

Abstract
The amide functional group plays a key role in the composition of biomolecules, including many clinically approved drugs. Bioisosterism is widely employed in the rational modification of lead compounds, being used to increase potency, enhance selectivity, improve pharmacokinetic properties, eliminate toxicity, and acquire novel chemical space to secure intellectual property. The introduction of a bioisostere leads to structural changes in molecular size, shape, electronic distribution, polarity, pKa, dipole or polarizability, which can be either favorable or detrimental to biological activity. This approach has opened up new avenues in drug design and development resulting in more efficient drug candidates introduced onto the market as well as in the clinical pipeline. Herein, we review the strategic decisions in selecting an amide bioisostere (the why), synthetic routes to each (the how), and success stories of each bioisostere (the implementation) to provide a comprehensive overview of this important toolbox for medicinal chemists.
Graphical Abstract
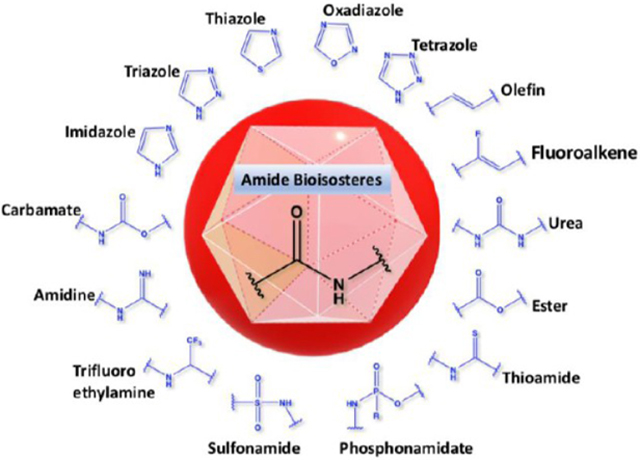
INTRODUCTION
The amide functional group plays a critical role in the composition of many biologically active molecules. This includes peptides and proteins, a wide variety of chemical probes and clinically approved and experimental synthetic and naturally derived drug molecules. Consequently, the amide functionality is of extreme interest to both medicinal chemists and chemical biologists.1,2 The amide group enjoys significant attention due to its unique ability to form relevant hydrogen bonding interactions. An amide typically (the keto tautomer predominating) consists of two types of hydrogen bonding sites, the carbonyl group and the amine group, which may act as a hydrogen bond acceptor (HBA) and hydrogen bond donor (HBD), respectively.3 The lone pair of the amine nitrogen is not available to accept a hydrogen bond due to its role in the resonance system of the functional group which installs significant conformational rigidity that further enhances or weakens the strength of the hydrogen bonds.
A peptide bond, a term erroneously used interchangeably with an amide bond, consists of an amide composed from the linkage of an α-amino nitrogen from one amino acid with the carbonyl carbon of a second amino acid. In peptide chemistry, the partial double-bond character of the amide group (due to delocalization of the lone pair of the nitrogen) results in planar geometry and thus allows adaptation of either the cis or trans conformation. In cis conformation, the two α-carbon atoms (Cα–C and N–Cα) lie on the same side of the peptide bond, whereas in the trans conformation these two carbon atoms are transposed. Nearly all proteins in their folded state adopt the trans configuration over cis to reduce steric hindrance between groups attached to the α-carbon atoms.4
The most common synthetic access to amides is provided by the reaction between a suitably functionalized carboxylic acid with a suitably functionalized amine, the same reaction that forms peptide bonds between amino acids to build proteins in biological systems.5 At the bench, to increase yields, shorten reaction times, and react sterically hindered or other low reactivity substrates, the most frequently employed method for the preparation of amides involves the reaction of an activated carboxylic acid derivative, such as acid chlorides, anhydrides, or esters, with amines or alternatively, the union of carboxylic acids with amines aided by stoichiometric amounts of dedicated coupling reagents, such as carbodiimides or 1H-benzotriazole derivatives, which achieve activation of the carboxylic acid in situ.6–9
The role of the amide bond has gained increasing prominence in drug design and development as more detailed knowledge about its properties has been revealed in the past century. Several top-selling drugs (Figure 1) such as lidocaine (1), paracetamol (2), the penicillins (embodied by the core structure 3), atorvastatin (4), chloramphenicol (5), moclobe-mide (6), captopril (7), acetazolamide (8), ponatinib (9), methotrexate (10), trimethobenzamide (11), among others contain an amide functional group as part of their pharmacophore or auxophore.
Figure 1.
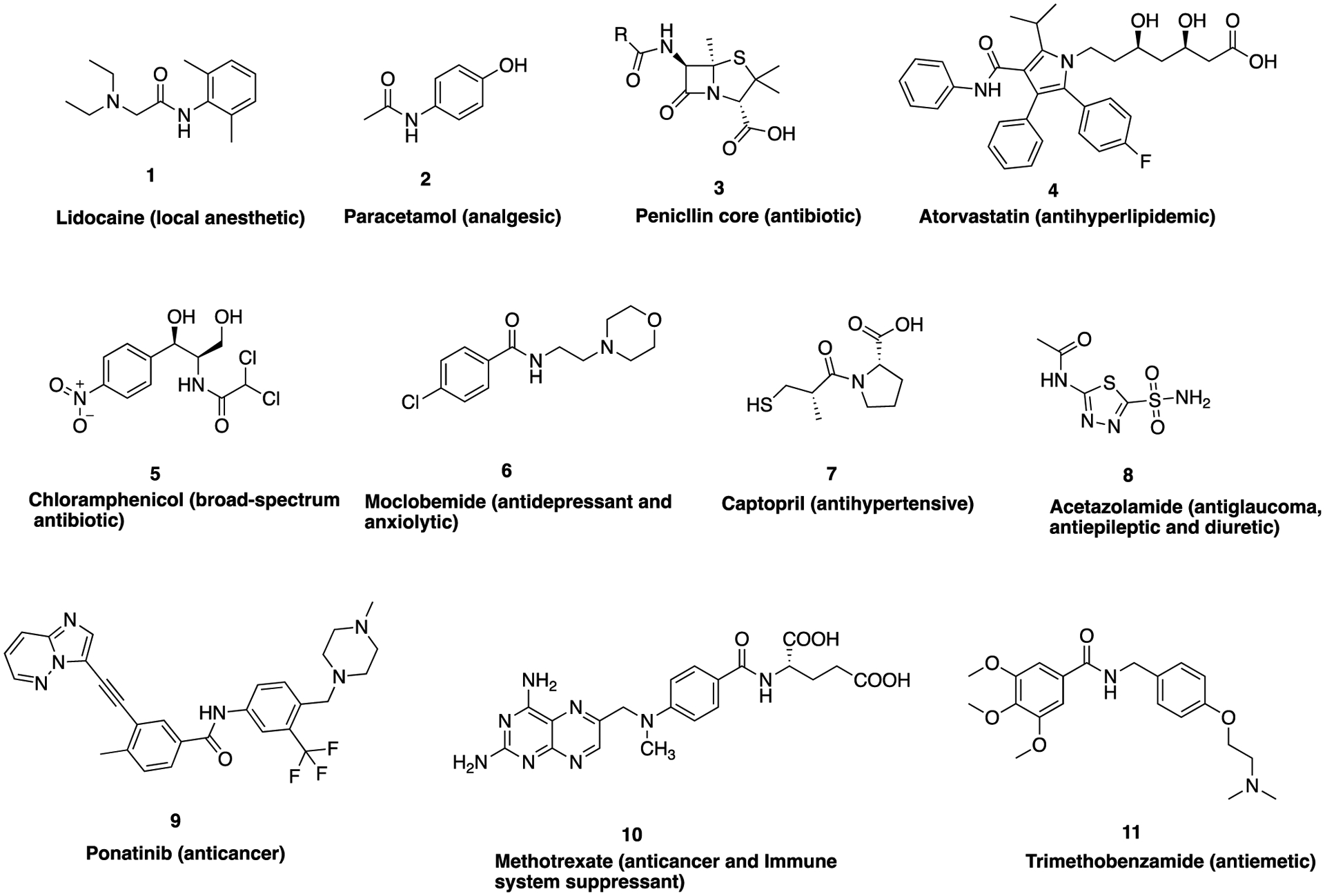
Structures of selected clinically approved drug molecules containing an amide bond and their biological function.
One of the initial hurdles to overcome in the drug discovery process is the identification of a lead compound that engages a validated target in a disease of interest. In most cases, the hit compound that forms the basic structural blueprint for hit-to-lead discovery and indeed the initial lead is deficient in one or several properties required for clinical success, the required level of potency, selectivity, suitable pharmacokinetic (PK), or pharmacodynamic (PD) parameters, that impart obstacles along the path to the identification of a clinical candidate.10 To counteract many of these issues, one of the most frequently employed strategies in hit-to-lead and lead optimization involves the replacement of a selected fragment within a compound of interest with another fragment that is known to closely mimic the properties of the original fragment or functional group. This process of replacement or modification of functional groups, having similar properties, is known as isosteric or bioisosteric replacement and has become a mainstay of the medicinal chemist’s toolbox.11–13
Bioisosterism is considered a qualitative technique to achieve the rational modification and optimization of a bioactive compound that can provide several beneficial effects including increased potency, enhanced selectivity, improvements in PK and PD properties, elimination or attenuation of toxicity and facilitate access to novel chemical space to attain patent protection.14 Bioisosteres can be categorized into classical and nonclassical subtypes. Classical bioisosteres are functional groups that satisfy Grimm’s hydride displacement law as well as Erlenmeyer’s concepts of isosteres and can be subdivided into five categories: (a) monovalent atoms or groups, (b) divalent atoms or groups, (c) trivalent atoms or groups, (d) tetravalent atoms or groups, and (e) ring equivalents (Table 1). On the other hand, nonclassical bioisosteres possess more advanced mimicry of their emulated counterparts and do not fulfill the criteria of steric and electronic factors required for classical isosteres (Table 1).15–17,11
Table 1.
Classification of Classical and Nonclassical Bioisosteres
| Classical bioisosteres | Nonclassical bioisosteres |
|---|---|
| a. Monovalent atoms or groups F and H; D and H; NH and OH; C and Si; RSH and ROH; -F; -Cl; -Br; -I; −OH; -OR; -SH; -PH2; -CH3; -CF3; -NH2 | a. Rings versus acyclic structures. Methyleneaminoxy methyl moiety (C=NOCH2, MAOMM) as a bioisostere of aryl and other aromatic scaffold. |
| b. Divalent atoms or groups -O-; -Se-; -CH2-; -C==N-; -C==S; -C==C-; -C==O; -C==NH; -COOR; -COSR; -CONHR; -COCH2R | b. Exchangeable groups, hydroxyl group bioisosteres, carbonyl group bioisosteres, amide group bioisosteres, thiourea group bioisosteres, halogen group bioisosteres, carboxylate group bioisosteres |
| c. Trivalent atoms or groups -As==; -N==; -P==; -CH== | |
| d. Tetravalent atoms or groups ==C==; ==Si== ; ==As+==; ==N+==; ==P+== | |
| e. Ring equivalents benzene; tetrahydrofuran; thiophene; pyridine |
The installation of a bioisostere leads to structural changes that can be either beneficial or deleterious to biological activity depending upon the effect on overall molecular size, shape, electronic distribution, lipophilicity, dipole moment, and polarizability.16 For example, the tetrazole heterocycle is a well-known bioisostere of a carboxylic acid.18 Several studies have demonstrated that the 1,2,3-triazole moiety is an amide surrogate.19 The utilization of this approach has opened new avenues in drug design and development, resulting in more efficient drug candidates that have reached the clinic as well as countless numbers in the pipeline.
The amide functional group is enzymatically very labile in vivo; therefore, amide bond bioisosteres that improve metabolic stability are of great interest, particularly for applications in peptide chemistry and the development of peptidomimetics.20 Peptide drugs represent a major class of pharmacologically active compounds, composed of small molecules and proteins that have been deployed to treat cancer, diabetes, infectious diseases, and cardiovascular diseases among other indications.21–23 The U.S. Food and Drug Administration (FDA) has approved over 60 peptide drugs, and approximately 140 new peptide therapeutics are currently being evaluated in clinical trials at the time of writing.24 Although peptides are endowed with high specificity and low toxicity and are generally well-tolerated in humans, many therapeutic peptides suffer from poor metabolic stability due to the rapid degradation of the amide bond by proteases.25,26 Thus, the introduction of structural motifs that mimic the physicochemical properties of the amide bond provides greater metabolic stability. The replacement of the amide bond by a bioisostere can afford new peptidomimetics with improved biological properties and retention of the desired therapeutic effect. To date, several amide bioisostere classes are known, including 1,2,3-triazole, oxadiazole, imidazole, tetrazole, pyrazole, indole, pyridine, pyrazine, retroinverted and reverse amide, urea, olefin, fluoroalkene, trifluoroethylamine, amidine, ester, sulfonamide, phosphonamidate, thioamide, and carbamate. The past decade has seen enormous success in the application of these amide bioisosteres in the construction of more sophisticated and often more active compounds. Choosing the most appropriate bioisostere is a complex task. Consideration of feasible synthetic routes, modification strategies, and bioactivity effects of prior successes in the field of amide bioisosteric replacement can provide valuable insights into drug discovery projects.
There are a number of prior articles available in the literature that discuss the application of several types of classical and nonclassical bioisosteres in drug design27–31 along with a more recent review focused on amide bioisosteres.32 This Perspective aims to be a reference guide for medicinal chemists to peruse a selection of the most common amide bond bioisosteres and make an informed choice on a potentially appropriate moiety to employ in their project, with the caveat that any hit or lead compound of interest will possess distinct properties that may preclude some or all of the highlighted bioisosteres from either a synthetic or biological perspective. We review a broad range of amide bioisosteres covering their successful design, application of modification strategies, bioisosteric relationships, and pharmacological advancements along with a summary of synthetic methodologies adopted to access more potent therapeutic agents. In this context, we have categorized amide bioisosteres into 13 categories: heterocycles, retroinverted and reverse amide, urea, carbamate, amidine, thioamide, trifluoroethylamine, sulfonamide, phosphonamidate, ester, olefin, fluoroalkene, and finally miscellaneous bioisosteres.
FIVE-MEMBERED RING HETEROCYCLES AS AMIDE BIOISOSTERES
A. Triazole.
The triazole moiety has repeatedly shown potential application in refining the therapeutic ability of amide bond-containing molecules and therefore is considered an excellent nonclassical bioisostere. Most importantly, the triazole motif is proficient in mimicking the configuration of the trans amide bond; either a trans or a cis conformation can be achieved around the heterocycle depending on the substitution pattern of the triazole.33 The 1,4-disubstituted 1,2,3-triazole scaffold is isosteric to the trans amide bond and has gained widespread use (Figure 2). The 1,5-disubstituted 1,2,3-triazole moiety mimics the cis amide bond, and its use is much less common.34 The distance between the substituents at the 1- and 4-position of the triazole ring is 5.0–5.1 Å, which is approximate to the 3.8–3.9 Å distance between trans amide substituents.35 Several fundamental properties, such as planarity, dipole moment and hydrogen bonding properties of the 1,2,3-triazole, are comparable to those of an amide.
Figure 2.
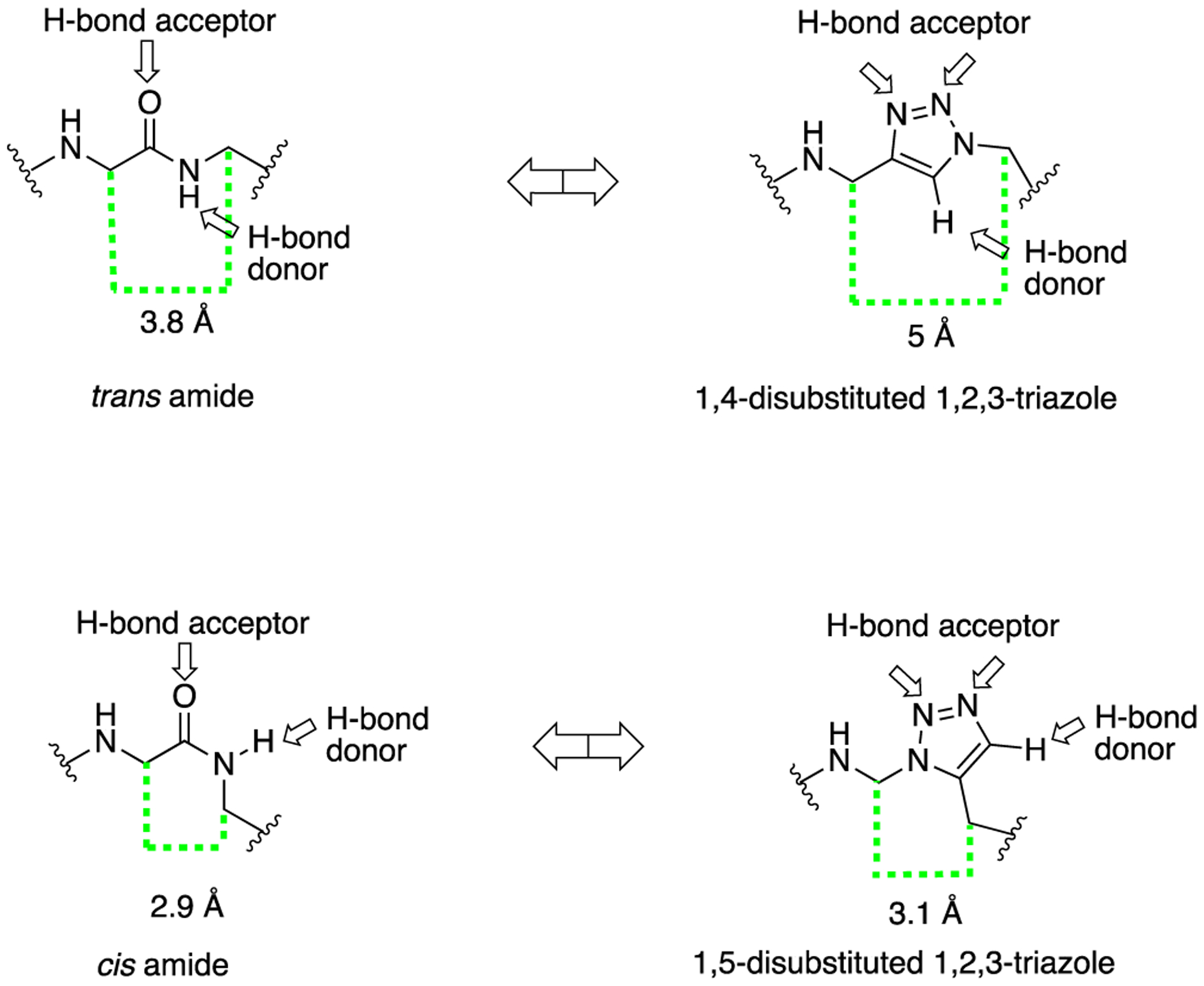
Representative structures of the trans and cis amide mimics 1,4-disubstitued 1,2,3-triazole and 1,5-disubstitued 1,2,3-triazole respectively, highlighting their distancing and hydrogen bonding capabilities.
Similarly, the dipole moment of the amide (~4 D) and 1,2,3-triazole (~5 D) is not significantly different.36 The two indicated nitrogen atoms (Figure 2) in the triazole moiety possess lone pairs of electrons that act as hydrogen bond acceptors, playing a similar role as the oxygen atom in the amide. The CH bond within the triazole exhibits a strong dipole moment and therefore can function as a hydrogen bond donor, analogous to the amide NH.37 The 1,2,3-triazole unit is particularly resistant to cleavage mediated by proteases, oxidation, and hydrolysis.38 In summary, the 1,2,3-triazole moiety possesses characteristics that could afford protease-resistant peptidomimetics with retained or even improved biological activity. Much literature has been published in recent years that have shown the advantages of the 1,2,3-triazole moiety.19 Successful replacement of the amide bond within bioactive scaffolds with a 1,2,3-triazole has led to the discovery of many potent bioactive molecules, including approved marketed drugs to combat human immunodeficiency virus (HIV) infection and acquired immune deficiency syndrome (AIDS), bacterial infections, cancer, and neurological disorders.
The Rana research group has investigated the RN-18 class of small molecules to target HIV-1 accessory protein viral infectivity factor (Vif) to identify novel antagonists.39–41 The 23 kDa Vif protein is critical for in vivo viral replication and has emerged as a validated target for HIV-1/AIDS drug development.42,43 Initially, the group had identified a lead compound RN-18 (12, Figure 3) that specifically antagonizes HIV-1 Vif protein function [in H9 (nonpermissive cells), IC50 = 6 μM] and obstructs viral replication. Compound 12 does not inhibit viral infectivity in permissive cells, MT4 (human T-lymphoid cells), even at 100 μM which highlights its Vif specificity.
Figure 3.
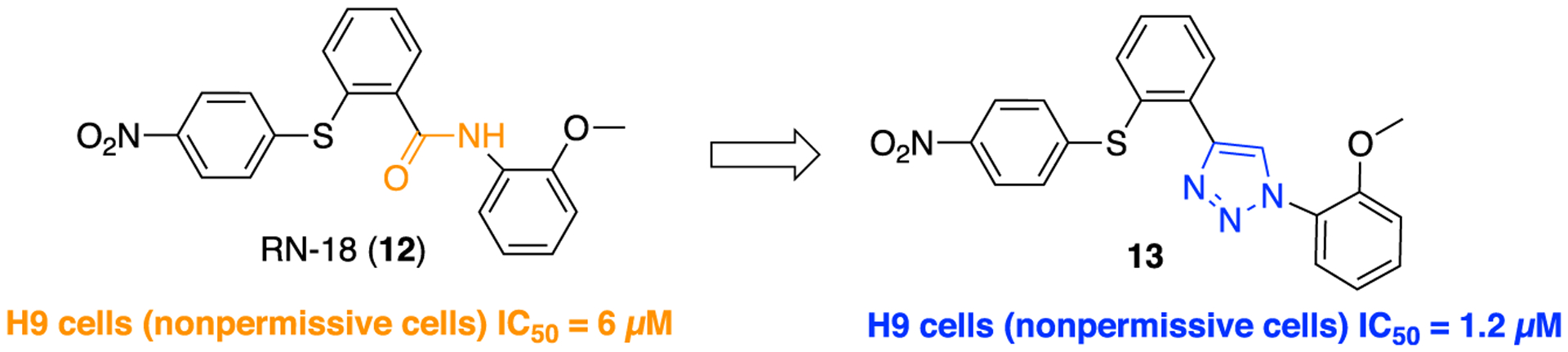
Replacement of the amide bond in RN-18 (12) with a 1,2,3-triazole moiety to afford compound 13 (1–2-methoxyphenyl)-4-(2-((4-nitrophenyl)thio)phenyl)-1H-1,2,3-triazole).
However, two significant concerns related to the potency and metabolic stability of RN-18-based Vif antagonists became an obstacle to further clinical development. In a follow-up study, the authors used the concept of bioisosterism and replaced the amide bond in 12 with 2,5-disubstituted-1,3,4-oxadiazole, 3,5-disubstituted-1,2,4-oxadiazole, 1,4-disubstituted-1H-1,2,3-triazole, or 1,5-disubstituted-1H-1,2,3-triazole moieties, aiming to improve the pharmacological profile. In the original paper, the authors did not comment on any structural role for the amide in 12. However, they synthesized new analogs that were tested in vitro using wild-type HIV-1 in both H9 and MT-4 cells. Results of this experiment have shown that the installation of only the 1,4-disubstituted-1,2,3-triazole moiety (compound 13; IC50 = 1.2 μM; H9 cells, Figure 3) as an amide bond bioisostere resulted in increased potency over the parent compound (IC50 = 6 μM; H9 cells). The general synthetic method (i) to access compound 13 through the application of copper catalyzed azide-alkyne cycloaddition (CuAAC) click chemistry is delineated in Scheme 1, i. Overall, compound 13 was determined to be a potent Vif antagonist. Inspired by the obtained results, a subseries of new triazole containing analogs (based on 13) was also synthesized that resulted in the discovery of more potent HIV-1 inhibitors than compound 13 itself.44
Scheme 1.
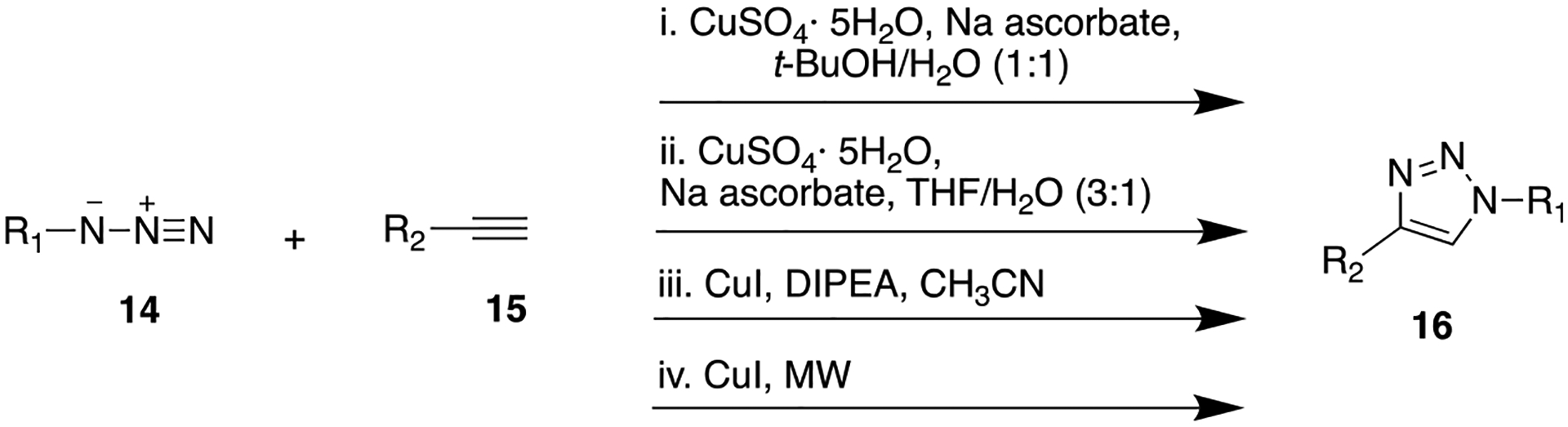
General Synthetic Pathway To Access Triazole Scaffold via Copper(I)-Catalyzed Azide-Alkyne Cycloaddition Click Chemistry
Vismodegib (GDC-0449, compound 17) is a highly specific small molecule synthetic smoothened (SMO) antagonist that inhibits the Hedgehog (Hh) signaling pathway, upregulated in basal-cell carcinoma (BCC).45 Compound 17 was approved by the FDA in 2012 to treat locally advanced and metastatic unresectable BCC. Several clinical studies indicated that many BCC patients experienced substantial benefits from 17; however, they developed side effects including muscle spasms, alopecia, dysgeusia, fatigue, and weight loss.46,47 Additionally, patients acquired resistance upon treatment with compound 17, which was associated with Hh pathway reactivation.48 Considering these problems, Passarella and colleagues modified the structural framework of compound 17, aiming to improve potency as well as other pharmacological features.49 Initially, a virtual library of 140 triazole-containing derivatives were screened. On the basis of the obtained docking results, several analogs were synthesized wherein a 1,2,3-triazole bioisostere was introduced in place of the amide of compound 17, along with additional alterations of the substituents on the phenyl rings (Figure 4).
Figure 4.
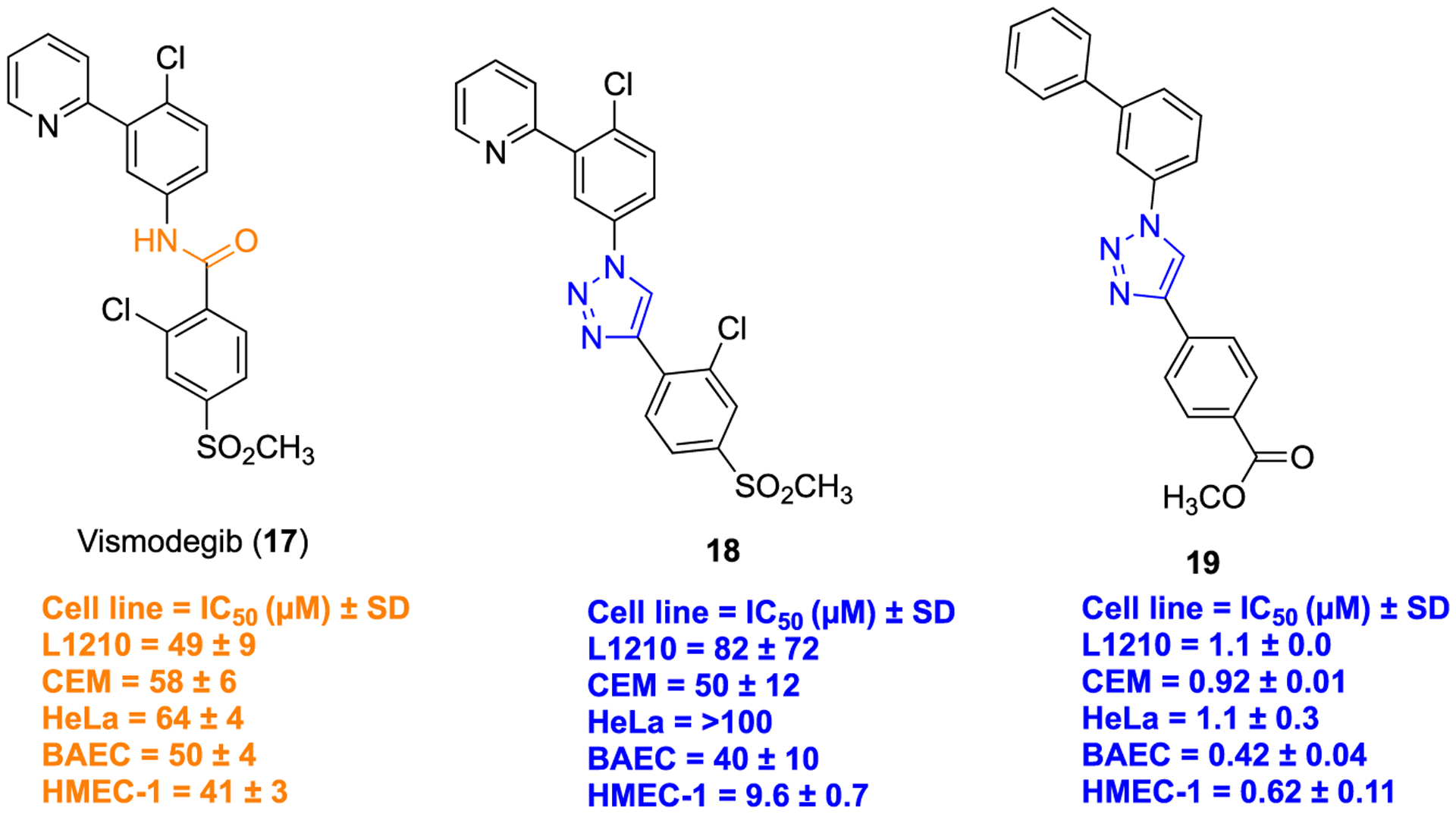
Example of amide versus 1,2,3-triazole bioisosterism in vismodegib (17), a potent Hh signaling pathway inhibitor.
The in vitro antiproliferative activity of all synthesized compounds was determined in three cancer lines (L1210, murine leukemia cells; CEM, human T-lymphocyte cells; HeLa, human cervix carcinoma), and two endothelial cell lines (HMEC-1, human microvascular endothelial cells; BAEC, bovine aortic endothelial cells). Compound 18 possessing the 1,2,3-triazole ring in place of the amide bond (17) displayed an appreciable activity benefit in HMEC-1 cells (IC50 = 9.6 ± 0.7 μM) as compared to parent 17 (IC50 = 41 ± 3 μM). Thus, it can be considered that the triazole ring is solely responsible for lowering IC50 in this context. The most potent compound of the series 19, containing further structural changes beyond the bioisostere, possessed greater influence on all tested cell lines compared with compound 17 (Figure 4). The general synthetic method (i) to obtain compounds 18 and 19 is depicted in Scheme 1, i. Overall, results suggested the 1,2,3-triazole scaffold provided favorable bioactivity outcomes on this pharmacophore.
In another study, the Passarella group synthesized triflorcas and a close analog containing a 1,2,3-triazole as an amide bond surrogate. In silico docking studies showed that the -NH-CO-function is involved in hydrogen bonding with Glu1127 and Lys1110 in the same way that the triazole ring is stabilized by polar interactions with Lys1110. Overall, docking results confirmed the binding of the 1,2,3-triazole bioisostere into the adenosine triphosphate (ATP) binding site of N-methyl-N′-nitrosoguanidine human osteosarcoma transforming gene (MET) in a similar fashion as triflorcas. Results of hepatocyte growth factor (HGF)-induced scattering of Madin-Darby canine kidney (MDCK) epithelial cells and in vitro tumorigenesis of H1437 cells (non-small-cell lung cancer) and GTL-16 cells (human gastric carcinoma) showed that the 1,2,3-triazole bioisostere was nearly equipotent with triflorcas. The overall results of this study suggest that the replacement of the amide bond with 1,2,3-triazole ring maintained the tumorigenesis inhibitory potential of the lead compound triflorcas.50–52
Imatinib is a tyrosine-kinase inhibitor that is approved for the treatment of chronic myelogenous leukemia (CML), certain types of gastrointestinal stromal tumors (GIST), and a number of other malignancies.53–57 The development of resistance to imatinib called for the need to develop small molecule inhibitors that could overcome imatinib resistant in CML patients.58 Passarella and colleagues reported the first example of amide bioisosterism in imatinib via a 1,2,3-triazole. Among all tested analogs, the most potent compound (FA030; structure not shown) possessing the 1,2,3-triazole motif in place of the amide bond of imatinib showed similar binding patterns in molecular modeling studies. FA030 displayed potent antienzymatic activity against recombinant Abl kinase but decreased activity in the imatinib-resistant mutant Abl T3151. In addition to this, FA030 decreased the phosphorylation of break point cluster region Abelson kinase (Bcr-Abl), c-Abl, signal transducer and activator of transcription 5 (STAT-5), and Src, a protooncogene encoding a non-receptor tyrosine kinase in a dose-dependent manner similar to imatinib.52
Modulation of the dopamine-3 subtype receptor (D3-R) influences neuronal activity in some parts of the brain; therefore, the D3-R has gained special attention in the development of therapeutics to treat neurological diseases such as schizophrenia, Parkinson’s disease (PD), and associated dyskinesias, as well as neuropsychiatric (particularly drug addiction) disorders.59,60 The development of selective D3-R ligands is an extremely challenging task due to their very close homology with dopamine-2 subtype receptor (D2-R).61 The Hauck Newman group discovered PG648 (20), OMO5–057 (21), and NGB2904 (22) as highly potent and selective D3-R ligands (Figure 5).62,63
Figure 5.
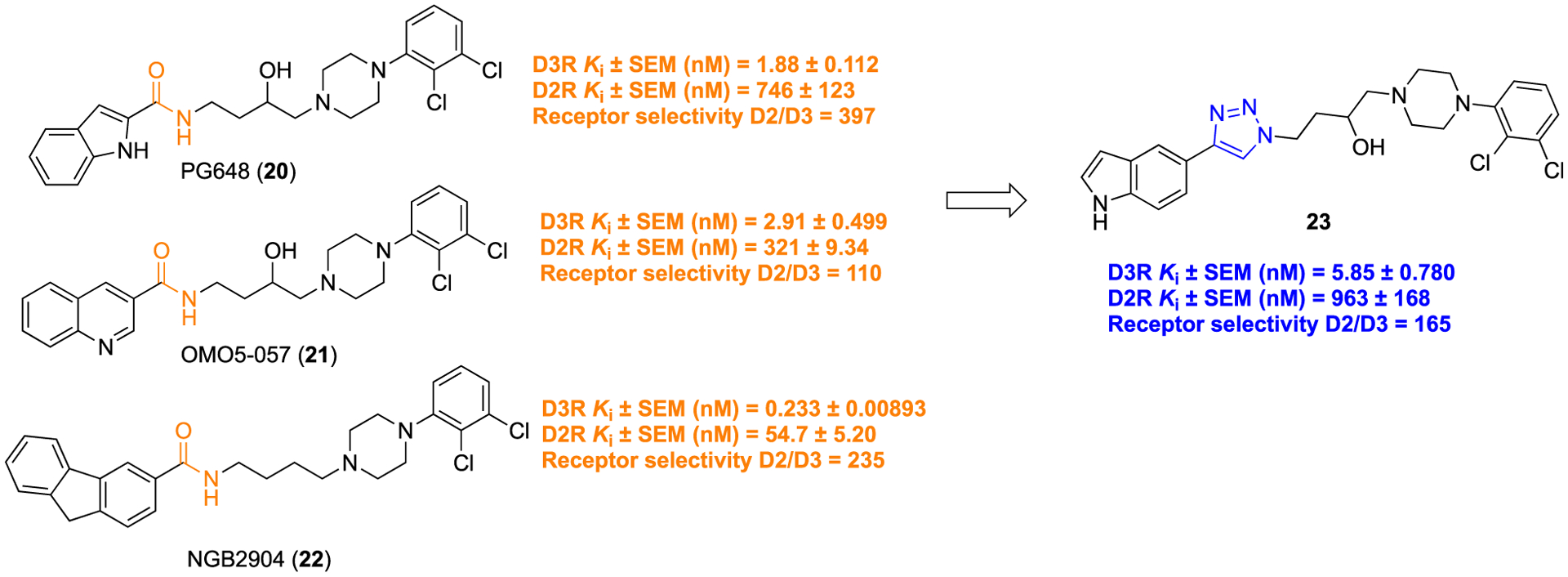
Molecular structures of highly potent D3-R ligands: 20, 21, 22 containing amide; 23, a 1,2,3-triazole analog.
Prior structure-activity relationship (SAR) studies disclosed a molecular sketch design that consists of an amalgamation of two pharmacophores, a phenylpiperazine moiety and an extended aryl ring unit, linked via a butyl chain essential to produce D3-R partial agonism/or antagonism.64 Moreover, the amide function provided a necessary element to impart maximum D3-R over D2-R selectivity.65 Although 20, 21, and 22 have shown potent D3-R binding affinity, their poor metabolic stability, mainly due to the presence of the amide, has impeded translation of these candidates to clinical use. To resolve this issue and in an effort to find more potent and D3-R selective ligands, a library of structurally modified analogs of 20, 21, and 22 were synthesized, in which the amide bond was replaced with a 1,2,3-triazole scaffold to generate a library of compounds.66
In this context, a 1,2,3-triazole was tested as a bioisosteric surrogate for the amide group in a series of compounds consisting of a substituted phenylpiperazine connected via an appropriate carbon linker to an aryl unit, with synthetic access provide by CuAAC click chemistry (Scheme 1, ii). This modification led to the discovery of compound 23, which showed promising D3-R binding affinity and 165-fold selectivity over the D2-R, evaluated by radioligand competition binding experiments using human embryonic kidney 293 (HEK293) cells, expressing human dopamine receptor subtypes. Compound 23 displayed better metabolic stability (35%) toward mouse liver microsomes (MLM) in phase 1 metabolic stability assay experiments than 20 (11%), remaining after 1 h incubation. Thus, it indicated that 1,2,3-triazole containing molecules could provide specific resistance to CYP450-mediated phase 1 metabolism over amide bioisosteres.
In a study aiming to replace an amide linker that would be capable of sustaining and modulating amide-related bioactivity, structural modifications to capsaicin (24) were explored in the context of a 1,2,3-triazole group as a bioisosteric element (Figure 6).67 The vanilloid and cannabinoid (CB) profiles of synthesized analogs were compared with compound 24 and other capsaicinoids. Success was achieved with compound 25, which presented antagonistic activity for vanilloid receptor 1, transient receptor potential cation channel subfamily V member 1 (TRPV1) with an IC50 of 0.69 ± 0.16 μM (found inactive as TRPV1 agonist with EC50 > 10000 nM and therefore tested as a TRPV1 antagonist). Compound 25 was synthesized by using copper iodide (CuI) catalyzed click chemistry as shown in the Scheme 1, iii. This compound also showed a significant submicromolar affinity for human cannabinoid 1 (hCB1) (Ki = 0.44 μM), the psychotropic cannabinoid receptor. In conclusion, the installation of a 1,2,3-triazole ring within the pharmacophore of compound 25 resulted in potent TRPV1 and CB1 selective antagonist activity. The authors in the original paper commented on the usefulness of compound 25 as a dual and unique TRPV1/CB1 antagonist; however, there was no mention that this was due solely to the 1,2,3-triazole bioisostere.
Figure 6.
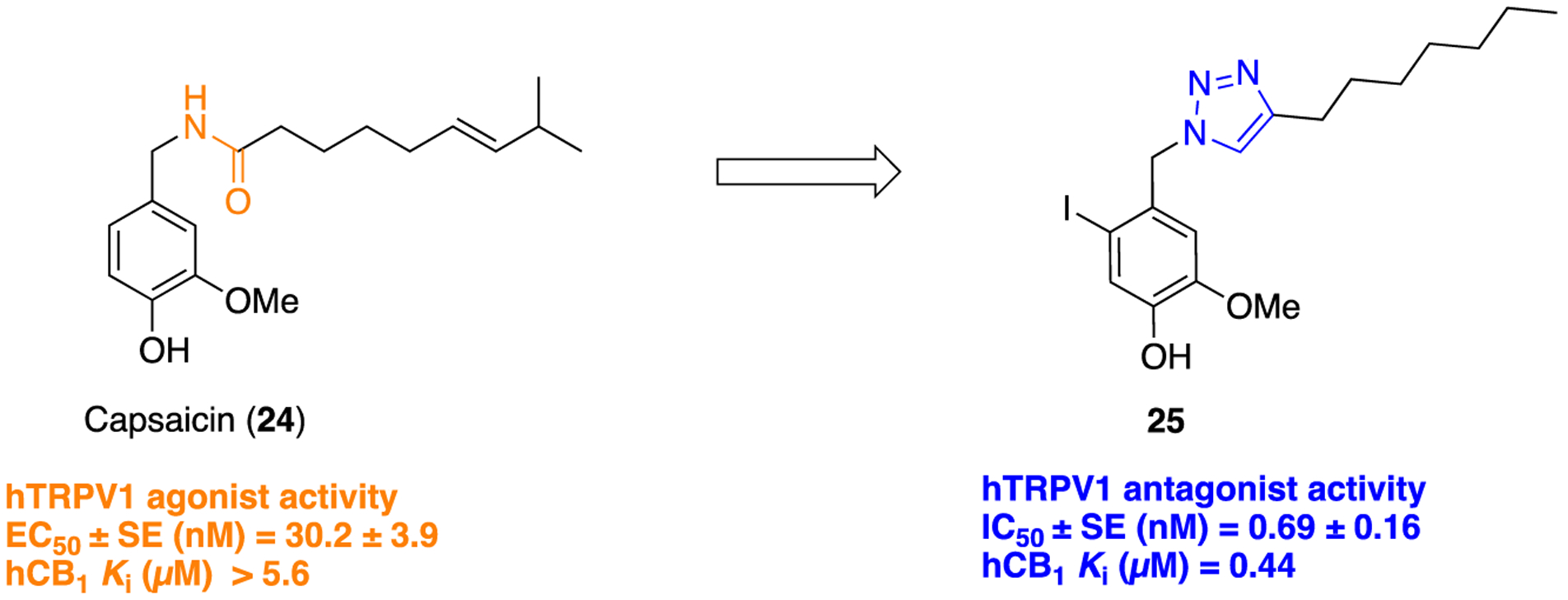
1,2,3-Triazole as an amide bond bioisostere in a TRPV1/CB1 antagonist.
An example of developing potent and selective cannabinoid-2 (CB2) receptor ligands using a bioisosteric approach was reported by Corelli and co-workers.68 Ligands selective to the CB2 receptor are desirable for treating pain associated with inflammation, cancer, and neuropathic conditions, which avoids the unwanted psychoactive effects typically induced by activation of the cannabinoid-1 (CB1) receptor.69–71 Aiming to optimize the physicochemical properties of a previously developed lead (26), the authors experimented with different heteroaromatic rings as amide surrogates in a series of 4-quinolone,3-carboxylic acid derivatives. The 1,2,3-triazole ring in 27 was prepared by microwave-assisted, CuI-catalyzed azide-alkyne 1,3-dipolar cycloaddition (Scheme 1, iv). The installation of a 1,2,3-triazole motif in place of the amide bond led to the development of the most potent compound among the series, 27 (Figure 7). This compound resolved solubility issues present in the lead compound (26) and also showed attractive results in in vitro CB1 and CB2 binding assays.
Figure 7.
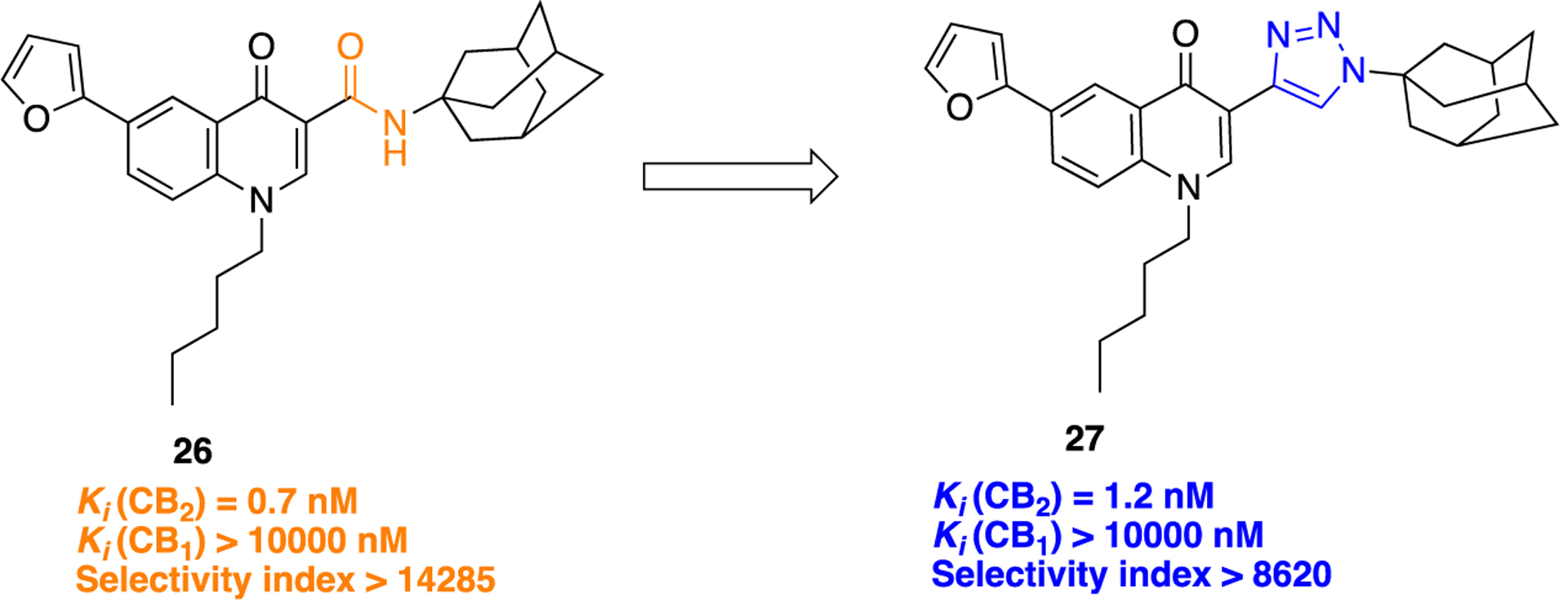
Molecular structure of lead compound 26 and its bioisostere 27.
A series of novel α-lipoic acid (LA) derivatives tethered to a chroman ring were designed, where the amide bond in previously synthesized analogs72 was replaced with several five-membered heterocyclic amide isosteres to study effects on the neuroprotective activity (Figure 8).73 Compound 29 possessing a 1,2,3-triazole was obtained by CuAAC click chemistry, as illustrated in Scheme 1, i.
Figure 8.
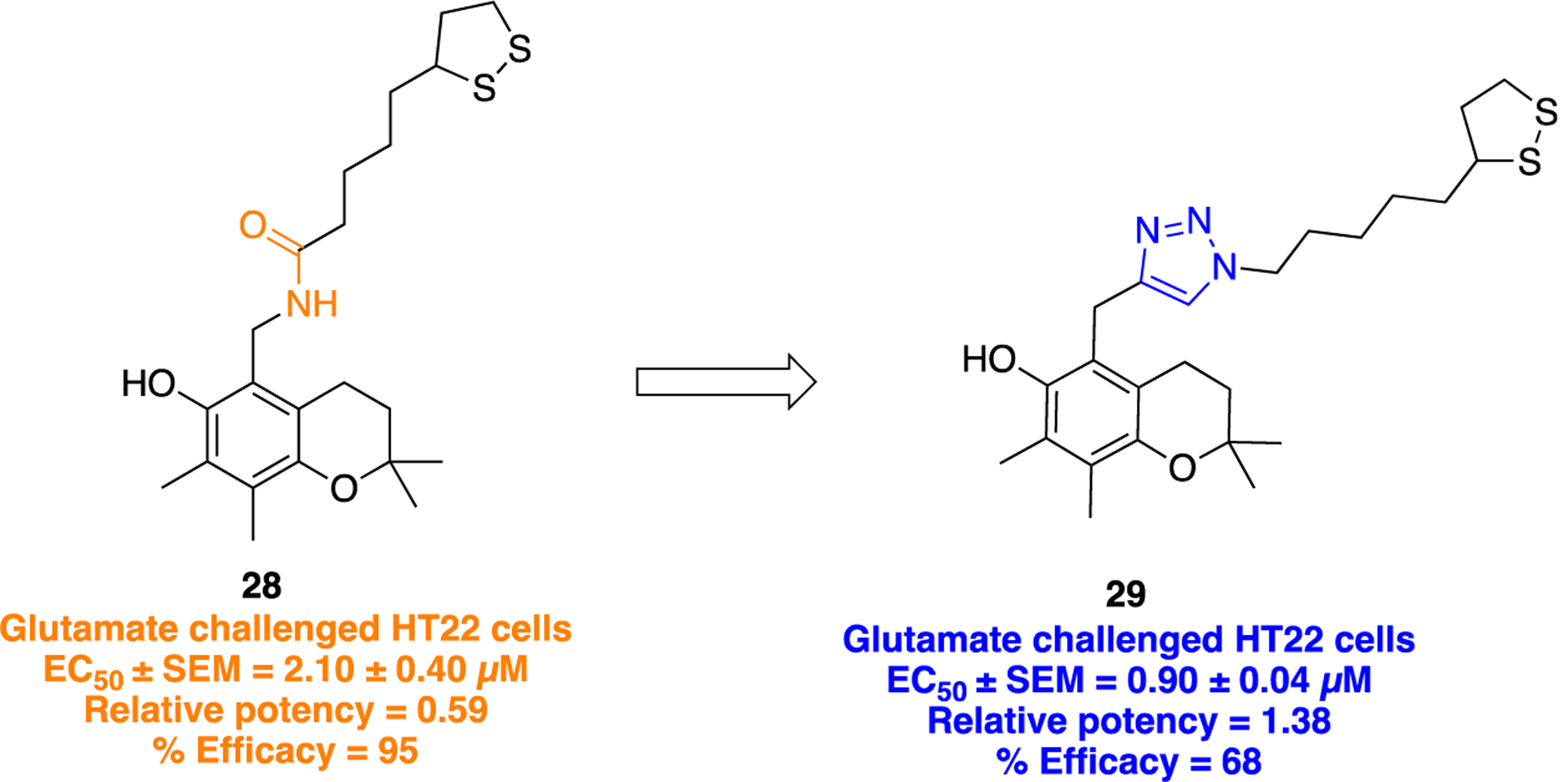
Structural transformation of parent compound 28 into 29 illustrating amide/triazole bioisosterism.
α-Lipoic acid or 1,2-dithiolane-3-pentanoic acid, reported as a therapeutic agent against oxidative stress models of death, and its structural analogs were found to be effective against reperfusion arrhythmias,74 acetylcholinesterase inhibition,75 butyrylcholinesterase inhibition,76 and nitric oxide synthase inhibition.77 Several studies also showed LA analogs as potent antiproliferative78–80 and anti-inflammatory agents.81 Results of neuroprotective activity experiments showed that bioisosteric replacement of the amide group in compound 28, by a 1,2,3-triazole (29), improved glutamate induced oxidative stress in mouse hippocampal neurons (HT22), EC50 = 2.10 ± 0.40 μM and EC50 = 0.90 ± 0.04 μM, respectively. In Figure 8, the relative potency was calculated by EC50 of standard/EC50 of tested compound, whereas the % efficacy was calculated at a fixed concentration of 10 μM (compounds were considered to be fully, partially, or weakly neuroprotective if % efficacy is >66–100%, >33–66%, and ≤33%, respectively). The SAR analysis revealed that the presence of a nitrogen heterocycle was mostly well tolerated and strongly influenced the neuroprotective activity of 5-substitued chroman derivatives, depending on the nature of the heterocycle.
An example wherein the 1,2,4-triazole was successfully installed as an amide bioisostere within a rigid ring system is found in the short-acting benzodiazepine alprazolam (31) (Figure 9). Benzodiazepines are a class of drugs used as anxiolytics, sedative-hypnotics, anticonvulsants, and muscle relaxants and have achieved significant economic success.82,83 This interesting journey started in 1963 when diazepam (30) was discovered and later approved for clinical use to treat anxiety, epilepsy, muscle spasms, and alcohol withdrawal symptoms.84–86 Compound 30 is a potent orally bioavailable central nervous system (CNS) penetrant small molecule, acting predominantly via positive allosteric modulation of γ-aminobutyric acid A (GABAA) chloride channels in the brain.87
Figure 9.
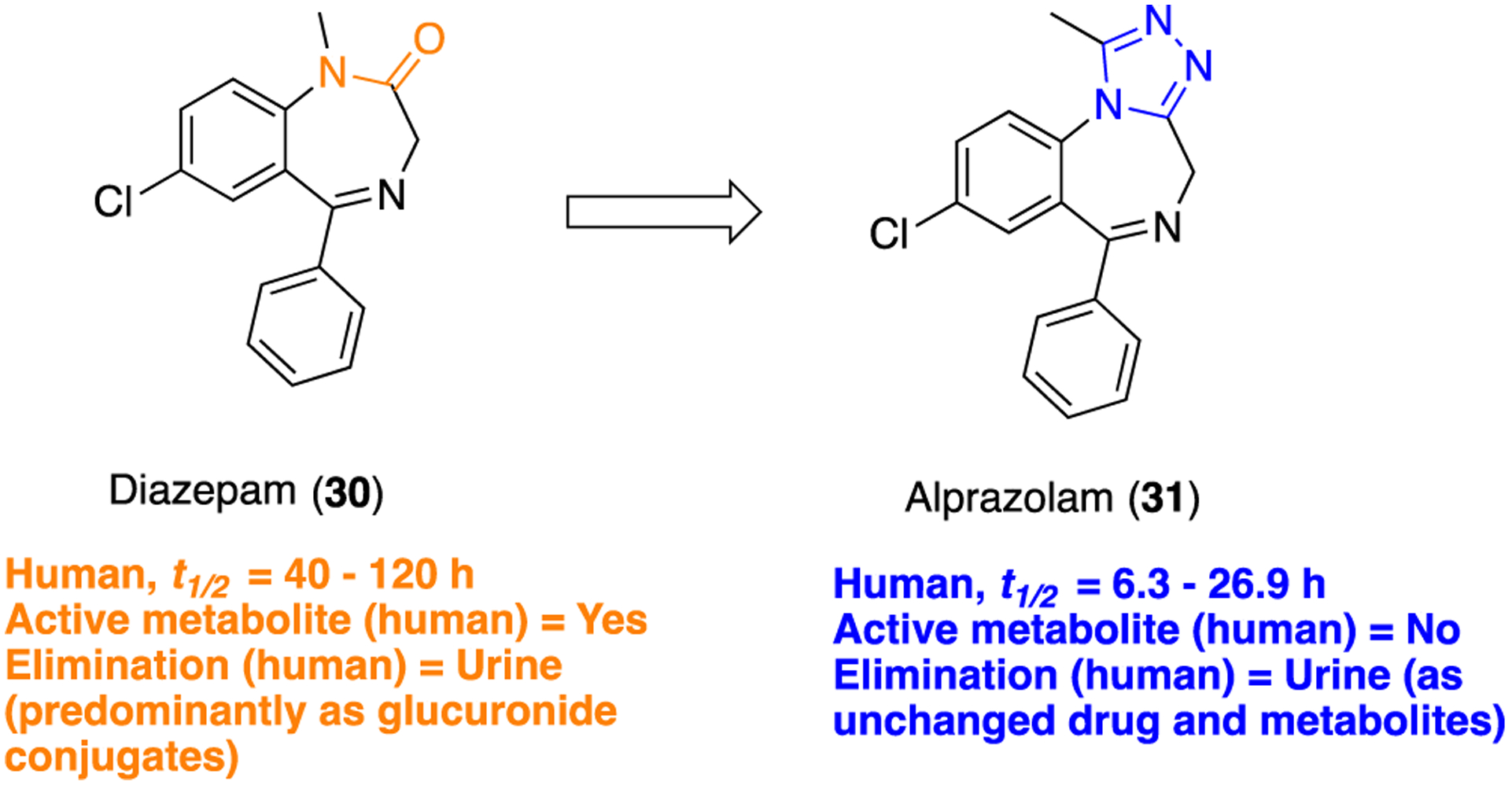
Replacement of an amide moiety with a 1,2,4-triazole: diazepam (30) to alprazolam (31).
In humans, 30 is metabolized by the liver cytochrome (CYP) P450 enzyme system into three pharmacologically active metabolites (Figure 10). Demethylation of 30 by CYP 2C9, 2C19, 2B6, 3A4, and 3A5 yields N-desmethyldiazepam/nordazepam (32), which is then oxidized to oxazepam (33). This metabolite 32 is a major circulating active metabolite (half-life of 50–120 h) equipotent to 30. As seen in Figure 10, the oxidation at C3 of 30 by CYP 3A4 and CYP 3A5 led to temazepam (34). The end product of 30 metabolism is 33 which is excreted in the urine. Each of these metabolites has a half-life of more than 24 h which in turn causes diazepam-induced toxicity. Subsequently, 33, 32, and 34 were developed and marketed as individual drugs.88
Figure 10.
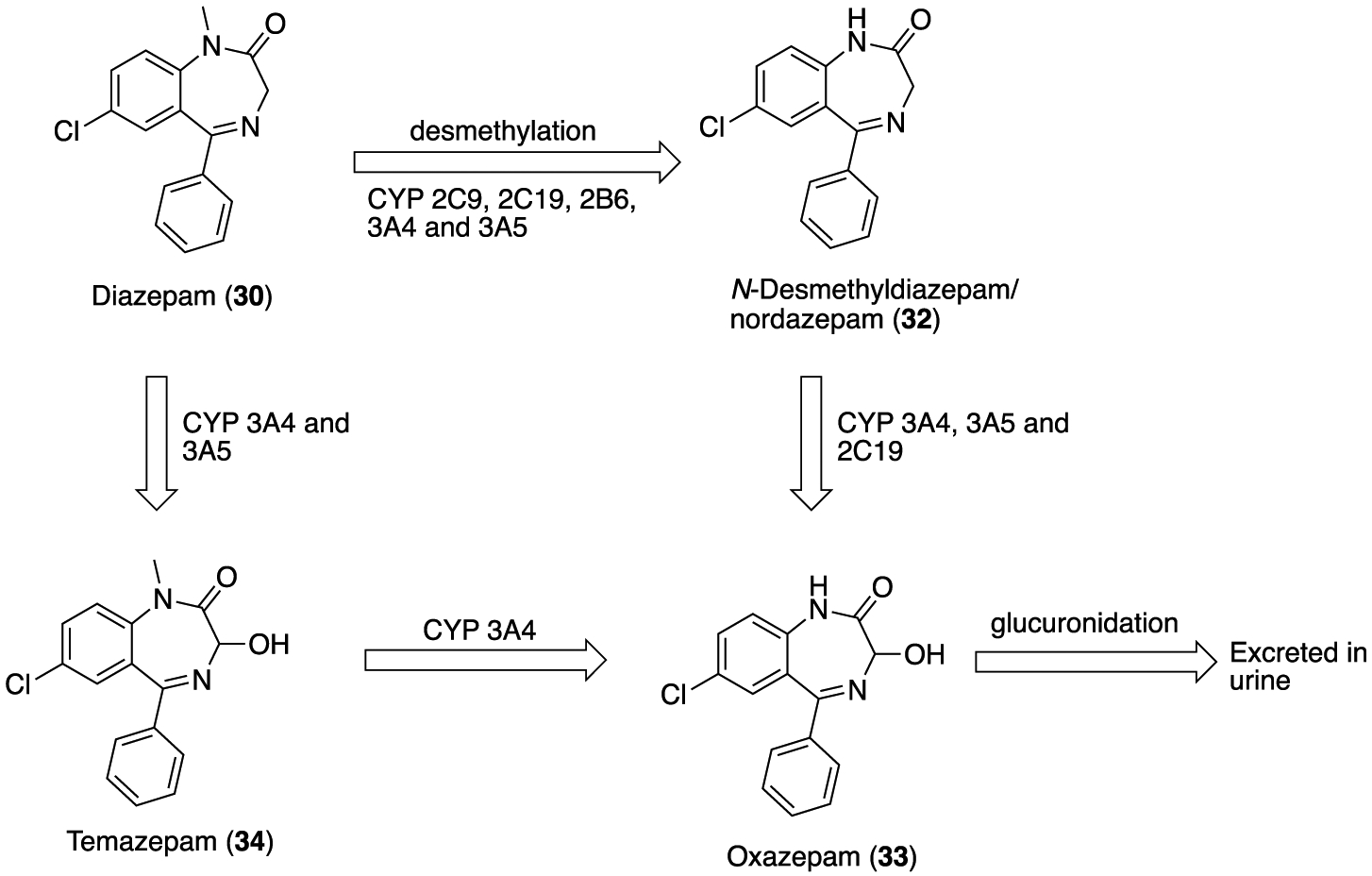
Metabolic transformation of diazepam (30) into three active metabolites: desmethyldiazepam/nordazepam (32), oxazepam (33), and temazepam (34).
In 1971, Hester and co-workers reported several close analogs of 30, including 31, wherein the amide functionality was replaced with a triazole moiety. Compound 31 was synthesized by refluxing 7-chloro-5-phenyl-1,3-dihydro-2H-benzo[e][1,4]diazepine-2-thione (35) with acethydrazide in n-butanol (Scheme 2) for 4 h.89 This compound was evaluated in vivo using CF-1 male albino mice and was found to be more potent than the parent, compound 30, in a variety of pharmacological assays. A few years later, 31 was approved by the FDA for the treatment of anxiety disorders. Human PD studies indicated that 31 did not produce active metabolites, resulting in significant suppression of diazepam-induced toxicity.90
Scheme 2.
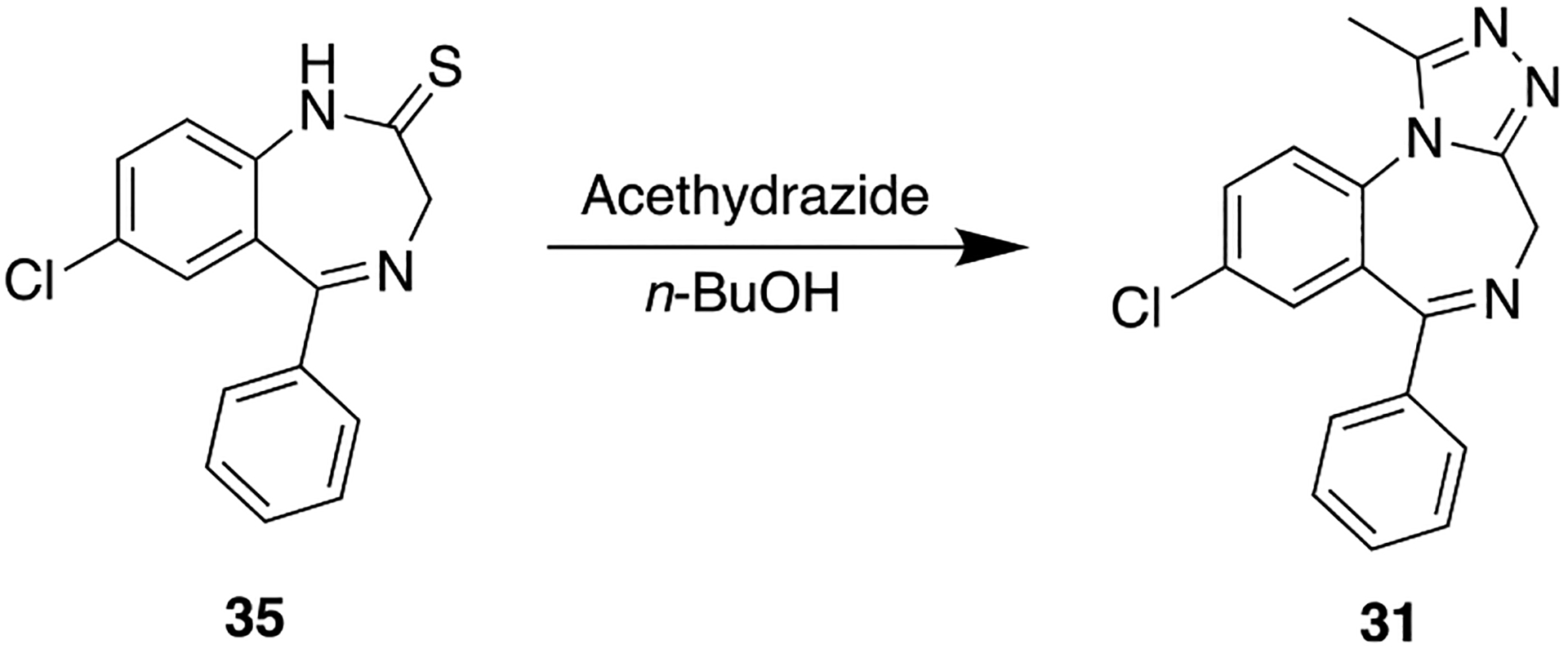
Synthesis of the Benzodiazepine Compound Alprazolam (31) from 7-Chloro-5-phenyl-1,3-dihydro-2H-benzo[e][1,4]diazepine-2-thione (35)
B. Oxadiazoles.
Oxadiazoles are members of the azole class of heterocycles composed of two carbons, two nitrogens, and one oxygen atom. The oxadiazole ring exists in several different regioisomeric forms: 1,2,4-isomers (two forms), 1,3,4-isomer (one form), and 1,2,5-isomer (one form) (Figure 11).91
Figure 11.
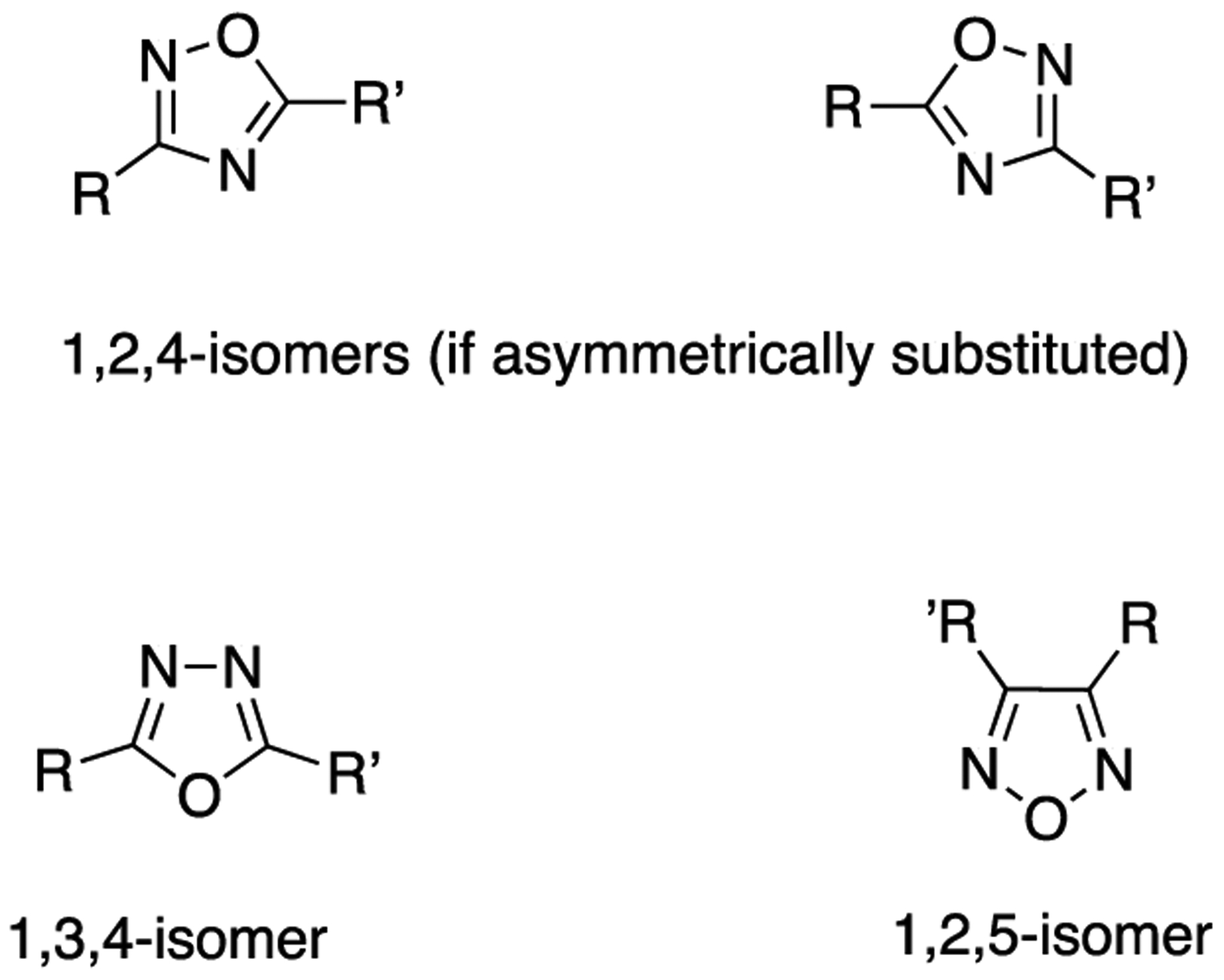
Different regioisomeric forms of the oxadiazole ring.
The oxadiazole moiety is a weak base because of the inductive effect of the extra heteroatom. Electrophilic substitutions at the carbon atom of the oxadiazole ring are difficult to accomplish due to the low electron density on the carbon atom, attributed to the electron withdrawing effect of the pyridine-type nitrogen. Electrophilic attack occurs at the nitrogen if the oxadiazole ring is substituted with an electron donating group (EDG). Oxadiazoles are largely resistant to nucleophilic attack but can undergo nucleophilic substitution in the same manner as an aliphatic sp2 carbon. These highlighted physicochemical properties of the oxadiazole motif impart versatility for its use in medicinal chemistry, and it has been utilized in the discovery of potent anti-HIV, anticancer, antimicrobial, anti-inflammatory, antidiabetic, and antiobesity agents.92 Moreover, several oxadiazole-containing molecules are known as D3-R ligands,93 benzodiazepine receptor agonists,94 muscarinic receptor agonists,95 mono-aminoxidase B inhibitors,96 selective adenosine A2B receptor antagonists,97 and 5-hydroxytryptamine (5-HT) receptor antagonists.98
Improved metabolic stability, membrane permeability, and bioavailability may be achieved with oxadiazoles, and they are often used as amide bioisosteres.92,99 Among the different regioisomeric forms, the 1,2,4-isomer and 1,3,4-isomer are most commonly employed in the bioisosteric replacement of an amide bond.100 Both of these isomers can mimic the molecular planarity and dipole moment of an amide, but being a nonclassical amide bioisostere, the heterocycle shows variation in aromatic, electrostatic, and hydrogen bonding character.101
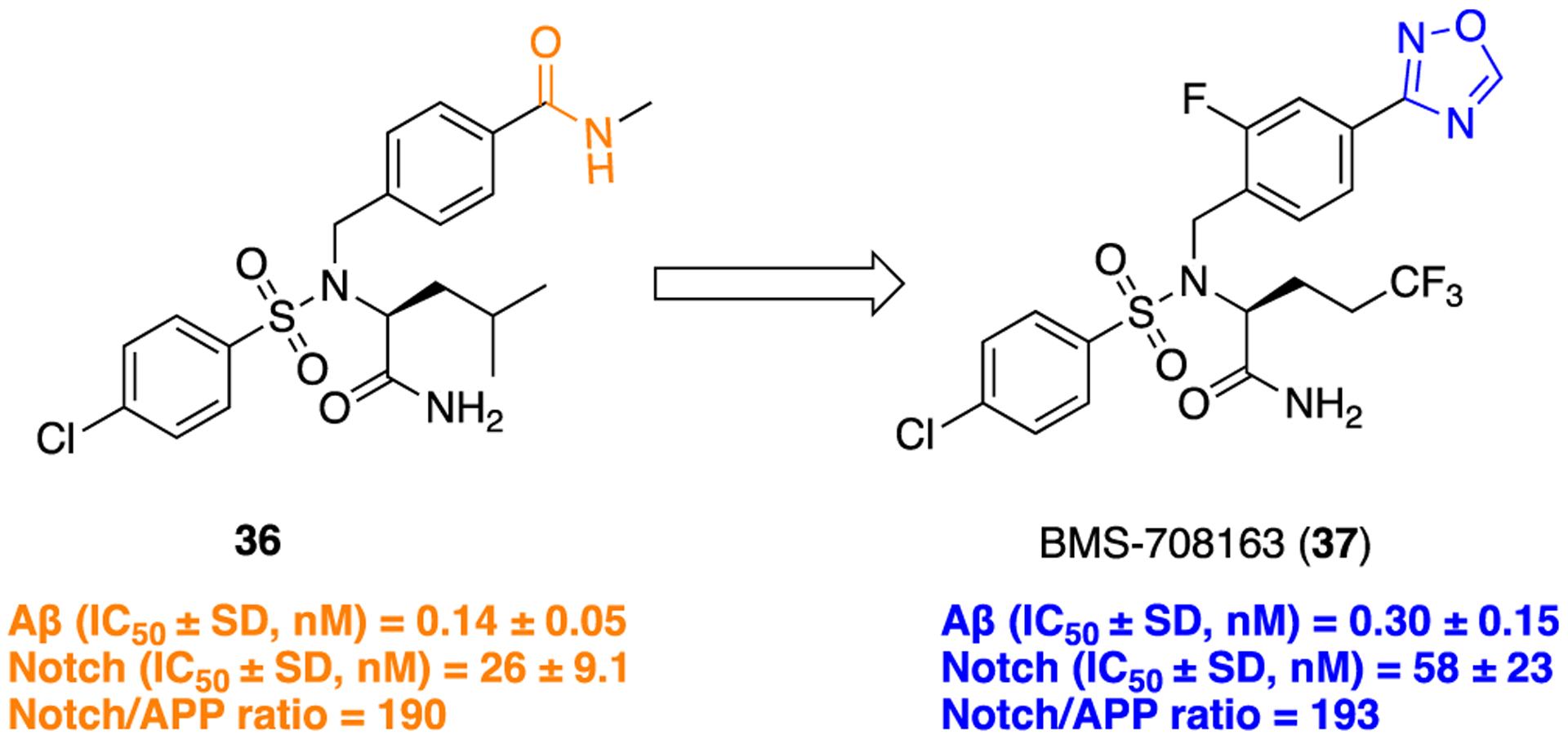
Researchers at Bristol-Myers Squibb evaluated the 1,2,4-oxadiazole ring as a bioisostere for the amide functionality within compound 36, which provided BMS-708163 (37), a compound that proved to have potent as well as selective inhibition for γ-secretase and possessed excellent PK properties in animal models as compared to the parent (Figure 12).
Figure 12.
Amide bioisosteric replacement with an oxadiazole ring leading to a potent and selective γ-secretase inhibitor.
The abnormal aggregation of the amyloid-β (Aβ) peptide is a known hallmark of Alzheimer disease (AD). Inhibition of γ-secretase is being explored to lower Aβ production in the AD patient’s brain.102 The γ-secretase enzyme also plays an essential role in the Notch signaling pathway, and the normal functioning of this pathway is important for proper development in all multicellular organisms.103,104 Therefore, the identification of a γ-secretase inhibitor with no effects at the Notch receptor is desirable.105
The oxadiazole moiety of compound 37 was introduced as depicted in Scheme 3, i. In vitro evaluation showed that compound 37 potently inhibited both Aβ40 (IC50 = 0.30 ± 0.15 nM (mean ± SD) and Aβ42 (IC50 = 0.27 ± 0.12 nM) cleavage in H4–8Sw cells. As designed, this compound demonstrated poor potency for Notch inhibition. Even though the parent amide-containing compound 36 showed potent γ-secretase inhibition, it possessed poor metabolic stability, indicating the potential for rapid in vivo clearance. In contrast, the replacement of the enzymatically labile amide bond with the oxadiazole ring significantly improved the metabolic stability of 37 [% remaining after 10 min of incubation is 32% (rat), 75% (dog), and 97% (human)]. Moreover, upon oral administration compound 37 notably reduced Aβ40 levels in the brain, plasma, and cerebrospinal fluid in both rats and dogs.
Scheme 3.
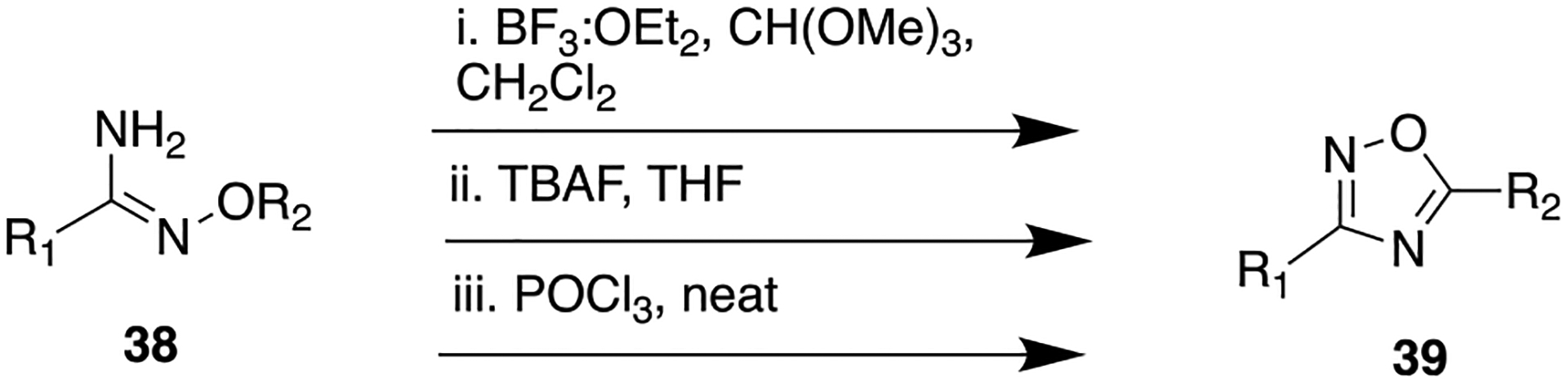
General Synthetic Route To Obtain the 1,2,4-Oxadiazole Moiety from an Amidoxime
In search of new structural lipoic acid (LA) analogs as potent neuroprotective agents, a set of 1,2-dithiolane/chroman hybrids was designed and synthesized by Koufaki et al.73 Compound 28 is an amide-containing LA structural analog which was previously reported (Figure 13).72 Initially, this compound was found to be effective against reperfusion arrhythmias and was included in this study to check its neuroprotective ability. To explore the SAR, the amide bond in 28 was replaced by different amide bioisosteres (triazole isostere 29 is discussed previously in the 1,2,3-triazole section). The general synthetic method to obtain a 1,2,4-oxadiazole bioisostere (40, Scheme 3, ii) was achieved by intermolecular cyclization of respective acyl amidoxime in the presence of tetrabutylammonium fluoride (TBAF). The neuroprotective potential of both compounds (28 and 40) was assessed in HT22 cells, challenged with glutamate to mediate excitotoxicity, which in turn could mimic cytotoxic conditions arising due to oxidative stress in AD and other neurodegenerative disorders. Compound 40 demonstrated substantial improvement in activity against glutamate-induced oxidative stress in HT22 cells (Figure 13) as compared to parent 28.
Figure 13.
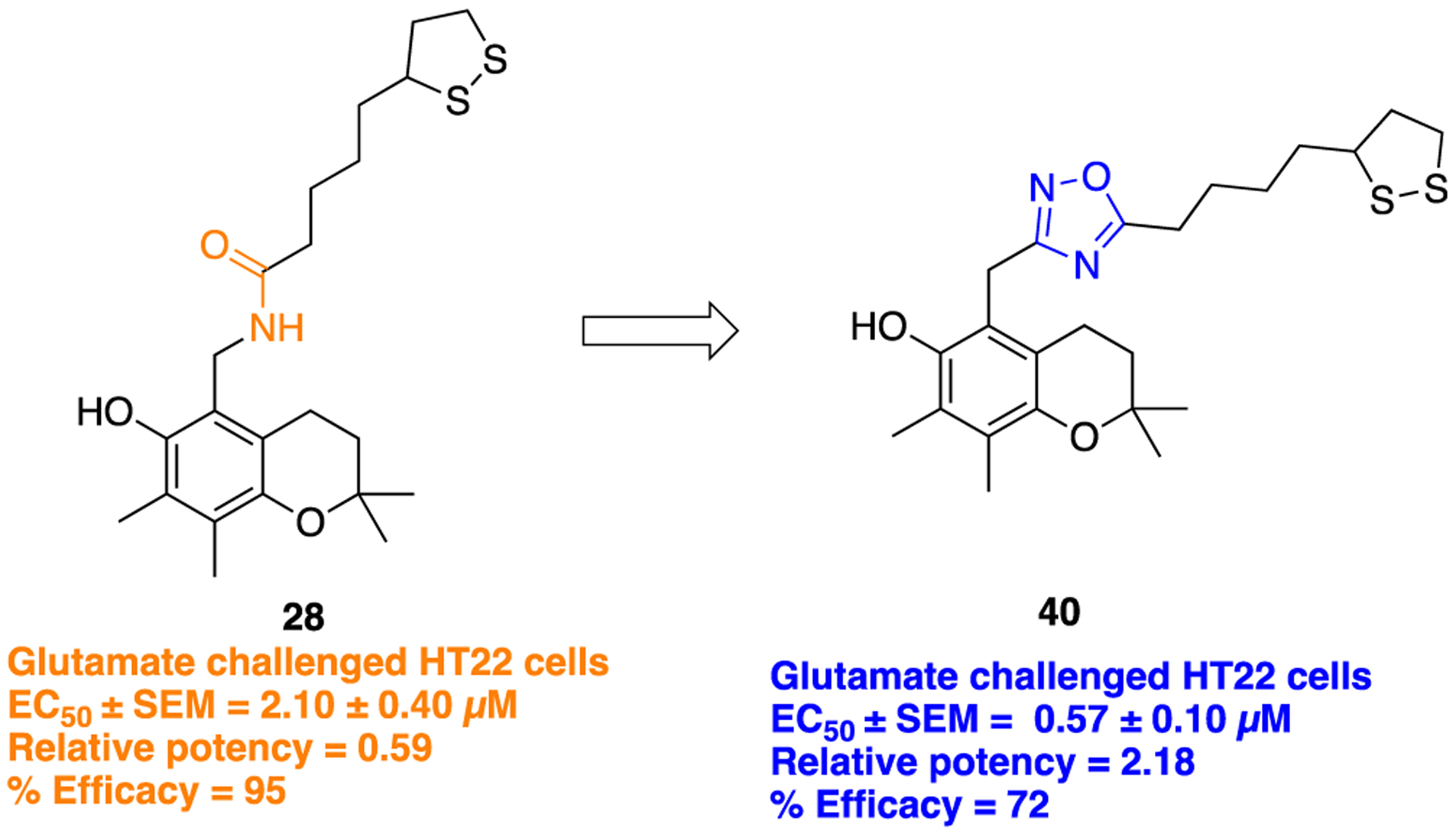
Structures of parent amide-containing α-lipoic acid analog 28 and its 1,2,4-oxadiazole bioisostere 40.
In another study reported by Koufaki et al., an amide bond replacement on chroman linked with two methylene carbons to a 3,4-dimethoxyphenyl ring (41) was used in the identification of a potent neuroprotective agent (Figure 14).106 Compound 42 bearing a 1,2,4-oxadiazole moiety was synthesized by following the general procedure as shown in Scheme 3, ii. Results of the in vitro evaluation of newly synthesized analogs, using glutamate-challenged HT22 cells, showed that several compounds displayed good neuroprotective ability, but among all, compound 42 demonstrated notable neuroprotection (EC50 = 254 ± 65 nM). Hence, the amide replacement with 1,2,4-oxadiazole proved to be successful in protecting neuronal cells from oxidative stress-induced cell death.
Figure 14.
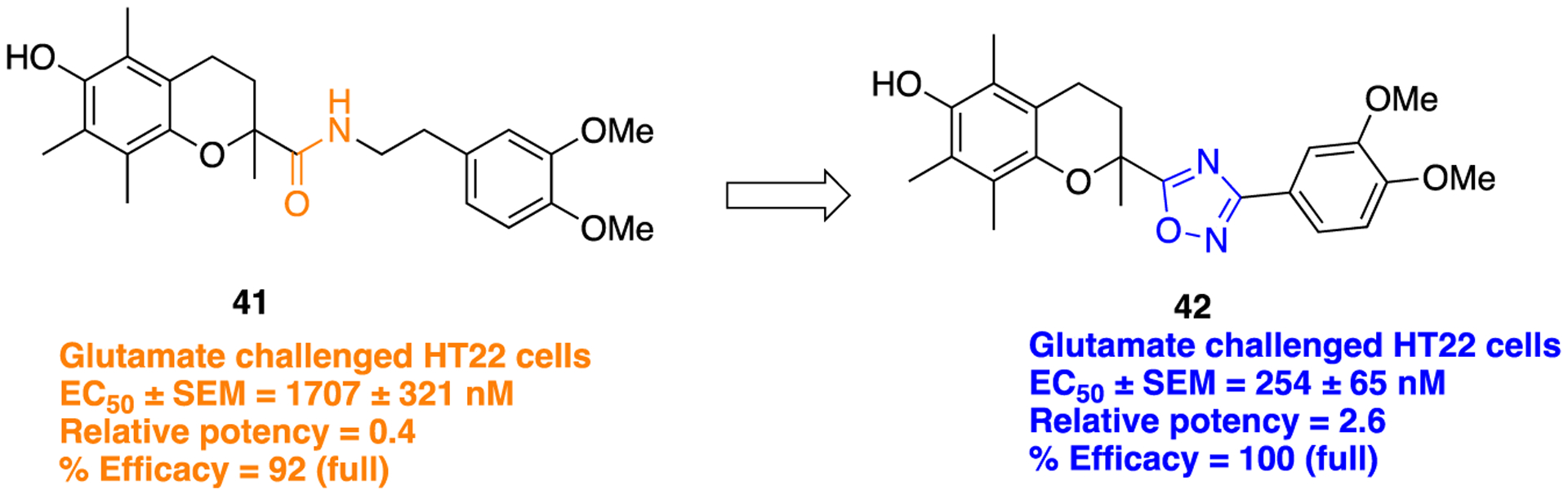
Structures of parent chroman analog 41 and its 1,2,4-oxadiazole bioisostere 42.
Another example describing the potential of the oxadiazole ring as an amide surrogate was published by the Lindsley group, aiming to discover potent metabotropic glutamate receptor subtype-7 negative allosteric modulators (mGlu7-NAMs).107 Initially, two compounds 43 and 44 were identified as potent mGlu7-NAMs.108,109 Both 43 and 44 displayed strong efficiency for the mGlu7-receptor, but their poor metabolic stability attributed to the amide linker hindered their further development (Figure 15). The authors decided to replace the amide linker with several amide bioisosteres. Success was achieved with the identification of a 1,3,4-oxadiazole in compound 45107 (Figure 15) that not only demonstrated potent mGlu7-NAM activity but also showed favorable plasma protein binding (rat fu = 0.10), low predicted hepatic clearance in microsomal incubations (rat CLhep = 27.7 mL min−1 kg−1), and high CNS penetration (rat Kp = 4.9, Kp,uu = 0.65); however, the authors did not mention the CNS penetration properties (rat fu and rat Kp) for the parent amides 43 and 44. Compound 45 exhibited a favorable selectivity index over another seven mGlu receptors (>10 μM vs mGlu1–6, 8). As shown in Scheme 4, 2-(1H-imidazol-1-yl)-5-(trifluoromethoxy)benzoic acid (46) undergoes a propylphos-phonic anhydride mediated microwave-assisted condensation of 3,4-dimethoxybenzohydrazide acid to furnish 45.
Figure 15.
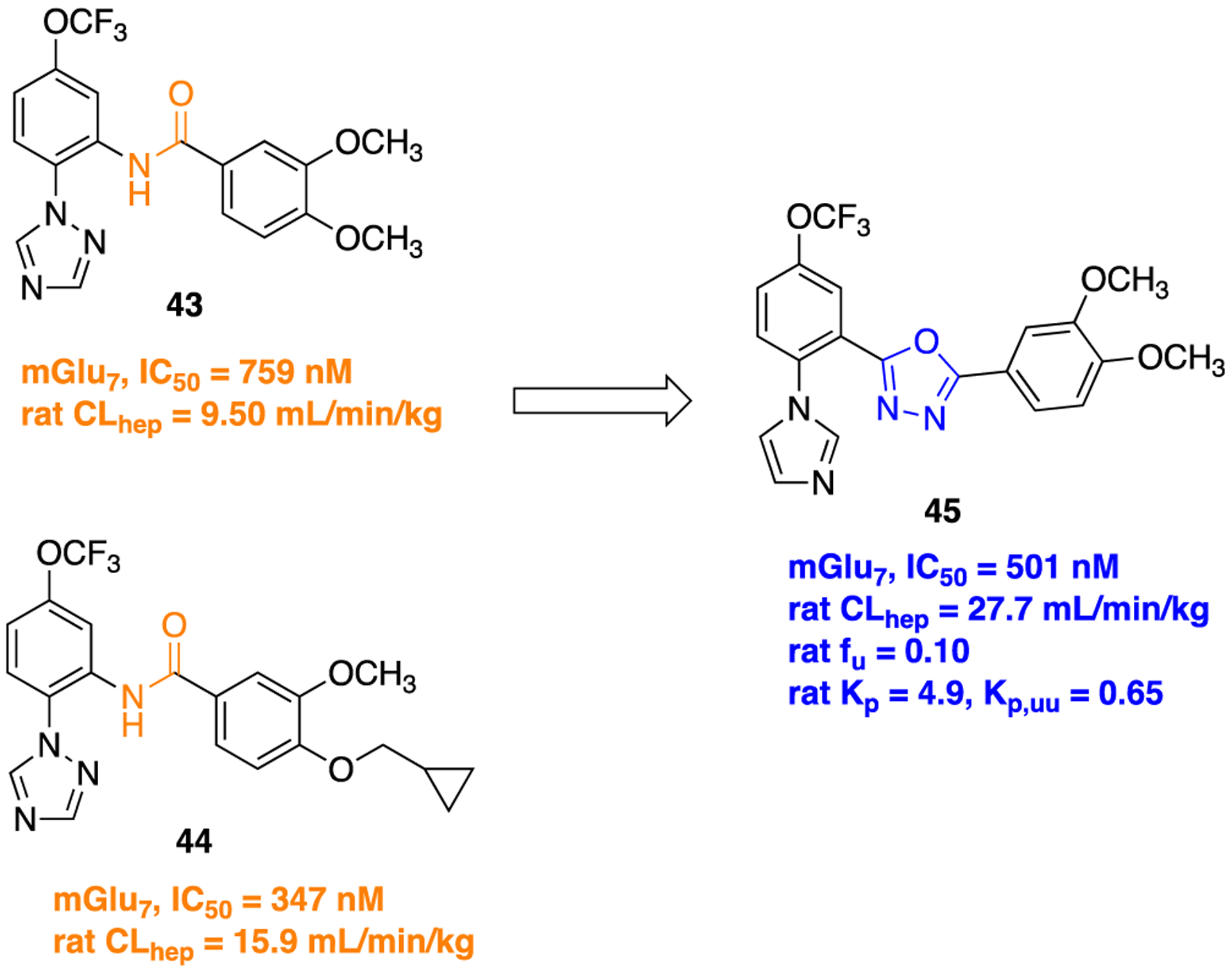
Structures of parent amides 43 and 44, potent mGlu7-NAMs and the 1,3,4-oxadiazole bioisostere 45.
Scheme 4.

