (i.e. better than 4 Å resolution) reconstructions of progressively smaller biological specimens have been reported in recent years (Figure 1). An unprecedented number of <200 kDa targets were determined in the past two years alone; a list of all such structures, with relevant details regarding sample preparation and imaging conditions, is summarized in Table S1. Among these is the 3.7 Å structure of the ~40 kDa S-adenosylmethionine-IV riboswitch RNA [5•], which is the smallest target resolved without a molecular scaffold to date, and on the cusp of the predicted 38 kDa limit of SPA [6]. Though these structures currently represent a minority (approximately <1%) of all SPA reconstructions deposited to the Electron Microscopy Data Bank (EMDB), this proportion is certain to change with further developments in sample preparation, data collection, and image processing algorithms. Here, we review recent achievements in pushing the lower size limits of cryo-EM SPA and emphasize special considerations for resolving targets in this size range, as well as insights into protein structure/function and drug discovery. We also discuss further areas for improvement and future prospects for high-resolution structure determination of small complexes.
Figure 1.
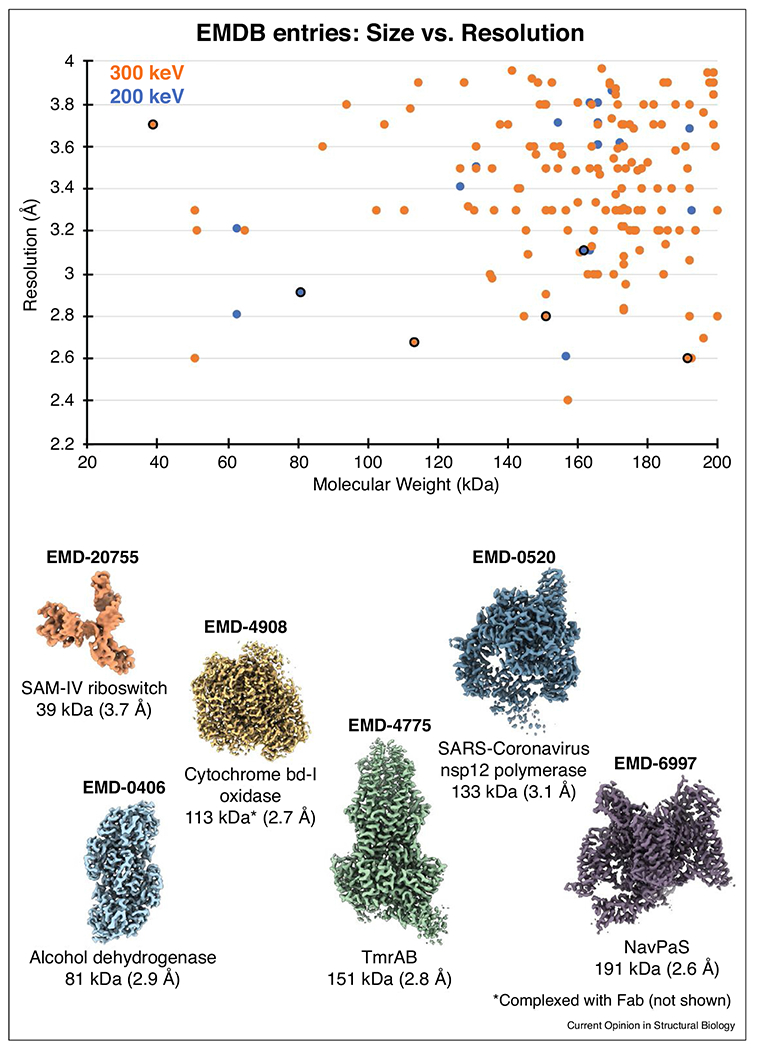
High-resolution sub-200 kDa structures determined by cryo-EM single-particle analysis. Top: Electron Microscopy Data Bank (EMDB) entries of complexes amassing below 200 kDa and resolved to better than 4 Å resolution, plotted by molecular weight and resolution. Data points are up to date to the point of this publication. Bottom: Cryo-EM density is shown for selected entries (outlined in black in the plot above) along with the corresponding EMDB identifier, molecule name, molecular mass, and reported resolution. Further details for each structure are listed in Table S1.
Methodologies and strategies for imaging small complexes
Considerations for the frozen specimen on the grid
Predictably, maximizing contrast and SNR is of paramount importance when imaging small biological targets. To do so, it is critical to prepare and image cryo-EM specimen grids containing non-overlapping macromolecules embedded in the thinnest possible layer of vitrified ice. Ideally, this would be ice that is only slightly thicker than the longest dimension of the targeted molecule.
