

Abstract
An increasing number of studies have reported that bacterial DNA methylation has important functions beyond their roles in restriction-modification systems, including the ability of affecting clinically relevant phenotypes such as virulence, host colonization, sporulation, biofilm formation, among others. Although insightful, such studies have a largely ad hoc nature, and would benefit from a systematic strategy enabling a joint functional characterization of bacterial methylomes by the Microbiology community. In this Opinion, we propose that highly conserved DNA methyltransferases (MTases) represent a unique opportunity for bacterial epigenomic studies. These MTases are rather common in Bacteria, span various taxonomic scales, and are present in multiple human pathogens. Apart from well characterized core DNA MTases, like those from Vibrio cholera, Salmonella enterica, Clostridioides difficile or Streptococcus pyogenes, multiple highly conserved DNA MTases are also found in numerous human pathogens including those belonging to the genera Burkholderia and Acinetobacter. We discuss why and how these MTases can be prioritized to enable a community-wide, integrative approach for functional epigenomic studies. Ultimately, we discuss how some highly conserved DNA MTases may emerge as promising targets for the development of novel epigenetic inhibitors for biomedical applications.
Keywords: restriction-modification systems, persistent / core genes, methylome, virulence, antimicrobials
Introduction
The information content of DNA is not limited to that contained within the primary nucleotide sequence. Instead, significant meaning can also be conveyed through the epigenetic states of DNA, e.g., by chemical modification such as DNA methylation. In Bacteria, DNA methylation is typically associated with restriction-modification (R-M) systems (see Glossary), which operate as key moderators of the flow of genetic information between cells by horizontal gene transfer (HGT) [1, 2]. R-M systems typically encode a DNA methyltransferase (MTase) that modifies particular DNA sequences in function of the presence of target recognition sites, and a restriction endonuclease (REase) that cleaves them when they are unmethylated [3] (Box 1). Some DNA MTases, known as solitary or orphan, were also identified as apparently lacking a cognate REase [4]. DNA methylation performed either by R-M MTases or orphan MTases were properly discussed in a few seminal works [5–7].
Box 1.
Restriction-Modification (R-M) types:
The three classical types of R-M systems differ in their molecular structure, sequence recognition, cleavage position and cofactor requirements [102–105]. Type I systems are complex hetero-oligomers either comprising one DNA sequence specificity (S), two REase and two MTase subunits with restriction and modification activities, or two MTase and one S subunits with modification activity only. Type II systems encoded on separate genes are composed of one homodimeric or homotetrameric REase and one monomeric MTase, and in most cases are able to operate separately and independently from each other at least in vitro. Some Type II systems, particularly Types IIB, IIG, IIL, and some IIH (collectively termed IIC) encode both restriction and modification domains within the same protein. Type III systems are heterotrimers or heterotetramers of products of two genes, res and mod, involved in restriction and modification, respectively. Both subunits are required for restriction, whereas Mod is sufficient to produce a modification. Finally, Type IV ‘restriction systems’, as opposed to R-M systems, are composed of one or two REases that cleave modified recognition sites.
Core genes:
Genes common to all genomes in a phylogenetically coherent group. They should contain the essential genes particular to that group as well as some non-essential ones.
Essential genes:
Typically involved in basic cellular processes such as translation, transcription, and replication. The concept of essentiality is not an intrinsic property of a gene, but instead a function of genetic and environmental factors. Essential genes can be essential in one species but not another, or under a defined growth condition but not in others.
Persistent genes:
Conserved above a predefined cutoff threshold of bacterial genomes. Although somewhat arbitrary, such threshold should take into consideration certain criteria, such as phylogenetic relatedness between organisms, and gene organization within genomes. By definition, persistent genes include core genes.
The bacterial genome has three major forms of DNA methylation: N6-methyladenine (6mA), N4-methylcytosine (4mC), and 5-methylcytosine (5mC), with 6mA being the most prevalent form. While 5mC may be detected with bisulfite sequencing, 6mA and 4mC events have been challenging to map at the genome-wide scale [8], limiting the comprehensive study of bacterial epigenomes. The study of bacterial methylomes entered a new era in 2012 when a new technology called single-molecule real-time (SMRT) sequencing [9] enabled the detection of all three major forms of bacterial DNA methylation. Since then, >2,350 (as of 03/2020) bacterial and archaeal methylomes [10, 11] have been determined at a quasi-exponential pace. Propelled by SMRT sequencing, an increasing number of studies documented the involvement of DNA methylation in often critical aspects of cell biology. Some examples include gene expression changes affecting cell motility [11], sporulation [12], virulence [13, 14], and in providing structural support for bacterial survival during antibiotic stress [15].
Previous bacterial epigenome studies have a largely ad hoc nature in that most have performed methylome mapping in one or few strains of the same species, and less frequently, across multiple species. A systematic examination of MTases across a large number of strains in a single species was only determined in few occasions, and in an even lower number of studies were MTase mutants constructed for phenotypic and molecular characterization [11, 16–19]. Such studies are insightful as they provide a comprehensive snapshot of MTase diversity, and some have been indeed capable of linking individual MTases to specific functions in the cell. But they face major challenges. For example, it is usually difficult for one single study to obtain sufficiently deep mechanistic insight, or comprehensively uncover phenotypes impacted by the loss of an MTase. It is also conceptually challenging to integrate epigenomic information stemming from different studies dealing with MTases present in few strains. More importantly, there have been limited attempts to identify specific methylation sites and mechanisms, underlying the epigenetic regulation of genes linked to defined phenotypes. Due to these limitations, some fundamental questions still remain unanswered: What phenotypes (in a particular species) are impacted by DNA MTases? Which specific methylation sites play important regulatory roles? What are the underlying epigenetic mechanisms regulating cellular phenotypes by specific methylation events?
Among all the diversity of DNA MTases in Bacteria [10, 20], some are highly conserved at the species level or at higher taxonomic ranks. Examples of well characterized ones include the Escherichia coli Dam enzyme (methylating at 5’-GATC-3’) and the Caulobacter crescentus CcrM enzyme (methylating at 5’-GANTC-3’). Dam and CcrM homologs are widespread in γ- and α-Proteobacteria respectively [21]. Both are encoded by core genes [22, 23] (Box 1), and recognized as conditionally essential for the viability of several species [19, 24–26], typically via mutation or overexpression approaches coupled to gene expression profile analyses. We recently witnessed a surge of studies focusing on less known conserved MTases belonging to different R-M types and operating the three major forms of DNA methylation [12, 14, 27–29]. Despite multiple evidence suggesting that R-M genes are frequently exchanged between species [30–32], and evolve very quickly [33, 34], the above-mentioned examples illustrate how certain MTases may endure strong selective pressure for retention in genomes. Several possibilities may account for such retention, including the involvement in epigenetic regulation of functionally relevant genes [12, 27], the ability of certain selfish R-M systems to induce post-segregational killing [7], or in shaping gene flux and host genome composition [35]. Whether Dam, CcrM, and the few other recent examples are merely outliers, or actually representatives of a broader set of conserved MTases (and eventually full R-Ms), is currently not clear.
In this Opinion, we summarized the landscape of DNA MTase conservation in the Bacterial kingdom. We observed that MTase conservation is more common than previously portrayed, spanning multiple phylogenetic levels, and being present in multiple human pathogens. We then propose that prioritizing conserved MTases can facilitate community-wide efforts for integrating experimental and multiple –omics data (e.g., genomic, transcriptomic, epigenomic) to more effectively address the fundamental questions laid above. Ultimately, we discuss how some of these targets may emerge as promising targets for the development of novel epigenetic inhibitors.