Synthetic Preparation of 45, a Potent and Selective mGlu7-NAM
The replacement of an amide with different isomeric forms of oxadiazole has been employed to improve metabolic stability of the parent compound in numerous bioactive compounds. One efficient example of this is found in β-homophenylalanine-based dipeptidyl peptidase enzyme inhibitors.110 The inhibition of dipeptidyl peptidase-4 (DPP-4) is linked to augmentation of the biological activity of the “incretin” hormones (glucagon-like peptide 1; GLP-1) and glucose-dependent insulinotropic polypeptide (GIP), which in turn is responsible for diminishing the pathophysiological symptoms of diabetes.111 Hence, DPP-4 inhibitors, or gliptins, emerged as a promising therapeutic target for novel oral antihyperglycemic agents. Initially, the Santhera group discovered β-homophenylalanine pyrrolidine-2-ylmethylamide derivative (47), a potent as well as selective DPP-4 inhibitor (Figure 16). Unfortunately, 47 suffered from high efflux ratio and low Caco-2 permeability, accounting for its poor PK properties. The bioisosteric replacement of the problematic amide employed a 1,2,4-oxadiazole and related heterocycle. Some success was achieved with the identification of two different isomeric oxadiazole containing compounds (48 and 49). Both compounds were found to be nearly equipotent with the parent (47) in DPP-4 inhibition assays, with slight improvement in vitro metabolic stability. Therefore, on the basis of these obtained biological results, new analogs containing an isomeric oxadiazole moiety with different substituents, which could provide long plasma half-lives to the molecule, were designed and synthesized. The adopted drug modification approach proved efficacious and led to the development of 50 and 51, which showed excellent DPP-4 inhibitory potential in vitro and improved oral glucose tolerance in ob/ob mice and also exhibited attractive PK profiles (Figure 16). The synthetic methods for the preparation of both 50 and 51 are depicted in Scheme 5.
Figure 16.

Structural optimization in the development of potent DPP-4 inhibitors to treat type 2 diabetes mellitus.
Scheme 5.
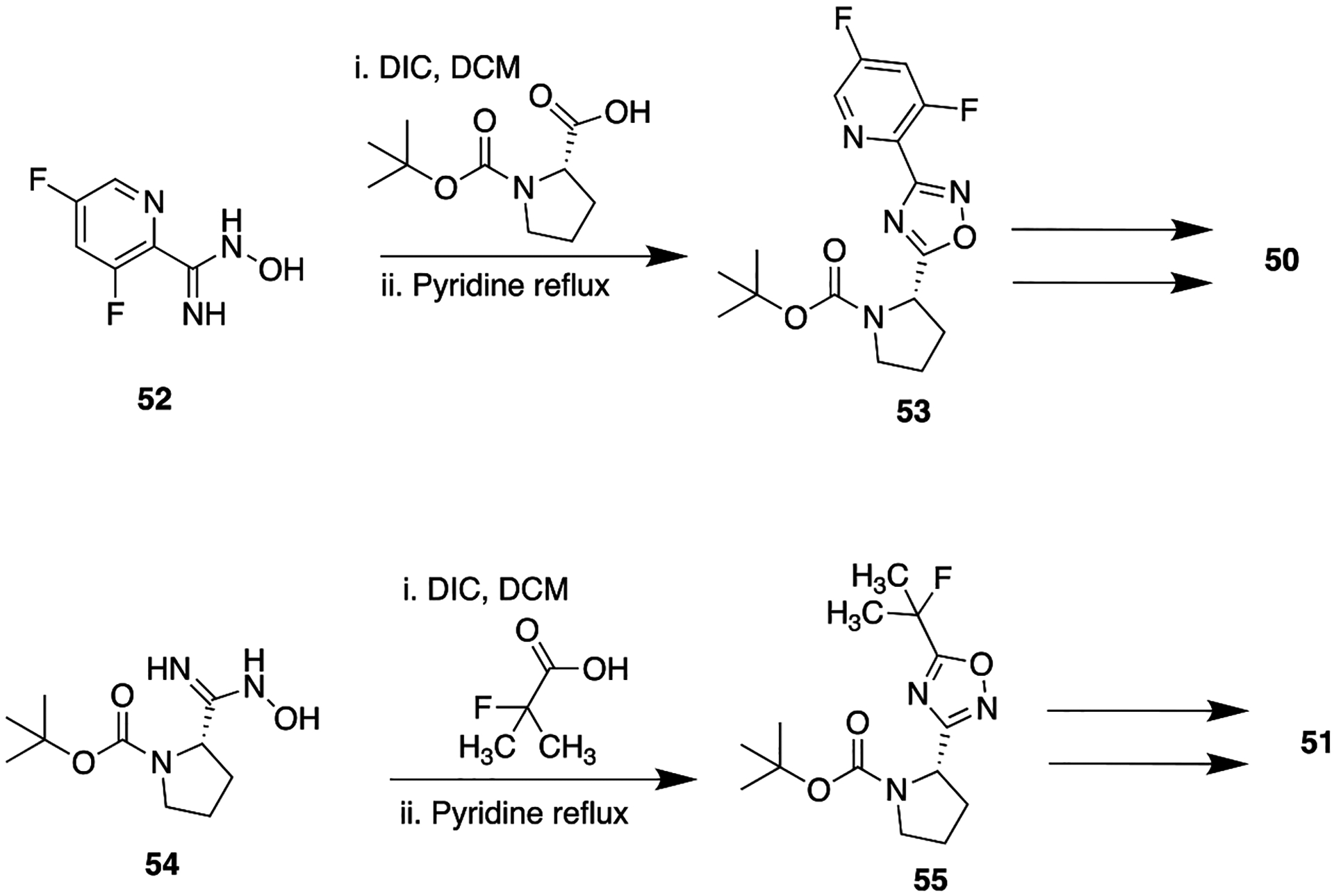
Synthetic Preparation of 50 and 51, Potent DPP Inhibitors
The final reaction in mammalian triglyceride synthesis is catalyzed by two acyl CoA:diacylglycerol acyltransferase (DGAT) enzymes (DGAT-1 and DGAT-2); out of these, inhibition of DGAT-1 is known as a feasible strategy for the treatment of diabetes and obesity.112 Recently, researchers at the Novartis Institutes of Biomedical Research have explored the 1,3,4-oxadiazole moiety as an amide bioisostere (56, Figure 17) in search of new potent DGAT-1 inhibitors.113
Figure 17.
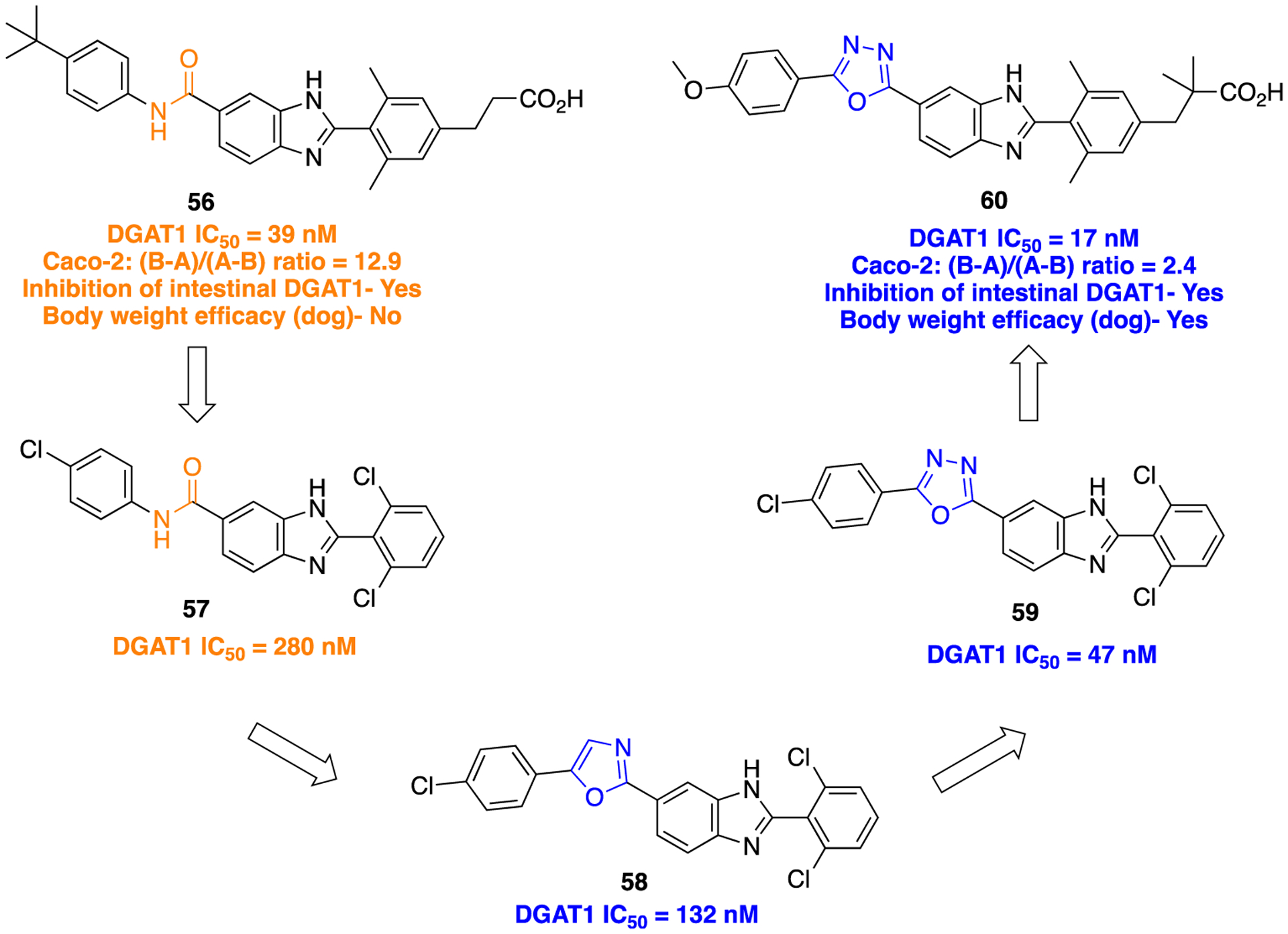
Lead optimization journey to discover a potent and orally bioavailable DGAT-1 inhibitor.
Initially compound 56, a benzimidazole derivative containing an amide linker joining the western phenyl ring, was determined to be a potent inhibitor of DGAT1.114 Besides its potent DGAT-1 inhibitory activity (IC50 = 39 nM; C2C12 mouse myoblast cellular assay), this compound has shown a low A–B value (0.7 × 10−6 cm/s) in the Caco-2 permeability assay and a low oral bioavailability profile in vivo. To improve membrane permeability and reduce efflux of the parent 56, the initial structural changes comprised bioisosteric replacement of the amide in 57 with oxazole (58) and oxadiazole (59), along with modifications on the carboxylic acid-containing fragment. The SAR studies revealed that replacement of the amide with oxazole and oxadiazole moieties, combined with removal of the propionic acid moiety, provided potent DGAT1 inhibition (Figure 17). On the basis of this SAR, the oxadiazole moiety was chosen as the amide surrogate to incorporate into this particular chemotype. The general synthetic method to furnish 1,3,4-oxadiazole bioisostere (as in 60) using Burgess reagent is shown in Scheme 6, i. Overall, these efforts lead to the development of a benzimidazole derivative containing a 1,3,4-oxadiazole moiety, 60, which displayed excellent DGAT-1 inhibition (Figure 17). Moreover, this compound desirably showed poor inhibition for DGAT-2 (IC50 > 10 μM) and a low risk of mutagenicity in the Ames assay. Compound 60 suppressed postprandial plasma triglycerides in a dose-dependent manner in healthy human subjects.
Scheme 6.
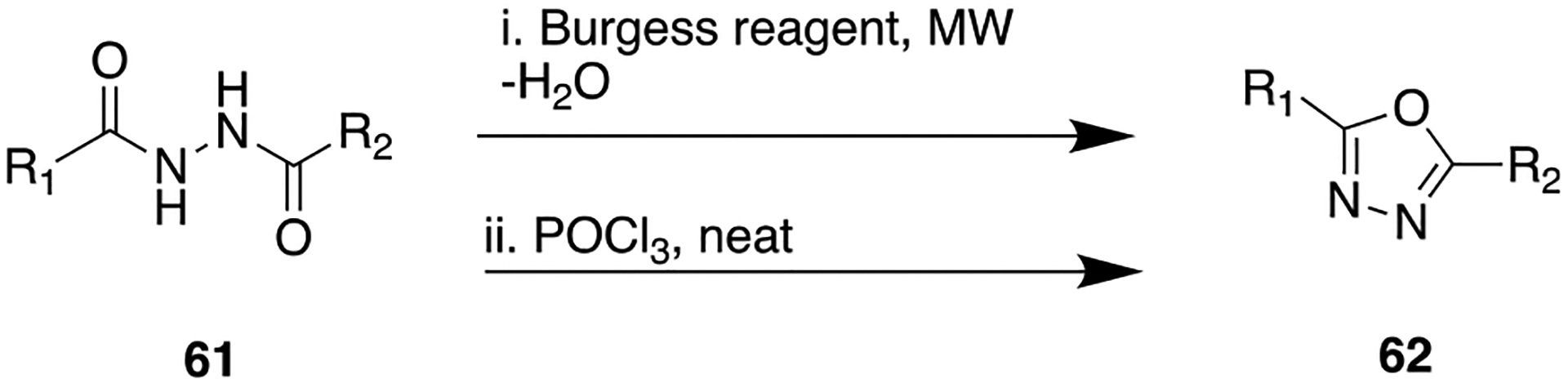
General Synthetic Route for Preparing 1,3,4-Oxadiazole from Diacylhydrazine
In the optimization of a series of previously identified 6-substituted 4-quinolone-3-carboxamides as highly selective and potent CB2 receptor ligands by Corelli and colleagues,115 the poor solubility profile of these compounds which hampered biological assays led to the use of a bioisosteric approach to give high-affinity CB2 receptor ligands with good selectivity, along with acceptable physicochemical properties. In a follow-up study, the amide bond present in the central core of the parent compounds116,117 63 and 64 was replaced by three different amide surrogates: a 1,2,3-triazole, a 1,3,4-oxadiazole, and a 1,2,4-oxadiazole. Second, the quinolone ring was also substituted with 2-furyl, 4-bromo, and 4-isopropoxyphenyl moieties to derive the SAR (Figure 18).68
Figure 18.
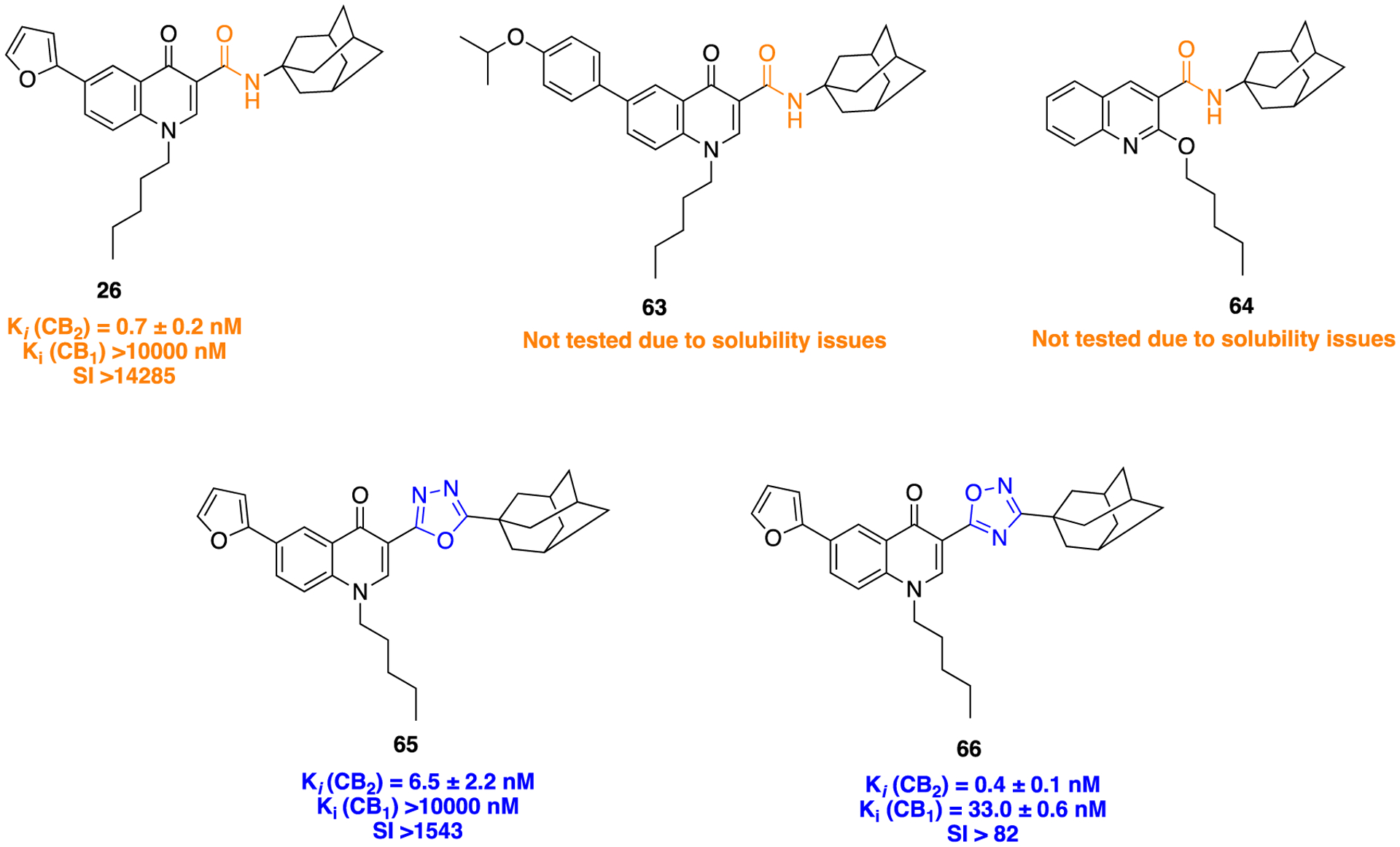
Bioisosteric amide replacement via 1,3,4-oxadiazole (65) and 1,2,4-oxadiazole (66) in a series of CB2 receptor ligands.
Results of this experiment showed that the 1,2,3-triazole-containing derivative 27 (discussed in the 1,2,3-triazole section above) emerged as the most potent CB2 receptor ligand with promising PD and physicochemical properties. Apart from this, the 1,3,4-oxadiazole (65) and 1,2,4-oxadiazole (66) derivatives showed notable CB2 receptor binding affinity of 6.5 ± 2.2 nM and 0.4 ± 0.1 nM, respectively (Figure 18). Unfortunately, the latter (66) exhibited poor selectivity over the CB1 receptor (80-fold), while compound 65 was nearly 1500-fold more selective to the CB1 receptor over CB2. Overall results were compared with lead compound 26, which showed potent CB2 receptor binding affinity with 14,000-fold selectivity over CB1 receptor. The general synthetic routes to access compounds 65 and 66 are shown in Scheme 6, ii, and Scheme 3, iii, respectively.
C. Imidazole.
Arguably the most successful example of amide versus imidazole bioisosterism is presented in the discovery of midazolam (67) at Hoffmann-LaRoche, Inc., U.S.118 Compound 67 or 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine is a structural analog of 30, in which the amide bond has been successfully replaced by an imidazole moiety (Figure 19). This structural modification increased the basicity of the molecule due to the imidazole ring nitrogen, which allowed the formation of water-soluble salts for pure aqueous injectables.119 Moreover, it undergoes rapid metabolic inactivation as compared to 30 and the other classical benzodiazepines which is attributed mainly to the presence of the methyl group appended to the imidazole ring. This results in a short window of action allowing greater clinical management options.
Figure 19.
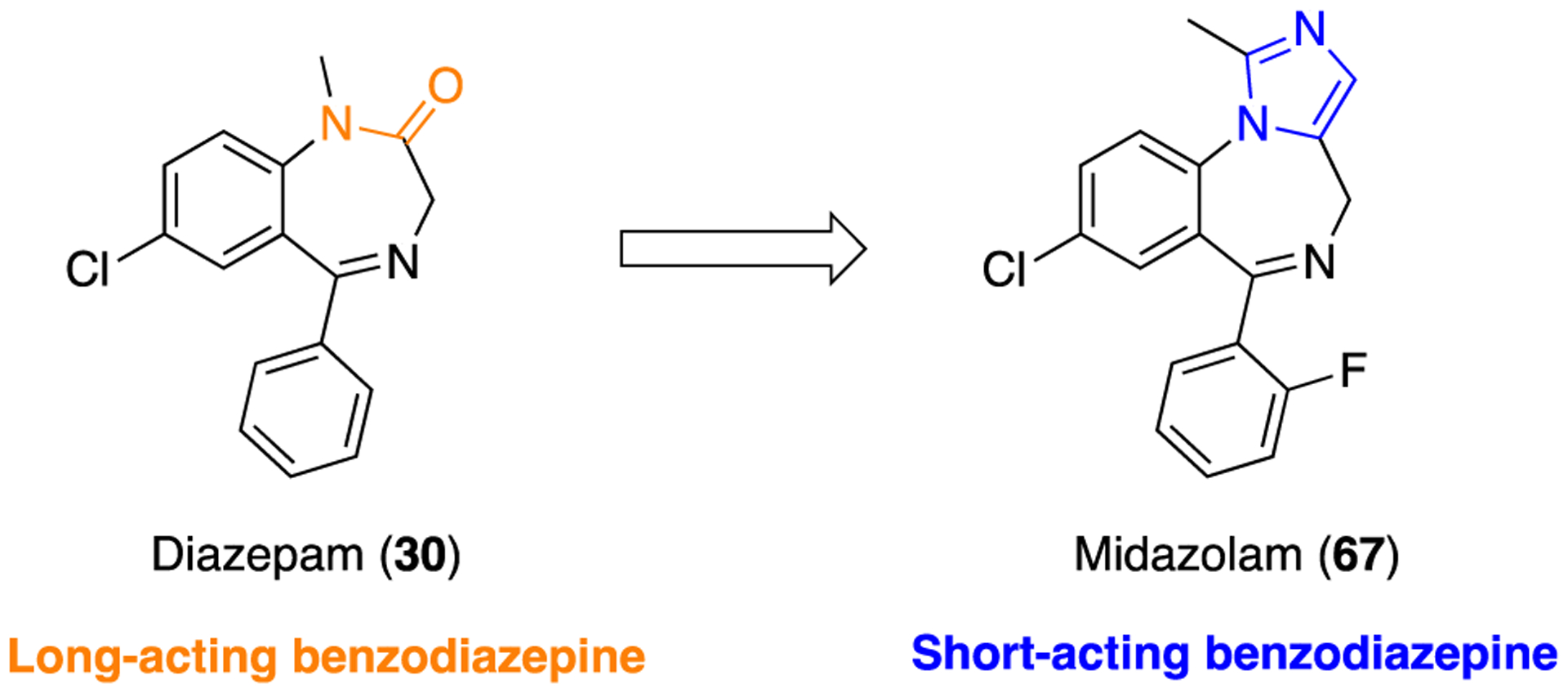
Structures of diazepam and the imidazole-containing analog midazolam.
Very recently, Taghizadeh et al. reported a one-pot condensation of benzodiazepines with the monoanion of tosylmethyl isocyanide or ethyl isocyanoacetate under mild conditions, which provides rapid synthetic access to 67 (Scheme 7).120 At present, midazolam is used for the treatment of seizures,121 irregular sleeping patterns,122 severe agitation, procedural sedation, and anesthesia.123
Scheme 7.
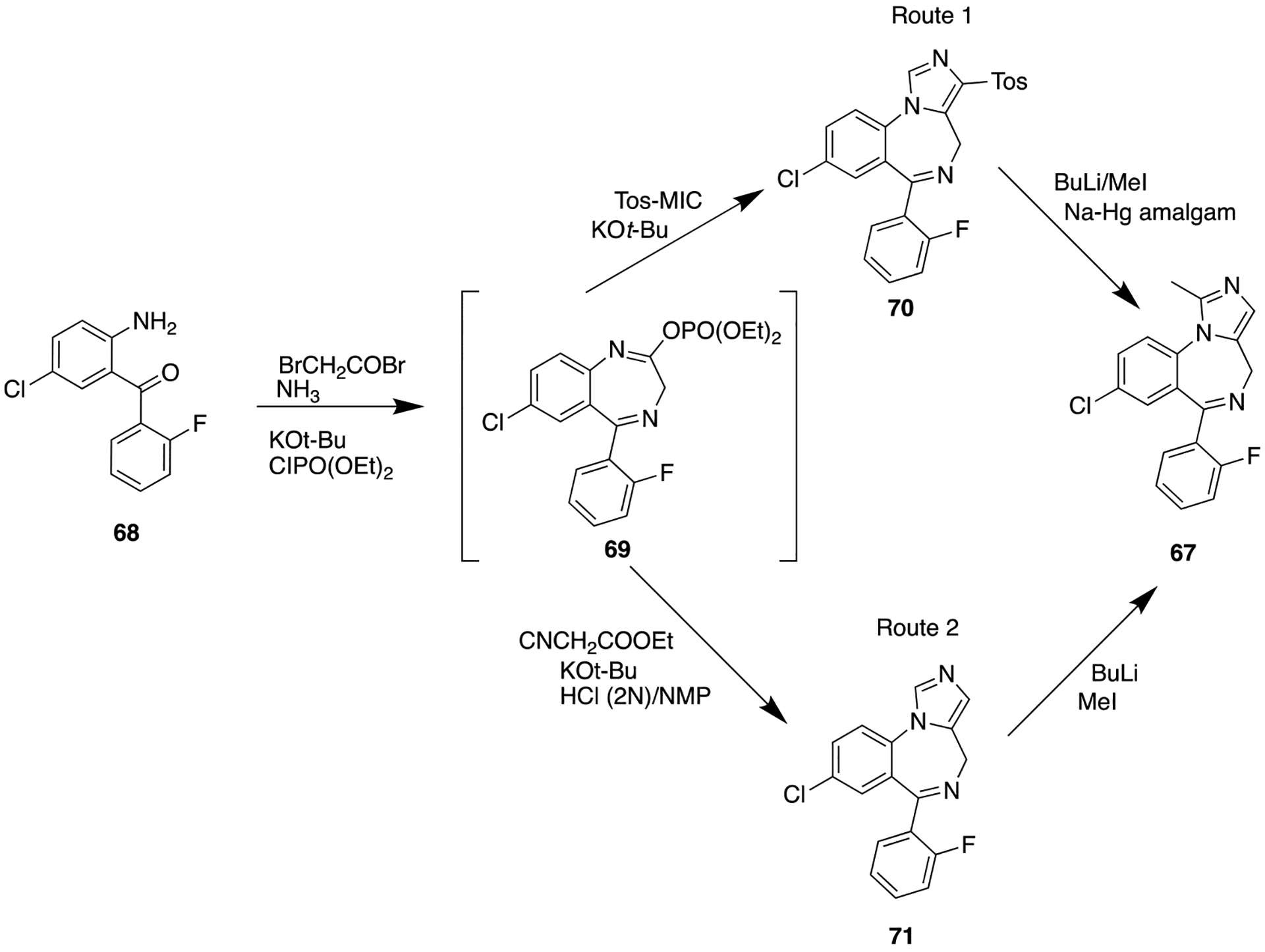
One-Pot Synthesis of Midazolam, 67, a Short Acting Benzodiazepine
Several preclinical and clinical studies have shown that blocking the calcitonin gene related peptide (CGRP) receptor could be beneficial to discover therapeutic agents for preventive treatment of migraine.124–126 Merck Research Laboratories previously reported a potent, orally bioavailable CGRP antagonist, telcagepant (MK-0974, compound 72), for the treatment of migraines.127,128 Further improvements in potency and PK properties of this lead compound 72 were sought via the isosteric replacement of the caprolactam core of 72 with several heterocyclic rings.129 It was assumed that the inclusion of the heterocyclic ring in place of the caprolactam core of 72 would improve solubility and reduce plasma protein binding, resulting in lower serum shifts. Initially, one fused triazole derivative (compound 73) showed comparable CGRP antagonistic potencies in CGRP binding assay and in related cell-based functional assays to 72 (Figure 20).
Figure 20.
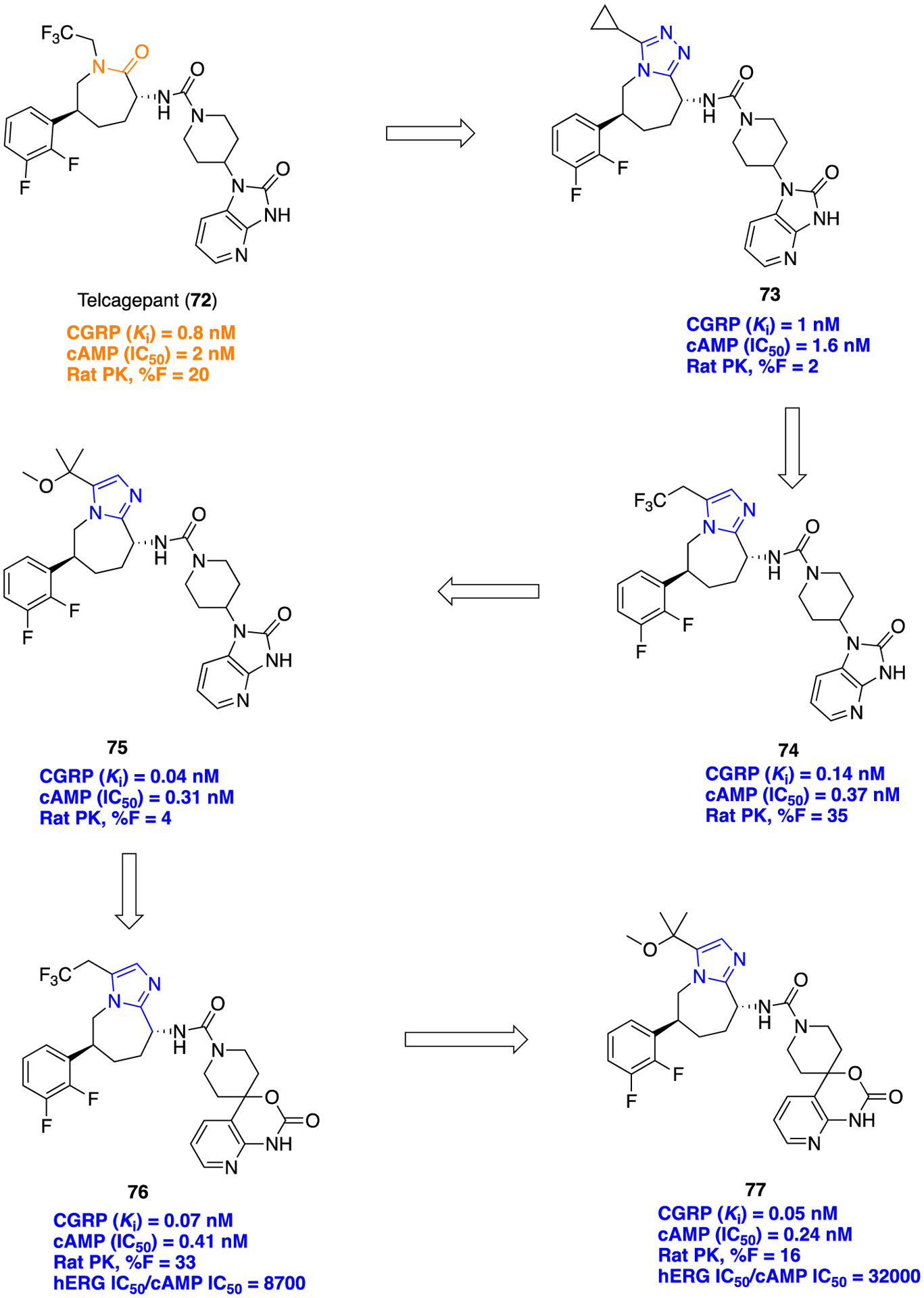
Lead optimization journey of telcagepant (72) to discover 77, a potent and orally bioavailable CGRP antagonist.
Unfortunately, 73 displayed poor PK properties in rats (F = 2%), and low permeability (Papp= 4 × 10−6 cm/s) compared to 72 (rat F = 20%; Papp = 21 × 10−6 cm/s). Upon further optimization, a fused imidazole containing molecule 74 showed the highest augmentation in permeability (Papp= 24 × 10−6 cm/s) and also improved rat oral bioavailability (F = 35%). On the basis of these encouraging results, several structural analogs of 74, having different substituents at both C5 and C4 on the imidazole ring, were synthesized and tested for their in vitro CGRP antagonistic activities. Several structural analogs of 74 have shown potent CGRP antagonistic activity in the nanomolar range (structures not shown); out of them, compound 75 (Figure 20) showed improved potency compared to 74. Despite excellent CGRP antagonistic binding affinity (Ki = 0.04 nM), 75 presented poor rat oral bioavailability (F = 2%). Persistent efforts in finding potent CGRP antagonist with desirable PK properties were continued, and consequently, azaoxindole and azabenzoxazinone-containing analogs were synthesized. Among this series of analogs, compound 76 maintained good potency and showed improved bioavailability in three species, with rat (F = 33%), dog (F = 28%), and rhesus (F = 17%). But in another in vivo model, rhesus capsaicin induced dermal vasodilation (CIDV) assay; 76 unexpectedly showed lower efficacy. Moreover, unfavorable human ether-a-go-go-related gene (hERG) activity (IC50 = 3.3 μM) of 76 impeded its development. The next challenge was to overcome hERG related undesirable activity, and therefore, structural analogs of 76 were prepared by varying different oxygenated substituents on the fused imidazole ring. The synthetic approach to provide 77 containing imidazoazepane scaffold is shown in Scheme 8. Among these tested analogs, compound 77 consists of a tertiary methyl ether which displayed excellent CGRP antagonistic activity, acceptable oral bioavailability, and diminished hERG activity. Overall, this fascinating lead optimization journey of 72 has discovered the most promising compound 77 as a preclinical candidate for the treatment of acute migraine.
Scheme 8.
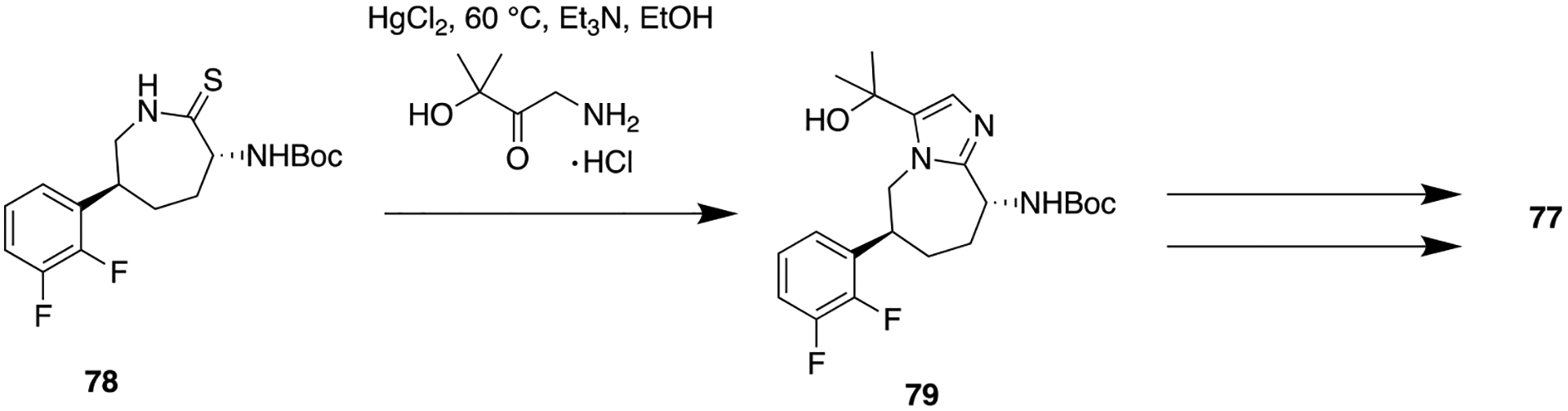
Synthetic Route for Compound 77
Very recently, a series of heterocyclic pyrimidinedione-based HIV-1 integrase inhibitors were prepared and tested by Peese and co-workers at Bristol-Myers Squibb, USA.130 The purpose of this study was to search for a potent HIV-1 integrase inhibitor that can resolve the issues for the development of resistance mutations associated with the first generation integrase inhibitors raltegravir and elvitegravir. Recently, two new integrase strand transfer inhibitors (INSTI’s), dolutegravir and bictegravir, gained FDA approval, and because of their reduced resistance mutation profile, they have been considered second generation integrase inhibitors.131 Raltegravir, 80, was discovered by Merck Research Laboratories132 in 2008, and BMS-707035, 81, was reported in 2018 by Bristol-Meyers Squibb as a potent HIV-1 strand transfer inhibitor.133 By considering the binding patterns of 80 and 81 into the integrase enzyme, Bristol-Meyers Squibb researchers decided to replace the amide within the pyrimidinedione chemotype (as present in 80 and 81) with different bioisosteres to improve activity against first-generation-resistant mutants while maintaining activity against wild-type integrase. New synthesized azole heterocyclic derivatives were initially screened for antiviral activity against the NL4–3 virus in a cellular assay. The SAR analysis revealed that oxazole, thiazole, and imidazole containing heterocycles showed potent activity against wild-type virus either equipotent or better than the parent amide, 81. However, with pyrazole, oxadiazole, and triazole, the antiviral activity was lost. These compounds were also tested for their activity toward the first-generation resistance mutations Q148R, G140S/Q148H, and N155H. The imidazole containing derivative 82 presented desirably increased potency compared with parent 81 (Figure 21). Modeling studies within the active site of the PFV (3S3N) wild-type integrase binding pocket revealed that the benzyl group of 81 is tightly positioned between Q148 and E152 (Figure 22). However, compound 82 showed a structural difference between the binding site of the wild-type integrase and the G140S/Q148H mutant. In order to provide better insights toward the altered potential of compound 82 to chelate Mg2+, bite angle (θ) and N-partial charge of the Mg2+-coordinating nitrogen were calculated and compared with 81. Overall, modeling results suggested that the wider geometry of imidazole 82 provided optimum Mg2+ binding which resulted in better steric occupancy of the mutant active site (Figure 22), and this can be correlated with the improved resistance toward G140S/Q148H.
Figure 21.
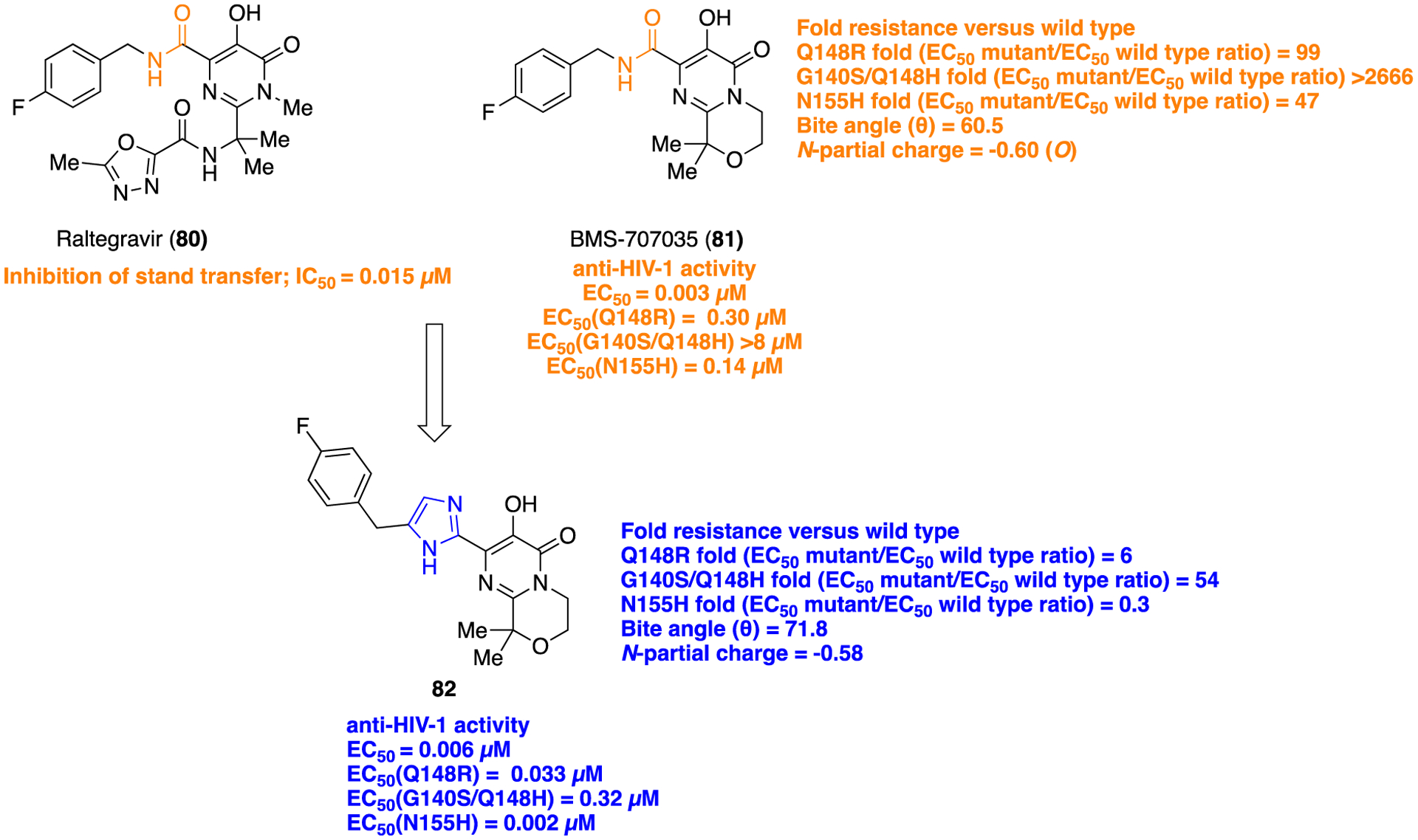
Structures of 80, 81, and imidazole bioisostere 82, potent HIV-1 integrase inhibitors.
Figure 22.
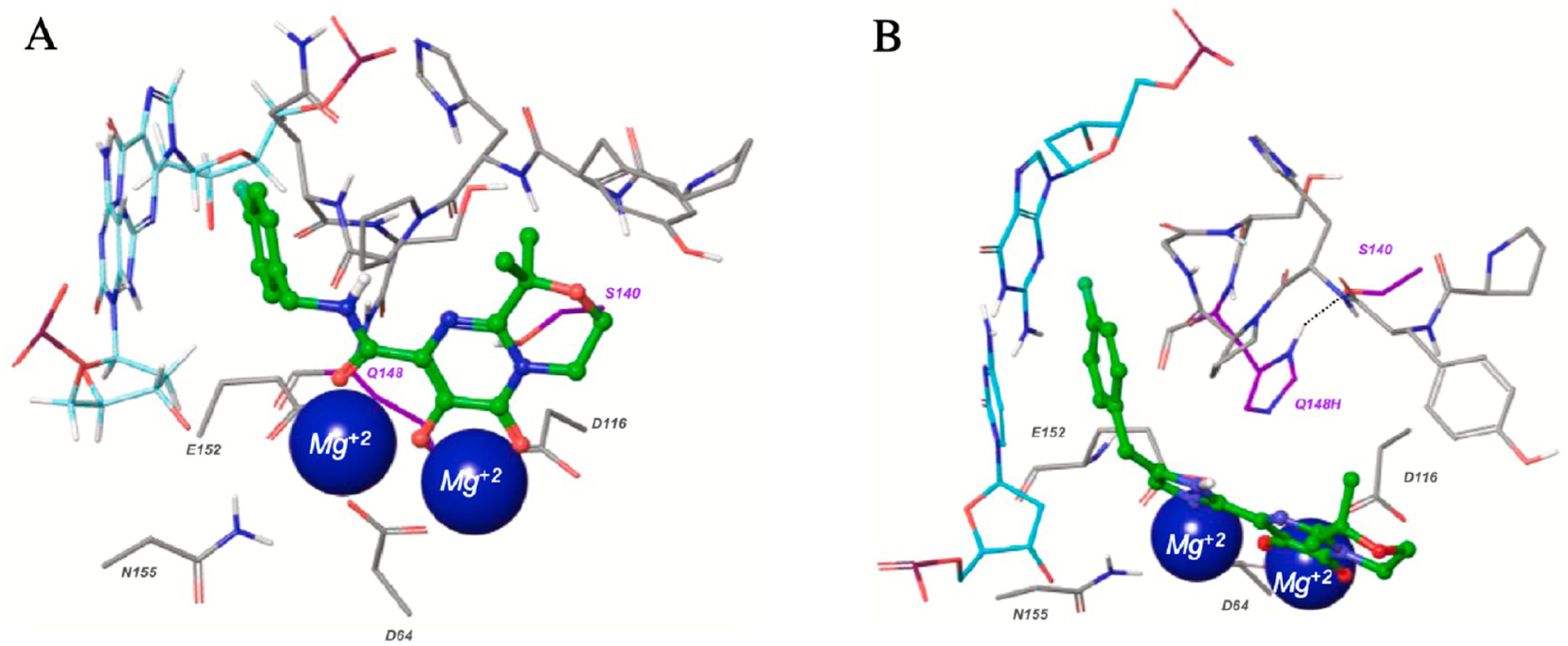
Binding patterns of 81 and 82 into the active sites of PFV integrase (3S3N) wild-type (A) and Q148H/G140S mutant (B). Reproduced with permission from Bioorganic & Medicinal Chemistry Letters (https://www.sciencedirect.com/journal/bioorganic-and-medicinal-chemistry-letters), Volume 30, 126784, Peese, K. M.; Naidu, B. N.; Patel, M.; Li, C.; Langley, D. R.; Terry, B.; Protack, T.; Gali, V.; Lin, Z.; Samanta, H. K.; Zheng, M.; Jenkins, S.; Dicker, I. B.; Krystal, M. R.; Meanwell, N. A.; Walker, M. A., Heterocycle amide isosteres: an approach to overcoming resistance for HIV-1 integrase strand transfer inhibitors.130 Copyright 2020 Elsevier.
The synthetic procedure to access 82 is delineated in Scheme 9. The authors assumed that an inhibitor with a wider geometry than parent 81 would provide better binding into the active site of first-generation resistant integrase. Results of fold resistance versus wild-type (EC50mutant/EC50wt) ratio, bite angle (θ), and N-partial charge, calculated using Schrodinger Maestro Elements, revealed that 82 improved resistance coverage due to its wide geometry and optimum Mg2+ binding into the mutant active site. In rat PK studies 82 displayed low clearance (CL (L min−1 kg−1) = 10) and very low volume of distribution (Vss (L/kg) = 0.3), resulting in moderate plasma exposure. Overall, the imidazole motif was identified to be a promising amide bioisostere, and the viability of this class could be used for further structural modification to the parent pyrimidinedione series.
Scheme 9.
Synthetic Route To Access Imidazole Pyrimidinedione-Containing Derivative 82
Stearoyl-CoA desaturase (SCD) has two isoforms, SCD1 and SCD5, in humans. Stearoyl-CoA desaturase 1 (SCD1) plays a critical role in regulating skeletal muscle fat metabolism.134 High SCD activity has been connected with several diseases, including diabetes, obesity, atherosclerosis, cancer, and viral infection. Therefore, inhibition of SCD1 emerged as a novel approach for the treatment of metabolic diseases, cancer, and viral infections. Shaoyi and colleagues reported thiazolylimidazolidinone-based SCD1 inhibitors and evaluated their potential for treating metabolic diseases.135 Initially, the Xenon group identified 2-aminothiazole-based SCD1 inhibitor 85 with moderate activity (mouse SCD1; IC50 = 3400 nM), and structural modification of 85 identified the more potent SCD1 inhibitor 86 with an IC50 (mouse) of 512 nM. Unfortunately, compound 86 showed poor human SCD1 activity determined by human liver cancer (HepG2) cell assay (IC50 = 2677 nM), and it was assumed that amide moieties at the C2- and C4-position of the thiazole core are the potential contributors for its poor permeability or stability in HepG2 cells. In order to improve selectivity and metabolic stability, as well as PK properties, the replacement of the amide bond at the C2-position in compound 86 (Figure 23) was undertaken by introduction of different five- or six-membered heterocycles.
Figure 23.
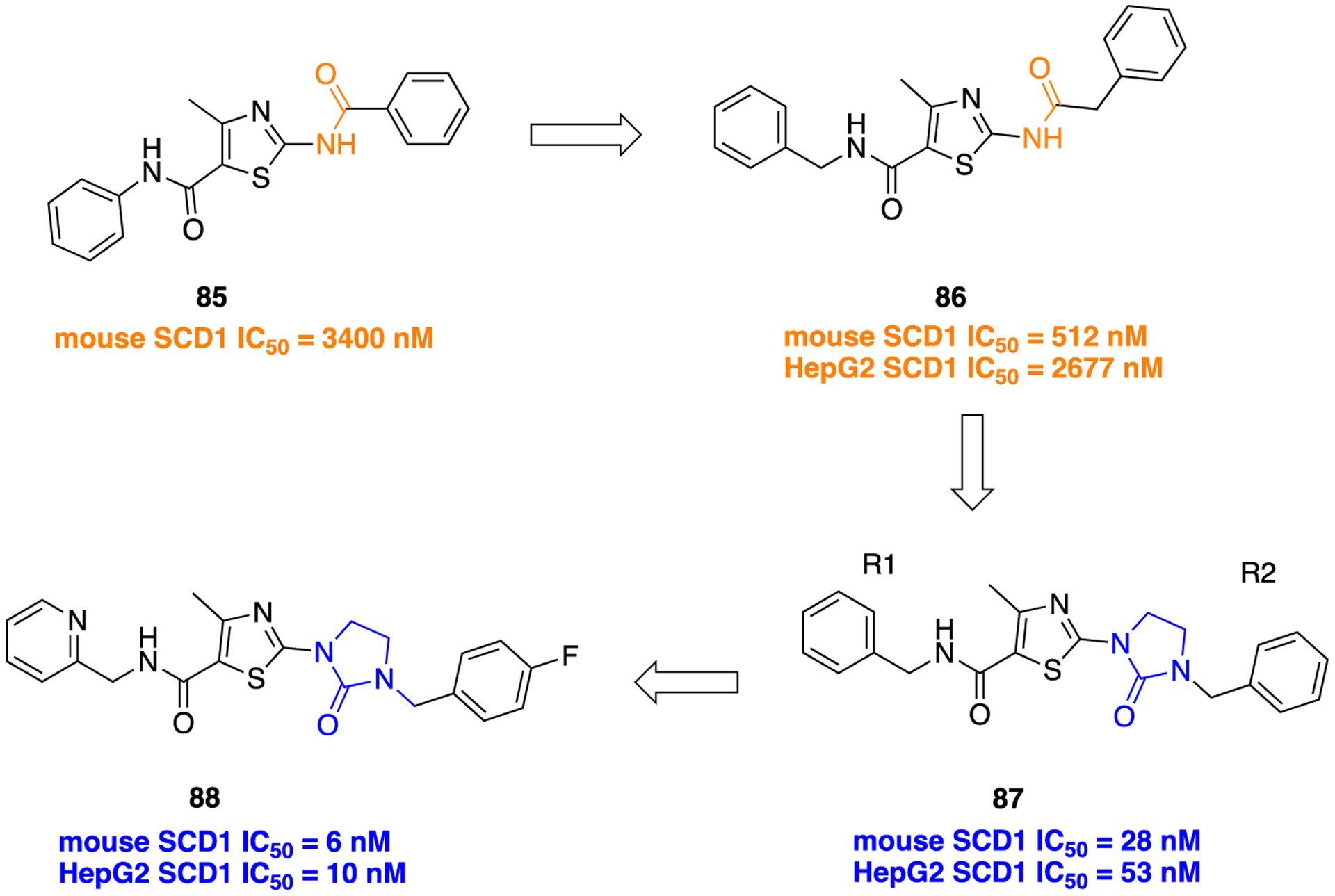
Bioisosteric replacement of the amide functional group in the identification of potent SCD1 inhibitors.
From this series of analogs, compound 87, an imidazolidinone-containing bioisostere, was identified as the most potent inhibitor in both the mouse SCD1 assay (IC50 = 28 nM) and the HepG2 SCD1 (IC50 = 53 nM) assay, but its metabolic stability was poor (10% remaining after 30 min incubation with 0.5 mg/mL rat liver microsomes). To overcome this, SAR studies continued with the thiazolylimidazolidinone (87) as the center core structure and variation of substituents on both the left (R1) and right (R2) side in the new analogs.
A new SAR was derived from these analogs that showed homologation at R1 by just one carbon led to drastically decreased SCD1 activity. In contrast, on the right side, a longer chain length up to propyl was able to maintain the SCD1 inhibitory activity. As illustrated in Scheme 10, imidazolidinone bioisostere was prepared by reacting ethyl 2-amino-4-methylthiazole-5-carboxylate (89) with 2-chlororethyl isocyanate followed by in situ cyclization in the presence of potassium carbonate. Next, further alkylation of 90 with 4-fluorobenzyl bromide afforded the substituted imidazolidinone moiety (88). Compound 88 (R1= pyridine-3-ylmethyl; R2 = 4-fluorobenzyl) resulted in the most potent SCD1 (mouse SCD1 IC50 = 6 nM; HepG2 SCD1 IC50 = 10 nM) activity along with acceptable rat liver microsomal stability (67% remaining after 30 min incubation with 0.5 mg/mL rat liver microsomes). However, in the resultant clinical studies of 88, adverse effects related to SCD1 inhibition were observed that impeded further development.
Scheme 10.
Synthetic Route for Compound 88
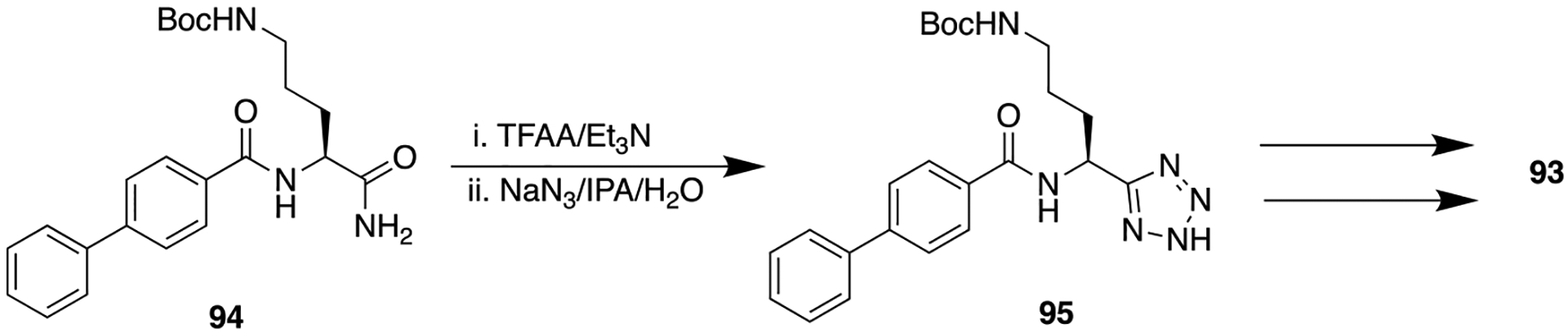
D. Tetrazole.
The tetrazole ring is frequently used as a metabolism-resistance bioisosteric replacement for carboxylic acids.18,136 However, several authors have also examined the capability of a tetrazole ring as an amide bioisostere, and subsequently, tetrazole gained attention due to its viability for amide bond replacement. In a first example from the protein arginine deiminases (PADs) inhibitor field, tetrazole Cl-amidine analogs were identified as potent PAD inhibitors that show better antiproliferative activities in U2OS (PAD-4 expressing osteosarcoma bone marrow) cells as compared to the parent 92, Cl-amidine (Figure 24).137
Figure 24.