Benchmarking experiments using the ~150 kDa enzyme aldolase (~10 nm) revealed that data collection efficiency and resolution are maximized when targeting over ice that is between 10–20 nm in thickness [7], such that the particles comprise a single monolayer within the vitreous ice. The use of gold grids may yield advantages to this end, as specimens frozen on gold substrates are typically thinner, and experience less beam-induced motion during exposures compared to those frozen on carbon [7,8••,9]. Ice thickness should be quantitatively assessed throughout data collection to ensure continuous imaging over the thinnest possible ice. Absolute thickness measurements can be performed using an energy filter [10] or through aperture limited scattering [8••], either of which can be incorporated into an automated data collection pipeline. Additionally, assessment of the Fourier transform of the acquired images is a particularly useful metric for ice thickness, as images containing optimally thin ice typically exhibit Thon rings that extend to high resolution, while the presence of a ring of increased intensities near 3.9 Å (often referred to as a ‘water ring’) is characteristic of ice that is thicker than ~50 nm [8••], which is substantially greater than the diameter of most small specimens. As such, performing on-the-fly estimation of the contrast transfer function (CTF) as part of an automated image acquisition scheme is particularly beneficial for maximizing the efficiency and quality of data collection.
While the ideal specimen grid is intuitive in theory, it is oftentimes not so straightforward in practice. We have observed that the smaller the complex, the thinner and therefore more fragile the ideal ice layer. Maintaining a high particle density in the grid holes seems to be necessary for supporting very thin layers of ice-embedded molecules, with the additional benefit of increasing the accuracy of subsequent image processing steps that utilize neighboring particles to facilitate alignment, such as the correction of beam-induced motion, local CTF estimation, and particle polishing [7,11–13]. Conversely, it is also important to ensure that the ice is not too thin. A detailed tomographic study of various single-particle specimen grids conducted by Noble et al. [14••] demonstrated that biological macromolecules commonly adsorb to and are damaged by the hydrophobic air-water interface, resulting in the adoption of a preferred orientation or even partial to complete sample denaturation [15–17]. As exposure to both air-water interfaces likely increases in thinner ice, the specimen may become more susceptible to these detrimental effects. Recently, grids overlaid with monolayer graphene were used to obtain a 2.6 Å structure of the smallest protein complex resolved to date, the ~52 kDa streptavidin [18••]. Graphene and graphene oxide monolayer supports [19–21] sequester macromolecules away from the air-water interface in addition to increasing particle density in the grid holes and are nearly electron-transparent, thereby enabling the imaging of small complexes without contributing additionally to background noise. With efforts towards functionalization of graphene monolayers already underway, the applicability of these supports for routine high-resolution structure determination of various small specimens is particularly promising.
Additional imaging devices can further increase image SNR
Microscope accessories such as phase plates and quantum energy filters can increase the SNR in cryo-EM images by introducing phase contrast and deflecting inelastically scattered electrons contributing to noise, respectively. Theoretically, both offer advantages over conventional imaging methods for resolving small biological targets, though their limitations have not systematically been tested. Several high-resolution structures have been determined using Volta phase plate (VPP) technology [22–25], including the first structure of the ~64 kDa human hemoglobin [26]. However, the VPP phase shift is an inconsistent phenomenon and must be constantly monitored and regenerated during data acquisition, making VPPs incompatible with automated imaging workflows. It is not clear whether phase plate technology in its current stage confers a significant advantage over conventional imaging for all small targets, as high-resolution structures of hemoglobin and streptavidin were recently obtained without a phase plate using conventional defocus imaging methodologies [18••,27•] (Figure 2 and Table S1). However, a more direct comparison using the adenosine A2A receptor coupled to an engineered G protein demonstrated that use of a VPP yielded improvements to resolution and the overall B-factor of the data [28]. Further comparisons coupled with advances to phase plate technology (see Conclusions and Future Prospects) are necessary to understand the benefits of these devices for resolving small complexes.
Figure 2.
