

Abstract
Damage-associated molecular patterns, such as HMGB1, play a well-recognized role in the development of pulmonary arterial hypertension (PAH), a progressive fatal disease of the pulmonary vasculature. However, the contribution of the particular type of vascular cells, type of cell death, or the form of released HMGB1 in PAH remains unclear. Moreover, although male PAH patients show a higher level of circulating HMGB1, its involvement in the severe PAH phenotype reported in males is unknown. In this study, we aimed to investigate the sources and active forms of HMGB1 released from damaged vascular cells and their contribution to the progressive type of PAH in males. Our results showed that HMGB1 is released by either pulmonary artery human endothelial (HPAEC) or human smooth muscle cells (HPASMC) that underwent necrotic cell death, although only HPASMC produce HMGB1 during apoptosis. Moreover, only HPASMC death induced a release of dimeric HMGB1, found to be mitochondrial reactive oxygen species dependent, and TLR4 activation. The modified Sugen/Hypoxia rat model replicates the human sexual dimorphism in PAH severity (right ventricle systolic pressure in males vs. females 54.7±2.3 vs. 44.6±2 mmHg). By using this model, we confirmed that necroptosis and necrosis are the primary sources of circulating HMGB1 in the male rats, although only necrosis increased circulation of HMGB1 dimers. Attenuation of necrosis but not apoptosis or necroptosis prevented TLR4 activation in males and blunted the sex differences in PAH severity. We conclude that necrosis, through the release of HMGB1 dimers, predisposes males to a progressive form of PAH.
Keywords: pulmonary hypertension, sex difference, apoptosis, necrosis, HMGB1
Graphical Abstract
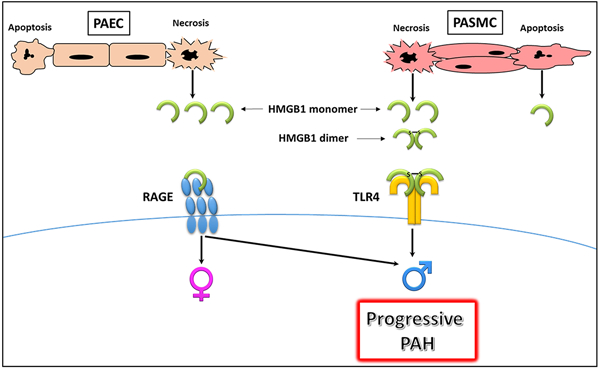
Summary:
HMGB1 release and activity depend on the type of vascular cells, type of cell death, and form of released HMGB1. HMGB1 released from necrotic cells promotes PAH severity in male rodents in the preclinical model of PAH, suggesting its involvement in progressive PAH and poor survival prognosis in male patients with PAH.
INTRODUCTION:
Pulmonary arterial hypertension (PAH) is a severe incurable disease that ultimately leads to the patient’s death due to the development of the right ventricle (RV) failure1. The damage of the pulmonary vascular cells is a well-recognized stimulus that initiates PAH development in the animal models2 and contributes to its progression through the entire course of the disease3. In humans, apoptosis and necrosis of the pulmonary vasculature were confirmed to present at different stages of the disease4, 5.
High mobility group box 1 (HMGB1) is a chromatin-associated protein that acts as an essential DNA chaperone. Once released from damaged cells, it also functions as a damage-associated molecular pattern (DAMP) and, by binding to several receptors, including toll-like receptor 4 (TLR4) and receptor for advanced glycation end products (RAGE), initiates inflammatory response and tissue repair. Multiple previous studies proposed the critical role of HMGB1 and its downstream signaling in PAH. Thus, it was found that PAH upregulates the extracellular levels of HMGB1 in the lungs and increases the levels of circulating HMGB1 in patients or preclinical animal models6–9. The therapeutic strategies that either neutralize HMGB1 or inhibit TLR4 and RAGE activity or expression, strongly attenuate PAH development and progression7–12.
There is a well-established sexual dimorphism in the prevalence of PAH and disease outcome. While females are more predisposed to PAH13, 14, males consistently show a poorer survival rate15, 16. Men present a significantly higher mean pulmonary artery pressure at diagnosis compared to women15. Male sex also associates with a worse functional class, higher brain natriuretic peptide levels, and shorter 6-min walk distances compared with female patients16. The progressive form of PAH in men corresponds with a worse response to standard PAH-specific therapies17 and the development of maladaptive right ventricular remodeling16. Thus, there is a strong necessity to utilize a sex-specific approach for handling and treating PAH. Nevertheless, the particular causes responsible for the development of a more aggressive form of PAH in men is still under debate and not well understood. Neither is it clear what type of therapy will be beneficial for men. The limited availability of adequate preclinical animal models that reproduce sex differences in the PAH severity additionally limits the research capabilities and prevents a better understanding of the mechanisms that drive PAH progression in males.
Recently we have discovered that male but not female PAH patients have significantly increased levels of circulating HMGB16. However, the mechanisms responsible for such sexual disparity in HMGB1 release remain unknown. There are numerous contradictions in the published literature regarding the type of cell death releasing HMGB1 or the biological activity of monomeric vs. oligomeric forms of HMGB1. Besides, it was proposed that the ability of cells to release HMGB1 depends on the cell type18, 19. Therefore, in this study, we aimed to investigate the contribution of different types of cell death (apoptosis vs. necrosis) in HMGB1 release from either human pulmonary artery endothelial (HPAEC) or human smooth muscle cells (HPASMC). We have also analyzed the contribution of monomeric and dimeric forms of HMGB1 in the activation of downstream signaling.
In this study, we also used an optimized Sugen/hypoxia model that allowed us to replicate a considerably more severe form of PAH in male rats. By using this model, we investigated the particular type of cell death responsible for the release of HMGB1 and manifestation of sex differences in PAH severity.
METHODS:
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Culturing pulmonary vascular cells
Human male pulmonary artery smooth muscle cells (HPASMCs), were cultured in Dulbecco’s modified Eagle’s medium (DMEM, GIBCO, ThermoFisher, Waltham, MA, cat#11965–092) containing 4.5g/L glucose and 5% FBS (GenClone, Genessee Scientific). Human male pulmonary artery endothelial cells (HPAEC) were cultured in Endothelial Cell Medium (ECM, ScienCell Research Laboratories Inc, cat #1001), containing 2% FBS.
Apoptosis and necrosis induction
One day before the experiment, HPASMC and HPAEC were plated at 70–80% of confluence into 12-well plates and grown overnight in the growth media described above. Apoptosis of HPASMC was induced by culturing the cells in DMEM, containing a reduced level of glucose (0.1g/L). In HPAEC, apoptosis was induced by culturing cells in serum-free ECM. Apoptosis in both cell types was measured 48 hours after induction. For necrosis induction, both cell types underwent 3–4 freeze/thawing cycles, as published6. Conditioned media collected from apoptotic or necrotic HPAEC or HPASMC was used to treat the corresponding naïve HPAEC or HPASMC for 0–6 hours. To confirm the relevance of the visualized monomeric and dimeric bands to HMGB1 as well as to validate the contribution of HMGB1 in the stimulation of TLR4 and RAGE, the necrotic conditioned media was mixed with 25u/ml of neutralizing anti-HMGB1 antibodies20 (clone 3E8, Biolegend, San Diego, CA, cat# 651402) and incubated for overnight. The next day, the protein A/G ultra link resin beads (Thermo Fisher, cat#53132) were added to bind HMGB1-IgG complex. The samples were rotated at 4°C for 2 hours and centrifuged (30sec at 1,000 g). The supernatant (conditioned media with depleted HMGB1) or original necrotic media were used to treat naïve HPASMC for 6 hours.
Isolation of rat pulmonary artery smooth muscle cells
Rat PASMC (RPASMC) were isolated from the pulmonary artery of male Sprague Dawley rats, as described before21. Briefly, the lung excised from the rat was placed in cold DMEM without glutamine and pyruvate (GIBCO, ThermoFisher, Waltham, MA, cat# 11960–044). The pulmonary artery vascular tree was separated from lung parenchyma, cut and transferred to 2 mL of 1 mg/mL collagenase-2 (Worthington Biochemical Corporation, Lakewood, NJ, cat# LS004176,) in DMEM without glutamine and pyruvate for 20 minutes at 37°C. After incubation, the supernatant was aspirated, and the cell suspension was mixed with 10 mL of warm DMEM with glutamine (GIBCO, ThermoFisher, Waltham, MA, cat# 11965–092) supplemented with 20% FBS (GenClone, Genessee Scientific cat# 25–5144). Cells were plated in a 10 cm dish covered with gelatin (cat# ES-006-B, Millipore). The media was changed every 2–3 days, and after 2–3 passages, the cells were stained for smooth muscle-actin (Anti-α-SM actin (1A4), Alexa-Fluor488 ab184675, Abcam, 1:100, Isotype IgG2a) using FACS performed in a Novocyte 2000 (ACEA Biosciences) instrument.
Protective role of low doses of ethanol
Isolated RPASMC were cultured in DMEM containing 10% FBS and 4.5g/l glucose. One day before the experiment, cells were and plated into a 12-well plate in density 1.8x105 cells/well. After 24 hours of cell stabilization, the culture medium was replaced by DMEM with 0.1% FBS, containing 0–10 mmol/l of ethanol or 0–5 ul/ml of DMSO. One hour later, cells were stimulated with 0.1 mM of sodium orthovanadate (MilliporeSigma, Burlington, MA, cat#50860500) for 20 minutes, followed by 0.5mM H2O2 (Fisher Chemical, cat#H325–100) for another 35 minutes to induce cell necrosis as previously published 22. At the end of the incubation period, the cells were collected, washed with DPBS (GIBCO, ThermoFisher, Waltham, MA, cat#14190–144) and used for apoptosis and necrosis quantification.
Quantification of apoptotic and necrotic cells
The level of apoptosis and necrosis was measured using Apoptosis and Necrosis Quantification Kit (Biotium Inc., Fremont, CA) according to the manufacturer’s protocol, as previously published23. Briefly, cells were collected with trypsin, washed with PBS, resuspended in 50µl of reaction mix containing buffer, FITC-Annexin V, and Ethidium homodimer. After 25 minutes of incubation in the dark at room temperature, samples were diluted with 200µl of a kit 1X buffer. The analysis was performed using the NovoCyte Flow Cytometer (ACEA Biosciences, San Diego, CA). Gating was done based on untreated cells.
Measuring the rate of hydrogen peroxide generation
The rate of hydrogen peroxide produced by HPASMC and HPAEC was measured in cells seeded at density 40000 cells per well in 96-well plate using the Amplex® Red Hydrogen Peroxide Assay Kit (Thermo Fisher Scientific, Burlington, ON, Canada, cat# A22188) as published24. Briefly, the cells were washed twice in Hanks’ Balanced Salt Solution (HBSS, Thermo Fisher Scientific, Burlington, ON, Canada, cat# 14025076) and 50 µM of Amplex® Red reagent and 0.1 U/mL of horseradish peroxidase were added. The total volume was adjusted to 100 µL with the provided reaction buffer, and the fluorescence was measured at excitation 540 nm and emission 590 nm using the plate reader (Synergy H1, Biotek, Winooski, VT). The kinetics of the reaction was recorded every 2 minutes for 60 minutes. The rate of H2O2 production was evaluated by subtracting the final fluorescence intensity from the initial fluorescence intensity, divided by time. The data were presented as a relative rate of peroxide generated per minute.
To evaluate the role of NADPH oxidases (NOX) enzymes in hydrogen peroxide production and HMGB1 dimerization, the cells were pre-treated with 25 nM of NOX1/NOX4 inhibitor, GKT137831 (Medkoo Biosciences, Morrisville, NC, cat # 522357) for 3 hours before measuring the H2O2 production.