Conserved DNA MTases are abundant in Bacteria
A total of 26,582 MTases are found in 5,568 complete bacterial genomes available in Genbank (considering only species with at least 10 complete genomes) (Figure 1a, Supplementary Tables 1–3, Supplementary Information). Type II MTases are present at the highest densities, in what is likely a consequence of Type II R-M systems’ ability to induce genetic addiction. Conversely, Types IIC and III are the least abundant. 52% of the species harbor persistent MTases (here defined as those conserved in at least 80% of each species’ genomes) (Box 1, Figure 1b, Supplementary Table 3, Supplementary Information). The frequency of persistent MTases varies widely among bacterial large phyla, and is unrelated with the density of total MTases (Figure 1a). For example, α-Proteobacteria harbors multiple persistent MTases, but show an overall low density of total MTases. On the other hand, phyla such as Fusobacteria and Chloroflexi are devoid of persistent MTases, but are rich in other MTases (Figure 1a). In 27% of the species, more than one persistent MTase is present (either belonging to the same or different Types) (Supplementary Tables 3, 4). 37% of the species harbor MTases consistently present across all genomes (core), the majority being of Type II (Figure 1c). These represent 8.5% from the total MTase dataset. The human obligate pathogen Neisseria gonorrhoeae stands out as the species harboring the most profuse arsenal of persistent / core MTases (n=10) spanning Types I and II / IIC. Since we have only included bacterial species with at least 10 complete genomes available at GenBank, we expect our estimate on the number and diversity of core / persistent MTases to increase in the future as more genomes get sequenced and novel MTases are found. On top of this, there is the possibility that small non-canonical MTases may have gone unnoticed, as recently pinpointed in a large-scale analysis in human microbiomes [36]. Certain core / persistent MTase genes may also undergo structural variations (e.g., at the level of the target recognition domain) capable of changing their recognition motif or rendering their products inactive in some genomes, while still being subtle enough to be classified in the same gene family. This is the case of, for example, the persistent Type II MTase from Mycobacterium tuberculosis recognizing CTGGAG. Hence, core and persistent MTases are abundant in Bacteria.
Figure 1.
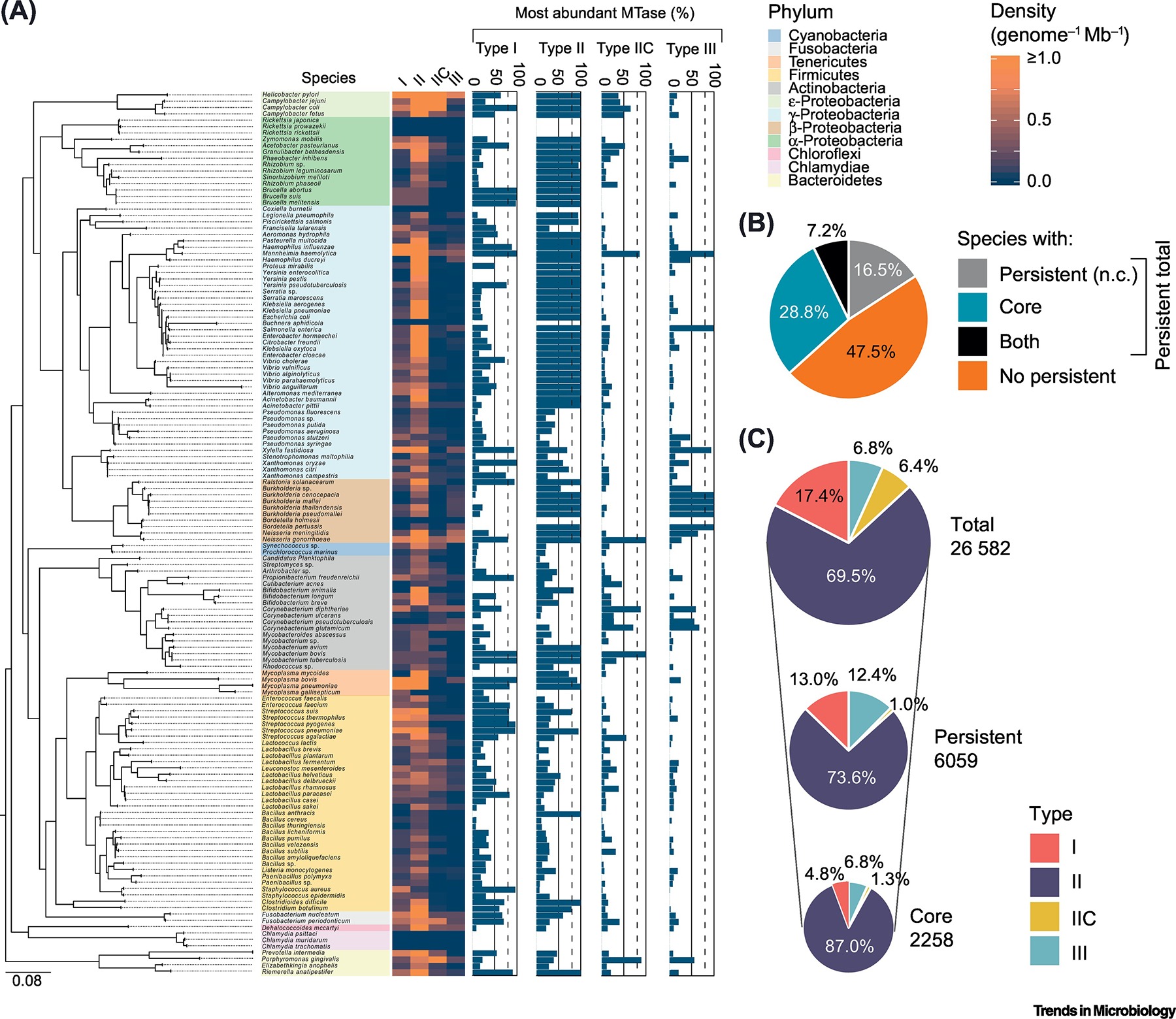
Summary of MTase conservation in bacterial genomes from Genbank. (a) Phylogenetic tree of the 139 bacterial species (colored by Phylum), for which at least 10 complete genomes were available at Genbank (corresponding to a total of 5,568 genomes). Heatmap corresponds to the density (per genome per Mb) of Types I, II, III MTases and Type IIC R-M systems for each species. Bar plots indicate the percentage of the most abundant MTase(s) found in each species, assuming as inclusion criteria a minimum of 80% similarity in amino-acid sequence and less than 20% difference in protein length. Stippled lines indicate a threshold of 80% above which an MTase can be considered persistent. 100% denotes a core gene. (b) Pie-chart summarizing the percentages of species analyzed containing either persistent non-core (n.c.) MTases, core MTases, both, or none. (c) Pie-charts showing the breakdown of total, persistent, and core MTases per Type.
Core and persistent DNA MTases differ substantially in their organization and sequence recognition
We next summarized the diversity of core and persistent MTases in terms of their organization (orphan versus part of an R-M system) and target sequence recognition (Supplementary Information). Across strains of the same species, MTases are found predominantly organized as part of complete R-M systems or as orphans, but less frequently as both (Figure 2). This suggests that for orphan MTases, loss of the cognate REase likely occurred early in the evolutionary history of these species. Alternatively, orphan MTases may have been acquired as such by HGT, and further kept under strong selective pressure [20]. The existence of multiple core and persistent complete R-M systems, suggests alternative roles such as gene expression regulation. For example, N. gonorrhoeae harbors a profuse arsenal of persistent / core complete systems (n=10) (Figure 2, Supplementary Table 4) belonging to Types I, II and IIC. Although it is not certain that all systems are active in all isolates, at least some of its Type II REases are known to be released in an active way during infection of host cells, and to enter the nucleus through nuclear pores, inducing double strand breaks in DNA during mitosis [37].
Figure 2.
