

Abstract
Background.
The treatment options for patients with peritoneal carcinomatosis (PC) of gastrointestinal and pancreaticobiliary origins are limited. The virus-like particle, CMP-001, composed of the Qβ bacteriophage capsid protein encapsulating a CpG-A oligodeoxynucleotide, activates plasmacytoid dendritic cells (pDCs) and triggers interferon alpha (IFNα) release, leading to a cascade of anti-tumor immune effects.
Methods.
To evaluate the ability of CMP-001 to trigger an immune response in patients with PC, peritoneal cells were isolated and stimulated ex vivo with CMP-001. Both IFNα release and percentage of pDC were quantified using enzyme-linked immunosorbent assay (ELISA) and flow cytometry, respectively. To evaluate the anti-tumor response in vivo, murine PC models were generated using mouse cancer cell lines (Panc02 and MC38) in immunocompetent mice treated with intraperitoneal CMP-001 or saline control. Survival was followed, and the immunophenotype of cells in the peritoneal tumor microenvironment was evaluated.
Results.
The pDCs accounted for 1% (range 0.1–3.9%; n = 17) of the isolated peritoneal cells. Ex vivo CMP-001 stimulation of the peritoneal cells released an average of 0.77 ng/ml of IFNα (range, 0–4700 pg/ml; n = 14). The IFNα concentration was proportional to the percentage of pDCs present in the peritoneal cell mixture (r = 0.6; p = 0.037). In murine PC models, intraperitoneal CMP-001 treatment elicited an anti-tumor immune response including an increase in chemokines (RANTES and MIP-1β), pro-inflammatory cytokines (IFNγ, interleukin 6 [IL-6], and IL-12), and peritoneal/tumor immune infiltration (CD4+/CD8+ T and natural killer [NK] cells). The CMP-001 treatment improved survival in both the Panc02 (median, 35 vs 28 days) and the MC38 (median: 57 vs 35 days) PC models (p < 0.05).
Conclusions.
As a novel immunotherapeutic agent, CMP-001 may be effective for treating patients with PC.
Colorectal and pancreatic cancers are the second and third most common causes of cancer-related deaths in the United States, respectively.1 Peritoneal spread, a common metastatic route in colorectal and pancreatic cancers, also is found in other adenocarcinomas of gastrointestinal origins.
The current treatment options are very limited. Whereas some patients can undergo aggressive surgery and systemic therapy, many patients are not candidates for any treatment. The median survival for patients with peritoneal carcinomatosis (PC) ranges from 2 to 6 months.2–6 Due to the aggressiveness of PC, the pursuit of novel therapies is essential.
The peritoneal tumor microenvironment is known to be immunosuppressive, and studies suggest that immature pDCs may play a critical role in maintaining this immunosuppressive environment.7,8 In ovarian cancer, malignant ascites has a higher proportion of pDCs among isolated mononuclear cells than in peripheral blood or tumor.7 Immature pDCs support regulatory T cells (Treg), leading to suppression of the anti-tumor immune response and tumor progression.9 When activated, particularly by toll-like receptor 9 (TLR9) agonists, pDCs can induce a robust anti-tumor T cell response via interferon alpha (IFNα).10,11 Peritoneal pDCs are therefore an attractive immunotherapeutic target for treating PC with TLR9-based therapeutics.
One such approach is to modify the suppressive peritoneal immune microenvironment through the use of immunostimulatory virus-like particles (VLPs). As a novel VLP, CMP-001 is composed of the Qβ bacteriophage capsid protein encapsulating an immunostimulatory CpG-A oligodeoxynucleotide (ODN) TLR9 agonist. Previous studies have demonstrated that intra-tumoral injection of CMP-001 activates pDCs, leading to type 1 interferon secretion that enhances anti-tumor immunity.10,12 In addition, intraperitoneal administration of TLR9 agonists has shown promise in models of ovarian cancer and peritoneal mesothelioma.13–15
This study quantified peritoneal pDCs in patients with PC of gastrointestinal and pancreatic origins and examined the ability of CMP-001 to induce release of IFNα from patient-derived peritoneal cells ex vivo. In addition, the impact of intraperitoneal CMP-001 treatment in a mouse PC model was evaluated.
MATERIALS AND METHODS
Patient Population and Sample Collection
Patients with PC were approached and consented to participate in the study under an institutional review board (IRB)-approved protocol (Gastrointestinal Molecular Epidemiology Resource, IRB#201202743) at the University of Iowa Hospitals and Clinics. Malignant ascites fluid was collected using a suction device during surgery or a percutaneous drainage catheter during ultrasound-guided paracentesis. Peritoneal lavage fluid was collected using a suction device during surgery after 500 ml of sterile saline had been instilled into the peritoneal cavity when only a minimal amount of ascites fluid was present.
CMP-001
Virus-like particles containing TLR9 agonists were provided by Checkmate Pharmaceuticals (Cambridge, MA, USA). These agonists were manufactured using the bacteriophage Qβ nanotechnology platform, which involves particle self-assembly upon mixing of the purified Qβ coat protein with nonmethylated CpG-A ODN (G10; 5′-GGGGGGGGGGGACGATCGTCGGGGGGGGGG-3′) at a 4:1 mass ratio. The resulting VLPs are approximately 30 nm in diameter.
Cell Lines
Panc02 mouse pancreatic adenocarcinoma cells were obtained from Dr. Xinhui Wang (Massachusetts General Hospital, MA, USA) and cultured in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Thomas Scientific, Swedesboro, NJ, USA), 100 U/ml of penicillin G, and 100 µg/ml of streptomycin (Gibco). From Dr. Lorenzo Ferri (McGill University, PQ, Canada), MC38 mouse colon carcinoma cells were obtained and cultured in the aforementioned culture media supplemented with 0.1 mmol of nonessential amino acids, 2 mmol of M L-glutamine, 1 mmol of sodium pyruvate, and 10 mmol of 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES; Gibco).
Mouse PC Model and CMP-001 Treatment
All animal studies were approved by the University of Iowa Institutional Animal Care and Use Committee (IACUC). To generate anti-Qβ antibodies, 6- to 8-week-old C57BL/6 N mice (Charles River Laboratories, Wilmington, MA, USA) were injected subcutaneously with either 12.5 µg of CMP-001 diluted in 100 µl of phosphate-buffered saline (PBS) or PBS alone 9 days before tumor challenge.