Assessment of mitochondrial superoxide production
To compare the level of superoxide production in HPASMC and HPAEC, both cell types seeded at density 40000 cells per well in 96-well plate were incubated with 7.5 µM of MitoSox red (Thermo Fisher Scientific, Burlington, ON, Canada, cat# M36008), mitochondrial superoxide indicator, for 30 min. Cells were washed twice with HBSS, and the fluorescence was measured on a plate reader (Synergy H1, Biotek, Winooski, VT) with the excitation at 510 nm and emission 580 nm.
The contribution of mitochondrial superoxide in HMGB1 dimerization was evaluated by incubation of HPASMC with mitochondrial-specific superoxide radical scavenger, Mito-Tempo (200 nM, Cayman Chemical, Ann Arbor, MI, cat # 16621), for 6 hours before MitoSox treatment.
Western blot analysis
The secreted HMGB1 was measured as described previously6, 23. Briefly, media collected from apoptotic or necrotic cells or rat plasma was mixed with either reduced or non-reduced 6X Laemmli sample buffer (Boston Bioproducts, Ashland, MA, cat #BP-111R or BP-111NR) and to measure total HMGB1 levels or dimeric/monomeric HMGB1 correspondingly. After 5 min of incubation at 95°C, the equal volume of sample (45ul of media or 10ul of 1:10 diluted plasma) was loaded on the 4–20% Mini-PROTEAN TGX Stain-Free gels (Bio-Rad Laboratories, Hercules, CA), electrophoretically separated using PowerPac Universal power supply and transferred using Trans-Blot Turbo transferring system (Bio-Rad Laboratories, Hercules, CA). The membranes were blocked with 5% BSA (Fisher Scientific, Fair Lawn, NJ, cat#9048–46-8) and incubated with anti-HMGB1 antibody (Abcam, Burlingame, CA, ab18256) diluted to 1:1000 for overnight at 4C. The next day, the membranes were washed three times with Tris-buffered saline (TBST) and incubated with anti-Rabbit IgG, HRP-Linked Antibody (Cell Signaling, Danvers, MA cat#7074S) diluted to 1:5000 for 1 hour at room temperature. The signal was recorded with the ChemiDoc MP Imaging System (Bio-Rad Laboratories, Hercules, CA) using a chemiluminescent protocol and analyzed using Image Laboratory software.
To measure TLR4 and RAGE expression in HPASMC or HPAEC, the cells were lysed in RIPA Buffer (Thermo Scientific, Rockford, IL, cat#89900). To prepare pulmonary tissue lysates, 20mg of freshly frozen lung tissue was homogenized in the lysis buffer containing 1M Tris, 1% Triton, 20% Glycerol, NaVO4, NaF and Phenylmethane Sulfonyl Fluoride (PMSF) and Halt Protease Inhibitor Cocktail (Thermo Scientific, Rockford, IL, cat#78438) as previously published 6, 25. The lysates were centrifuged at 10,000g for 10 minutes at 4°C, and the protein concentration in the collected supernatant was measured using the Pierce BCA Protein Assay Kit (Thermo Scientific, Rockford, IL, cat#23225) according to manufacturer’s instructions. The samples were electrophoretically separated as described above and probed with anti-TLR4 (Santa Cruz Biotechnology, Dallas, TX, cat#293072) or anti-RAGE (A-9) (Santa Cruz Biotechnology, Dallas, TX, cat#365154) diluted to 1:1000. The protein loading was normalized per total sample protein using free stain gels, as published26.
Model of progressive pulmonary hypertension in males
Sprague Dawley rats purchased from Charles River (Wilmington, MA) were bred in-house. The colony was kept in a 12-hour light-dark cycle and received standard rodent food and water ad libitum. All experimental procedures were approved by the University of Arizona’s IACUC. At 10 weeks of age, male and female rats (N=8 in each group) were randomized to receive either vehicle (Control group) or a single dose of the VEGF receptor 2 antagonist SU5416, 50 mg/kg s.c., as published25 (Su/Hx group). After injection of SU5416, rats were placed in a hypoxic chamber (BioSpherix, Redfield, NJ) with oxygen levels maintained at the level of 10% ± 0.5% during the entire duration of the study (1 week). The O2 and CO2 levels were continuously monitored using oxygen controller PROOX 110 (BioSpherix) and LB-2 CO2 analyzer (Sensormedics). The Control rats were kept under normoxic conditions. The advanced stage of PAH was induced by using the same dose of SU5416 and analyzing rats after exposure to hypoxia (10% ± 0.5% of O2) for three weeks and keeping them in normoxia for another two weeks (5 weeks total) as previously published25, 27.
Anti-apoptotic, anti-necroptosis and anti-necrosis therapy in rats
To investigate the contribution of cell death on HMGB1 release and PAH severity, male PAH rats were treated with anti-apoptotic, anti-necroptosis, or anti-necrosis drugs or corresponding vehicles (N=6 in each group). To inhibit apoptosis, rats were injected with a cell-permeable irreversible pan-caspase inhibitor Z-VAD(OMe)-FMK (Biorbyt Ltd, Cambridge, UK, cat# orb181387) in the dose 1 mg/kg i.v. as previously reported28. For necroptosis inhibition, rats were treated with a cell-permeable, potent, and selective blocker of necroptosis Necrostatin-1 (Cayman Chemical, Ann Arbor, MI, cat # 11658), 1 mg/kg i.v.29 Necrosis was attenuated by a cell-permeable inhibitor of necrosis, Necrox-5 methanesulfonate (Cayman Chemical, Ann Arbor, MI, cat# 17278) at the dose 1 mg/kg i.p. as previously published30. Additional two groups of rats were treated with either dimethyl sulfoxide (DMSO, Sigma-Aldrich, St. Louis, MO, cat# D2438) in the dose 50 µl of 20% solution per 100g of body weight i.v. as a vehicle for Z-VAD(OMe)-FMK and Necrostatin-1, or with ethanol (Decon Labs., King of Prussia, PA, cat# 2701) 100 µl of 40% solution per 100g of body weight i.p. as a vehicle for Necrox-5. All treatments were initiated on the day of Sugen injection (day 0) and were given daily at the same time of the day during the entire period of study (1 week).
Hemodynamic measurements
One or five weeks after Su/Hx injection, untreated or treated PAH rats and healthy controls were anesthetized with Inactin (Sigma-Aldrich, T133, 100 mg/kg i.p.), and instrumented for the measurement of right ventricle (RV) hemodynamics as described31. A customized pressure transducer catheter (SPR-513, Millar Instruments, Houston, TX) connected to a Millar Transducer Control Unit TC-510 and a PL3504 PowerLab 4/35 data acquisition system (ADInstruments, Colorado Springs, CO) was inserted into the jugular vein and advanced to the RV. After RV pressure was fully stabilized, it was continuously recorded for 15–30 min. At the end of pressure recording, a PE-240 polyethylene tube was inserted into the trachea to facilitate breathing and connected to a Harvard Rodent Ventilator (Model 683; Harvard Apparatus, South Natick, MA). The thorax was opened, the left atrium was cut, and the lungs were flushed with saline (0.9% sodium chloride) via a needle inserted into RV. Animals were euthanized by removing heart/lung block. Lungs, RV, and left ventricle + septum (LV+S) were dissected, weighed, and quickly frozen in the liquid nitrogen. The analysis of the RV function was performed using the Blood Pressure Software module (AD Instruments, Colorado Springs, CO).
Statistical analysis:
Statistical analysis was performed using the GraphPad Prism software. Data were expressed as mean value (± SEM), and significance was determined by the analysis of variance (ANOVA) or unpaired t-test. For ANOVA, Bonferroni multiple comparison tests were used to compare the multiple or selected pairs of columns. A value of P < 0.05 was considered significant. The Grubbs’ test (extreme studentized deviate) was used to determine the significant outliers. This criterion was pre-determined before the initiation of the data analysis.
RESULTS:
Induction of pulmonary vascular cell death
To evaluate the contribution of apoptosis of HPASMC or HPAEC in HMGB1 release, we first elaborated on the protocol that induces a reproducible and comparable level of apoptotic cell death in either type of pulmonary vascular cells. We discovered that HPASMC underwent apoptosis when we restricted the level of glucose in the media (Fig. 1A). In contrast, HPAEC responded by apoptosis when they were cultured in the serum-free media (Fig. 1B). Both conditions induced a similar level of apoptosis in either HPASMC or HPAEC 48 hours after the treatment initiation and did not stimulate any significant increase in necrotic cell death. Importantly, we found these conditions to be cell-specific, as HPAEC growing in glucose-free media or HPASMC exposed to serum-free media did not show an increase in apoptotic cell death (Fig. S1A, B). We also confirmed that hypoxic conditions produced no additional cell death in starved HPAEC (Fig. S1C), or even induce protection of HPASMC restricted by glucose (Fig. S1D). This observation is in high agreement with the previously published data showing that hypoxia-induced activation of HIF1α and other pro-survival factors increase proliferation and vitality of PASMC32, 33, or PAEC34, 35.
Figure 1. Apoptosis induction in HPASMC and HPAEC.
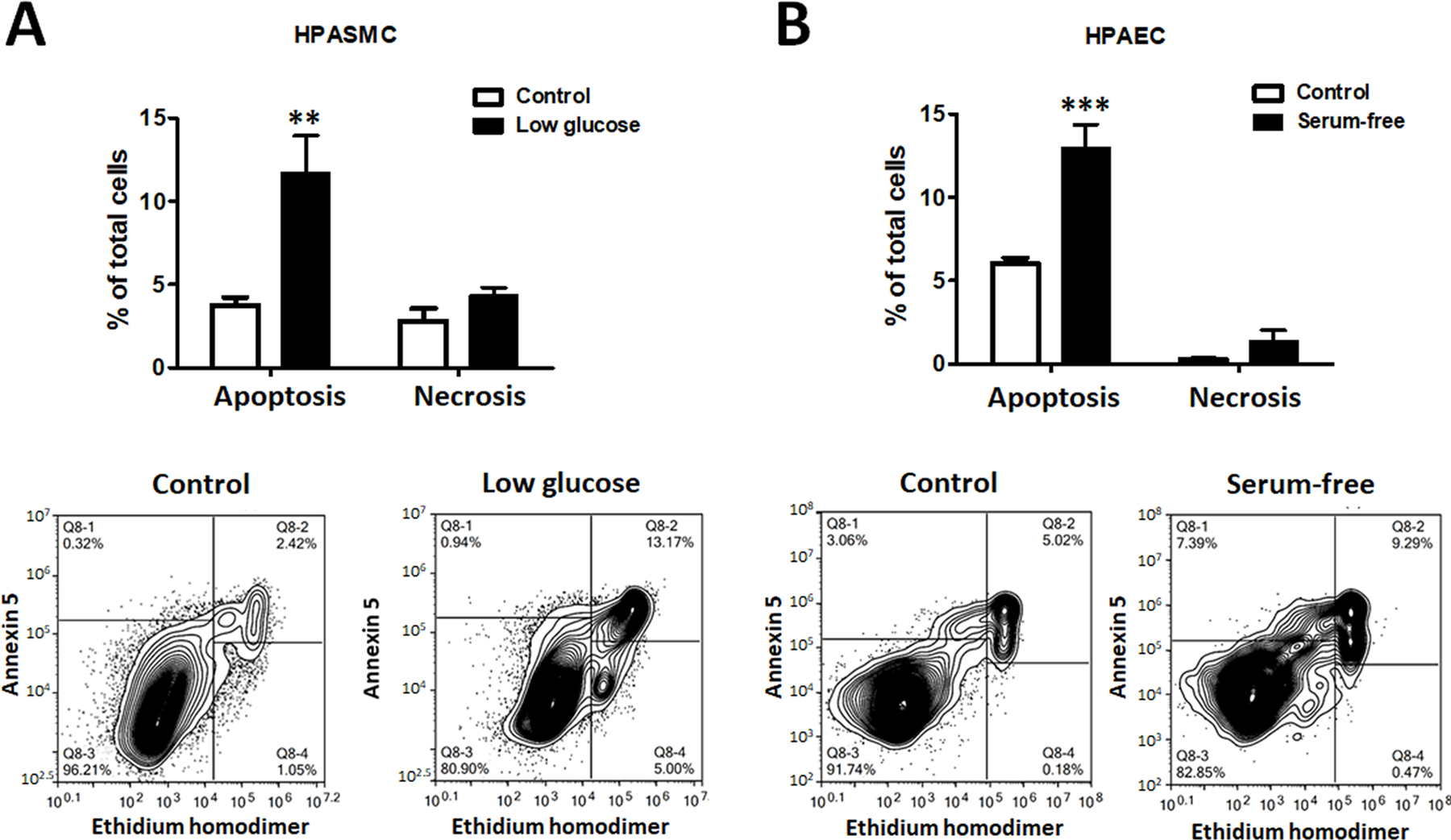
The comparable level of apoptosis was induced in human pulmonary artery smooth muscle cells (HPASMC) by culturing them in a low-glucose media ((0.1g/L, A) and in human pulmonary artery endothelial cell (HPAEC) by culturing them in a serum-free media (B). The treatments did not induce significant necrotic cell death in any cell type. Data presented as Mean±SEM, **P<0.01, ***P<0.001 vs. untreated Control. N=6 in all groups.