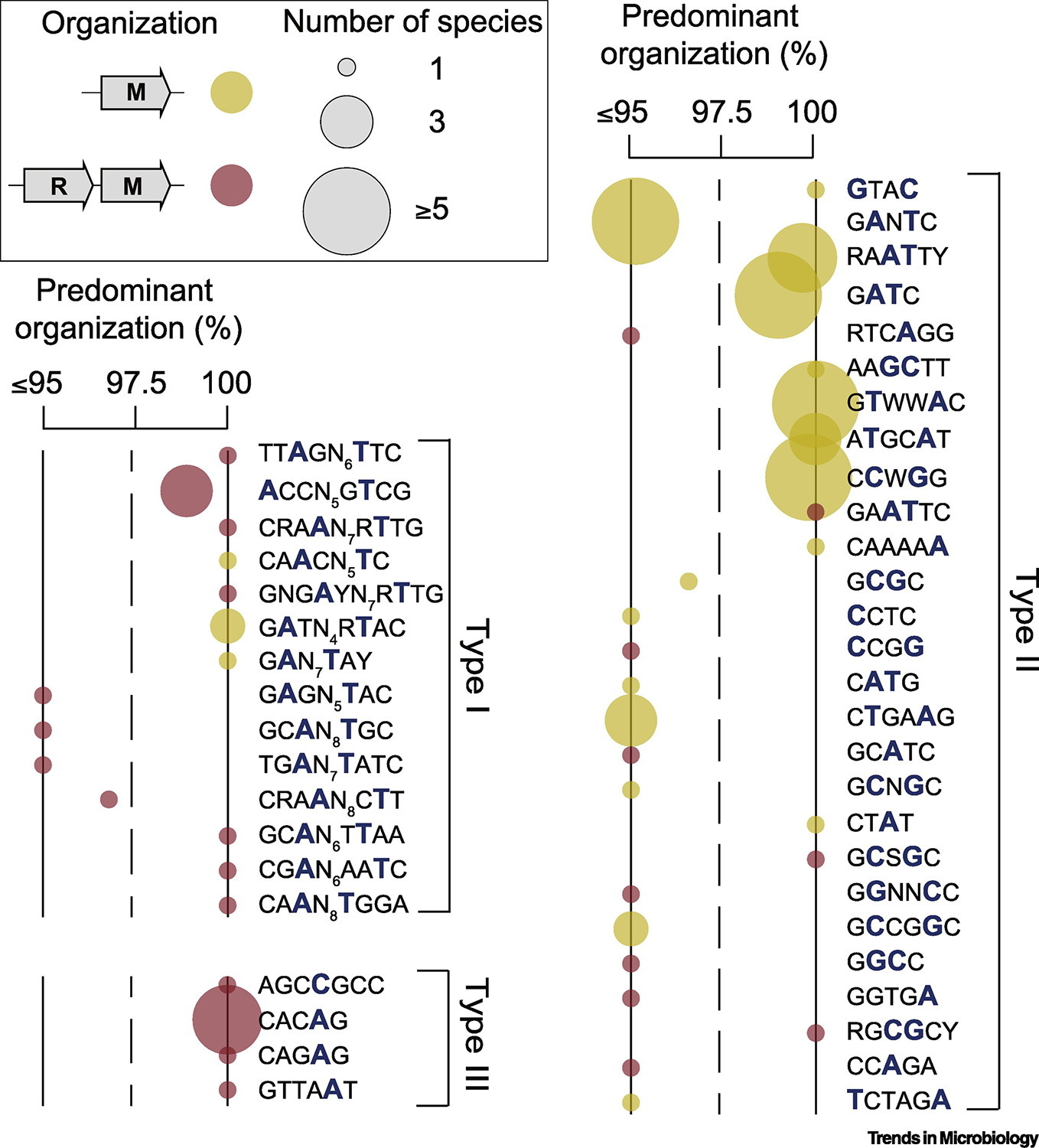
Summary of the organization and target recognition motifs of persistent MTases based on the REBASE database. Yellow circles represent solitary MTases, whereas red ones represent complete systems. 100% means that the MTase is always found either as solitary (without a cognate endonuclease) or as part of a complete R-M system. Values below 100% indicate that both organizations are present, being the most predominant highlighted. For example, Type II GTWWAC-recognizing MTases are exclusively solitary, whereas Type III CACAG-recognizing MTases are exclusively found within complete R-M systems. GATC-recognizing MTases are found as solitary in 98.9% of the species analyzed, and the remaining 1.1% in complete systems. Target recognition motifs shown are based on the REBASE database. Circle radius is proportional to the number of species in which the MTase is present
Persistent MTases are also very diverse in terms of sequence recognition (Figure 2, Supplementary Table 4). We observed a total of 48 different methylation motifs belonging to the three major R-M Types, among which 73% methylate at 6mA. These observations are expected to be conservative as they correspond solely to MTases whose recognition sequence has been confirmed by SMRT-seq [38] and some of these MTases recognize variable motifs. As expected, the MTases for which more functional studies have been published [39] (namely Dam, Dcm, CcrM) also correspond to those most widespread across a higher number of species (Figure 2). Hence, core and persistent MTases are diverse in terms of organization and target recognition sequence.
Core and persistent DNA MTases are found at multiple taxonomic scales
Genes can differ significantly in their taxonomic distributions, with more broadly conserved genes having ‘housekeeping’ functions and less conserved genes being responsible for the phenotypic differences observed between organisms. In this regard, persistent genes can be restricted to any taxonomic level (e.g.: domain-, family-, genus-, species- or strain-specific). Once persistent genes have been defined to identify a related group of organisms, the biological roles performed by these genes’ products can provide insights into functions and phenotypes that may be characteristic (and even critical) to those groups. One example, is that of Dam MTase, conserved in a large subset of γ-proteobacteria (Figure 3), including the clinically relevant genera Escherichia, Salmonella, Vibrio, and Yersinia. Its acquisition might have been the key evolutionary moment that created a new mechanism capable of DNA strand discrimination based on the hemi-methylated state of newly replicated DNA [40]. Such mechanism is critical for the regulation of multiple cellular processes. For example, during DNA mismatch repair in E. coli, the MutH protein recognizes hemi-methylated DNA and cuts the non-methylated daughter strand, ensuring that the methylated parental strand will be used as template for repair-associated DNA synthesis [41]. In addition, hemi-methylated GATC sites can activate gene expression upon passage of the replication fork [42, 43], and coordinate the initiation of replication within cell cycle in E. coli [44].
Figure 3.