On day 0, the mice were injected intraperitoneally with 1 × 105 of Panc02 or MC38 cells in 200 µl of PBS. On days 5, 9, and 13 after the tumor challenge, the mice were injected intraperitoneally with either 100 µg of CMP-001 diluted in 100 µl of PBS or 100 µl of PBS alone. The mice were either monitored for survival or euthanized on day 26 after tumor challenge for further analysis.
Detection of pDC in Human Peritoneal Fluids
Using peritoneal fluid obtained from the patients with PC, peritoneal cells were pelleted by centrifugation and stained for pDC using Lin1-FITC (lineage cocktail 1, BD, Franklin Lakes, NJ, USA) and BDCA-2 (clone REA693, Miltenyi Biotec, Bergisch Gladbach, Germany). The cells were fixed in 0.5% formaldehyde in PBS and resuspended in fluorescence-activated cell sorting (FACS) buffer (1 × PBS, 1% FBS, 1 mmol of ethylenediaminetetraacetic acid [EDTA], 0.01% NaN3). Samples were run on the BD LSR II flow cytometer, and all data were analyzed using FlowJo software (BD).
Immune Profiling of Mouse Peritoneal Fluid and Tumors
Peritoneal cells were isolated from ascites fluids by centrifugation at 1200 rpm and lysing of red blood cells. For peritoneal tumors, single-cell suspensions of tumors were prepared using the mouse tumor dissociation kit and gen-tleMACS tissue dissociator following the manufacturer’s instructions (Miltenyi Biotec). For cell-surface staining, 2 × 106 cells were plated in a 96-well round-bottom plate (Corning, Corning, NY, USA). Live-cell staining was performed using the Zombie Aqua Fixable Viability Kit (BioLegend, San Diego, CA, USA). Viability staining was performed in PBS for 30 min at room temperature in the dark. After viability staining, the cells were incubated in the presence of CD16/CD32 TruStain FcX (BioLegend) to block Fc receptors, then stained with combinations of antibodies for the following cell surface proteins: CD3 (clone 17A2), CD4 (GK1.5), CD8α (53–6.7), CD11b (M1/70), CD11c (N418), CD25 (PC61), CD45.2 (104), CD49b (DX5), F4/80 (BM8), I-A/I-E (M5/114.15.2) Ly-6C (HK1.4), and Ly-6G (1A8). All the antibodies were purchased from BioLegend. The cells were stained in FACS buffer (1 × PBS, 1% FBS, 1 mmol of EDTA, 0.01% NaN3) at 4 °C for 30 min at room temperature in the dark. The cells then were fixed in Fixation Buffer (BioLegend) for 20 min at room temperature in the dark, washed, and resuspended in FACS buffer.
For intracellular staining, the cells were first stained for viability and surface proteins, as described earlier. The cells then were incubated with the Foxp3/Transcription Factor Staining Buffer Set (eBioscience/Invitrogen, Waltham, MA, USA) for 2 h at 4 °C to fix the cells. The cells then were stained with Foxp3 (clone MF-14; BioLegend) in the presence of CD16/CD32 TruStain FcX (BioLegend) in permeabilization buffer for 30 min at room temperature. The cells were resuspended in FACS buffer. All flow cytometry samples were collected on a BD LSR II flow cytometer, and all data were analyzed using FlowJo software (Franklin Lakes, NJ, USA).
Multiplex Immunoassay
Cytokine and chemokine levels in mouse ascites fluid were quantified using the Bio-Plex Pro Mouse Cytokine 23-Plex Immunoassay (Bio-Rad, Hercules, CA, USA) with the addition of vascular endothelial growth factor (VEGF). The Bio-Plex was performed according to the manufacturer’s instructions. Ascites fluid was collected and centrifuged at 1200 rpm for 5 min at 4 °C to pellet the red blood cells. The supernatant was removed and centrifuged again at 10,000 rpm for 5 min at 4 °C. The supernatant then was used for the multiplex assay.
Ex Vivo Stimulation of Human Peritoneal Cells With CMP-001
From peritoneal fluid in patients with PC, 2.5 × 105 cells were isolated and plated per well in a 96-well round-bottom plate. The cells were plated in RPMI 1640 (Gibco) supplemented with 10% heat-inactivated FBS (Thomas Scientific), 100 U/ml of penicillin G, 100 µg/ml of streptomycin, and 2 mmol of L-glutamine (Gibco). The cells then were stimulated with 20 µg/mL of CMP-001 in the presence of recombinant immunoglobulin G (IgG) anti-Qβ (Cytos Biotechnology, Schlieren, Switzerland; 10-µg/mL final concentration). After 2 days, the conditioned media were collected, and levels of IFNα were determined via ELISA (VeriKineTM Human IFNα ELISA kit; PBL Assay Science, Piscataway, NJ, USA).
Histology
Tumor tissues from mice were fixed in 10% neutral buffered Formalin solution. Samples were routinely processed, embedded, sectioned at 4-µm thickness, and stained with hematoxylin and eosin. Tumor morphology and necrosis were assessed in a blinded fashion, and tumor necrosis was scored semi-quantitatively by a board-certified veterinary pathologist (K.N.G.).
Statistical Analysis
Statistical analysis was performed using GraphPad Prism software (GraphPad, San Diego, CA, USA). Statistical significance was determined by the unpaired two-tailed Student’s t test for continuous variables, the log-rank test for Kaplan Meier survival analysis, or linear regression for correlation. All p values lower than 0.05 were considered significant.
RESULTS
pDCs Are Present in the Peritoneal Cavity of Patients with PC of Gastrointestinal and Pancreaticobiliary Origins
Peritoneal fluids were obtained from 17 patients with PC of various gastrointestinal and pancreaticobiliary origins (Table 1). By flow cytometry, pDCs were detected in all 17 peritoneal fluid samples. The pDC population in the peritoneal cells ranged from 0.01% to 4.73% (Table 1). The pancreatic cancer patients had a higher percentage of pDCs in the peritoneal cavity than the patients with cancers of the gastrointestinal tract (2.6 vs 0.7%; p = 0.01). The chemotherapy-naїve patients had a higher percentage of pDCs in the peritoneal cavity (2.3 vs 0.7%; p = 0.01). Patient GI1034 had both pre-chemotherapy and postchemotherapy malignant ascites collected and analyzed. The percentage of pDCs dropped from 2.05 to 0.58% after a single dose of FOLFOX chemotherapy. These results suggest that cancer type and systemic treatment may have an impact on the number of pDCs in the peritoneal tumor microenvironment.