To induce necrosis, we subjected both cell type to several cycles of cell freezing and thawing as published6, which produced cell rupture similar to necrosis without any contribution of the controlled type of cell death. Since the conditioned media collected from dying cells was used to stimulate the naïve cells, we avoided using any chemical reagents for the induction of either type of cell death.
Role of apoptosis and necrosis cell death in HMGB1 release and downstream signaling
We found that necrosis of either HPAEC or HPASMC induces a severe and comparable level of HMGB1 accumulation in cell media (Fig. 2A). However, only apoptotic HPASMC but not HPAEC contributed to HMGB1 release. The amount of HMGB1 released by apoptotic HPASMC was considerably lower compared to HMGB1 produced by necrosis, although apoptosis was induced in only ~12% of cells, while freeze/thaw protocol stimulated almost complete cell rapture. Given the inconsistency in the literature about apoptotic cells as a potential source of HMGB1, we consider our discovery to be very important. Indeed, by providing the evidence of a cell-specific contribution of apoptotic cells in HMGB1 release, we could explain the source of the present contradictions.
Figure 2. Necrosis and apoptosis of HPASMC and HPAEC in HMGB1 release and receptor activation.
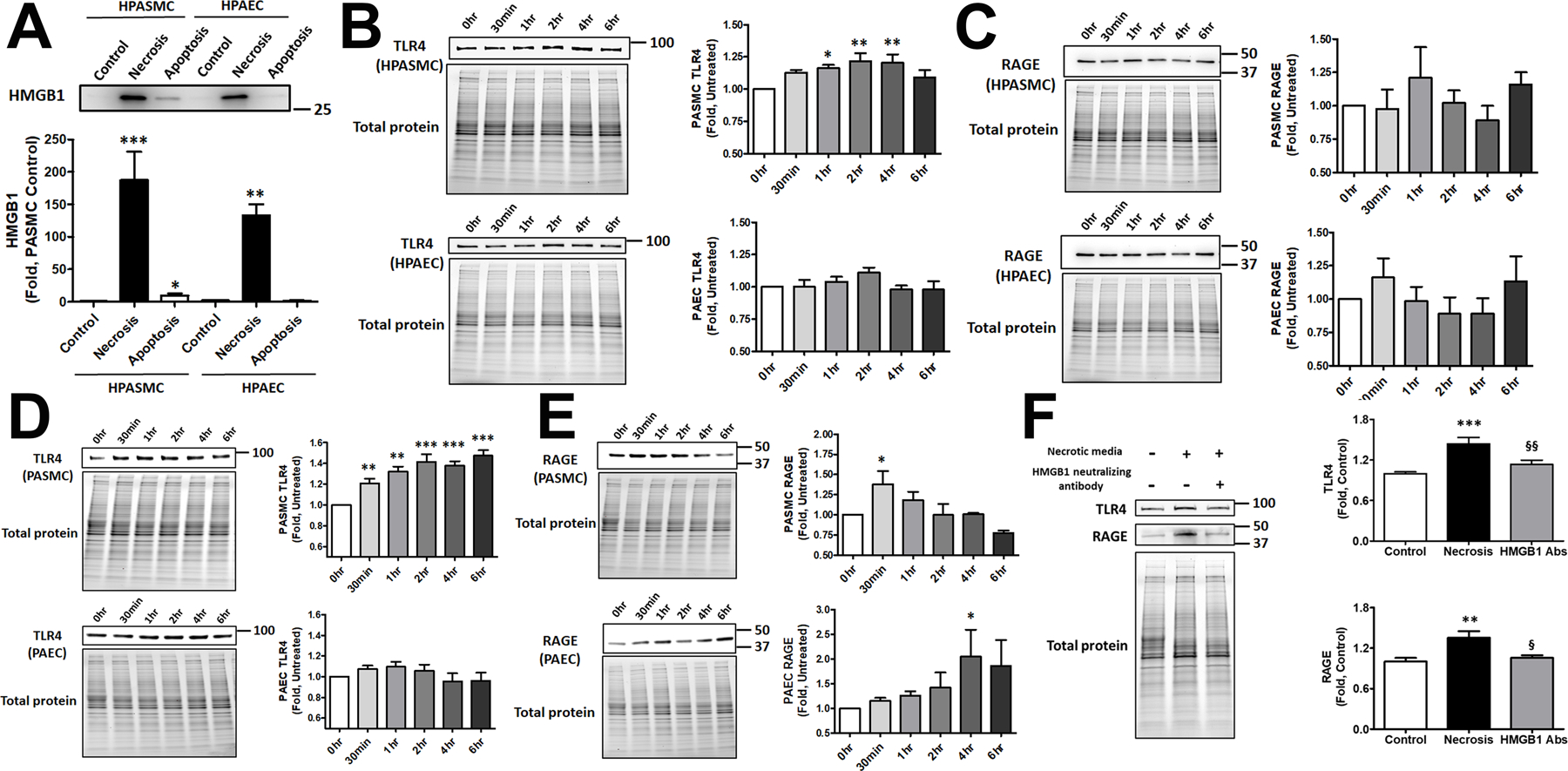
Apoptotic HPASMC but not HPAEC release HMGB1. Necrosis produces a strong and comparable increase of the extracellular HMGB1 in both cell types (A, N=5). Conditioned media collected from apoptotic cells and used to treat the corresponding naïve cells induced transient activation of TLR4 in HPASMC but not HPAEC (B, N=5). Apoptotic media did not change the activity of RAGE in either cell type (C, N=5). Media collected from necrotic cells induced prolonged activation of TLR4 in HPASMC (D upper panel, N=5), while the activity of TLR4 in HPAEC was not changed (D lower panel, N=5). The stimulation of RAGE by necrotic media was early and transit in HPASMC (E upper panel, N=4) and delayed in HPAEC (E lower panel, N=4). Neutralization of HMGB1 prevents TLR4 or RAGE activation by conditioned media collected from necrotic HPASMC, confirming the importance of HMGB1 in receptor stimulation (F, N=13). Data presented as Mean±SEM, *P<0.05, **P<0.01, ***P<0.001 vs. untreated Control.
To examine the ability of HMGB1 released from apoptotic or necrotic cells to activate TLR4 and RAGE receptors, naïve HPASMC and HPAEC were stimulated with conditioned media collected from corresponding cells and used to measure the level of TLR4 and RAGE expression. Consistent with our previous results, we found that apoptotic HPASMC but not HPAEC activated TLR4 1–4 hours after cell stimulation by apoptotic media (Fig. 2B). There was no effect of apoptotic media on RAGE in either type of vascular cells (Fig. 2 C). The conditioned media collected from necrotic cells also stimulated TLR4 in HPASMC, although this stimulation started earlier and was more prolonged and intense compared to apoptotic media (Fig. 2D, upper panel). Surprisingly, even necrotic HPAEC media did not activate TLR4 (Fig. 2D, lower panel). However, necrotic media activated RAGE in both cell types, although the profile of this activation was different – a transient early activation in HPASMC (Fig. 2E, upper panel) and a late prolonged activation in HPAEC (Fig. 2E, lower panel). These results confirm a significant difference between two primary vascular cell types not only in their ability to release HMGB1 but also in their responses to the neighboring apoptosis and necrosis, with HPASMC primarily activating TLR4 and HPAEC – RAGE receptors.
Although both, necrotic and apoptotic cells, release multiple factors that could act as DAMPs and activate TLR4 and RAGE, we have confirmed that the conditioned media depleted by HMGB1 does not activate TLR4 or RAGE (Fig. 2 F), suggesting the primary role of HMGB1 in the activation of both receptors.
Cell difference in HMGB1 dimerization
Since HMGB1 has several cysteine residues, in the presence of oxidative conditions, it readily forms a dimer, although the functional role of such dimerization is not established. We discovered that the necrotic and apoptotic media collected from HPASMC but not HPAEC contains dimeric HMGB1 (Fig. 3A, left graph), and HPASMC necrosis produces more HMGB1 dimers compared to HPASMC apoptosis. Importantly, the necrotic media depleted by HMGB1 with anti-HMGB1 neutralizing antibodies showed a strongly attenuated signal from either monomeric or dimeric form, thus confirming the relevance of both signals to HMGB1 (Fig. S2A). The separation of HMGB1 signal on monomers and dimers in HPASMC but not HPAEC cells reduces the level of monomeric HMGB1 in HPASMC that now became significantly lower compared to monomeric HMGB1 in necrotic HPAEC (as compared to non-separated HMGB1 presented in Fig. 2A). Taken together, these results suggest that in HPASMC the total HMGB1 signal consists of both monomeric and dimeric forms.
Figure 3. The release of HMGB1 dimer from HPASMC is mitochondrial ROS dependent.