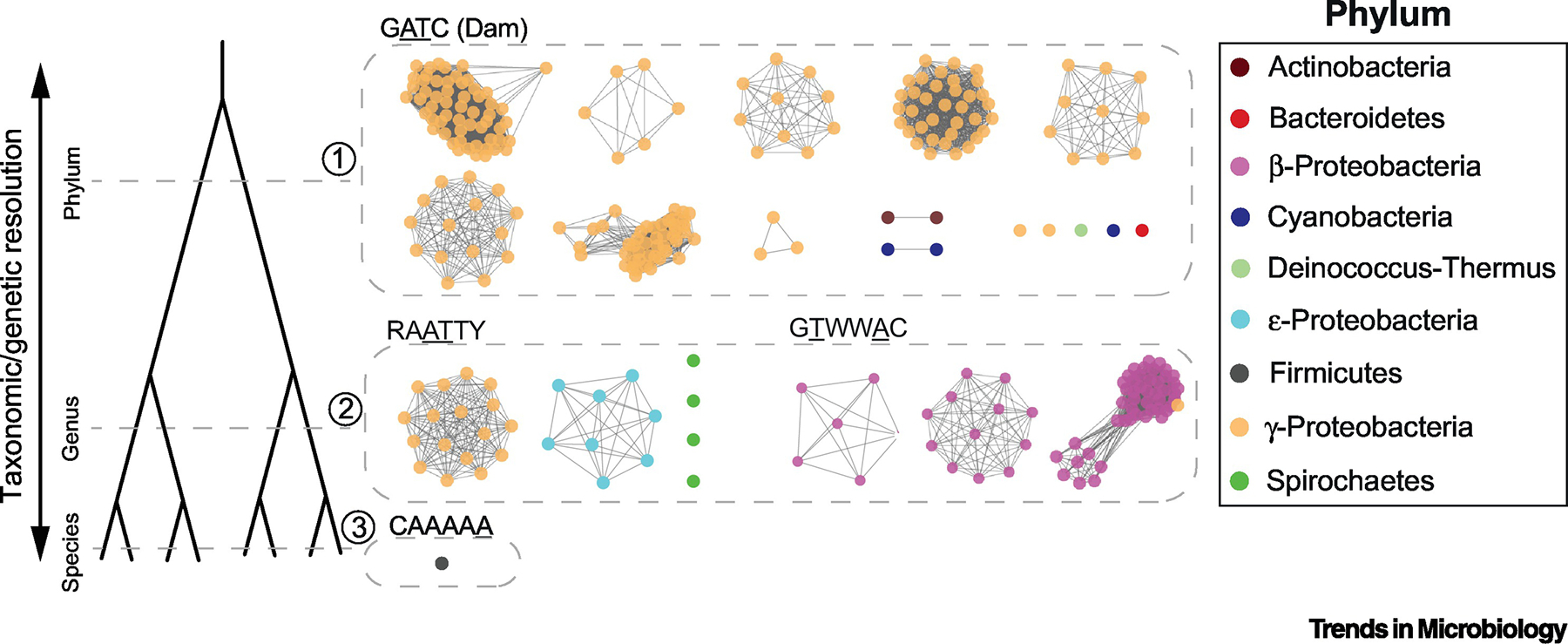
Illustrative examples of sequence similarity networks of persistent MTases conserved at different taxonomic resolutions (Supplementary Table 5). Each node represents one protein. To avoid redundancy and improve visualization, only one genome per species is shown (typically the reference/representative genome). Edges correspond to pairwise protein sequence identity >60%. Node colors correspond to different phyla.
Closer to the genus level conservation, we can highlight as illustrative examples, those of RAATTY and GTWWAC-recognizing MTases. The former are pervasive in Acinetobacteriales and Campylobacteriales, while the latter are often found in Burkholderiales. Information on the functional relevance of these MTases is virtually inexistent, but they are expected to play specific roles that help maintain the identity of these genera. In line with this hypothesis, is the recent observation that RAATTY methylation is required for efficient transformation in Campylobacter jejuni [45]. Genes mainly preserved at the species- and strain-level are also of interest, as they may be involved in exclusive ecological adaptations to particular niches. One example is that of CamA, a 6mA persistent MTase recognizing CAAAAA involved in the sporulation and biofilm formation in C. difficile [12] (Figure 3), and also the most species-specific persistent MTase currently known.
The acquisition of a new functional R-M system by a bacterial clone may significantly reduce its ability of engaging in genetic exchanges with conspecific bacteria [46]. This may help carving preferential routes of DNA exchange between its offspring (which inherited this R-M system), favor the maintenance of cohesive population structures, and eventually give rise to a new lineage in the population [47]. Specific lineages of important pathogens that have recently changed their R-M repertoires and show higher sexual isolation include Burkholderia pseudomallei, E. coli, Neisseria meningitidis, Staphylococcus aureus, and Streptococcus pneumoniae [48–51]. A Type I R-M system for example, decreased transfer to and from a major methicillin-resistant S. aureus lineage [52]. Hence, core and persistent MTases are found at multiple taxonomic scales, where they are expected to play roles that help shape phylogenetic structure.
Core and persistent MTases as an opportunity for integrative studies of bacterial epigenomes
Persistent genes, as orthologs shared by all (or almost all) members of an evolutionarily coherent group, likely reflect the important functions positively selected over time [53, 54]. They are also more likely to facilitate standardization and extrapolation from well-studied bacterial strains to newly sequenced ones using systems-level approaches, rendering possible direct comparisons of findings from different laboratories (Figure 4). In this regard, core and persistent MTases appear as particularly attractive targets to be prioritized in bacterial epigenomic studies, as they allow the integration and analysis of multi-dimensional omics data to retrieve meaningful information from bacterial epigenomes, and to ultimately address the questions laid down in the introductory section.
Figure 4.
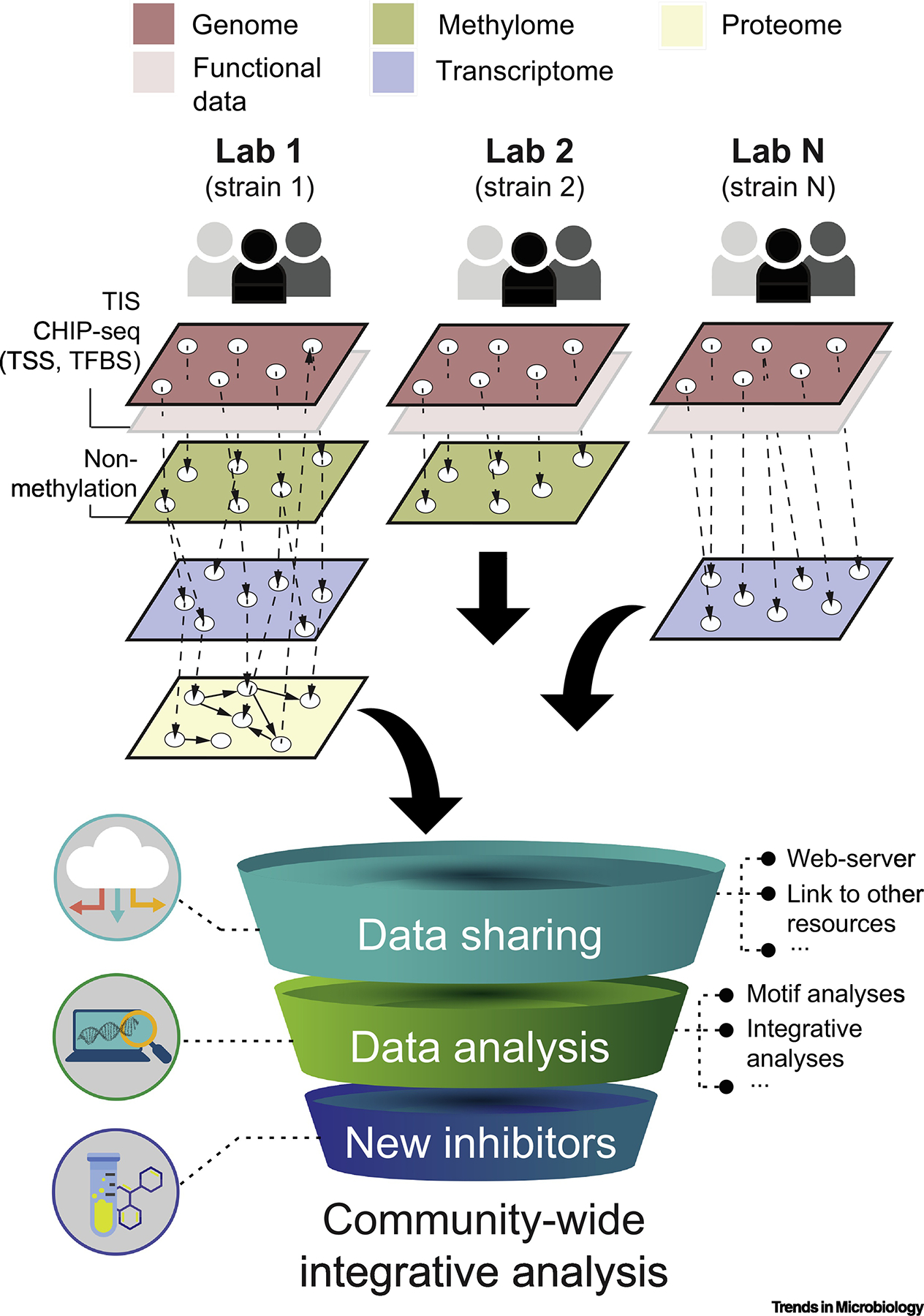
Overview of a large-scale community-wide integrative approach for bacterial methylome analyses. Core and persistent MTases can be prioritized to build a trans-omic network across multiple laboratories merging multiple functional data gathered at different experimental conditions. The latter may build upon MTase mutants generated by Transposon Insertion mutagenesis coupled with deep Sequencing (TIS), site-directed mutagenesis of methylation sites, genome-wide profiling of DNA binding proteins (ChIP-seq), transcription start site (TSS) mapping, and identification of methylation-sensitive transcription factors. Multiple layers of omics data may ultimately be commonly shared, linked to other resources (e.g., REBASE), allow for an in-depth analysis of, for example, methylation motif conservation, phase-variable DNA methyltransferases, and accelerate the research of novel epigenomic inhibitors.