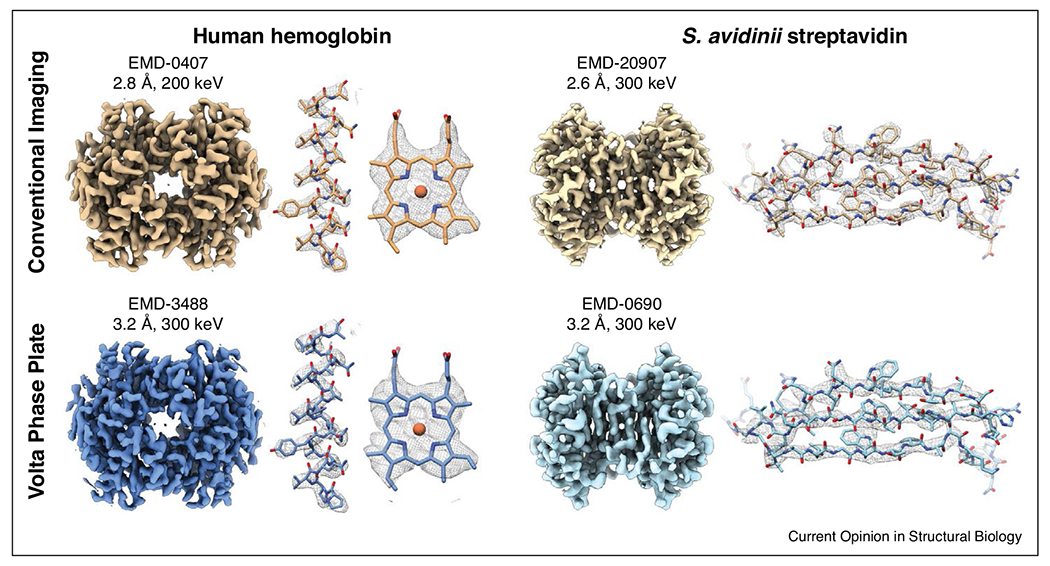
Cryo-EM reconstructions of 64 kDa hemoglobin and 52 kDa streptavidin imaged with and without a Volta Phase Plate (VPP). Left: Hemoglobin determined to 2.8 Å resolution using conventional defocus-based imaging (top) and to 3.2 Å using a VPP (bottom). Right: Streptavidin determined to 2.6 Å resolution using conventional defocus-based imaging (top) and to 3.2 Å using a VPP (bottom). The full map and density features (with the corresponding atomic model docked in) are shown for each structure.
Inaccurate image alignment of small complexes is a major limitation to successful structure determination
Despite efforts to increase image contrast and SNR, structure determination of small complexes that are asymmetric, conformationally flexible, or particularly lacking in distinguishing structural features may be thwarted due to misalignment by 3-dimensional (3D) image reconstruction programs. Signal-subtracted datasets of subtetrameric streptavidin have demonstrated that high-quality 2D class averages and 3D reconstructions could be obtained for particles as small as the ~13 kDa monomer when the correct angular information (previously determined from the high-resolution tetramer structure) was supplied; however, when global angular searches were performed on the datasets, the accuracy of image alignment decreased significantly with particle size [29]. We have also encountered similar particle misalignment issues in our attempts to reconstruct the asymmetric ~43 kDa catalytic subunit of protein kinase A bound to inhibitor (iPKAcat) [27•]. Although particles were readily visible in the high-contrast images, and the 2D class averages contained discernible structural details, the resolution of the final 3D reconstruction was limited by gross misalignment of the particle images. Structure determination of small membrane proteins with few or no extramembrane domains is also challenging, as the bulk signal from the disordered detergent micelle or lipid nanodisc commonly confounds image alignment. In some cases, partial signal subtraction of disordered regions from the particle images can yield some improvements to the reconstruction [30,31]; however, the likelihood of success will depend on proper alignment of the original (non-subtracted) images [32] and, as forewarned above, whether the remaining signal in the subtracted images is sufficient for accurate angular assignment.
Further improvements to cryo-EM SPA image reconstruction algorithms that facilitate more robust alignment of low-SNR particle images will be required to accommodate more routine structural studies of smaller and conformationally variable biological targets. In the meantime, the addition of rigid and easily identifiable fiducial markers (e.g. monoclonal antigen-binding fragments (Fabs), nanobodies, or megabodies) to small targets otherwise lacking in distinct structural features can facilitate more accurate image alignment with the added benefit of stabilizing the target complex and overcoming preferred orientation [33–35]. If desired, the fiducial density can then be excluded in silico during the final steps of 3D refinement through focused masking or signal subtraction. Structures of the ~61 kDa human serotonin transporter and of the ~49 kDa Plasmodium falciparum CQ-resistance transporter complexed with Fab were recently determined to 3.6 Å and 3.2 Å, respectively, demonstrating the value of this approach for resolving complexes well below 100 kDa [30,36]. Additionally, attachment of small protein targets to a symmetric molecular scaffold via Designed Ankyrin Repeat Protein (DARPin) adaptors has been successfully implemented for structure determination of complexes as small as the ~26 kDa green fluorescent protein [37•,38,39]. The modularity of these scaffolds allows for customization to suit a broad range of targets, provided flexibility and self-association of the attached complex are not issues [38].
Small complexes can be visualized at 200 and 300 keV
While the vast majority of sub-200 kDa targets were obtained using microscopes operating at 300 keV, a small subset of structures was determined using comparatively cheaper 200 keV instruments [40–48], the limits of which were explored by Herzik et al. [11,27•]. These results are significant because they expand the utility and accessibility of cryo-EM SPA for high-resolution studies of complexes of various sizes. However, there does not yet seem to be an obvious choice of electron energy for imaging smaller complexes. The increase in image contrast arising from the higher elastic scattering cross-section at lower energies is offset by the concomitant increase in inelastic scattering events [49], lens aberrations [50], and the fact that existing direct detectors perform optimally with higher keV electrons. However, certain optical aberrations can now be estimated and corrected for in silico [51,52], and efforts to develop a detector for imaging 100 keV electrons are currently underway [53••] (see Conclusions and Future Prospects).