Structure of 92 and its bioisostere 93, potent PAD inhibitors.
Human proteins undergo a range of post-translational modifications (PTMs) such as citrullination which is catalyzed by PADs, involving the hydrolysis of arginine residues to form citrulline. Abnormal PAD activity is linked with onset and progression of ulcerative colitis, inflammatory bowel disease, rheumatoid arthritis, lupus, and certain cancers.138 Compound 92 belongs to the haloacetamidine class that showed efficacy in a variety of preclinical models of autoimmune diseases and cancer. As 92 is a peptide-based compound; it primarily suffers from metabolic degradation by proteolysis. In this context, structural modifications of the parent compound (92) by replacing the C-terminal amide bond with a tetrazole ring to improve potency, selectivity, and bioavailability were performed. Tetrazole analogs of compound 92 were initially screened for their potency and selectivity by determining kinact/KI values (a measure of potency for an irreversible inhibitor) for PAD1, −2, −3, and −4. Compound 93, a biphenyl tetrazole tert-butyl Cl-amidine (the synthetic route of 93 is given in Scheme 11), showed the best potency among all tested analogs and therefore was evaluated in an antiproliferative activity assay using U2OS cells at the fixed concentration of 20 μM. Results displayed that 93 (EC50 = 10 ± 2.5 μM) has 16-fold better activity than the parent 92 (EC50 = 160 ± 20 μM), which could be attributed to its increased hydrophobicity that enhanced cell penetration.
Scheme 11.
Synthetic Route To Access the Biphenyl Tetrazole tert-Butyl Cl-amidine-Containing PAD Inhibitor 93
Previously it was shown that PAD-4 isozyme-mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap (NET) which causes direct organ damage and can trigger endothelial as well as epithelial cell death.139 Therefore, in this case, inhibiting PAD-4 would be helpful to reduce NET formation. Compound 92 has been shown to block NET formation and modulate the lupus phenotype in animal models.140 Although compound 93 had very poor in vitro inhibitory values against the PAD-4 isozyme compared to parent 92, it was checked for its potential to block NET formation by using a DNA/neutrophil elastase overlap assay. Strikingly, 93 was significantly more potent than 92 in the NET assay, possibly due to its hydrophobic nature, which enhanced cellular uptake.
A similarly successful tactic has been described for the identification of potent neuroprotective agents in a series of 1,2-dithiolane-based analogs. Many studies reported that LA is a potent antioxidant and it can regenerate intracellular glutathione (GSH), vitamin C, and vitamin E levels.141,142 Also, it has beneficial effects in a number of pathological conditions such as inflammation, neurodegeneration, ischemia-reperfusion injury, diabetes, etc. related to oxidative stress.143 Koufaki and co-workers designed a small library of LA (or 1,2-dithiolane) based analogs where the amide bond present in LA amide-dopamine conjugate (96) was bioisosterically replaced by several heterocyclic five-membered rings including triazole, oxadiazole, tetrazole, and thiazole to improve neuroprotective activity with that of parent 96.144 The authors did not comment on why they decided to make conformational changes from the natural trans-amide (96) to the cis-amide-like tetrazole (97). Results of in vitro neuroprotection assay using glutamate challenged HT22 cells revealed that the tetrazole containing analog (97) displayed the strongest neuroprotective activity compared to the parent 96 and also among all tested compounds (Figure 25).
Figure 25.
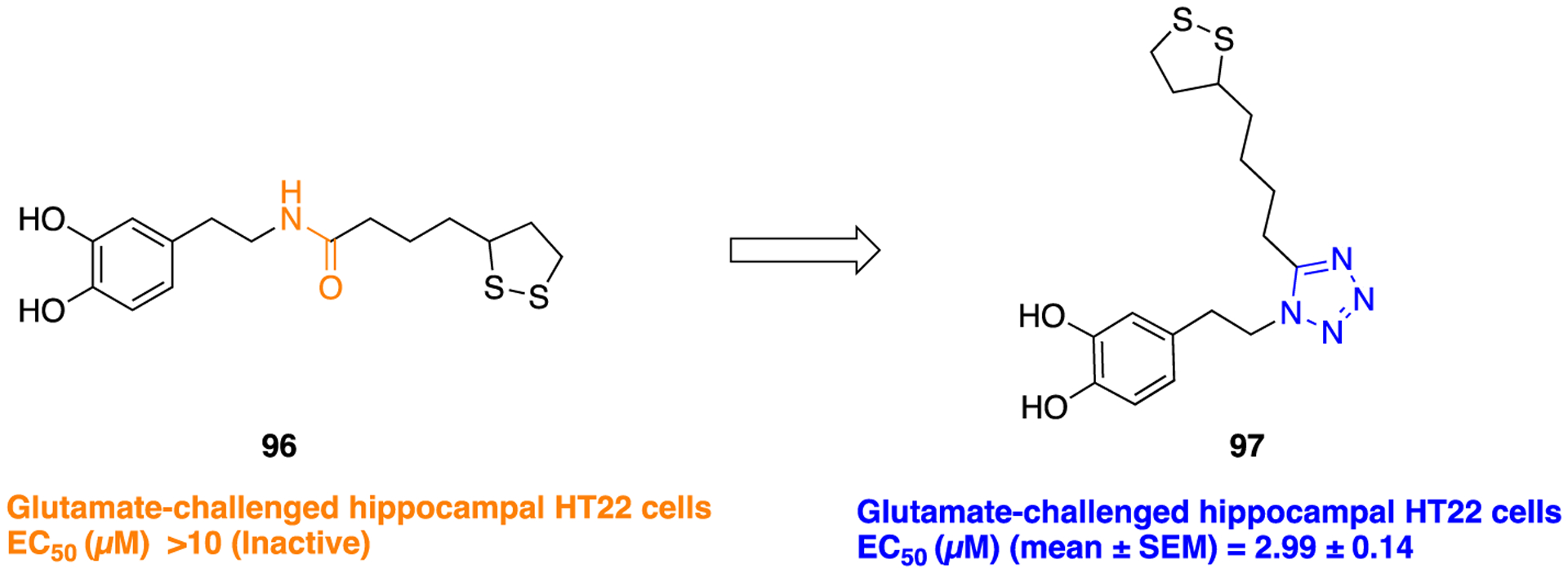
Structures of LA amide-dopamine conjugate 96 and its tetrazole containing bioisostere 97.
The synthetic route to access 97 is delineated in Scheme 12, i, in which trimethylsilyl azide (TMSA) in the presence of triphenylphosphine and diisopropyl azodicarboxylate (DIAD) was used to obtain the desired tetrazole intermediate. The primary SAR analysis on this series of compounds showed that potent neuroprotective activity could be achieved by insertion of the heteroaromatic head in the alkyl-1,2-dithiolane chemotype in conjunction with other antioxidant moieties such as the catechol.
Scheme 12.
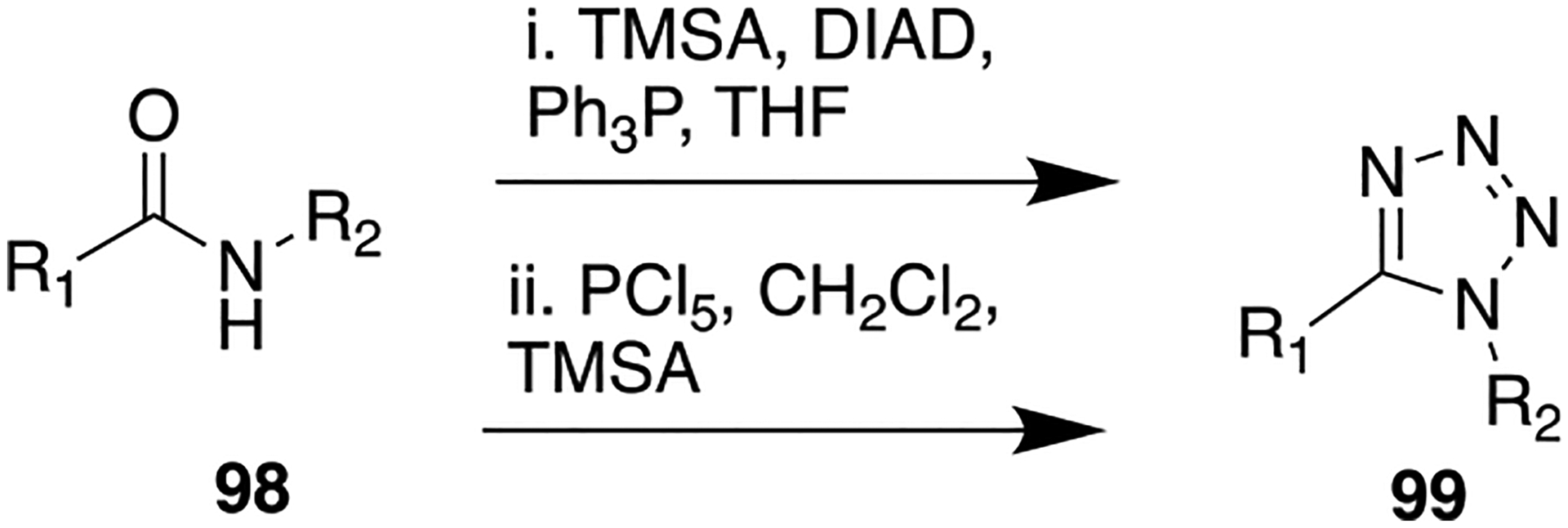
General Synthetic Route To Access Tetrazole, Nonclassical Amide Bioisostere
In 2009, Koufaki and co-workers again conducted a similar type of study in which novel 1,2-dithiolane/chroman hybrids were designed and evaluated for their neuroprotective potential in glutamate-challenged HT22 cells.73 Several nonclassical bioisosteres such as triazole (discussed in the triazole section, 29), oxadiazole (discussed in the oxadiazole section, 40), and tetrazole were employed to replace the amide bond in the parent compound 28. Here, we highlight the in vitro neuroprotection assay results obtained upon tetrazole installation in compound 100 (Figure 26). This compound was synthesized according to the general synthetic procedure depicted in Scheme 12, i. In this study, the authors did not discuss effects on activity related to the restricted conformation obtained with the cis-amide-like tetrazole (100) compared with the trans-amide (28). Being less flexible in comparison with other dithiolane/chroman hybrids of the same series, 100 exhibited a slight reduction in activity (EC50 = 3.04 ± 1.15 μM) compared to the parent amide 28 (EC50 = 2.10 ± 0.40 μM). However, the percentage efficacy of both 100 (100%) and 28 (95%) was almost equal, which signifies its potent neuroprotective ability.
Figure 26.
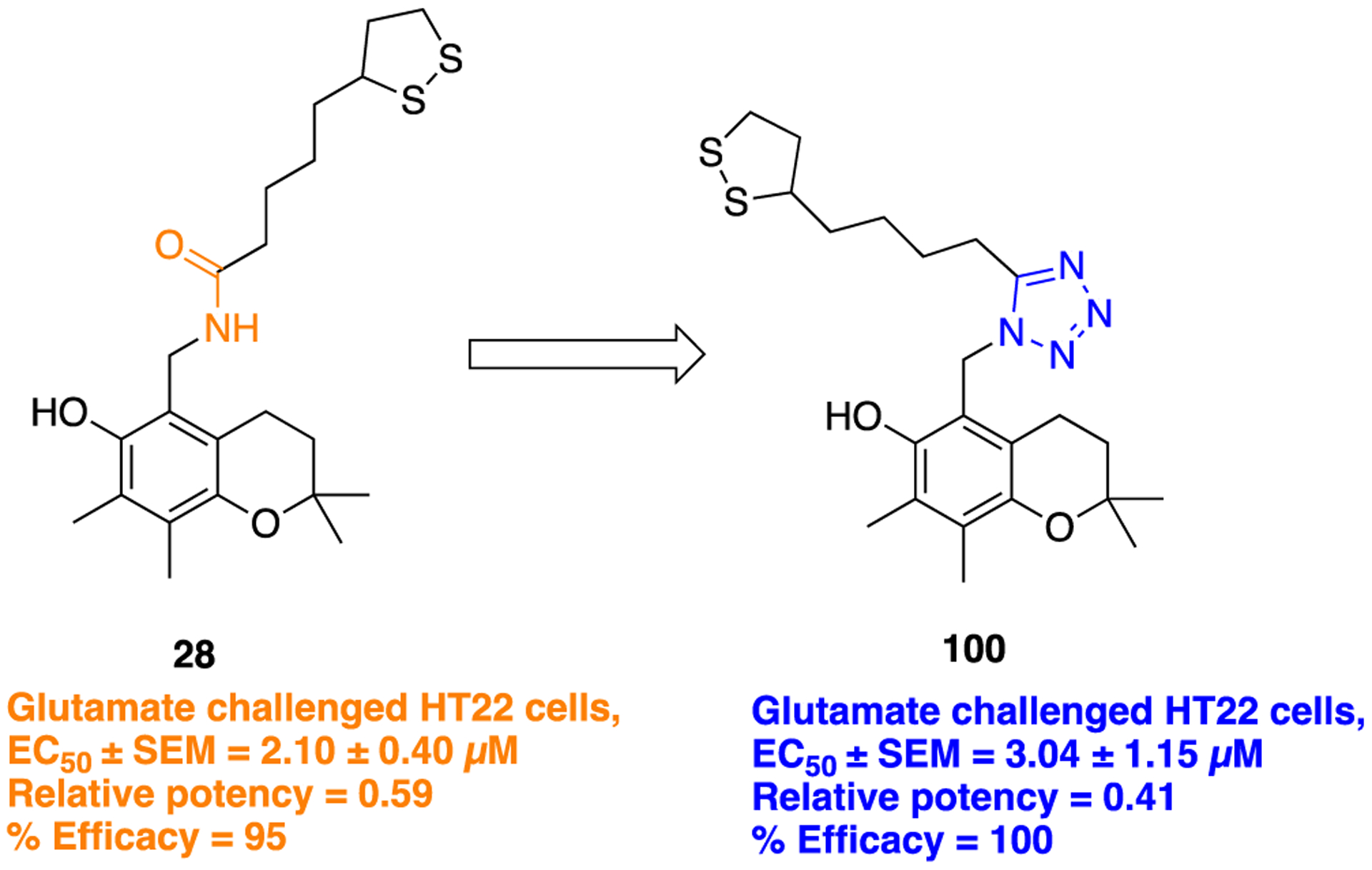
Molecular structures of parent LA-amide analog 28 and its tetrazole bioisostere 100.
Neuronal nicotinic acetylcholine receptors (nAChRs) belong to the “Cys-loop” superfamily of ligand-gated ion channels, widely expressed throughout the brain, and are suggested to be a potential therapeutic target for the treatment of cognitive disorders.145,146 Several studies also indicate that α7-AChRs share a target for therapeutic intervention in AD and schizophrenia.147,148 In an effort to identify more potent α7-AChRs agonist with desirable drug-like properties, Beinat and colleagues149 explored the SAR of SEN12333 (101), a potent α7-AChR agonist (Ki = 670 ± 119 nM) developed by another group at Siena Biotech and Wyeth (Figure 27).150 Due to limited SAR studies for this chemotype (101), these authors have previously identified that the four-carbon chain linker is essential for optimal binding and functional activity at α7-AChRs.151 In the present work, the authors investigated the role of the amide bond in 101, particularly its connectivity and bioisosteric replacement (Figure 27).
Figure 27.
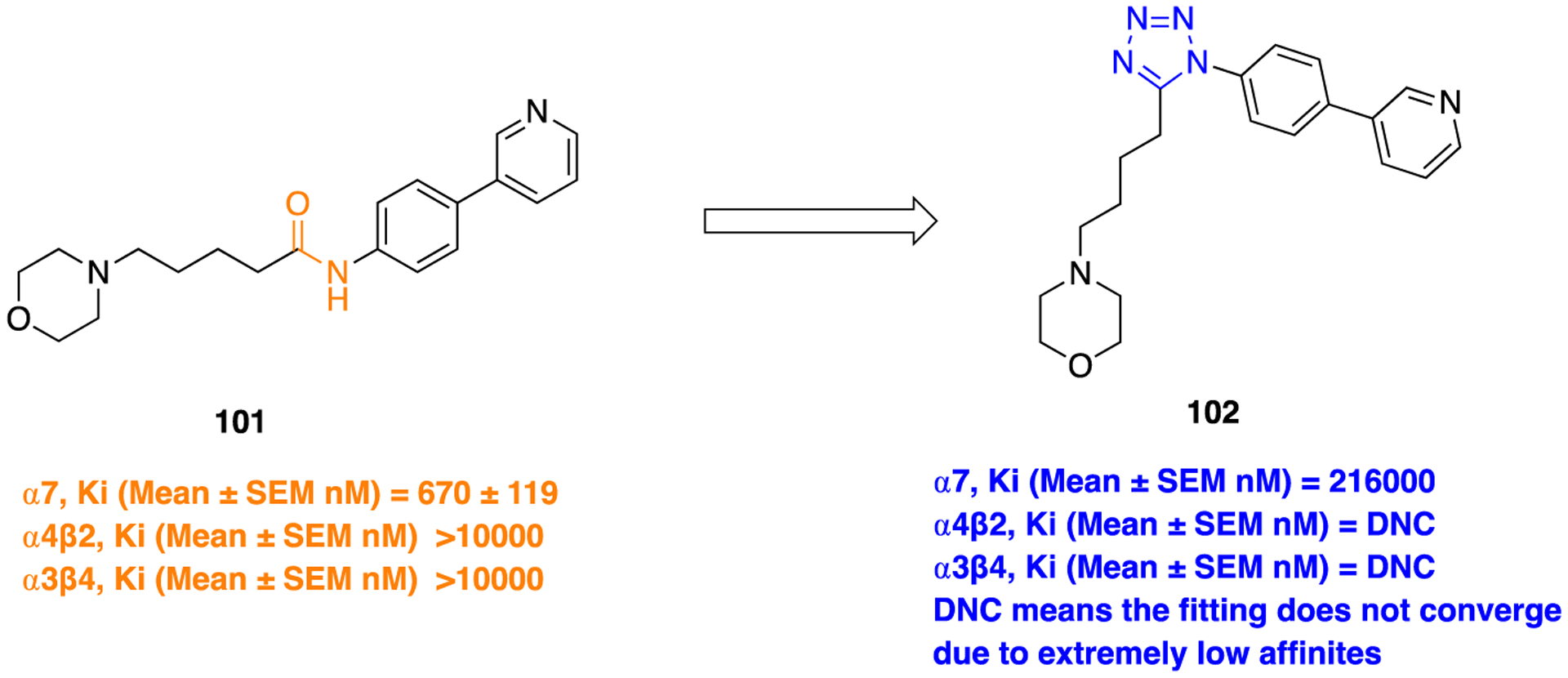
Amide bond bioisosteric modification in 101 with a tetrazole functionality (102).
Several compounds were synthesized and tested for their agonistic activity for α7-AChRs. The transposition of amide 101 to the reverse amide (structure not shown here) resulted in a marked decrease of affinity at α7-AChRs over that of 101. The bioisosteric replacement of the amide in 101 with a tetrazole ring (a cis amide isostere) was undertaken to provide further mechanistic details into the binding mode of 101 at α7-AChR and potentially reduce the number of rotatable bonds within the molecule. X-ray crystallographic studies were performed to identify the solid-state conformations for both 101 and a tetrazole containing bioisostere (102). Compound 102 was readily synthesized from a corresponding amide using phosphorus pentachloride to initially afford the respective imidoyl chloride derivative followed by the addition of trimethylsilylazide to furnish the tetrazole moiety (Scheme 12, ii). The obtained Oak Ridge thermal ellipsoid plots (ORTEPs) of 101 and 102 are shown in Figure 28. These plots revealed that amide (101) adopts a trans-conformation at the lowest energy. However, the tetrazole ring (102) locks the amide bond into cis-conformation and resides in a bent form. If the binding conformation of 101 is similar to the low energy (i.e., trans-conformation), these studies suggest the binding mode of 101 and 102 would be vastly different. Contrary to expectations of the tetrazole bioisostere (102), affinity to α7-AChR was completely abolished (Figure 27), which may be due to increased steric hindrance or electronics of the tetrazole ring at the receptor-binding site. Thus, the obtained SAR results provided valuable insights about the essential molecular requirements of this particular chemotype and concluded that the original amide bond connectivity, as in 101, is favorable for binding affinity as well as agonistic activity at α7-AChRs.
Figure 28.
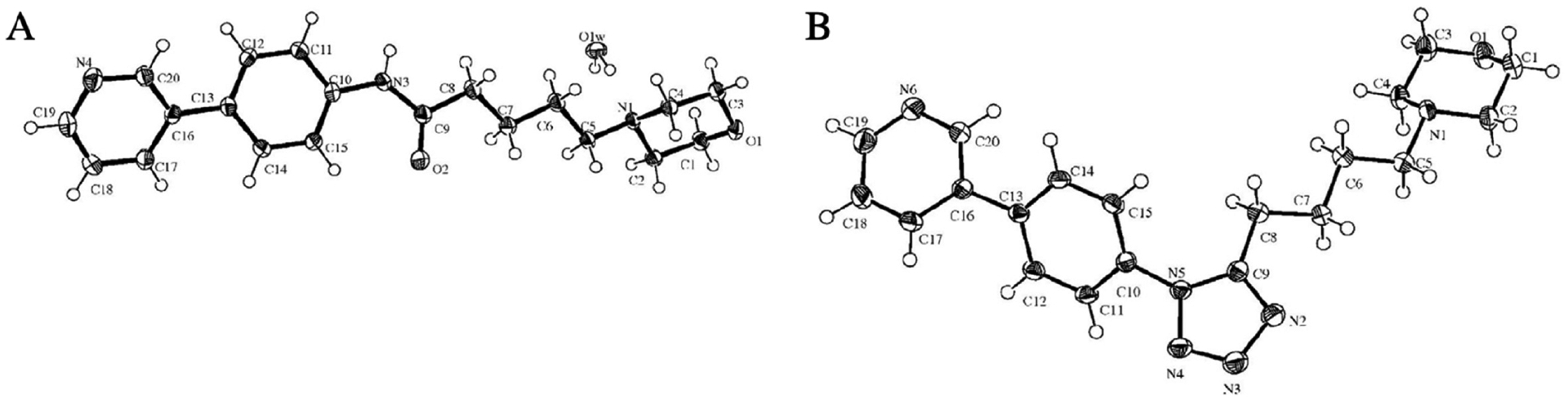
Oak Ridge thermal ellipsoid plots of 101 (A) and 102 (B). Reproduced with permission from European Journal of Medicinal Chemistry (https://www.sciencedirect.com/journal/european-journal-of-medicinal-chemistry), Volume 84, pp 200–205, Beinat, C.; Reekie, T.; Hibbs, D.; Xie, T.; Olson, T. T.; Xiao, Y.; Harvey, A.; O’Connor, S.; Coles, C.; Tsanaktsidis, J.; Kassiou, M., Investigations of amide bond variation and biaryl modification in analogues of α7 nAChR agonist SEN12333.149 Copyright 2014 Elsevier.
E. Pyrazole.
Prolylcarboxypeptidase (PrCP) or prolinecar-boxypeptidase represents a potential therapeutic target for the treatment of cardiovascular, inflammatory, and metabolic diseases, wherein substantial efforts have been made to generate potent PrCP inhibitors.152–154 A medicinal chemistry campaign to discover novel inhibitors of PrCP at Merck Research Laboratories investigated the use of a pyrazole scaffold as an amide bioisostere in a class of previously reported PrCP inhibitors 103 and 104 to modify their PK properties while maintaining potency for both human (h) and mouse (m) PrCPs.155 Compounds 103 and 104 showed potent PrCP in vitro inhibitory activity of IC50 (h, m) = 3.5 and 5 nM and IC50 (h, m) = 2.5 and 1.4 nM, respectively (Figure 29). In mouse PK studies, 104 showed a long half-life, which results in improved oral bioavailability as compared to 103, which suggested that the morpholine substituent replacement could be afforded without disturbing potency. Next, when the pyrazole ring was installed (compound 105) in place of the amide function on this chemotype, it brought a significant loss in PrCP inhibitory activity. Further optimization was made to regain the potency. In due course, compound 106 was identified as the most potent PrCP inhibitor containing a pyrazole bioisostere (Figure 29).
Figure 29.
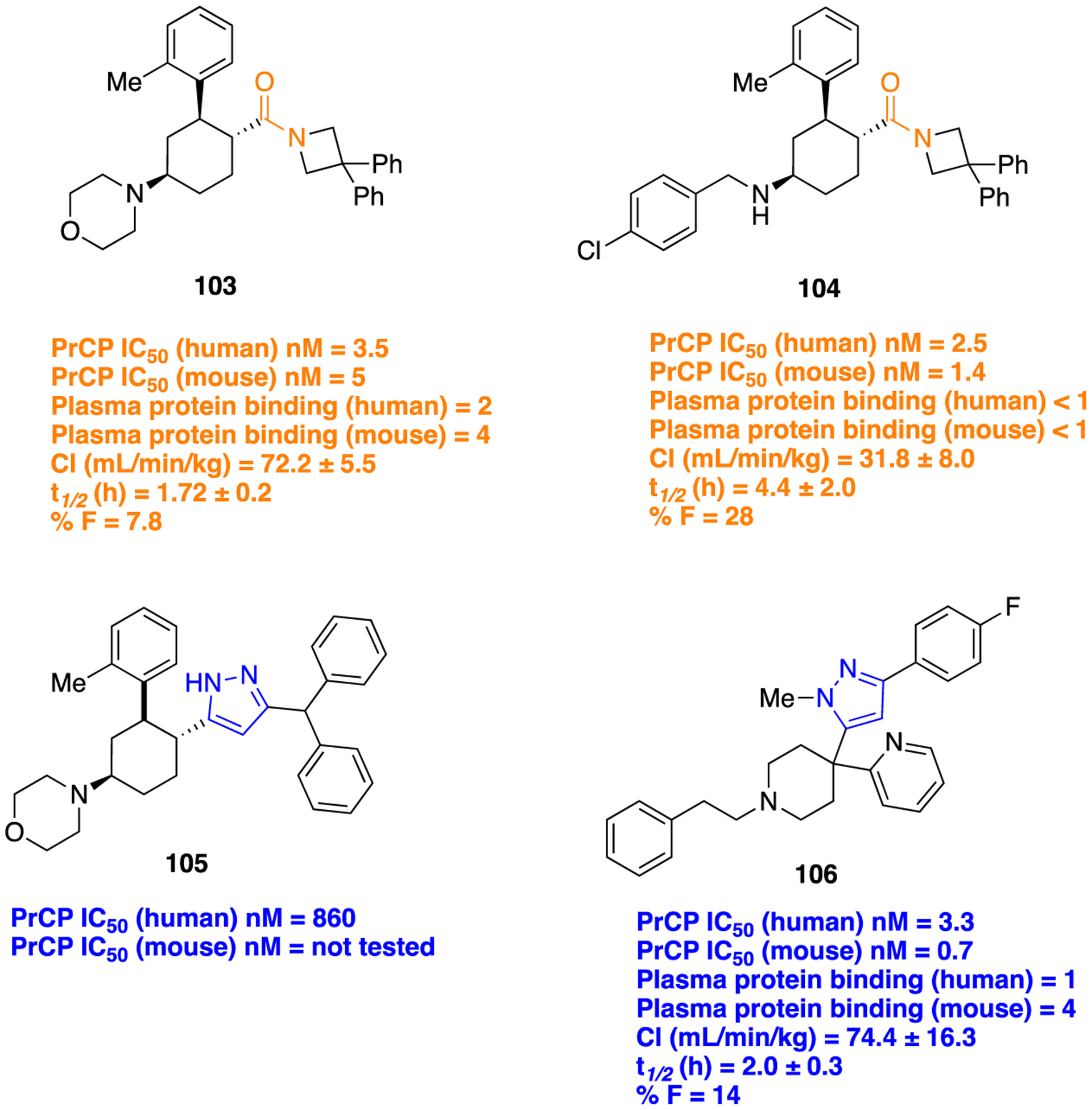
Bioisosteric replacement of amide (103 and 104) with pyrazole (105 and 106) to discover potent PrCP inhibitors.
Moreover, this compound also showed a modest half-life and oral bioavailability in the mouse comparable to the original amides, i.e., 103 and 104. In summary, the preliminary installation of the pyrazole (105) as an amide surrogate attenuated the in vitro PrCP potency; however, an exhaustive SAR analysis showed that the exchange of an amino-cyclohexane core with a piperidine motif was beneficial in restoring the in vitro PrCP activity. Moreover, simplified analogs with no stereogenic centers including 106 were also potent PrCP inhibitors.156
F. Miscellaneous Heterocyclic Amide Bioisosteres.
Several studies have shown that the CB1 receptor, a member of the G-protein-coupled receptor (GPCRs) family, is a promising therapeutic target against a wide range of pathological conditions including mood and anxiety disorders, neuropathic pain, Huntington’s disease (HD), PD, multiple sclerosis, traumatic brain and spinal cord injury, glaucoma, obesity, myocardial infarction, stroke, and hypertension.157,158 Previously, the indole-3-carboxamide derivatives 107 and 108 were reported as CB1 agonists which showed potent antinociceptive activity in the mouse tail-flick test after intravenous (iv) administration, and both demonstrated a fast onset and short duration of action.159,160 Efforts were made at improving metabolic stability through a bioisosteric approach, and thus, introduction of conformationally constrained and steric blocking groups in place of the piperazine amide (107 and 108) was evaluated (Figure 30).161
Figure 30.
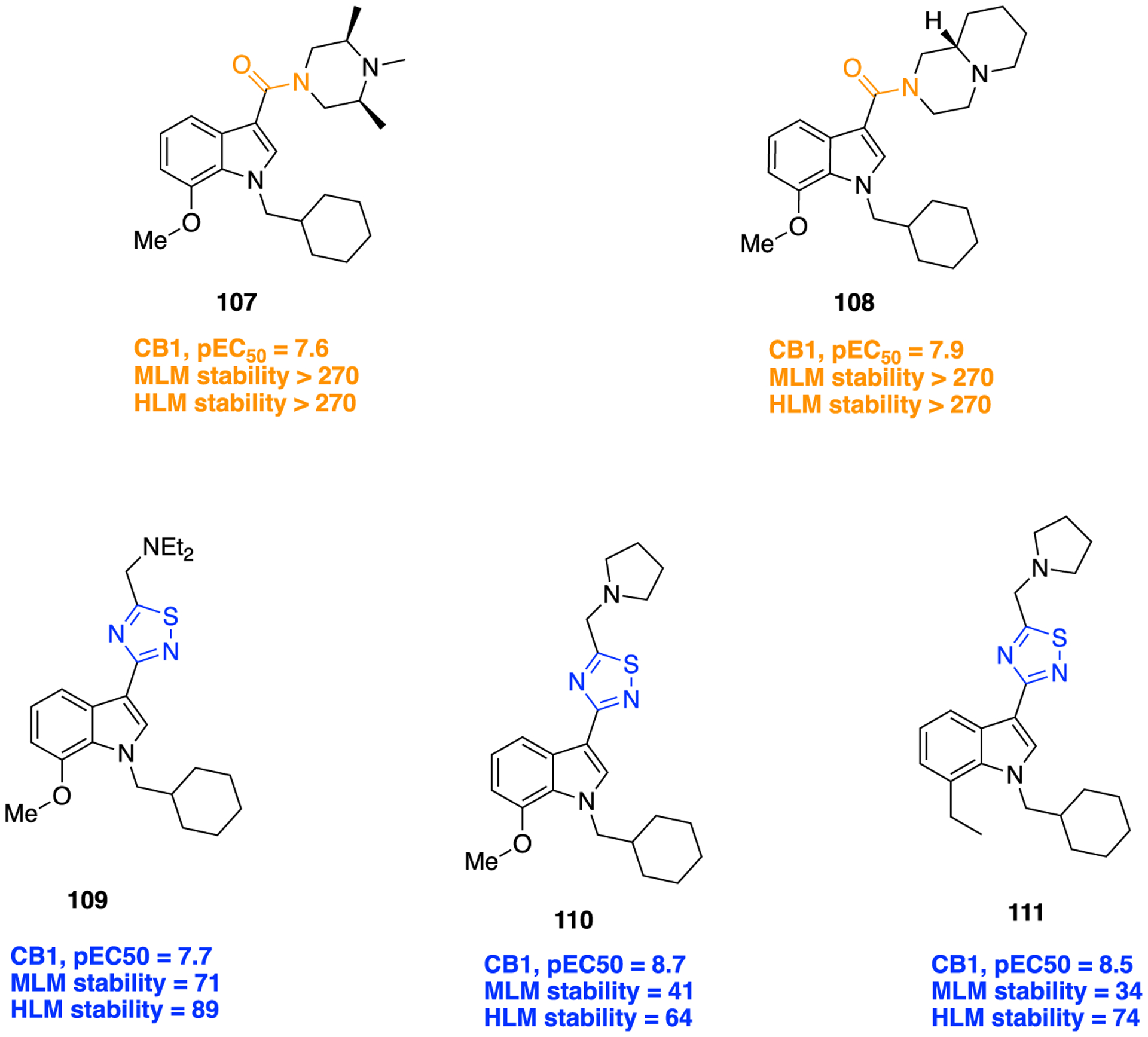
Amide versus thiadiazole bioisosterism in search of potent CB1 agonist.
New compounds consisting of different bioisosteric heterocycles (oxazole, 1,2,4-oxadiazole, 1,3,4-oxadiazole, thiazole, and thiadiazole) with a simple tertiary amine were synthesized and evaluated for in vitro CB1 agonist activity by using Chinese hamster ovary (CHO) cells doubly transfected with human CB1 and luciferase. The SAR analysis of this series revealed that all new compounds retained CB1 agonistic activity. However, the replacement of the piperazine amide by a 1,2,4-thiadiazole motif (109) provided the best combination to retain CB1 agonist potency (pEC50 = 7.7) and metabolic stability [MLM = 71; human liver microsome (HLM) = 89] within the series (Figure 30).
Next, SAR was explored around the amine moiety and several compounds were prepared and evaluated for CB1 receptor agonistic activity and metabolic stability. These compounds showed CB1 agonistic activities comparable with 109. Success was achieved upon constraining the diethyl fragment into a pyrrolidine ring (compound 110) and with compound 111 containing a similar sized ethyl group in place of the 7-methoxy group (Figure 30). This compound displayed comparable potency and stability to 110 and was therefore selected for advanced in vitro experiments. Results demonstrated that 111 showed high binding affinities for both human CB1 (pKi = 8.2) and CB2 (pKi = 8.5), tested by radioligand competition binding assay using [3H] CP 55940 binding to either human CB1 or CB2 receptors expressed in insect Sf9 membranes. Figure 31 shows the superimposed images of 111 and 107 suggesting appropriate biomimicry by the thiadiazole moiety. Moreover, in the mouse tail-flick test, a preclinical model of nociception, 111 showed improved duration of action after iv administration compared to the parent 107.161
Figure 31.
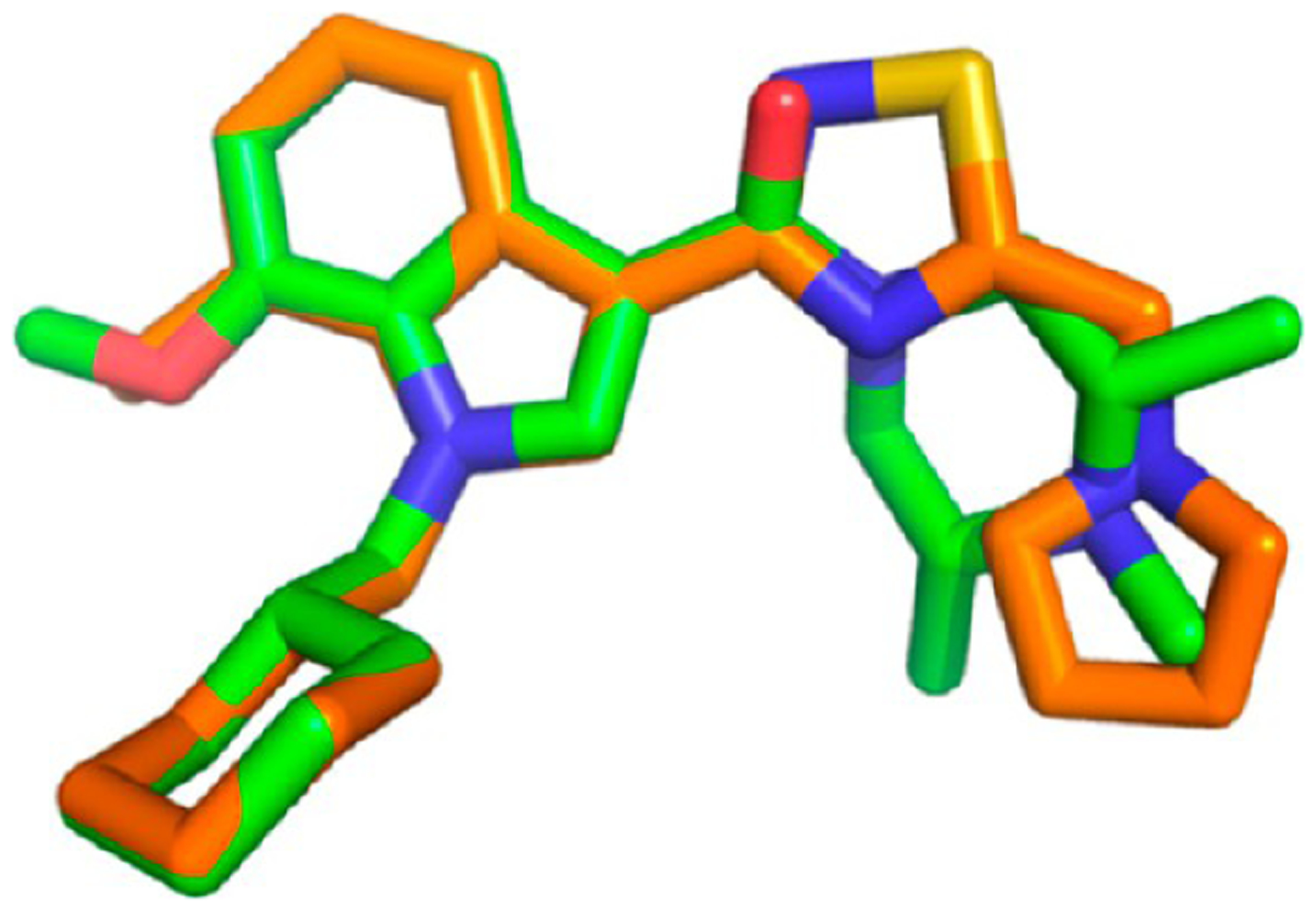
Superimposition modeling image of 107 (green) and 111 (orange). Reproduced with permission from Bioorganic & Medicinal Chemistry Letters (https://www.sciencedirect.com/journalbioorganic-and-medicinal-chemistry-letters/), Volume 21, pp 506–509, Morrison, A. J.; Adam, J. M.; Baker, J. A.; Campbell, R. A.; Clark, J. K.; Cottney, J. E.; Deehan, M.; Easson, A. M.; Fields, R.; Francis, S.; Jeremiah, F.; Keddie, N.; Kiyoi, T.; McArthur, D. R.; Meyer, K.; Ratcliffe, P. D.; Schulz, J.; Wishart, G.; Yoshiizumi, K., Design, synthesis, and structure-activity relationships of indole-3-heterocycles as agonists of the CB1 receptor.161 Copyright 2011 Elsevier.
The matrix metalloproteinases (MMPs), also called matrixins, belong to the metzincin group of proteases, which possess a conserved zinc-binding motif in their catalytic site. These MMPs play a crucial role in many physiological processes, and their expression is increased in various pathological conditions such as inflammation, angiogenesis, metabolic bone disease, and cancer. Therefore, inhibitors of MMPs are valuable compounds for therapeutic development in these diseases.162–164 A series of succinyl hydroxamate matrix metalloproteinase inhibitors were prepared by incorporating both aromatic and aliphatic substituents in place of an amide moiety present in general structure 112 (Figure 32).165
Figure 32.
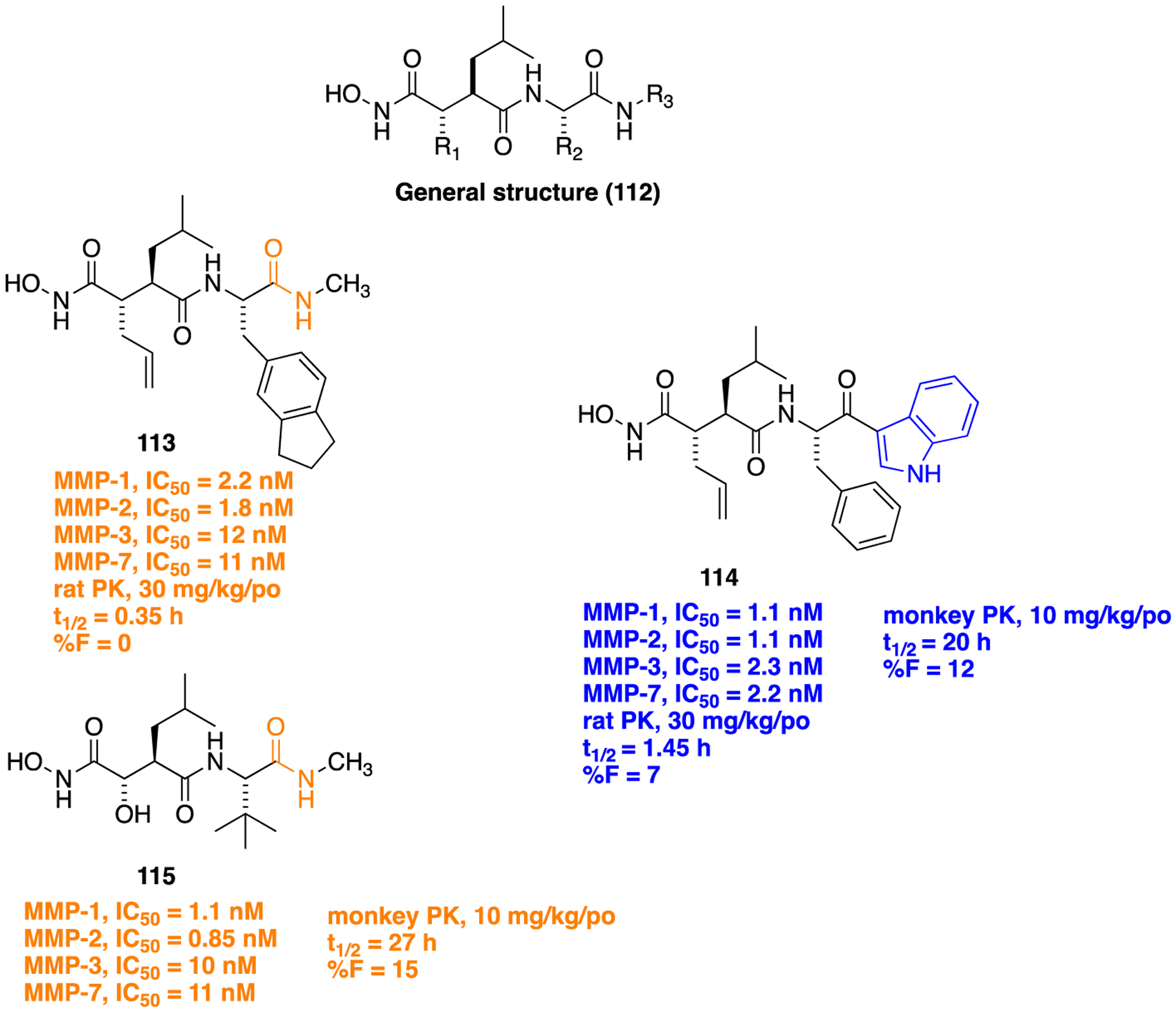
General structure (112) showing the molecular template for designing MMP inhibitors, isosteric replacement of the C-terminal amide bond in 113 with 3-acyl indole (114), and the structure of marimastat, (115), a broad-spectrum MMP inhibitor.
Previously, the 3-acyl indole ring had been employed as an isosteric replacement for an aryl amide moiety in developing platelet-activating factor (PAF) antagonists. This modification enhanced the binding energy of the series and stabilized a hydrolysis-prone amide function which resulted in the identification of several PAF antagonists with good oral bioavailability.166 In this study, the authors replicated this strategy and replaced the C-terminal amide (113) with 3-acyl indole (114) to ascertain its effects on MMP inhibitory potency and oral bioavailability. Results were encouraging, and as expected, 114 was found to be a potent broad-spectrum MMP inhibitor (Figure 32). The synthetic route for 114 is delineated in Scheme 13. This compound showed modest oral bioavailability in rats similar to 113. Furthermore, in monkeys, 114 at an oral dose of 10 mg/kg exhibited a half-life of 20 h, which was comparable to marimastat (115) (oral dose, 10 mg/kg; 27 h), a broad-spectrum MMP inhibitor taken as reference (Figure 32).
Scheme 13.
Synthesis of Indole Bioisostere 114, a Potent MMP Inhibitor
An example where an acetamide functional group was replaced with a trifluoroethyl, pyridine, pyrimidine, pyrazine, or thiazole ring was reported in the optimization of antagonists of the adenosine A2A (A2A) receptor, a GPCR mediating the physiological actions of adenosine, involved in coordinating motor functions.167–169 Previously, the authors have reported a series of 2,4,6-trisubstituted pyrimidines that showed potent antagonism at the A2A receptor and confirmed their efficacy in haloperidol-induced catalepsy. The molecular structures of all these compounds possess an acetamide group, due to which they showed varied stability under acidic conditions; therefore, this acetamido fragment was bioisosterically replaced from compound 119 (a preclinical candidate), chosen as the starting point (Figure 33).
Figure 33.
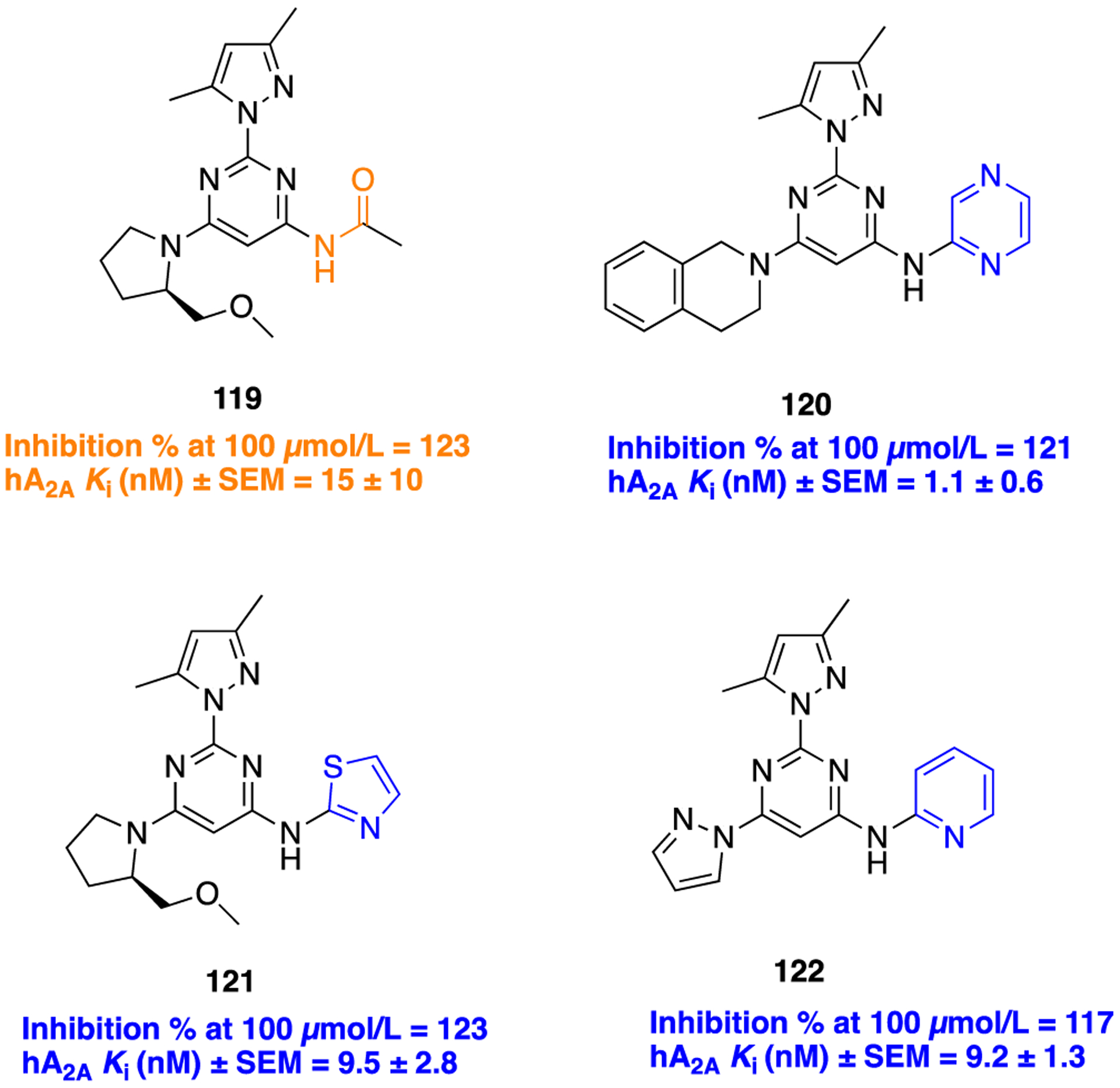
Molecular structures of the parent amide 119 and pyrazine (120), thiazole (121), and pyridine (122) bioisosteres.
Preladenant was selected as the standard reference compound in vitro human A2A receptor binding assays.170 Merck introduced preladenant (SCH 420814) as a potent A2A receptor antagonist for the treatment of PD. Results from clinical trials suggested the discontinuation of preladenant due to adverse events.169,171 The preliminary SAR analysis showed that the installation of pyrazine with 1,2,3,4-tetrahydroisoqui-noline (compound 120) improved potency more than 10-fold compared to the parent 119. It exhibits superior antagonistic potency of 1.1 nM at human A2A receptor comparable to that of preladenant (1 nM). The synthetic route to obtain the pyrazine bioisostere 120 is given in Scheme 14. On the other hand, two more compounds consisting of a thiazole with (R)-2-(methoxymethyl)pyrrolidine (121) and a pyridine with pyrazole (122) also displayed superior potency compared to parent 119 (Figure 33). Thus, the observed SAR trend on this series was unclear, warranting further detailed investigations.
Scheme 14.
Synthetic Route for Obtaining the Pyrazine Bioisostere 120, a Potent A2A Receptor Antagonist
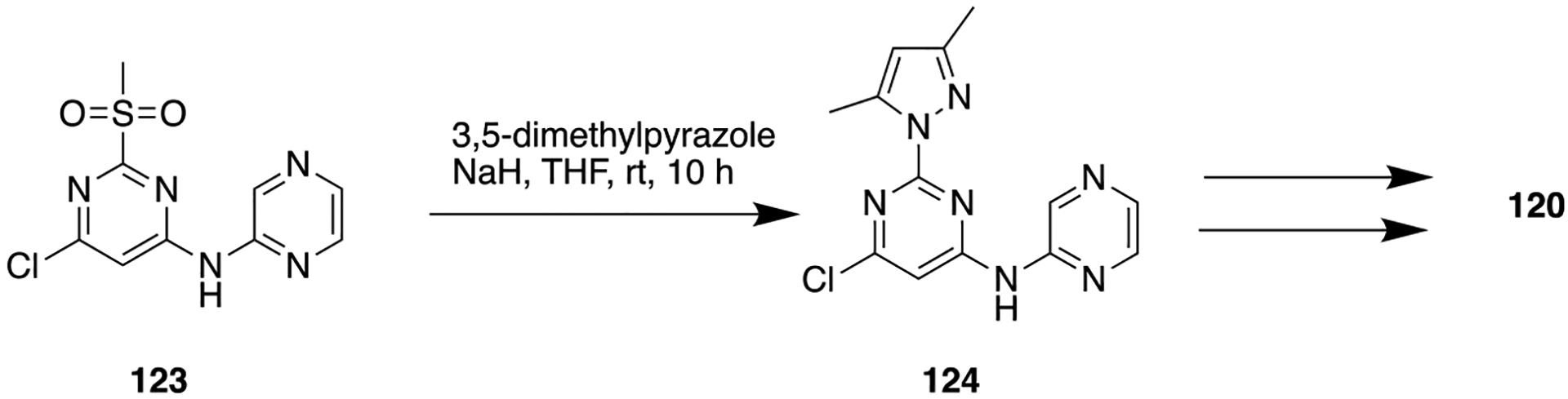
Tyrosine kinase 2 (TYK2) belongs to the Janus kinase (JAK) family and plays a critical role in driving interleukin 12 (IL-12) and interleukin 23 (IL-23) pathways, and these pathways are correlated with the pathogenesis of autoimmune diseases such as psoriasis, lupus, multiple sclerosis, and inflammatory bowel disease.172 Therefore, TYK2 appeared as a therapeutic target for various human autoimmune diseases. The challenging task is to develop a selective TYK2 inhibitor over other JAK family members (i.e., JAK1, −2, −3) because of their high sequence homology to TYK2.173 Lead 125 was identified as a TYK-2 inhibitor, and its subsequent optimization to a fluorocyclopropane analog 126 was reported. To improve TYK2 potency, the authors prepared several structurally constrained analogs derived from lead 126, where the amide bond was cyclized onto the pyridine ring to form fused heterocycles (Figure 34).174 The prepared analogs were evaluated for their TYR2 and JAK2 inhibition and resulted in some active scaffolds including imidazopyridine, oxazolopyridine, thiazolopyridine, and pyrazolopyridine. Among them, the imidazopyridine containing analog, 127, showed an IC50 value of 1.7 nM for TYR2 along with increased selectivity over JAK-2 compared to parent 125.
Figure 34.
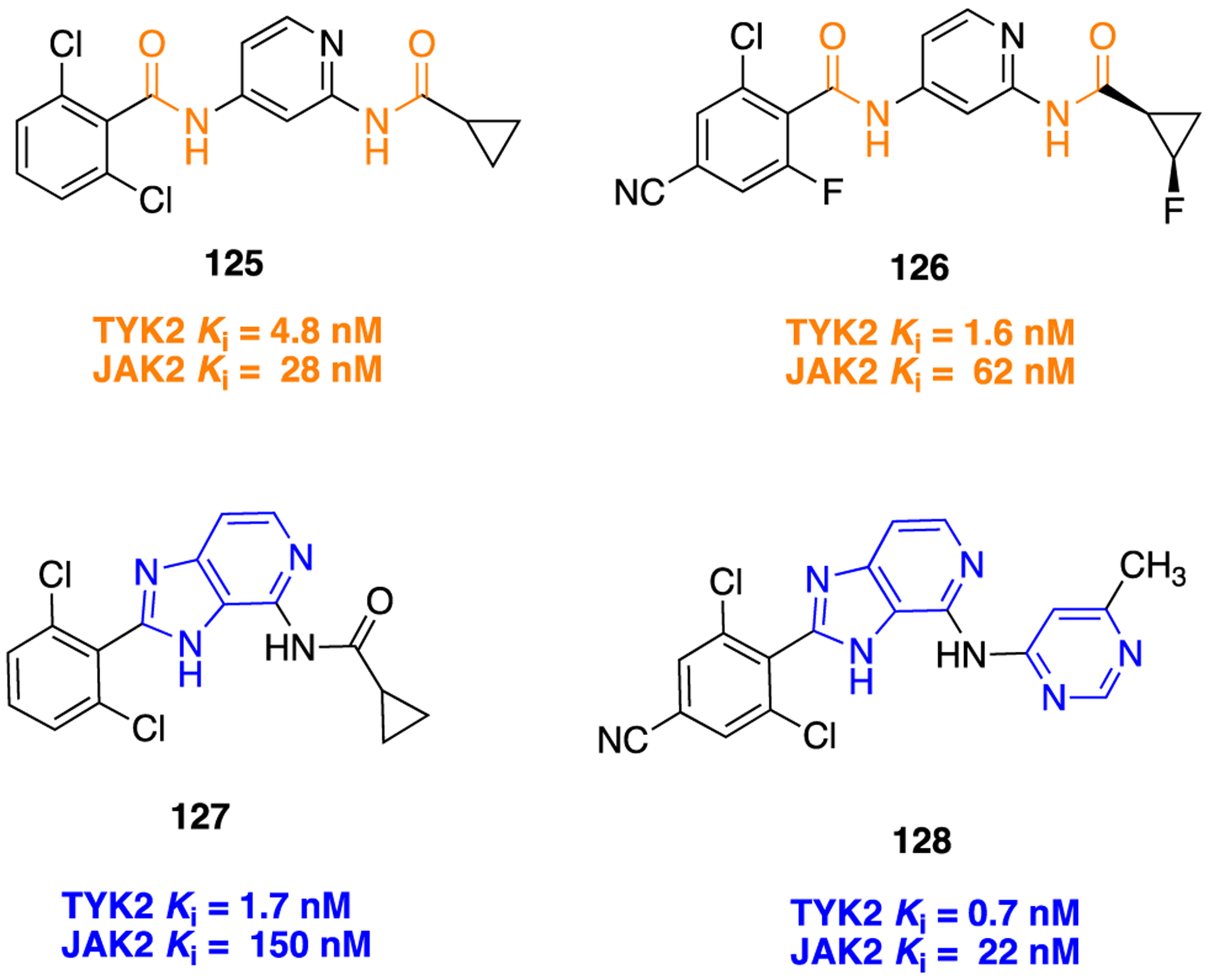
Structures of lead TYR2 inhibitor 125 and its optimized analogs 126–128.
These results show that the constrained core (127) is a better choice over the unconstrained amide core (125) in terms of improving the selectivity for JAK2. Further optimization of 127 by replacement of the cyclopropylamide portion with an aminopyrimidine moiety and appending a cyano group was performed. Earlier, it was observed, as in the case of unconstrained amide series (126), that the introduction of a cyano group at the fourth position of the dichlorophenyl ring led to significant improvement in in vivo clearance in rodents. Of these new analogs, compound 128 bearing a 6-methylpyrimidine showed potent inhibition of TYK2 (Ki = 0.7 nM) and the IL-23 pathways in cells (EC50 = 66 nM) along with high selectivity over JAK2. As described in Scheme 15, compound 128 was synthesized by reacting 2,6-dichloro-4-iodobenzoic acid (129), 2-chloropyridine-3,4-diamine, and polyphosphoric acid. The resulting isolated product was treated with POBr3 solution to give the desired compound 130 (4-bromo-2-(2,6-dichloro-4-iodophenyl)-3H-imidazo[4,5-c]pyridine). Next, the mixture of compound 130, Zn(CN)2, and Pd(PPh3)4 was heated to obtain compound 131. Finally, microwave-assisted Pd-catalyzed coupling reaction of compound 131 and 6-methylpyrimidine-4-amine was done to obtain 128. This compound demonstrated high permeability and exhibited moderate stability in liver microsomes. Moreover, in mice, 128 displayed a dose-dependent reduction of interleukin 17 (IL-17) production in a PK/PD model and in an imiquimod-induced psoriasis model.174
Scheme 15.
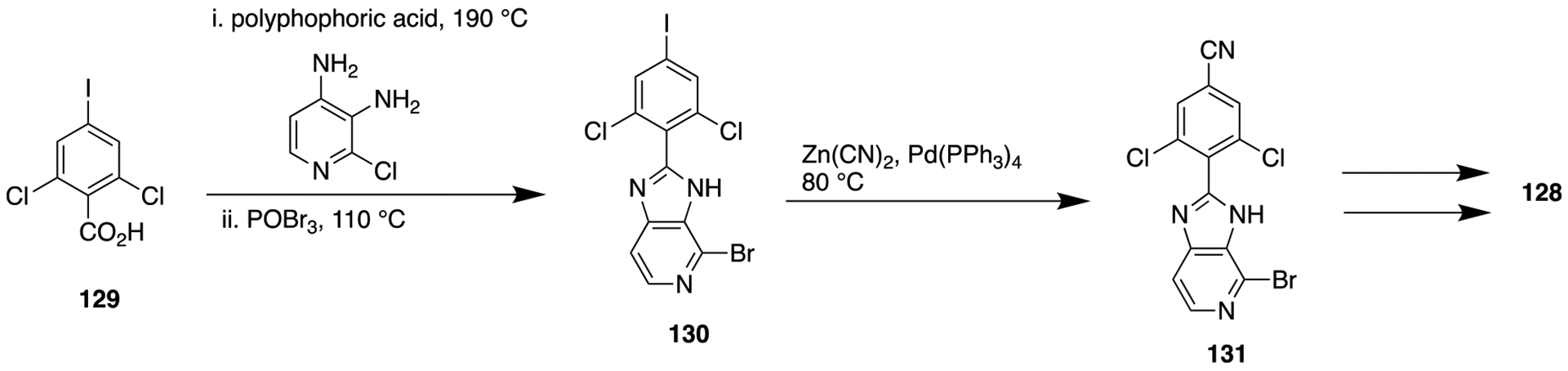
Synthetic Route To Obtain Compound 128, a Potent TYR2 Inhibitor
During research to identify new dopamine D2 receptor modulating agents, Baures and co-workers demonstrated the bioisosteric relationship existing between the amide bond (l-prolyl-l-leucylglycinamide 132) and diketopiperazine (DKP) functionalities (133, 134, and 135) (Figure 35).175 Earlier, these authors have reported that 132 modulates the dopamine D2 receptor by increasing the affinity of the receptor for agonists.176,177 Compound 133 was designed previously178 to mimic an N-terminal “C5” conformation involving an intermolecular hydrogen bond between the prolyl nitrogen and the lactam NH in an analog of parent 132 (structure not shown), which possessed better activity in a number of pharmacological assays.179,180 In order to further explore SAR, the DKP containing compounds 134 and 135 were designed in which a “C5” conformational mimic was incorporated into the parent structure (132) and a bicyclic lactam moiety was assimilated (present in a potent 132 analog, structure not shown), respectively.181 The dopamine receptor modulation ability of 134 and 135 was assessed by measuring activity in a [3H]spiro-peridol/N-propylnorapomorphine (NPA) D2 receptor competitive binding assay in the presence as well as absence of 5′-guanylyl imidodiphosphate (Gpp(NH)p); KH and KL represent the inhibitor constant (Ki) of agonist calculated for the high- and low-affinity components of the [3H]spiroperidol binding, respectively (Figure 35).
Figure 35.
Bioisosteric replacement of the amide within 132, with diketopiperazine (133, 134, and 135), to develop potent dopamine D2 receptor modulators.
The obtained results suggested that both 134 and 135 increased the affinity of the dopamine receptor for agonists and the percentage of dopamine D2 receptors which existed in the high-affinity state. However, the potency seen with 134 and 135 was greater than the parent 132, whereas it was not as great as observed for 133. Furthermore, compound 134 was found to be 10 times more potent than 135 which in turn was 10 times more potent than the parent 132 in an in vivo assay measuring apomorphine-induced rotational behavior in a 6-hydroxydopamine-lesioned rat model of PD. The synthesis of a diketopiperazine scaffold was accomplished by reacting N-chloroacetyl derivative (136) with l-leucine benzyl ester to obtain 137, which further gave compound 134 over several steps (Scheme 16). Many authors have also shown the bioisosteric relationship between amide and DKP which has resulted in the identification of a number of peptidomimetics and β-turn mimetics.182
Scheme 16.
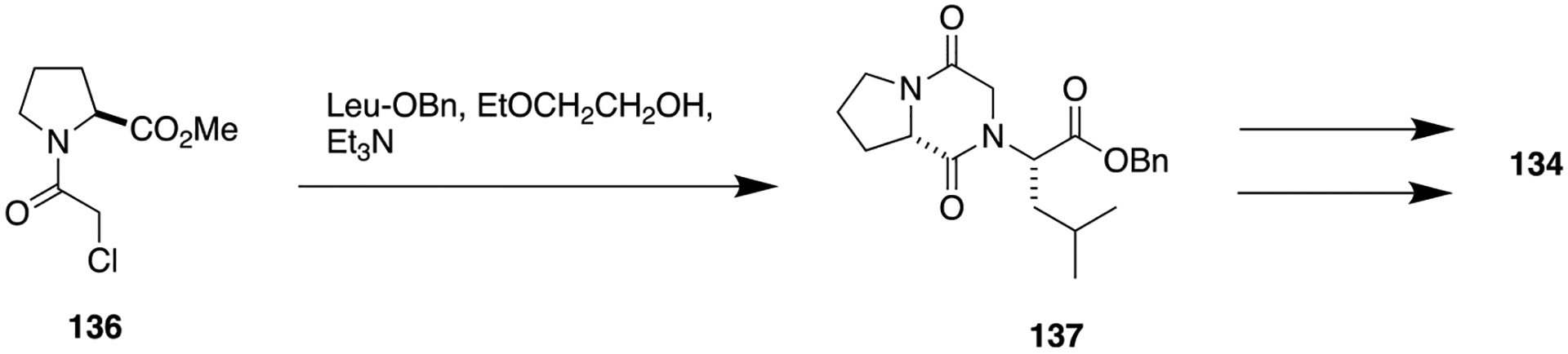
Diketopiperazine Bioisostere Synthesis from l-Proline To Obtain Compound 134
RETROINVERTED AND REVERSE AMIDES AS AMIDE BIOISOSTERES
The retroinverted or retroinverso alteration (NH–C(O)) is a popular approach for improving the resistance of an amide-containing molecule toward enzymatic cleavage, wherein the amide bond geometry and topology are maintained. This modification can be achieved by retroinversion from N → C to C → N, which does not alter the conformational flexibility of the molecule but improves the in vivo stability and prevents amide bond hydrolysis by proteases. On the other hand, a reverse amide, also known as a retro-amide (−C(O)NH−), retains the hydrogen bonding properties of an amide but is diametrically opposed.11 Several lines of experimental evidence suggest that the change in the location of the hydrogen bond donor and acceptor influences bioactivity.149,183,184 Here, examples where the use of a retroinverted or reverse amide produced beneficial as well as detrimental effects are discussed.
An interesting example where the installation of the retroinverted amide as an amide isostere significantly affected target specificity is a novel aldo-keto reductase 1C3 (AKR1C3) inhibitor for the potential treatment of acute myeloid leukemia (AML) and T-cell acute lymphoblastic leukemia (T-ALL).183,185–187 Several structural chemical classes, such as steroids, flavones, jasmonates, and nonsteroidal anti-inflammatory drugs (NSAIDs), have been tested to identify and develop potent AKR1C3 inhibitors.183 Unfortunately, the lack of inhibitory potency and isoform selectivity had no observable clinical efficacy in castration-resistant prostate cancer (CRPC) patients,188–190 preventing further clinical development. It has been reported that baccharin, a natural product isolated from honeybee propolis, is a highly selective inhibitor (500-fold selectivity over AKR1C2) of AKR1C3, with an IC50 value of 0.11 μM.191 This discovery has made baccharin a promising hit compound for the development of more potent and selective AKR1C3 inhibitors. Similarly, we previously reported the preliminary SAR of several baccharin analogs as AKR1C3 inhibitors.192 In another study, we reported the design, synthesis, and evaluation of potent AKR1C3 inhibitors containing a more stable amide bioisostere. However, their selectivity over AKR1C1 and AKR1C2 isoforms was relatively low.193
A recent report describes a library of compounds that were designed utilizing the concept of bioisosterism to obtain an extensive SAR map for AKR1C3 inhibition based on a baccharin pharmacophore.183 The results of in vitro enzyme inhibition assays indicated that several compounds produced a marked inhibition of AKR1C3 and had moderate to significant isoform selectivity for AKR1C3 over the other isoforms. Compound 138, a meta-amide baccharin analog, had potent AKR1C3 inhibition (AKR1C3 IC50 = 0.066 μM), with a 109-fold selectivity for AKR1C3 (Figure 36). Alteration of the central ring substitution pattern to an all meta arrangement and installation of a retroamide bioisostere yielded compound 139, which was equipotent with 138 (AKR1C3 IC50 = 0.07 μM). Most importantly, this compound had >2800-fold selectivity for C3 over C1 and C2, thus making it the most selective AKR1C3 inhibitor across all known classes of inhibitors reported thus far (Figure 36). Modeling experiments confirmed that 139 showed better binding affinity through the formation of strong hydrogen bonds to the carbonyl of the retroinverted amide, due to the reduced distance between the amino acid hydrogen bond donors and the amide carbonyl acceptor (Figure 37).
Figure 36.
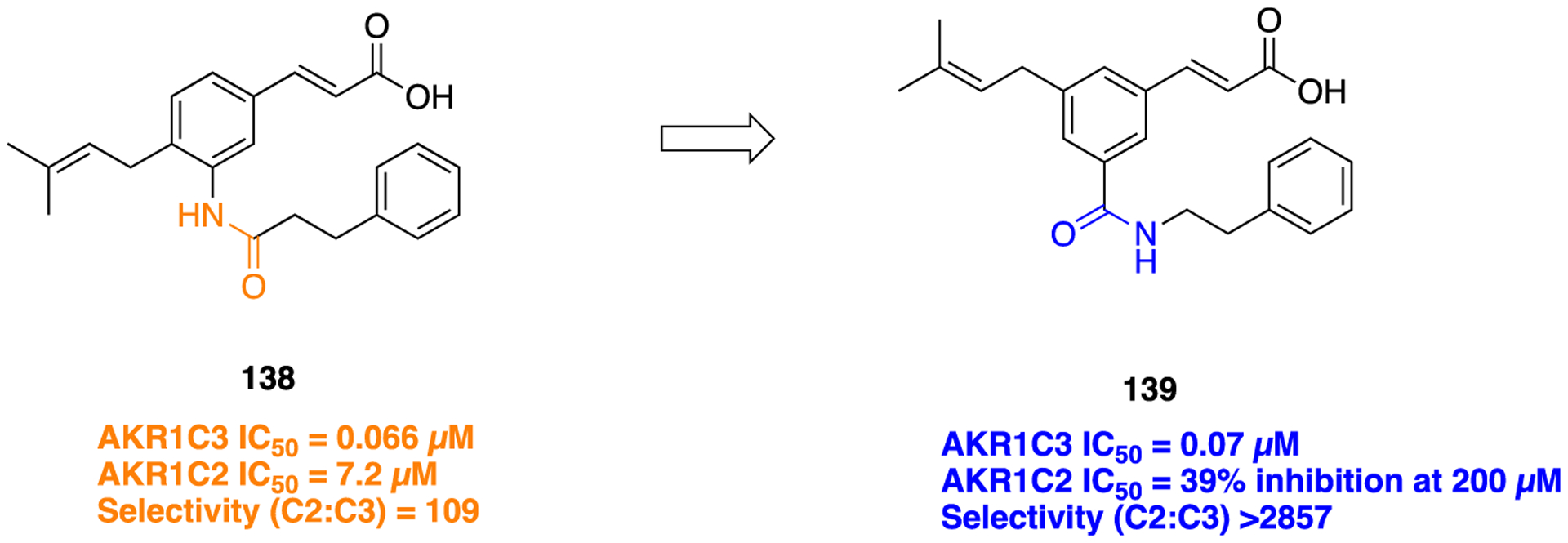
Structures of an amide-containing baccharin analog 138, a potent but low selective AKR1C3 inhibitor, and 139, a potent and highly selective retroinverted amide bioisostere.
Figure 37.