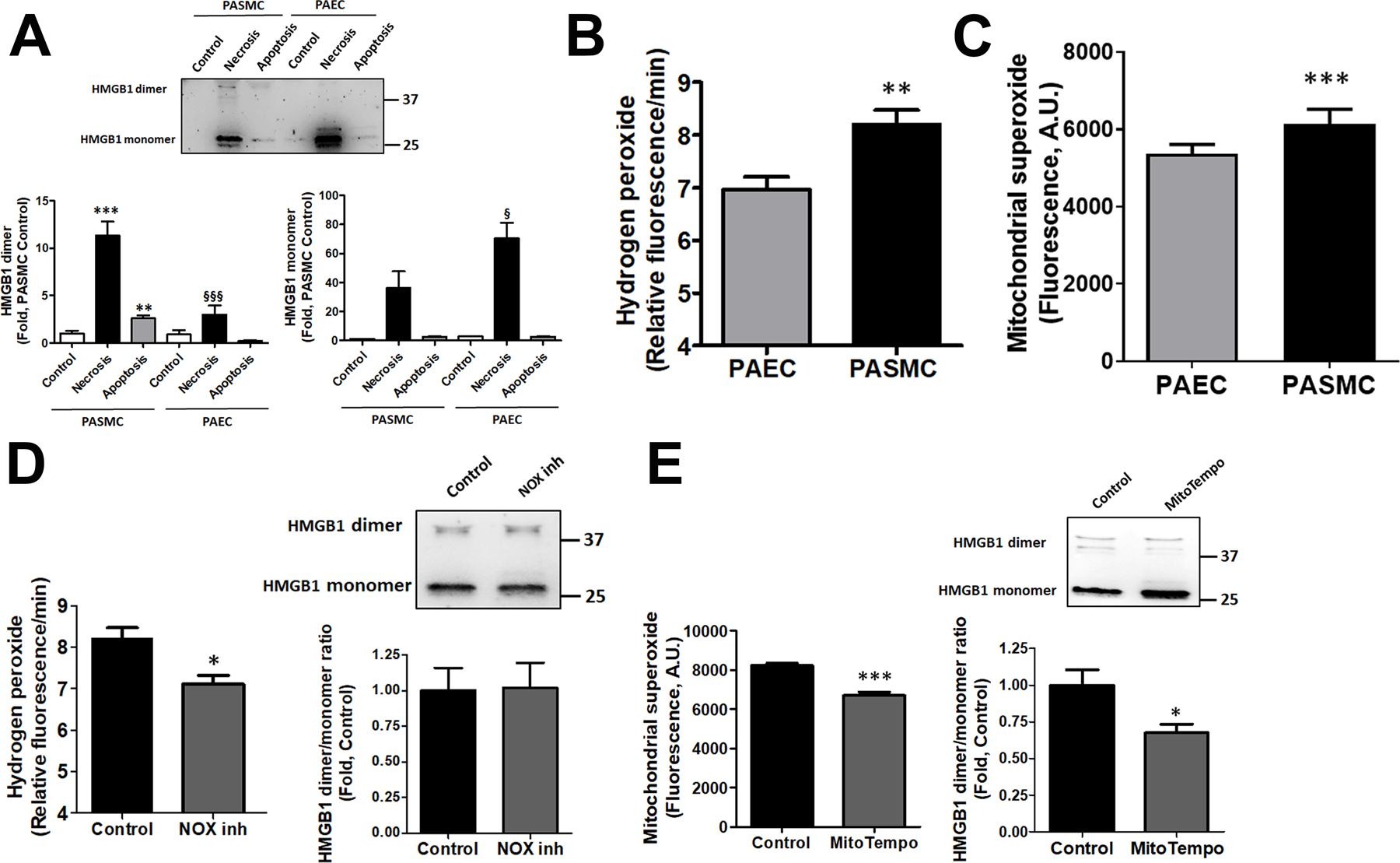
HPASMC but not HPAEC death induces a release of HMGB1 in a dimeric form (A, N=5). The elevated production of reactive oxygen species (ROS), such as hydrogen peroxide (H2O2, B, N=8) or mitochondrial superoxide (O2−, C, N=8) found in HPASMC versus HPAEC, could be responsible for the oxidation of HMGB1 into dimers. However, attenuation of H2O2 levels by pretreatment HPASMC with NOX1/4 inhibitor, GKT137831, did not alter HMGB1 dimer/monomer levels in cells (D, N=6–8). In contrast, incubation of HPASMC in mitochondria-specific O2− scavenger, MitoTempol, reduced not only the levels of mitochondrial ROS but also HMGB1 dimer/monomer ratio (E, N=6–8). Data presented as Mean±SEM, *P<0.05, **P<0.01, ***P<0.001 vs. untreated Control; §P<0.05, §§§P<0.001 vs. necrosis in HPASMC.
To investigate the mechanism responsible for HMGB1 dimerization in HPASMC, we measured the levels of reactive oxygen species (ROS) production in untreated HPASMC and HPAEC. We found that the levels of both primary cellular ROS, hydrogen peroxide (H2O2), and superoxide (O2−), were increased in HPASMC compared to HPAEC (Fig. 3 B, C). The role of hydrogen peroxide in HMGB1 dimerization was further investigated by measuring the ratio of HMGB1 dimer and monomers in the presence of GKT137831, an inhibitor of two primary H2O2 producing NADPH oxidase isoforms, NOX1 and NOX4. Indeed, NOX1/4 inhibitor reduced the production of H2O2 in HPASMC, although it failed to attenuate the HMGB1 dimerization (Fig. 3D). In contrast, treatment of HPASMC with mitochondria-specific antioxidant, MitoTempol, attenuated the levels of mitochondrial superoxide and reduced HMGB1 dimer/monomer ratio (Fig. 3E). These results propose a critical role of mitochondrial ROS in HMGB1 dimerization.
Progressive form of pulmonary hypertension in males
Based on our previous findings6, 23, we hypothesized that HMGB1 release and signaling plays a more important role in males compared to females. Moreover, we proposed that HMGB1 could be responsible for the more severe form of PAH in males15. However, the classical preclinical models of PAH, including chromic hypoxia, monocrotaline and Sugen/hypoxia, show no difference in the levels of the right ventricle (RV) pressure or hypertrophy, providing no opportunity to replicate and study the mechanisms responsible for the sex difference in PAH severity. Therefore, to evaluate the relevance of our in vitro findings to the pathobiology of pulmonary hypertension in males, we first elaborated on a preclinical animal model that nicely simulated the sex difference in PAH severity described in patients. Thus, we discovered that PAH induced by a high dose of Sugen (50 mg/kg, s.c.) followed by one week of exposure to hypoxia, reproduces sex differences in PAH severity reported in humans. Indeed, in this model, the male rats showed a significantly higher RV systolic pressure (RVSP, Fig. 4A) and RV hypertrophy (Fig. 4B) compared to females. Moreover, despite the higher RVSP in males, which places males under a higher demand in RV performance, and despite the significantly upregulated RV contractility and relaxation in male Controls vs females Controls (Fig. 4C), PAH males had a similar level of RV performance compare to PAH females. This loss of the initial sex difference in RV function and failing to adequately respond to the pressure demand suggests the development of RV dysfunction in male rats similar to male PAH patients16. Indeed, male but not female PAH rats showed a significant drop in RV contractility index (Fig. 4E). We propose this novel model to be an important tool to investigate the mechanisms responsible for this sex difference.
Figure 4. Model of progressive PAH in males.
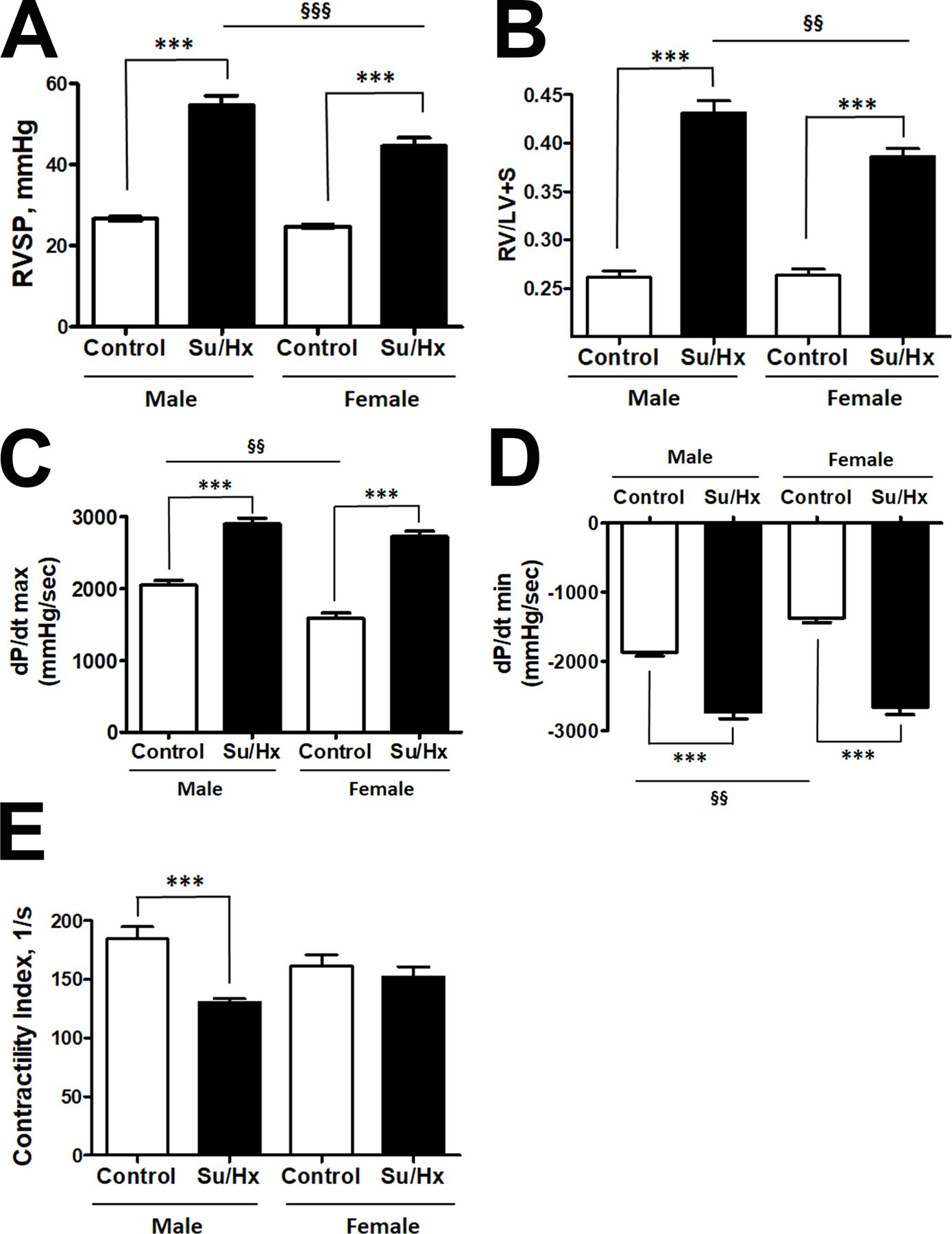
To mimic the sex difference in PAH severity, rats were injected with a high dose of SU5416 (50 mg/kg s.c.) followed by 1 week of exposure to hypoxia (10% of O2). There was a significant elevation of right ventricle (RV) systolic pressure (RVSP, A) and RV hypertrophy (B) in both sexes, although the changes in males were significantly more severe compared to females. At the same time, the initial sex difference in RV contractility (C) and relaxation function (D) was eliminated by PAH, suggesting that RV in males did not respond adequately to the demand. The contractility index (E) as an additional measure of RV performance was decreased in male but not female PAH rats. Data presented as Mean±SEM, ***P<0.001 vs. sex-matched Controls; §§P<0.01, §§§P<0.001 vs. Male Control. N=8 in all groups.
In agreement with the previously published reports from our group and others36–38, we did not find a significant sex difference in RVSP and RV hypertrophy when untreated rats were analyzed at a developed stage of PAH (Fig. S3 A, B). The RV contractility (Fig. S3C) was also similar in both sexes, while RV relaxation was lower in females compared to males (Fig. S3D). However, since female’s RVSP was not significantly lower anymore, the decrease in female’s RV performance suggest the loss of protection seen in females at the earlier stage. The same conclusion could be made based on the analysis of RV contractility index, which was not changed in females at the early stage (Fig. 4E) but was found to be significantly different vs. healthy controls in both sexes at an advanced stage (Fig. S3E). Although a more detailed study is required to conclude about the mechanisms responsible for the loss of sex difference at the developed stage of PAH, the loss of protection in females secondary to PAH-mediated misbalance between protective and disease-promoting female sex hormones is pretty well described39, 40.