Novel and drug-bound structures of small membrane protein complexes
Cryo-EM SPA has revolutionized the structure determination of membrane proteins in particular, as membrane proteins are notoriously recalcitrant to crystallization efforts due to their amphipathic nature. It is perhaps unsurprising, then, that membrane proteins stabilized in detergents or lipid nanodiscs comprise over 60% of all high-resolution sub-200 kDa structures deposited into the EMDB to date, with nearly all entries obtained within the past two years alone. Among these were de novo structures of the human lipid exporter ABCB4 [54], the otopetrin proton channels OTOP1 [55] and OTOP3 [55,56], several members of the TMEM16 scramblase family [40,57,58], as well as of the structurally homologous OSCA mechanosensitive ion channel family [46,59]. It is interesting to note that the majority of structures were obtained using a microscope equipped with an energy filter, which may have provided some gain in SNR particularly around the transmembrane region, which is surrounded by disordered detergent or lipid molecules. This may especially be the case for the complexes determined using 200 keV, all of which were imaged using an energy filter with the exception of the OSCA 1.2 channel [46]. Finally, a number of high-resolution structures of agonist-bound channels, in which protein-ligand interactions within the binding site were resolved with high fidelity [60–64], have provided valuable insights to the molecular bases for toxin binding and selectivity. Considering the multitude of disease-relevant small membrane protein complexes (e.g. G protein-coupled receptors, which comprise over one-third of all small-molecule drug targets) and the immense pharmacological efforts towards developing novel therapeutics against these targets, these findings highlight the exciting potential of cryo-EM for modulating channel activity through structure-aided drug design.
Conclusions and future prospects
The visualization of small biological macromolecules by cryo-EM SPA had long been dismissed as an insurmountable task, but recent advancements in sample preparation and imaging methodologies are rapidly pushing the frontiers of specimen sizes that can be resolved to high resolution. Numerous groundbreaking studies from the past few years have demonstrated that high-resolution reconstructions of diverse biological targets ranging from membrane proteins to RNA complexes, and as small as 40–50 kDa, are attainable by cryo-EM SPA. A summary of the strategies and approaches mentioned in this review is presented in Figure 3. Looking ahead, novel technologies such as automated sample vitrification instruments (i. e. Spotiton (commercially Chameleon) [65,66], Shakeitoff [67], and VitroJet [68]) may mitigate sample adsorption to the air-water interface by shortening the sample dispense-to-plunge time and permit users to reproducibly control for thin ice. Ongoing development of a continuous-wave focused laser phase plate [69•] that can produce a constant phase shift with no information loss may further push the envelope for high-resolution structure determination of small complexes. Additionally, the potential for routine specimen screening and structure determination of small targets at 100 keV or lower is an exciting prospect from both financial and practical standpoints, and it will be particularly interesting to explore the resolving capabilities of lower electron energies when paired with an appropriately optimized detector. Given that half of all proteins in the human proteome are <50 kDa in molecular weight [70], these collective advances will greatly expand the breadth of new and previously unanswered biological questions that can be investigated using cryo-EM SPA.
Figure 3.
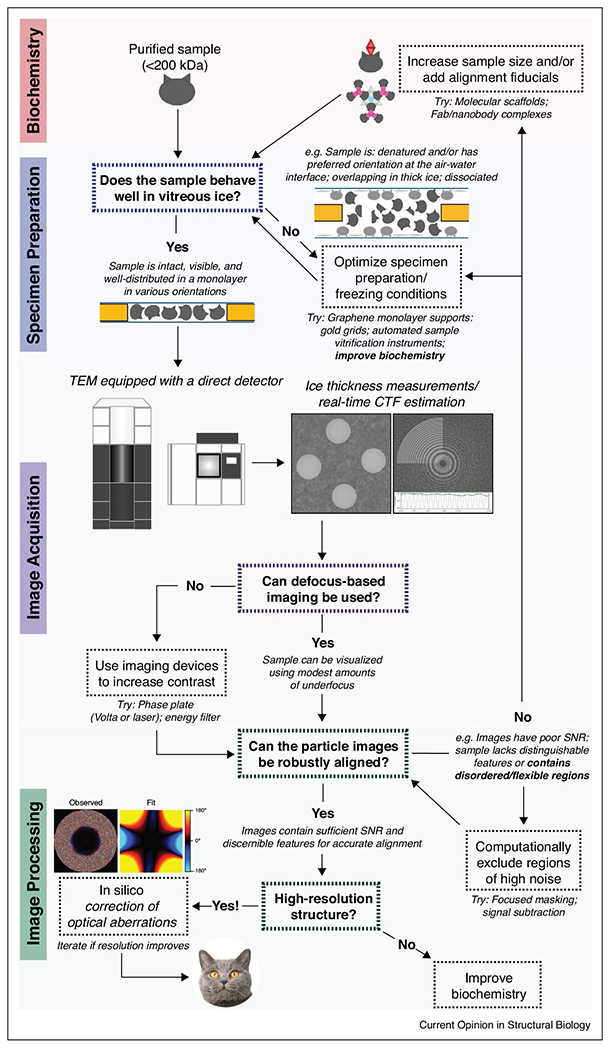
Flowchart for cryo-EM SPA analysis of small biological complexes. Important considerations for structure determination of small targets at each stage of frozen specimen preparation, data acquisition, and image processing are highlighted. Potential pitfalls and possibilities for overcoming them (as demonstrated by published work) are also indicated.