Compound 139 docked within the active site of AKR1C3 (gray, PDB code 3UG8): strong predicted hydrogen bond (green dotted line) and weak predicted hydrogen bond (white dotted line). Reproduced with permission from Journal of Medicinal Chemistry,183 Copyright 2019 American Chemical Society.
Compound 139 was synthesized in several synthetic steps, starting from 3-bromo-5-iodobenzoyl chloride (Scheme 17). The combination of compound 139 and the antileukemic drugs daunorubicin and cytarabine produced synergism against AML and patient derived T-ALL cells. Furthermore, 139 had a >100-fold dose reduction index in a daunorubicin-resistant AML cell line and >100-fold dose reduction of cytarabine in both AML cell lines and primary T-ALL cells.
Scheme 17.

Synthetic Route To Access the Potent and Highly Selective AKR1C3 Inhibitor 139
In the search for a potent melatonergic receptor agonist, a series of melatonin-based compounds has been described by de la Fuente Revenga and co-workers, where the acetamido group of melatonin (143) was replaced with several amide bioisosteres such as a reversed amide, oxazole, 1,2,4-oxadiazole, 1,3,4-oxadiazole, 1,3,4-oxadiazol(thio)ones, 1,3,4-triazol(thio)-ones, and 1,3,4-thiadiazole.184 Compound 143, N-acetyl-5-methoxytryptamine, is a neurohormone secreted primarily by the pineal gland during the night.194 The physiological and pharmacological effects of 143 are primarily mediated by the activation of the high-affinity GPCRs, melatonin receptor 1 (MT1R) and melatonin receptor 2 (MT2R).195 Numerous studies have shown that 143 and its ligands have the potential to treat pathological conditions such as sleep disturbances, type 2 diabetes, obesity, major depression, mood disorders, neurodegeneration, severe pain, inflammation, and certain forms of cancer.196–198 Earlier attempts involving detailed SAR exploration indicated that modifications on the acetamido group of 143 were detrimental as they caused either a substantial decrease or complete loss of binding affinity and potency for melatonin receptors (MTRs).199–203 Furthermore, in silico studies indicated that the acetamido group of 143 can form captodative hydrogen bonds at the binding site of MTRs.204 Therefore, the authors installed different amide bioisosteres to replace the N-acetyl fragment in 143 in order to ascertain the structural binding pocket of this fragment in terms of its size, shape, electronic distribution, and lipophilicity. The binding and functional activity studies of the prepared compounds were determined using human MT1R or MT2R stably transfected in the Chinese hamster ovary cells. The results indicated that the replacement of the acetamido group of the parent 143 by retroamides (144 and 145) produced the highest binding affinities, with Ki values in the low nanomolar and subnanomolar range (Figure 38).
Figure 38.
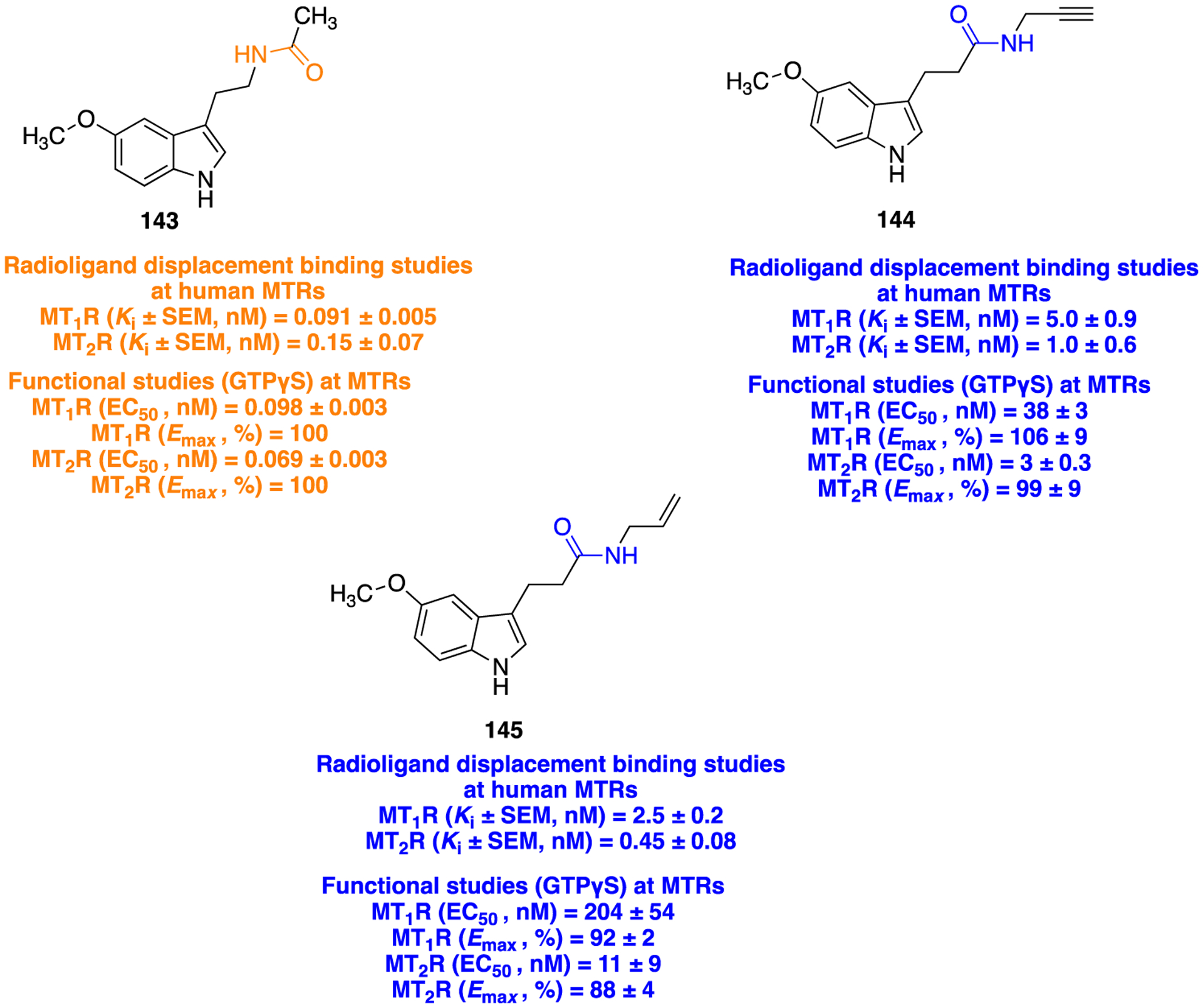
Structures of the parent amide 143 and the reverse amide bioisosteres 144 and 145, MTR ligands.
The SAR analysis of retroamides with unsaturated side chains indicated that alkenyl or alkynyl substitution was well tolerated but the length of the spacer affected the potency of the compounds. For example, retroamides with one methylene unit (structures not shown here) did not produce favorable results. In contrast, both 144 and 145, which had an ethylene spacer, retained potency at MTRs. The synthetic route to access 144 and 145 is delineated in Scheme 18 where commercially available 2-(5-methoxy-1H-indol-3-yl)alkanoic acid was activated with carbonyldiimidazole (CDI) and then reacted with propargylamine or allylamine to obtain 144 and 145, respectively. Furthermore, compound 144 was analyzed for brain repairing efficacy, i.e., neurogenic potential, by using rat neural stem cells as an in vitro system. The results of this experiment indicated that 144 induces moderate cell differentiation but failed to stimulate neural maturation. Overall, the bioisosteric replacement of an amide with a retroamide yielded compounds that retained MTRs binding affinity, suggesting that the acetamido group could be transformed without a significant loss of binding affinity.184
Scheme 18.
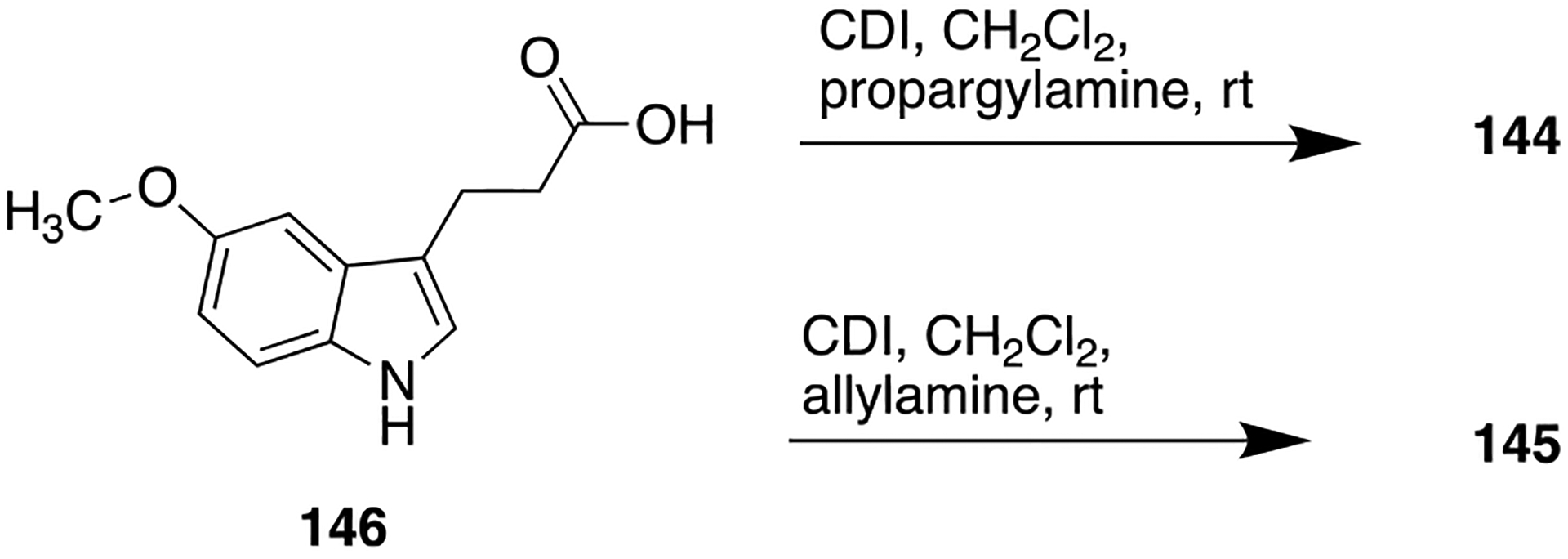
Synthetic Preparation of the Melatonin-Based Compounds 144 and 145
The above examples illustrate that despite their relatively simple structural differences, reverse amide bioisosteres can overcome issues related to the potency and selectivity of amide-containing counterparts. There are of course many examples where reverse amide surrogation does not produce beneficial pharmacological effects, and one such case is discussed in the tetrazole section of this review above, where Beinat and colleagues installed reverse amide and tetrazole isosteres to develop potent α7-AChRs agonists.149
UREA BIOISOSTERES
Urea has the ability to form multiple stable hydrogen bonds with a variety of protein and receptor targets, a very important element for its molecular recognition and bioactivity. The central carbonyl functions as a nucleophile and hydrogen bond acceptor with the ability to accept two hydrogen bonds. The protons on the two nitrogens serve as hydrogen bond donors, capable of providing two to four hydrogen bonds dependent on substitution.205 These interactions allow for specificity in drug action, biological activity, and manipulation of physicochemical properties, which is why urea derivatives are widely used in medicinal chemistry. Ghosh and co-workers proposed that substitution on the urea nitrogen atoms significantly affects the conformational preferences of urea derivatives due to the delocalization of nonbonded electrons α to the carbonyl atom.206 Urea is hydrotropic, meaning it can be used to promote the solubility of poorly soluble drugs and improve the permeability of molecules it is introduced into.205 In some cases, urea compounds can have poor solubility or permeability. However, various techniques can be used to address this issue, for instance, modulating the hydrogen bonding ability of ureas by introducing electron donating or electron withdrawing groups (EWGs) on the substituents, disruption of molecular planarity to enhance solubility by introducing substituents on one urea nitrogen to make it nonsymmetrical and break planarity, or introducing a substituent at the ortho position of N-aryl groups of arylureas.206 Overall, solubility and permeability depend on other substitutions within the molecule. There are several FDA approved drugs containing the urea moiety for use in a variety of human diseases such as antidiabetics, antibacterials, anticancer, and other therapeutic agents. Urea derivatives play a vital role in the regulation of various pharmacological activities such as the ability to improve potency and target selectivity, modulation of physiochemical properties, and contribution to metabolic liability.205
In order to identify the necessary structural motifs for potent acyl-coenzyme A:cholesterol acyltransferase (ACAT) inhibition in vitro and hypocholesterolemic agents in vivo, Roark and colleagues employed a peptidomimetic approach. Initially, a series of oleic and fatty acid analogs were synthesized wherein the peptide amide bonds in N-(2,4,6-trimethoxyphenyl)- and N-(2,6-dimethylphenyl)oleamides, and stearamides underwent various bioisosteric replacements to examine effect on biological activity. Results showed that only two urea bioisosteres 148 and 149 (Figure 39) retained considerable inhibitory activity in vitro (IC50 = 0.32 μM and 0.16 μM, respectively). Although 149 showed a 4-fold decrease in potency in vitro (IC50 = 0.16 μM) compared to its amide counterpart 147 (IC50 = 0.043 μM) (Figure 39), it was more potent in vivo. When 149 was dosed at 0.05% in the diet of cholesterol-fed rats, it lowered total cholesterol by 55% versus a 38% decrease in total cholesterol with the amide-based compound at the same dose. As a result, only bioisosteres having both hydrogen bond donor and acceptor functionalities yielded compounds that retained ACAT inhibitory potency activity in vitro as shown by sulfonamide, thioamide, retroamides, urea, carbamate, and thiocarbamate bioisosteres. Additionally, the location of the hydrogen bond donor and acceptor was crucial as retroinverted amides were less potent than the amides. Bioisosteric analogs with no, or one, hydrogen bond donor or acceptor functionality, as shown by ester, oxymethylene, thiomethylene, methyleneamino, sulfoxide, ketomethylene, and hydroxyethylene, led to uniformly inactive compounds. Therefore, the urea moiety was an acceptable bioisosteric replacement for the amide.207 Later the same group extended this original fatty acid amide series to encompass β-ketoamides.208 A simple series of N,N′-diphenylurea derivatives were also found to be potent ACAT inhibitors.209 Further evaluation led to the identification of a novel series of potent urea derivatives and a new ACAT inhibitors. Structure-activity relationship studies in this series revealed that a certain spatial arrangement, five atom spacer, between two phenyl rings is critical, lipophilic bulk is well tolerated, the 2,6-positions of the 2,6-diisopropylphenyl are essential to orient the required carbonyl group out of plane of the phenyl ring in order to obtain optimal enzyme interactions. Isosteric replacement of the urea moiety resulted in significantly less active compounds (10- to 1000-fold). From this series of analogs, a potent inhibitor of ACAT (IC50 = 0.017 μM) (150) (Figure 39) was discovered; it dose-dependently lowered plasma cholesterol in cholesterol-fed rats, but further investigations showed the compound was poorly absorbed. To overcome this, SAR studies continued with N-phenyl-N′-[(1-phenylcyclopentyl)methyl]ureas and compound 151 (Figure 39) was discovered (Scheme 19, i) which did not produce adrenal toxicity in guinea pigs and had substantial lipid-modulating activity in a chronic model of preestablished dyslipidemia in rats. The general synthetic route to obtain compound 151 is depicted in Scheme 19, i, where the urea moiety was furnished by reacting the corresponding isocyanate and amine. Overall, subsequent evaluation in animal models of hypercholesterolemia suggested that their physical properties were important determinants of their pharmacokinetics and efficiency.
Figure 39.
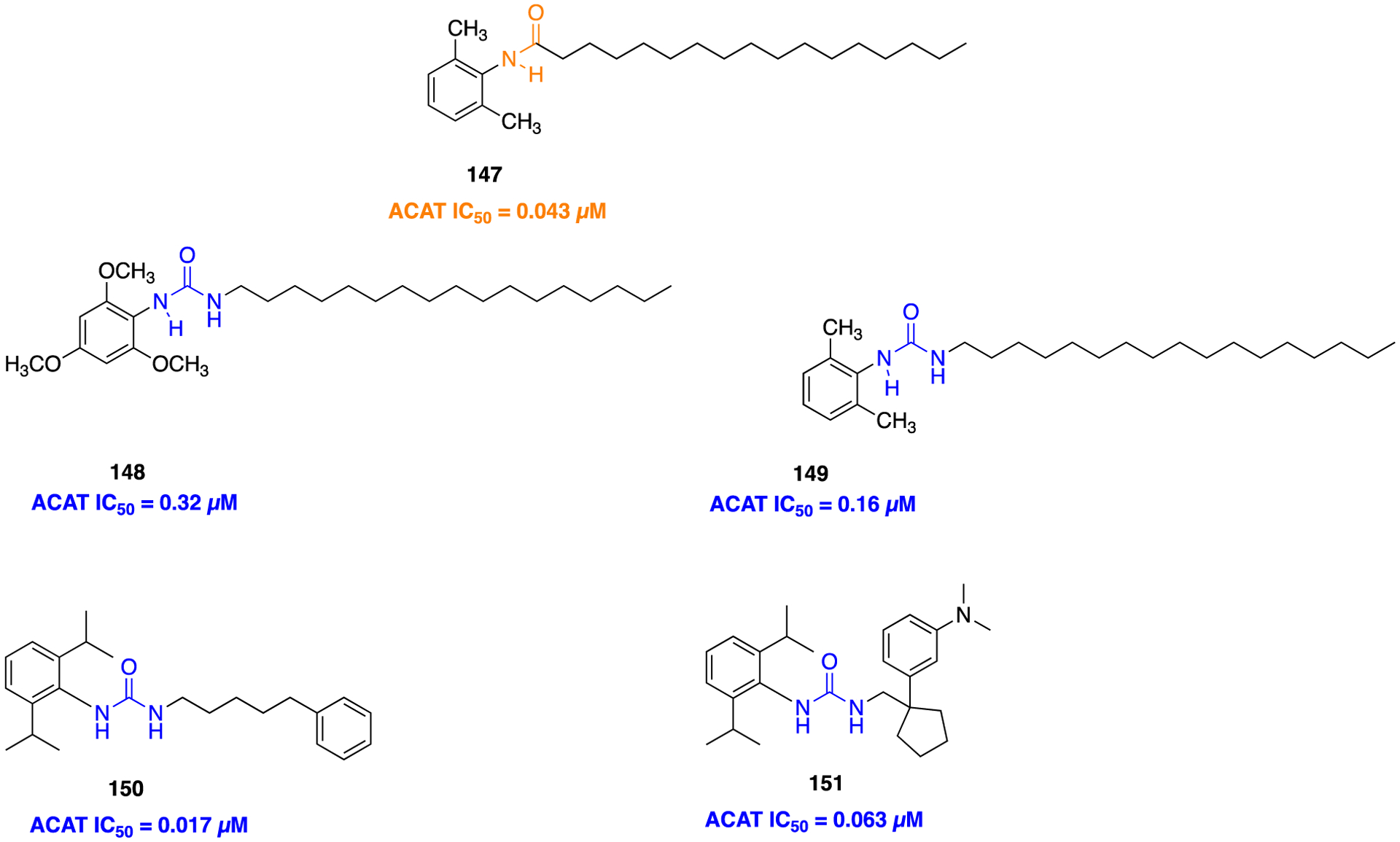
Structures of parent amide 147, urea bioisosteres 148, 149, and 150, and 151, a potent urea ACAT inhibitor.
Scheme 19.
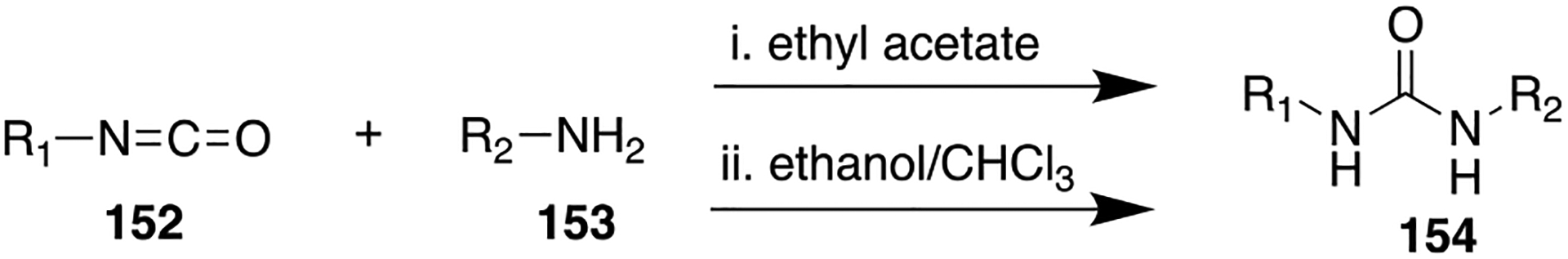
General Synthetic Route To Prepare Urea Bioisostere via Isocyanate
Bioisosteric replacement of an amide bond in (±)-15-deoxyspergualin (DSG, 155) (Figure 40), a synthetic derivative of spergualin, a product isolated from Bacillus laterosporous, was explored by Lebreton and co-workers.210 Previously, 155 has been used as an anticancer agent and in animal transplantation either in allografts, xenografts, or graft-versus-host disease (GVHD) animal models. In 1994, 155 was commercialized in Japan as an immunosuppressive agent. However, 155 has three drawbacks limiting its use: low chemical stability in aqueous solution leading to hydroxylation, presence of the inactive (+)-155 isomer in a racemic mixture which is not reported to be immunosuppressive and can contribute to acute toxicity, and low oral bioavailability (<5%) preventing oral administration. New analogs of 155 were synthesized and tested in a GVHD model in mice. By use of a bioisosteric replacement approach, variations were implemented at the hydroxyglycine central carbon atom to prevent issues associated with chirality and hydrolytic sensitivity. Initially, a malonic derivative was identified as the first analog of a new series of potent immunosuppressive agents where the retro-inverted amide bond was linked to a hexylguanidino moiety, 156 (Figure 40). Structure-activity relationship studies where the “western-amide” was reversed compared to 155 led to active analogs at 3 and 1 mg/kg. However, other “western-amide” replacements such as a two-methylene unit, carbamate, or urea led to inactive compounds. The “easternamide” of the malonic derivative was varied and led to more analogs wherein a urea derivative, 157 was active at 3 mg/kg. Replacing the NH atoms of the urea by oxygen led to compound 158 which was prepared via acid-amine coupling using HOBt and DCC in dichloromethane (Scheme 20). Compound 158 was found to be very active at 3 mg/kg in a GVHD model (Figure 40). However, when compound 158 was tested in a more demanding model for acute organ rejection after heart allotransplantation, the urea derivative (157) was only slightly active, whereas compound 158 was found to be the most potent and slightly more active than compound 155 in the heart allotransplantation model in rats. This was due to the absence of chirality leading to improved chemical stability when compared to compound 158 (Figure 40); therefore, compound 158 was selected as a drug candidate.
Figure 40.
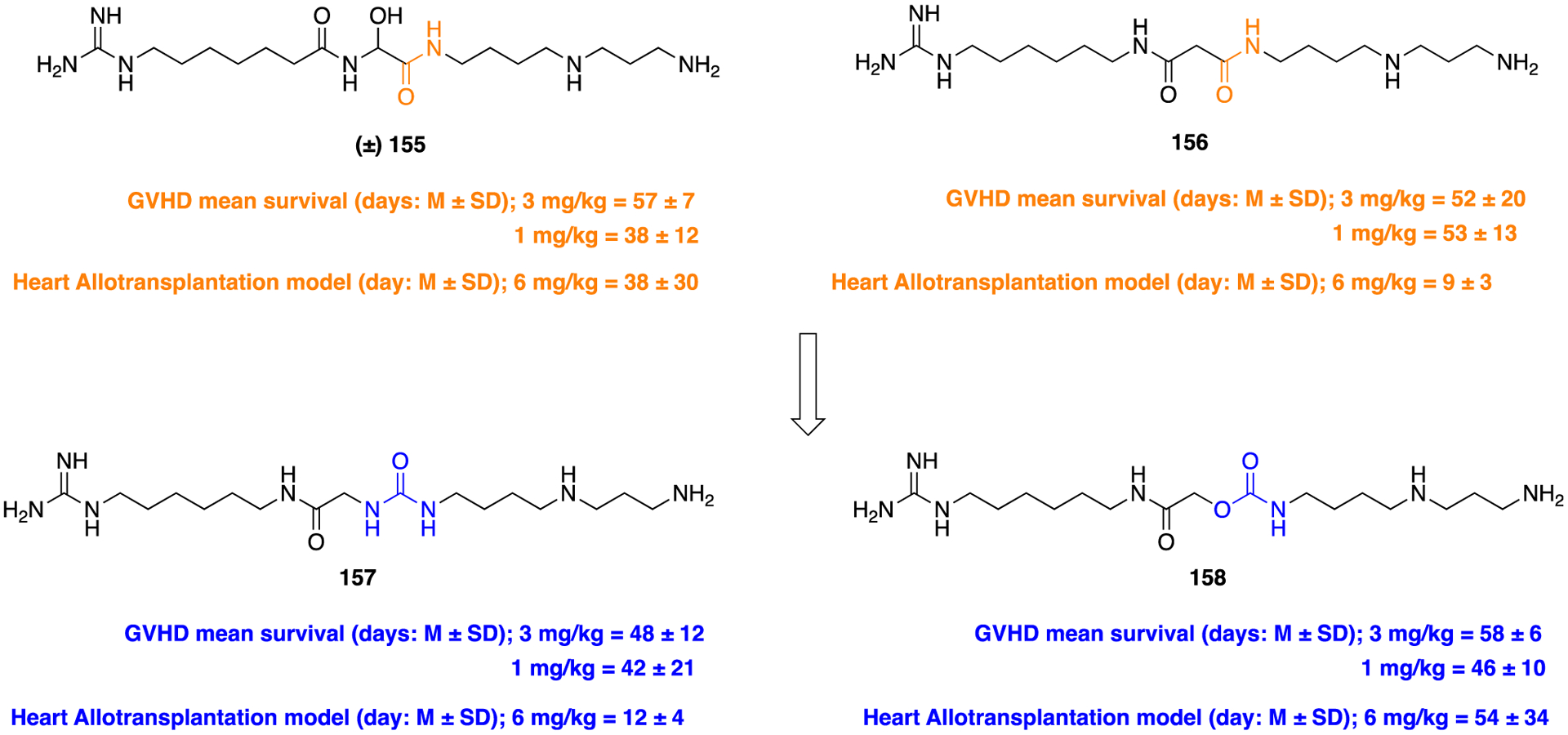
Structures of DSG (155) and bioisosteric analogs.
Scheme 20.
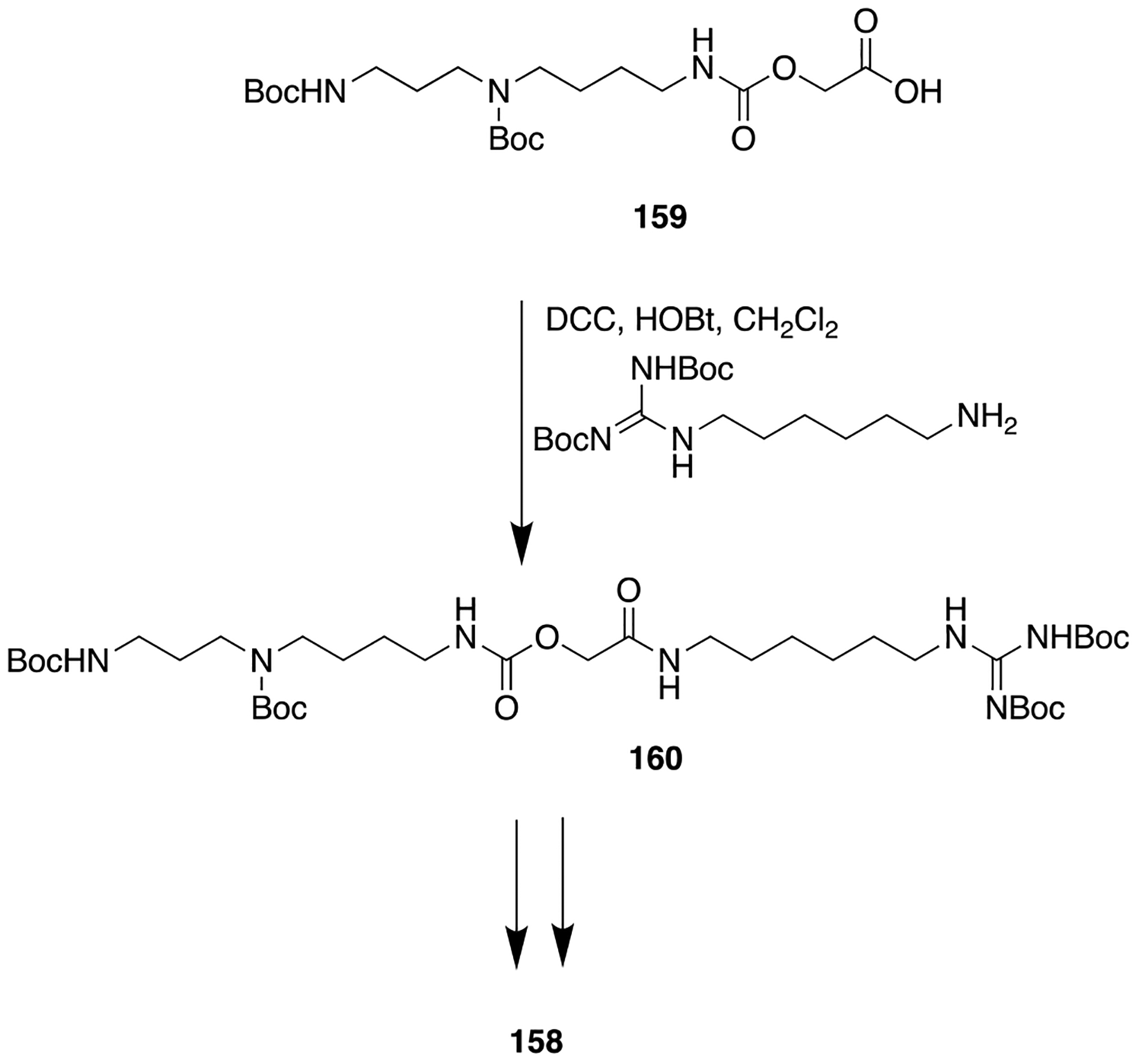
Synthetic Procedure To Access 158, a Potent Immunosuppressor
Successful bioisosteric replacement of an amide with a urea moiety was achieved based on 1,6-naphthyridine moieties in which the lead compound 161 had similar activity as ganciclovir, an antiviral drug for human cytomegalovirus (HCMV). Previous SAR studies revealed that the optimal positions for nitrogen atoms on the naphthyridine ring are at the 1- and 6-positions.211 Additionally, substitution at C-8 and a bulky alkoxy group at the 2′-position are beneficial for activity (Figure 41).212 It was also discovered that the internal hydrogen bond between the NH of the amide and heteroatoms labeled N1 and O2′ (Figure 41) are key to maintaining the active conformation of the molecule. Chan and co-workers evaluated new napthyridines, wherein bioisosteric replacement of the amide group with other groups can maintain hydrogen bonds, thioamides, and urea as new anti-HCMV compounds. First, thioamide derivatives were studied due to their structural similarity to the amide motif and potential ability for in vivo stability. Results showed that methoxybenzylamine substitution was 3 times less active than the parent amide. There was only slight improvement with an increase in the bulk of alkoxy substituent to 2′-ethoxybenzylamine or 2′-isopropoxybenzylamine. Replacing the amide with a longer thioamide resulted in the loss of some desirable interaction at this position; therefore, thioamide derivatives were not further studied. Next, urea derivatives were evaluated since they have the potential to form two internal hydrogen bonds. More specifically, benzyl and phenylureas were studied due to previous SAR studies revealing that substitution on naphthyridine can enhance potency leading to substitutions at carbons 3 and 8 also being evaluated. The benzylureas revealed that unsubstituted analogs resulted in nonselective activity; the 2′ methoxy analog was active, and increasing the bulk of the substituent to isopropoxy resulted in enhanced activity. Similar results were seen in the phenylurea derivatives where an unsubstituted analog gave an inactive derivative, an alkoxy group substituent led to active compounds, and an increase in the bulk of the alkoxy group resulted in enhanced activity. The 2′ isopropoxy derivative 162 was the most active compound (Figure 41) with IC50 = 30 ng/mL in this series. Substitution on 1,6-napthyridine indicated that a cyano or methyl group at C-3 eliminated activity, whereas introducing a bromine atom at C-8 did not improve potency in urea derivatives. These novel anti-HCMV compounds revealed that ureas and amides are maintained in similar active conformation through internal hydrogen bonding. The lack of 2′-alkoxy substituents led to toxicity or no activity due to the presence of only one internal hydrogen bond and more degrees of freedom being present around the amide bond. Overall, internal hydrogen bonding is an important feature in 1,6-napthyridine urea derivatives for potent activity as HCMV inhibitors.213
Figure 41.
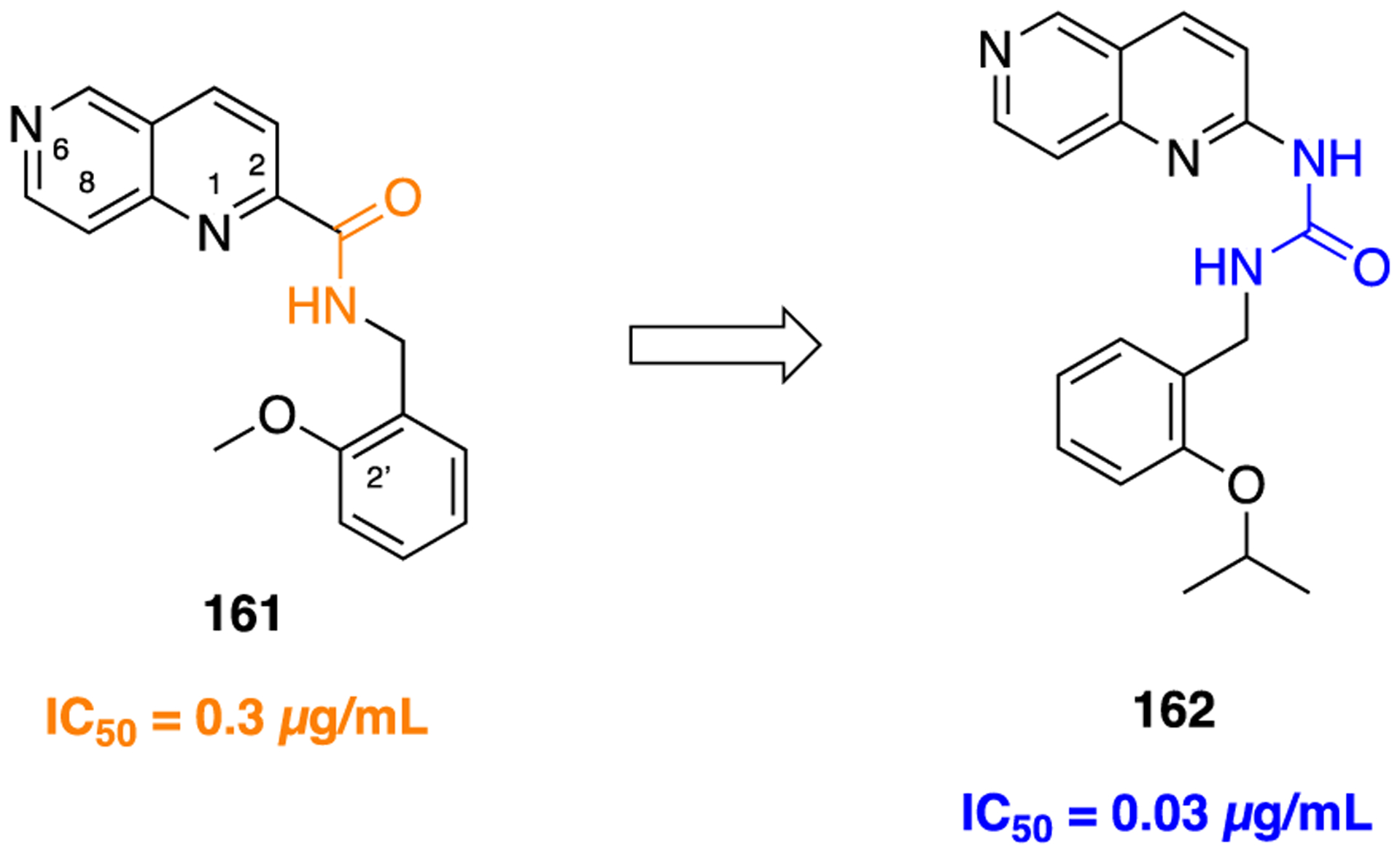
Structures of lead compound 161 for anti-human cytomegalovirus (HCMV) and 2′-isopropoxyurea bioisostere 162.
Bioisosteric replacement of an amide with a urea moiety was compared in ceramide analogs; B13, compound 163, a ceramide analog consisting of a phenyl group instead of an alkenyl chain and (1R, 2R) configuration versus the (2S, 3R) configuration of natural ceramides, was synthesized.214 Previously, 163 was shown to have potent cytotoxic activity in human colon cancer cells that underwent apoptosis.215 It also displayed selective toxicity toward malignant cells over nonmalignant cells. Moreover, it prevented metastasis in liver colon cancer cells of a nude mouse model. This led to the synthesis and evaluation of 3-alkyl-1-(1-methyl-2-phenylethyl)-ureas, novel analogs of 163.216 The amide group of 163 underwent a bioisosteric replacement with urea in the presence or absence of a p-nitro group on the phenyl ring, two hydroxyl groups on the backbone, varying stereochemistry on the backbone along with varying chain length of the alkyl chain with octyl and dodecyl groups. These derivatives were evaluated for their cytotoxic activity in five human tumor cell lines (Caki-2, human renal cancer; HT-29, human colon cancer; A549, human lung cancer; PC-3, human prostate cancer; HL-60, human leukemia). Thus, compound 163, displayed modest activity in the HL-60 cell line (IC50 = 28 μM) and weak activity in each of the other four cell lines (Figure 42). A urea analog, 164, was synthesized by following the general synthetic procedure as shown in Scheme 19, ii. Compound 164 had comparable cytotoxicity in Caki-2, A549, and HL-60 but showed significantly potent cytotoxicity in HT-29 and PC-3 when compared to 163 (Figure 42). Therefore, urea serves as a suitable amide bioisostere. When the p-nitro group was excised, it resulted in more than a 3-fold decrease in cytotoxicity. When octyl and dodecyl groups were added to the urea moiety, the dodecyl analogs were more active compared to octyl. This is consistent with other studies of ceramide where biological activity was enhanced with increasing acyl chain length.217,218 This is a known phenomenon in medicinal chemistry wherein enhanced lipophilicity increases activity to a certain limit.219 Results showed that retention of stereochemistry on the backbone may not be essential for potent cytotoxic activity. However, this may not be certain because the 163 urea analogs containing (1R, 2R) and (1S, 2S) enantiomers displayed similar activity, while enantiomers of unsubstituted phenyl rings and 1-hydroxymethyls were inactive, making a conclusive comparison challenging. Overall, the urea motif appeared to serve as a suitable bioisosteric replacement of an amide for novel ceramide analogs.216
Figure 42.
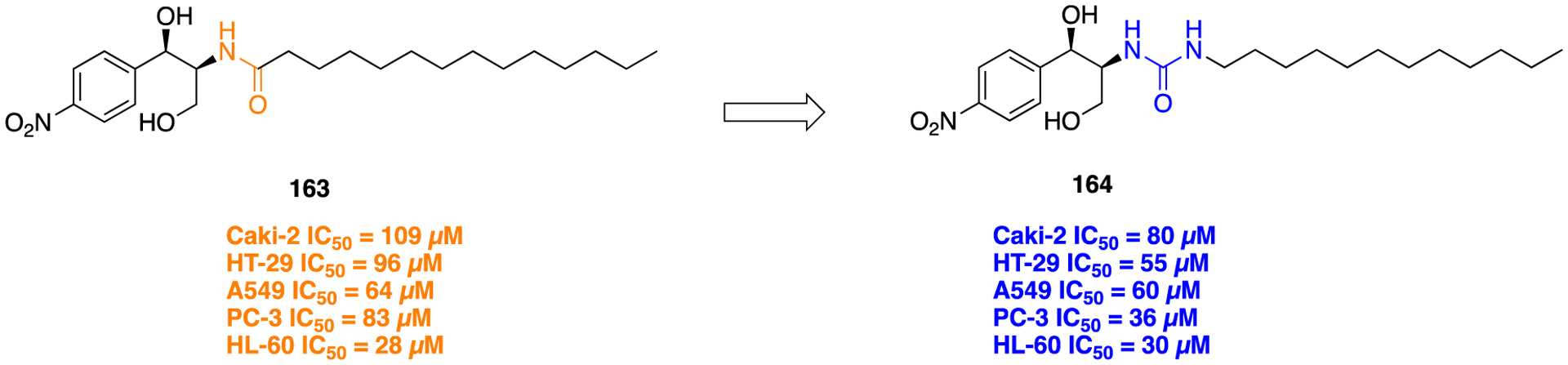
Structures of 163, B13, a ceramide analog tested for in vitro cytotoxicity against five human tumor cell lines, and 164, a more potent urea bioisostere.
The antitrypanosomal activities of novel derivatives against Trypanosoma brucei, the causative agent of human African trypanosomiasis (HAT), were explored.220 Human African trypanosomiasis occurs in 36 nations of sub-Saharan Africa and is an insect vectored disease caused by Trypanosoma brucei gambiense which progresses slowly from early stage to late stage or can progress rapidly when caused by Trypanosoma brucei rhodesiense. In late-stage HAT, the central nervous system is infected and the disease becomes fatal if patients are left untreated regardless of the contributing parasite.221 The compound 2-(2-benzamido)ethyl-4-phenylthizaole 165 (Figure 43) was one of 1035 compounds from a phenotypic high-throughput screen of 700,000 compounds that inhibited the growth of T. brucei in vitro at concentrations below 3.6 μM and was nontoxic to mammalian cells (Huh7). A total of 82 novel analogs were synthesized by Patrick and colleagues and tested against T. brucei rhodesiense STIB900 and L6 rat myoblast cells for cytotoxicity in vitro. It was found that 44 derivatives were more potent than 165 with antiparasitic IC50 values ranging from 9 nM to >300 μM in which eight of them displayed IC50 values below 100 nM. Overall, ureas were significantly more potent than amides in addition to being more selective for the parasite. From these, the most potent and selective for T. brucei rhodesiense was a piperidyl urea analog 166 with an IC50 = 0.0090 μM. Thirty-three compounds with IC50 values below 0.2 μg/mL were tested in mice infected with T.b. rhodesiense STIB900. Despite these compounds displaying in vitro potencies, none of them cured any infected mice, although seven compounds did cause a reduction in parasitemia by at least 97% compared to the untreated control group. Relapses occurred in all cases. Further analysis was taken in which the stability of 15 compounds in MLM and HLM, with and without the nicotinamide adenine dinucleotide phosphate (NADPH) cofactor, was evaluated to determine if metabolic instability was the contributing factor for the lack of in vivo efficacy. The SAR studies revealed that the incorporation of a piperidyl urea moiety (166) significantly improved the potency against T.b. brucei when compared to 165. However, compound 166 suffered from low metabolic stability in MLM (t1/2 = 1.6 min) and also was the least stable to HLM (t1/2 = 4.9 min) in comparison to 165 (Figure 43). These microsomal studies revealed that compound 166 collectively had poor metabolic stability and therefore was not further evaluated in vivo.220
Figure 43.
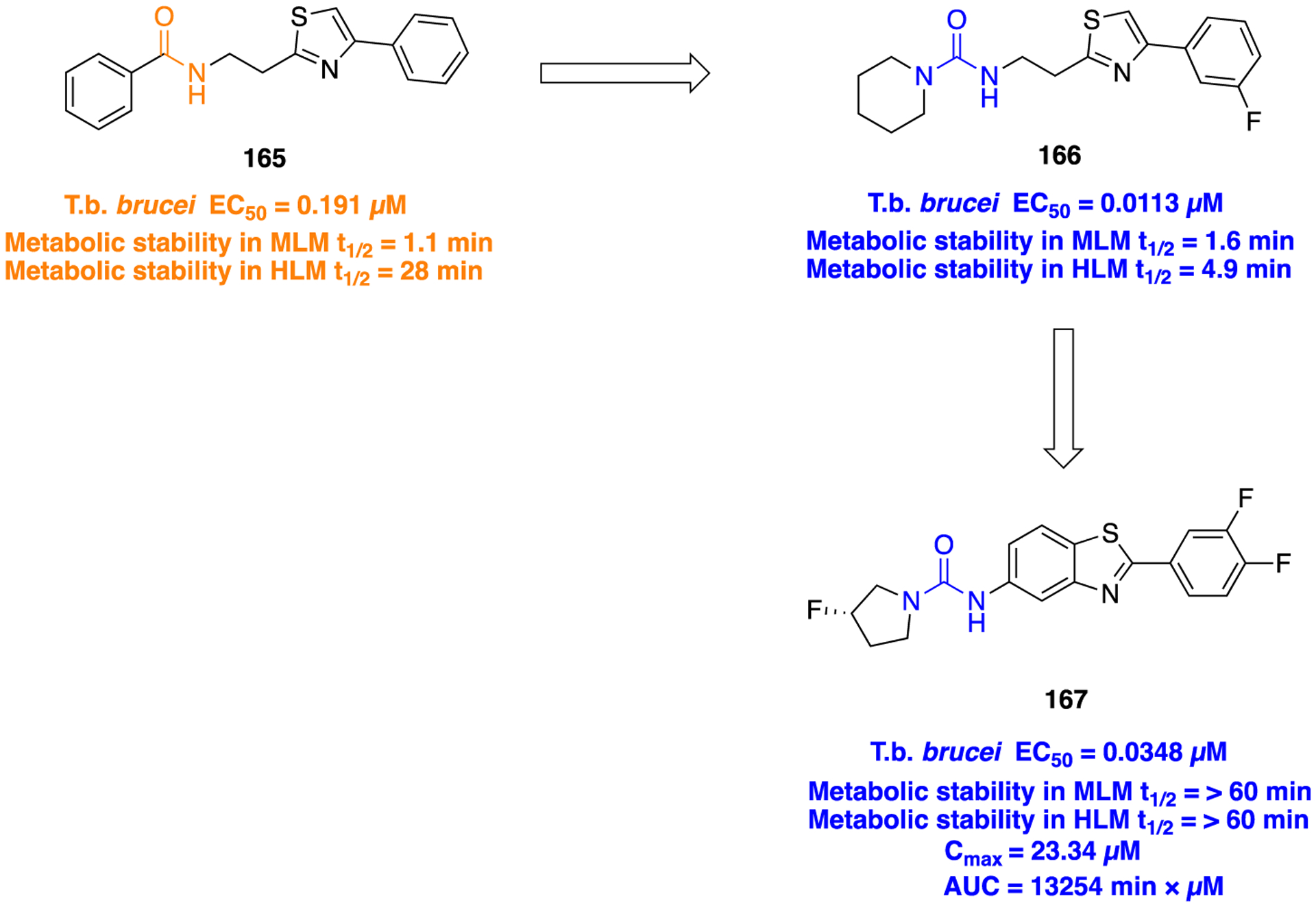
Antitrypanosomal agent 165, an amide derivative, was further optimized to urea derivatives 166 and 167.
In order to improve metabolic stability while maintaining potency, further optimization was attempted. This led to more promising compounds based around urea derivatives of 2-arylbenzothiazol-5-amines, more specifically (S)-2-(3,4-difluor-ophenyl)-5-(3-fluoro-N-pyrrolidylamido)benzothiazole 167 (Figure 43). Herein modifications in other sites of 165 were performed, and in almost all cases except three analogs, the three-carbon bridge between the amide and thiazole nitrogen atoms of 165 was retained. Modifications were performed via replacement of the carbonyl oxygen of the amide with sulfur, various substituents on the piperidine ring of 166, and alkylation or unsaturation of the ethylene linker. Results showed that none of these provided any advantage regarding the antitrypanosomal activity or metabolic stability. Enhanced metabolic stability was observed only in the presence of benzoxazole- or benzothiazole-5-amino moiety in fused ring analogs. However, antitrypanosomal activity was only retained in benzothiazole derivatives. Thus, the ethylene linker played a role in the poor metabolic stability of initial thiazole analogs. From this series, 167 was discovered which exhibited good activity in vitro (T.b. brucei; EC50 = 0.0348 μM), had no detectable toxicity when tested in two human cell lines [human lymphoblast cells (CRL-8155) and HepG2], and possessed a half-life of >60 min in both MLM and HLM (Figure 43). The target compound 167 was prepared by reacting a primary amine (168) with triethylamine in dichloromethane. Triphosgene was added followed by the addition of (S)-3-fluoropyrrolidine hydrogen chloride (Scheme 21). Compound 167 was also tested in mice; oral administration of 5 mg/kg intraperitoneal (ip) dose resulted in Cmax = 23.34 μM and AUC = 13,254 min·μM with excellent brain penetration (brain concentration of 9.07 μM and brain/plasma ratio of 4.00). The compound was advanced to in vivo efficacy studies in which oral dosing cured 5/5 acute and chronic models of HAT, making it a representative lead for the future development of drugs combating this disease.221
Scheme 21.
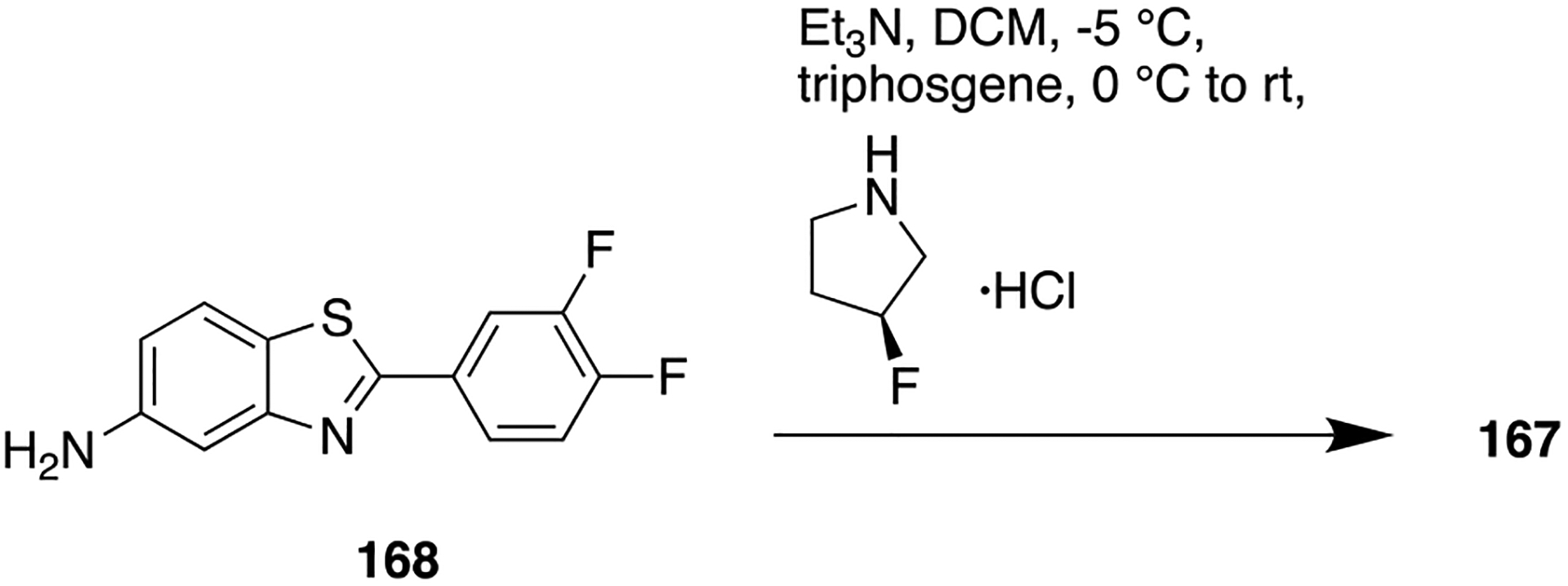
Synthesis of 167, a Novel and Potential Drug for Human African Trypanosomiasis
In order to target the ThDP binding site of Escherichia coli pyruvate dehydrogenase complex E1 component (PDHc-E1), two novel “open-chain” linkers, comprising amide and urea functionality, were studied for their effect on E. coli PDHc-E1 inhibition. The pyruvate dehydrogenase complex (PDHc) catalyzes the conversion of pyruvate into acetyl-CoA, while PDHc-E1 catalyzes the first irreversible step of this multistep process. The PDHc-E1 protein has been reported to be a promising target for the development of fungicide and herbicide agents.222 Several studies on small molecule inhibitors of PDHc-E1 have been reported, but they suffer from structural complexity and contain a highly charged pyrophosphate. Therefore, there is a need for the development of small molecules as inhibitors of PDHc-E1 that overcome these limitations. First, SAR studies revealed that placing iodine in the 5-position of the 1,2,3-triazole moiety of compounds under investigation had the potential to increase inhibitory and antifungal activity against E. coli PDHc-E1. Additionally, the linker was essential for biological activity, thus leading to the optimization of 1,2,3-triazole derivatives with amide and urea moieties as “open-chain” linker analogs of 169 (Figure 44). Amide and urea derivatives were evaluated for their in vitro inhibition of E. coli PDHc-E1. Most compounds had a good inhibitory activity with IC50 < 20 μM. These studies revealed that compounds with an amide linkage possessed inhibitory activity highly correlated to substituents on the benzene ring; electron-withdrawing groups led to more potent inhibitory activity, as seen in 170 (Figure 44). This compound possessing a 4-NO2 substituent resulted in the most potent inhibitory activity, IC50 = 3.58 ± 0.52 μM (Figure 44). Overall, the amide linkage has higher inhibitory activity than parent triazole 169. As for the urea linkage, inhibitory activity was improved compared to the corresponding amide derivatives with the same substituents, thus resulting in compound 171, which had the same potency as 170. The synthetic method utilized for the preparation of compound 171 is outlined in Scheme 22. He and co-workers tried to synthesize a urea derivative with a 4-NO2 in the benzene ring; however, they were unsuccessful. Next, they performed molecular docking studies that showed that the binding mode of 171 is similar to 170 in the active site of E. coli PDHc-E1 (Figure 45). However, the two NH groups of the urea form strong hydrogen bonds with Glu522; the oxygen of the phenoxy also hydrogen bonds with Leu264 which could be due to the increase in chain length afforded by the urea moiety. Overall, the amide and urea linkages have a beneficial effect on E. coli PDHc-E1 inhibition versus the 1,2,3-triazole derivatives.222
Figure 44.
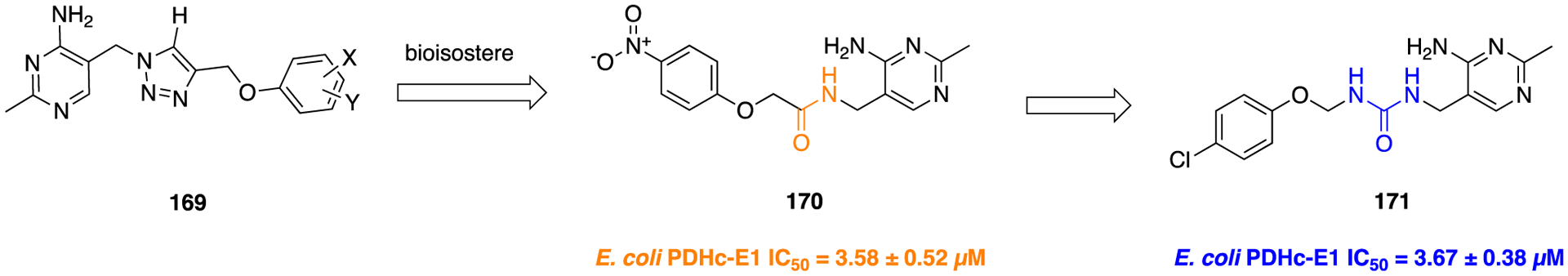
Structures of the amide (169) and urea (171) bioisostere derivatives of the triazole-based Escherichia coli PDHc-E1 inhibitor 169.
Scheme 22.
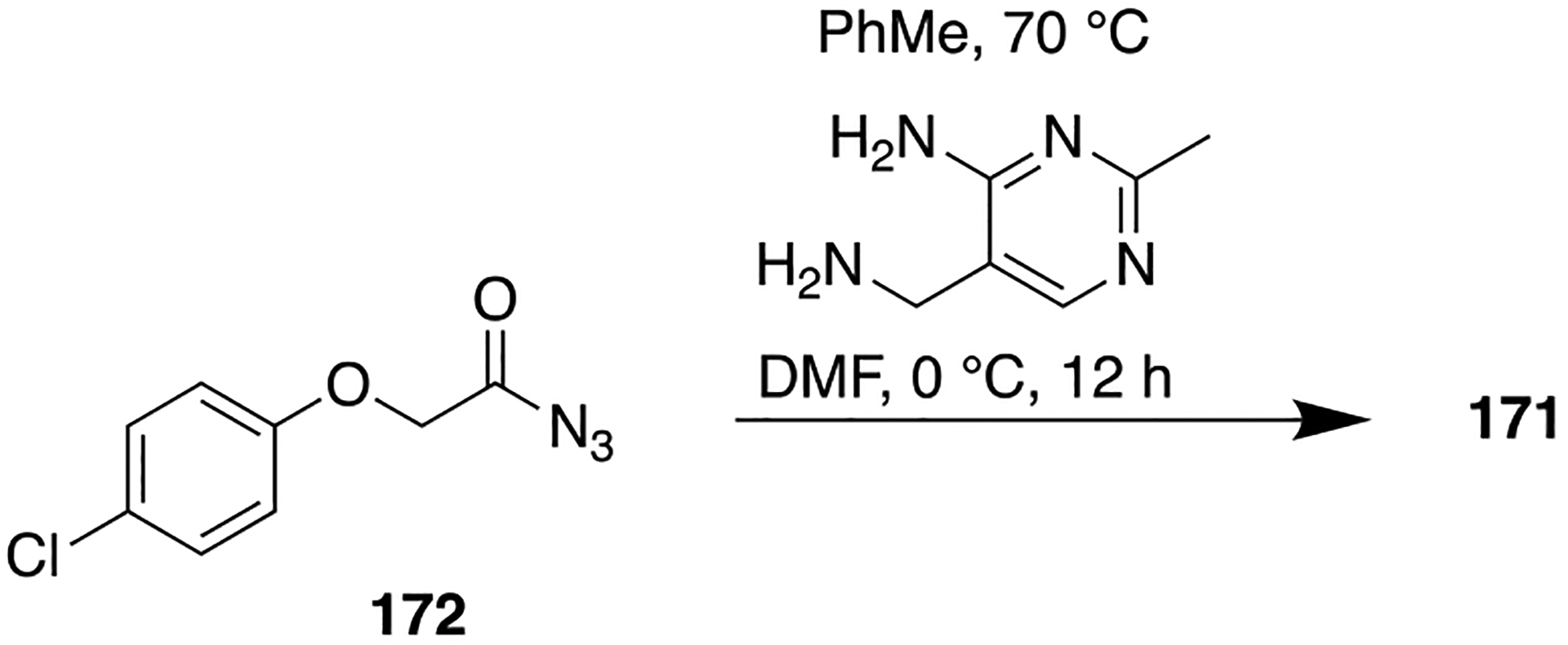
Synthetic Route To Access the Potent Urea Derivative 171, an E. coli PDHc-E1 Inhibitor
Figure 45.
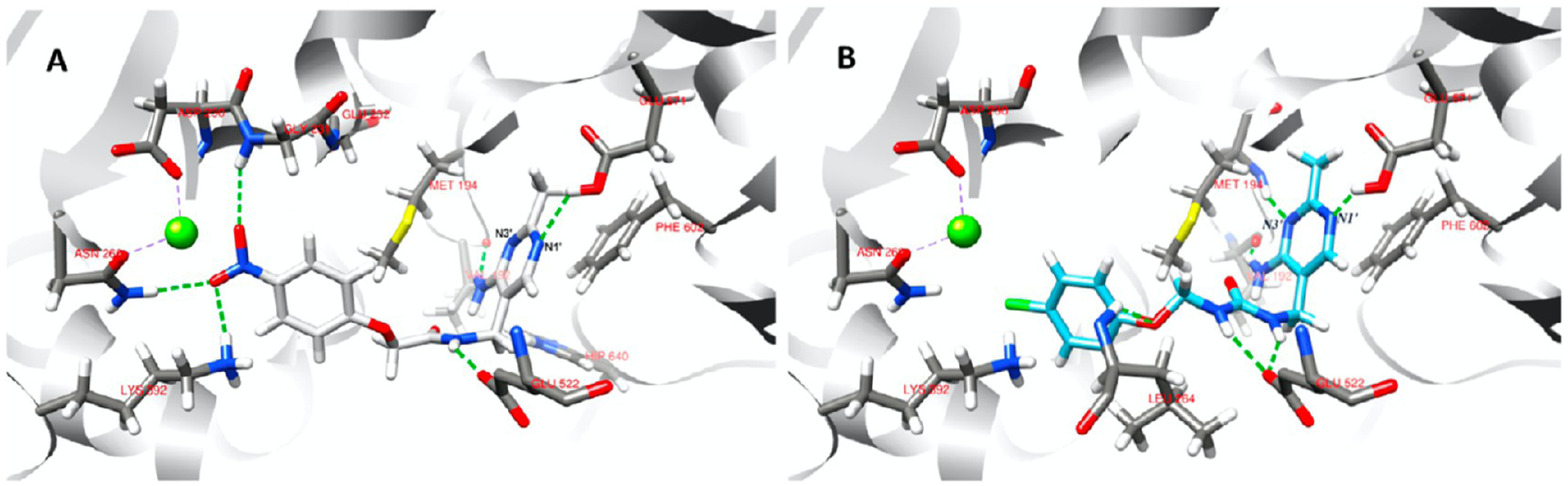
Graphical representation of the binding modes of 170 (A) and 171 (B) into the active site of E. coli PDHc-E1 displayed in ribbon. Ligands and residues are depicted by stick, while the hydrogen and coordination bonds are shown with dashed lines (green). Reproduced with permission from Bioorganic & Medicinal Chemistry (https://www.sciencedirect.com/journal/bioorganic-and-medicinal-chemistry), Volume 22, pp 3180–3186, He, J. B.; Ren, Y. L.; Sun, Q. S.; You, G. Y.; Zhang, L.; Zou, P.; Feng, L. L.; Wan, J.; He, H. W., Design, synthesis and molecular docking of amide and urea derivatives as Escherichia coli PDHc-E1 inhibitors.222 Copyright 2014 Elsevier.
CARBAMATE BIOISOSTERES
Ghosh and co-workers have described that a carbamate motif can be considered as structurally related to an amide-ester hybrid. Carbamates can modulate inter- and intramolecular interactions with target enzymes or receptors. Additionally, they induce a degree of conformational restriction as a result of the delocalization of nonbonded electrons on the nitrogen atom. Hydrogen bonding also occurs via the carboxyl group and NH backbone. Carbamates possess a pseudo double bond; thus, two isomers, syn and anti, can coexist. They display close similarity to amides; however, they prefer the anti-isomer. Ultimately solvent, concentration, salts, and pH influence the adaptation of syn and anti isomers in carbamates. Overall, carbamates possess very good chemical and proteolytic stabilities. They serve as amide bioisosteres in peptidomimetics due to their chemical stability and ability to permeate cell membranes. Carbamate-based prodrugs must undergo extensive hydrolysis in order to release the active drug at a suitable rate to achieve the desired activity profile.223
The global pandemic of HIV/AIDS was first identified in 1983–1984. The protease enzyme of HIV cleaves the kh and gag-pol polyprotein into its essential structural parts. Protease inhibitors block this enzyme, thereby preventing the development of the HIV virus. Saquinavir 173 (Figure 46) was approved in 1995 as the first HIV protease inhibitor.224,225 Although 173 is a potent protease inhibitor, which is effective against both HIV-1 and HIV-2 proteases, this can also lead to reduced susceptibility to clinical resistance due to these enzymes being the most genetically divergent strains. Ghosh and co-workers investigated the replacement of the P2-asparagine of 173 with (2S,3′R)-tetrahydrofuranylglycine 174 which led to increased enzyme affinities for HIV-1 and HIV-2 proteases and significant enhancement of antiviral potency.226 These compounds prevented HIV-1 in MT4 infected with 111b isolate; 174 was 3-fold more potent (CIC95 = 8 ± 4 nM) than 173 (CIC95 = 22 ± 7 nM). In HIV-2, 174 was 2-fold more potent (IC50 = 0.24 nM) than 173 (IC50 = 0.5 nM) (Figure 46).227 An X-ray crystal structure of 173 bound to the HIV-1 protease showed that the carbonyl oxygen of the asparagine is within hydrogen bonding distance to the NH group within both Asp29 and Asp30, present in the S2 binding domain of the HIV-1 protease. On the basis of this, the authors hypothesized that the ether oxygen would conformationally constrain and lend its lone pairs for hydrogen bonding to the residues in the S2 region of the active site. A model for 174 was superimposed on the structure of 173, which revealed that the tetrahydrofuran oxygen of 174 is indeed in close proximity to the carbonyl oxygen of asparagine. Overall, replacing the P2-asparagine of 173 with (2S,3′R)-tetrahydrofuranylglycine 174 led to an over 4-fold increase in enzyme inhibition for HIV-1 (IC50 = 0.23 ± 0.1 nM) and HIV-2 (IC50 = 0.054 ± 0.027 nM), respectively.228
Figure 46.
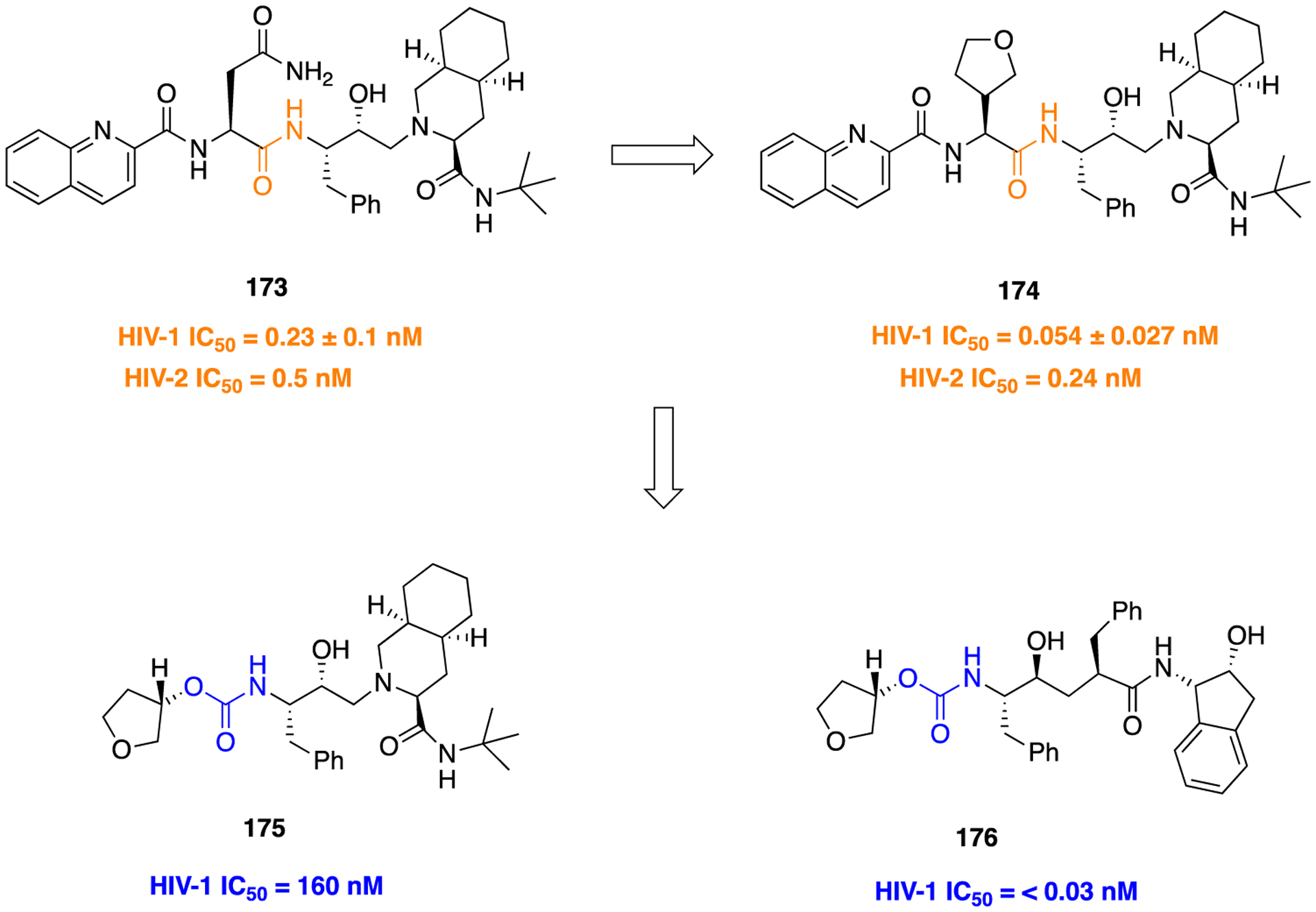
Potent HIV protease inhibitors incorporating carbamate bioisosteres.
To continue their studies of designing novel, cyclic ligands for the HIV-1 protease substrate binding site, Ghosh and co-workers introduced urethanes instead of N-terminus acyclic tert-butylurethane.226 Incorporation of various 3-tetrahydrofuranyl- or 3-tetrahydropyranylurethanes as P2 ligands in hydroxyethylamine and hydroxyethylene series was developed. Compound 175 (IC50 = 160 nM) (Figure 46) bearing a tetrahydrofuranylurethane moiety exhibited more than an 18-fold potency increase relative to its BOC derivative (structure not shown) (IC50 ≥ 3000 nM). Replacing a BOC urethane with 3(S)-tetrahydrofuranylurethane (176) resulted in over a 10-fold increase in inhibitory potency (IC50 ≤ 0.03 nM) and prevented the spread of HIV-1 (CIC95 = 3 nM) (structure not shown). The synthetic route leading to compound 176 is delineated in Scheme 23, where 3-(S)-tetrahydrofuranyl succinimidyl carbonate was reacted with amine 177 in the presence of triethylamine to afford 176. It was concluded from this study that 3-tetrahydrofurans and pyrans are high-affinity ligands for the S2 binding domain of HIV-1 protease resulting in potent inhibitors.
Scheme 23.
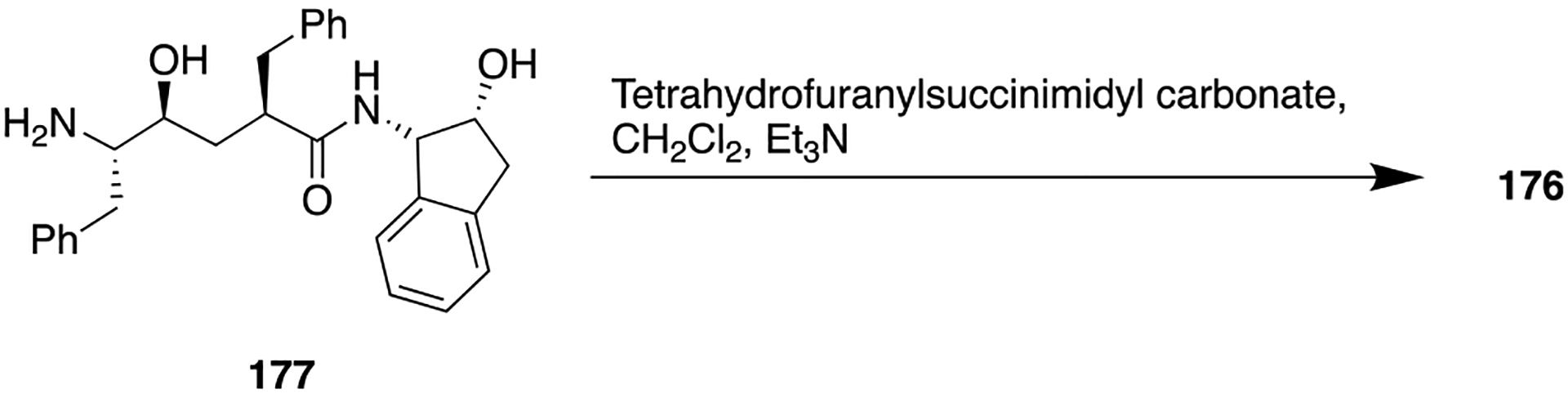
Synthetic Route To Access 3-(S)-Tetrahydrofuranylurethane-Containing Analog 176, a Potent HIV-1 Protease Inhibitor
Inhibiting β-secretase may provide a promising target for AD.229 On the basis of a β-secretase inhibitor 178 (Aβ40 (cell) IC50 = 0.082 nM) (Figure 47),230 Peters and colleagues designed a series of carbamate derivatives of dibenzazepinone as potent and metabolically stable γ-secretase inhibitors.231 First, they evaluated the potential of 178 as a lead structure by preparing close analogs, one of which was the carbamate analog 179 (Figure 47). However, 179 displayed poor activity (IC50 = 1500 nM). Despite this, it was found that a carbamate in place of an amide was tolerated in further synthesized compounds. Moreover, although 179 was a weak γ-secretase inhibitor, it gave rise to the possibility of deriving a stereogenic center from the chiral pool such as lactic acid. Thus, it could possibly prevent the separation of diastereomers in the process of inhibitor synthesis. Changing a malanomide analog to a carbamate 180 (IC50 = 1.7 nM) (Figure 47) gave an improvement in solubility and microsomal stability and led to reduced clearance in vivo. The synthetic procedure to obtain compound 180 is shown in Scheme 24. Arguably, a carbamate motif is biodegradable; however, in this case it was stable with plasma and buffer at various pH values (1.7, 4.7, and 7.8). At pH 12.8, slow degradation occurred (t1/2 = 57 h). In an in vivo AD model, both the malanomides and 180 dose-dependently increased the ratio of C-terminal fragment (AβPP) after oral administration with a minimal effective dose of three doses (3 mg/kg) over 2 days. Both the malanomide and carbamate analogs were potent γ-secretase inhibitors with single digit nanomolar inhibitory activity in vivo and were also efficacious in a mouse model of AD after oral administration.
Figure 47.
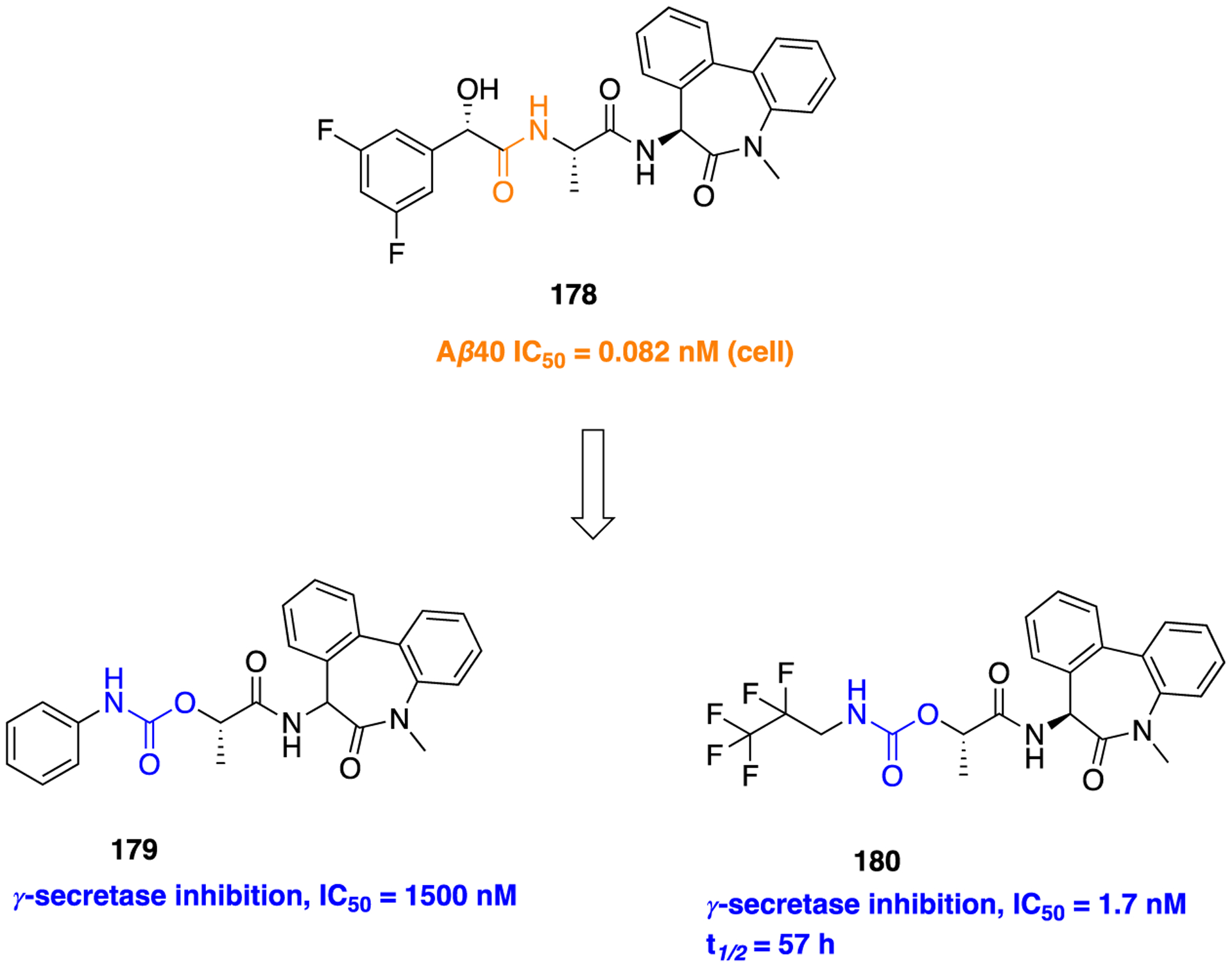
Structures of parent amide 178, a γ-secretase inhibitor, and potent carbamate-based derivatives 179 and 180 for the treatment of AD.
Scheme 24.