Sex difference in PAH mediated HMGB1 release and function
As expected, the more severe PAH discovered in males corresponded with significantly higher levels of circulating HMGB1 in male rats (Fig. 5A). Moreover, we found that PAH significantly increased the expression of TLR4 in the lungs of male rats, while females showed similar levels of TLR4 in Control and PAH groups (Fig. 5B). In contrast, the levels of pulmonary RAGE were strongly elevated in the lungs of both sexes (Fig. 5C). We concluded that activation of HMGB1/TLR4 axis could be responsible for the progressive form of PAH in males. In contrast, the expression of RAGE elevated in both sexes, while known for being an essential contributor in PAH pathogenesis25, 41, is unlikely to contribute to the sex difference in PAH severity.
Figure 5. An increased HMGB1 release in male PAH rats is fully attenuated only by anti-necrotic therapy.
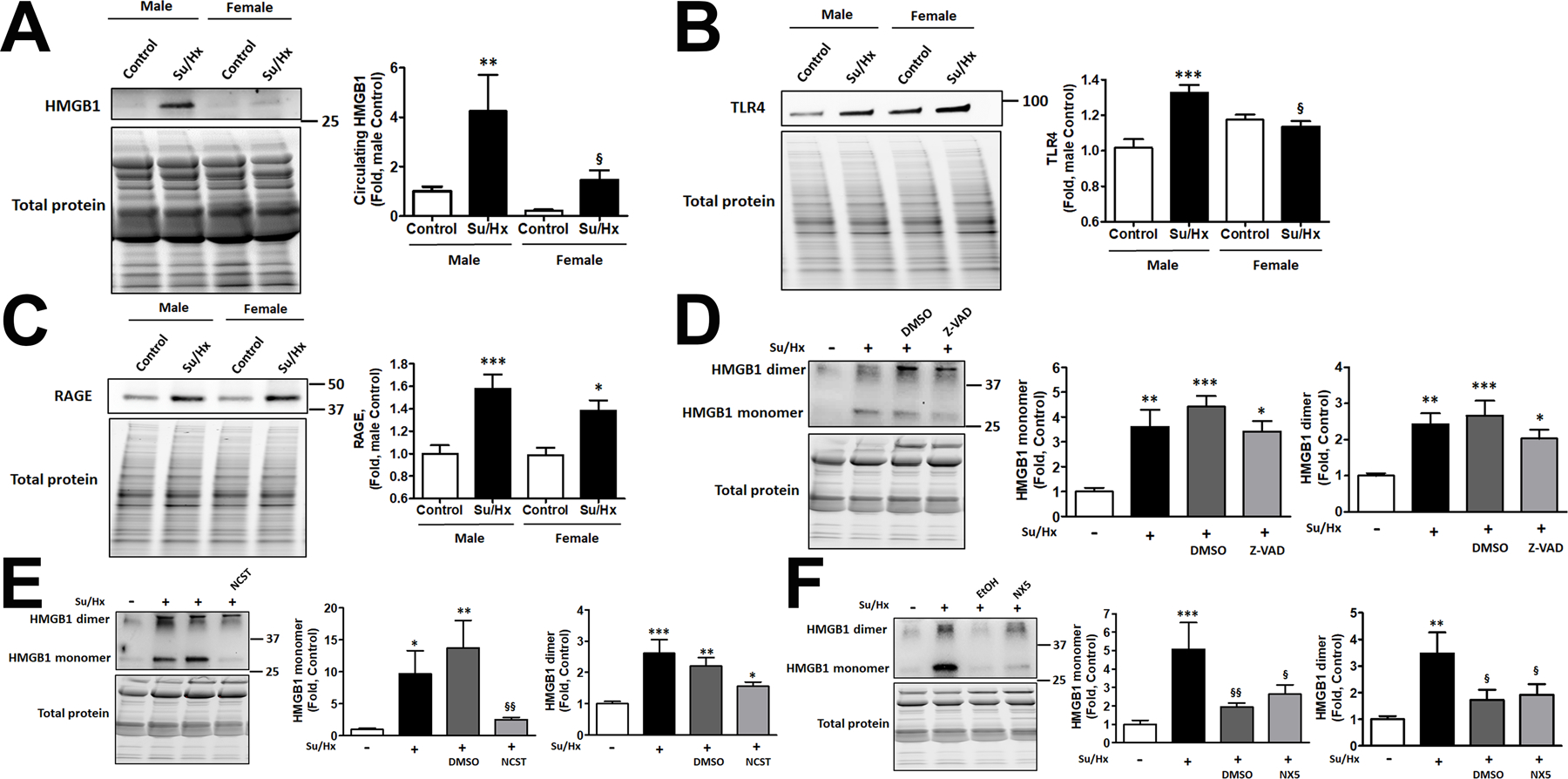
PAH mediated a substantial increase in the levels of circulating HMGB1 in males, while in females the changes were significantly less pronounced and did not reach statistical significance compared to female Controls (A, N=6–8). The increased release of HMGB1 in males corresponded with significant activation of TLR4 in the lungs of male PAH rats, while females showed no changes in TLR4 in response to PAH (B, N=8). Both sexes had a significantly elevated expression of RAGE in the lungs of PAH rats (C, N=8). To evaluate the contribution of different types of cell death on the elevated HMGB1 release, male rats were treated with pan-caspase inhibitor Z-VAD(OMe)-FMK (Z-VAD), the selective blocker of necroptosis Necrostatin-1 (NCST), the inhibitor of necrosis Necrox-5 (NX-5), or vehicles (DMSO, used as vehicle in Z-VAD and NCST and ethanol (EtOH), a vehicle for NX-5). All treatments were given daily starting from the day of SU5416 injection and continued throughout the study (1 week). There were no significant changes in the amount of circulating HMGB1 monomers or dimers in Z-VAD treated rats (D, N=6). NCST significantly reduced the plasma levels of HMGB1 monomers but not dimers (E, N=6). NX-5 and its vehicle (EtOH) strongly attenuated the release of either HMGB1 monomers or dimers (F, N=6). Data presented as Mean±SEM, *P<0.05, **P<0.01, ***P<0.001 vs. sex-matched Controls; §P<0.05 vs. Male Su/Hx (A, B) or vs. untreated Su/Hx rats (F); §§P<0.01 vs. vehicle-treated rats (E, F).
Release of HMGB1 in the PAH male rats treated by anti-apoptotic, anti-necroptotic and anti-necrotic drugs
In addition to the validation of HMGB1 monomeric/dimeric signals by HMGB1 neutralizing antibodies as described above (Fig. S2A), the formation of dimers in vivo was first visualized in control and diseased samples by running them in non-reduced and reduced conditions (Fig. S2B). Next, we investigated which particular type of cell death – apoptosis, controlled necrosis (necroptosis), or passive necrosis - is responsible for the increased levels of the circulating HMGB1 monomers and dimers and PAH progression in males. For this purpose, we treated PAH male rats with pan-caspase inhibitor Z-VAD(OMe)-FMK (Z-VAD group), the selective blocker of necroptosis Necrostatin-1 (NCST group), an inhibitor of necrosis induced by increased mitochondrial ROS production, Necrox-5 (NX5 group). The collected plasma was used to measure the levels of circulating HMGB1 in rats that underwent one of these therapies and compared them to the healthy controls, untreated diseased animals, or rats treated with corresponding vehicles (DMSO for Z-VAD or NCST; ethanol (EtOH) for NX5). We found that anti-apoptotic therapy, although induced a slight decrease in circulating HMGB1 monomers and dimers, did not significantly reduce any HMGB1 form in plasma of male rats (Fig. 5D). In contrast, necroptosis inhibition significantly decreased the amount of circulating HMGB1 monomer compared to either untreated Su/Hx rats or rats treated with vehicle (Fig. 5E), although HMGB1 dimer remained elevated. Finally, an inhibitor of necrosis significantly attenuated the levels of both - HMGB1 monomers and dimers - in plasma compared to untreated Su/Hx rats (Fig. 5F). Surprisingly, NX5 vehicle, ethanol, has also produced a similar effect. We found a previously published report that a low dose of ethanol protects hepatocytes from necrotic damage42. To understand whether the pulmonary vascular cells could undergo similar protection, we reproduced the experimental conditions reported for hepatocytes. Indeed, RPASMC isolated from untreated SD rats showed a similar to hepatocytes dose-depended effect in response ethanol treatment. At a low dose, ethanol was protective against the necrotic cell death (Fig. S4A), while increased ethanol concentrations lost this protection. Importantly, the concentration of ethanol that showed protection (0.1 mM) was precisely the same concentration we used in the animals as a vehicle for NX5. There was no protection from ethanol against apoptotic cell death. We conclude that a low dose of ethanol protects the pulmonary vascular cells from necrotic cell death and attenuates the release of HMGB1 monomers or dimers. In contrast, the low dose of DMSO (1 µl/ml), used as a vehicle for Z-VAD or NCST, produced no changes in the level of necrosis and only mild increase of apoptosis (Fig. S4B). However, since chronic injections of DMSO did not alter plasma HMGB1 monomers or dimers (Fig. 5D, E) or activation of TLR4 (Fig. 6E) and RAGE (Fig. 6F) and hemodynamic parameters (Fig. 6A–D), we concluded that this vehicle does not contribute to HMGB1 release, activation of its receptors or PAH development.
Figure 6. Hemodynamic changes and expression of TLR4 and RAGE in rats received anti-apoptotic, anti-necroptotic, and anti-necrotic therapy.