TABLE 1.
Patients’ characteristics and peritoneal pDC content
| Subject ID | Age (years) | Sex | Primary site | Histologic type | Collection timea | Collection method | % pDC |
|---|---|---|---|---|---|---|---|
| GI0791 | 62 | F | Colon | Adenocarcinoma | Post-chemotherapy | OR | 1.41 |
| GI0794 | 66 | M | Appendix | Adenocarcinoma | Pre-chemotherapy | OR | 0.79 |
| GI0812 | 51 | F | Pancreas | Adenocarcinoma | Pre-chemotherapy | US | 3.94 |
| GI0827 | 68 | F | Appendix | Adenocarcinoma | On chemotherapy | OR | 0.17 |
| GI0828 | 69 | M | Pancreas | Mucinous adenocarcinoma | Pre-chemotherapy | OR | 4.73 |
| GI0830 | 64 | F | Pancreas | Adenocarcinoma | On chemotherapy | US | 0.62 |
| GI0863 | 62 | M | Appendix | Mucinous adenocarcinoma | Pre-chemotherapy | US | 1.32 |
| GI0866 | 67 | F | Appendix | Signet ring cell carcinoma | Post-chemotherapy | OR | 0.41 |
| GI0896 | 57 | F | Appendix | Goblet cell carcinoma | Post-chemotherapy | OR | 0.39 |
| GI0949 | 56 | M | Colon | Adenocarcinoma | Post-chemotherapy | OR | 0.53 |
| GI0970 | 23 | M | Rectum | Adenocarcinoma | Post-chemotherapy | OR | 0.87 |
| GI0955 | 40 | F | Small bowel | Adenocarcinoma | Post-chemotherapy | OR | 2.60 |
| GI1005 | 44 | F | Colon | Adenocarcinoma | On pembrolizumab | OR | 0.01 |
| GI1034 | 56 | F | Appendix | Adenocarcinoma | On chemotherapy | US | 0.58 |
| GI1050 | 60 | F | Pancreas | Adenocarcinoma | Pre-chemotherapy | OR | 1.17 |
| GI1095 | 54 | M | Rectum | Adenocarcinoma | On chemotherapy | OR | 0.06 |
| GI1107 | 47 | F | Colon | Adenocarcinoma | Post-chemotherapy | OR | 0.06 |
OR intraoperative ascites drainage or peritoneal lavage, US ultrasound-guided paracentesis
“Postchemotherapy” (patients received their last chemotherapy dose within 4 weeks after fluid collection); “Pre-chemotherapy” (patients did not receive chemotherapy or other therapy for at least 6 months); “On chemotherapy” (patients received active chemotherapy within 4 weeks after fluid collection; On pembrolizumab (patient received pembrolizumab after fluid collection)
CMP-001 Stimulates IFNα Production from Patient-Derived Peritoneal Cells
Peritoneal cells isolated from 14 patients with PC were incubated with CMP-001 in the presence of anti-Qβ antibodies for 48 h, and levels of IFNα in the culture media were determined via ELISA (Fig. 1a). A significant positive correlation was found between the levels of IFNα released in response to CMP-001 plus anti-Qβ antibodies and the percentage of pDCs present in the peritoneal cell populations (Fig. 1b). The levels of IFNα release were negligible in the absence of anti-Qβ antibodies, as previously reported with pDCs isolated from the peripheral blood.12 No correlation was found between IFNα release and frequency of other peritoneal cell types (data not shown). These data suggest that CMP-001 activates peritoneal pDCs, leading to IFNα release in the presence of anti-Qβ antibodies.
FIG. 1.

Interferon alpha (IFNα) production by peritoneal cells isolated from patients with peritoneal carcinomatosis. a Levels of IFNα in culture media after incubation of peritoneal cells from 14 different patients with CMP-001 and anti-Qβ antibodies for 48 h were determined via enzyme-linked immunosorbent assay (ELISA). b Correlation of IFNα levels and percentages of pDCs. A positive correlation exists between IFNα levels and percentages of pDCs (p = 0.037; R2 = 0.32)
Intraperitoneal CMP-001 Enhances Survival in Mouse PC
Previous studies have determined that anti-Qβ antibodies are critical for CMP-001-mediated activation of a robust immune response.12 In addition, minimal IFNα release from pDCs isolated from patient peritoneal fluid was observed after the ex vivo stimulation with CMP-001 in the absence of anti-Qβ antibodies. Therefore, in the mouse PC model, immunocompetent mice were first treated subcutaneously with CMP-001 to induce development of antibodies to the Qβ capsid protein of CMP-001, which typically required 10–14 days, before the first intraperitoneal CMP-001 treatment in order to test their potential therapeutic effects in mouse tumor models with very rapid tumor growth and short survival.
On day 9 after priming, the mice were challenged intraperitoneally with Panc02 or MC38 cancer cells followed by intraperitoneal treatment with CMP-001 on days 5, 9 and 13 after the tumor challenge (Fig. 2a). An overall increase in survival of the mice that received CMP-001 treatment compared with PBS control was observed with both Panc02 (median, 35 vs 28 days; p = 0.003) and MC38 (median, 57 vs 35 days; p = 0.039) PC models (Fig. 2b, c).
FIG. 2.