Supplementary Material
Acknowledgements
M.W. is supported as a National Science Foundation Graduate Student Research fellow. G.C.L. is supported by a young investigator award from Amgen, and by the National Institutes of Health (NIH) grants R21AR072910 and R21AG061697.
Footnotes
Conflict of interest statement
Nothing declared.
Appendix A.: Supplementary data
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.sbi.2020.05.007.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Yan C, Wan R, Bai R, Huang G, Shi Y: Structure of a yeast activated spliceosome at 3.5 A resolution. Science 2016, 353:904–911. [DOI] [PubMed] [Google Scholar]
- 2.Liu Z, Gutierrez-Vargas C, Wei J, Grassucci RA, Ramesh M, Espina N, Sun M, Tutuncuoglu B, Madison-Antenucci S, Woolford JL et al. : Structure and assembly model for the Trypanosoma cruzi 60S ribosomal subunit. Proc Natl Acad Sci USA 2016, 113:12174–12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Costa TR, Ignatiou A, Orlova EV: Methods in molecular biology. Methods Mol Biol Clifton N J 2017, 1615:377–413. [DOI] [PubMed] [Google Scholar]
- 4.Merk A, Bartesaghi A, Banerjee S, Falconieri V, Rao P, Davis MI, Pragani R, Boxer MB, Earl LA, Milne JL et al. : Breaking cryo-EM resolution barriers to facilitate drug discovery. Cell 2016, 165:1698–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.•.Zhang K, Li S, Kappel K, Pintilie G, Su Z, Mou T-C, Schmid MF, Das R, Chiu W: Cryo-EM structure of a 40 kDa SAM-IV riboswitch RNAat 3.7 Å resolution. Nat Commun 2019, 10:5511. [DOI] [PMC free article] [PubMed] [Google Scholar]; Presents the first high-resolution cryo-EM structures of a pure RNA specimen, the SAM-IV riboswitch, which is the smallest biological target resolved without a molecular scaffold by cryo-EM SPA to date.
- 6.Henderson R: The potential and limitations of neutrons, electrons and X-rays for atomic resolution microscopy of unstained biological molecules. Q Rev Biophys 1995, 28:171–193. [DOI] [PubMed] [Google Scholar]
- 7.Kim LY, Rice WJ, Eng ET, Kopylov M, Cheng A, Raczkowski AM, Jordan KD, Bobe D, Potter CS, Carragher B: Benchmarking cryo-EM single particle analysis workflow. Front Mol Biosci 2018, 5:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.••.Rice WJ, Cheng A, Noble AJ, Eng ET, Kim LY, Carragher B, Potter CS: Routine determination of ice thickness for Cryo-EM grids. J Struct Biol 2018, 204:38–44 [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes methods for measuring the ice thickness of frozen SPA specimens as an integrated part of data collection to determine optimal areas for image acquisition and enable specimen characterization.
- 9.Russo C, Passmore L: Ultrastable gold substrates for electron cryomicroscopy. Science 2014, 346:1377–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brydson R: A brief review of quantitative aspects of electron energy loss spectroscopy and imaging. Mater Sci Tech Ser 2000, 16:1187–1198. [Google Scholar]
- 11.Jr MA, Wu M, Lander GC: Achieving better-than-3-A resolution by single-particle cryo-EM at 200 keV. Nat Methods 2017, 14:1075–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Su M: goCTF: geometrically optimized CTF determination for single-particle cryo-EM. J Struct Biol 2018, 205:22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scheres S: Methods in enzymology. Methods Enzymol 2016, 579:125–157. [DOI] [PubMed] [Google Scholar]
- 14.••.Noble AJ, Dandey VP, Wei H, Brasch J, Chase J, Acharya P, Tan Y, Zhang Z, Kim LY, Scapin G et al. : Routine single particle CryoEM sample and grid characterization by tomography. eLife 2018, 7:e34257. [DOI] [PMC free article] [PubMed] [Google Scholar]; Characterizes particle behavior and distribution in vitreous ice across a broad range of frozen SPA specimens using fiducial-less tomography. Reveals that the vast majority of particles adsorb to the air-water interface, with varying consequences of protein denaturation and preferred orientation, and also highlights the potential for using tomography on SPA grids to guide data collection.