Synthetic Route To Access the Dibenzazepinone-Based γ-Secretase Inhibitor 180
Substance P (SP) is an 11 amino acid molecule that preferentially binds to the neurokinin-1 (NK-1) receptor, where it acts as a neurotransmitter and neuromodulator in the central and peripheral nervous system. Substance P is broken down into several bioactive fragments; substance P 1–7 (SP1–7), a heptapeptide, is a major N-terminal metabolite that has been comprehensively evaluated. It has been shown to have high affinity to specific binding sites in the spinal cord and brain. Numerous reports propose that it may have its own putative receptor. Fransson and colleagues were able to demonstrate that it could induce antihyperalgesia in diabetic mice.232 It is thereby suggested that SP1–7 peptidomimetics could have the potential to serve as therapeutic agents for neuropathic pain. Initially, alanine and subsequent N- and C-terminal modifications of SP1–7 and endomorphin-2, and subsequent truncation gave rise to the lead compound 182 (Figure 48). This compound had a more pronounced effect in diabetic mice than SP1–7 when administered intrathecally. Structure-activity relationship studies of dipeptide analogs and rigid analogs of H-Phe-Phe-NH2 revealed that two benzyl moieties in the (S, S) configuration and a primary amine at the N-terminal with a crucial primary amide function in the C-terminal were the structural motifs optimal in order to achieve high binding affinity. However, this pharmacophore led to high efflux of the compound. Therefore, it was investigated what pharmacophore was essential through the exploration of compounds with fewer hydrogen bond donors. The N-terminal of the basic phenylalanine in 182 (Ki = 1.5 nM) was exchanged for a more lipophilic benzyloxycarbonyl moiety giving rise to 183 (Figure 48). The synthesis of compound 183 was accomplished through the reaction of C-terminal activated benzyloxycarbonyl protected phenylalanine (184) with ammonia (Scheme 25). Compound 183 was then evaluated in an SP1–7 binding assay, which gave rise to a good binding affinity (Ki = 5.2 nM) while still maintaining interactions with the target. The in vitro profile of 182 displayed poor “drug-like” properties as a result of low membrane permeability, high in vitro clearance (Clint = 97.3 μL mim−1 mg−1) (Figure 48) and no measurable plasma protein binding. This could be due to rapid degradation in plasma; therefore, this compound was not further investigated in vivo. Compound 183, in which the N-terminal amine was replaced with a carbamate motif, resulted in improved membrane permeability (Papp = 31 × 10−6 cm/s), low efflux, and moderate in vitro clearance (Clint = 22.6 μL mim−1 mg−1) (Figure 48). Despite these promising results, compound 183 experienced hydrolytic cleavage in rat blood/plasma, inducing extrahepatic clearance, thus preventing good exposure after a single dose in rats. When tested in human plasma, this was not an issue. Compound 183 was further evaluated with different C-terminal groups (hydroxamic acid, acyl cyanamide, acyl sulfonamide, acyl hydrazine, and 1,3,4-oxadiazole) using a bioisosteric approach. Overall, these analogs showed moderate to good binding affinities for the SP1–7 binding site (IC50 = 4–60 nM). These C-terminally modified compounds were also evaluated for their uptake, metabolic stability, and permeability to determine the effect of bioisosterism on pharmacokinetic properties. When compared to 183, the rat plasma stabilities were improved for all analogs except two. Additionally, three out of the five showed a low risk of oxidative metabolism and comparable Clint to 183. Overall, isosteres provided compounds with increased permeability, low efflux, and good metabolic stability.232
Figure 48.
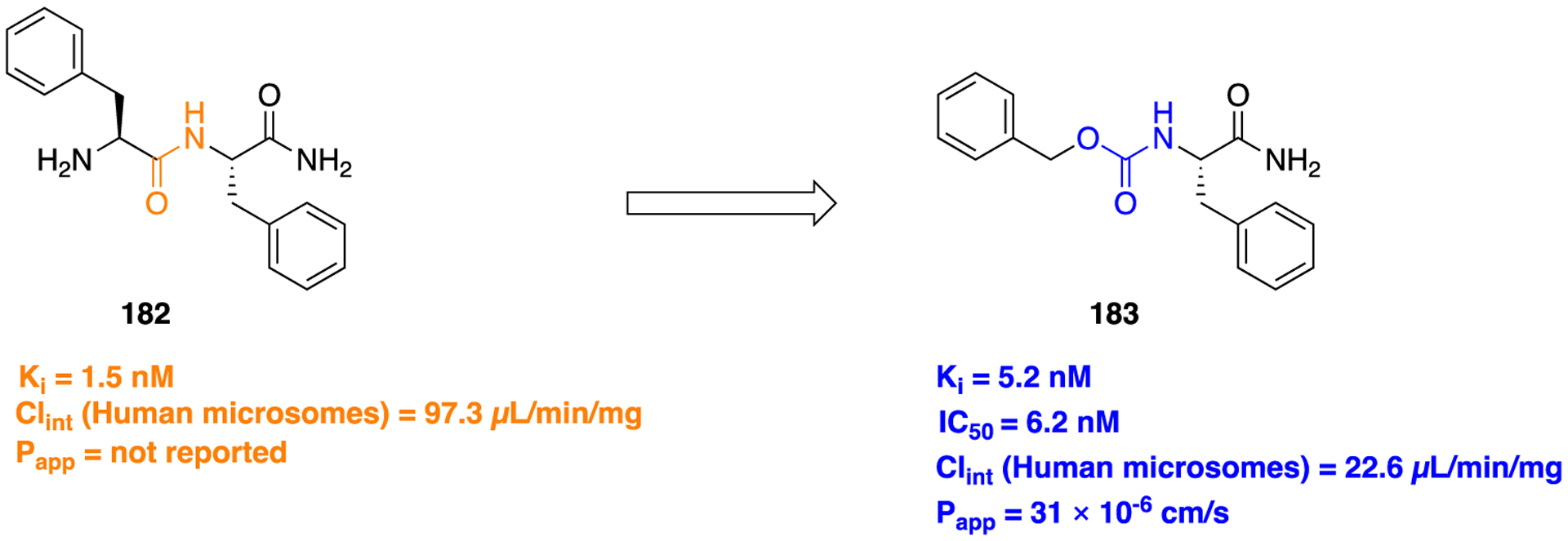
Replacement of the amide bond in 182 with a carbamate moiety to afford compound 183, novel SP1–7 analogs as potential therapeutic agents for neuropathic pain.
Scheme 25.

Synthetic Route To a Carbamate-Based Dipeptide SP1–7 Analog (183)
AMIDINE BIOISOSTERES
Very late activating antigen-4 (VLA-4), a key cell surface integrin, is expressed in circulating leukocytes. It plays a critical role in inflammation via the promotion of leukocyte attachment and extravasation from the vasculature into peripheral tissues. Thus, VLA-4 antagonists could be useful for the treatment of multiple sclerosis, rheumatoid arthritis, and asthma.233 Kamenecka and co-workers previously reported potent and specific amide VLA-4 antagonists 185 and 186 (Figure 49).234 These N-phenylsulfonylproline biphenylalanine derivatives suffered from poor pharmacokinetic properties such as low oral bioavailability (F < 10%) and high plasma clearance in rat (Clp > 90 mL min−1 kg−1) (Figure 49). This raised speculation of whether these deficiencies were a result of the amide bond, and so this group was either removed or modified. Bioisosteric replacement of the amide bond with an ester caused a 200-fold decrease in potency while N-methylation gave rise to a 4000-fold drop. Therefore, it was concluded that the NH was essential for potency. Removal of the amide carbonyl motif resulted in a 2000-fold reduction in potency. This could be the result of the secondary amine being too basic to engage in essential hydrogen bonding or as a result of a conformational change in the molecule leading to a reduction in potency. It was still unclear if the oxygen atom was needed, so Kamenecka and co-workers sought amidines as amide bond bioisosteres to help attenuate the basicity on the nitrogen group, thus giving rise to improved potency and pharmacokinetics. Results showed that changing the oxygen atom in 185 to an NH (187) resulted in almost equipotent VLA-4 inhibitory activity (Figure 49), thus confirming that the amide bond oxygen is not necessary for potency, but the amide oxygen or amide nitrogen can still act as a hydrogen bond acceptor. Next, the NH bond of 187 was modified by amalgamation with an EWG on the nitrogen linked with one methylene group (188). This modification did not produce significant improvement in antagonizing VLA-4; however, both compounds (187 and 188) displayed slightly better pharmacokinetic profiles than 185 and 186 (Figure 49). The synthetic procedure of amidine bioisosteres 187 and 188 is outlined in Scheme 26.233
Figure 49.
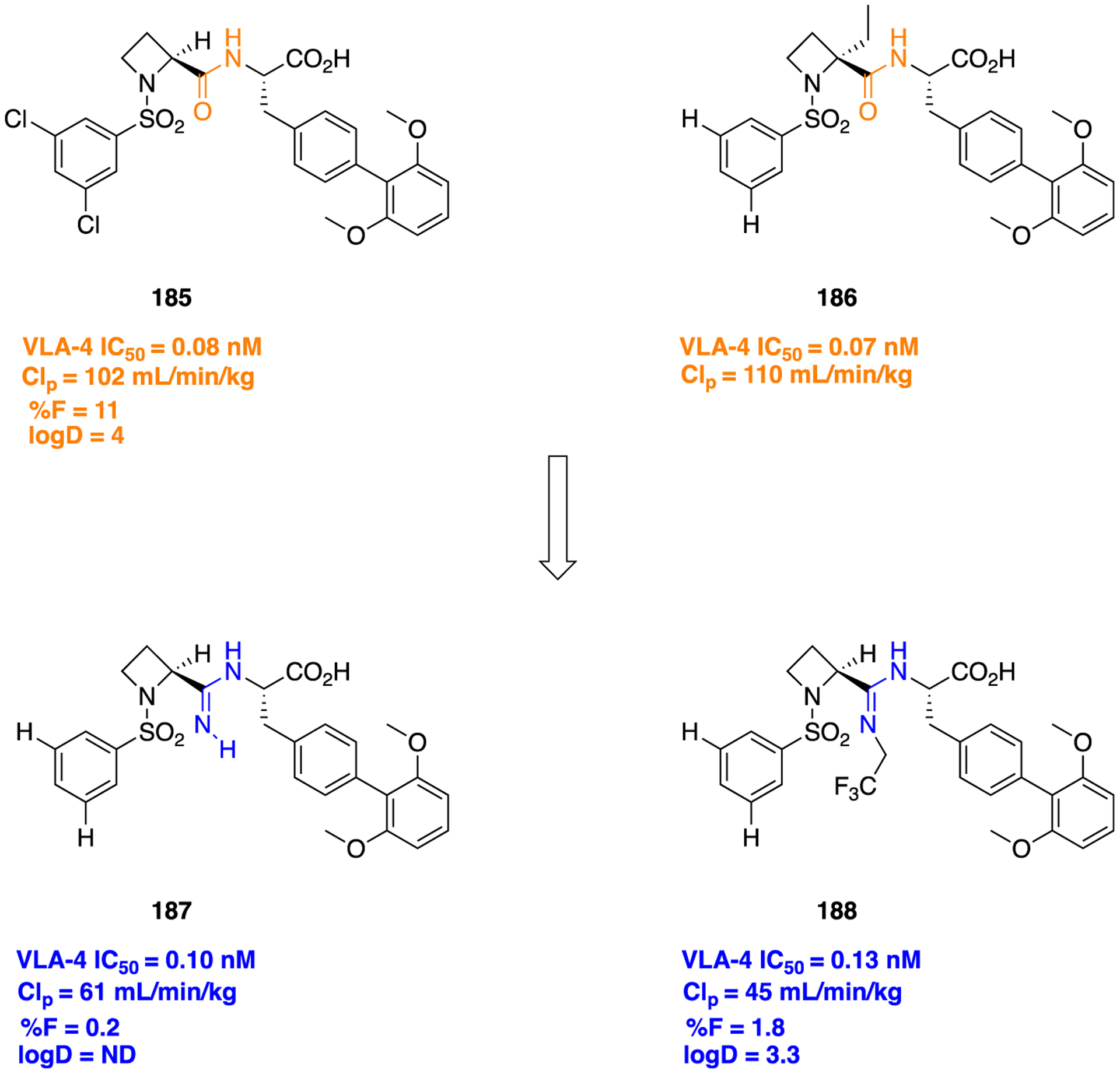
Amidine as a bioisostere of the amide functionality in VLA-4 antagonists.
Scheme 26.

Synthetic Route To Access Amidine Derivatives as VLA-4 Antagonists
THIOAMIDE BIOISOSTERES
The thioamide and amide functional groups share the same number of atoms and valence electrons, which are arranged similarly. The thiocarbonyl bond is longer than a carbonyl bond. Additionally, the C–N bond in the thioamide is shorter in comparison to the amide due to the larger contribution of resonance. Two structural deviations of the thioamide from the amide do exist: there is a more rigid out-of-plane bending vibration mode due to the nitrogen in the thioamide, and there is an increase in rotational barrier across the C–N bond in thioamides over amides by 2–3 kcal mol−1. Thioamides, like amides and esters, can exist as cis and trans isomers. The proton affinity of sulfur in a thioamide (203 kcal mol−1) is greater than that of oxygen (199 kcal mol−1) in an amide. Despite this, thioamides are weak hydrogen bond acceptors compared to amides since this is negated by the unfavorable entropies that go with hydrogen bond formation in thioamides.235
Bacterial resistance is an emerging issue when combating bacterial infections and has led to the continuous search for novel antibiotics. Previously, Thorarensen and colleagues reported the optimization of anthranilic acid hit compound 190, which led to the discovery of lead compound 191 displaying potent broad-spectrum antibacterial activity (Figure 50).236 This group sought to explore the role of the linker connecting the A- and B-rings in 191. They decided to take the amide bioisostere approach based on the following two hypotheses: the amide linker was involved in an essential interaction with the target and disruption of this would be detrimental to activity, and the purpose of the amide linker was to act as a conformational control element thereby linking essential pharmacophores. They proceeded to select functional groups that were successful amide bioisosteres through their ability to maintain essential recognition elements of an amide via incorporation of hydrogen bond acceptor or hydrogen bond donor properties. Some of their previous work revealed that electron-withdrawing groups (−Cl, −Br, −NO2, and −CN) at the 5-position on the A ring were interchangeable and resulted in similar antibacterial activity. Additionally, 3- versus 4-substituted B-rings resulted in analogs with similar activity; however, 3-substitution had a slight improvement in potency. The selection of a B-ring substituent was influenced by the fact that the corresponding amide illustrated reasonable antibacterial activity. Next, substitution patterns were picked on the sole basis of the ease of synthesis. The thioamide moiety as in 192 was readily available via reflux in toluene with compound 193 and Lawesson’s reagent. The hydrolysis of the ester fragment with treatment of lithium hydroxide gave rise to compound 192 (Scheme 27). The ability of the new linker to serve as an amide bioisosteric replacement was monitored based on its activity against Staphylococcus aureus. Results showed that the thioamide 192 (Scheme 27) was equipotent with its corresponding amide. The minimal inhibitory concentrations (MIC) were tested across five different bacterial strains, but improvements were seen in only three strains (Staphylococcus aureus UC 9218 (SAUR), Staphylococcus epidermidis UC 12084 (SEPId), and Haemophilus influenzae 30063 (HINF)) for the thioamide 192 in comparison to the amide 191 (Figure 50). However, the improvement in activity for the thioamide was not clear. Conversion from an amide to a thioamide is only a small change in linker construction. Nevertheless, results did show that removing this linker resulted in analogs devoid of activity. Replacing the linker with a five-membered ring heterocycle results in the abolishment of activity. Using an alkane caused faint antibacterial activity, but an alkene, more specifically an E-alkene, provided equipotent activity to its corresponding amide, suggesting a critical role in retaining bioactive conformation. It can be concluded that an amide is not necessary for activity but plays a role as a recognition element and adequately positions the aryl rings. Therefore, an amide can be replaced without significantly affecting MIC antibacterial activity of novel linkers.
Figure 50.
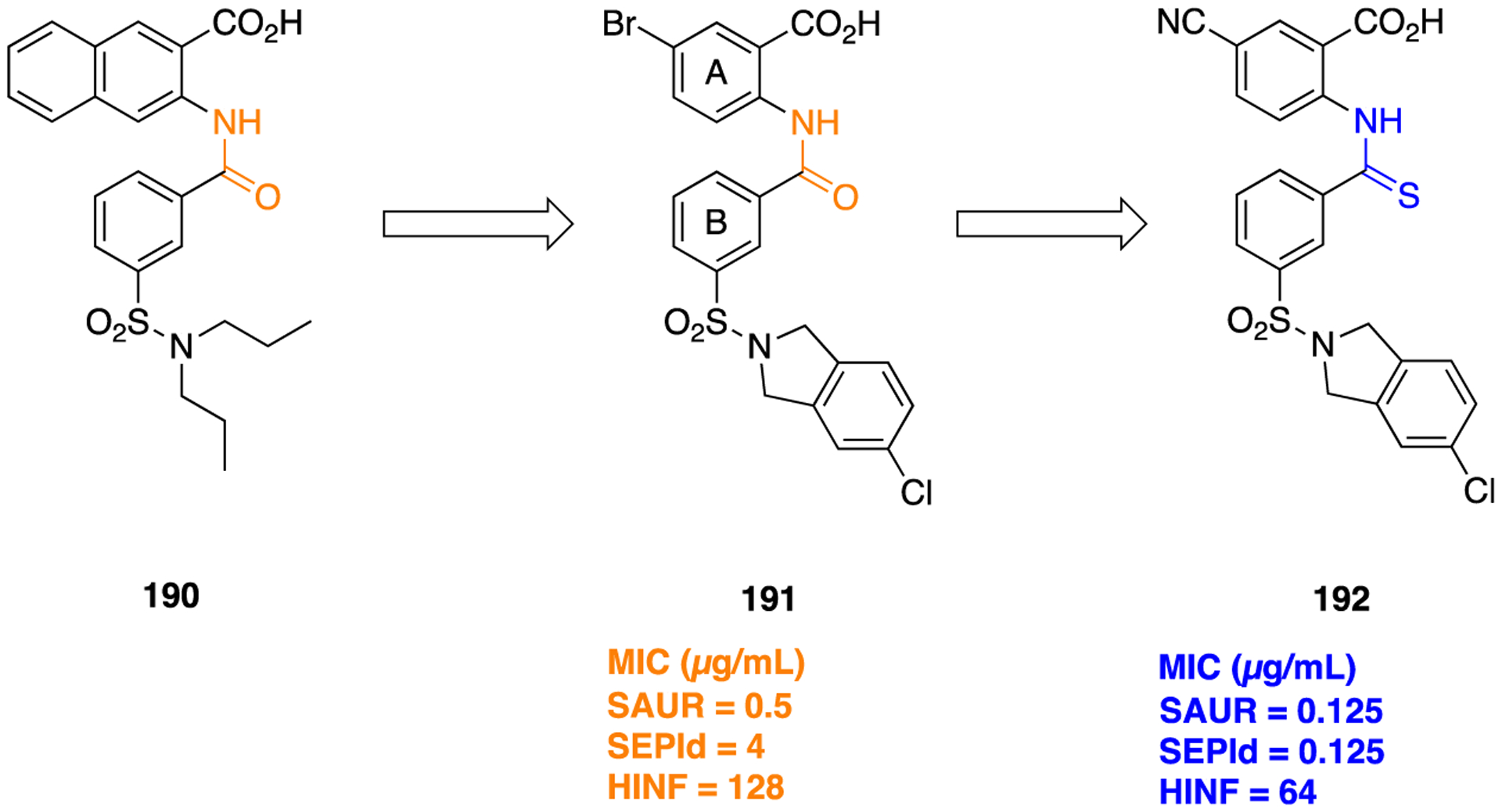
Anthranilic acid lead 190, advanced lead 191, and alternative thioamide bioisostere 192 as antibacterial agents.
Scheme 27.
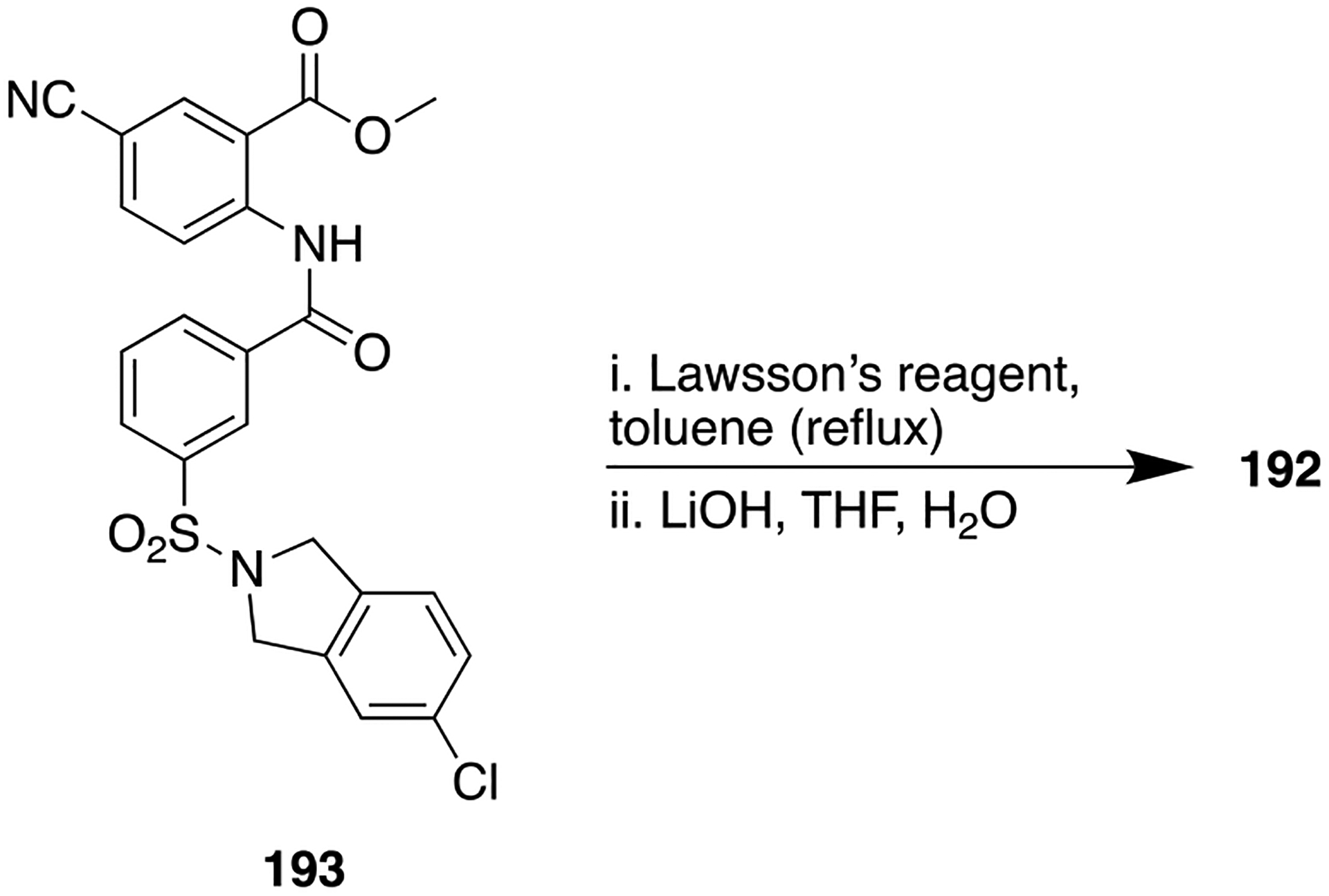
Synthetic Route To Access Thioamide 192
TRIFLUOROETHYLAMINE BIOISOSTERES
The introduction of a trifluoroethylamine group [CH(CF3)-NH] is an emerging mimic of the natural parent amide bond in a number of peptidomimetic compounds. In 2000, Zanda and co-workers published the first example of peptide mimics that incorporated a trifluoroethylamine group as a replacement for the amide moiety.237 The trifluoroethylamine motif has the ability to reduce the basicity of the NH while preserving hydrogen bond donating properties, retains the backbone geometry of the amide bond (bond angle close to 120°), and has structural similarity with a tetrahedral proteolytic transition state (TS), and the C–CF3 bond is isopolar with the carbonyl bond.238 An additional benefit is the sp3 hybridization of the trifluoroethylamine functional group that can influence the conformation of substituent groups into a target receptors’ active site, enhancing hydrogen bond strength. Zanda and co-workers replaced both a glycine amide bond and a malonamide to a partially modified retro-Ψ[NHCH(CF3)] peptide (Figure 51).239 They also synthesized a Ψ[NHCH(CF3) retrothiorphan analog 197 (Figure 51) over the course of several steps starting with the reaction of N-trifluorocrotonoyloxazolidin-2-one (198) and l-phenyl alaninol (Scheme 28). Compound 197 was determined to be a potent and selective inhibitor of metalloproteinase neutral endopeptidase (NEP) which does not inhibit the related zinc proteinase, angiotensin-converting enzyme (ACE), and plays a key role in controlling blood pressure.238 Compound 197 was evaluated for its inhibitory capacity toward NEP 24.11 achieving IC50 values greater than the thiophan parent scaffold, with Ki values over 4 M. Results proved that there was loss of NEP inhibition capacity when comparing compound 197 and its stereoisomers with (R)- and (S)-retrothiorphan (Ki = 2.3 nM and 210 nM, respectively). Despite these results, it was found that the trifluoroethylamine unit possesses high metabolic stability and restricts the molecule into accessible conformational states, thereby resulting in well-defined conformational motifs. As a result, it is only an effective replacement if the carbonyl group of the parent peptide is not involved in key hydrogen bonding with the receptor.238
Figure 51.
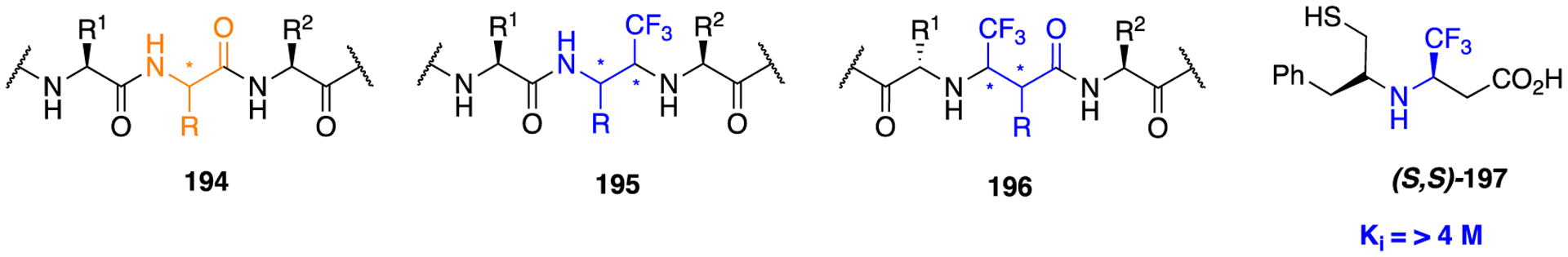
Molecular structures of natural 194 and bioisosteric Ψ[CH(CF3)NH]Gly 195 and 196 peptides and (S,S) [NHCH(CF3)]-retrothiorphan diastereomer 197.
Scheme 28.
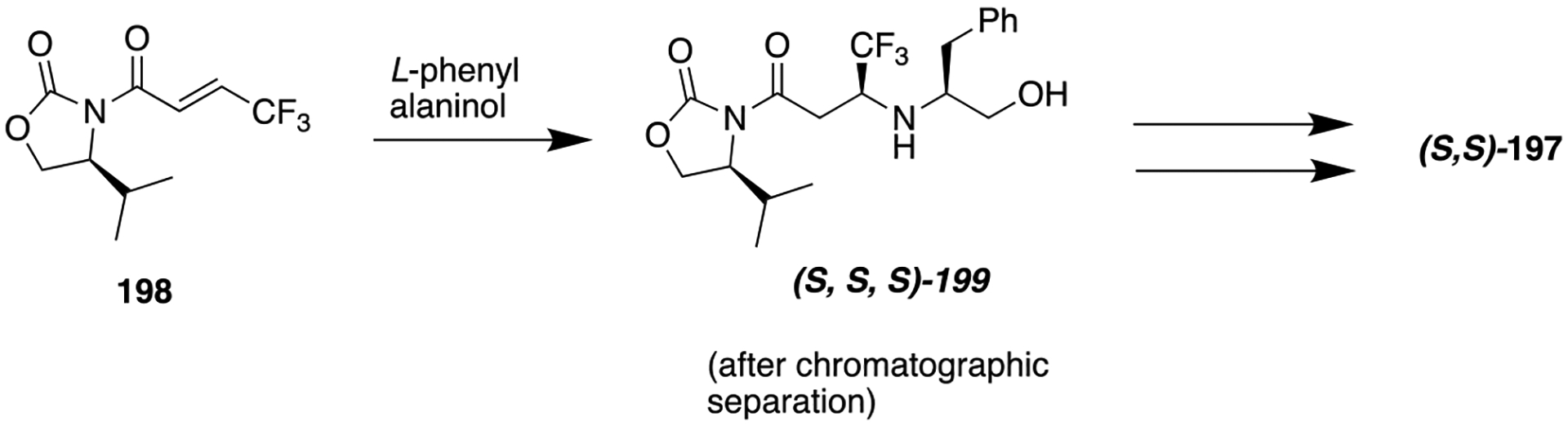
Synthesis Route To Access Ψ[NHCH(CF3)]-retro-thiorphan 197
Successful replacement of an amide with a trifluoroethylamine isostere has progressed beyond peptidomimetics; it has also been used in the lead optimization of odanacatib, 202 (Figure 52), a highly potent and selective drug candidate for the inhibition of cathepsin K (Cat K).240 Cathepsin K, a lysosomal cysteine protease, is abundantly expressed in osteoclasts where it is responsible for degradation of the bone protein matrix. Blocking secreted cathepsin K can help prevent bone loss without affecting the ability of osteoblasts to build bone and thereby enhancing bone strength and quality.241 Thus, Cat K inhibitors can be used as a therapeutic treatment for osteoporosis. Initially, X-ray crystallography showed that in an irreversible dipeptide inhibitor of Cat K, the P1–P2 amide hydrogen bonds with the enzyme backbone involving the NH and carbonyl groups. However, the P2–P3 amide bond only forms one hydrogen bond to Gly66 with the NH while the oxygen points toward the solvent. Thus, the P2–P3 amide bond can be replaced with an isostere without having detrimental effects on potency. In order to identify a potent and selective inhibitor of Cat K, the initial lead 200 was synthesized (Figure 52). It proved highly selective over cathepsins B, L, and S in enzyme assays and displayed good PK properties.242 However, further studies revealed that its lysosomotropic properties caused the loss of selectivity in cell-based assays, more specifically in a Cat S-dependent B cell line assay where it blocked antigen presentations. Overall, this initial lead compound resulted in reduced effective selectivity in whole-cell assays which could cause an increase in potential adverse effects in the clinic.243 Black and co-workers found that not only an analog of compound 200 containing a trifluoroethylamine moiety at the P2–P3 amide enhanced potency and selectivity over other cathepsins but the resulting compounds were also stable to P1–P2 amide bond cleavage similar to dipeptide inhibitors. Introduction of a trifluoroethylamine bioisostere was found to be a viable substitution at the P2–P3 amide for inhibitors of Cat K. Optimization studies led to the replacement of the P2 amide bond with a trifluoroethylamine isostere; compound 201 showed nearly comparative Cat K inhibition as 200. Additionally, 201 was nearly 26,000-fold more selective for Cat K over Cat B (Figure 52).244 In vivo studies of this compound showed it was orally bioavailable to rat, dog, and rhesus monkey; however, it had metabolic liabilities such as a short half-life (2 h) and clearance (7.5 mL min−1 kg−1) in monkey (Figure 52).243 In order to minimize leucine hydroxylation, amide hydrolysis, and lactonization, the P1 and P2 residues were modified and led to the final drug candidate 202. The general synthetic route to obtain the trifluoroethylamine bioisostere 202 is outlined in Scheme 29, i. Compound 202 is a potent, selective, and neutral cathepsin K inhibitor that maintains excellent selectivity and activity in both enzymatic and cell-based assays. It has minimal effects on intracellular collagen accumulation compared to other cathepsin K inhibitors, balicatib and relacatib, with long half-lives in preclinical species.240 The compound continued into clinical development for the treatment of postmenopausal osteoporosis and bone metastasis. It progressively increased bone mineral density over 5 years and reduced the risk of fractures in patients. However, development was discontinued before the filing of a new drug application (NDA) due to an increased risk of cardiovascular events and risk of stroke.245
Figure 52.
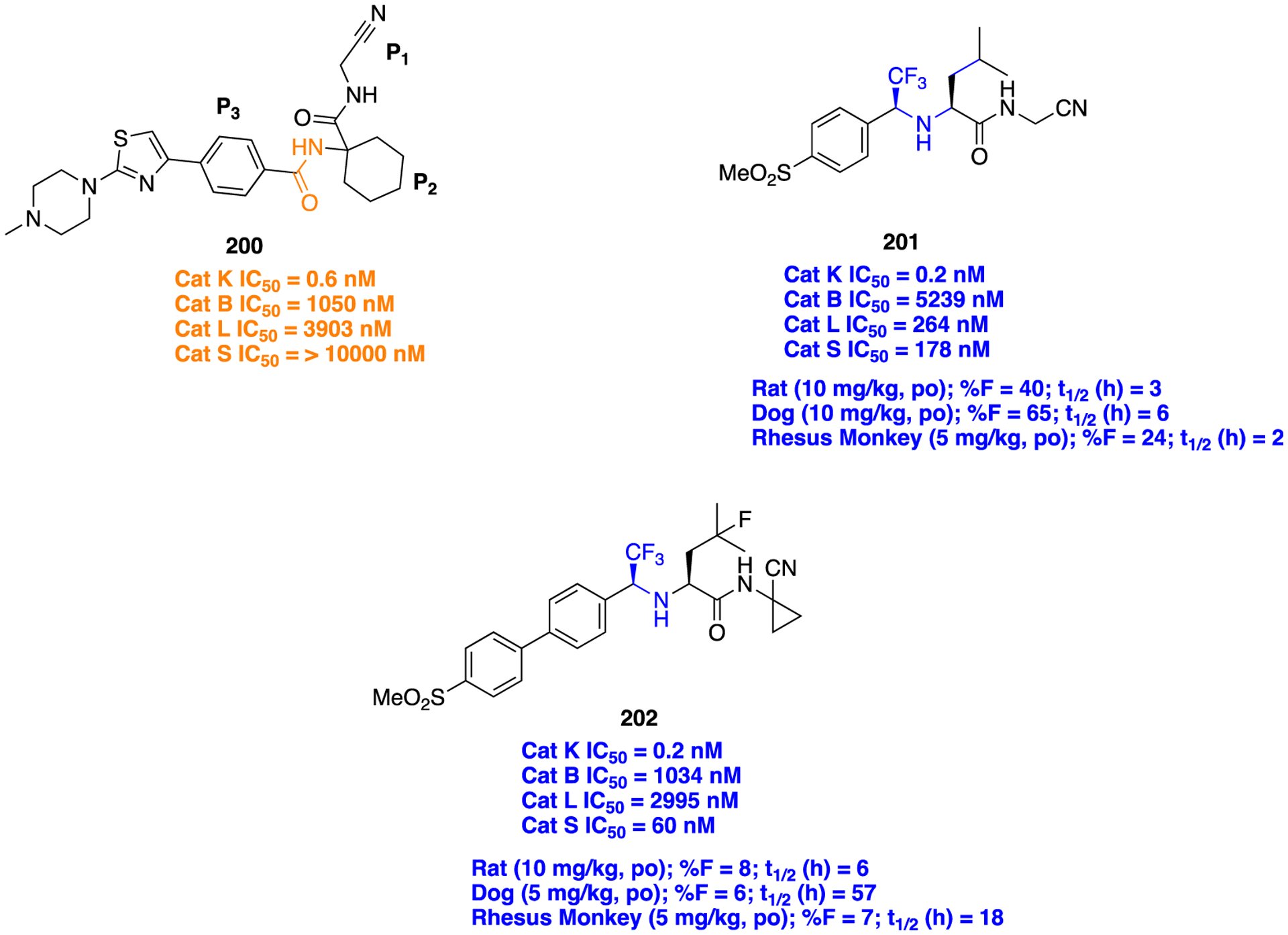
Structures of 200 a reversible dipeptide inhibitor of cathepsin K, 201 a neutral cathepsin K inhibitor, and 202 a potent cathepsin K inhibitor containing the trifluoroethylamine bioisostere.
Scheme 29.
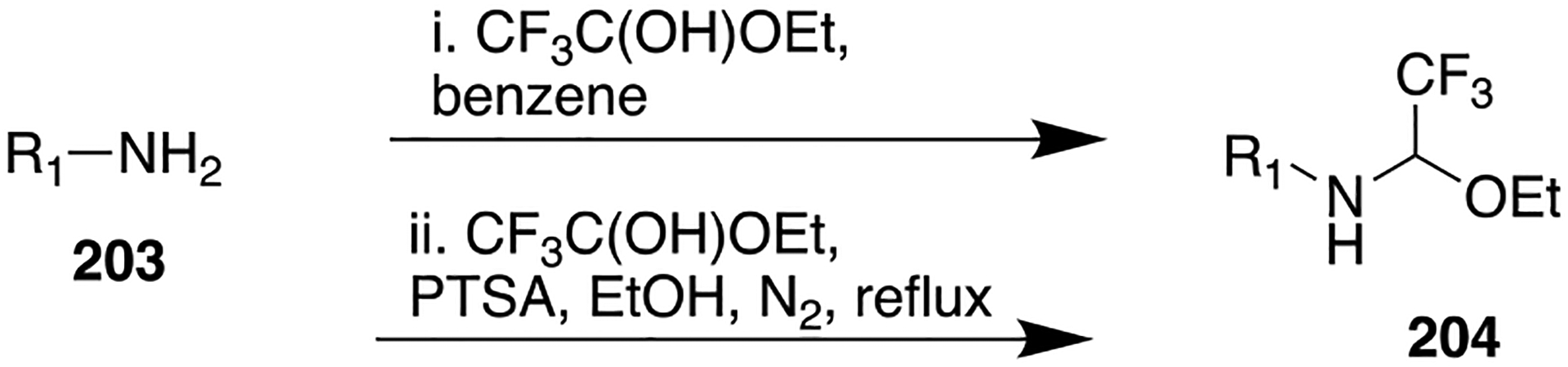
General Synthetic Route To Generate Trifluoroethylamine Bioisostere via a Trifluoroacetaldehyde Ethyl Hemiacetal
An example of where an amide replacement with the trifluoroethylamine moiety affects selectivity of a drug toward different targets can be appreciated with telaprevir, 205. It is a first-generation peptidomimetic and potent hepatitis C virus (HCV) NS3 protease inhibitor with IC50 = 70 nM (Figure 53). Moreover, 205 has the ability of inhibiting other cysteine proteases such as cathepsin B with an IC50 of approximately 210 nM and is also an antiviral agent, EC50 = 210 nM in the replicon. Replacing the P4 amide moiety with a trifluoroethylamine surrogate identified two active inhibitors of cathepsin S with IC50 = 2 nM and 0.6 nM for compounds 206 and 207, respectively (Figure 53). However, in contrast to the unfluorinated parent compound 205, compounds 206 and 207 only exhibited a weak inhibition of HCV NS3 with 34% and 29% inhibition at 25 μM in the HCV replicon, respectively, despite the structural changes being remote from the active site.16
Figure 53.
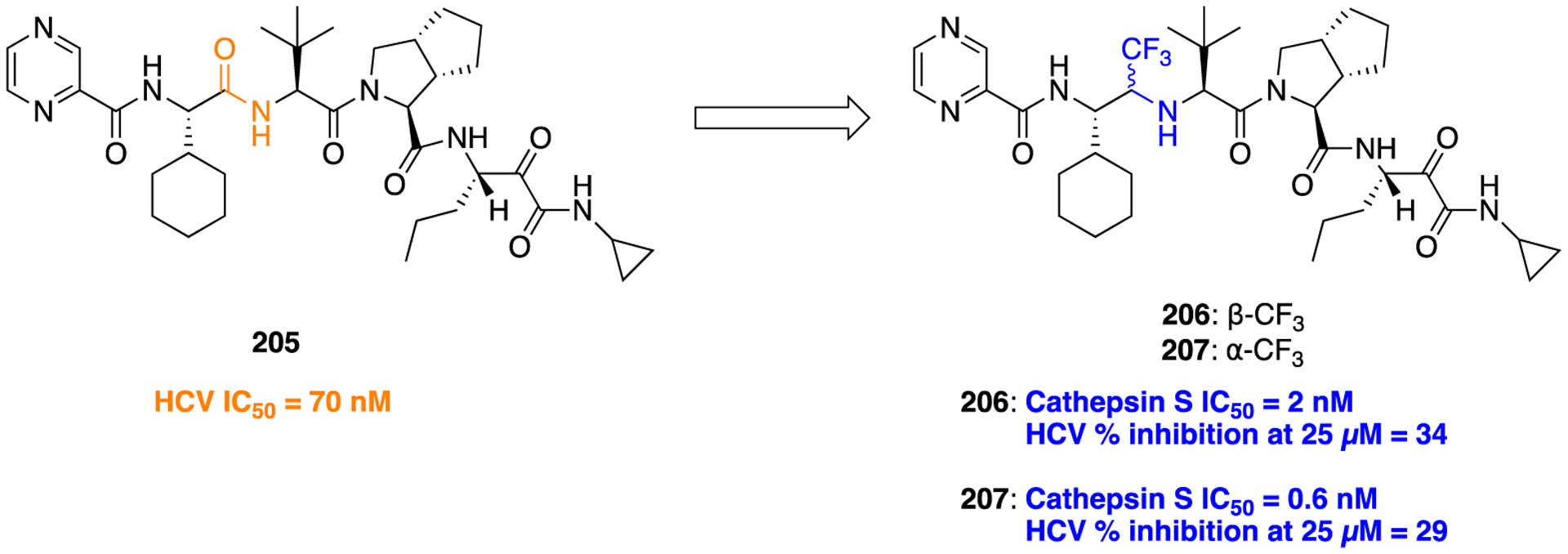
Structures of 205, a potent hepatitis C virus (HCV) NS3 protease inhibitor, and trifluoromethylated-containing cathepsin S inhibitors 206 and 207.
Bioisosteric replacement of an amide with a trifluoroethylamine moiety was examined in the design of β-site amyloid precursor protein cleaving enzyme 1 (BACE-1) inhibitors. Inhibitors containing a P1 or P2 amide motif exhibit high efflux transporter liability along with metabolic lability.246 This was attributed to the release of aniline upon amide cleavage in vivo, thereby potentiating toxicity after metabolic activation. Butler and co-workers sought to identify an amide replacement that provided excellent potency but avoided the aniline structural motif and its toxicity. The X-ray cocrystal structure of a related BACE-1 inhibitor analog, compound 208 (Figure 54), showed that the amide NH is positioned in such a way that it engages with the backbone carbonyl of the Gly230 residue in order to partake in a critical hydrogen bonding interaction. Therefore, preservation of the NH of the aniline is necessary. It was found that the planar aniline moiety has sp3 nature on the benzylic carbon resulting in an orthogonal arrangement of the C–N bond relative to the fluorophenyl P1 ring, facilitating an optimal geometry for hydrogen bonding to Gly230. Due to uncertainty of what structural motifs were necessary, a parallel synthesis was performed in order to evaluate the SAR for BACE-1 which identified an ethylamine analog. This analog showed modest improvement in BACE-1 cell-free assay potency with IC50 = 13.9 μM. However, it has a more potent increase in whole-cell assay potency of about 1000-fold. This was attributed to the basicity of the analog which increases the tendency for accumulation into the endosome, an acidic intracellular compartment where BACE-1 resides. Further optimization focused on utilization of electron-withdrawing groups and incorporation of branched amines to further enhance potency. In due course, a CF3-cyclopropane derivative, compound 209 (Figure 54), was synthesized via reductive amination with 1-(trifluoromethyl)cyclopropan-1-amine, followed by the removal of the amidine protecting group (Scheme 30). Compound 209 maintained good potency in both BACE-1 cell-free assay (IC50 = 0.0069 μM) and whole-cell assay (IC50 = 0.018 μM) and overall an optimal balance of properties. A cocrystal structure of compound 209 in BACE-1 was obtained, and results showed that the thioamidine and difluorophenyl ring are orientated similarly to previously described BACE-1 inhibitor structures. The difluorophenyl occupies the S1 pocket as predicted, and the benzylic amine substituent adopts an orthogonal orientation toward the plane of the difluorophenyl P1 group in order to involve the carbonyl of Gly230. Results showed that improvements in BACE-1 cell-free assay potency using the CF3-containing lead was due to the CF3 group pointing toward and filling the S3 pocket while the cyclopropyl moiety fills a lipophilic pocket at the interface of the S1 and S3 pockets. In the end, compound 209 possessed significant inhibition activity toward cytochrome P450 subtype 2D6 (CYP 2D6) and development was discontinued.246
Figure 54.
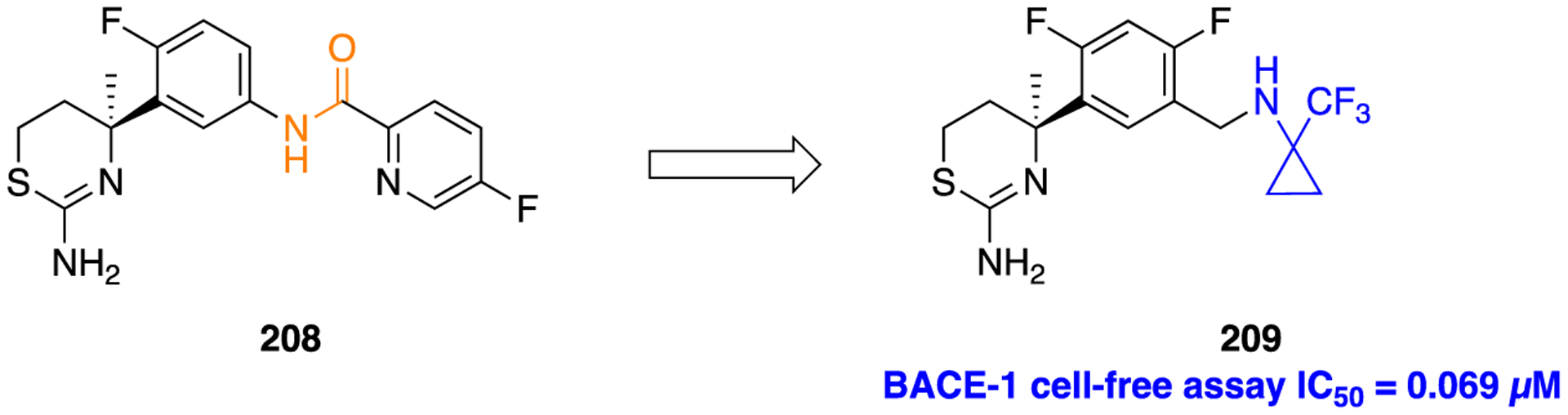
Structures of 208, an amide-based BACE-1 inhibitor, and CF3-cyclopropane bioisostere BACE-1 inhibitor 209.
Scheme 30.
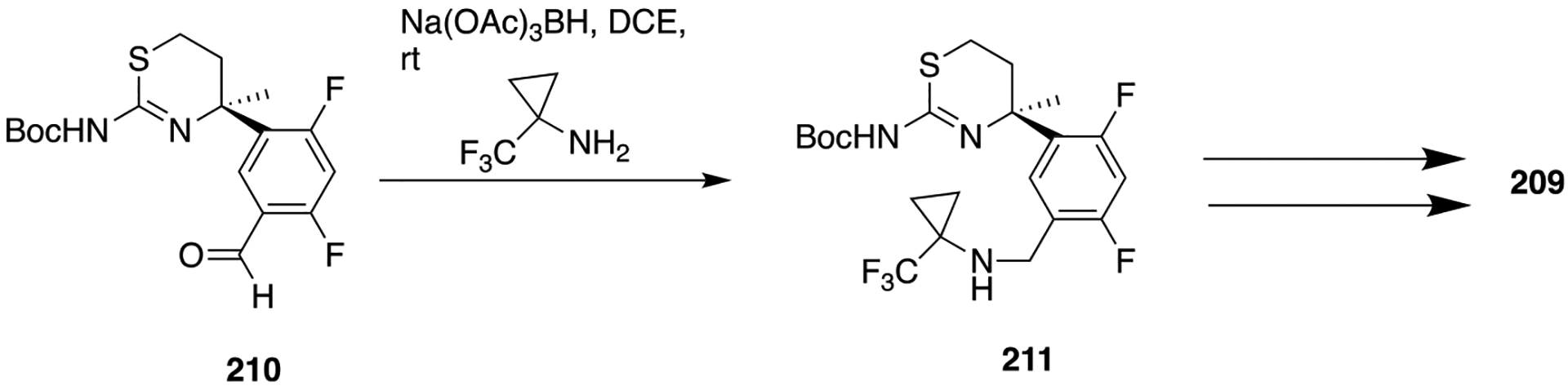
Synthetic Route To Access Compound 209, a BACE-1 Inhibitor
An amide bond replacement on a malonyl moiety by an amide surrogate trifluoroethylamine was employed in the identification of novel receptor tyrosine kinases (RTKs) inhibitors, more specifically malonamide-type dual inhibitors of the vascular endothelial growth factor receptor-2 (VEGFR2) and c-Met receptors. Gaudette and colleagues initially investigated what changes were suitable for VEGFR2 and c-Met enzyme inhibition via initial SAR studies of compound 212, the parent malonamide (Figure 55).247 First, the left-hand side of compound 212 was left unchanged and one amide moiety of the malonamide head group was replaced by a trifluoroethylamine moiety on the eastern side. Results showed that replacement of the eastern amide bond of the parent malonamide by the trifluoroethylamine moiety led to 23-fold and 11-fold decrease in activity for c-Met (IC50 = 0.70 μM) and VEGFR2 (IC50 = 0.21 μM), respectively, compared to compound 212 (IC50 = 0.03 μM and IC50 = 0.02 μM for c-Met and VEGFR2, respectively). The decrease in activity in VEGFR2 and c-Met could be due to the steric clash between the CF3 group and hydrophobic back pocket of the kinase active site and/or the loss of the carbonyl group, a strong hydrogen bond acceptor in compound 212. Replacement of the western amide bond by a trifluoroethylamine group caused a 4-fold decrease in activity against c-Met (IC50 = 0.12 μM). However, the compound showed similar VEGFR2 inhibitory activity (IC50 = 0.01 μM) as compound 212. The same observations were seen in a pyrimidine-based series, compound 213 which was found to be of highly selective and potent for VEGFR2 (IC50 = 0.04 μM). It was also profiled against a set of eight kinases where it was also highly selective for VEGFR3. Lastly, there was a significant effect on VEGF-dependent angiogenic activity where there was inhibition of VEGF-dependent proliferation of human umbilical vein endothelial cells (HUVEC). Replacing an amide moiety of a malonamide head group by a trifluoroethylamine led to the identification of novel, potent, and selective inhibitors of VEGFR2 enzyme. Compound 213 was synthesized over the course of several steps starting with the coupling of a corresponding amine with trifluoroacetaldehyde ethyl hemiacetal (Scheme 29, ii).247
Figure 55.