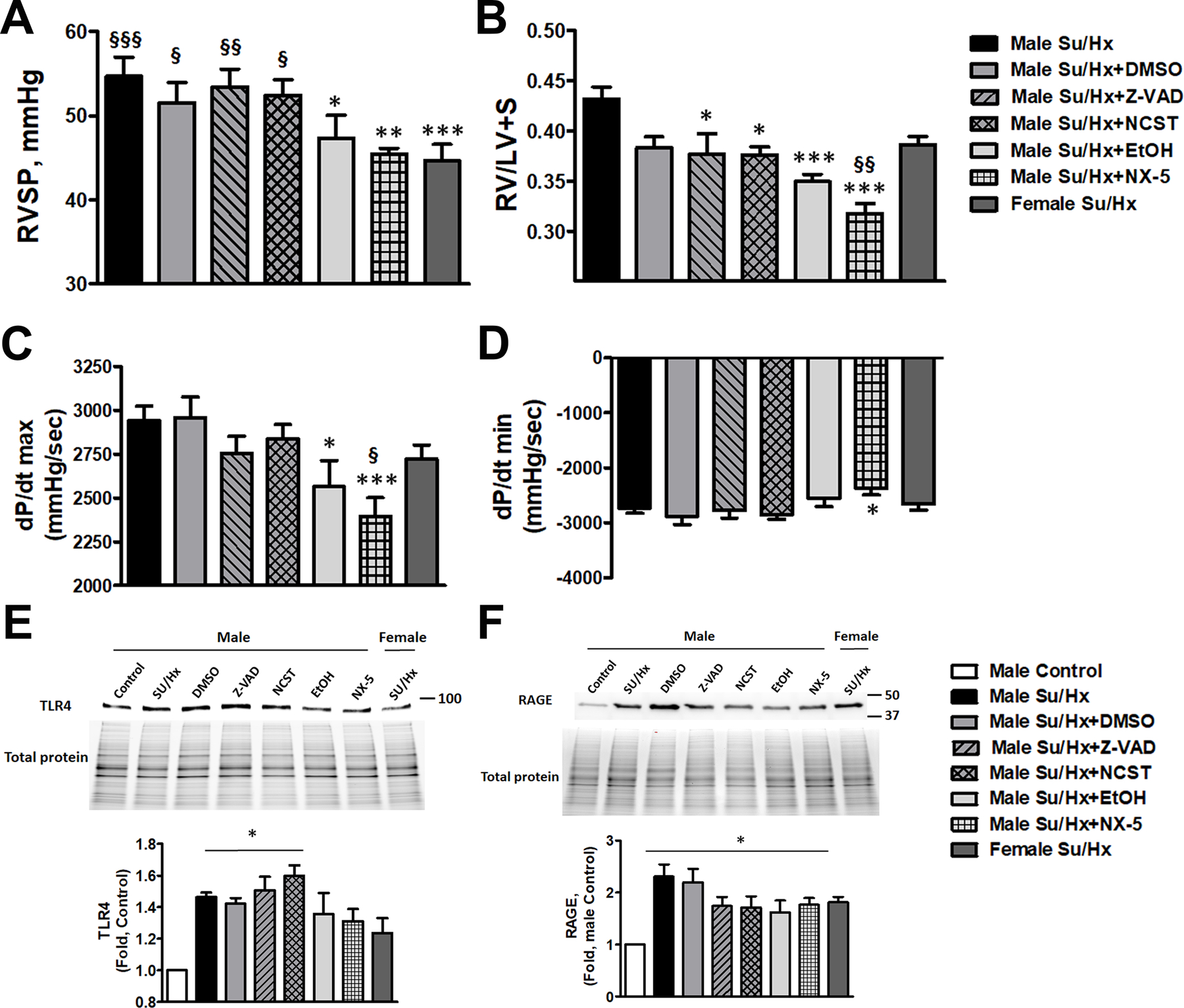
Treatment approaches that decrease necrotic cell death (Necrox-5 (NX-5) or its vehicle, ethanol (EtOH)) significantly attenuated RVSP in male PAH rats down to female level, thus eliminating the sex differences (A, N=6–8). RVSP in rats received anti-apoptotic (Z-VAD(OMe)-FMK (Z-VAD)), anti-necroptotic (Necrostatin-1 (NCST)) treatments or their vehicle, DMSO, remained unchanged. NX-5 and EtOH significantly attenuated RV hypertrophy (B, N=6–8) compared to untreated Su/Hx male rats and even below Su/Hx female level (NX-5). Although Z-VAD and NCST treatment slightly reduced RV/LV+S ratio compared to untreated Su/Hx male rats, the changes were not significantly different versus the group treated by the vehicle (DMSO). NX-5 and EtOH but not any other interventions significantly reduced RV contractility (dP/dt max) in male PAH rats (C, N=6–8). NX-5 attenuated RV dP/dt min (D, N=6–8), suggesting a reduced RV load in these rats. Induction of PAH in male rats corresponded with a significant upregulation of TLR4 and RAGE expression in the lungs. The expression of TLR4 (E, N=5) maintained significantly high in the rats treated by anti-apoptotic ((Z-VAD(OMe)-FMK (Z-VAD)), anti-necroptotic (Necrostatin-1 (NCST)) agents or their vehicle (DMSO), but stopped being significant and was found to be reduced down to female levels in the lung of rats treated by an anti-necrotic agent (Necrox-5 (NX-5)) or low dose of ethanol (EtOH). The expression of RAGE (F, N=6) remained significantly upregulated in all treatment groups and was increased in PAH females. Data presented as Mean±SEM, *P<0.05, **P<0.01, ***P<0.001 vs. untreated Su/Hx male rats (A-D) or vs. healthy male Controls (E, F); §P<0.05 vs. §§P<0.01, §§§P<0.001 vs. Su/Ha female rats (A-D).
The critical role of necrosis in the manifestation of sex difference in PAH severity
To understand whether any type of cell death is responsible for a more progressive form of PAH in males, we measured the primary hemodynamic parameters – RVSP, RV hypertrophy, RV contractility and relaxation in male rats treated with Z-VAD, NCST, NX5 or vehicles and compared them with untreated Su/Hx male and female rats. We found that only EtOH and NX5 treated male rats showed a significant decrease in RV pressure down to the female levels (Fig. 6A). Thus, attenuation of necrosis by either NX5, a known necrosis inhibitor30, or by ethanol that at low doses was found to be protected against necrotic cell death in hepatocytes42 and RPASMC (Fig. S4A), abolishes the sex difference in PAH severity. In contrast, rats treated with anti-apoptotic, anti-necroptotic therapy or their vehicle, DMSO, show a preserved sexual dimorphism in RV pressure increase. Although anti-apoptotic and anti-necroptotic therapies slightly attenuated RV hypertrophy when compared to the untreated Su/Hx male rats, they failed to do so compared to vehicle control (Fig. 6B). EtOH and NX5 again showed pronounced protection, with NX5 decreasing RV remodeling even significantly below the female’s level (Fig. 6B). Moreover, attenuation of necrosis significantly reduced RV load, since RV contractility (dP/dt max) was attenuated only in treatment groups associated with necrosis reduction (Fig. 6C). There were also slight changes in dP/dt min in the NX5 group (Fig. 6D), suggesting that necrosis could also affect RV relaxation.
Reduced necrosis but not apoptosis or necroptosis attenuates TLR4 signaling in the lungs of male rats with PAH
Since the increase of TLR4 signaling was found to be male-specific (Fig.5 B, C), we studied whether TLR4 expression was attenuated in rats treated by the therapy that blunted the sex difference in PAH severity. In agreement with the RVSP, TLR4 expression was not altered in the rats treated with Z-VAD, NCST or their vehicle compared to untreated male Su/Hx rats and remained significantly elevated vs. healthy male Controls (Fig. 6E). In contrast, rats treated by EtOH or NX5 showed a decreased TLR4 expression down to female levels and stopped being different from healthy male Controls. We conclude that increased necrosis in males with PAH activates TLR4 signaling, which, in turn, could be responsible for the progressive form of PAH in males.
Analysis of RAGE expression (Fig. 6F) revealed that PAH induces a significant upregulation of its protein levels in the lungs, although, as in Fig. 5C, it was elevated in both sexes. None of the treatments significantly attenuated the expression of RAGE. Taken together, our in vitro and in vivo results suggest that necrosis could induce a transient activation of RAGE (Fig. 2E). However, chronically its expression is not regulated by any type of cell death in males and is not contributing to the sex difference in PAH severity.
DISCUSSION:
The initial damage of pulmonary vascular cells was introduced in PAH pathogenesis paradigm long ago2. It considers being one of the primary causes of pulmonary vascular remodeling due to an uncontrolled vascular over-repair43. However, the particular molecular mechanisms initiated by cell death are not fully understood. Recently, several simultaneous studies confirmed the critical role of HMGB1 in PAH development and progression. HMGB1 is one of the most extensively studied DAMPs that stimulates two types of damage-mediated responses – activation of inflammatory and immune cells to clean the site of damage and initiation of tissue repair. Different research groups, including ours, discovered that in PAH patients and preclinical PAH animal models, the signal from an extra-nuclear HMGB1 in the pulmonary vascular wall and the levels of circulated HMGB1 are strongly elevated6–9. HMGB1-neutralizing antibody7–9, HMGB1 inhibitors11, 12, or peptide therapeutics that alter HMGB1/TLR4 interaction10 significantly attenuated the development of preclinical PAH.
Nevertheless, there are multiple contradictions in current knowledge. Thus, although the passive release of HMGB1 from cells that died by necrosis is well confirmed, the ability of apoptotic cells to release HMGB1 is under debate. Indeed, some previous research points toward the nonimmunogenic nature of apoptotic cell death, in particular, due to the inability to release HMGB1 bound to apoptotic chromatin44, 45. In contrast, it was shown that apoptosis inducers stimulate HMGB1 release from cancer cell lines (Jurkat, U937, HeLa S6, and Panc-1 cells)46. Whether the ability of apoptotic cells to secrete HMGB1 is cell-specific and whether apoptotic cell death is contributing to HMGB1 release in PAH remains unknown. To answer these questions, we identified the cell-specific conditions that induce a comparable level of apoptotic cell death in HPAEC and HPASMC and measured the level of HMGB1 release from apoptotic pulmonary vascular cells with necrotic death served as a positive control. We discovered that, indeed, the ability of apoptotic cells to release HMGB1 depends on the cell type. Thus, HPAEC did not produce any significant changes in the levels of extracellular HMGB1, confirming our previous observations43, while HPASMC secreted a substantial amount of HMGB1. To our knowledge, this is the first evidence of the cell-specific capacity to secrete HMGB1 upon apoptosis induction. The particular mechanisms responsible for this cell difference yet remained to be elucidated. HPAEC and HPASMC may have a different level of HMGB1 distribution between nuclear and cytosolic fractions. Indeed, it seems highly probable that cytosolic HMGB1 that is not bound to chromatin could be released in the event of apoptotic cell death.
It is important to mention that the conditions used in our study to induce apoptosis (glucose restriction and cell starvation) are highly relevant to PAH. Thus, found a significant decrease in the levels of plasma glucose in Su/Hx rats (Fig. S5), which could correspond to a higher demand for glucose in multiple tissues switched to aerobic glycolysis47. The severe or complete vasooclusion due to the formation of concentric lesions or microtrombosis in small pulmonary arteries reduces or prevents the supply of blood to the area distal to the occlusion. Moreover, the insufficiency of vasa vasorum, which becomes leaky, dysfunctional, and fails to supply nutrition to pulmonary artery cells, is also described48. Thus, although these restrictions could induce changes in cellular metabolism, the similar changes are occurring in the pulmonary vascular cells during the course of the disease and could contribute to the PAH progression.
The conditioned media collected from apoptotic or necrotic cells showed a cell-specific profile in the activation of the primary HMGB1 receptors, TLR4, and RAGE, which also was never reported before. This discovery is of high importance since the induction of HPASMC apoptosis was proposed to be an effective strategy to attenuate the increased media thickness of pulmonary arteries and reduce pulmonary pressure. However, in light of our discovery, the positive effect of apoptosis inducers could be limited by simultaneous activation of TLR4-mediated pro-inflammatory response49, 50.
Another contradiction present in the field is related to the biological activity of oligomeric forms of HMGB1. HMGB1 contains three cysteine residue, which could form intra-molecular or cross-molecular disulfide bonds. However, the role of HMGB1 dimers or higher oligomers in HMGB1 interaction with TLR4 or RAGE is not clear. Previous studies confirmed the importance of HMGB1 Cys106 in binding to TLR4 since mutation of this residue completely abolished HMGB1-mediated TLR4 activation51. It was also described that in the presence of oxidants, HMGB1 could dimerize through Cys106, and such dimers were found to bind to TLR4/MD2 complex with higher affinity than monomeric HMGB152. In our study, we confirmed that the formation of HMGB1 dimers occurs specifically in HPASMC that have a higher rate of ROS production compared to HPAEC. We also verified that mitochondrial ROS, rather than cytosolic ROS produced by NOX enzymes, are responsible for HMGB1 oligomerization. The importance of mitochondrial ROS in the regulation of the redox environment in HPASMC could reflect the high levels of mitochondrial respiration known for smooth muscle cells compared to endothelial cells that appear to be primarily glycolytic.
The elevated levels of HMGB1 dimerization in HPASMC could explain TLR4 activation in cells treated with conditioned media collected from HPASMC and the absence of such TLR4 stimulation in cells treated by media collected from HPAEC. Moreover, HMGB1 dimerization was found to play a critical role in the manifestation of sex difference in PAH severity, since only therapy that significantly attenuated the levels of circulating HMGB1 dimers reduced RVSP and TLR4 activation in lungs of male rats down to female levels.