Intraperitoneal CMP-001 treatment in mouse models of peritoneal carcinomatosis. a Representative schematic of CMP-001 treatment. In C57BL/6 N mice, 12.5 µg of CMP-001 or PBS was injected subcutaneously 9 days before intraperitoneal injections with Panc02 mouse pancreatic cancer cells or MC38 mouse colon carcinoma cells. On days 5, 9, and 13 after the tumor challenge, the mice were treated intraperitoneally with either PBS or 100 µg of CMP-001. b, c Kaplan-Meier curves showing significant survival benefit of CMP-001 treatment (gray dotted lines) versus PBS control (black solid lines) of Panc02 (b 10 mice per group; P = 0.003) and MC38 (c 9 mice per group; P = 0.039) tumor-bearing mice. d–h Necropsy of mice 26 days after Panc02 tumor challenge. The d total tumor weight and e volume of ascites of the mice show significant reduction in the CMP-001-treated mice. Each dot represents an individual mouse (11 or 12 mice per group; p < 0.001). f Representative gross pictures of CMP-001-and PBS-treated mice. The red arrows are pointing at peritoneal tumors. Notably, the peritoneal tumors in the CMP-001-treated mice are hemorrhagic. g Representative photomicrographs of peritoneal tumors from three CMP-001- or PBS-treated mice (hematoxylin and eosin staining). The dotted lines outline the zones of necrosis (*). Scale bars: 100 µm. h Semi-quantitative scoring of tumor necrosis (0 [no necrosis], 1 [0–10% necrosis], 2 [11–25% necrosis], 3 [≥ 25% necrosis]) showing CMP-001-treated mice with a significant amount of tumor necrosis (6 mice per group; p < 0.0001)
CMP-001 Treatment Induces Tumor Necrosis and Reduces Peritoneal Tumor Burden
On day 26 after the tumor challenge with Panc02 pancreatic cancer cells, total tumor weight and ascites volume were significantly decreased in the mice treated with CMP-001 compared to the PBS control (Fig. 2d, e). The CMP-001-treated peritoneal tumors appeared more hemorrhagic (Fig. 2f). Histologically, marked areas of necrosis were observed in tumors from the CMP-001-treated mice and little to no necrosis in tumors from the PBS control mice (Fig. 2g, h).
Intraperitoneal CMP-001 Treatment Alters the Innate Immune Cell Populations in the Ascites and Peritoneal Tumors
On day 26 after the tumor challenge, peritoneal cells were collected by peritoneal lavage, as previously described.16 Peritoneal tumors were collected. Using flow cytometry, a trending or significant increase in the numbers and frequencies of monocytes (Fig. 3a, f; Fig. S1), dendritic cells (Fig. 3c, h; Fig. S1), and natural killer (NK) cells (Fig. 3d, i; Fig. S1) were observed in both ascites fluid and peritoneal tumors of the mice treated with CMP-001. In contrast, no difference in the numbers of granulocytic cells (Fig. 3b, g) or macrophages (Fig. 3e, j) were observed. The frequencies of granulocytes and macrophages were decreased in ascites and peritoneal tumors, respectively (Fig. S1). Together, these data demonstrate that CMP-001 treatment increases NK cells, which are known to have anti-tumor activity, and dendritic cells, which are important for activating the adaptive immune response. Further studies are needed to characterize the phenotype of monocytes present in the ascites and peritoneal tumors of CMP-001-treated mice.
FIG. 3.

Impact of CMP-001 on peritoneal innate immunity. Peritoneal ascites fluid and peritoneal tumors were collected 13 days after the third CMP-001 treatment (day 26 after the tumor challenge). Innate immune cell populations were stained for flow cytometric analysis and enumerated as a–e the total number per milliliter of ascites or f–j the total number per gram of tumors. a, f Ly6C+Ly-6G–CD11b+F4/80– monocytes, b, g Ly6C+Ly6G+CD11b+F4/80– granulocytes, c, h CD11c+MHCII+ dendritic cells, d, i CD49b+CD3– NK cells, and e, j F40/80+CD11b+ macrophages. a–e Each group for ascites had 10–11 mice. f–j Each group for tumors had 7–9 mice. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
CMP-001 Treatment Enhances T Cell Infiltration in Ascites and Peritoneal Tumors
In antigen-presenting cells (APC), CMP-001 activates TLR9.17 Activation of APCs, including pDCs, leads to their migration and subsequent activation of immune cells including CD4+ and CD8+ T cells, which are a critical part of the anti-tumor response.18 On day 26 after the tumor challenge, a notable increase in total numbers of CD4+ and CD8+ T cells in both the ascites fluid (Fig. 4a, b) and peritoneal tumors (Fig. 4d, e) of mice treated with CMP-001 were observed. A comparable trend in the frequencies of these cell populations also was noted, but the frequency of CD8+ T cells in the ascites of CMP-001-treated mice (Fig. S2) was not significantly increased.
FIG. 4.

Impact of CMP-001 on peritoneal adaptive immunity. Peritoneal ascites fluid and peritoneal tumors were collected 13 days after the third CMP-001 treatment (day 26 after the tumor challenge). a–c Peritoneal fluid cells were stained for a CD3+CD4+ T cells, b CD3+CD8+ T cells, and c Foxp3+CD4+CD3+ regulatory T cells. Then total numbers were determined via flow cytometric analysis and enumerated as total number per milliliter of ascites (10–11 mice per group). d–f Tumor-infiltrating cells were stained for d CD3+CD4+ T cells, e CD3+CD8+ T cells, and f Foxp3+CD4+CD3+ regulatory T cells. Then total numbers were determined via flow cytometric analysis and enumerated as the total number per gram of tumor (7–9 mice per group for tumors). *p < 0.05; **p < 0.01
The tumor microenvironment often is filled with suppressive cell populations, including Regulatory T cells (Tregs), leading to enhanced tumor growth.19 Moreover, in response to a robust immune response, there often is an anti-inflammatory or regulatory response to prevent aberrant inflammation. Therefore, the presence of Tregs in the ascites and peritoneal tumors of the mice was examined. The total number of Tregs was increased in the ascites and peritoneal tumors of the mice treated with CMP-001 (Fig. 4c, f). Interestingly, the frequency of Tregs among CD4+ T cells was increased in the ascites, but decreased in the peritoneal tumors of the CMP-001-treated mice (Fig. S2). These data suggest that CMP-001 treatment stimulates a robust immune response, but also may alter the regulatory immune response.