1. How to identify phenotypes that are impacted by DNA MTases?
Our understanding of the genetic mechanisms that underlie biological processes has relied extensively on loss-of-function approaches that reduce or ablate gene function. Through the analysis of the phenotypes caused by such perturbations, one can elucidate the wild-type function of a given gene. For example, non-targeted DNA mutagenesis approaches such as large scale random Transposon Insertion mutagenesis coupled with deep Sequencing (TIS) has become a powerful tool to simultaneously assess the essentiality of genes under defined experimental conditions and to rapidly connect genotype to phenotype in a wide range of bacteria [55]. Several variants of TIS have been independently developed [56–59], and applied to a variety of bacteria, allowing to assess the role of certain DNA MTases as controllers of critical cellular processes [60] and / or as conditionally essential genes [61–63]. Relevant functional information has also been obtained by targeted mutagenesis or overexpression of DNA MTases [11, 16–19]. A comprehensive global transcriptome and functional profiling by RNA-seq offers the opportunity to further dissect the range of differentially expressed genes in a methylation-free strain. Integrative analyses that incorporate RNA-seq data and other omics experiments are also becoming prevalent. For example, pairwise integration of RNA-seq and DNA methylation is typically performed by the analysis of correlation between differentially expressed genes and methylation patterns (e.g. using linear models, logistic regression, or empirical Bayes models), or alternatively, through the identification of sets of genes that have coordinated differential expression and methylation [64]. For an in-depth understanding of the complex relationships between multiple omics sets, tools such as MultiDataSet [65], CNAMet [66], and SuperExactTest [67] can be used. The latter, for example, has been recently used to aid in the identification of novel functional roles of a bacterial MTase [12].
2. How to identify specific methylation sites playing important regulatory roles?
Another outstanding question concerns the different regulatory roles played by distinctive subsets of methylation sites in a genome. Two approaches, based on intra- and inter-genome analyses, can be considered. The former, stems from the observation of a positive correlation between the number of methylation sites in a gene and the fold change of expression between wild type and MTase mutants [11, 60], suggesting that epigenetic regulation of expression may be driven by multiple methylation sites particularly in promoter regions. In this case, genomic regions with significant high density of methylation sites should be targeted for site-directed mutagenesis or genetic editing in order to gauge the impact of each methylation site mutation [27, 68–70]. A second orthogonal approach inspired on phylogenetic footprinting, deduces functional relevance based on the degree of conservation of orthologous methylation motifs across multiple genomes. By comparing multiple methylomes associated to a persistent MTase, one can distinguish between strictly conserved orthologous target methylation sites, and variable ones (e.g., harboring SNPs or indels). While the former are likely to preferentially play housekeeping roles, at least some of the latter are expected to serve as ON / OFF regulators through phase variation. An additional benefit of an orthogonal approach conducted across a substantial number of same-species genomes, is to gain sufficient statistical power to perform a systematic interrogation of non-methylated motifs sites. Such approach has recently allowed for a more systematic detection and analysis of both highly conserved and non-methylated sites in methylomes associated with persistent MTases [12, 71].
3. How to identify epigenetic mechanisms regulating cellular phenotypes by methylation?
Finally, we are left with the question of the mechanisms of epigenetic regulation. Here, more comprehensive studies will be necessary to fully characterize the precise mechanisms by which DNA methylation modulates gene expression and alters bacterial phenotypes. Such studies would benefit from the integration of methylome information with other assays, such as high-confidence genome-wide transcriptional landscape inference and transcription start site calling [72, 73], or mapping of transcription factor binding sites (TFBSs). Our understanding of the latter for example, has been mainly achieved by means of chromatin immunoprecipitation (ChIP) [74] assays eventually coupled to next-generation sequencing (ChIP-seq) [75]. The increasingly growing number of available bacterial epigenomes, has not only spurred a surge in comparative epigenomic studies, but also calls for additional integration with fine-resolution TFBS maps, which in Bacteria is still limited to a few species, namely E. coli [76, 77], Bacillus subtilis [78], and M. tuberculosis [79]. While an alternative strategy would be to use comparative genomics across a large genomic dataset to identify putative TFBSs [12], the generation of additional CHIP-seq data would provide valuable insight and stimulate sharing across laboratories.
Overlaying comprehensive TFBS and methylation maps becomes critical for elucidating complex transcriptional networks, and in few cases, has allowed characterizing multiple ON / OFF methylation-dependent phase variation systems [80–82]. The variable expression of MTases via, for example, slipped-strand mispairing of simple sequence repeats (SSRs), may lead to genome-wide methylation changes, and to altered expression of multiple genes (commonly termed phasevarions) [83]. In an appraisal of the potential for phase-variation in bacterial methyltransferases, two recent studies revealed the presence of SSRs in as much as 2 and 17.4% of Type I hsdM and Type III mod genes respectively [84, 85]. Such type of systematic studies, coupled with information provided by long-read sequencing technologies, will likely set the stage for further large-scale analyses of whole bacterial phasomes and development of controllable toggle switches.
Another interesting point would be to test the hypothesis that the thermodynamic effect of DNA methylation induces conformational changes to a bacterial chromosome, increasing gene accessibility to the transcriptional machinery [86, 87]. Generation of methylation-induced non-B topologies [86, 88], is likely to take place at higher methylation densities [89], and should provide key insight on how structural changes can alter the repertoire of genes exposed to the cellular transcriptional machinery. Techniques such as circular dichroism and chromatin conformation capture (e.g.: Hi-C), can be used to elucidate the effects of bacterial DNA methylation on DNA conformation and, consequently, on gene expression [90, 91]. Additionally, it would be worth testing the extent to which non-canonical (non-B) DNA conformations contribute to the occurrence of non-methylated sites, particularly for those cases that cannot be explained by protein competitive binding.
Hence, all the three above-mentioned questions would strongly benefit from a community-wide analysis of core and persistent MTases.
Concluding remarks and future perspectives
In this Opinion we propose that core and persistent DNA MTases should be prioritized in community-wide integrative studies to better understand bacterial epigenomes as well as the drivers behind MTase conservation. To illustrate this, we provided a comprehensive summary of the MTase conservation landscape in Bacteria, and highlight a catalog of 145 core and persistent MTases across 72 unique species, as well as a framework to guide future methylome analyses. These core and persistent MTases include not only well characterized ones, but also multiple previously unknown ones in human and animal pathogens. These observations open a new window to more effectively study the basic science and translational aspects of epigenetic regulation in bacteria and call for a community-wide integrative effort using a data and knowledge sharing strategy such as the one we outlined in this Opinion.