The contribution of sex to PAH prevalence and outcomes is very well established and consists of two aspects - a higher incidence of PAH in women53, and a more progressive and severe PAH development15, which contributes to a poorer survival prognosis in men16. Our previous research provides strong evidence of the sex dimorphism in the type of cell death activated in response to stress and the amount of released HMGB123. Thus, we found that male sex is associated with a higher level of necrotic cell death and substantial HMGB1 release in response to stress. In contrast, females are prone to respond to stressful conditions by apoptosis and do not secrete any significant amounts of HMGB1. This effect was confirmed in PAEC isolated from mice or human donors23, in Su/Hx treated rats and PAH patients6, and in kidney, spleen, and aorta of spontaneously hypertensive rats30, suggesting that it is common for different cell types. It was also found to be independent of sex hormones since it presents in cells isolated and cultured outside of the body23. In these previous studies, we suggested that sex difference in the HMGB1 signaling could be responsible for the activated TLR4 pathway specifically in PAH males6, and for the pro-inflammatory phenotype of PAH found to be directly associated with the male sex36.
Our group has recently discovered that the chronic mitochondrial dysfunction induced by a point mutation in Fe-S scaffold protein, NFU1, causes a spontaneous PAH that fully reproduces the male:female ratio (1:3) in PAH prevalence31. However, there is a very limited number of pre-clinical studies that reproduce the sexual dimorphism in PAH severity. In this study, we first discovered that PAH induced by a high dose of Sugen 5416 (50 mg/kg) as previously described25, 27 and characterized at the early but relatively established stage of disease (mean RVSP 54.7±2.3 mmHg in males and 44.6±2 in females), produces a significantly more severe form of PAH in males compared to females. Since this model reproduces the reported sex difference in mean pulmonary artery pressure (mPAP) at baseline15, we used it to study the contribution of different types of cell death in the manifestation of this sex difference. As expected, only anti-necrotic therapies, which significantly decreased circulated HMGB1 monomers and dimers, reduced RVSP to the female level and strongly attenuated RV hypertrophy and contractility. We conclude that apoptosis and necroptosis do not contribute to the progressive PAH in males.
The necrosis of pulmonary arteries, although much less studied compared to apoptosis, was described in several studies published in the middle of last century as one of the common histopathological changes in patients with PAH5, 54, 55. It has been noticed that necrotic changes have been especially evident in patients with a rapidly progressive course of the disease and the highest pulmonary artery blood pressure5. The authors speculated that the high pressure in the pulmonary circulation could significantly reduce the blood supply to the pulmonary vessels through vasa vasorum; thus, impairs the nutritive supply and induces vascular wall ischemia. Coupled with an insufficient adaptation that involves an increased number of opened bronchopulmonary anastomoses, these patients with a highly progressive form of PAH developed severe arterial necrosis. We believe that these mechanisms described for the patients with an aggressive form of PAH could be relevant to the discovered in this study activation of necrosis in males. Indeed, the higher pulmonary pressure and more progressive form of PAH in male rats compared to females predispose them to necrotic damage of the pulmonary vascular wall.
A release of HMGB1 could mediate downstream signaling through activation of either TLR4 or RAGE receptors. The potential contribution of each of these receptors has been proposed to contribute to PAH pathobiology by the previous research, although there is a disagreement in the literature on which receptor - TLR4 or RAGE - is a more critical contributor to PAH. Interestingly, among the studies that reported the sex of the animal models used, the majority that proposed the importance of TLR4 were done on male rats or mice7, 11. In contrast, studies showing the importance of RAGE were done on females, patient cohorts that consist mostly of females, or both sexes25, 41, 56. The additional research is required to confirm whether the contradictions in the contribution of TLR4 vs. RAGE present in the literature are due to insufficient consideration of sex. Nevertheless, based on the current results, we propose that activation of HMGB1/TLR4 axis rather than HMGB1/RAGE is responsible for the development of the more progressive form of PAH in males. Indeed, TLR4 activation was found to occur specifically in male PAH rats and became significantly attenuated by anti-necrotic therapy. In contrast, the levels of RAGE activation were comparable between the sexes. Moreover, male-specific activation of TLR4 signaling in PAH was already reported by our group6. In this previous study, we used a model of advanced PAH model that was analyzed 14 weeks after Su/Hx injection. Taken together, our previous and current results propose the importance of TLR4 signaling in males on different stages of PAH development and could be responsible for the male-specific inflammatory response36 that predispose males to accelerated PAH progression. In contrast, RAGE that also found to be significantly upregulated by PAH becomes activated in both sexes, and while its activation in females well correlates with apoptotic cell death25, the particular mechanisms responsible for its activation in males yet to be elucidated.
In conclusion, our study provides novel insight on HMGB1 release and action. It also confirms the essential role of necrotic cell death in HMGB1 release, activation of TLR4, and the manifestation of sex difference in PAH severity. This finding suggests that a considerably more active HMGB1/TLR4 axis in males could represent an important therapeutic target to reduce the PAH progression and improve the survival prognosis in male patients.
Supplementary Material
PERSPECTIVES:
Activation of the inflammatory and immune system plays an essential role in the pathogenesis of PAH57, 58. However, while blocking inflammation remains an attractive approach and could become an important part of the complex PAH therapy, the previous attempts of targeting inflammation were not successful59. It has been proposed that a more precise approach for treating inflammation could produce higher benefits58, 59. Based on our discovery, there is a strong possibility that the responsiveness to anti-inflammatory therapy could depend on patient sex. Although women are more predisposed to PAH, the results reported in this study and our previous research6, 23, provide strong evidence of more activated inflammatory pathways in men that could predispose them to accelerated PAH progression and poor survival prognosis. Thus, this study highlights the importance of taking sex into consideration in future research and in the treatment of PAH and suggests that male patients may benefit from the implementation of anti-inflammatory drugs as a part of PAH therapy.
NOVELTY AND SIGNIFICANCE:
What Is New?
Apoptosis of human pulmonary artery smooth muscle cells (HPASMC) but not human endothelial cells (HPAEC) release pro-inflammatory alarming HMGB1, suggesting that secretion of HMGB1 from apoptosis cells depends on a cell type.
HMGB1 released from HPASMC consists of monomeric and dimeric HMGB1; necrotic HPAEC release only HMGB1 monomers.
The formation of HMGB1 dimers in HPASMC depends on the presence of mitochondrial reactive oxygen species (ROS) and correlates with TLR4 activation in cells and in a rat model of pulmonary arterial hypertension (PAH).
Necrosis but not other types of cell death releases monomeric and dimeric HMGB1 and accelerates the progression of PAH in males, producing a sex difference in PAH severity described in PAH patients.
What Is Relevant?
PAH is hypertension occurring in the pulmonary circulation due to the remodeling of the pulmonary artery wall that leads to cardiac failure and death.
This study addresses an important role of necrotic cell death and HMGB1 release in sexual dimorphism of PAH progression.
SOURCES OF FUNDING:
This work was supported by NIH grants R01HL133085 (OR), R01HL132918 (RR), and R01HL151447 (RR).
Footnotes
DISCLOSURES:
None.
REFERENCES:
- 1.Hemnes AR and Humbert M. Pathobiology of pulmonary arterial hypertension: understanding the roads less travelled. European respiratory review : an official journal of the European Respiratory Society. 2017;26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF and Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2001;15:427–38. [DOI] [PubMed] [Google Scholar]
- 3.Jurasz P, Courtman D, Babaie S and Stewart DJ. Role of apoptosis in pulmonary hypertension: from experimental models to clinical trials. Pharmacology & therapeutics. 2010;126:1–8. [DOI] [PubMed] [Google Scholar]
- 4.Levy M, Maurey C, Celermajer DS, Vouhe PR, Danel C, Bonnet D and Israel-Biet D. Impaired apoptosis of pulmonary endothelial cells is associated with intimal proliferation and irreversibility of pulmonary hypertension in congenital heart disease. Journal of the American College of Cardiology. 2007;49:803–10. [DOI] [PubMed] [Google Scholar]
- 5.Heath D and Whitaker W. Hypertensive pulmonary vascular disease. Circulation. 1956;14:323–43. [DOI] [PubMed] [Google Scholar]
- 6.Rafikov R, Nair V, Sinari S, Babu H, Sullivan JC, Yuan JX, Desai AA and Rafikova O. Gender Difference in Damage-Mediated Signaling Contributes to Pulmonary Arterial Hypertension. Antioxidants & redox signaling. 2019;31:917–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bauer EM, Shapiro R, Zheng H, Ahmad F, Ishizawar D, Comhair SA, Erzurum SC, Billiar TR and Bauer PM. High mobility group box 1 contributes to the pathogenesis of experimental pulmonary hypertension via activation of Toll-like receptor 4. Molecular medicine. 2013;18:1509–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dai M, Xiao R, Cai L, Ge T, Zhu L and Hu Q. HMGB1 is mechanistically essential in the development of experimental pulmonary hypertension. American journal of physiology Cell physiology. 2019;316:C175–C185. [DOI] [PubMed] [Google Scholar]
- 9.Sadamura-Takenaka Y, Ito T, Noma S, Oyama Y, Yamada S, Kawahara K, Inoue H and Maruyama I. HMGB1 promotes the development of pulmonary arterial hypertension in rats. PloS one. 2014;9:e102482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldenberg NM, Hu Y, Hu X, Volchuk A, Zhao YD, Kucherenko MM, Knosalla C, de Perrot M, Tracey KJ, Al-Abed Y, Steinberg BE and Kuebler WM. Therapeutic Targeting of High-Mobility Group Box-1 in Pulmonary Arterial Hypertension. American journal of respiratory and critical care medicine. 2019;199:1566–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J, Tian XT, Peng Z, Li WQ, Cao YY, Li Y and Li XH. HMGB1/TLR4 promotes hypoxic pulmonary hypertension via suppressing BMPR2 signaling. Vascular pharmacology. 2019;117:35–44. [DOI] [PubMed] [Google Scholar]
- 12.Yang PS, Kim DH, Lee YJ, Lee SE, Kang WJ, Chang HJ and Shin JS. Glycyrrhizin, inhibitor of high mobility group box-1, attenuates monocrotaline-induced pulmonary hypertension and vascular remodeling in rats. Respiratory research. 2014;15:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Badesch DB, Raskob GE, Elliott CG, Krichman AM, Farber HW, Frost AE, Barst RJ, Benza RL, Liou TG, Turner M, Giles S, Feldkircher K, Miller DP and McGoon MD. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest. 2010;137:376–87. [DOI] [PubMed] [Google Scholar]
- 14.Mair KM, Johansen AK, Wright AF, Wallace E and MacLean MR. Pulmonary arterial hypertension: basis of sex differences in incidence and treatment response. British journal of pharmacology. 2014;171:567–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shapiro S, Traiger GL, Turner M, McGoon MD, Wason P and Barst RJ. Sex differences in the diagnosis, treatment, and outcome of patients with pulmonary arterial hypertension enrolled in the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Chest. 2012;141:363–373. [DOI] [PubMed] [Google Scholar]