Elevated Pro-Inflammatory Cytokines and Chemokines in the Ascites of CMP-001-Treated Mice
The impact of CMP-001 treatment on the cytokine and chemokine milieu in the peritoneal cavity of the tumor-bearing mice 26 days after the tumor challenge was determined. A significant increase in RANTES (CCL5), a T cell chemoattractant, in the ascites of the CMP-001-treated mice was observed (Fig. 5a). Levels of RANTES have previously been correlated with CD8+ T cell infiltration, which corresponds to our flow cytometry data.20 Furthermore, RANTES also induces proliferation of NK cells.21 In addition, MIP-1β was elevated in the ascites of the CMP-001-treated mice compared with the PBS control (Fig. 5b). As a chemoattractant for NK cells and monocytes,22 MIP-1β (CCL4) correlates with the increase in NK cells and monocytes observed in the CMP-001-treated mice.
FIG. 5.
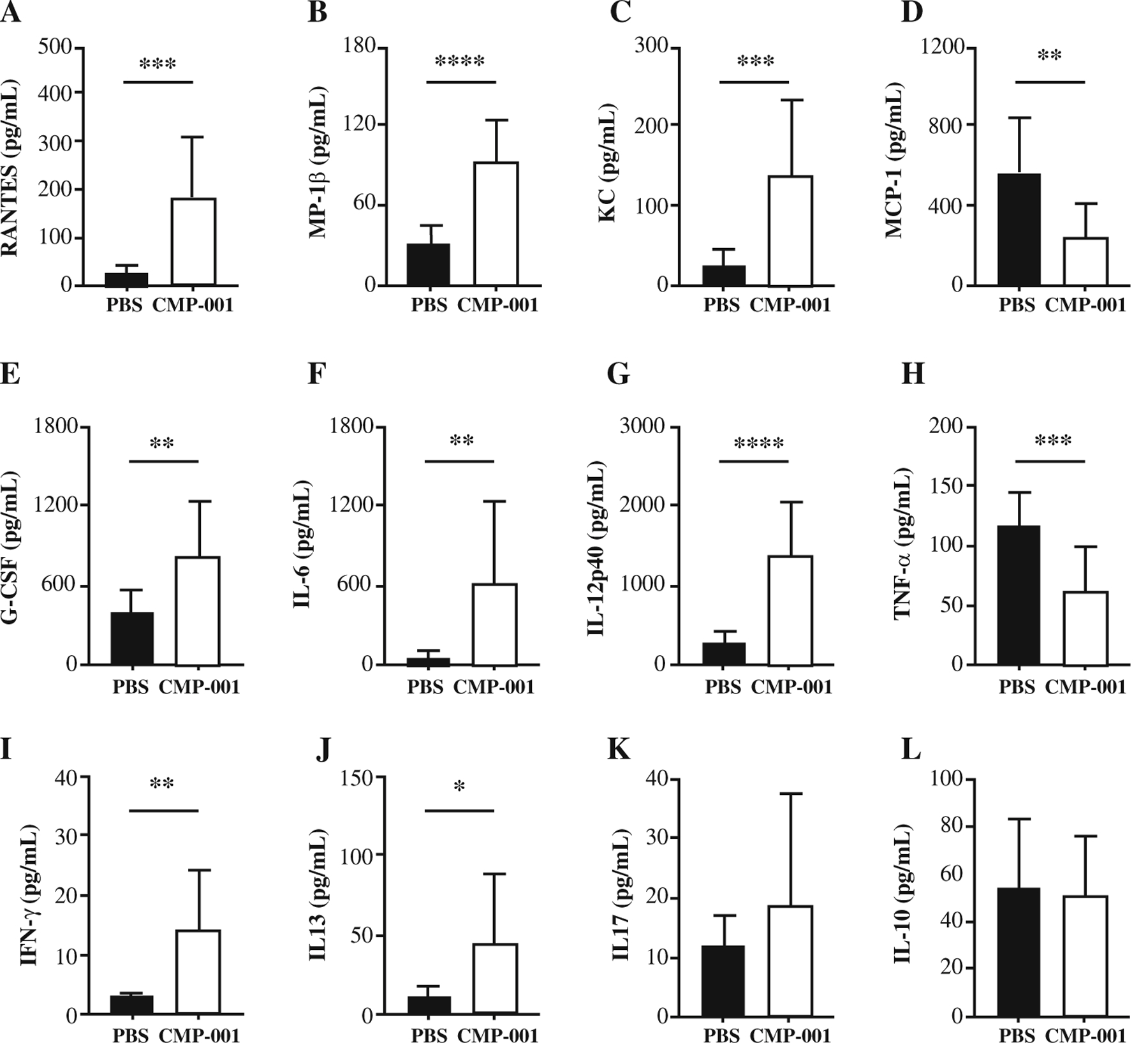
Treatment with CMP-001 enhances pro-inflammatory cytokine production in the ascites fluid of tumor-bearing mice. a–l Mice were injected subcutaneously with CMP-001 or PBS. Then 9 days later, the mice were challenged intraperitoneally with Panc02 mouse pancreatic cancer cells. On days 5, 9, and 13 after the tumor challenge, the mice were treated intraperitoneally with either PBS or CMP-001. On day 26 after the tumor challenge, ascites fluid was collected, and a Bio-Plex immunoassay was performed to determine levels of cytokines and chemokines. a RANTES. b MIP-1β. c KC. d MCP-1. e G-CSF. f IL-6. g IL-12p40. h TNFα. i IFN-γ. j IL-13. k IL-17. l IL-10. a–l Each group had 11 mice. *p < 0.05. **p < 0.01. ***p < 0.001. ****p < 0.0001
A significant increase in the chemokine KC (CXCL1), a potent neutrophil chemoattractant,23 was found in the ascites of the CMP-001-treated mice (Fig. 5c). Although no difference in neutrophils between the CMP-001 and PBS control mice was observed, neutrophils are short-lived cells, so the differences could have been missed by looking at this particular time point. In support of this hypothesis, an increase in neutrophil recruitment 24 h after CMP-001 treatment and 14 days after the tumor challenge in the CMP-001-treated mice was observed (data not shown). The findings showed an overall decrease in monocyte chemotactic protein-1 (MCP-1), also known as CCL2, in the ascites of the mice treated with CMP-001 (Fig. 5d). As the name suggests, MCP-1 is a major monocyte chemoattractant.24 In contrast to the decrease in MCP-1, an increase in monocytes in the tumors and ascites of the CMP-001-treated mice was observed (Fig. 3a, f). However, as mentioned earlier, MIP-1β also is a monocyte chemoattractant, which could explain the increase in monocytes observed even with a decrease in the MCP-1 level. A significant increase in granulocyte colony-stimulating factor (G-CSF) in the CMP-001-treated mice compared with the PBS control also was noted (Fig. 5e).