Structures of parent malonamide 212 and selective and potent VEGFR2 inhibitor 213.
SULFONAMIDE BIOISOSTERES
Sulfonamide functional groups are structurally related to amides in which the carbonyl moiety is replaced with an isosteric SO2 group. As in amide bioisosteres, sulfonamides are known to increase water solubility and could provide additional HBAs. Lim et al. reported a urea analog of B13, an aromatic ceramide analog with a phenyl ring in lieu of an alkenyl chain, that exhibited slightly more potent cytotoxicity compared to 163 (Figure 56). As previously mentioned in the urea bioisosteres section of this paper, results indicated that the urea appears to serve as a bioisostere of the amide.216 A series of novel d-erythro ceramide analogs with alkylsulfonamido chains were synthesized and evaluated for their cellular toxicity against human tumor cell lines HeLA, Caki-2, PC-3, and HL-60. Modest cytotoxicity (IC50 = 48 μM) was displayed by 163 against HL-60 leukemic cells. An aromatic sulfonamide ceramide analog, 214, was synthesized via the coupling of the acid (216) and amine (215) with HOBt and EDCl (Scheme 31). In vitro evaluation showed that compound 214 had comparable cytotoxicity in Caki-2 (IC50 = 103 μM) and HeLa cancer cells (IC50 = 65 μM) but the greatest potent cytotoxicity in HL-60 leukemic cells (IC50 = 28 μM) (Figure 56). Further comprehensive analysis of the sulfonamide ceramides is needed; however, their cytotoxic properties appear to be cancer cell specific due to their structural similarities to 163. The introduction of a para-halogen onto the phenyl ring of d-erthyro ceramide analogs resulted in compounds with comparable or somewhat lower activity than unsubstituted phenyl ceramide analogs. Variations in the alkyl sulfonyl chain length had a more significant influence on the activity of aromatic ceramide analogs, more specifically, an aromatic sulfonamido ceramide analog.248
Figure 56.
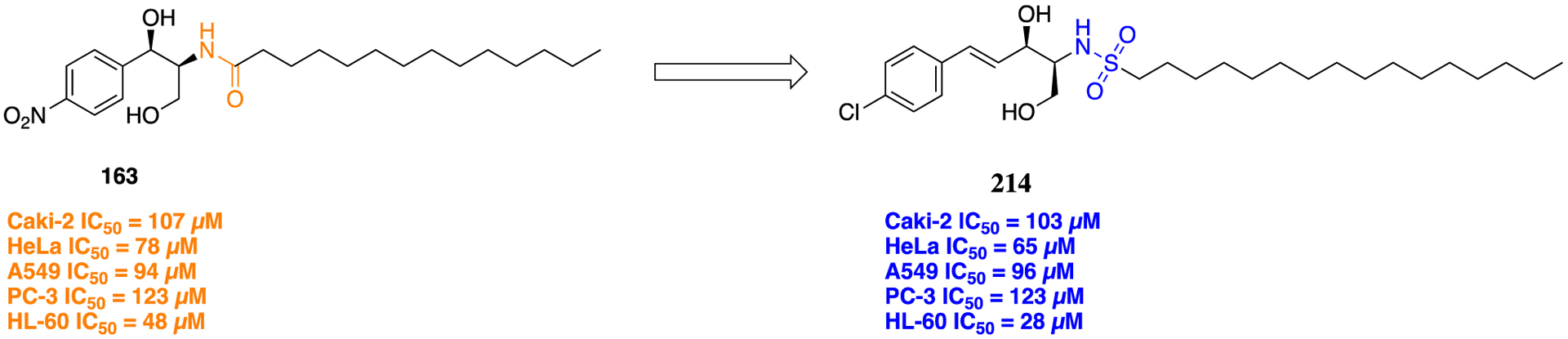
Structures of amide 163 and an aromatic sulfonamido ceramide bioisostere with more potent cytotoxic activity.
Scheme 31.
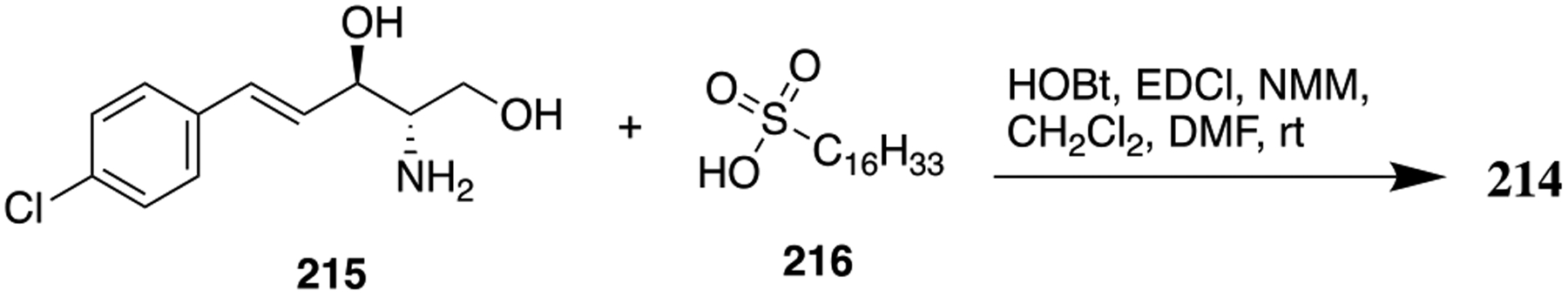
Synthetic Route To Access the Aromatic Alkyl Sulfonamido Ceramide Analog 214
The human isoprenylcysteine carboxyl methyltransferase (hIcmt) is a promising anticancer target. Proteins contain a C-terminal sequence motif called Caax box (where C is cysteine, aa is an aliphatic residue, and X is one of several amino acid residues). These motifs are post-translationally modified for cellular targeting, most common being membrane association. The protein K-Ras is one of the >120 proteins containing a Caax box; it undergoes farnesylation, endoproteolytic cleavage of the -aaX residues, and methyl esterification of the free carboxylate of the prenylated cysteine by isoprenylcysteine carboxyl methyltransferase (Icmt). When mutant K-Ras is in the constitutively active form in the GTP-bound conformation, it results in tumorigenesis. Inhibition of human Icmt has been thought of as blocking the activity of oncogenic Ras. N-Acetyl-S-farnesyl-l-cysteine (AFC) is a substrate for hIcmt, and previous work by Majmudar and co-workers has shown that amide modified farnesyl cysteine analogs (AMFCs) are low-micromolar inhibitors of hIcmt. No structural information on hIcmt exists, thereby possessing a challenge in designing Icmt inhibitors. It was hypothesized by Majmudar and co-workers that a bioisosteric replacement of an amide bond in AMFC would give rise to molecules with retained interaction with hIcmt and provide more insight on inhibition requirements. Compound 217 (Figure 57) represents one of the most potent AMFCs (IC50 = 4.3 μM) synthesized by Donelson et al.249 The goal was to determine the structural requirements for hIcmt inhibitors by prenylated cysteine derivatives with a sulfonamide bioisostere. Sulfonyl chlorides with varying electronic character, steric bulk, and alkyl aromatic scaffolds were incorporated. These new analogs were evaluated as substrates and inhibitors of human Icmt using a vapor diffusion assay (VDA). Results showed that no sulfonamide modified farnesyl cysteine analogs (SMFCs) are effective substrates but rather serve as moderate inhibitors of hIcmt. Compound 218 was obtained via reaction of a corresponding primary amine and sulfonyl chloride in the presence of sodium carbonate using dioxane as a solvent (Scheme 32, i). Compound 218 displayed 64% hIcmt inhibition at 10 μM using 25 μM AFC as the substrate with an IC50 = 8.8 ± 0.5 μM (Figure 57). It was hypothesized that the reason why SMFCs are not substrates for hIcmt is due to the reason that the amide bond or the carbonyl carbon is necessary for substrate activity. Additionally, SMFCs do inhibit Icmt but have varying strength of inhibition. Poor hIcmt inhibitors were analogs containing highly bulky or rigid motifs. Compounds with electron-withdrawing groups were more potent than electron-donating groups; however, some exceptions do exist. A planar bulk next to the sulfonamide bond enhances inhibitory activity. The effect of the prenyl group was also evaluated, but none of these compounds exhibited substrate activity at 25 μM and were poor inhibitors of hICmt compared to their farnesyl analogs. As a result, the farnesyl group is highly important for hIcmt inhibition by SMFCs. Overall, the determining factor for hICmt inhibition by SMFCs is related to the presence of the farnesyl chain, resulting in a specific binding interaction rather than just acting as hydrophobic bulk. Stereochemistry at the α carbon did not play a significant role in hIcmt inhibition by SMFCs. When deviating from the carboxylate group, there is a reduction in hICmt inhibitory activity; however, this needs to be evaluated further with more analogs. Overall, the sulfonamide bond is a viable bioisosteric replacement for the amide bond in AFC derived hICmt inhibitors.250
Figure 57.
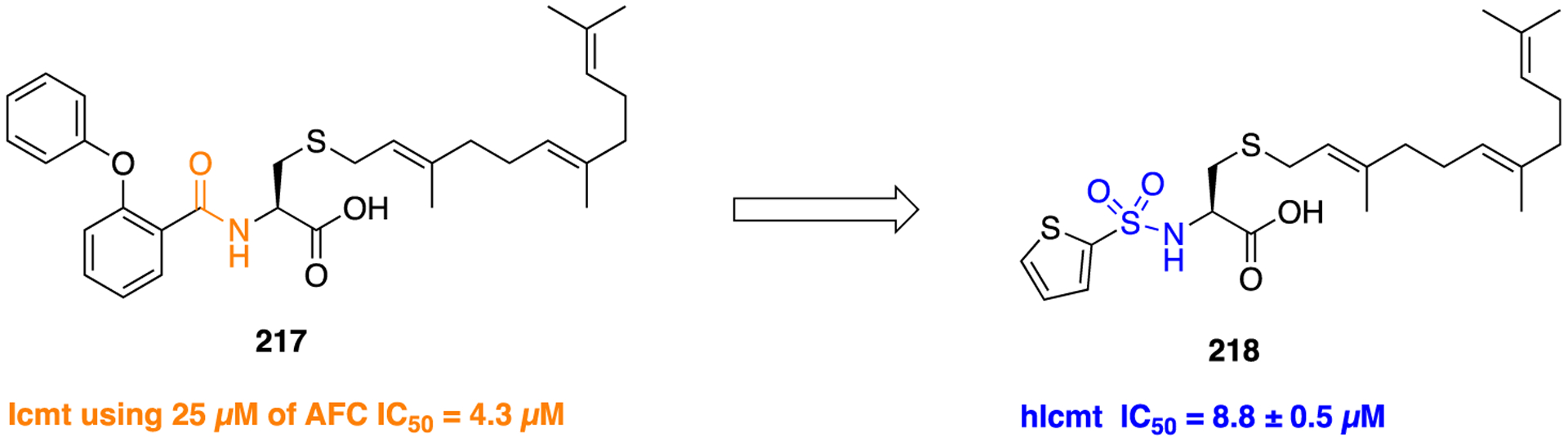
Phenoxyphenyl farnesylcysteine 217 and sulfonamide-modified farnesylcysteine (SMFC) analog 218, as hIcmt inhibitors.
Scheme 32.
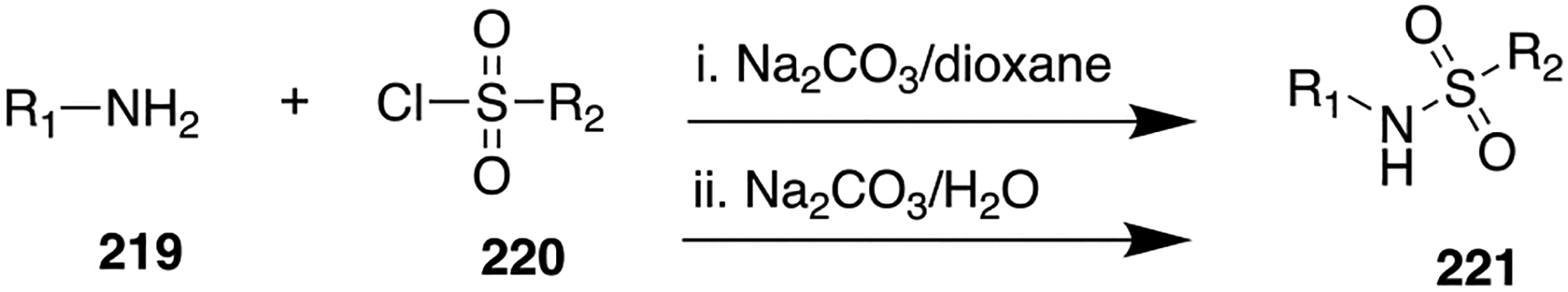
General Synthetic Procedure To Obtain Sulfonamide Bioisostere
Natural products have a successful history of being a source of new therapeutics as a result of their high chemical diversity and biochemical specificity.251 Capsaicin 24, the main pungent and irritating component of chili peppers, was reported to selectively inhibit the growth of various tumor cell lines. Early work gave rise to controversy regarding its mutagenic and carcinogenic abilities; however, recent studies have proven that it induces apoptosis in a large number of tumor cell types and that capsaicinoids possess in vitro and in vivo antitumor activity. Thus, 24 has the potential of serving as a lead against malignant tumors. However, some limitations such as its poor stability, attributed to the liable amide motif, and strong pungency limit its use and application in drug discovery.252 The pungency of 24 is related to its ability to directly promote stimuli on transient receptor potential vanilloid subtype 1, thus causing Ca2+ influx and subsequently nociceptive hyper-polarization. The molecular mechanisms of capsaicin-induced cell death are still uncertain. Despite this, the capsaicinoid scaffold gave rise to molecules exhibiting improved physiochemical and pharmacological profiles. Ferreira and colleagues have investigated the optimization of capsaicin-like analogs as potential anticancer agents. The development of these compounds began with structural changes to the 24 structure (Figure 58), which was divided into three parts: the methylcatechol group (region A), the amide bond (region B), and an alkyl side chain (region C). In region A, cyclization was investigated which converted the methylcatechol group to the 1,3-benzodioxole bicyclic motif. This modification could give rise to important advantages since the benzodioxole system is a privileged structure for substances containing antitumor activity.253 In the first analog series, the amide bond linker (region B) was maintained or underwent a bioisosteric replacement by an ester, and the alkyl side chain (region C) was replaced by several alkyl/aryl groups to preserve the hydrophobic nature of this region. In the first series, amide (222) or ester analogs (223) (Figure 58) revealed that some of these analogs had equipotent cytotoxicity to 24 against Mus musculus skin melanoma cells (B16F10) and M. D. Anderson metastasis breast adenocarcinoma (MDA-MB-231). Despite this, these analogs suffered from poor chemical stability, as did 24. The sulfonamide group, known to be very stable against chemical hydrolysis and other physiochemical degradation processes, was used as a bioisosteric replacement of the amide and ester linkers to afford 224–226 (Figure 58).
Figure 58.
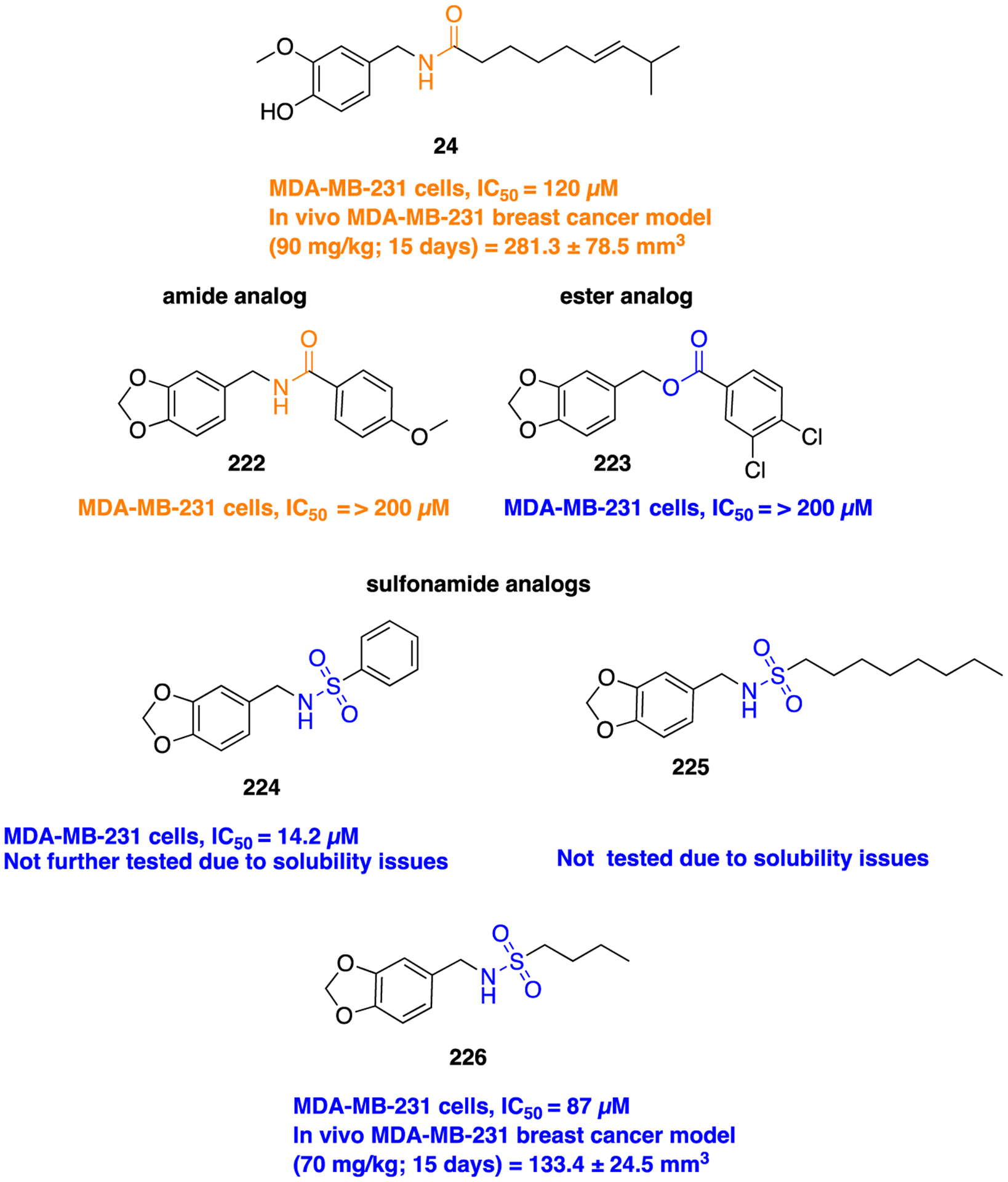
Capsaicin (24) and step by step development of novel capsaicin-like bioisosteric analogs 222–226.
Additionally, the use of the sulfonamide group was supported by the results of exploratory data analysis wherein the designed sulfonamide analogs clustered with the same set of compounds that had previously shown activity. Thus, aryl and alkyl sulfonamide bioisosteres were synthesized, giving rise to 224 and 225 (Figure 58). These analogs were a success because they resulted in slightly superior cytotoxic activity compared to 24, displayed improved stability, but had poor solubility. In order to overcome this pitfall, a less hydrophobic sulfonamide analog was designed in which the eight-methylene unit of 225 was truncated to contain a four-carbon alkyl chain 226 leading to increased aqueous solubility. Compound 226 was synthesized by reacting a corresponding primary amine and sulfonyl chloride in the presence of sodium carbonate and water (Scheme 32, ii). A theoretical approach was taken in order to find more energetically favorable conformations for 24 and 226, calculate molecular properties, and gain insight on SAR.252 Next, the cytotoxic effects of 226 on MDA-MB-231 and human mammary epithelial cells (MCF10A) were evaluated. Results illustrated that both 24 and 226 both decreased the viability of MDA-MB-231 cells with IC50 values of 120 and 87 μM, respectively; however, 226 is potently more cytotoxic. Additionally, MCF10A cells were more resistant to treatment with 226 (IC50 = 198 μM) and also induced a change in cell morphology. Results indicated that 226 is an antitumor candidate with a cytotoxic mechanism. This new capsaicin analog was capable of interrupting cell-cycle progression via S-phase arrest and inducing cell death in MDA-MB-231 cells and apoptosis via the downregulation of the antiapoptotic Bcl-2 protein. Results also showed that 226 leads to apoptosis through caspase-3 cleavage in the death-inducing signaling complexes, TRAIL-R1/DR4 and TRAIL-R2/DR-5. In vivo results also indicated that 226 has antitumor properties as it was more effective (133.4 ± 24. 5 mm3) than 24 (281.3 ± 78.5 mm3) in reducing mean tumor volume and impairing tumor progression in the MDA-MB-231 model of breast cancer (Figure 58). Overall, the activity of 226 against MDA-MB-231, a well-established breast cancer model, and its lower pungency suggest its promising role as an anticancer agent for breast cancer.252
Novel irreversible HIV-1 protease inhibitors containing sulfonamide and sulfone groups as amide bond bioisosteres were discovered.254 Biotech Research Institute, Korea, reported an irreversible inhibitor 227 (Figure 59) with IC50 = 20 nM containing an epoxide moiety as an active site isostere. The authors designed inhibitors with improved potency and fewer peptide bonds.254 First, the replacement of the amide carbonyl group in the C-terminus in compound 227 with sulfonamide occurred since the hydrogen bonding between inhibitor and water in the enzyme was reported. The sulfonamide group would be able to maintain this hydrogen bonding as well as improve stability against amide hydrolysis by proteolytic enzymes. Next, the P2 site was taken into account for the design of new inhibitors since the pocket size of P2 is large in order to fit asparagine and valine. Therefore, replacing asparagine with suitable amphiphilic groups could increase the potency of inhibitors. Thus, the following modifications were made: inserting a sulfonamide for the P1–P2 amide bond and a sulfone group for the P2 asparagine, as seen in 228. The sulfonamide bioisostere (228) installation was achieved through the use of 3-chloropropane-1-sulfonyl chloride which is commercially available. Two sulfonamide bioisosteres of 227 were designed (229 and 230) (Figure 59). However, both of these bioisosteres were not the most potent of the series (IC50 = 30 nM and 20.2 nM, respectively). Results revealed that 228 was an inhibitor with high affinity, irreversibility, and high potency (IC50 = 6.6 nM) which could serve as a potential chemotherapeutic agent for the treatment of HIV.
Figure 59.
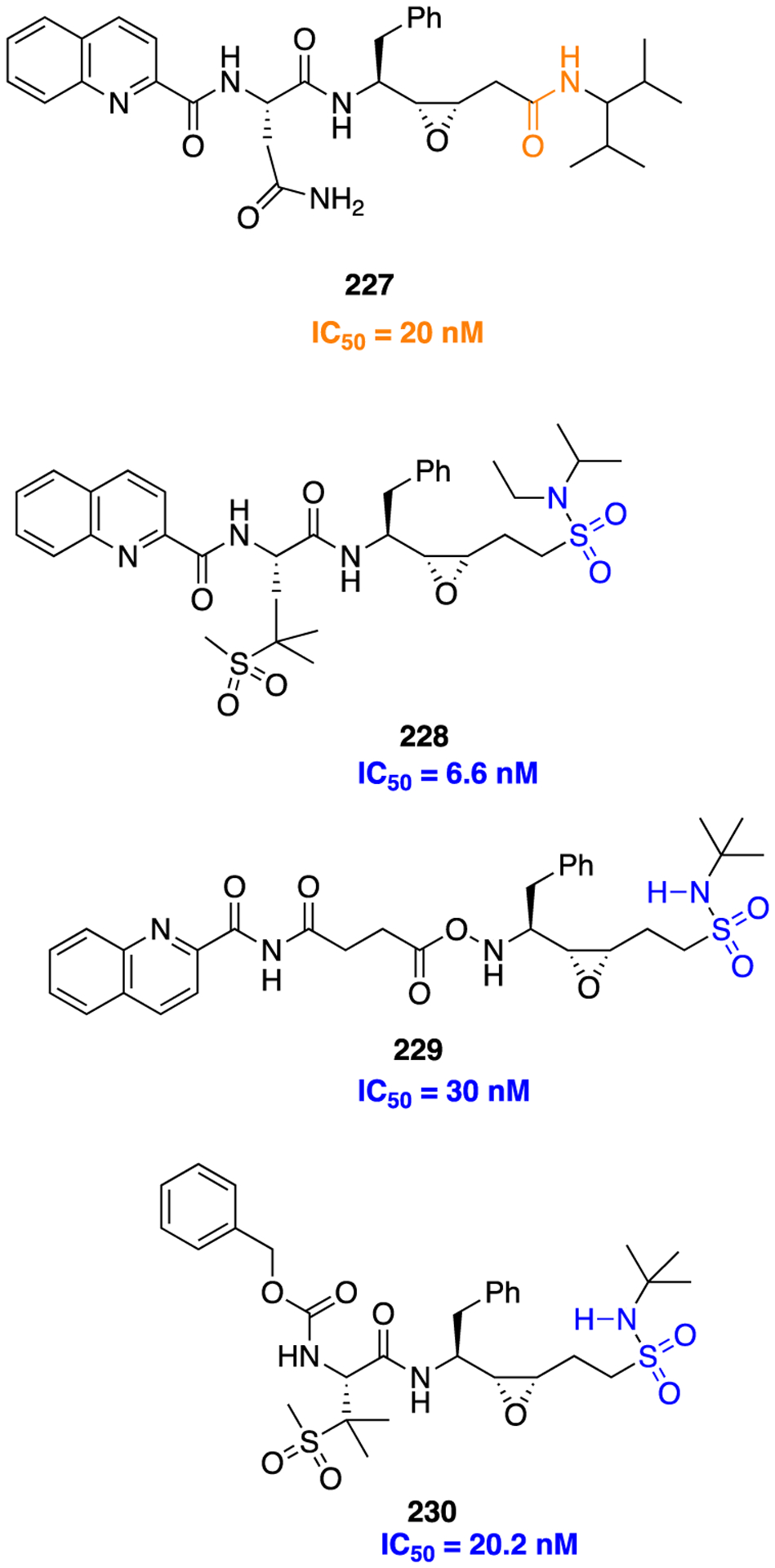
Novel irreversible HIV-1 protease inhibitors encompassing sulfonamide and sulfone as amide bioisosteres.
PHOSPHONAMIDATE BIOISOSTERES
The RNA-dependent RNA polymerase (RdRp) of the hepatitis C virus, also known as nonstructural protein 5B (NS5B), is essential for the synthesis of viral RNA.255 The protein has no functional equivalent in the mammalian system, and thus it is a target for inhibition by both nucleoside and non-nucleoside inhibitors. The 3D structure of RdRp suggests that NS5B has a cupped right-hand-like structure with a finger, palm, and thumb domain.256 Several drugs such as filibuvir and lomibuvir bind to the thumb domain, whereas dasabuvir, GSK5852, HCV-796 bind to the palm domain.257,258 Phosphonamidate bioisosterism has been reported to represent a new class of non-nucleoside inhibitors of HCV NS5B. The sulfonamide and amide groups were modified in the small molecules, 231 and 232 (Figure 60), respectively, which are non-nucleoside NS5B inhibitors that bind to the allosteric site 2 of the thumb domain of the HCV viral polymerase (Figure 61).259
Figure 60.
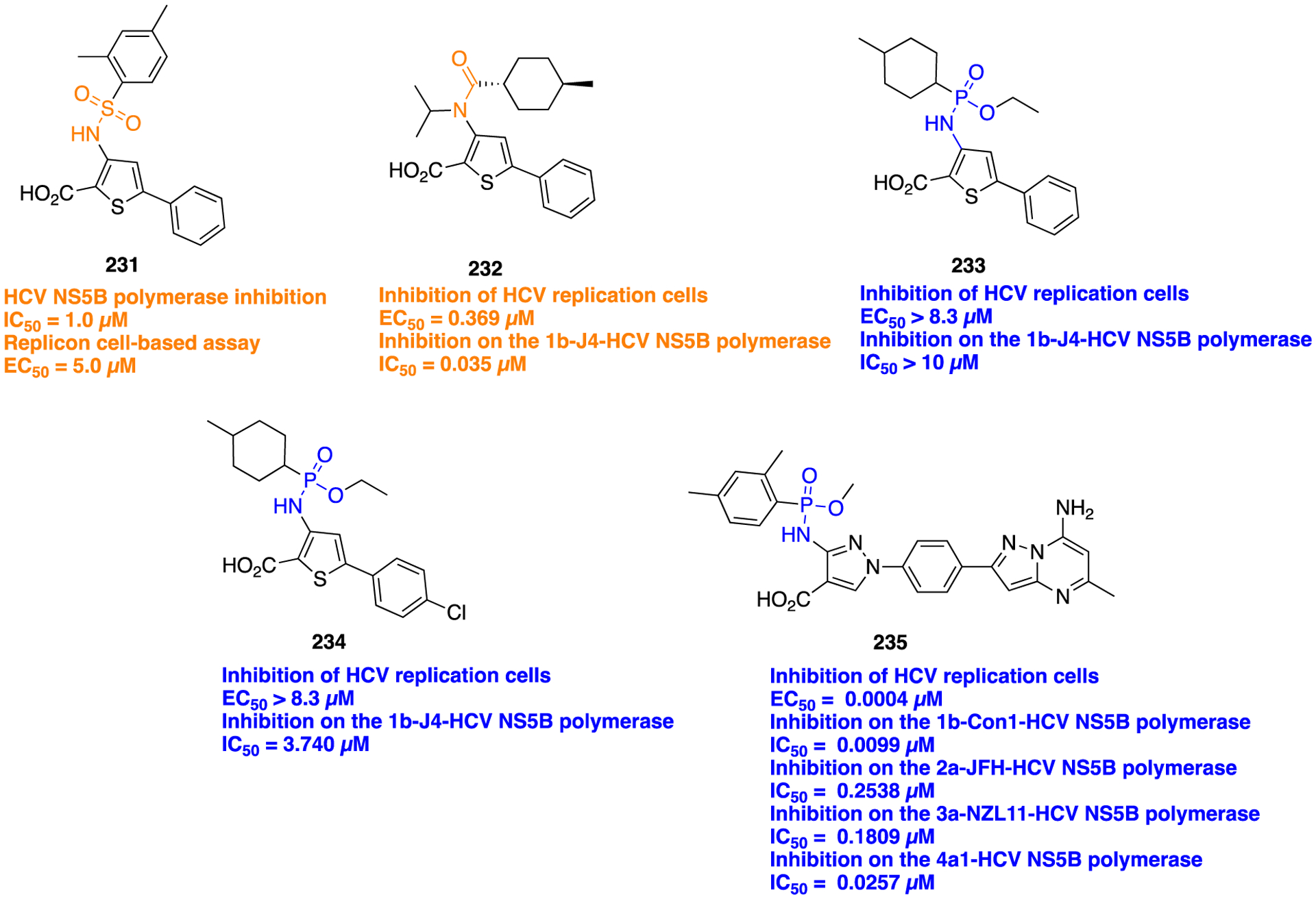
Non-nucleoside NS5B inhibitors (except 233), containing the phosphonamidate group as an amide bioisostere (233, 234, and 235).
Figure 61.
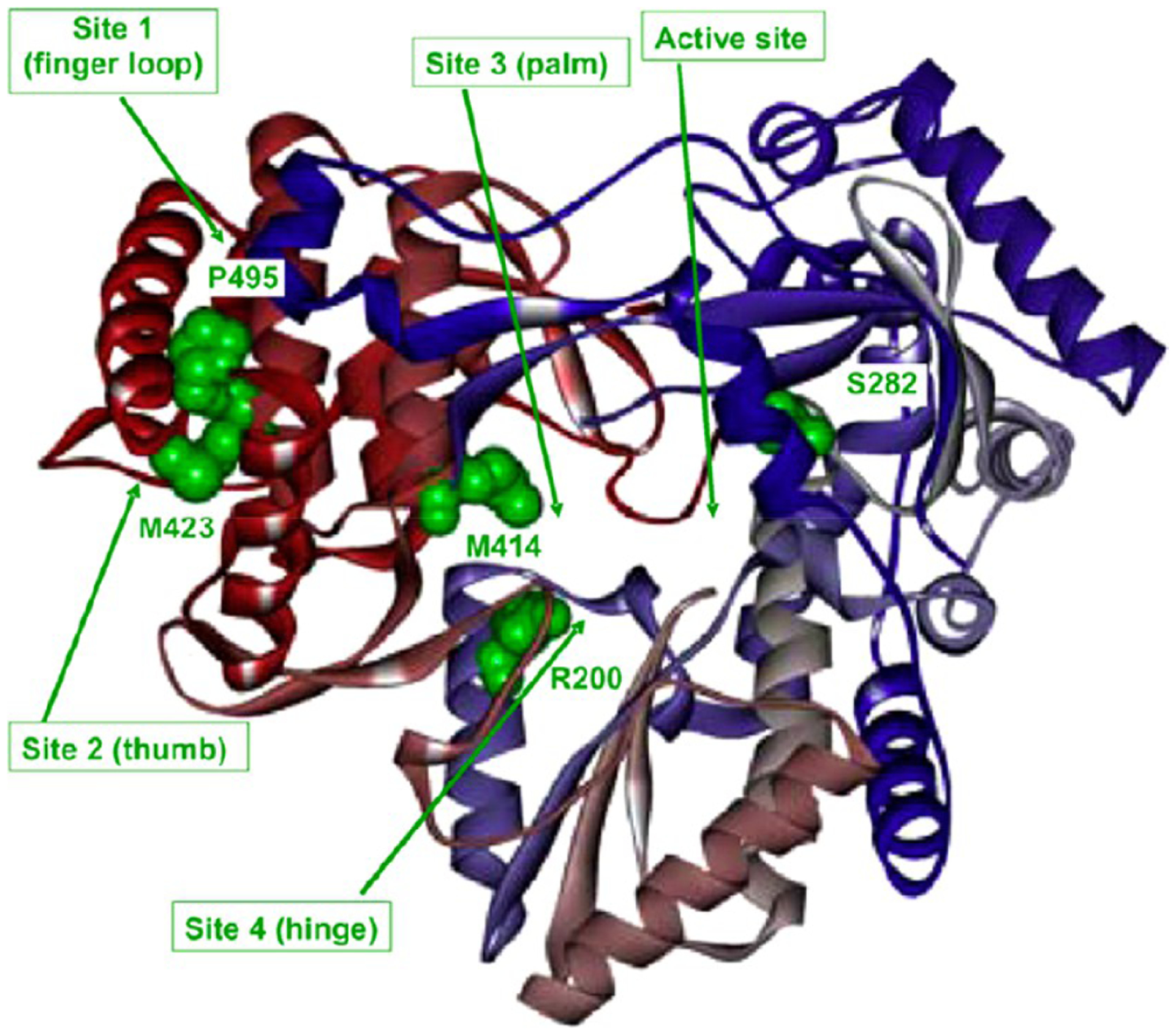
Ribbon structure of NS5B HCV viral polymerase. Reproduced with permission from Bioorganic & Medicinal Chemistry Letters (https://www.sciencedirect.com/journal/bioorganic-and-medicinal-chemistry-letters), Volume 26, pp 4536–4541, Pierra Rouviere, C.; Amador, A.; Badaroux, E.; Convard, T.; Da Costa, D.; Dukhan, D.; Griffe, L.; Griffon, J. F.; LaColla, M.; Leroy, F.; Liuzzi, M.; Loi, A. G.; McCarville, J.; Mascia, V.; Milhau, J.; Onidi, L.; Paparin, J. L.; Rahali, R.; Sais, E.; Seifer, M.; Surleraux, D.; Standring, D.; Dousson, C., Synthesis of potent and broad genotypically active NS5B HCV non-nucleoside inhibitors binding to the thumb domain allosteric site 2 of the viral polymerase.259 Copyright 2016 Elsevier.
Compound 232 had potent activity in genotypes 1a and 1b infections in preclinical trials. In order to test the hypothesis of phosphonamidate bioisosterism, two compounds, 233 and 234, were synthesized and evaluated for inhibition of HCV replication in the HCV replicon luciferase assay and inhibition of the 1b-J4 HCV NS5B polymerase.259 Only compound 234 produced a modest inhibition of the NS5B polymerase (IC50 = 3.74 μM). Subsequently, detailed SAR investigations either on R1-R2-R3-R4-substitutions or on the thiophene moiety were conducted to determine the effect on the potency of this new phosphonamidate class of compounds. Many analogs, with varying substituents, were synthesized and evaluated, and ultimately, compound 235 was identified as the most active analog, with efficacy greater than that of the parent compound, 232. Scheme 33 describes the key synthetic route to generate the phosphonamidate bioisostere as in compound 235. Compound 235 had broad genotype profile: IC50 = 0.0099 μM, 0.2538 μM, 0.1809 μM, and 0.0257 μM for genotypes 1b, 2a, 3a, and 4a, respectively. Furthermore, 235 had high selectivity index with subnanomolar potency in the replicon assay (EC50 = 0.0004 μM) without producing cytotoxicity. Cocrystallization studies of 235 complexed with the viral NS5B indicated that the inhibitor is hydrogen bonded through the carboxylate functional group to the main chain of Ser476 and Tyr477 and to the side chain of Arg 501 with the oxo function of the phosphonamidate, confirming its interactions with the viral protein in the thumb 2 domain of NS5B (Figure 62). Compound 235, designated IDX17119, was selected for further preclinical and PK studies. Overall, the phosphonamidate proved to be a viable bioisosteric replacement for the amide function in developing potent non-nucleoside NS5B inhibitors.
Scheme 33.
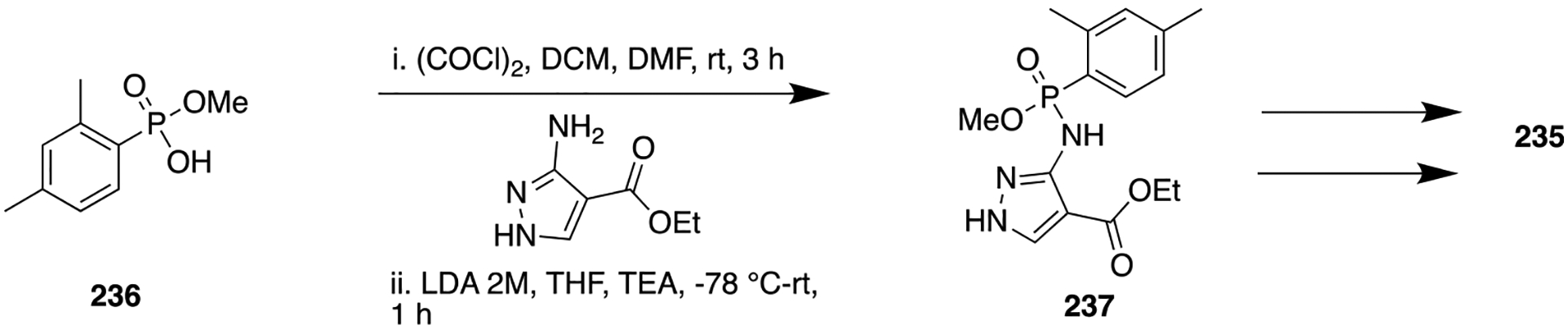
Synthetic Route To Access 235
Figure 62.
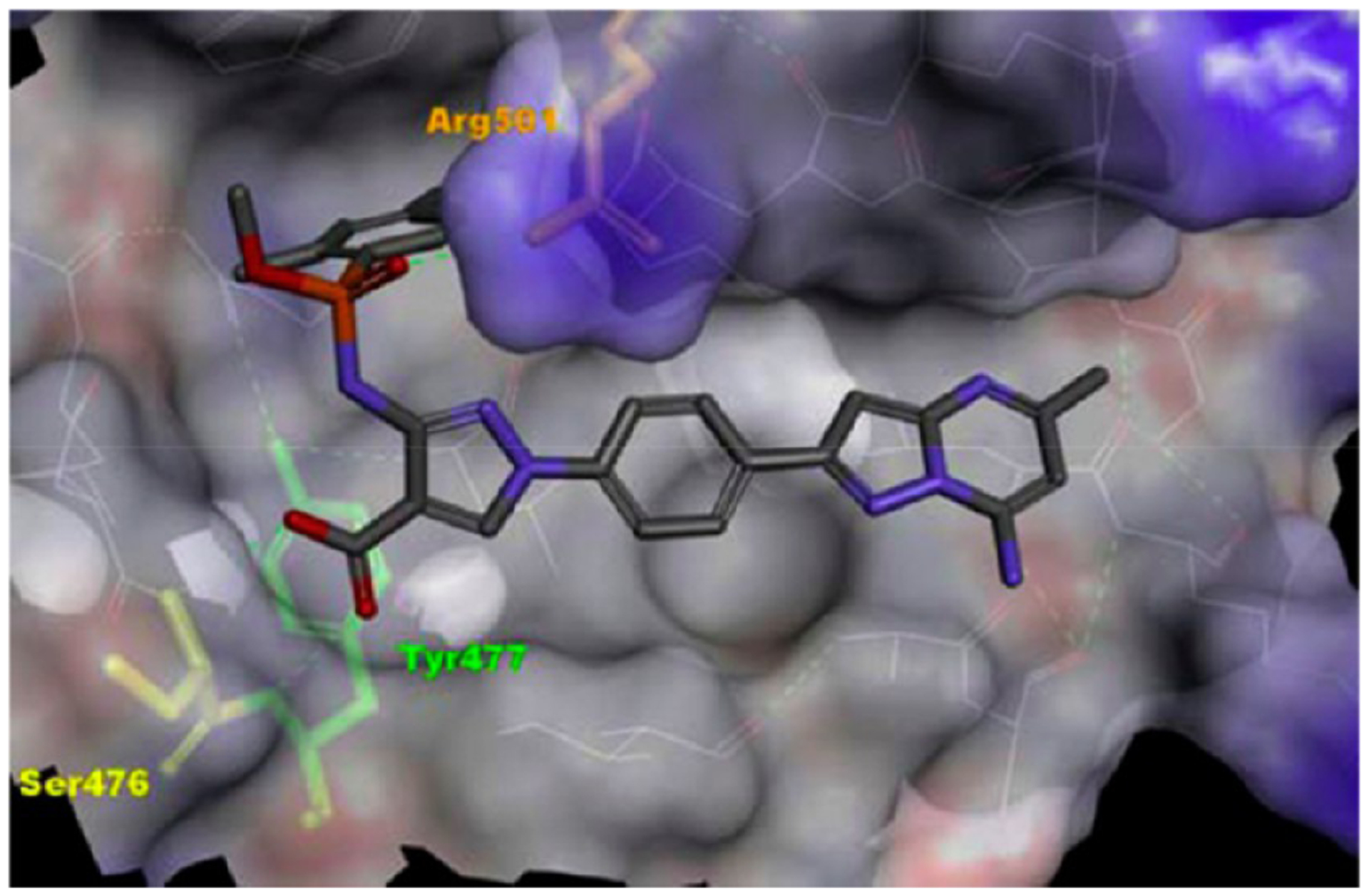
Cocrystallization image of 235 complexed with the viral NS5B (PDB code 5CZB). Reproduced with permission from Bioorganic & Medicinal Chemistry Letters (https://www.sciencedirect.com/journal/bioorganic-and-medicinal-chemistry-letters), Volume 26, pp 4536–4541, Pierra Rouviere, C.; Amador, A.; Badaroux, E.; Convard, T.; Da Costa, D.; Dukhan, D.; Griffe, L.; Griffon, J. F.; LaColla, M.; Leroy, F.; Liuzzi, M.; Loi, A. G.; McCarville, J.; Mascia, V.; Milhau, J.; Onidi, L.; Paparin, J. L.; Rahali, R.; Sais, E.; Seifer, M.; Surleraux, D.; Standring, D.; Dousson, C., Synthesis of potent and broad genotypically active NS5B HCV non-nucleoside inhibitors binding to the thumb domain allosteric site 2 of the viral polymerase.259 Copyright 2016 Elsevier.
ESTER BIOISOSTERES
Geometrically an ester and amide are comparable. As with the resonance of an amide, in an ester, the lone pair of electrons on the oxygen delocalizes into the antibonding orbital of the ester carbonyl group. Similar to what happens with amides, the electron delocalization results in cis and trans isomers. The trans conformation of an ester is more stable than the cis conformation, as seen with amides. Additionally, esters and amides differ based on hydrogen bond formation, thus affecting protein folding and stability. Esters are weak hydrogen bond acceptors and cannot hydrogen bond-donate. In addition to this, esters are vulnerable to hydrolysis in vivo.235 Amide isosteres can modulate polarity and bioavailability, while esters are used to address metabolism issues due to their vulnerability to cleavage in vivo, most often forming the cleavable component of prodrugs.16
Opioids are a common drug for the treatment of moderate to severe pain. The three major subtypes of opioid peptide receptors are μ (MOPr), δ (DOPr), and κ (KOPr). Leu-enkephalin (238) (Figure 63) is an agonist for the DOPr. However, 238 suffers from short half-life and high hydro-philicity, thereby hampering its ability to serve as a druggable compound. Additionally, enkephalins are rapidly metabolized by peptidases in the body as a result of their amide bonds and therefore cannot cross the blood-brain barrier (BBB) and reach opioid receptors in the central nervous system.260 Previously Rochon and co-workers have reported the effects of replacing the amide bonds in 238 with E-alkenes. When doing so, the replacement of the first amide bond by E-alkene did not affect the biological activity of 238. Therefore, it was concluded that this bond is not involved in intramolecular or intermolecular interactions with the DOPr.260
Figure 63.
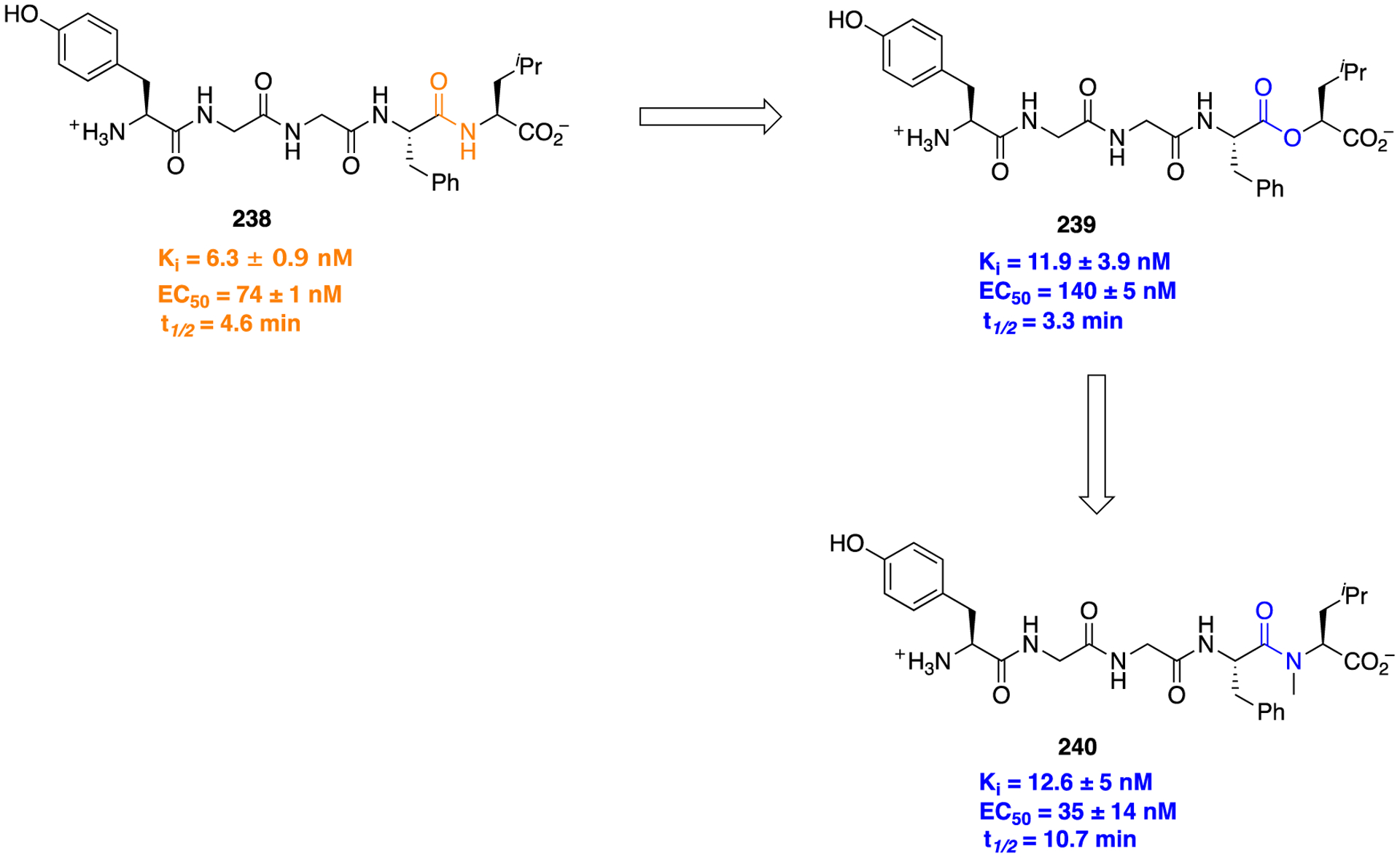
Structures of peptide 238 and analogs in which the amide bond was replaced by an ester 239 or N-methylamide bond 240.
Next, they sought to employ other amide bioisosteres including esters and N-methyl amides. The reason for the choice of these motifs, in particular, is their potential to act as hydrogen bond acceptors and not as hydrogen bond donors. The N-methyl group introduces a steric bulk, thereby distorting the planarity of the amide and restricting the conformation around the modified amide. An ester functional group is a better mimic of a peptide bond since it does not undergo many geometric alterations; however, the partial double bond nature of the ester bond is not as pronounced as the amide bond. Thus, esters are more structurally flexible. Rochon and co-workers replaced all of the amide bonds of 238 with an ester or an N-methyl amide. To evaluate the ability of each compound to bind the DOPr, competitive binding assays using GH3/DOPr cell membrane penetration were used. Results showed that replacing the fourth amide bond with an ester (239) or N-methyl amide (240) gave compounds with similar affinities to that of 238 (Figure 63). Compound 240 was synthesized by a multistep peptide approach where N-methylated leucine amino acid starting material was commercially available. When the third amide bond of 238 was replaced by an ester, most of the affinity for the DOPr was retained (Ki = 34 ± 17). The phosphorylation of extracellular signal-regulated kinases 1 and 2 (ERK1/2) is used as an indicator of a compound’s ability to act as an agonist. The activation of the DOPR by an agonist stimulates the rapid and transient phosphorylation of ERK1/2. However, the maximal effect of 238 is 5 min after stimulation; therefore, compounds were evaluated after 5 min of stimulation with varying concentrations (10−9 to 10−5 M). Compounds 239 and 240 at concentrations of 10−7 M produced significant phosphorylation of ERK1/2 similar to that of 238.
Previously, this group reported the internalization of fluorescent chimeric DOPr (DOPr-GFp) in transfected DRGF11 cells in order to screen for active compounds. Compounds 239 and 240 were measured for their ability to induce internalization of DOPr-GFR. At 1 μM, 238 produced an internalization that was visually seen within 5 min and complete by 30 min. The level of internalization by 239 and 240 was similar to that of 238. These compounds were also tested for their ability to inhibit the electric-field induced contractions of the mouse vas deferens. Compound 240 has the greatest activity (EC50 = 35 ± 14 nM) compared to 240 and 238 (EC50 = 140 ± 5 nM and EC50 = 74 ± 1 nM), respectively. A goal of these 238 analogs was their ability to cross the blood-brain barrier; therefore, their lipophilicity was assessed. On the basis of all the modifications, the N-methyl amide resulted in compounds with the highest lipophilicity. Compounds 239 and 240 had high total polar surface area (tPSA) values in comparison to CNS acting drugs. Results also indicated that compound 240 had an increase in half-life (t1/2 = 10.7 min) compared to 238 (t1/2 = 4.6 min), which is due to the replacement of the fourth amide bond with an N-methyl amide function, thereby increasing the stability of the third amide bond. On the other hand, when an ester is introduced to 239, there was surprisingly little effect on the half-life (t1/2 = 3.3 min). These results showed that the systematic replacement of amides with bioisosteres such as an ester and N-methyl amide was an efficient way for designing novel peptide analogs of 238.260
OLEFIN BIOISOSTERES
The bioisosteric replacement of a peptide bond with a nonhydrolyzable alkene functional group has been used to improve the “drug-like” properties of many parent compounds. The alkene bioisostere has been shown to mimic the planar geometrical features, but not the electronic and electrostatic properties, of an amide bond.261,262 The bond lengths and angles of an alkene are virtually identical to an amide. In contrast, an alkene does not have hydrogen bonding capacity due to the absence of heteroatoms.235 Furthermore, the value of the dipole moment is higher for an amide bond (3.6 D) than for an alkene (0.1 D).263 Several studies have reported that the replacement of an amide bond with an alkene bioisostere produces molecules that are efficacious. For example, dihydrouracil analog 241 is a potent inhibitor of the RNA-dependent RNA polymerase, NS5B protein of the hepatitis C virus genotypes GT1a (EC50 = 51 nM) and GT1b (EC50 = 19 nM).264 However, this compound had inherent pharmacokinetic issues; for example, 241 was rapidly cleared from plasma and thus had poor oral bioavailability (F = 1.4%) in rats. These results could be explained by the low aqueous solubility (<1.5 μM at pH 7.2) and poor membrane permeability of 241. Modification of the amide linker in 241 with one that lacked one or both hydrogen bonding groups enhanced the overall membrane permeability potential.265 In order to identify a more potent HCV NS5B polymerase inhibitor with improved PK properties, the amide linkage was successfully replaced with a trans olefin, yielding compound ABT-072 (242) (Figure 64). The olefination of aldehyde group in 243 with diethyl (4-nitrobenzyl)phosphonate followed by the nitro reduction and methanesulfonylation gave 242 (Scheme 34). In the HCV genotype 1 (GT1) replicon assay, 242 had a greater potency for both the GT1a (EC50 = 1 nM) and GT1b (EC50 = 0.3 nM) polymerase compared to the parent compound 241.
Figure 64.
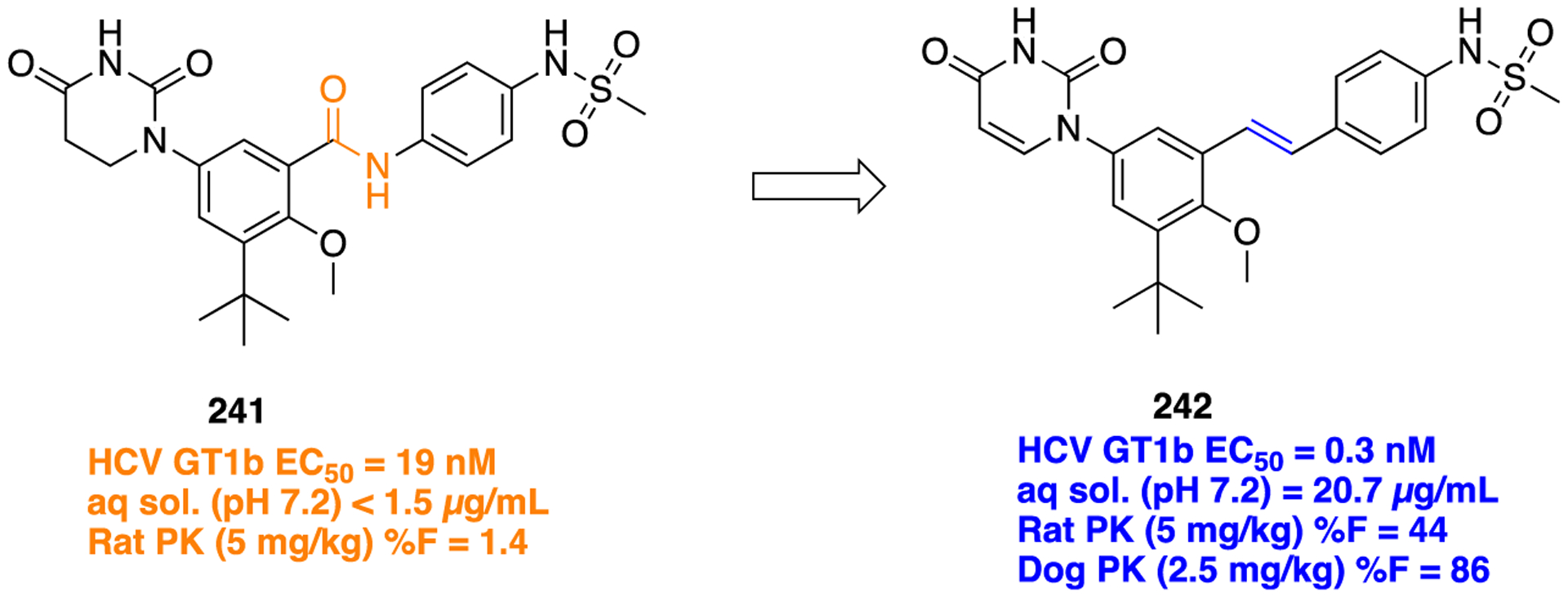
Amide to trans olefin bioisosteric replacement leading to the clinical candidate 242.
Scheme 34.
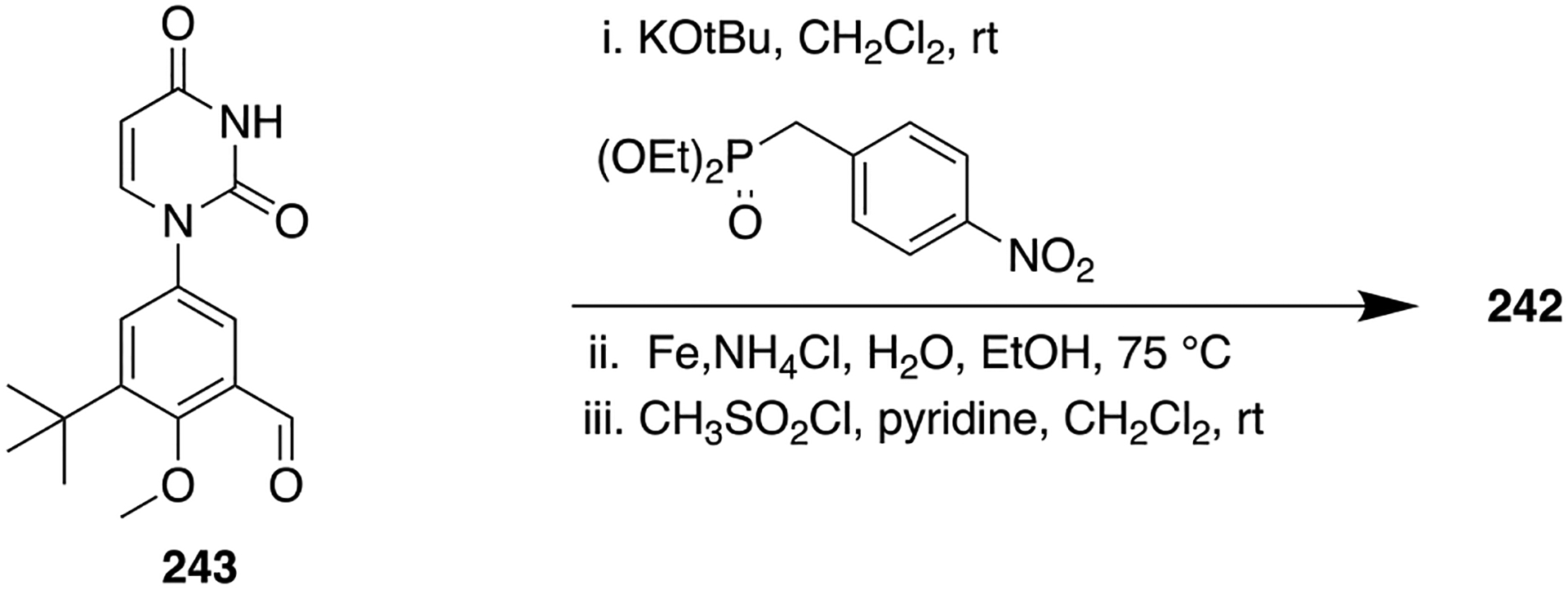
Synthetic Route To Access 242, a Non-Nucleoside HCV NS5B Polymerase Inhibitor
Moreover, in PK studies, the plasma clearance of 242 was significantly decreased, thereby producing greater oral bioavailability in both rat (5 mg/kg, per oral (po), t1/2 = 3.1 h, and F = 44%) and dog (2.5 mg/kg, po, t1/2 = 5.4 h, and F = 86%). On the basis of these results, compound 242 was nominated for clinical studies. Pharmacokinetic studies in healthy human subjects indicated that 242 achieved maximum concentration within 5–9 h in plasma, with a mean terminal phase half-life of 7–9 h. Subsequently, phase II trials indicated that 242 was efficacious for treating genotype 1 infected HCV patients in combination with the other HCV protease inhibitor such as ritonavir, as well as ribavirin.
The calcitonin gene-related peptide is a 37 amino acid neuropeptide that has been shown to mediate various biological functions such as vasodilation and nociception.266 The CGRP is released from sensory nerves and plays an integral role in the pathophysiology of migraine.267 (E)-Alkene bioisosterism was used to develop acyclic CGRP antagonists, where the metabolically unstable central amide bond in a previously developed compound, 244 was replaced with an E-alkene linked to a substituted pyridyl moiety (245) (Figure 65).268,269 Compound 245 was synthesized via palladium-catalyzed Heck coupling reaction with an unsaturated halide (246) and an alkene (247). The general synthetic procedure is delineated in Scheme 35. These molecular structural changes substantially improved the bioavailability and pharmacokinetic profile of compound 245 in rats compared to 244 while maintaining antagonist activity at the CGRP receptor.
Figure 65.
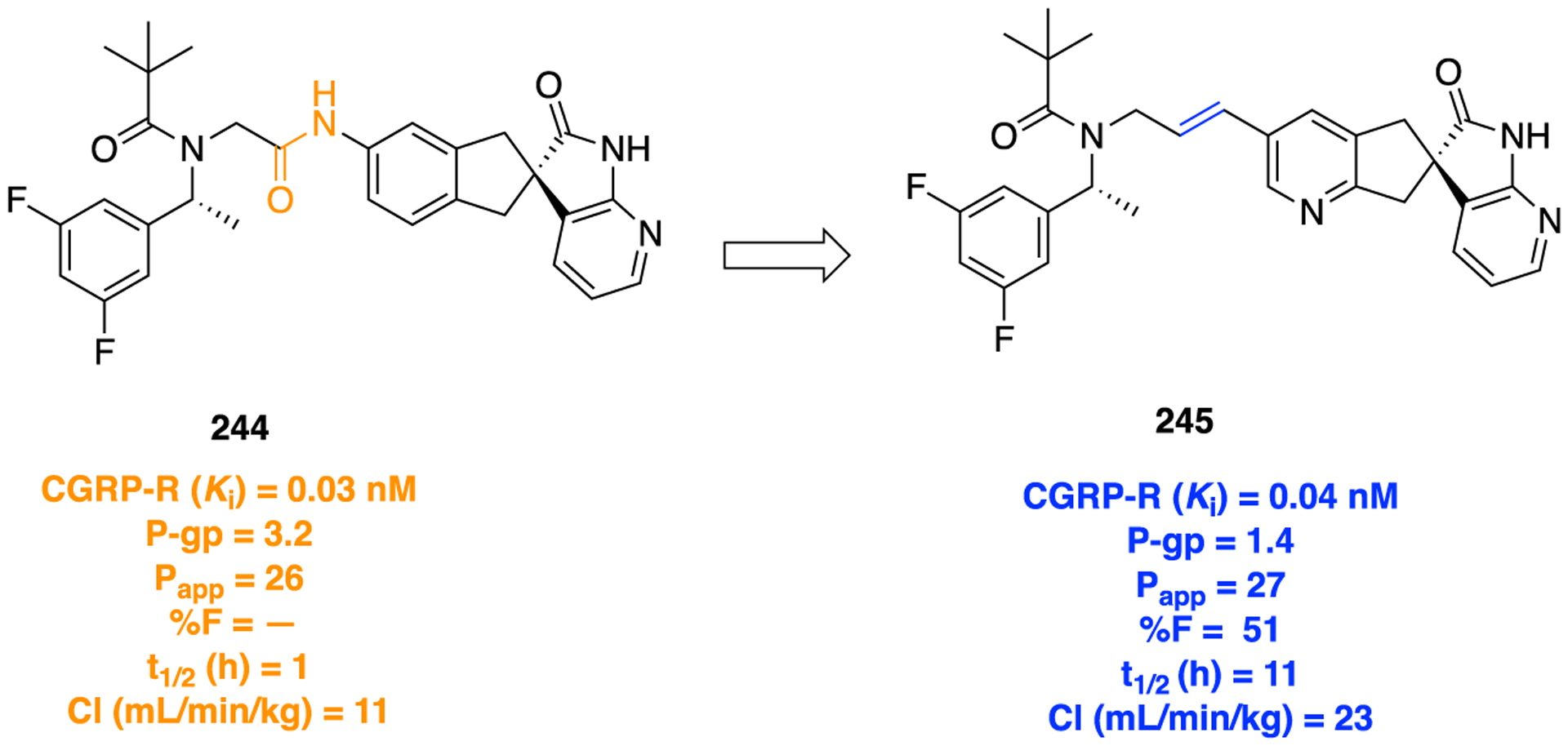
Example of the replacement of a central amide of an azaoxindole derivative (244) with an E-alkene moiety 245.
Scheme 35.
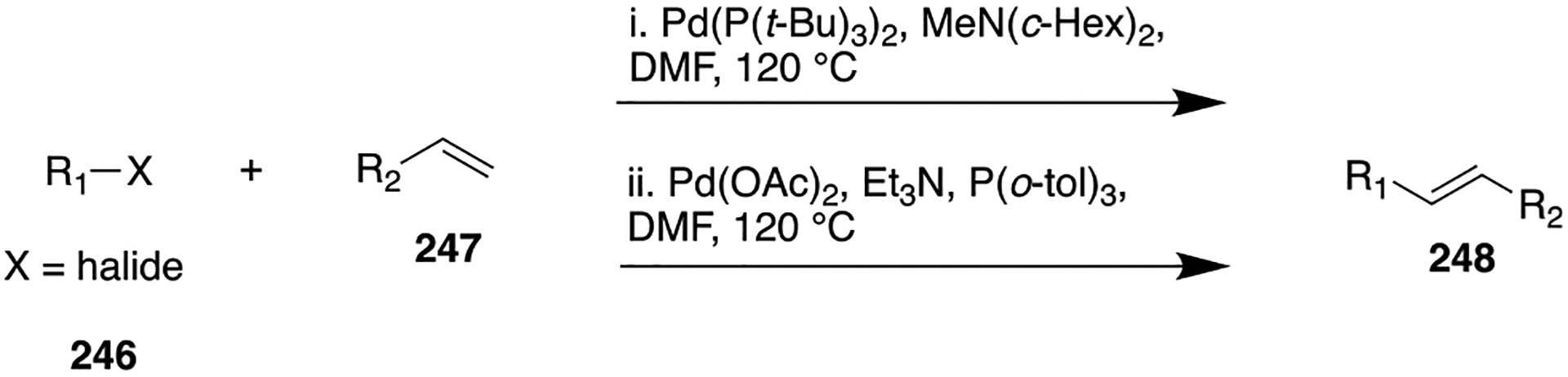
General Synthetic Route To Access Alkene Bioisosteres
In 2007, Thorarensen et al. explored the SAR of the previously discovered lead compound, 249, and newly synthesized anthranilic acid derivatives270 to elucidate the role of the linker connecting the two important pharmacophoric aryl A and B rings for antibacterial efficacy.236 The amide linker in 249 was successfully replaced by several amide bioisosteres, and the E-alkene-containing analog 250 was found to have significantly lower MIC values, particularly in Staphylococcus aureus UC9218 and Staphylococcus epidermidis UC 12084 (SEPId) (Figure 66), than the parent compound 249. The general synthetic method showing Wittig olefination of an Wittig reagent (methyl 2-((bromotriphenyl-λ5-phosphaneyl)methyl)-5-cyanobenzoate) with an aldehyde (251) is described in Scheme 36, i. Overall, results suggest that the amide may not be essential for bioactivity, and it perhaps only keeps the pharmacophoric elements at the defined distances. Thus, an amide can be dispensable without causing a significant decrease in MIC values.
Figure 66.
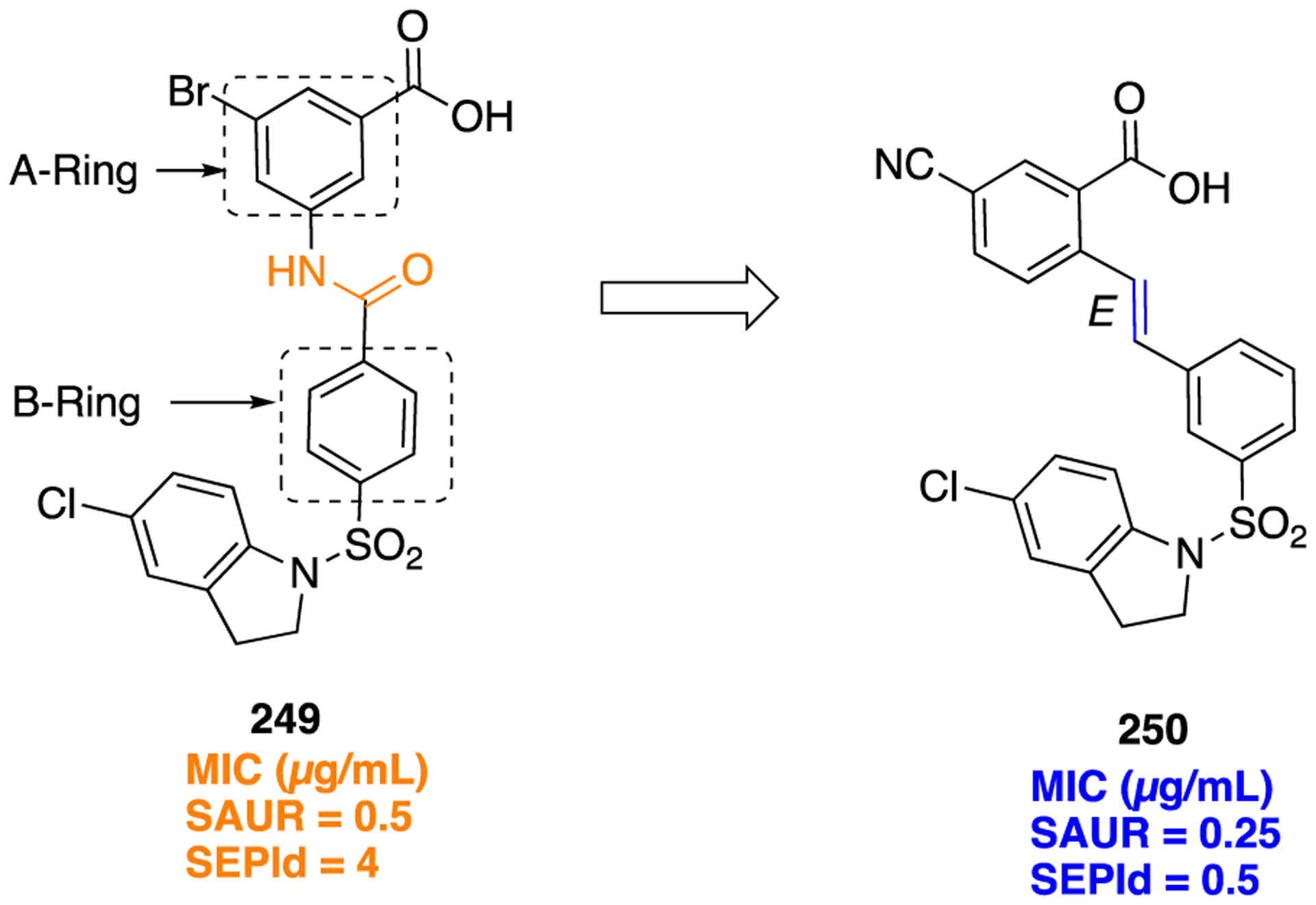
Structural modification of an amide linker in 249 to an E-alkene in 250 that joins A and B rings.
Scheme 36.
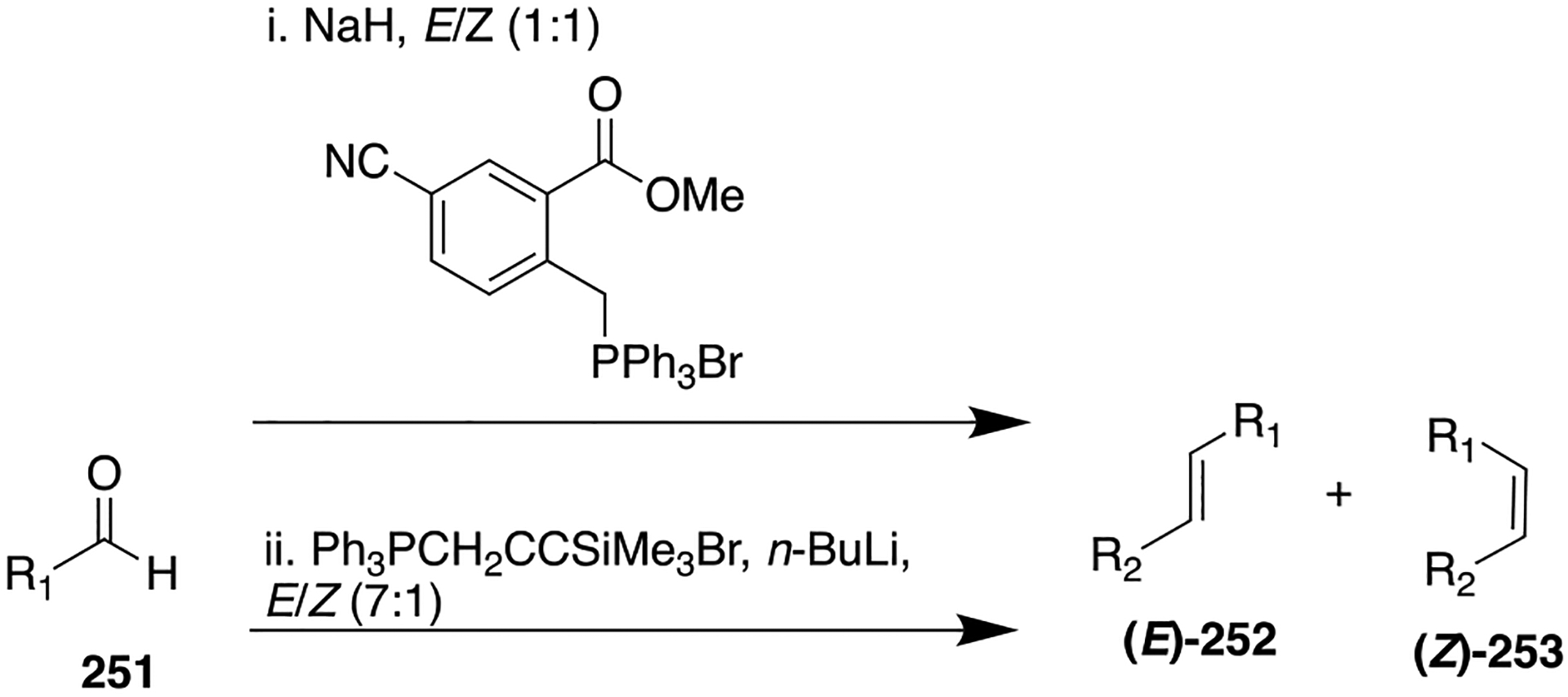
General Synthesis of E-Alkene Bioisosteres from Respective Aldehyde
In another study exploring the modification of amide to alkene, utilizing amylin (20–29) as a parent structure, Brouwer and co-workers reported the synthesis of alkene dipeptide/alkene dipeptidosulfonamide isosteres using a solid phase cross metathesis method.271 Amylin, or human islet amyloid polypeptide, is a peptide hormone composed of 37 amino acids, where amylin (20–29) refers to the highly amyloido-genic region that is involved in the pathogenesis of type 2 diabetes. The modification in amylin (20–29) to yield compound 254 was postulated to be beneficial in determining the peptide backbone interactions and peptide stabilization in enzymatic degradation. Given that the alkene moiety closely resembled the geometry of the amide bond and that the amalgamation of a sulfonamide bond may increase the flexibility of the backbone, the authors transformed the amide function (at positions 27 and 28) of 254 into the alkene derivative 255 and the alkenesulfonamide (256) dipeptide analogs (Figure 67). The adopted synthetic route for 255 and 256, depicted in Scheme 37, used resin bound H-Ser(OtBu) as a starting material.
Figure 67.
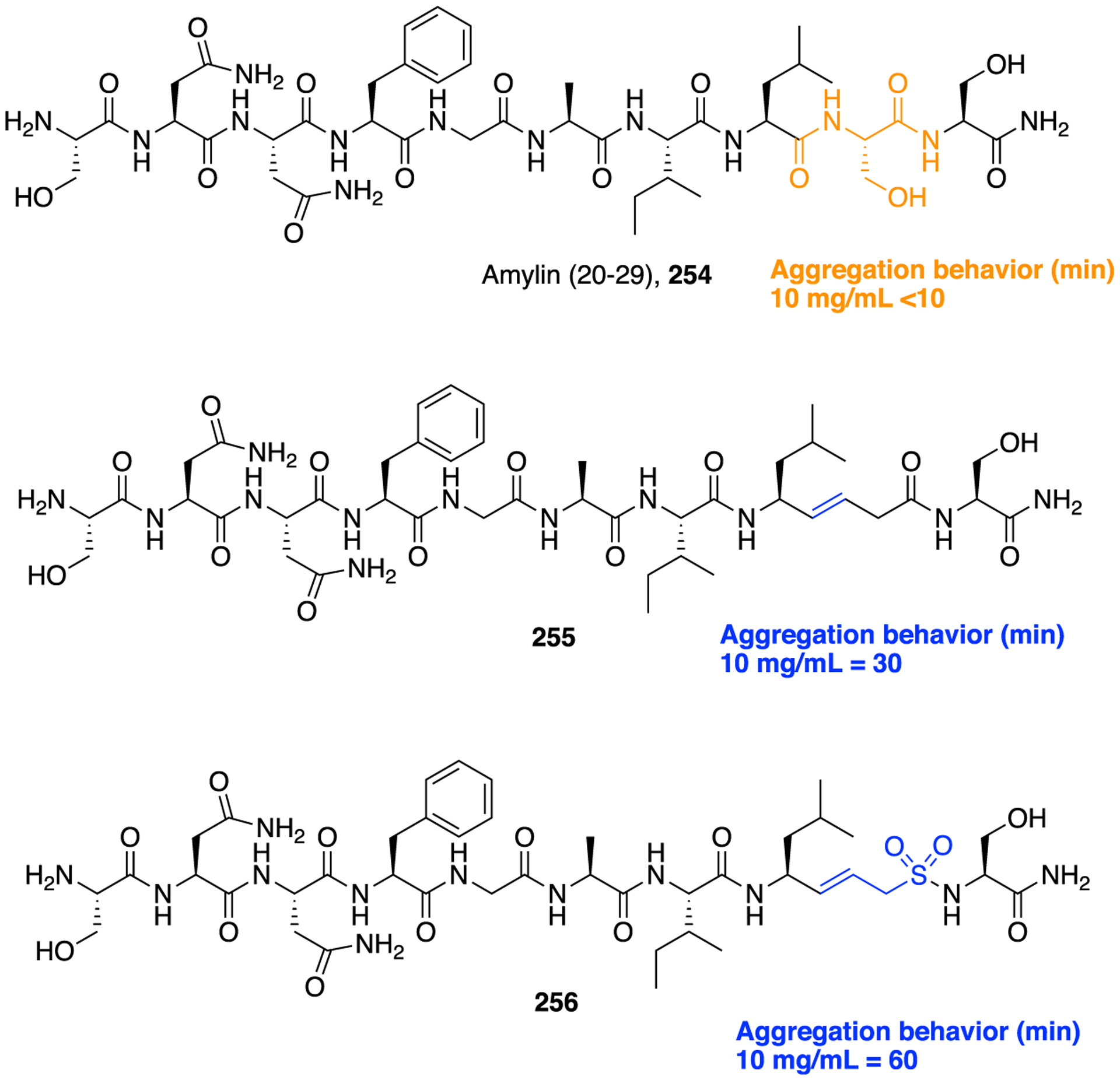
Molecular structures of amylin (20–29) 254 and its modified bioisosteric analogs.
Scheme 37.
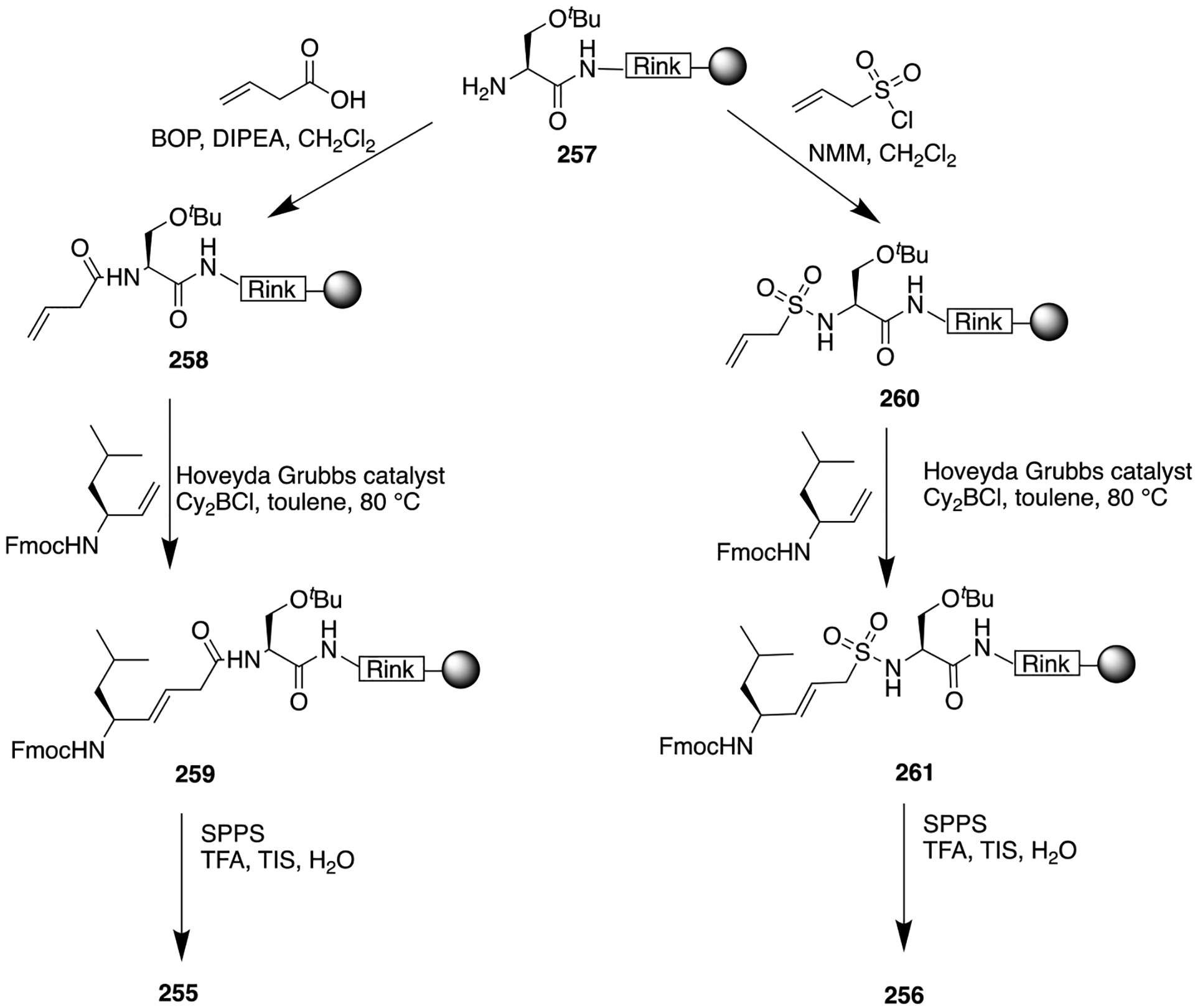
Solid Phase Synthesis of an Alkene Dipeptide (255) and Alkene Dipeptidosulfonamide Isostere (256)
Both compounds were characterized by electrospray mass spectrometry. Rapid gel formation, which was used to measure aggregation behavior, indicated that 255 and 256 decrease gel formation time by 30 and 60 min, respectively, compared to parent amylin (<10 min). Compared to 254, both 255 and 256 displayed a significantly low number of amyloid fibrils in the transmission electron microscopy images. This retardation in fibril formation by both bioisosteres could be due to a lack of hydrogen bonding capabilities in the peptide backbone. Fourier transform infrared spectra (FTIR) and circular dichroism (CD) studies also validated that the modified dipeptide peptidomimetics exhibited superior activity in the retardation of fibril formation and altered β-sheet secondary structures when compared to the parent, 254.
Another instance of application of amide bond replacement and amide bioisosterism is seen in Yamamoto et al. studies to find an efficient DNA minor groove binding agent where the group synthesized distamycin related structural analogs.272 Distamycin (262) is a naturally occurring tripeptide antibiotic that binds to the nonintercalative minor groove of DNA, with high selectivity toward AT-rich sequences.273 The structural modification of the parent compound 262 at the distal C-terminal, by replacing the amidine group with a dimethylamino group, yielded compound 263, and further alteration at the N-terminal in 263 yielded 264, which had a nitro group. Compounds 263 and 264 were altered so as not to contain any amide bioisostere. One of the inter-pyrrole linkers (amide bond) of 264 was replaced by a trans-olefin to produce compound 265 to determine the effect of amide bond replacement or amide bioisosterism on its DNA binding properties (Figure 68).
Figure 68.
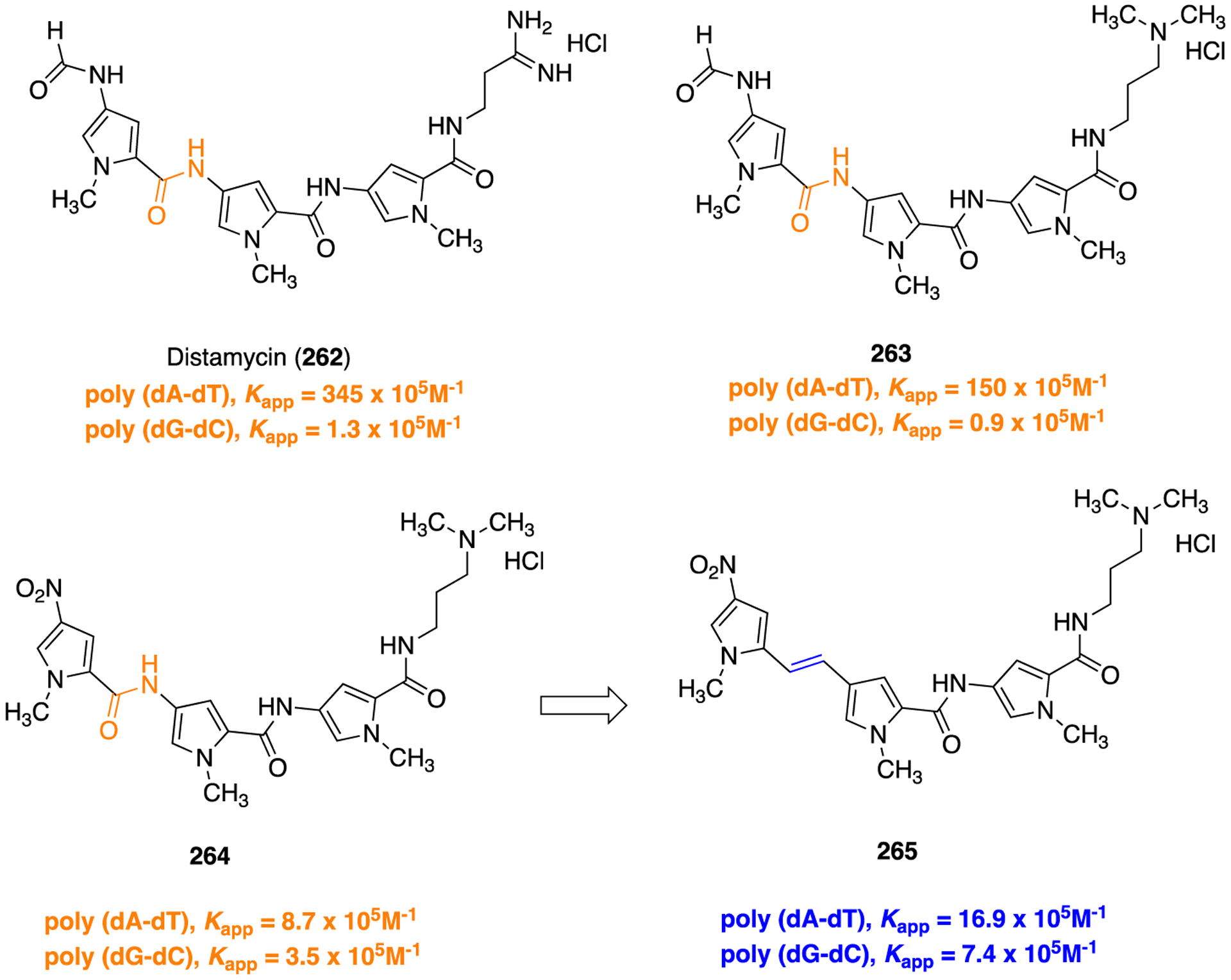
Distamycin (262) and its bioisosteric analogs.
The general synthetic route to access compound 265 is shown in Scheme 35, ii. The results of the ethidium displacement assay indicated that the modified compounds 263, 264, and 265 had a lower DNA binding affinity for poly(dA-dT) compared to 262. However, the replacement of the amide bond with a trans olefin, as in compound 265, produced the highest DNA binding for poly(dG-dC); Kapp = 7.4 × 105 M−1 compared with 262, 263, and 264. Overall, the structural modification related to amide function could produce compounds that may have different DNA recognition patterns.
An interesting observation regarding the effect of replacing a peptide bond with an E-alkene is provided by the bombesin receptor antagonist 268, derived from compound 266, with speculation that the E-alkene bond could provide high lipophilicity and enhanced resistance to biodegradation.274 The biological results were compared to a short chain pseudopeptide 267, which is also a bombesin analog. Both peptides 267 ([d-Phe6,Leu13Ψ(CH2NH)Leu14]bombesin(6–14)) and 268 ([d-Phe6,Leu13Ψ[(E)-CH=CH]Leu14]-bombesin(6–14)) have similar structures except that 268 has a E-alkene bond inserted between the 13th and 14th position in bombesin (see cross-reference for synthetic procedure) (Figure 69).
Figure 69.