Due to their indispensability in bacteria, essential MTases (which are often core) are potential targets for the development of epigenetic inhibitors capable of, for example, enhancing the therapeutic activity of antimicrobials. For instance, Dam inhibition reportedly weakens bacterial pathogenicity in vivo, as GATC methylation controls virulence gene expression in various organisms [92–96]. GATC methylation was also found to play a role in drug potentiation, by curbing the therapeutic activity of the β-lactam and quinolone classes of antibiotics [15]. Indeed, Dam represents an attractive target for epigenetic inhibition of the multiple biological processes it regulates (e.g., virulence), as it lacks mammalian homologs while being conserved in several enteric pathogens [97–99]. Unlike Dam, CcrM has been found to be essential for viability in multiple bacteria [25, 100, 101], thus raising the possibility that inhibitors of methylation may be bactericidal in some cases. Although very promising, Dam, CcrM and other similar MTases are prevalent across multiple bacterial species. From the point of view of the development of more targeted epigenetic inhibitors, other core / persistent MTases specific to only one of few species, may hold greater interest. One example is that of the CAAAAA MTase of C. difficile, involved in the sporulation and biofilm formation in C. difficile [12].
We should emphasize that by proposing the prioritization of core and persistent MTases in methylome studies, we are by no means devaluing research focusing on ad hoc MTases. Such studies should be encouraged as they provide important contributes towards the understanding of MTase diversity and their specific roles in, for example, the emergence / maintenance of genetic cohesion of particularly virulent lineages [48–51], and genetic regulation in their natural (i.e. non-experimentally perturbed) environment. Under such circumstances, recently acquired MTases may also represent good candidates for inhibitor development.
We anticipate that in the next few years, advances on existing and forthcoming long-read sequencing technologies, concurrently with additional progress in the understanding of multiple functional roles of core / persistent MTases, will offer unprecedent opportunities for achieving a more complete snapshot of bacterial methylomes, especially in human pathogens. These current and future advances make the present times an exciting period for studying and harnessing the bacteria epigenomics for medical and clinical impact.
Supplementary Material
Outstanding questions.
To what extent additional DNA chemical modifications (beyond 6mA, 5mC and 4mC) play regulatory roles in bacteria?
How to identify the functions of persistent MTases?
What targets of these MTases have a regulatory nature?
How to disentangle between different epigenetic pathways?
Highlights.
DNA methylation is the epigenetic mark most commonly found throughout the living world. In Bacteria, it is responsible for a variety of functional roles, including defense against foreign DNA, regulation of chromosome replication and segregation, mismatch repair, control of virulence gene expression, among others.
DNA methyltransferases (MTases) are responsible for transferring a methyl group from an S-adenosyl-L-methionine (AdoMet) donor to DNA. Dam, Dcm, and CcrM are examples of bacterial DNA MTases that have been comprehensively characterized for their roles in gene regulation.
Here we summarized the landscape of DNA MTase conservation in Bacteria and observed that MTase conservation is more common than previously portrayed, spanning several phylogenetic levels, and being present in multiple human and animal pathogens. Information on the functional relevance of these MTases is virtually inexistent, but they are expected to play key functional roles.
We also discuss why and how these MTases can be prioritized to enable a community-wide, integrative approach for functional epigenomic studies. Ultimately, we discuss how some highly conserved DNA MTases may emerge as promising targets for the development of novel epigenetic inhibitors for biomedical applications.
Acknowledgements
We acknowledge Eduardo P.C. Rocha (Institut Pasteur, Paris, France), and Mi Ni, Yangmei Li from Fang lab for critical reading and for providing helpful comments / suggestions. We would also like to acknowledge the anonymous reviewers for their constructive comments. The work was funded by R01 GM114472 (G.F.) and R01 GM128955 (G.F.) from the National Institutes of Health. G.F. is an Irma T. Hirschl / Monique Weill-Caulier Trust Research Scholar. This work was also supported in part through the computational resources and staff expertise provided by the Department of Scientific Computing at the Icahn School of Medicine at Mount Sinai.
Glossary
- DNA methyltransferase
Family of enzymes that catalyze the transfer of a methyl group from an S-adenosyl-L-methionine (AdoMet) donor to DNA
- Epigenome
Complete record of all chemical modifications to DNA. Together with the epitranscriptome (chemical modifications of RNA) and epiproteome (chemical modifications of proteins), makes up the epi-ome
- Methylome
Complete record of all methyl modifications to either DNA, RNA, or proteins in a particular cell or organism
- Restriction-Modification systems
Almost ubiquitous in Prokaryotes, these systems consist of a DNA methyltransferase that methylates a specific target sequence in the host genome, and a cognate restriction endonuclease that cleaves unmethylated or inappropriately methylated targets from exogenous DNA. They are thus typically regarded as innate defense systems, and, depending on Type, as molecular parasites
- Single Molecule Real Time (SMRT) sequencing
Third generation long-read sequencing-by-synthesis technology, based on the real-time imaging of fluorescently tagged nucleotides as they are synthesized along individual DNA template molecules. The duration between consecutive pulses of light directly reflects the DNA polymerase kinetics, including the impact caused by DNA modification events
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Financial Interests
No competing financial interests.
References
- 1.Thomas CM and Nielsen KM (2005) Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat. Rev. Microbiol 3 (9), 711–21. [DOI] [PubMed] [Google Scholar]
- 2.Labrie SJ et al. (2010) Bacteriophage resistance mechanisms. Nat. Rev. Microbiol 8 (5), 317–27. [DOI] [PubMed] [Google Scholar]
- 3.Mruk I and Kobayashi I (2014) To be or not to be: regulation of restriction-modification systems and other toxin-antitoxin systems. Nucleic Acids Res 42 (1), 70–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murphy J et al. (2013) Bacteriophage orphan DNA methyltransferases: insights from their bacterial origin, function, and occurrence. Appl. Environ. Microbiol 79 (24), 7547–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rocha EP et al. (2001) Evolutionary role of restriction/modification systems as revealed by comparative genome analysis. Genome Res 11 (6), 946–58. [DOI] [PubMed] [Google Scholar]
- 6.Pingoud A et al. (2005) Type II restriction endonucleases: structure and mechanism. Cell Mol. Life. Sci 62 (6), 685–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobayashi I (2001) Behavior of restriction-modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res 29 (18), 3742–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beaulaurier J et al. (2019) Deciphering bacterial epigenomes using modern sequencing technologies. Nat. Rev. Genet 20 (3), 157–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flusberg BA et al. (2010) Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 7 (6), 461–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blow MJ et al. (2016) The epigenomic landscape of Prokaryotes. PLoS Genet 12 (2), e1005854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fang G et al. (2012) Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nat. Biotechnol 30 (12), 1232–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oliveira PH et al. (2020) Epigenomic characterization of Clostridioides difficile finds a conserved DNA methyltransferase that mediates sporulation and pathogenesis. Nat. Microbiol 5 (1), 166–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar S et al. (2018) N4-cytosine DNA methylation regulates transcription and pathogenesis in Helicobacter pylori. Nucleic Acids Res 46 (7), 3429–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nye TM et al. (2019) DNA methylation from a Type I restriction modification system influences gene expression and virulence in Streptococcus pyogenes. PLoS Pathog 15 (6), e1007841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cohen NR et al. (2016) A role for the bacterial GATC methylome in antibiotic stress survival. Nat. Genet 48 (5), 581–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kahramanoglou C et al. (2012) Genomics of DNA cytosine methylation in Escherichia coli reveals its role in stationary phase transcription. Nat. Commun 3, 886. [DOI] [PubMed] [Google Scholar]