Significant increases in the classic pro-inflammatory cytokines, IL-6 and IL-12, in the ascites fluid of mice treated with CMP-001 compared with PBS control were observed (Fig. 5f, g). Both IL-6 and IL-12 are produced by macrophages, monocytes, and dendritic cells, usually in response to pattern recognition receptor (PRR) activation.25 Because CMP-001 activates the PRR TLR9, it likely is responsible for the increased production of these cytokines. Among its several functions, IL-12 contributes to CD8+ T cell and NK cell function.26–29 This fits with the increased presence of CD8+ T cells and NK cells in mice treated with CMP-001. Although IL-6 has dual roles in cancer, both pro- and anti-tumor, it does have a role in effector T cell development,30 which also fits with the data in Fig. 4. In contrast, significantly decreased levels of TNFa in the ascites of mice treated with CMP-001 compared with the PBS control was noted (Fig. 5h).
Cytokines associated with T cell activation and various T helper cell subsets were examined.31 Levels of the Th1-associated cytokine IFNγ were increased in the ascites of the mice treated with CMP-001 (Fig. 5i). The levels of IL-13, a Th2 cytokine, were significantly increased in the ascites fluid of the mice treated with CMP-001 (Fig. 5j). However, the levels of other Th2 related cytokines, including IL-4 and IL-5, were extremely low in both groups (data not shown). No difference in the Th17-associated cytokine IL-17 was observed (Fig. 5k). Finally, no difference in the Treg-associated cytokine IL-10 between CMP-001 and PBS control was noted (Fig. 5l).
DISCUSSION
Peritoneal carcinomatosis is a deadly metastatic form of gastrointestinal and pancreaticobiliary cancer. The efficacy of current chemotherapeutic agents is unfortunately limited, and patients often die within 2 to 6 months after peritoneal disease is apparent on imaging studies. Current treatments include cytoreductive surgery and chemotherapy, but not all patients are candidates for these treatments.32
Intraperitoneal immunotherapy is a growing field, with various studies examining checkpoint blockade, CAR T cell therapy, and catumaxomab treatment.33–36 In this study, a novel immunotherapeutic agent targeting TLR9 and its potential as an intraperitoneal treatment for PC were assessed.
The use of TLR9 agonists for cancer immunotherapy, as a monotherapy or combination with other therapies, has been explored for various types of cancers in both murine models and clinical trials.11,37–42 These TLR9 agonist studies have focused heavily on systemic, subcutaneous, and intra-tumoral routes of administration. However, the presence of multiple peritoneal tumors makes subcutaneous and intra-tumoral routes of injection less than ideal. Moreover, systemic administration does not take advantage of the local immune environment inside the peritoneal cavity.
Previous studies have examined the efficacy of intraperitoneal TLR9 agonist monotherapy in mouse models of ovarian carcinoma and diffuse malignant peritoneal mesothelioma.13–15 With both tumor models, intraperitoneal TLR9 agonist administration led to increased survival and was superior to intravenous administration with enhanced pro-inflammatory cytokine production and NK cell and granulocyte recruitment.13 An intraperitoneal TLR9 agonist also has been used in combination with cetuximab and cisplatin, showing promising results in a mouse model of ovarian carcinoma.43 However, intraperitoneal therapy with TLR9 agonists has not been studied for pancreatic or colorectal peritoneal carcinomatosis.
As a TLR9 agonist-containing VLP, CMP-001 has immunologic properties distinct from those of soluble TLR9 agonists. The CMP-001 VLP consists of a Qβ bacteriophage capsid protein encapsulating a CpG-A ODN, which serves as the TLR9 agonist. This capsid protein prevents the rapid degradation of the TLR9 agonist, leading to enhanced stability.12,44 Previous studies also suggest that encapsulation of the TLR9 agonist leads to decreased systemic side effects.44 Moreover, the Qβ capsid protein likely alters the pathway by which the TLR9 agonist is taken up by phagocytic cells. Previous studies have demonstrated that the presence of anti-Qβ antibodies is necessary for enhanced survival of CMP-001-treated mice.12 In addition, pDCs from human ascites secrete IFNα only in response to treatment with CMP-001 in the presence of anti-Qβ antibodies. For these reasons, we postulate that CMP-001 and anti-Qβ antibodies form immune complexes that are taken up through receptor-mediated phagocytosis. This route of uptake leads to enhanced pDC activation and cytokine production. Future studies will aim to determine the mechanistic uptake of CMP-001 by phagocytic cells.
In this study, the role of CMP-001 in ex vivo stimulation of human peritoneal cells and in an in vivo mouse PC model was examined. We found that patients with PC have pDCs present and that these cells secret IFNα in response to CMP-001 plus anti-Qβ antibody treatment. This secretion of IFNα is directly proportional to the percentage of pDCs present in the peritoneal fluid.
In addition, we examined the therapeutic efficacy of CMP-001 in a pre-clinical mouse PC model. In two different mouse PC models, we found an overall increase in survival of mice treated with CMP-001 compared with PBS control. In addition, CMP-001 treatment significantly delayed disease progression, as indicated by the reduction in volume of ascites and tumor burden present in the peritoneal cavity. The reduction in tumor burden likely is due to the robust immune response triggered by CMP-001 treatment. Mice treated with CMP-001 displayed an increase in pro-inflammatory cytokine production and in immune cell infiltration including NK and CD4+/CD8+ T cells. The TLR9 agonist CpG-A, present in CMP-001, is a potent activator of pDCs and has previously been shown to elicit a robust adaptive immune response.40,45,46 We postulate that CMP-001 is activating pDCs in the peritoneal cavity of mice, leading to a release of IFNα, which subsequently activates the IFNα/β receptor complex on neighboring immune cells, such as monocytes and macrophages. These activated innate immune cells in turn secrete chemokines and cytokines for recruiting and activating anti-tumor NK cells and CD4+/CD8+ T cells. In addition, the pro-inflammatory cytokines released in response to CMP-001 treatment promote an anti-tumor “hot” tumor microenvironment, leading to decreased tumor growth. Further studies are warranted to dissect the mechanism of peritoneal immune trafficking and anti-tumor immune response induced by CMP-001.