Amino acid sequences of bombesin (266), pseudopeptide 267, and E-alkene isostere 268.
The antagonistic activity of 268 for the bombesin receptor was 10 times greater than 267 in terms of its inhibition of bombesin-stimulated amylase release in rat pancreatic acini. Furthermore, additional pharmacological experiments revealed that 268 efficiently blocked bombesin-stimulated amylase release; however, 267, even at the highest concentration used, failed to completely block amylase release, indicating that compound 268 is a very potent and specific bombesin antagonist in rat pancreatic acini that could be a useful probe for studying the biological actions of bombesin and its related peptides.
Another example of an E-olefin bioisostere as an amide bond replacement are the mutants of Aβ(1–40) Phe19-Phe20 which inhibit amyloid Aβ peptide amyloidogenesis and have been implicated in the formation of many distinct quaternary structures involved in the pathogenesis of AD.275–277 The Aβ fibril structure is stabilized by various inter- and intramolecular hydrophobic interactions, salt bridges, and intermolecular hydrogen bonds. Therefore, interrupting intermolecular backbone-backbone hydrogen bonding in one of the hydrophobic subsequences, i.e., LVFFA (17–21) in Aβ(1–40), could prevent many peptides from forming cross-β-sheets-based amyloid fibrils. Subsequently, Kelly et al. in 2005 substituted a trans alkene moiety for an amide in the key Phe19-Phe20 subsequence, thereby eliminating a hydrogen bond donor and acceptor from the desired site (Figure 70).
Figure 70.
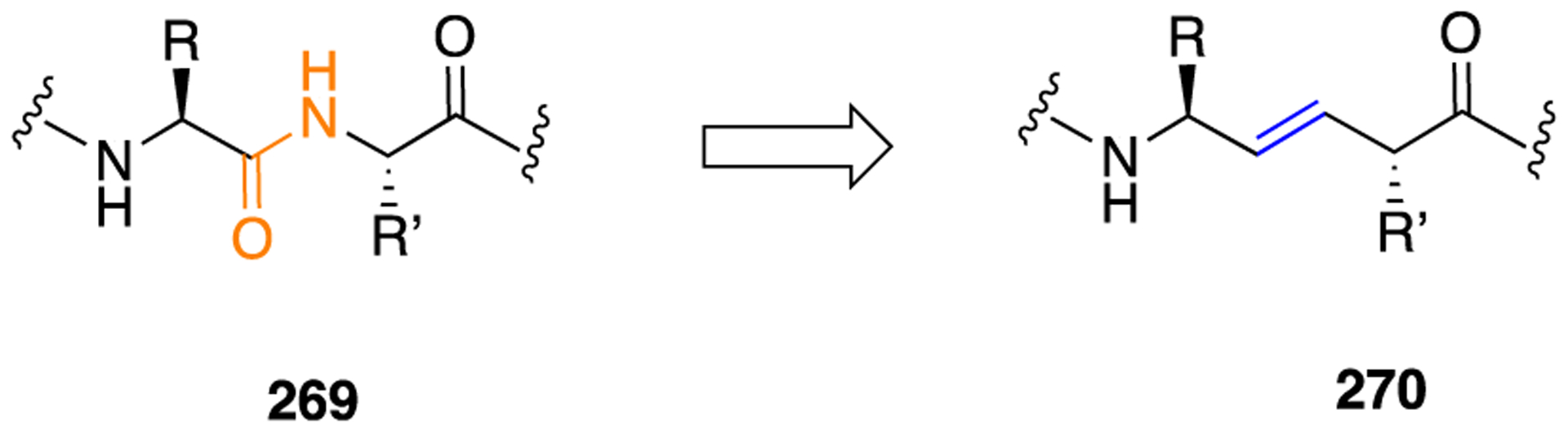
Graphical representation of the modification of peptide backbone from an amide to E-olefin, which is neither a hydrogen bond donor nor acceptor.
Initially, the stereoselective E-olefin dipeptide isostere (270) was synthesized according to the general synthetic method shown in Scheme 36, ii, and then incorporated into the Aβ(1–40) peptide in place of phenylalanine 19 and 20 (abbreviated as Phe19-Phe20) by solid phase peptide synthesis using hexafluorophosphate azabenzotriazole tetramethyl uronium (HATU) activation. The final Phe19-Phe20 E-olefin analog of Aβ(1–40) was referred to as EOAβ, and its efficacy to inhibit fibrillar aggregation into protofilaments and fibrils having a cross β-sheet structure was confirmed by circular dichroism and atomic force microscopy (AFM) methods.278
Researchers at the Neuroscience Research Pharmaceutical Product Division of Abbott Laboratories, prepared E-alkene bioisosteres of the amide bond in cholecystokinin (CCK) peptides, with the aim of developing potent and selective cholecystokinin brain (CCK-B) ligands.279 The systematic replacement of the amide linkage via an E-alkene was done to mimic the size, geometry, bond angle, and bond length of the amide. While the amide bond undergoes restricted rotation, it does still rotate, which is important for hydrogen bonding at the receptor binding site. In contrast, the E-alkene cannot form hydrogen bonds, and thus, the amide linkages at different positions of CCK peptides were replaced by an E-alkene and tested to determine if they were bioactive. This modification strategy was used as it was postulated that it would provide important information about the role of each amide bond in the CCK peptide. Among six pseudopeptides synthesized, the potency and functional activity of the 273 (Boc-Leu[E-CH=CH]Gly-Trp-Leu-Asp-PheNH2) isostere was similar to that of the parent peptides, Boc-protected CCK-6 hexapeptide (Boc-Leu-Gly-Trp-Leu-Asp-PheNH2; 272) and Boc-protected (CCK-4) tetrapeptide (Boc-Trp-Met-Asp-PheNH2; 271) (Figure 71).
Figure 71.

Bioisosteric replacement of amide linkages from parent CCK peptides 271 and 272 to develop E-alkene isostere 273, a potent CCK-B receptor ligand.
The target peptide, 273, was synthesized by incorporating an E-alkene dipeptide isostere (i.e., Boc-LeuΨ[E-CH=CH]Gly-OH) and a CCK residue using a standard peptide coupling procedure (Scheme 38). Overall, the results of this study indicated that the bioisosteric replacement of an amide bond at the Leu-Gly region of CCK-6 is acceptable for retaining the bioactive conformation required for receptor activation.279
Scheme 38.

Synthetic Route Showing the Peptide Coupling of the Dipeptide Isostere with CCK Fragment To Obtain Target Pseudopeptide 273
FLUOROALKENE BIOISOSTERES
Fluoroalkene, also known as monofluoroalkene, has generated interest as a surrogate of the amide functional group. Geometrically, both the amide and fluoroalkene are similar; for example, the length of the C=O bond of the amide is 1.228 Å and the C–N bond is 1.368 Å, whereas the C–F bond is 1.376 Å and C=C bond is 1.333 Å. Furthermore, the fluorine atom mimics the oxygen atom of the carbonyl to a certain extent because both have similar van der Waals radii (1.47 Å for fluorine and 1.52 Å for oxygen).280 The double bond of the fluoroalkene imparts rigidity to the molecule. The high electronegative fluorine atom mimics the carbonyl oxygen of the amide and thus provides fluoroalkene with a dipole moment of 1.4 D (3.6 D in the case of an amide). Finally, the fluoroalkene isostere could serve as a weak hydrogen bond acceptor to make either intermolecular or intramolecular hydrogen bonds. Furthermore, the (Z)-fluoroalkene mimics the s-trans amide bond, whereas the (E)-fluoroalkene mimics the s-cis form, and it could be explained because no equilibrium exists between them. Also, the fluoroalkene moiety is more lipophilic than an amide and therefore could be potentially used as a metabolically stable amide bioisostere.280 In this context, several investigators have shown that the replacement of an amide bond with the fluoroalkene moiety produced superior metabolic stability, lipophilicity, and binding selectivity.
The search for a selective DOPr agonist is of interest for the development of novel medications to treat chronic pain, inflammation, and cancer pain. Compound 238 (Tyr1-Gly2-Gly3-Phe4-Leu6) is a pentapeptide that is an endogenous ligand for the δ opioid receptor, but its therapeutic application is limited due to its extremely poor selectivity with respect to the μ opioid receptor and hydrophilic character.281 Also, the in vivo administration of 238 is problematic due to poor PK properties, including the rapid cleavage of Tyr1-Gly2 by aminopeptidase N in human plasma (t1/2 = 0.69 min) and Gly3-Phe4 by the angiotensin-converting enzyme present in the blood-brain barrier.282,283 To improve these pharmacological attributes of 238, Altman and colleagues tactfully replaced the amide linkage of the Tyr1-Gly2 with a fluoroalkene (275) and trifluoroethylamine moiety to yield compounds (S)-276 and (R)-277, respectively (Figure 72).284,285 The key synthetic steps to generate a fluoroalkene isostere in compound 275 are shown in Scheme 39.
Figure 72.
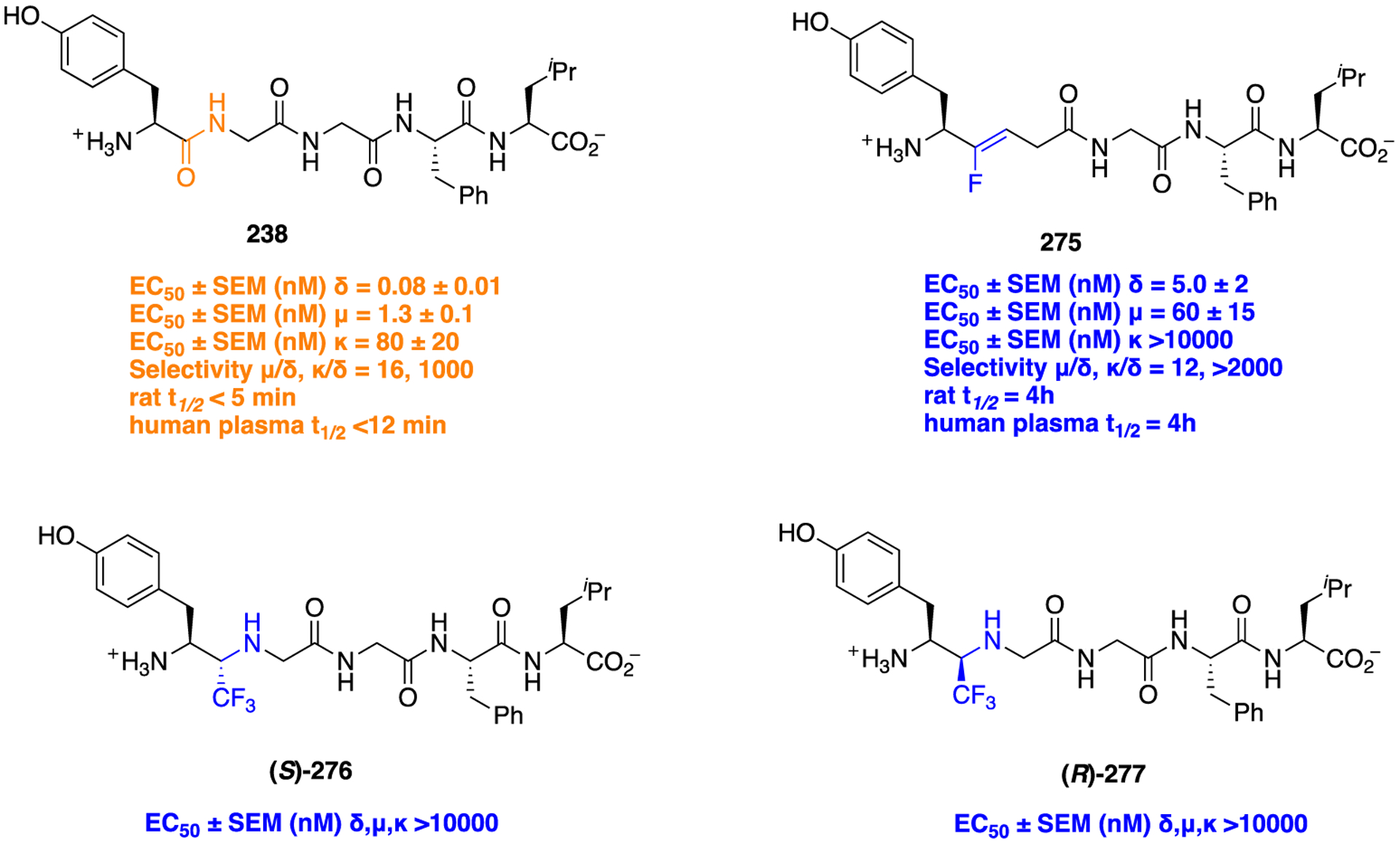
Bioisosteric replacement of an amide bond at the Tyr1-Gly2 position in 238 with a fluoroalkene to produce 275 and a trifluoroethylamine moiety to produce (S)-276 and (R)-277.
Scheme 39.
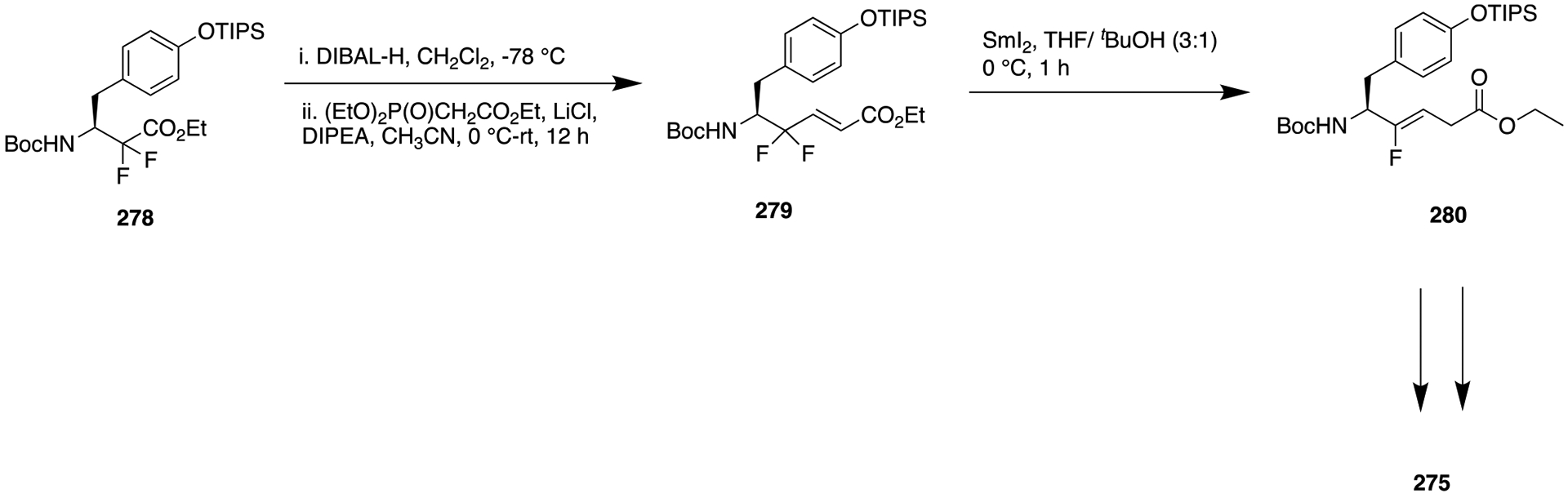
Synthetic Route To Access Leu-enkephalin Analog 275
The results indicated that compound 275 activated δ-receptors stably expressed in CHO cells (EC50 = 5 nM), whereas the progenitor 238 had a 60-fold higher activity (EC50 = 0.08 nM). However, 275 had a similar selectivity for μ versus δ as the parent peptide 238. Most importantly, 275 had excellent stability in both Sprague-Dawley rat (76% remaining of 275 at 4 h versus t1/2 < 5 min for 238) and human plasma (68% remaining of 275 at 4 h versus t1/2 < 12 min for 238) compared to 238. Furthermore, 275 had improved in vivo distribution in a BALB/c mouse model with 50 mg/kg intravenous dose and the plasma concentrations of 275 were detected for 2 h, whereas 238 was completely metabolized within 5 min. Also, 275 was orally bioavailable and plasma concentrations were detected up to 30 min after an oral dose of 400 mg/kg in the BALB/c mouse model.
These results suggested that 275 has significantly improved in vivo distribution (in both plasma and CNS), which was negligible for 238. This improvement of 275 over 238 could be explained by the σ-electron withdrawing effects of the fluorine atom, which decreased the basicity of the neighboring functional groups, making them less prone to hydrogen bond with H2O, as well as the catalytic domains of the metabolizing enzymes. Neither (S)-276 nor (R)-277 were active at δ or μ receptors and therefore were not assessed for stability, metabolism, and distribution.
In another study by a different research group, the bioisosteric replacement of the Gly3-Phe4 amide bond was conducted by Nadon et al.286 Previously, these authors systematically replaced the Gly3-Phe4 amide linkage with a thioamide (Ki = 47 ± 17 nM)287 and ester bioisostere (Ki= 34 ± 17 nM) (Figure 73).260 These isosteres retained δ-opioid receptor activity, which suggested that a hydrogen bond donor at this site of 238 was not essential, and this led to the design of a fluoroalkene bioisostere (283). This compound was obtained by first synthesizing fmoc-protected benzyl fluoro-pentenoic acid intermediate (286) which was later used in solid-phase synthesis (SPPS) utilizing 2-chlorotrityl chloride resin (CTS). The synthetic route of 283 is shown in Scheme 40.
Figure 73.
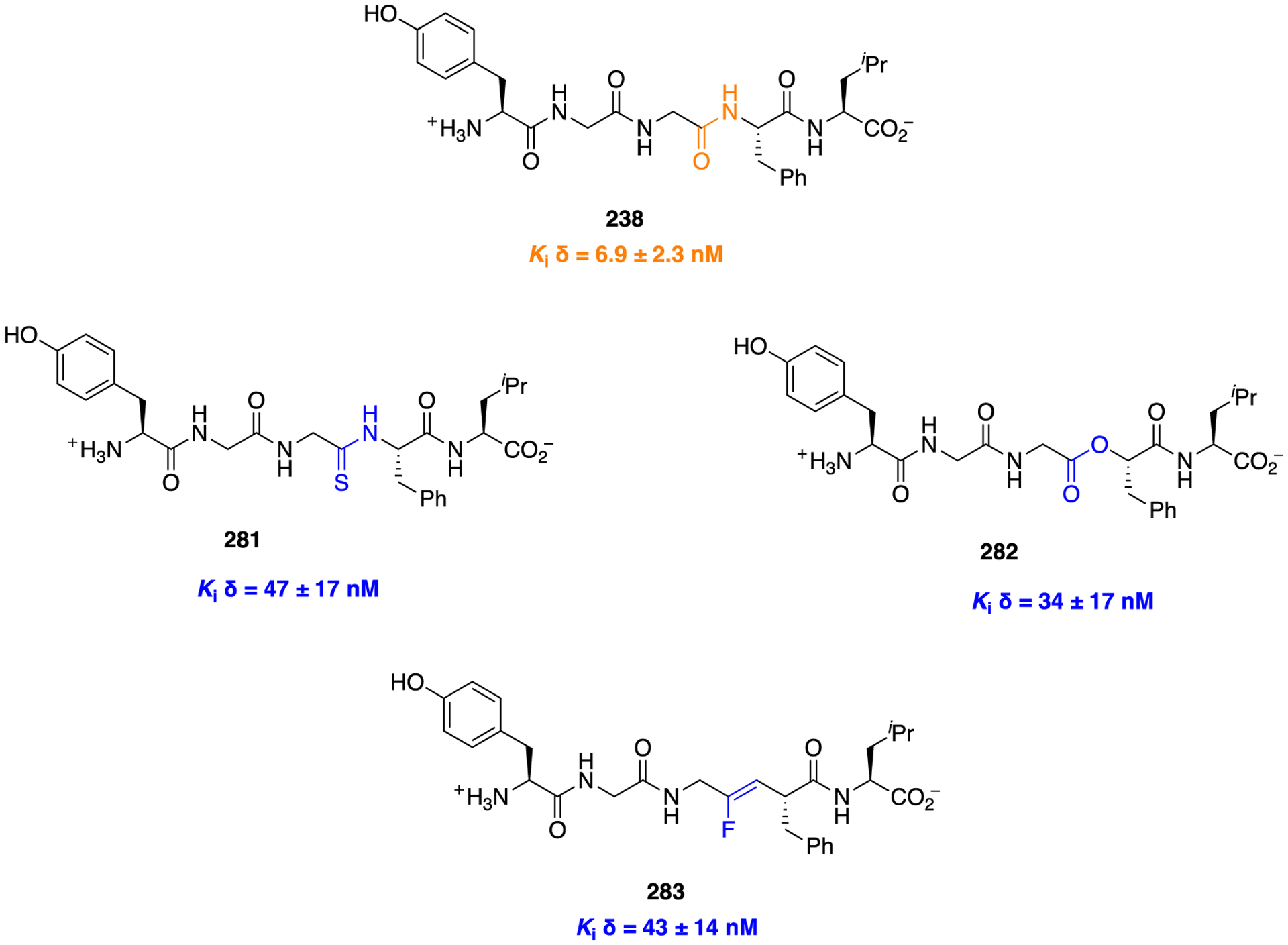
Structures of the parent peptide 238 and its fluoroalkene isostere 283.
Scheme 40.

Synthetic Pathway To Obtain the Fluoroalkene Peptidomimetic Compound 283
The δ-opioid receptor binding affinity of 238 and 283 was calculated by competitive binding assays using iodinated deltorphin I ([125I]-deltorphin I) in membrane preparations obtained from HEK293 cells expressing a FLAG-tagged version of the mouse δ-opioid receptor. The results indicated that 283 had a high affinity for δ-opioid receptor (Ki = 43 ± 14 nM), although its affinity was lower than the parent peptide 238 (Ki = 6.9 ± 2.3 nM) (Figure 73). However, its affinity for the δ-opioid receptor was significantly greater than the trans-alkene isostere (Ki = 587 ± 19 nM).288 This weak activity of the trans-alkene isostere highlighted the significant role of the fluorine atom as an effective functional mimicry of the hydrogen bond accepting feature of the amide.
Daclatasvir (formerly known as BMS-790052) (290) (Figure 74) is a small molecule inhibitor of HCV NS5A that has potent in vitro anti-HCV efficacy in the picomolar range in HCV replicons from various genotypes.289 In order to obtain an improved structural understanding of this chemotype template for designing novel NS5A inhibitors, Chang and co-workers synthesized fluoro-olefin-based structures, as they hypothesized they may provide a new peptidomimetic approach for developing potent and structurally unique inhibitors of HCV NS5A.290 The intermolecular hydrogen bonded amide group in 290 was successfully replaced with a fluoro-olefin motif to obtain compound 291, and the synthetic route is depicted in Scheme 41.
Figure 74.