- 17.Kwiatek A et al. (2015) Type III methyltransferase M.NgoAX from Neisseria gonorrhoeae FA1090 regulates biofilm formation and interactions with human cells. Front. Microbiol 6, 1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stephenson SA and Brown PD (2016) Epigenetic Influence of Dam methylation on gene expression and attachment in uropathogenic Escherichia coli. Front. Public Health 4, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Payelleville A et al. (2017) DNA adenine methyltransferase (Dam) overexpression impairs Photorhabdus luminescens motility and virulence. Front. Microbiol 8, 1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oliveira PH et al. (2014) The interplay of restriction-modification systems with mobile genetic elements and their prokaryotic hosts. Nucleic Acids Res 42 (16), 10618–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mouammine A and Collier J (2018) The impact of DNA methylation in Alphaproteobacteria. Mol Microbiol 110 (1), 1–10. [DOI] [PubMed] [Google Scholar]
- 22.Payelleville A et al. (2018) The complete methylome of an entomopathogenic bacterium reveals the existence of loci with unmethylated adenines. Sci. Rep 8 (1), 12091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Christen B et al. (2011) The essential genome of a bacterium. Mol. Syst. Biol 7, 528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Julio SM et al. (2001) DNA adenine methylase is essential for viability and plays a role in the pathogenesis of Yersinia pseudotuberculosis and Vibrio cholerae. Infect. Immun 69 (12), 7610–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stephens C et al. (1996) A cell cycle-regulated bacterial DNA methyltransferase is essential for viability. Proc. Natl. Acad. Sci. USA 93 (3), 1210–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gonzalez D et al. (2014) The functions of DNA methylation by CcrM in Caulobacter crescentus: a global approach. Nucleic Acids Res 42 (6), 3720–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Estibariz I et al. (2019) The core genome m5C methyltransferase JHP1050 (M.Hpy99III) plays an important role in orchestrating gene expression in Helicobacter pylori. Nucleic Acids Res 47 (5), 2336–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gartner K et al. (2019) Cytosine N4-methylation via M.Ssp6803II is involved in the regulation of transcription, fine-tuning of DNA replication and DNA repair in the Cyanobacterium Synechocystis sp. PCC 6803. Front. Microbiol 10, 1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hagemann M et al. (2018) Identification of the DNA methyltransferases establishing the methylome of the cyanobacterium Synechocystis sp. PCC 6803. DNA Res 25 (4), 343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kita K et al. (1999) Evidence of horizontal transfer of the EcoO109I restriction-modification gene to Escherichia coli chromosomal DNA. J. Bacteriol 181 (21), 6822–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kobayashi I et al. (1999) Shaping the genome--restriction-modification systems as mobile genetic elements. Curr. Opin. Genet. Dev 9 (6), 649–56. [DOI] [PubMed] [Google Scholar]
- 32.Rocha EP et al. (1999) Analysis of long repeats in bacterial genomes reveals alternative evolutionary mechanisms in Bacillus subtilis and other competent prokaryotes. Mol. Biol. Evol 16 (9), 1219–30. [DOI] [PubMed] [Google Scholar]
- 33.Lauster R (1989) Evolution of type II DNA methyltransferases. A gene duplication model. J. Mol. Biol 206 (2), 313–21. [DOI] [PubMed] [Google Scholar]
- 34.Jeltsch A and Pingoud A (1996) Horizontal gene transfer contributes to the wide distribution and evolution of Type II restriction-modification systems. J. Mol. Evol 42 (2), 91–6. [DOI] [PubMed] [Google Scholar]
- 35.Seshasayee AS et al. (2012) Context-dependent conservation of DNA methyltransferases in bacteria. Nucleic Acids Res 40 (15), 7066–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sberro H et al. (2019) Large-scale analyses of human microbiomes reveal thousands of small, novel genes. Cell 178 (5), 1245–1259 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weyler L et al. (2014) Restriction endonucleases from invasive Neisseria gonorrhoeae cause double-strand breaks and distort mitosis in epithelial cells during infection. PLoS One 9 (12), e114208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roberts RJ et al. (2015) REBASE--a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res 43, D298–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sanchez-Romero MA and Casadesus J (2020) The bacterial epigenome. Nat. Rev. Microbiol 18 (1), 7–20. [DOI] [PubMed] [Google Scholar]
- 40.Putnam CD (2016) Evolution of the methyl directed mismatch repair system in Escherichia coli. DNA Repair 38, 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kunkel TA and Erie DA (2005) DNA mismatch repair. Annu. Rev. Biochem 74, 681–710. [DOI] [PubMed] [Google Scholar]
- 42.Roberts D et al. (1985) IS10 transposition is regulated by DNA adenine methylation. Cell 43 (1), 117–30. [DOI] [PubMed] [Google Scholar]
- 43.Camacho EM and Casadesus J (2005) Regulation of traJ transcription in the Salmonella virulence plasmid by strand-specific DNA adenine hemimethylation. Mol. Microbiol 57 (6), 1700–18. [DOI] [PubMed] [Google Scholar]
- 44.Slater S et al. (1995) E. coli SeqA protein binds oriC in two different methyl-modulated reactions appropriate to its roles in DNA replication initiation and origin sequestration. Cell 82 (6), 927–36. [DOI] [PubMed] [Google Scholar]
- 45.Beauchamp JM et al. (2017) Methylation-dependent DNA discrimination in natural transformation of Campylobacter jejuni. Proc. Natl. Acad. Sci. USA 114 (38), E8053–E8061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jeltsch A (2003) Maintenance of species identity and controlling speciation of bacteria: a new function for restriction/modification systems? Gene 317 (1–2), 13–6. [DOI] [PubMed] [Google Scholar]
- 47.Oliveira PH et al. (2016) Regulation of genetic flux between bacteria by restriction-modification systems. Proc. Natl. Acad. Sci. USA 113 (20), 5658–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Budroni S et al. (2011) Neisseria meningitidis is structured in clades associated with restriction modification systems that modulate homologous recombination. Proc. Natl. Acad. Sci. USA 108 (11), 4494–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Croucher NJ et al. (2014) Diversification of bacterial genome content through distinct mechanisms over different timescales. Nat. Commun 5, 5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nandi T et al. (2015) Burkholderia pseudomallei sequencing identifies genomic clades with distinct recombination, accessory, and epigenetic profiles. Genome Res 25 (1), 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Forde BM et al. (2015) Lineage-specific methyltransferases define the methylome of the globally disseminated Escherichia coli ST131 clone. MBio 6 (6), e01602–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roberts GA et al. (2013) Impact of target site distribution for Type I restriction enzymes on the evolution of methicillin-resistant Staphylococcus aureus (MRSA) populations. Nucleic Acids Res 41 (15), 7472–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maddamsetti R et al. (2017) Core genes evolve rapidly in the long-term evolution experiment with Escherichia coli. Genome Biol. Evol 9 (4), 1072–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oliveira PH et al. (2017) The chromosomal organization of horizontal gene transfer in bacteria. Nat Commun 8 (1), 841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chao MC et al. (2016) The design and analysis of transposon insertion sequencing experiments. Nat. Rev. Microbiol 14 (2), 119–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Opijnen T et al. (2009) Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat Methods 6 (10), 767–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goodman AL et al. (2009) Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe 6 (3), 279–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gawronski JD et al. (2009) Tracking insertion mutants within libraries by deep sequencing and a genome-wide screen for Haemophilus genes required in the lung. Proc. Natl. Acad. Sci. USA 106 (38), 16422–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Langridge GC et al. (2009) Simultaneous assay of every Salmonella typhi gene using one million transposon mutants. Genome Res 19 (12), 2308–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chao MC et al. (2015) A cytosine methytransferase modulates the cell envelope stress response in the Cholera pathogen. PLoS Genet 11 (12), e1005739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sternon JF et al. (2018) Transposon sequencing of Brucella abortus uncovers essential genes for growth in vitro and inside macrophages. Infect. Immun 86 (8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Phan MD et al. (2013) The serum resistome of a globally disseminated multidrug resistant uropathogenic Escherichia coli clone. PLoS Genet 9 (10), e1003834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dembek M et al. (2015) High-throughput analysis of gene essentiality and sporulation in Clostridium difficile. MBio 6 (2), e02383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jiao Y et al. (2014) A systems-level integrative framework for genome-wide DNA methylation and gene expression data identifies differential gene expression modules under epigenetic control. Bioinformatics 30 (16), 2360–6. [DOI] [PubMed] [Google Scholar]