Additional studies including dosing and toxicity are needed to optimize CMP-001 treatment before a clinical trial. Systemic toxicity in response to intraperitoneal CMP-001 treatment currently is under investigation. None of the mice demonstrated signs of distress or died from intraperitoneal CMP-001 treatment in this study. Overall, our current results lay an exciting foundation for further investigation into both the mechanism of CMP-001 and its therapeutic potential for treating PC.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the Holden Comprehensive Cancer Center through funds from the “Mezhir Awards for Collaborative Research” and the National Cancer Institute of the National Institutes of Health under award number P30 CA086862 for supporting the Molecular Epidemiology Resource Core and Flow Cytometry Facility. Ann M. Miller was supported by the National Institutes of Health Free Radical and Radiation Biology T32 CA078586 training grant. The investigational agent CMP-001 was kindly provided by Checkmate Pharmaceuticals. The Panc02 and MC38 cells were kind gifts from Dr. Xinhui Wang (Massachusetts General Hospital, MA) and Dr. Lorenzo Ferri (McGIll University, PQ, Canada), respectively.
Footnotes
DISCLOSURE Caitlin Lemke-Miltner holds stock options in Checkmate Pharmaceuticals. Sue Blackwell owns stocks and holds stock options in Checkmate Pharmaceuticals. George J. Weiner received research funding from Checkmate Pharmaceuticals, but not for this work. Carlos H. F. Chan received the study compound CMP-001 from Checkmate Pharmaceuticals, but did not receive any research funding for this work.
Publisher’s Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material The online version of this article (https://doi.org/10.1245/s10434-020-08591-7) contains supplementary material, which is available to authorized users.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. [DOI] [PubMed] [Google Scholar]
- 2.Elias D, Gilly F, Boutitie F, et al. Peritoneal colorectal carcinomatosis treated with surgery and perioperative intraperitoneal chemotherapy: retrospective analysis of 523 patients from a multicentric French study. J Clin Oncol. 2010;28:63–8. [DOI] [PubMed] [Google Scholar]
- 3.Franko J, Shi Q, Goldman CD, et al. Treatment of colorectal peritoneal carcinomatosis with systemic chemotherapy: a pooled analysis of North Central Cancer Treatment Group phase III trials N9741 and N9841. J Clin Oncol. 2012;30:263–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Franko J, Shi Q, Meyers JP, et al. Prognosis of patients with peritoneal metastatic colorectal cancer given systemic therapy: an analysis of individual patient data from prospective randomised trials from the Analysis and Research in Cancers of the Digestive System (ARCAD) database. Lancet Oncol. 2016;17:1709–19. [DOI] [PubMed] [Google Scholar]
- 5.Thomassen I, van Gestel YR, Lemmens VE, de Hingh IH. Incidence, prognosis, and treatment options for patients with synchronous peritoneal carcinomatosis and liver metastases from colorectal origin. Dis Colon Rectum. 2013;56:1373–80. [DOI] [PubMed] [Google Scholar]
- 6.van Oudheusden TR, Razenberg LG, van Gestel YR, Creemers GJ, Lemmens VE, de Hingh IH. Systemic treatment of patients with metachronous peritoneal carcinomatosis of colorectal origin. Sci Rep. 2015;5:18632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Labidi-Galy SI, Sisirak V, Meeus P, et al. Quantitative and functional alterations of plasmacytoid dendritic cells contribute to immune tolerance in ovarian cancer. Cancer Res. 2011;71:5423–34. [DOI] [PubMed] [Google Scholar]
- 8.Wertel I, Polak G, Bednarek W, Barczynski B, Rolinski J, Kotarski J. Dendritic cell subsets in the peritoneal fluid and peripheral blood of women suffering from ovarian cancer. Cytometry B Clin Cytom. 2008;74:251–8. [DOI] [PubMed] [Google Scholar]
- 9.Mitchell D, Chintala S, Dey M. Plasmacytoid dendritic cell in immunity and cancer. J Neuroimmunol. 2018;322:63–73. [DOI] [PubMed] [Google Scholar]
- 10.Makkouk A, Joshi VB, Wongrakpanich A, et al. Biodegradable microparticles loaded with doxorubicin and CpG ODN for in situ immunization against cancer. AAPS J. 2015;17:184–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krieg AM. CpG still rocks! Update on an accidental drug. Nucleic Acid Ther. 2012;22:77–89. [DOI] [PubMed] [Google Scholar]
- 12.Lemke-Miltner CD, Blackwell SE, Yin C, Krug AE, Morris AJ, Krieg AM, Weiner GJ. Antibody opsonization of a TLR9-agonist-containing virus-like particle enhances in situ immunization. J Immunol. 2020;204:1386–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Cesare M, Calcaterra C, Pratesi G, et al. Eradication of ovarian tumor xenografts by locoregional administration of targeted immunotherapy. Clin Cancer Res. 2008;14:5512–8. [DOI] [PubMed] [Google Scholar]
- 14.De Cesare M, Sfondrini L, Campiglio M, et al. Ascites regression and survival increase in mice bearing advanced-stage human ovarian carcinomas and repeatedly treated intraperitoneally with CpG-ODN. J Immunother. 2010;33:8–15. [DOI] [PubMed] [Google Scholar]
- 15.De Cesare M, Sfondrini L, Pennati M, et al. CpG-oligodeoxynucleotides exert remarkable antitumor activity against diffuse malignant peritoneal mesothelioma orthotopic xenografts. J Transl Med. 2016;14:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ray A, Dittel BN. Isolation of mouse peritoneal cavity cells. J Vis Exp. 2010;35:1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hemmi H, Takeuchi O, Kawai T, et al. A toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–5. [DOI] [PubMed] [Google Scholar]