- 15.Glaeser RM, Han B-G: Opinion: hazards faced by macromolecules when confined to thin aqueous films. Biophys Rep 2017, 3:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Jongh HH, Kosters HA, Kudryashova E, Meinders MB, Trofimova D, Wierenga PA: Protein adsorption at air-water interfaces: a combination of details. Biopolymers 2004, 74:131–135. [DOI] [PubMed] [Google Scholar]
- 17.Glaeser RM: Proteins, interfaces, and cryo-EM grids. CurrOpin Colloid In 2018, 34:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.••.Han Y, Fan X, Wang H, Zhao F, Tully CG, Kong J, Yao N, Yan N: High yield monolayer graphene grids for near-atomic resolution cryo-electron microscopy. Biorxiv 2019. 10.1101/827089 [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes a straightforward and economical procedure for preparing graphene monolayer grids for cryo-EM, and demonstrates the utility of graphene grids for resolving small complexes to high resolution. Presents the 2.6A structure of the 52-kDa streptavidin, which is the highest-resolution structure of a sub-100 kDa target to date.
- 19.Palovcak E, Wang F, Zheng SQ, Yu Z, Li S, Betegon M, Bulkley D, Agard DA, Cheng Y: A simple and robust procedure for preparing graphene-oxide cryo-EM grids. J Struct Biol 2018, 204:80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu N, Zhang J, Chen Y, Liu C, Zhang X, Xu K, Wen J, Luo Z, Chen S, Gao P et al. : Bioactive functionalized monolayer graphene for high-resolution cryo-electron microscopy. J Am Chem Soc 2019, 141:4016–4025. [DOI] [PubMed] [Google Scholar]
- 21.Naydenova K, Peet MJ, Russo CJ: Multifunctional graphene supports for electron cryomicroscopy. Proc National Acad Sci USA 2019, 116 201904766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li K, Sun C, Klose T, Irimia-Dominguez J, Vago FS, Vidal R, Jiang W: Sub-3 Å apoferritin structure determined with full range of phase shifts using a single position of volta phase plate. J Struct Biol 2019, 206:225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang Y-L, Khoshouei M, Deganutti G, Glukhova A, Koole C, Peat TS, Radjainia M, Plitzko JM, Baumeister W, Miller LJ et al. : Cryo-EM structure of the active, Gs-protein complexed, human CGRP receptor. Nature 2018, 561:492–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gong X, Qian H, Cao P, Zhao X, Zhou Q, Lei J, Yan N: Structural basis for the recognition of Sonic Hedgehog by human Patched1. Science 2018, 361:eaas8935. [DOI] [PubMed] [Google Scholar]
- 25.Liang Y-L, Khoshouei M, Glukhova A, Furness SG, Zhao P, Clydesdale L, Koole C, Truong TT, Thal DM, Lei S et al. : Phase-plate cryo-EM structure of a biased agonist-bound human GLP-1 receptor-Gs complex. Nature 2018, 555:121–125. [DOI] [PubMed] [Google Scholar]
- 26.Khoshouei M, Radjainia M, Baumeister W, Danev R: Cryo-EM structure of haemoglobin at 3.2 Å determined with the Volta phase plate. Nat Commun 2017, 8 ncomms16099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.•.Herzik MA, Wu M, Lander GC: High-resolution structure determination of sub-100 kDa complexes using conventional cryo-EM. Nat Commun 2019, 10:1032. [DOI] [PMC free article] [PubMed] [Google Scholar]; Demonstrates that high-resolution structures of sub-100 kDa targets can be obtained at 200 keV using conventional defocus imaging.