- 65.Hernandez-Ferrer C et al. (2017) MultiDataSet: an R package for encapsulating multiple data sets with application to omic data integration. BMC Bioinformatics 18 (1), 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Louhimo R and Hautaniemi S (2011) CNAmet: an R package for integrating copy number, methylation and expression data. Bioinformatics 27 (6), 887–8. [DOI] [PubMed] [Google Scholar]
- 67.Wang M et al. (2015) Efficient test and visualization of multi-set intersections. Sci. Rep 5, 16923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van der Woude M et al. (1996) Epigenetic phase variation of the pap operon in Escherichia coli. Trends Microbiol 4 (1), 5–9. [DOI] [PubMed] [Google Scholar]
- 69.Wallecha A et al. (2002) Dam- and OxyR-dependent phase variation of agn43: essential elements and evidence for a new role of DNA methylation. J. Bacteriol 184 (12), 3338–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lim HN and van Oudenaarden A (2007) A multistep epigenetic switch enables the stable inheritance of DNA methylation states. Nat. Genet 39 (2), 269–75. [DOI] [PubMed] [Google Scholar]
- 71.Erill I et al. (2017) Comparative analysis of Ralstonia solanacearum methylomes. Front. Plant Sci 8, 504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mirauta B et al. (2014) Parseq: reconstruction of microbial transcription landscape from RNASeq read counts using state-space models. Bioinformatics 30 (10), 1409–16. [DOI] [PubMed] [Google Scholar]
- 73.Jorjani H and Zavolan M (2014) TSSer: an automated method to identify transcription start sites in prokaryotic genomes from differential RNA sequencing data. Bioinformatics 30 (7), 971–4. [DOI] [PubMed] [Google Scholar]
- 74.Solomon MJ et al. (1988) Mapping protein-DNA interactions in vivo with formaldehyde: evidence that histone H4 is retained on a highly transcribed gene. Cell 53 (6), 937–47. [DOI] [PubMed] [Google Scholar]
- 75.Mikkelsen TS et al. (2007) Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448 (7153), 553–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gama-Castro S et al. (2016) RegulonDB version 9.0: high-level integration of gene regulation, coexpression, motif clustering and beyond. Nucleic Acids Res 44 (D1), D133–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ishihama A et al. (2016) Transcription profile of Escherichia coli: genomic SELEX search for regulatory targets of transcription factors. Nucleic Acids Res 44 (5), 2058–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sierro N et al. (2008) DBTBS: a database of transcriptional regulation in Bacillus subtilis containing upstream intergenic conservation information. Nucleic Acids Res 36, D93–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Minch KJ et al. (2015) The DNA-binding network of Mycobacterium tuberculosis. Nat. Commun 6, 5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Casadesus J and Low DA (2013) Programmed heterogeneity: epigenetic mechanisms in bacteria. J. Biol. Chem 288 (20), 13929–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.De Ste Croix M et al. (2017) Phase-variable methylation and epigenetic regulation by type I restriction-modification systems. FEMS Microbiol Rev 41 (Supp_1), S3–S15. [DOI] [PubMed] [Google Scholar]
- 82.Atack JM et al. (2018) Phasevarions of Bacterial Pathogens: Methylomics Sheds New Light on Old Enemies. Trends Microbiol 26 (8), 715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Phillips ZN et al. (2019) Phasevarions of bacterial pathogens - phase-variable epigenetic regulators evolving from restriction-modification systems. Microbiology 165 (9), 917–928. [DOI] [PubMed] [Google Scholar]
- 84.Atack JM et al. (2018) A survey of Type III restriction-modification systems reveals numerous, novel epigenetic regulators controlling phase-variable regulons; phasevarions. Nucleic Acids Res 46 (7), 3532–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Atack JM et al. (2020) DNA sequence repeats identify numerous Type I restriction-modification systems that are potential epigenetic regulators controlling phase-variable regulons; phasevarions. FASEB J 34 (1), 1038–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Polaczek P et al. (1998) GATC motifs may alter the conformation of DNA depending on sequence context and N6-adenine methylation status: possible implications for DNA-protein recognition. Mol. Gen. Genet 258 (5), 488.–. [DOI] [PubMed] [Google Scholar]
- 87.Ngo TT et al. (2016) Effects of cytosine modifications on DNA flexibility and nucleosome mechanical stability. Nat. Commun 7, 10813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Diekmann S (1987) DNA methylation can enhance or induce DNA curvature. EMBO J 6 (13), 4213.–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Moller A et al. (1981) 7-Methylguanine in poly(dG-dC).poly(dG-dC) facilitates z-DNA formation. Proc. Natl. Acad. Sci. U. S. A 78 (8), 4777.–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hognon C et al. (2019) Cooperative effects of cytosine methylation on DNA structure and dynamics. J. Phys. Chem. B 123 (34), 7365.–. [DOI] [PubMed] [Google Scholar]
- 91.Lee DS et al. (2019) Simultaneous profiling of 3D genome structure and DNA methylation in single human cells. Nat. Methods 16 (10), 999.–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Garcia-Del Portillo F et al. (1999) DNA adenine methylase mutants of Salmonella typhimurium show defects in protein secretion, cell invasion, and M cell cytotoxicity. Proc. Natl. Acad. Sci. USA 96 (20), 11578.–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pucciarelli MG et al. (2002) Envelope instability in DNA adenine methylase mutants of Salmonella enterica. Microbiology 148 (Pt 4), 1171.–. [DOI] [PubMed] [Google Scholar]
- 94.Heithoff DM et al. (1999) An essential role for DNA adenine methylation in bacterial virulence. Science 284 (5416), 967.–. [DOI] [PubMed] [Google Scholar]
- 95.Watson ME Jr. et al. (2004) Inactivation of deoxyadenosine methyltransferase (dam) attenuates Haemophilus influenzae virulence. Mol. Microbiol 53 (2), 651.–. [DOI] [PubMed] [Google Scholar]
- 96.Robinson VL et al. (2005) A dam mutant of Yersinia pestis is attenuated and induces protection against plague. FEMS Microbiol. Lett 252 (2), 251.–. [DOI] [PubMed] [Google Scholar]
- 97.Luo N et al. (2018) DNA methyltransferase inhibition upregulates MHC-I to potentiate cytotoxic T lymphocyte responses in breast cancer. Nat. Commun 9 (1), 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stresemann C et al. (2006) Functional diversity of DNA methyltransferase inhibitors in human cancer cell lines. Cancer Res 66 (5), 2794.–. [DOI] [PubMed] [Google Scholar]
- 99.Brueckner B and Lyko F (2004) DNA methyltransferase inhibitors: old and new drugs for an epigenetic cancer therapy. Trends Pharmacol. Sci 25 (11), 551.–. [DOI] [PubMed] [Google Scholar]
- 100.Kahng LS and Shapiro L (2001) The CcrM DNA methyltransferase of Agrobacterium tumefaciens is essential, and its activity is cell cycle regulated. J. Bacteriol 183 (10), 3065.–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Robertson GT et al. (2000) The Brucella abortus CcrM DNA methyltransferase is essential for viability, and its overexpression attenuates intracellular replication in murine macrophages. J. Bacteriol 182 (12), 3482.–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Roberts RJ et al. (2003) A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res 31 (7), 1805.–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rao DN et al. (2014) Type III restriction-modification enzymes: a historical perspective. Nucleic Acids Res 42 (1), 45.–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Murray NE (2000) Type I restriction systems: sophisticated molecular machines (a legacy of Bertani and Weigle). Microbiol. Mol. Biol. Rev 64 (2), 412.–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pingoud A et al. (2005) Type II restriction endonucleases: structure and mechanism. Cell Mol. Life Sci 62 (6), 685.–. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.