- 16.Jacobs W, van de Veerdonk MC, Trip P, de Man F, Heymans MW, Marcus JT, Kawut SM, Bogaard HJ, Boonstra A and Vonk Noordegraaf A. The right ventricle explains sex differences in survival in idiopathic pulmonary arterial hypertension. Chest. 2014;145:1230–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gabler NB, French B, Strom BL, Liu Z, Palevsky HI, Taichman DB, Kawut SM and Halpern SD. Race and sex differences in response to endothelin receptor antagonists for pulmonary arterial hypertension. Chest. 2012;141:20–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He SJ, Cheng J, Feng X, Yu Y, Tian L and Huang Q. The dual role and therapeutic potential of high-mobility group box 1 in cancer. Oncotarget. 2017;8:64534–64550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martinotti S, Patrone M and Ranzato E. Emerging roles for HMGB1 protein in immunity, inflammation, and cancer. ImmunoTargets and therapy. 2015;4:101–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Narayanaswamy PB, Tkachuk S, Haller H, Dumler I and Kiyan Y. CHK1 and RAD51 activation after DNA damage is regulated via urokinase receptor/TLR4 signaling. Cell death & disease. 2016;7:e2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rafikov R, Sun X, Rafikova O, Louise Meadows M, Desai AA, Khalpey Z, Yuan JX, Fineman JR and Black SM. Complex I dysfunction underlies the glycolytic switch in pulmonary hypertensive smooth muscle cells. Redox biology. 2015;6:278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shin EY, Min DS, Shin JC, Shin KS, Hyun MS, Ha KS, Kim HS, Ahn HY and Kim EG. Involvement of phospholipase D in oxidative stress-induced necrosis of vascular smooth muscle cells. FEBS letters. 2001;508:277–81. [DOI] [PubMed] [Google Scholar]
- 23.Zemskova M, Kurdyukov S, James J, McClain N, Rafikov R and Rafikova O. Sex-specific stress response and HMGB1 release in pulmonary endothelial cells. PloS one. 2020;15:e0231267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rafikova O, Rafikov R, Kangath A, Qu N, Aggarwal S, Sharma S, Desai J, Fields T, Ludewig B, Yuan JX, Jonigk D and Black SM. Redox regulation of epidermal growth factor receptor signaling during the development of pulmonary hypertension. Free radical biology & medicine. 2016;95:96–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rafikov R, McBride ML, Zemskova M, Kurdyukov S, McClain N, Niihori M, Langlais PR and Rafikova O. Inositol monophosphatase 1 as a novel interacting partner of RAGE in pulmonary hypertension. American journal of physiology Lung cellular and molecular physiology. 2019;316:L428–L444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rivero-Gutierrez B, Anzola A, Martinez-Augustin O and de Medina FS. Stain-free detection as loading control alternative to Ponceau and housekeeping protein immunodetection in Western blotting. Analytical biochemistry. 2014;467:1–3. [DOI] [PubMed] [Google Scholar]
- 27.Rafikova O, Williams ER, McBride ML, Zemskova M, Srivastava A, Nair V, Desai AA, Langlais PR, Zemskov E, Simon M, Mandarino LJ and Rafikov R. Hemolysis-induced Lung Vascular Leakage Contributes to the Development of Pulmonary Hypertension. American journal of respiratory cell and molecular biology. 2018;59:334–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu Y, Cui H, Xia Y and Gan H. RIPK3-Mediated Necroptosis and Apoptosis Contributes to Renal Tubular Cell Progressive Loss and Chronic Kidney Disease Progression in Rats. PloS one. 2016;11:e0156729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Q, Wei S, Lu J, Fu W, Chen H, Huang Q, Chen Z and Zeng Z. Necrostatin-1 accelerates time to death in a rat model of cecal ligation and puncture and massively increases hepatocyte caspase-3 cleavage. American journal of physiology Gastrointestinal and liver physiology. 2019;316:G551–G561. [DOI] [PubMed] [Google Scholar]
- 30.Abdelbary M, Rafikova O, Gillis EE, Musall JB, Baban B, O’Connor PM, Brands MW and Sullivan JC. Necrosis Contributes to the Development of Hypertension in Male, but Not Female, Spontaneously Hypertensive Rats. Hypertension. 2019;74:1524–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niihori M, Eccles CA, Kurdyukov S, Zemskova M, Varghese MV, Stepanova AA, Galkin A, Rafikov R and Rafikova O. Rats with a Human Mutation of NFU1 Develop Pulmonary Hypertension. American journal of respiratory cell and molecular biology. 2020;62:231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raghavan A, Zhou G, Zhou Q, Ibe JC, Ramchandran R, Yang Q, Racherla H, Raychaudhuri P and Raj JU. Hypoxia-induced pulmonary arterial smooth muscle cell proliferation is controlled by forkhead box M1. American journal of respiratory cell and molecular biology. 2012;46:431–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith KA and Yuan JX. Hypoxia-inducible factor-1alpha in pulmonary arterial smooth muscle cells and hypoxia-induced pulmonary hypertension. American journal of respiratory and critical care medicine. 2014;189:245–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu W and Erzurum SC. Endothelial cell energy metabolism, proliferation, and apoptosis in pulmonary hypertension. Comprehensive Physiology. 2011;1:357–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Porter KM, Kang BY, Adesina SE, Murphy TC, Hart CM and Sutliff RL. Chronic hypoxia promotes pulmonary artery endothelial cell proliferation through H2O2-induced 5-lipoxygenase. PloS one. 2014;9:e98532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rafikova O, Rafikov R, Meadows ML, Kangath A, Jonigk D and Black SM. The sexual dimorphism associated with pulmonary hypertension corresponds to a fibrotic phenotype. Pulmonary circulation. 2015;5:184–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lahm T, Frump AL, Albrecht ME, Fisher AJ, Cook TG, Jones TJ, Yakubov B, Whitson J, Fuchs RK, Liu A, Chesler NC and Brown MB. 17beta-Estradiol mediates superior adaptation of right ventricular function to acute strenuous exercise in female rats with severe pulmonary hypertension. American journal of physiology Lung cellular and molecular physiology. 2016;311:L375–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Docherty CK, Nilsen M and MacLean MR. Influence of 2-Methoxyestradiol and Sex on Hypoxia-Induced Pulmonary Hypertension and Hypoxia-Inducible Factor-1-alpha. Journal of the American Heart Association. 2019;8:e011628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.White K, Johansen AK, Nilsen M, Ciuclan L, Wallace E, Paton L, Campbell A, Morecroft I, Loughlin L, McClure JD, Thomas M, Mair KM and MacLean MR. Activity of the estrogen-metabolizing enzyme cytochrome P450 1B1 influences the development of pulmonary arterial hypertension. Circulation. 2012;126:1087–98. [DOI] [PubMed] [Google Scholar]
- 40.Chen X, Talati M, Fessel JP, Hemnes AR, Gladson S, French J, Shay S, Trammell A, Phillips JA, Hamid R, Cogan JD, Dawson EP, Womble KE, Hedges LK, Martinez EG, Wheeler LA, Loyd JE, Majka SJ, West J and Austin ED. Estrogen Metabolite 16alpha-Hydroxyestrone Exacerbates Bone Morphogenetic Protein Receptor Type II-Associated Pulmonary Arterial Hypertension Through MicroRNA-29-Mediated Modulation of Cellular Metabolism. Circulation. 2016;133:82–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meloche J, Courchesne A, Barrier M, Carter S, Bisserier M, Paulin R, Lauzon-Joset JF, Breuils-Bonnet S, Tremblay E, Biardel S, Racine C, Courture C, Bonnet P, Majka SM, Deshaies Y, Picard F, Provencher S and Bonnet S. Critical role for the advanced glycation end-products receptor in pulmonary arterial hypertension etiology. Journal of the American Heart Association. 2013;2:e005157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Castilla R, Gonzalez R, Fouad D, Fraga E and Muntane J. Dual effect of ethanol on cell death in primary culture of human and rat hepatocytes. Alcohol and alcoholism. 2004;39:290–6. [DOI] [PubMed] [Google Scholar]
- 43.Rafikova O, Al Ghouleh I and Rafikov R. Focus on Early Events: Pathogenesis of Pulmonary Arterial Hypertension Development. Antioxidants & redox signaling. 2019;31:933–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bianchi ME and Manfredi A. Chromatin and cell death. Biochimica et biophysica acta. 2004;1677:181–6. [DOI] [PubMed] [Google Scholar]
- 45.Anggayasti WL, Mancera RL, Bottomley S and Helmerhorst E. The self-association of HMGB1 and its possible role in the binding to DNA and cell membrane receptors. FEBS letters. 2017;591:282–294. [DOI] [PubMed] [Google Scholar]
- 46.Bell CW, Jiang W, Reich CF 3rd and Pisetsky DS. The extracellular release of HMGB1 during apoptotic cell death. American journal of physiology Cell physiology. 2006;291:C1318–25. [DOI] [PubMed] [Google Scholar]
- 47.Frump ALLT. The Basic Science of Metabolism in Pulmonary Arterial Hypertension. Advances in Pulmonary Hypertension 2018;17:95–102. [Google Scholar]
- 48.Davie NJ, Crossno JT Jr., Frid MG, Hofmeister SE, Reeves JT, Hyde DM, Carpenter TC, Brunetti JA, McNiece IK and Stenmark KR. Hypoxia-induced pulmonary artery adventitial remodeling and neovascularization: contribution of progenitor cells. American journal of physiology Lung cellular and molecular physiology. 2004;286:L668–78. [DOI] [PubMed] [Google Scholar]
- 49.Gurbanov E and Shiliang X. The key role of apoptosis in the pathogenesis and treatment of pulmonary hypertension. European journal of cardio-thoracic surgery : official journal of the European Association for Cardio-thoracic Surgery. 2006;30:499–507. [DOI] [PubMed] [Google Scholar]
- 50.Suzuki YJ, Ibrahim YF and Shults NV. Apoptosis-based therapy to treat pulmonary arterial hypertension. Journal of rare diseases research & treatment. 2016;1:17–24. [PMC free article] [PubMed] [Google Scholar]
- 51.Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, Lu B, Chavan S, Rosas-Ballina M, Al-Abed Y, Akira S, Bierhaus A, Erlandsson-Harris H, Andersson U and Tracey KJ. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:11942–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kwak MSJW, Youn JH, Kwon MK, Shin JS. Cys106 Oxidation of High Mobility Group Box 1 (HMGB1) Promotes Self-Oligomerization and Cytokine Release Paper presented at: J Immunol; 2012. [Google Scholar]
- 53.Hoeper MM and Simon RGJ. The changing landscape of pulmonary arterial hypertension and implications for patient care. European respiratory review : an official journal of the European Respiratory Society. 2014;23:450–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Symmers WS. Necrotizing pulmonary arteriopathy associated with pulmonary hypertension. Journal of clinical pathology. 1952;5:36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aitchison JD and Richmond HG. Pulmonary hypertension associated with necrotizing pulmonary arteritis. British heart journal. 1955;17:312–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nakamura K, Sakaguchi M, Matsubara H, Akagi S, Sarashina T, Ejiri K, Akazawa K, Kondo M, Nakagawa K, Yoshida M, Miyoshi T, Ogo T, Oto T, Toyooka S, Higashimoto Y, Fukami K and Ito H. Crucial role of RAGE in inappropriate increase of smooth muscle cells from patients with pulmonary arterial hypertension. PloS one. 2018;13:e0203046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goldenberg NM, Rabinovitch M and Steinberg BE. Inflammatory Basis of Pulmonary Arterial Hypertension: Implications for Perioperative and Critical Care Medicine. Anesthesiology. 2019;131:898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Voelkel NF, Tamosiuniene R and Nicolls MR. Challenges and opportunities in treating inflammation associated with pulmonary hypertension. Expert review of cardiovascular therapy. 2016;14:939–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kumar R and Graham B. How does inflammation contribute to pulmonary hypertension? The European respiratory journal. 2018;51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.