- 28.Garcia-Nafria J, Lee Y, Bai X, Carpenter B, Tate CG: Cryo-EM structure of the adenosine A2A receptor coupled to an engineered heterotrimeric G protein. eLife 2018, 7 e35946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fan X, Wang J, Zhang X, Yang Z, Zhang J-C, Zhao L, Peng H-L, Lei J, Wang H-W: Single particle cryo-EM reconstruction of 52kDa streptavidin at 3.2Angstrom resolution. Nat Commun 2019, 10:2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim J, Tan Y, Wicht KJ, Erramilli SK, Dhingra SK, Okombo J, Vendome J, Hagenah LM, Giacometti SI, Warren AL et al. : Structure and drug resistance of the Plasmodium falciparum transporter PfCRT. Nature 2019, 576:315–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reid MS, Kern DM, Brohawn SG: Cryo-EM structure of the potassium-chloride cotransporter KCC4 in lipid nanodiscs. Biorxiv 2019. 10.1101/805267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bai X, Rajendra E, Yang G, Shi Y, Scheres SH: Sampling the conformational space of the catalytic subunit of human γ-secretase. eLife 2015, 4:e11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu S, Avila-Sakar A, Kim J, Booth DS, Greenberg CH, Rossi A, Liao M, Li X, Alian A, Griner SL et al. : Fabs enable single particle cryoEM studies of small proteins. Structure 2012, 20:582–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen AH, Thomsen AR, Cahill TJ, Huang R, Huang L-Y, Marcink T, Clarke OB, Heissel S, Masoudi A, Ben-Hail D et al. : Structure of an endosomal signaling GPCR–G protein–β-arrestin megacomplex. Nat Struct Mol Biol 2019, 26:1123–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uchański T, Masiulis S, Fischer B, Kalichuk V, Wohlkönig A, Zögg T, Remaut H, Vranken W, Aricescu RA, Pardon E et al. : Megabodies expand the nanobody toolkit for protein structure determination by single-particle cryo-EM. Biorxiv 2019. 10.1101/812230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coleman JA, Yang D, Zhao Z, Wen P-C, Yoshioka C, Tajkhorshid E, Gouaux E: Serotonin transporter-ibogaine complexes illuminate mechanisms of inhibition and transport. Nature 2019, 569:141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.•.Liu Y, Huynh DT, Yeates TO: A 3.8 Å resolution cryo-EM structure of a small protein bound to an imaging scaffold. Nat Commun 2019, 10:1864. [DOI] [PMC free article] [PubMed] [Google Scholar]; Demonstrates the utility of designed symmetrical molecular scaffolds with DARPin adaptors for imaging small cargo proteins and presents the 3.8 A structure of the 26 kDa green fluorescent protein determined using this strategy.
- 38.Liu Y, Gonen S, Gonen T, Yeates TO: Near-atomic cryo-EM imaging of a small protein displayed on a designed scaffolding system. Proc National Acad Sci USA 2018, 115:201718825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yao Q, Weaver SJ, Mock J-Y, Jensen GJ: Fusion of DARPin to aldolase enables visualization of small protein by Cryo-EM. Structure 2019. 10.1016/j.str.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kalienkova V, Mosina V, Bryner L, Oostergetel GT, Dutzler R, Paulino C: Stepwise activation mechanism of the scramblase nhTMEM16 revealed by cryo-EM. eLife 2019, 8:e44364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kirchdoerfer RN, Ward AB: Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat Commun 2019, 10:2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garaeva AA, Guskov A, Slotboom DJ, Paulino C: A one-gate elevator mechanism for the human neutral amino acid transporter ASCT2. Nat Commun 2019, 10:3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang C, Konermann S, Brideau NJ, Lotfy P, Wu X, Novick SJ, Strutzenberg T, Griffin PR, Hsu PD, Lyumkis D: Structural basis for the RNA-guided ribonuclease activity of CRISPR-Cas13d. Cell 2018, 175:212–223.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oosterheert W, van Bezouwen LS, Rodenburg RN, Granneman J, Förster F, Mattevi A, Gros P: Cryo-EM structures of human STEAP4 reveal mechanism of iron(III) reduction. Nat Commun 2018, 9:4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stock C, Hielkema L, Tascón I, Wunnicke D, Oostergetel G, Azkargorta M, Paulino C, Hänelt I: Cryo-EM structures of KdpFABC suggest a K+ transport mechanism via two inter-subunit half-channels. Nat Commun 2018, 9:4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jojoa-Cruz S, Saotome K, Murthy SE, Tsui C, Sansom MS, Patapoutian A, Ward AB: Cryo-EM structure of the mechanically activated ion channel OSCA1.2. eLife 2018, 7:e41845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fuchsbauer O, Swuec P, Zimberger C, Amigues B, Levesque S, Agudelo D, Duringer A, Chaves-Sanjuan A, Spinelli S, Rousseau GM et al. : Cas9 allosteric inhibition by the anti-CRISPR protein AcrllA6. Mol Cell 2019. 10.1016/j.molcel.2019.09.012. [DOI] [PubMed] [Google Scholar]
- 48.Itskanov S, Park E: Structure of the posttranslational Sec protein-translocation channel complex from yeast. Science 2018, 363:84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Glaeser R: Methods in enzymology. Methods Enzymol 2016, 579:19–50. [DOI] [PubMed] [Google Scholar]
- 50.Saxton OW: Observation of lens aberrations for high resolution electron microscopy II: simple expressions for optimal estimates. Ultramicroscopy 2015, 151:168–177. [DOI] [PubMed] [Google Scholar]
- 51.Zivanov J, Nakane T, Scheres SH: Estimation of high-order aberrations and anisotropic magnification from Cryo-EM datasets in RELION-3.1. Biorxiv 2019. 10.1101/798066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, Scheres SH: New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 2018, 7:e42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.••.Naydenova K, McMullan G, Peet M, Lee Y, Edwards P, Chen S, Leahy E, Scotcher S, Henderson R, Russo C: CryoEM at 100 keV: a demonstration and prospects. IUCRJ 2019, 6:1086–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]; Proof-of-principle that specimens of varying sizes can be resolved at 100 keV using a hybrid pixel camera. Also offers perspectives on future prospects for microscope and detector optimization for the routine imaging of targets at lower electron energies.