- 18.Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol. 2015;15:471–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression: implications for anticancer therapy. Nat Rev Clin Oncol. 2019;16:356–71. [DOI] [PubMed] [Google Scholar]
- 20.Negus RP, Stamp GW, Hadley J, Balkwill FR. Quantitative assessment of the leukocyte infiltrate in ovarian cancer and its relationship to the expression of C-C chemokines. Am J Pathol. 1997;150:1723–34. [PMC free article] [PubMed] [Google Scholar]
- 21.Maghazachi AA, Al-Aoukaty A, Schall TJ. CC chemokines induce the generation of killer cells from CD56 + cells. Eur J Immunol. 1996;26:315–9. [DOI] [PubMed] [Google Scholar]
- 22.Menten P, Wuyts A, Van Damme J. Macrophage inflammatory protein-1. Cytokine Growth Factor Rev. 2002;13:455–81. [DOI] [PubMed] [Google Scholar]
- 23.Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–50. [DOI] [PubMed] [Google Scholar]
- 24.Yoshimura T, Robinson EA, Tanaka S, Appella E, Kuratsu J, Leonard EJ. Purification and amino acid analysis of two human glioma-derived monocyte chemoattractants. J Exp Med. 1989;169:1449–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–20. [DOI] [PubMed] [Google Scholar]
- 26.Ardolino M, Azimi CS, Iannello A, et al. Cytokine therapy reverses NK cell anergy in MHC-deficient tumors. J Clin Invest. 2014;124:4781–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Athie-Morales V, Smits HH, Cantrell DA, Hilkens CM. Sustained IL-12 signaling is required for Th1 development. J Immunol. 2004;172:61–9. [DOI] [PubMed] [Google Scholar]
- 28.Murphy KM, Ouyang W, Farrar JD, et al. Signaling and transcription in T helper development. Annu Rev Immunol. 2000;18:451–94. [DOI] [PubMed] [Google Scholar]
- 29.Ruffell B, Chang-Strachan D, Chan V, et al. Macrophage IL-10 blocks CD8 + T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. 2014;26:623–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okada M, Kitahara M, Kishimoto S, Matsuda T, Hirano T, Kishimoto T. IL-6/BSF-2 functions as a killer helper factor in the in vitro induction of cytotoxic T cells. J Immunol. 1988;141:1543–9. [PubMed] [Google Scholar]
- 31.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu Rev Immunol. 2010;28:445–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morano WF, Aggarwal A, Love P, Richard SD, Esquivel J, Bowne WB. Intraperitoneal immunotherapy: historical perspectives and modern therapy. Cancer Gene Ther. 2016;23:373–81. [DOI] [PubMed] [Google Scholar]
- 33.Heiss MM, Murawa P, Koralewski P, et al. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: results of a prospective randomized phase II/III trial. Int J Cancer. 2010;127:2209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hong H, Brown CE, Ostberg JR, et al. L1 Cell Adhesion molecule-specific chimeric antigen receptor-redirected human T cells exhibit specific and efficient antitumor activity against human ovarian cancer in mice. PLoS ONE. 2016;11:e0146885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma Z, Li W, Yoshiya S, et al. Augmentation of immune checkpoint cancer immunotherapy with IL18. Clin Cancer Res. 2016;22:2969–80. [DOI] [PubMed] [Google Scholar]
- 36.Strohlein MA, Heiss MM. The trifunctional antibody catumaxomab in treatment of malignant ascites and peritoneal carcinomatosis. Future Oncol. 2010;6:1387–94. [DOI] [PubMed] [Google Scholar]
- 37.Hofmann MA, Kors C, Audring H, Walden P, Sterry W, Trefzer U. Phase 1 evaluation of intralesionally injected TLR9-agonist PF-3512676 in patients with basal cell carcinoma or metastatic melanoma. J Immunother. 2008;31:520–7. [DOI] [PubMed] [Google Scholar]
- 38.Kim YH, Girardi M, Duvic M, et al. Phase I trial of a toll-like receptor 9 agonist, PF-3512676 (CPG 7909), in patients with treatment-refractory, cutaneous T-cell lymphoma. J Am Acad Dermatol. 2010;63:975–83. [DOI] [PubMed] [Google Scholar]
- 39.Li J, Song W, Czerwinski DK, et al. Lymphoma immunotherapy with CpG oligodeoxynucleotides requires TLR9 either in the host or in the tumor itself. J Immunol. 2007;179:2493–500. [DOI] [PubMed] [Google Scholar]
- 40.Molenkamp BG, Sluijter BJ, van Leeuwen PA, et al. Local administration of PF-3512676 CpG-B instigates tumor-specific CD8 + T-cell reactivity in melanoma patients. Clin Cancer Res. 2008;14:4532–42. [DOI] [PubMed] [Google Scholar]
- 41.Wang S, Campos J, Gallotta M, et al. Intratumoral injection of a CpG oligonucleotide reverts resistance to PD-1 blockade by expanding multifunctional CD8 + T cells. Proc Natl Acad Sci U S A. 2016;113:E7240–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Witzig TE, Wiseman GA, Maurer MJ, et al. A phase I trial of immunostimulatory CpG 7909 oligodeoxynucleotide and 90 yttrium ibritumomab tiuxetan radioimmunotherapy for relapsed B-cell non-Hodgkin lymphoma. Am J Hematol. 2013;88:589–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sommariva M, de Cesare M, Meini A, et al. High efficacy of CpG-ODN, cetuximab and cisplatin combination for very advanced ovarian xenograft tumors. J Transl Med. 2013;11:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Storni T, Ruedl C, Schwarz K, Schwendener RA, Renner WA, Bachmann MF. Nonmethylated CG motifs packaged into viruslike particles induce protective cytotoxic T cell responses in the absence of systemic side effects. J Immunol. 2004;172:1777–85. [DOI] [PubMed] [Google Scholar]
- 45.Schlecht G, Garcia S, Escriou N, Freitas AA, Leclerc C, Dadaglio G. Murine plasmacytoid dendritic cells induce effector/memory CD8 + T-cell responses in vivo after viral stimulation. Blood. 2004;104:1808–15. [DOI] [PubMed] [Google Scholar]
- 46.Krug A, Rothenfusser S, Hornung V, et al. Identification of CpG oligonucleotide sequences with high induction of IFN-alpha/beta in plasmacytoid dendritic cells. Eur J Immunol. 2001;31:2154–63. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.