- 54.Olsen JA, Alam A, Kowal J, Stieger B, Locher KP: Structure of the human lipid exporter ABCB4 in a lipid environment. Nat Struct Mol Biol 2019. 10.1038/s41594-019-0354-3. [DOI] [PubMed] [Google Scholar]
- 55.Saotome K, Teng B, Tsui C, Lee W-H, Tu Y-H, Kaplan JP, Sansom MS, Liman ER, Ward AB: Structures of the otopetrin proton channels Otopl and Otop3. Nat Struct Mol Biol 2019, 26:518–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen Q, Zeng W, She J, Bai X, Jiang Y: Structural and functional characterization of an otopetrin family proton channel. eLife 2019, 8:e46710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bushell SR, Pike AC, Falzone ME, Rorsman NJ.Ta CM, Corey RA, Newport TD, Christianson JC, Scofano LF, Shintre CA et al. : The structural basis of lipid scrambling and inactivation in the endoplasmic reticulum scramblase TMEM16K. Nat Commun 2019, 10:3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alvadia C, Lim NK, Mosina V, Oostergetel GT, Dutzler R, Paulino C: Cryo-EM structures and functional characterization of the murine lipid scramblase TMEM16F. eLife 2019, 8:e44365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang M, Wang D, Kang Y, Wu J-X, Yao F, Pan C, Yan Z, Song C, Chen L: Structure of the mechanosensitive OSCA channels. Nat Struct Mol Biol 2018, 25:850–858. [DOI] [PubMed] [Google Scholar]
- 60.Koehl A, Hu H, Maeda S, Zhang Y, Ou 0, Paggi JM, Latorraca NR, Hilger D, Dawson R, Matile H et al. : Structure of the μ-opioid receptor-Gi protein complex. Nature 2018, 558:547–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yan R, Zhao X, Lei J, Zhou C: Structure of the human LAT1–4F2hc heteromeric amino acid transporter complex. Nature 2019, 568:127–130. [DOI] [PubMed] [Google Scholar]
- 62.Shen H, Li Z, Jiang Y, Pan X, Wu J, Cristofori-Armstrong B, Smith JJ, Chin YK, Lei J, Zhou C et al. : Structural basis for the modulation of voltage-gated sodium channels by animal toxins. Science 2018, 362:eaau2596. [DOI] [PubMed] [Google Scholar]
- 63.Clairfeuille T, Cloake A, Infield DT, Llongueras JP, Arthur CP, Li Z, Jian Y, Martin-Eauclaire M-F, Bougis PE, Ciferri C et al. : Structural basis of «-scorpion toxin action on Na v channels. Science 2019, 363:eaav8573. [DOI] [PubMed] [Google Scholar]
- 64.Liu F, Zhang Z, Levit A, Levring J, Touhara KK, Shoichet BK, Chen J: Structural identification of a hotspot on CFTR for potentiation. Science 2019, 364:1184–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Razinkov I, Dandey VP, Wei H, Zhang Z, Melnekoff D, Rice WJ, Wigge C, Potter CS, Carragher B: A new method for vitrifying samples for cryoEM. J Struct Biol 2016, 195:190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dandey VP, Wei H, Zhang Z, Tan Y, Acharya P, Eng ET, Rice WJ, Kahn PA, Potter CS, Carragher B: Spotiton: New features and applications. J Struct Biol 2018, 202:161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rubinstein JL, Guo H, Ripstein ZA, Haydaroglu A, Au A, Yip CM, Trani JM, Benlekbir S, KwokT: Shake-it-off: a simple ultrasonic cryo-EM specimen-preparation device. Acta Crystallogr Sect D 2019, 75:1063–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ravelli R, Nijpels F, Henderikx R, Weissenberger G, Thewessem S, Gijsbers A, Beulen B, Lopez-lglesias C, Peters PJ: Automated cryo-EM sample preparation by pin-printing and jet vitrification. Biorxiv 2019. 10.1101/651208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.•.Schwartz O, Axelrod JJ, Campbell SL, Turnbaugh C, Glaeser RM, Müller H: Laser phase plate for transmission electron microscopy. Nat Methods 2019, 16:1016–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes the design and implementation of a laser phase plate within a transmission electron microscope and presents initial proof-of-principle that phase contrast can be generated when imaging an amorphous carbon film.
- 70.Brocchieri L, Karlin S: Protein length in eukaryotic and prokaryotic proteomes. Nucleic Acids Res 2005, 33:3390–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.