Bioisosteric replacement of the amide function in daclatasvir, 290, with a fluoro-olefin moiety to yield 291.
Scheme 41.
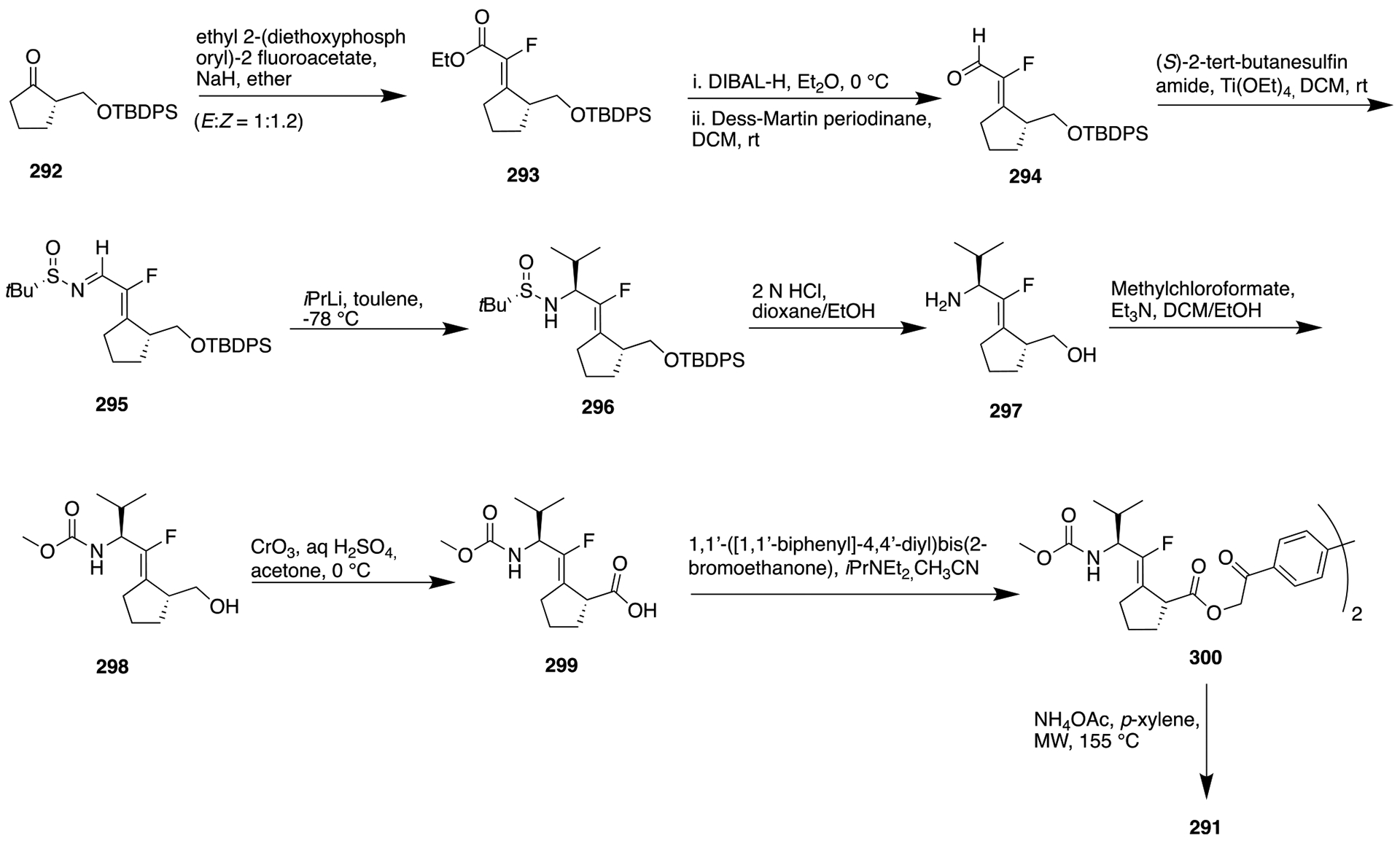
Diastereoselective Synthesis of Fluoro-olefin Analog 291 by Chang and Co-workers
The anti-HCV efficacy was measured in a clone A replicon whole cell assay, and the results indicated that 291 inhibited HCV genotype 1b replication in the picomolar range (EC50 = 79 pM), similar to the parent peptide 290 (Figure 74). Furthermore, the computational modeling studies indicated that this fluoro-olefin isostere favored a γ-turn substructure which is necessary for the interaction with the NS5A protein. Finally, 291 did not have any cytotoxicity up to the highest concentration tested (CC50 > 10 μM).
MISCELLANEOUS AMIDE BIOISOSTERES
The signaling of RhoaA/C is important for metastases and invasion of several cancers. Knockout and siRNA data have revealed an important in vivo role for RhoC in breast cancer metastasis as well as for megakaryoblastic leukemia transcriptional coactivator proteins (MKL1) and serum response factor (SRF) in breast and cancer metastases. These results reveal that Rho signaling and, more specifically, Rho-regulated gene transcription can be targets for cancer.291 Evelyn and colleagues identified compound 301 (Figure 75), which blocks SRE-luciferase gene transcription when RhoA and RhoC signaling pathways are activated. Compound 301 potently inhibited LPA-induced DNA synthesis in PC-3 cells (<1 μM). Additionally, it inhibited the growth of RhoC-overexpressing melanoma cells at nanomolar concentrations but was less active in cell lines expressing lower levels of RhoC. Compound 301 inhibited spontaneous PC-3 prostate cancer cell Rho-dependent invasion through a Matrigel matrix but failed to do so in vitro in a Gαi-dependent lysophosphatidic acid (LPA-stimulated) SKOV-3 ovarian cancer cell invasion model. On the basis of these favorable results on cancer cell function, it was decided that 301 would be a promising lead. Compound 301 also showed acute cellular toxicity toward PC-3 cells at 24 h and consequently decrease in cell viability.
Figure 75.
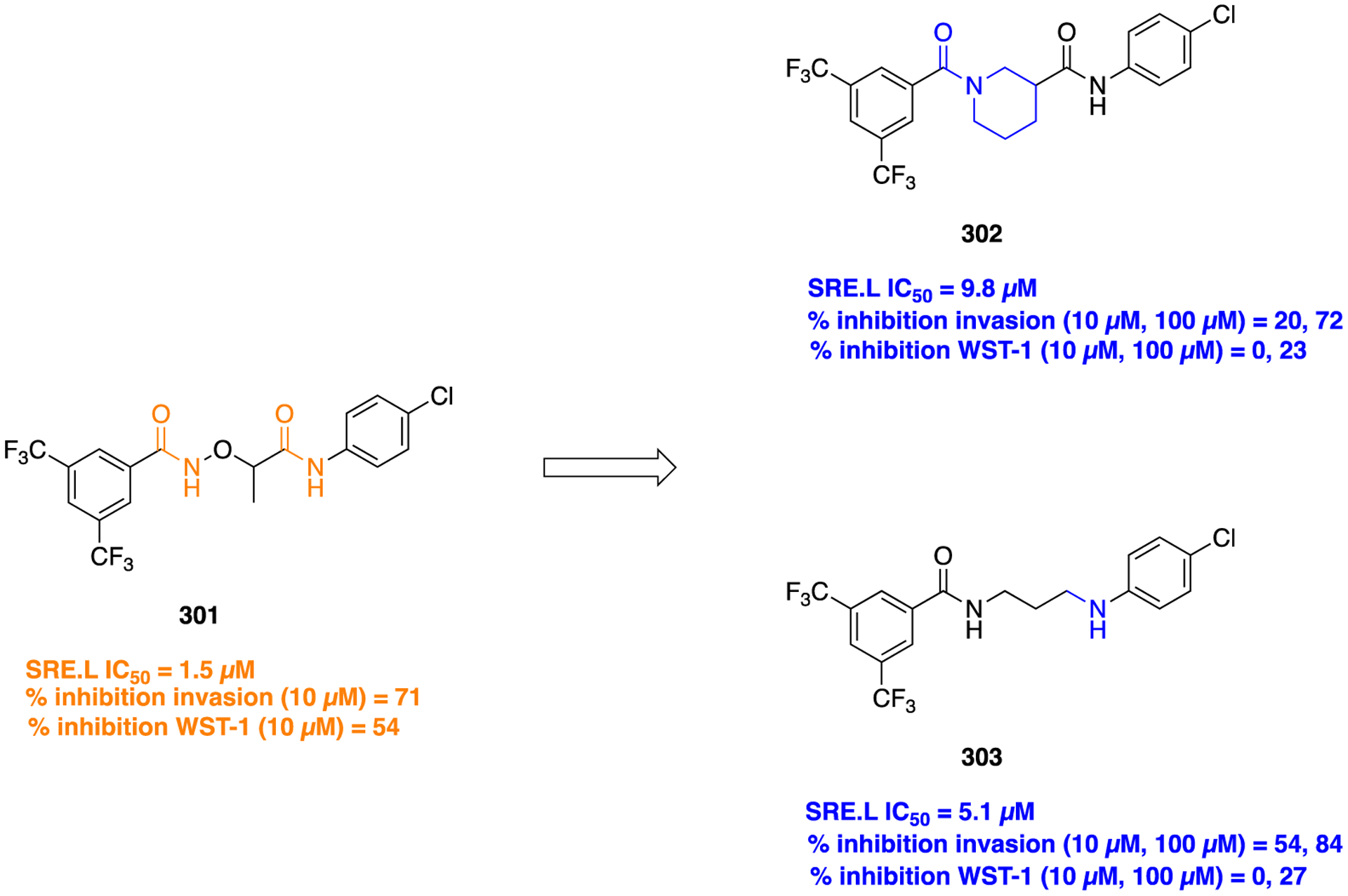
Inhibitor of Rho/MLK1 transcriptional pathway 301 and analogs with amide bioisosteric replacements 302 and 303.
Therefore, the next goal was to make modifications in this initial lead to improve potency and or selectivity while diminishing its cytotoxicity. The three areas of concern from this initial lead were the nitrogen-oxygen bond between the two carboxamides being susceptible to cleavage by thiols, the two carboxamides limiting potency and interfering with cell penetration, and the two aromatic rings not being optimal motifs for potency and selectivity. The initial strategy to modify these areas included removing the nitrogen-oxygen bond and employing a bioisosteric replacement of the amides, and conformational restriction imposed between the two aromatic rings. Making changes on the tether between the two carboxamides of 301 was first evaluated. Results indicated that removing the methyl group led to little effect on activity or selectivity. Replacing the oxygen atom with a carbon atom eliminated acute toxicity even at higher doses. Next, modifications were made on the secondary amides. When the hydrogen bond donor was removed to improve cell permeability, this gave rise to a dramatic loss in activity. When the anilides were replaced, it had a detrimental effect, but reducing the amides to amines was more successful. A monoamine (303) (Figure 75) retained moderate selectivity with little cytotoxicity compared to another monoamine and diamine analogs. The bioisosteric replacement of the amide with triazoles, thiazole, and oxazole led to completely inactive compounds indicating “traditional” heterocylic amide bioisosteres would be ineffective.
Several analogs were tested for their ability to inhibit PC-3 prostate cancer cell invasion in a Matrigel matrix (% inhibition invasion at 10 and 100 μM) as well as their ability to inhibit mitochondrial metabolism when using WST-1 (% inhibition WST-1 at 10 and 100 μM) in comparison to 301 (Figure 75). All conformationally restricted analogs had equivalent efficacies against SRE.L at 10 and 100 μM and had similar activity in a Matrigel assay. Compound 302 was slightly less toxic at 100 μM (23%) with an efficacy:toxicity profile at 100 μM higher than that of 301 (Figure 75). The monoamine bioisostere 305 was synthesized by alkylation of 4-chloroaniline with 3-bromopropylamine which led to compound 303 over several steps (Scheme 42). Compound 303 had the best efficacy:cytotoxicity ratio in transfected cell SRE-luciferase studies. Additionally, it inhibits invasion at 10 μM (54%), similar to that of 301 (71%), with no apparent toxicity. At 100 μM concentration, there is almost complete inhibition of invasion (84%) with less toxicity (27%) compared to 301 at 10 μM (54%). Overall the nipecotic amide 302 and monamine 303 were able to inhibit Gα12QL-stimulated SRE.L-luciferase expression with efficacy equal to 301 but with reduced cytotoxicity. These studies revealed the correlation between inhibition of Rho-mediated gene expression to cell invasion in a Matrigel matrix model of metastasis.291
Scheme 42.

Synthetic Access To Amide Bioisostere Analog 303 of a Rho/MLK1 Transcriptional Pathway Inhibitor 301
A thiophenecarboxylic acid series is one of several chemical classes that has been reported to inhibit the function of NS5B in genotype la and lb HCV at an allosteric thumb site near the active site.292 Exploration of this series began with 3-arylsulfonamidothiophene-2-carboxylic acids and then proceeded to the application of aromatic and heteroaromatic amide isostere analogs. The problem with these amide bioisosteres was that they resulted in weaker enzyme inhibitors with reduced cellular potency. Therefore, Canales and colleagues turned to other structural motifs in order to identify novel NS5B inhibitors of the thiophenecarboxylic acid series.292 They decided to use substituted N-acylhydrazines as tertiary amide replacements. Similar to amides, N-acylhydrazines are conformationally restricted due to resonance between the α-nitrogen and the carbonyl. As a result, trisubstituted N-acylhydrazines were synthesized, and a direct hydrazine analog (compound 307) of the protypical amide 306 (IC50 = 0.086 μM) (Figure 76) led to a compound with similar NS5B inhibitory potency (IC50 = 0.088 μM). A cocrystal structure of this direct hydrazine analog bound to NS5B proved the conformational similarity of the N-acylhydrazine to the amide. Additionally, the physiochemical properties were not significantly changed with the N-acylhydrazine replacement. It was then determined that the N-acylhydrazine was a reasonable tertiary amide bioisostere, thus resulting in the evaluation of a range of Nβ substituents having increased steric bulk to further rigidify the orthogonal conformation along with adding polar substituents in order to engage the solvent-exposed region next to the binding pocket. Linear and cyclic alcohols resulted in similar binding affinities to that of 306. Nonaromatic heterocycles gave rise to excellent binding affinities; however, inhibition of enzymatic activity was lower for these analogs in comparison to 306. The synthetic method for a 4-Me-piperidinyl analog 308 is shown in Scheme 43. In vitro evaluation of compound 308 (IC50 = 0.063 μM) (Figure 76) showed significant improvement in cellular activity for both genotypes. This study showed that trisubstituted acylhydrazines could serve as tertiary amide bioisosteres.
Figure 76.
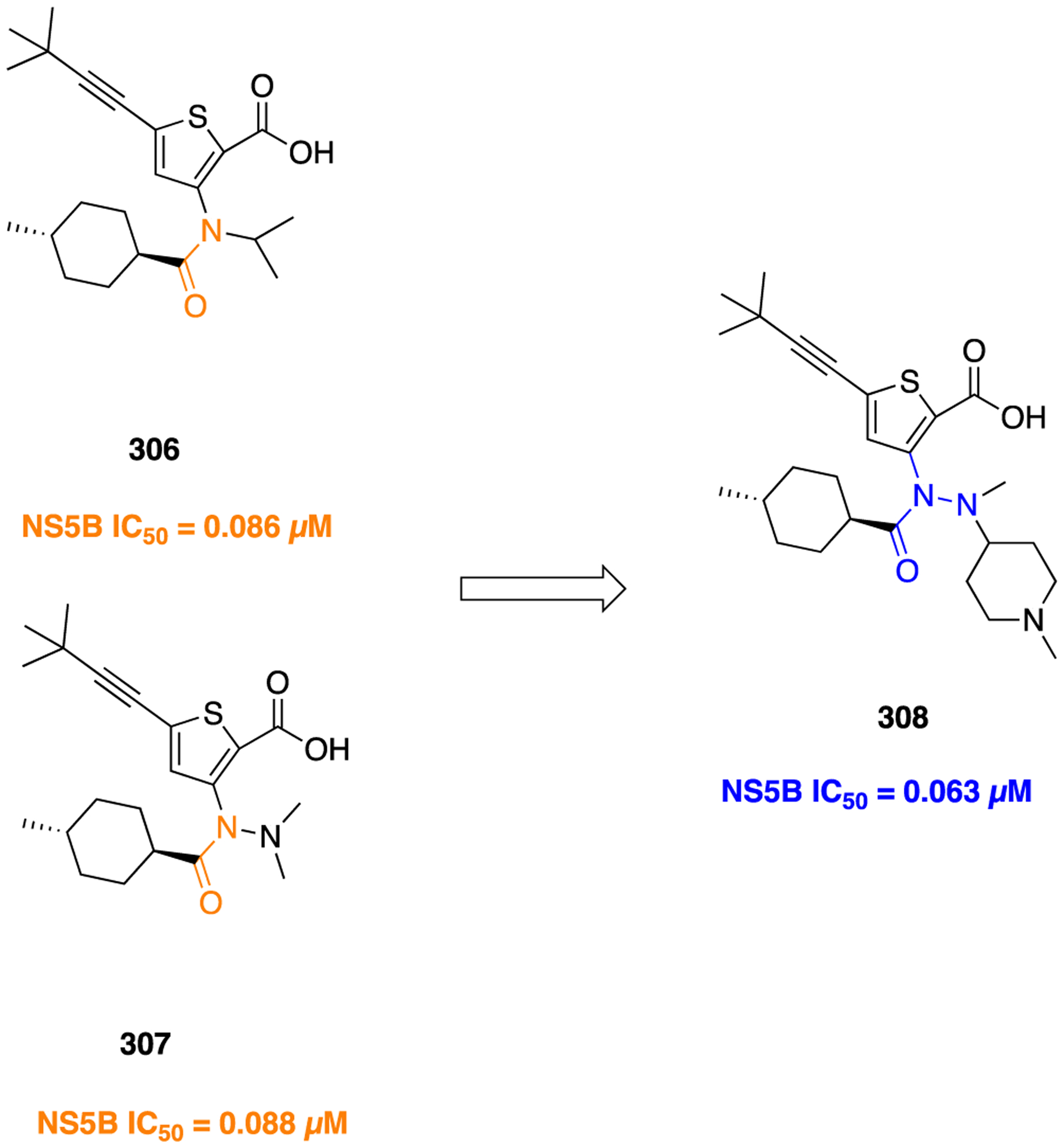
Trisubstituted acylhydrazine as a tertiary amide bioisostere.
Scheme 43.
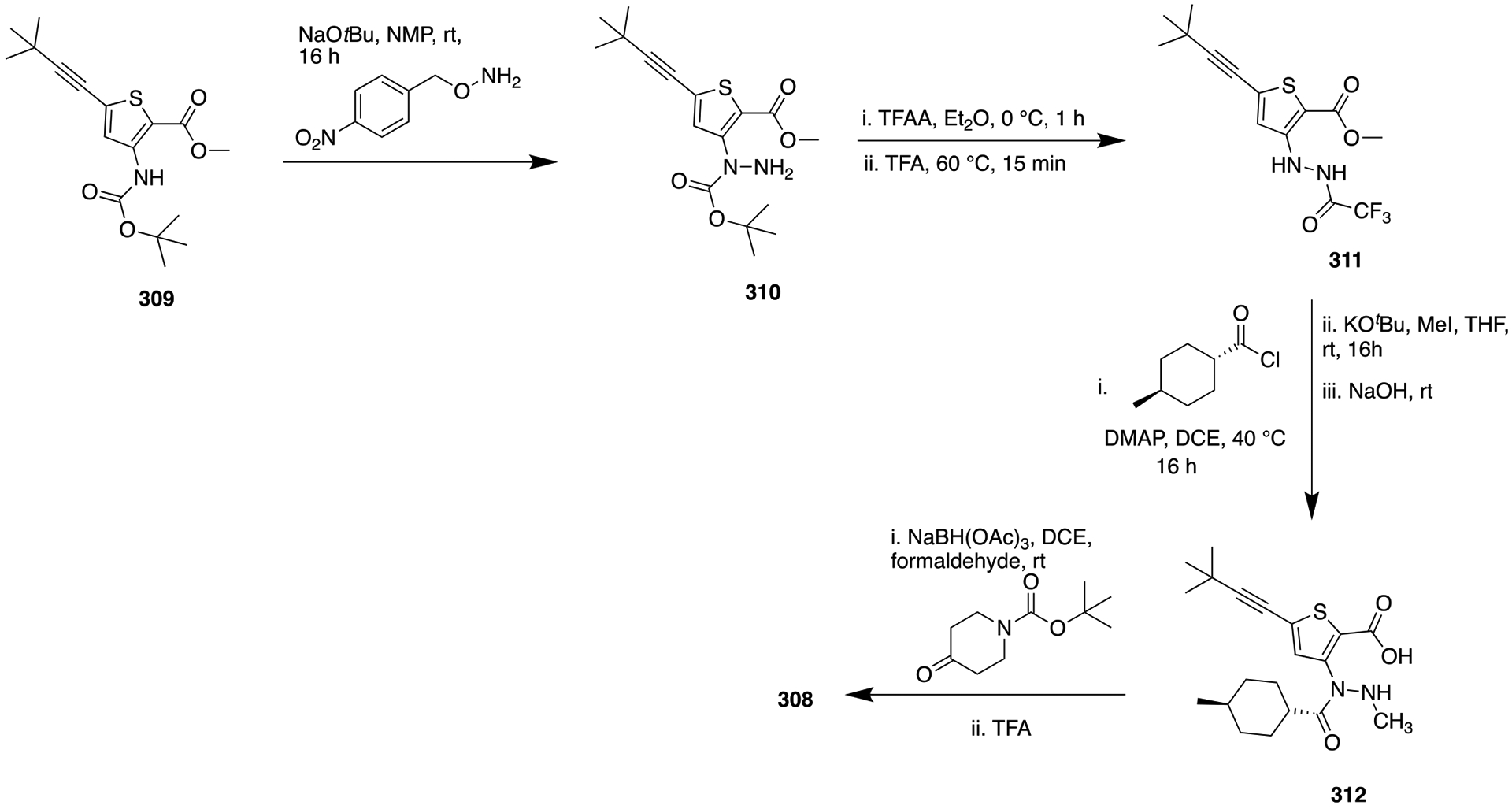
Synthetic Route To Access 308, a HCV NS5B Thumb Site II Inhibitor
CONCLUSION
The amide bond, beyond its critical function in protein structures, is most often encountered in hit, lead, and candidate compounds for therapeutic discovery across a wide range of disease indications. The resonance structure of the amide group gives way to restricted configuration engendering trans geometry of substituents, hydrogen bond accepting ability of the oxygen but not the nitrogen atom and hydrogen bond donating ability (if the nitrogen is not trisubstituted). These collective features provide for the often-critical role the amide plays in the pharmacophore or auxophore of bioactive compounds, providing either target binding interactions, positioning of bioactive configuration, or both. Successful bioisosteres of the amide bond are required to closely mimic these features while overcoming the Achilles heel of the group: its metabolic lability. Thus, amide bioisosteres tend to see employment in lead compound optimization to improve absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties.
Bioisosteres, by nature of their differing size, shape, and hydrogen bond donating and accepting profile, can convey additional target binding interactions to enhance (or degrade) activity depending on the interactions the amide bond makes with the target protein. However, this is often seen in combination with extensive SAR studies guided by in silico approaches that is often not directly attributed to the bioisostere itself but more a combination of differing substituents in differing configuration. While many potential bioisosteres of the amide group have been evaluated, there are a select few, summarized herein (Table 2), that provide a blend of similar hydrogen bonding pattern, restricted rotation, lipophilicity, and metabolic stability. For example, the ethane moiety has been employed as a bioisostere of the amide group that provides for significant metabolic stability but fails to mimic the hydrogen bonding, configurational, and hydrophilic character of the amide. Thus, ethane’s use is limited to compounds wherein the amide is not present in the pharmacophore or auxophore. The ability of amide bioisosteres to confer additional properties beyond PK/PD modulation, such as engendering blood-brain barrier penetration, has been reported (see methylamide 240 as an example), although it is worth noting that this is not an inherent property of any bioisostere to date but more a design feature to remove penetration hindering molecular features of the amide bond, such as hydrogen bond donors and charged acids generated by metabolic cleavage, or simply to increase lipophilicity, three strategies widely employed in CNS drug discovery.293
Table 2.
Summary of the Most Common Amide Bond Bioisosteres Discussed Herein, Their Physicochemical Properties, and Representative Examples of Pharmacokinetic Effectsa
| Bioisostere | Physicochemical Properties Relative to Amide Bond | Representative Examples1,2 | Design Considerations |
|---|---|---|---|
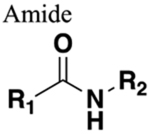 |
Naturally occupies trans configuration. Substituent distance = 3.8–3.9 Å. Dipole moment = approx. 4 Debye. Functions as HBD and HBA. Susceptible to cleavage mediated by proteases, oxidation and hydrolysis (rate of cleavage depends on steric environment). |
||
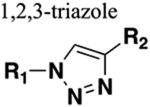 |
Mimics trans configuration upon 1,4-disubstitution (can mimic cis configuration upon 1,5-disubstitution). Substituent distance = 5.0–5.1 Å. Dipole moment = approx. 5 Debye. Functions as HBD and HBA. Resistant to cleavage mediated by proteases, oxidation and hydrolysis. |
Removal of reactive metabolites but substantial decrease in human half-life (compare amide 30 to triazole bioisostere 31). Three-fold increase in metabolic stability (compare amide 20 with triazole bioisostere 23) |
Increased sterics. If carbonyl of amide is a hydrogen bond acceptor the orientation of the nitrogens within the triazole alter distance and angle between donor and acceptor. |
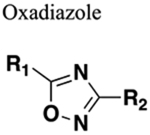 |
Weak base. Functions as HBA only. Electrophilic substitution at carbon difficult. Electrophilic substitution at nitrogen possible with electron-donating group substituents. Resistant to nucleophilic attack but undergo nucleophilic substitution. Generally resistant to cleavage mediated by proteases, oxidation and hydrolysis. |
Two-three-fold increase in clearance in a rat model (compare amide 43 to oxadiazole bioisostere 45). Reduced metabolic stability (compare amide 47 to oxadiazole bioisosteres 48 and 49). |
Increased sterics. If amine of amide is a hydrogen bond donor this will be lost upon replacement. |
 |
Mimics trans configuration. Functions as HBD (if nitrogen unsubstituted) and HBA. Mimics charge of amide nitrogen. Generally resistant to cleavage mediated by proteases, oxidation and hydrolysis. |
Increased bioavailability (rat) (compare amide 72 to imidazole bioisostere 74). Retained bite angle and N-partial charge (compare amide 81 to imidazole bioisostere 82). |
Increased sterics. If carbonyl of amide accepts two hydrogen bonds one will be lost upon replacement. |
 |
Functions as HBA only. Can mimic trans or cis configuration depending on substitution pattern. Generally resistant to cleavage mediated by proteases, oxidation and hydrolysis. |
102 | Increased sterics. If carbonyl of amide is a hydrogen bond acceptor or the amine a hydrogen bond donor these will be lost upon replacement. |
 |
Functions as a HBA and HBD (if nitrogen unsubstituted). Distance between substituents greater than in amide. Generally resistant to cleavage mediated by proteases, oxidation and hydrolysis. |
106 | Increased sterics. Increased lipophilicity. If carbonyl of amide is a hydrogen bond acceptor or the amine a hydrogen bond donor these will be lost upon replacement. |
 |
Functions as HBA only. Distance between substituents greater than in amide. Generally resistant to cleavage mediated by proteases, oxidation and hydrolysis. |
111 | Increased sterics. If carbonyl of amide is a hydrogen bond acceptor or the amine a hydrogen bond donor these will be lost upon replacement. |
 |
Bioisosteric with aryl amide. Much larger. HBD if nitrogen not substituted. Not a HBA as lone pair electrons involved in aromaticity. |
40-fold increase in half-life and seven-fold increase in %F (compare amide 113 with 3-acyl indole bioisostere 114). | Increased sterics. Increased lipophilicity. If carbonyl of amide is a hydrogen bond acceptor this will be lost upon replacement. |
 |
Generally only bioisosteric with terminal amides. HBA only. Much larger size. Greater rigidity. Greater rigidity. Generally resistant to cleavage mediated by proteases, oxidation and hydrolysis. Provide locking of substituents into bioactive configuration. |
120 Increase in target selectivity (compare amide 125 to constrained bioisostere 127). |
Increased sterics. Increased charge. If carbonyl of amide accepts two hydrogen bonds one will be lost upon replacement. Increased sterics. |
 |
Greater rigidity. Generally resistant to cleavage mediated by proteases, oxidation and hydrolysis. Provide locking of substituents into bioactive configuration. Ability to mimic β-turns. |
134 | Increased sterics. Chain length is increased by a methylene. If amine of amide is a hydrogen bond donor this will be lost upon replacement. Addition of hydrogen bond donor and acceptors maybe advantageous or deleterious to binding. |
 |
Very similar properties to amide but reversed orientation of HBD and HBA. | 139 | No improvement of PK properties. |
 |
Additional HBA and HBDs. Increases solubility over equally substituted amide, especially when non-symmetrical. Extends chain length by one nitrogen group. |
148 | Increased length by one atom. |
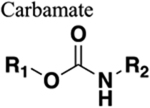 |
Functions as HBD (if nitrogen unsubstituted) and HBA. Some degree of rigidity. Undergo rapid hydrolysis. |
Exceptional long half-life example 180. Reduced clearance (human microsomes) (compare amide 182 to carbamate bioisostere 183). |
Little to no improvement in PK. |
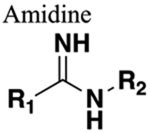 |
One additional HBD (if nitrogen not substituted). Greater charge. Similar distance and rotational constriction. |
Reduced plasma clearance but limited improvement of %F (amidine bioisostere 188). | Increased charged and likely reduction of BBB penetration. |
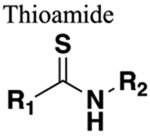 |
Thiocarbonyl bond is longer than carbonyl. C-N bond in thioamide is shorter. Increased rotational constriction. Weak HBA, strong HBD. |
192 | Longer C-S bond may induce steric clash. |
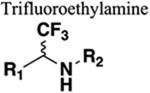 |
Reduced basicity of NH, but can HBD. Similar bond angle (120°). C-CF3 is isopolar with carbonyl group. Increased lipophilicity. Stereochemistry can be manipulated to enhance target binding interactions. |
197 | Steric effects of CF3 compared with O. If carbonyl of amide is a hydrogen bond acceptor this will be diminished or lost entirely upon replacement. |
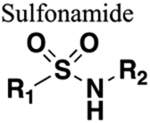 |
Increased water solubility. Additional HBAs. No increase in chain length. |
214 | Increased steric bulk and chain length. Increased water solubility. |
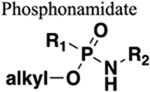 |
Increased water solubility and charge. Stereochemistry may increase (or decrease) interactions with target binding site. Additional HBAs. Metabolically labile. |
233 | Increased charge. Reduced cell permeability. Metabolically labile to different enzymes than amides. |
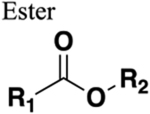 |
Decreased metabolic stability. Can mimic cis or trans conformation. Cannot HBD, weak HBA. |
>Three-fold decrease of half-life (compare amide 239 with ester bioisostere 240). | Deleterious to metabolic stability |
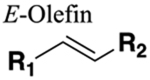 |
Mimic geometry of trans amides. No HBAs or HBDs. Dipole moment much lower (0.1 Debye). Resistant to cleavage mediated by proteases, oxidation and hydrolysis. |
Significantly increased bioavailability (compare amide 241 with E-olefin bioisostere 242). Significantly increased half-life (compare amide 244 with E-olefin bioisostere 245). |
Geometric mimic only. If amide bond is engaged in binding interactions these will be lost upon replacement. |
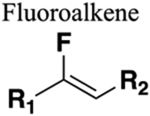 |
(Z)-fluoroalkene mimics the s-trans amide. (E)-fluoroalkene mimics the s-cis amide. Length of C-F bond (1.376 Å) is similar to carbonyl (1.228 Å). C-N and C=C bond length both approx. 1.3 Å. Fluorine has similar van der Waals radius to oxygen. Lower dipole moment (1.4 Debye). Resistant to cleavage mediated by proteases, oxidation and hydrolysis. Increased lipophilicity. |
Significant increase in half-life (>20-fold) in human plasma (compare amide 238 to fluoroalkene bioisostere 275). | Geometric mimic. If carbonyl of amide is a hydrogen-bond acceptor or the amine a hydrogen bond donor these will be lost upon replacement. |
 |
Functions as a HBD and acceptor. Free rotation around carbon and nitrogen. Same chain length. |
303 | Increased charge (depending on R2). If carbonyl of amide forms hydrogen bonds these are lost upon replacement. |
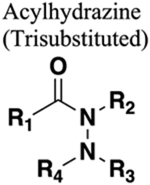 |
Trisubstituted acylhydrazine is bioisosteric to tertiary amide. Similar conformation. Functions as a HBA only. Increased chain length by one nitrogen. Less restricted rotation around N-N bond compared with amide. |
308 | Increased sterics. Increased chain length. If nitrogen of amide is a hydrogen bond donator this will be lost upon replacement. |
 |
Influences basicity of proximal amine. Generally resistant to cleavage mediated by proteases, oxidation and hydrolysis. May increase steric bulk without increasing lipophilicity.294 Decreases planarity. |
Increases metabolic stability. Little details on effect on physicochemical, biological or PIOPD properties. For an overview see reference.295 |
Increased sterics. Loss of trans geometry. |
Footnotes:
Improvements to potency and selectivity are generally excluded as they are most likely due to additional substituent modifications and/or increased target binding and cannot necessarily be attributed to the bioisostere alone.
Comparisons are provided for compounds with the same or very similar substituents to the bioisostere where possible.
Novel bioisoteres continue to be developed with new heterocyclic rings and inorganic elements being investigated for their suitability as amide bioisosteres. The aminooxetane moiety, wherein the amine is bound directly to the heterocycle, imparts increased metabolic and chemical stability to peptidomimetic derivatives; however, the impact of this novel bioisostere on biological activity remains to be fully elucidated.296,297 The loss of the rigidity of the amide bond and the increased separation of the oxygen from the backbone structure may have critical roles to play in retaining activity. Additionally, the oxetane ring has been utilized as a bioisostere for the methylene and gem-dimethyl groups16 and carboxylic acids,298 which may complicate its effects as a bioisostere for the amide group. The synthesis of oxetane-containing diketopiperazine bioisosteres, wherein the oxetane (or an azetidine) ring replaces the constrained amide carbonyl constituent, thus affording a spirocyclic system and combining the beneficial effects of two amide bioisosteres to maximize metabolic protection, has been reported.299 However, no biological activity associated with these structures has been published to the best of our knowledge, making the suitability of this spirocycle as a bioisostere of the amide bond unclear. In both cases the ability of the oxetane ring to introduce three-dimensionality over the two-dimensionality of a carbonyl benefits in moving away from planar “drug-like” structures and into more novel chemical space.300
Boron-containing structures continue to gain traction in medicinal chemistry.301,302 The boronamide structure B(OH)-NH is a theoretical bioisostere for the amide moiety. Initial modeling studies of energy barriers to rotation performed on N-methyl methylboronamide illustrate the double bond character of the boron-nitrogen bond and single bond character of the B–O bond mimicking the natural amide bond.303 However, given synthetic challenges in accessing this particular functional group with the desired variety of substituents, the theory, to the best of our knowledge, has not been reduced to practice.
Silicon is gaining attention in medicinal chemistry for the role it can play in expanding chemical space.304 The reported use of silanols to mimic the transition state of protease inhibitors305 and to act as a biosiostere of terminal N-(hydroxy)amides in histone deacetylases (HDAC) inhibitors (albeit with reduced activity)306 is pointing the way for the direct investigation of silanols as amide bioisosteres. The pioneering work of Chris McGuigan in the development of phosphonamidate protide technology307 has led to a large amount of literature on the synthesis and biological stability of these moieties. These data can perhaps be mined to discover potential improvements on the traditional C–P bond-containing phosphonamidite bioisostere described above or to introduce the phosphonamidate moiety as an amide bioisostere itself, albeit with intracellular metabolic lability.
These few selected examples provide a glimpse of emerging novel amide bioisosteres that are under evaluation for adding to the medicinal chemist’s toolbox. The examples illustrated in this Perspective were chosen to highlight the power of amide bond bioisosteres in hit-to-lead and lead optimization to preserve, or enhance, activity and selectivity and to enhance PK/PD properties. Included are many examples of clinical candidates and clinically approved drugs reflecting the importance of this approach to drug discovery.
ACKNOWLEDGMENTS
We thank the National Cancer Institute of the National Institutes of Health under Award R01CA226436 and the National Institute for Neurological Disorders and Stroke of the National Institutes of Health under Award R01NS106879 and the University of Nebraska Medical Center for funding support. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATIONS USED
- Aβ
amyloid-β
- A2A
adenosine A2A
- A549
human lung cancer
- ACAT
A:cholesterol acyltransferase
- ACE
angiotensin-converting enzyme
- ADMET
absorption, distribution, metabolism, excretion, and toxicity
- AD
Alzheimer’s disease
- AFC
N-acetyl-S-farnesyl-l-cysteine
- AFM
atomic force microscopy
- AIDS
acquired immune deficiency syndrome
- AKR1C3
aldo-keto reductase 1C3
- AML
acute myeloid leukemia
- AMFC
amide modified farnesyl cysteine analog
- ATP
adenosine triphosphate
- BBB
blood-brain barrier
- B16F10
Mus musculus skin melanoma cells
- BACE-1
β-site amyloid precursor protein cleaving enzyme 1
- BAEC
bovine aortic endothelial cell
- BCC
basal-cell carcinoma
- Bcr-Abl
break point cluster region Abelson kinase
- Caki-2
human renal cancer
- CB
cannabinoid
- CB1
cannabinoid-1
- CB2
cannabinoid-2
- CCK
cholecystokinin
- CCK-B
cholecystokinin brain
- CD
circular dichroism
- Cat K
cathepsin K
- CDI
carbonyldiimidazole
- CEM
human T-lymphocyte cell
- CGRP
calcitonin gene related peptide
- CHO
Chinese hamster ovary
- CIDV
capsaicin induced dermal vasodilation
- CML
chronic myelogenous leukemia
- CuAAC
copper(I)-catalyzed azide-alkyne cycloaddition
- CuI
copper iodide
- CNS
central nervous system
- CRL-8155
human lymphoblast cell
- CRPC
castration-resistant prostate cancer
- CTC
2-chlorotrityl chloride resin
- CyP
liver cytochrome
- D2-R
dopamine-2 subtype receptor
- D3-R
dopamine-3 subtype receptor
- DGAT
acyl CoA:diacylglycerol acyltransferase
- DKP
diketopiperazine
- DOPr
δ opioid receptor
- DPP-4
dipeptidyl peptidase-4
- DIAD
diisopropyl azodicarboxylate
- DSG
15-deoxyspergualin
- EDG
electron donating group
- ERK1/2
extracellular signal-regulated kinases
1
and
2
- EWG
electron withdrawing group
- FDA
Federal Drug Administration
- FTIR
Fourier transform infrared
- GABAA
γ-aminobutyric acid A
- GPCR
G-protein-coupled receptor
- GIST
gastrointestinal stomatal tumor
- GLP-1
glucagon-like peptide 1
- GIP
glucose-dependent insulinotropic polypeptide
- Gpp(NH)p
5′-guanylyl imidodiphosphate
- GSH
glutathione
- GT1
genotype 1
- GTL-16
human gastric carcinoma
- GVHD
graft-versus-host disease
- h
human or hour
- HATU
hexafluorophosphate azabenzotriazole tetramethyluranium
- hCB1
human cannabinoid 1
- H1437
non-small-cell lung cancer
- HAT
human African trypanosomiasis
- HBA
hydrogen bond acceptor
- HBD
hydrogen bond donor
- HCV
hepatitis C virus
- HCMV
human cytomegalovirus
- HD
Huntington’s disease
- HDAC
histone deacetylase
- HGF
hepatocyte growth factor
- Hh
Hedgehog
- hIcmt
human isoprenylcysteine carboxyl methyltransferase
- HeLa
human cervix carcinoma
- hERG
human ether-a-go-go-related gene
- HEK293
human embryonic kidney 293
- HepG2
human liver cancer
- HINF
Haemophilus influenzae 30063
- HIV
human immunodeficiency virus infection
- HMEC-1
human microvascular endothelial cells
- HL-60
human leukemia
- HLM
human liver microsome
- HT22
mouse hippocampal neuron
- HT-29
human colon cancer
- 5-HT
5-hydroxytryptamine
- HUVEC
human umbilical vein endothelial cell
- Icmt
isoprenylcysteine carboxyl methyltransferase
- ip
intraperitoneal
- iv
intravenous
- IL-12
interleukin 12
- IL-17
interleukin 17
- IL-23
interleukin 23
- INSTI
integrase strand transfer inhibitor
- JAK
Janus kinase
- KOPr
κ opioid receptor
- LA
α-lipoic acid
- L1210
murine leukemia cell
- m
mouse
- MCF10A
human mammary epithelial cell
- mGlu7-NAM
metabotropic glutamate receptor subtype-7 negative allosteric modulator
- MDCK
Madin-Darby canine kidney
- MDA-MB-231
M. D. Anderson metastasis breast adenocarcinoma
- Met
N-methyl-N′-nitrosoguanidine human osteosarcoma transforming gene
- MIC
minimal inhibitory concentration
- MKL1
megakaryoblastic leukemia transcriptional coactivator protein
- MLM
mouse liver microsome
- MMP
matrix metalloproteinase
- MOPr
μ opioid receptor
- MT4
human T-lymphoid cell
- MTR
melatonin receptor
- MT1R
melatonin receptor 1
- MT2R
melatonin receptor 2
- MW
microwave
- nAChR
neuronal nicotinic acetylcholine receptor
- NADPH
nicotinamide adenine dinucleotide phosphate
- NDA
new drug application
- NEP
metalloproteinase neutral endopeptidase
- NET
neutrophil extracellular trap
- NK-1
neurokinin-1
- NPA
[3H]spiro-peridol/N-propylnorapomorphine
- NS5B
nonstructural protein 5B
- NSAID
nonsteroidal anti-inflammatory drug
- ORTEP
Oak Ridge thermal ellipsoid plot
- PAD
protein arginine deiminase
- PAF
platelet-activating factor
- PC-3
human prostate cancer
- PD
pharmacodynamic
- PD
Parkinson’s disease
- PDHc
pyruvate dehydrogenase complex
- PDHc-E1
pyruvate dehydrogenase complex-E1component
- PK
pharmacokinetic
- PrCP
prolylcarboxypeptidase
- po
per oral
- PTM
post-translational modification
- RdRp
RNA-dependent RNA polymerase
- RTK
receptor tyrosine kinase
- SAR
structure-activity relationship
- SAUR
Staphylococcus aureus UC 9218
- SCD
stearoyl-CoA desaturase
- SCD1
stearoyl-CoA desaturase-1
- SEP1d
Staphylococcus epidermidis UC 12084
- SEPId
Staphylococcus epidermidis (UC 12084)
- SMO
smoothened
- SMFC
sulfonamide modified farnesylcysteine analog
- SP
substance P
- SRF
serum response factor
- SPPS
solid-phase synthesis
- STAT-5
signal transducer and activator of transcription 5
- T-ALL
T-cell acute lymphoblastic leukemia
- TBAF
tetrabutylammonium fluoride
- TMSA
trimethylsilyl azide
- tPSA
total polar surface area
- TRPV1
transient receptor potential cation channel subfamily V member 1
- TS
transition state
- TYK2
tyrosine kinase 2
- U2OS
PAD-4 expressing osteosarcoma bone marrow
- VEGFR2
vascular endothelial growth factor receptor-2
- VDA
vapor diffusion assay
- VLA-4
very late activating antigen-4
- Vif
viral infectivity factor
Biographies
Shikha Kumari received her B.S. from the University of Delhi, India, and perused an M.S. in Pharmaceutical Chemistry from Banasthali University, India. In 2016, she received her Ph.D. in Biomedical Sciences from ACBR, University of Delhi, India, and worked in the area of medicinal chemistry that includes design, synthesis, and development of novel heterocyclic compounds against neurological disorders and cancer. She carried out her postdoctoral research work (2016–2018) in Dr. Manisha Tiwari’s (ACBR, University of Delhi) and Professor Nasimul Hoda’s laboratories (Jamia Millia Islamia, India). In 2019, she joined Dr. Paul C. Trippier’s lab at the University of Nebraska Medical Center as a postdoctoral member and is working on the development of novel treatments for ischemic stroke and cancer.
Angelica V. Carmona obtained her B.S. in Biochemistry from Mount Saint Mary’s University Los Angeles, CA, U.S.. In 2018, she joined Dr. Paul C. Trippier’s lab at Texas Tech University Health Sciences Center and in 2019 moved to the University of Nebraska Medical Center as a Ph.D. scholar. Her current research work focuses on synthesizing novel neuroprotective compounds as potential therapeutics for the treatment of the rare pediatric disorder Batten disease.
Amit K. Tiwari obtained his B.Pharm. from the Ram-Eesh Institute of Technology, India. He received his M.S. and Ph.D. in Pharmaceutical Sciences from the St. John’s University, Queens, NY (2008 and 2011, respectively). He then carried out postdoctoral research in Professor James Gallo’s laboratory at Icahn School of Medicine (2011–2013) in the area of pharmacokinetics. He joined Tuskegee University as an Assistant Professor prior to joining the Department of Pharmacology and Experimental Therapeutics at the University of Toledo, Ohio, as an Assistant Professor in 2015. In 2019, he was promoted to Associate Professor with tenure. His research interests are in broad areas of cancer pharmacology, drug development, and medicinal chemistry.
Paul. C. Trippier gained his M.Chem. from the University of Hull (U.K.) and his D.Phil. in Synthetic Organic Chemistry from the University of Oxford under the mentorship of Professor Mark Moloney. After postdoctoral positions with Prof. Chris McGuigan (Cardiff University) and Prof. Richard Silverman (Northwestern University) he began his independent career at Texas Tech University Health Sciences Center where he was promoted to Associate Professor with tenure. In 2019, he moved to the College of Pharmacy at the University of Nebraska Medical Center where his research group investigates small molecule probe and drug discovery for cancer, ischemic stroke, and neurodegenerative diseases among other indications.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.0c00530
The authors declare no competing financial interest.
Contributor Information
Amit K. Tiwari, Department of Pharmacology and Experimental Therapeutics, College of Pharmacy and Pharmaceutical Sciences, The University of Toledo, Toledo, Ohio 43614, United States
Paul C. Trippier, Department of Pharmaceutical Sciences, College of Pharmacy, Fred & Pamela Buffett Cancer Center, and UNMC Center for Drug Discovery, University of Nebraska Medical Center, Omaha, Nebraska 68198, United States.
REFERENCES
- (1).Pattabiraman VR; Bode JW Rethinking amide bond synthesis. Nature 2011, 480, 471–479. [DOI] [PubMed] [Google Scholar]
- (2).de Figueiredo RM; Suppo JS; Campagne JM Nonclassical routes for amide bond formation. Chem. Rev 2016, 116, 12029–12122. [DOI] [PubMed] [Google Scholar]
- (3).Johansson A; Kollman P; Rothenberg S; McKelvey J Hydrogen bonding ability of the amide group. J. Am. Chem. Soc 1974, 96, 3794–3800. [Google Scholar]
- (4).Kovacs E; Rozsa B; Csomos A; Csizmadia IG; Mucsi Z Amide activation in ground and excited states. Molecules 2018, 23, 2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Scheidt K Organic chemistry: amide bonds made in reverse. Nature 2010, 465, 1020–1022. [DOI] [PubMed] [Google Scholar]
- (6).Valeur E; Bradley M Amide bond formation: beyond the myth of coupling reagents. Chem. Soc. Rev 2009, 38, 606–631. [DOI] [PubMed] [Google Scholar]
- (7).Dunetz JR; Magano J; Weisenburger GA Large-scale applications of amide coupling reagents for the synthesis of pharmaceuticals. Org. Process Res. Dev 2016, 20, 140–177. [Google Scholar]
- (8).Leggio A; Belsito EL; De Luca G; Di Gioia ML; Leotta V; Romio E; Siciliano C; Liguori A One-pot synthesis of amides from carboxylic acids activated using thionyl chloride. RSC Adv. 2016, 6, 34468–34475. [Google Scholar]
- (9).Mahjour B; Shen Y; Liu W; Cernak T A map of the amine-carboxylic acid coupling system. Nature 2020, 580, 71–75. [DOI] [PubMed] [Google Scholar]
- (10).Hefti FF Requirements for a lead compound to become a clinical candidate. BMC Neurosci. 2008, 9 (Suppl. 3), S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Patani GA; LaVoie EJ Bioisosterism: a rational approach in drug design. Chem. Rev 1996, 96, 3147–3176. [DOI] [PubMed] [Google Scholar]
- (12).Lima LM; Barreiro EJ Bioisosterism: a useful strategy for molecular modification and drug design. Curr. Med. Chem 2005, 12, 23–49. [DOI] [PubMed] [Google Scholar]
- (13).Langmuir I Isomorphism, isosterism and covalence. J. Am. Chem. Soc 1919, 41, 1543–1559. [Google Scholar]
- (14).Papadatos G; Brown N In silico applications of bioisosterism in contemporary medicinal chemistry practice. WIREs Comput. Mol. Sci 2013, 3, 339–354. [Google Scholar]
- (15).Dick A; Cocklin S Bioisosteric replacement as a tool in anti-HIV drug design. Pharmaceuticals 2020, 13, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Meanwell NA Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem 2011, 54, 2529–2591. [DOI] [PubMed] [Google Scholar]
- (17).Meanwell NA The Influence of Bioisosteres in Drug Design: Tactical Applications to Address Developability Problems In Tactics in Contemporary Drug Design; Meanwell NA, Ed.; Springer: Berlin, 2015; pp 283–381. [Google Scholar]
- (18).Herr RJ 5-Substituted-1H-tetrazoles as carboxylic acid isosteres: medicinal chemistry and synthetic methods. Bioorg. Med. Chem 2002, 10, 3379–3393. [DOI] [PubMed] [Google Scholar]
- (19).Bonandi E; Christodoulou MS; Fumagalli G; Perdicchia D; Rastelli G; Passarella D The 1,2,3-triazole ring as a bioisostere in medicinal chemistry. Drug Discovery Today 2017, 22, 1572–1581. [DOI] [PubMed] [Google Scholar]
- (20).Lenci E; Trabocchi A Peptidomimetic toolbox for drug discovery. Chem. Soc. Rev 2020, 49, 3262–3277. [DOI] [PubMed] [Google Scholar]
- (21).Fosgerau K; Hoffmann T Peptide therapeutics: current status and future directions. Drug Discovery Today 2015, 20, 122–128. [DOI] [PubMed] [Google Scholar]
- (22).Henninot A; Collins JC; Nuss JM The current state of peptide drug discovery: back to the future? J. Med. Chem 2018, 61, 1382–1414. [DOI] [PubMed] [Google Scholar]
- (23).Lau JL; Dunn MK Therapeutic peptides: historical perspectives, current development trends, and future directions. Bioorg. Med. Chem 2018, 26, 2700–2707. [DOI] [PubMed] [Google Scholar]
- (24).Lee AC; Harris JL; Khanna KK; Hong JH A comprehensive review on current advances in peptide drug development and design. Int. J. Mol. Sci 2019, 20, 2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).He R; Finan B; Mayer JP; DiMarchi RD Peptide conjugates with small molecules designed to enhance efficacy and safety. Molecules 2019, 24, 1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kaspar AA; Reichert JM Future directions for peptide therapeutics development. Drug Discovery Today 2013, 18, 807–817. [DOI] [PubMed] [Google Scholar]
- (27).Cramer RD; Clark RD; Patterson DE; Ferguson AM Bioisosterism as a molecular diversity descriptor: steric fields of single “topomeric” conformers. J. Med. Chem 1996, 39, 3060–3069. [DOI] [PubMed] [Google Scholar]
- (28).Ertl P In silico identification of bioisosteric functional groups. Curr. Opin. Drug Discovery Dev 2007, 10, 281–288. [PubMed] [Google Scholar]
- (29).Devereux M; Popelier PLA In silico techniques for the identification of bioisosteric replacements for drug design. Curr. Top. Med. Chem 2010, 10, 657–668. [DOI] [PubMed] [Google Scholar]
- (30).Olesen PH The use of bioisosteric groups in lead optimization. Curr. Opin. Drug Discovery Dev 2001, 4, 471–478. [PubMed] [Google Scholar]
- (31).Thornber CW Isosterism and molecular modification in drug design. Chem. Soc. Rev 1979, 8, 563–580. [Google Scholar]
- (32).Sun S; Jia Q; Zhang Z Applications of amide isosteres in medicinal chemistry. Bioorg. Med. Chem. Lett 2019, 29, 2535–2550. [DOI] [PubMed] [Google Scholar]
- (33).Bachl J; Mayr J; Sayago FJ; Cativiela C; Díaz Díaz D Amide-triazole isosteric substitution for tuning self-assembly and incorporating new functions into soft supramolecular materials. Chem. Commun 2015, 51, 5294–5297. [DOI] [PubMed] [Google Scholar]
- (34).Tron GC; Pirali T; Billington RA; Canonico PL; Sorba G; Genazzani AA Click chemistry reactions in medicinal chemistry: applications of the 1,3-dipolar cycloaddition between azides and alkynes. Med. Res. Rev 2008, 28, 278–308. [DOI] [PubMed] [Google Scholar]
- (35).Hou J; Liu X; Shen J; Zhao G; Wang PG The impact of click chemistry in medicinal chemistry. Expert Opin. Drug Discovery 2012, 7, 489–501. [DOI] [PubMed] [Google Scholar]
- (36).Prasher P; Sharma M Tailored therapeutics based on 1,2,3–1H-triazoles: a mini review. MedChemComm 2019, 10, 1302–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Chrysina ED; Bokor É; Alexacou K-M; Charavgi M-D; Oikonomakos GN; Zographos SE; Leonidas DD; Oikonomakos NG; Somsák L Amide-1,2,3-triazole bioisosterism: the glycogen phosphorylase case. Tetrahedron: Asymmetry 2009, 20, 733–740. [Google Scholar]
- (38).Angell YL; Burgess K Peptidomimetics via copper-catalyzed azide-alkyne cycloadditions. Chem. Soc. Rev 2007, 36, 1674–1689. [DOI] [PubMed] [Google Scholar]
- (39).Nathans R; Cao H; Sharova N; Ali A; Sharkey M; Stranska R; Stevenson M; Rana TM Small-molecule inhibition of HIV-1 Vif. Nat. Biotechnol 2008, 26, 1187–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ali A; Wang J; Nathans RS; Cao H; Sharova N; Stevenson M; Rana TM Synthesis and structure-activity relationship studies of HIV-1 virion infectivity factor (Vif) inhibitors that block viral replication. ChemMedChem 2012, 7, 1217–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Mohammed I; Parai MK; Jiang X; Sharova N; Singh G; Stevenson M; Rana TM SAR and lead optimization of an HIV-1 Vif-APOBEC3G axis inhibitor. ACS Med. Chem. Lett 2012, 3, 465–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Miller JH; Presnyak V; Smith HC The dimerization domain of HIV-1 viral infectivity factor Vif is required to block virion incorporation of APOBEC3G. Retrovirology 2007, 4, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Ribeiro AC; Maia e Silva A; Santa-Marta M; Pombo A; Moniz-Pereira J; Goncalves J; Barahona I Functional analysis of Vif protein shows less restriction of human immunodeficiency virus type 2 by APOBEC3G. J. Virol 2005, 79, 823–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Mohammed I; Kummetha IR; Singh G; Sharova N; Lichinchi G; Dang J; Stevenson M; Rana TM 1,2,3-Triazoles as amide bioisosteres: discovery of a new class of potent HIV-1 Vif antagonists. J. Med. Chem 2016, 59, 7677–7682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Rudin CM Vismodegib. Clin. Cancer Res 2012, 18, 3218–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Cirrone F; Harris CS Vismodegib and the hedgehog pathway: a new treatment for basal cell carcinoma. Clin. Ther 2012, 34, 2039–2050. [DOI] [PubMed] [Google Scholar]
- (47).Koelblinger P; Lang R New developments in the treatment of basal cell carcinoma: update on current and emerging treatment options with a focus on vismodegib. OncoTargets Ther. 2018, 11, 8327–8340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Sharpe HJ; Pau G; Dijkgraaf GJ; Basset-Seguin N; Modrusan Z; Januario T; Tsui V; Durham AB; Dlugosz AA; Haverty PM; Bourgon R; Tang JY; Sarin KY; Dirix L; Fisher DC; Rudin CM; Sofen H; Migden MR; Yauch RL; de Sauvage FJ Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 2015, 27, 327–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Christodoulou MS; Mori M; Pantano R; Alfonsi R; Infante P; Botta M; Damia G; Ricci F; Sotiropoulou PA; Liekens S; Botta B; Passarella D Click reaction as a tool to combine pharmacophores: the case of vismodegib. ChemPlusChem 2015, 80, 938–943. [DOI] [PubMed] [Google Scholar]
- (50).Colombo F; Tintori C; Furlan A; Borrelli S; Christodoulou MS; Dono R; Maina F; Botta M; Amat M; Bosch J; Passarella D ‘Click’ synthesis of a triazole-based inhibitor of Met functions in cancer cells. Bioorg. Med. Chem. Lett 2012, 22, 4693–4696. [DOI] [PubMed] [Google Scholar]
- (51).Furlan A; Roux B; Lamballe F; Conti F; Issaly N; Daian F; Guillemot JF; Richelme S; Contensin M; Bosch J; Passarella D; Piccolo O; Dono R; Maina F Combined drug action of 2-phenylimidazo[2,1-b]benzothiazole derivatives on cancer cells according to their oncogenic molecular signatures. PLoS One 2012, 7, No. e46738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Arioli F; Borrelli S; Colombo F; Falchi F; Filippi I; Crespan E; Naldini A; Scalia G; Silvani A; Maga G; Carraro F; Botta M; Passarella D N-[2-methyl-5-(triazol-1-yl)phenyl]-pyrimidin-2-amine as a scaffold for the synthesis of inhibitors of Bcr-Abl. ChemMedChem 2011, 6e, 2009–2018. [DOI] [PubMed] [Google Scholar]
- (53).Buchdunger E; O’Reilley T; Wood J Pharmacology of imatinib (STI571). Eur. J. Cancer 2002, 38 (Suppl. 5), S28–S36. [DOI] [PubMed] [Google Scholar]
- (54).Hernandez-Boluda JC; Cervantes F Imatinib mesylate (gleevec, glivec): a new therapy for chronic myeloid leukemia and other malignancies. Drugs Today (Barc) 2002, 38, 601–613. [DOI] [PubMed] [Google Scholar]
- (55).Moen MD; McKeage K; Plosker GL; Siddiqui MA Imatinib: a review of its use in chronic myeloid leukaemia. Drugs 2007, 67, 299–320. [DOI] [PubMed] [Google Scholar]
- (56).Waller CF Imatinib mesylate. Recent Results Cancer Res. 2010, 184, 3–20. [DOI] [PubMed] [Google Scholar]
- (57).Iqbal N; Iqbal N Imatinib: a breakthrough of targeted therapy in cancer. Chemother. Res. Pract 2014, 2014, 357027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Schenone S; Bruno O; Radi M; Botta M New insights into small-molecule inhibitors of Bcr-Abl. Med. Res. Rev 2011, 31, 1–41. [DOI] [PubMed] [Google Scholar]
- (59).Leggio GM; Bucolo C; Platania CB; Salomone S; Drago F Current drug treatments targeting dopamine D3 receptor. Pharmacol. Ther 2016, 165, 164–177. [DOI] [PubMed] [Google Scholar]
- (60).Joyce JN Dopamine D3 receptor as a therapeutic target for antipsychotic and antiparkinsonian drugs. Pharmacol. Ther 2001, 90, 231–259. [DOI] [PubMed] [Google Scholar]
- (61).Maramai S; Gemma S; Brogi S; Campiani G; Butini S; Stark H; Brindisi M Dopamine D3 receptor antagonists as potential therapeutics for the treatment of neurological diseases. Front. Neurosci 2016, 10, 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Newman AH; Grundt P; Cyriac G; Deschamps JR; Taylor M; Kumar R; Ho D; Luedtke RR N-(4-(4-(2,3-dichloro-or 2-methoxyphenyl)piperazin-1-yl)butyl)heterobiarylcarboxamides with functionalized linking chains as high affinity and enantioselective D3 receptor antagonists. J. Med. Chem 2009, 52, 2559–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Boateng CA; Bakare OM; Zhan J; Banala AK; Burzynski C; Pommier E; Keck TM; Donthamsetti P; Javitch JA; Rais R; Slusher BS; Xi ZX; Newman AH High affinity dopamine D3 Receptor (D3R)-selective antagonists attenuate heroin self-administration in wild-type but not D3R knockout mice. J. Med. Chem 2015, 58, 6195–6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Heidbreder CA; Newman AH Current perspectives on selective dopamine D(3) receptor antagonists as pharmacotherapeutics for addictions and related disorders. Ann. N. Y. Acad. Sci 2010, 1187, 4–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Banala AK; Levy BA; Khatri SS; Furman CA; Roof RA; Mishra Y; Griffin SA; Sibley DR; Luedtke RR; Newman AH N-(3-fluoro-4-(4-(2-methoxy or 2,3-dichlorophenyl)piperazine-1-yl)butyl)arylcarboxamides as selective dopamine D3 receptor ligands: critical role of the carboxamide linker for D3 receptor selectivity. J. Med. Chem 2011, 54, 3581–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Keck TM; Banala AK; Slack RD; Burzynski C; Bonifazi A; Okunola-Bakare OM; Moore M; Deschamps JR; Rais R; Slusher BS; Newman AH Using click chemistry toward novel 1,2,3-triazole-linked dopamine D3 receptor ligands. Bioorg. Med. Chem 2015, 23, 4000–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Appendino G; Bacchiega S; Minassi A; Cascio MG; De Petrocellis L; Di Marzo V The 1,2,3-triazole ring as a peptido- and olefinomimetic element: discovery of click vanilloids and cannabinoids. Angew. Chem., Int. Ed 2007, 46, 9312–9315. [DOI] [PubMed] [Google Scholar]
- (68).Mugnaini C; Nocerino S; Pedani V; Pasquini S; Tafi A; De Chiaro M; Bellucci L; Valoti M; Guida F; Luongo L; Dragoni S; Ligresti A; Rosenberg A; Bolognini D; Cascio MG; Pertwee RG; Moaddel R; Maione S; Di Marzo V; Corelli F Investigations on the 4-quinolone-3-carboxylic acid motif part 5: modulation of the physicochemical profile of a set of potent and selective cannabinoid-2 receptor ligands through a bioisosteric approach. ChemMedChem 2012, 7, 920–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Cabral GA; Griffin-Thomas L Emerging role of the cannabinoid receptor CB2 in immune regulation: therapeutic prospects for neuroinflammation. Expert Rev. Mol. Med 2009, 11, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Dhopeshwarkar A; Mackie K CB2 Cannabinoid receptors as a therapeutic target-what does the future hold? Mol. Pharmacol 2014, 86, 430–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Turcotte C; Blanchet MR; Laviolette M; Flamand N The CB2 receptor and its role as a regulator of inflammation. Cell. Mol. Life Sci 2016, 73, 4449–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Koufaki M; Detsi A; Theodorou E; Kiziridi C; Calogeropoulou T; Vassilopoulos A; Kourounakis AP; Rekka E; Kourounakis PN; Gaitanaki C; Papazafiri P Synthesis of chroman analogues of lipoic acid and evaluation of their activity against reperfusion arrhythmias. Bioorg. Med. Chem 2004, 12, 4835–4841. [DOI] [PubMed] [Google Scholar]
- (73).Koufaki M; Kiziridi C; Alexi X; Alexis MN Design and synthesis of novel neuroprotective 1,2-dithiolane/chroman hybrids. Bioorg. Med. Chem 2009, 17, 6432–6441. [DOI] [PubMed] [Google Scholar]
- (74).Koufaki M; Calogeropoulou T; Detsi A; Roditis A; Kourounakis AP; Papazafiri P; Tsiakitzis K; Gaitanaki C; Beis I; Kourounakis PN Novel potent inhibitors of lipid peroxidation with protective effects against reperfusion arrhythmias. J. Med. Chem 2001, 44, 4300–4303. [DOI] [PubMed] [Google Scholar]
- (75).Rosini M; Andrisano V; Bartolini M; Bolognesi ML; Hrelia P; Minarini A; Tarozzi A; Melchiorre C Rational approach to discover multipotent anti-Alzheimer drugs. J. Med. Chem 2005, 48, 360–363. [DOI] [PubMed] [Google Scholar]
- (76).Maczurek A; Shanmugam K; Munch G Inflammation and the redox-sensitive AGE-RAGE pathway as a therapeutic target in Alzheimer’s disease. Ann. N. Y. Acad. Sci 2008, 1126, 147–151. [DOI] [PubMed] [Google Scholar]
- (77).Harnett JJ; Auguet M; Viossat I; Dolo C; Bigg D; Chabrier PE Novel lipoic acid analogues that inhibit nitric oxide synthase. Bioorg. Med. Chem. Lett 2002, 12, 1439–1442. [DOI] [PubMed] [Google Scholar]
- (78).Dozio E; Ruscica M; Passafaro L; Dogliotti G; Steffani L; Marthyn P; Pagani A; Demartini G; Esposti D; Fraschini F; Magni P The natural antioxidant alpha-lipoic acid induces p27-(Kip1)-dependent cell cycle arrest and apoptosis in MCF-7 human breast cancer cells. Eur. J. Pharmacol 2010, 641, 29–34. [DOI] [PubMed] [Google Scholar]
- (79).Zhang SJ; Ge QF; Guo DW; Hu WX; Liu HZ Synthesis and anticancer evaluation of α-lipoic acid derivatives. Bioorg. Med. Chem. Lett 2010, 20, 3078–3083. [DOI] [PubMed] [Google Scholar]
- (80).Dorsam B; Fahrer J The disulfide compound α-lipoic acid and its derivatives: a novel class of anticancer agents targeting mitochondria. Cancer Lett. 2016, 371, 12–19. [DOI] [PubMed] [Google Scholar]
- (81).Melagraki G; Afantitis A; Igglessi-Markopoulou O; Detsi A; Koufaki M; Kontogiorgis C; Hadjipavlou-Litina DJ Synthesis and evaluation of the antioxidant and anti-inflammatory activity of novel coumarin-3-aminoamides and their alpha-lipoic acid adducts. Eur. J. Med. Chem 2009, 44, 3020–3026. [DOI] [PubMed] [Google Scholar]
- (82).Chouinard G Issues in the clinical use of benzodiazepines: potency, withdrawal, and rebound. J. Clin. Psychiatry 2004, 65 (Suppl. 5), 7–12. [PubMed] [Google Scholar]
- (83).Balon R Benzodiazepines revisited. Psychother. Psychosom 2013, 82, 353–354. [DOI] [PubMed] [Google Scholar]
- (84).Hershon HI; Parsonage M Comparative trial of diazepam and pheneturide in treatment of epilepsy. Lancet 1969, 294, 859–862. [DOI] [PubMed] [Google Scholar]
- (85).Robinson GM; Sellers EM Diazepam withdrawal seizures. Can. Med. Assoc. J 1982, 126, 944–945. [PMC free article] [PubMed] [Google Scholar]
- (86).Devenyi P; Harrison ML Prevention of alcohol withdrawal seizures with oral diazepam loading. Can. Med. Assoc. J 1985, 132, 798–800. [PMC free article] [PubMed] [Google Scholar]
- (87).Griffin CE 3rd; Kaye AM; Bueno FR; Kaye AD Benzodiazepine pharmacology and central nervous system-mediated effects. Ochsner J. 2013, 13, 214–223. [PMC free article] [PubMed] [Google Scholar]
- (88).Calcaterra NE; Barrow JC Classics in chemical neuroscience: diazepam (valium). ACS Chem. Neurosci 2014, 5, 253–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Hester JB Jr.; Rudzik AD; Kamdar BV 6-phenyl-4H-s-triazolo[4,3-a][1,4]benzodiazepines which have central nervous system depressant activity. J. Med. Chem 1971, 14, 1078–1081. [DOI] [PubMed] [Google Scholar]
- (90).Verster JC; Volkerts ER Clinical pharmacology, clinical efficacy, and behavioral toxicity of alprazolam: a review of the literature. CNS Drug Rev. 2004, 10, 45–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).de Oliveira CS; Lira BF; Barbosa-Filho JM; Lorenzo JG; de Athayde-Filho PF Synthetic approaches and pharmacological activity of 1,3,4-oxadiazoles: a review of the literature from 2000–2012. Molecules 2012, 17, 10192–10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Bostrom J; Hogner A; Llinas A; Wellner E; Plowright AT Oxadiazoles in medicinal chemistry. J. Med. Chem 2012, 55, 1817–1830. [DOI] [PubMed] [Google Scholar]
- (93).Cao Y; Min C; Acharya S; Kim KM; Cheon SH Design, synthesis and evaluation of bitopic arylpiperazinephenyl-1,2,4-oxadiazoles as preferential dopamine D3 receptor ligands. Bioorg. Med. Chem 2016, 24, 191–200. [DOI] [PubMed] [Google Scholar]
- (94).Mashayekh S; Rahmanipour N; Mahmoodi B; Ahmadi F; Motaharian D; Shahhosseini S; Shafaroodi H; Banafshe HR; Shafiee A; Navidpour L Synthesis, receptor affinity and effect on pentylenetetrazole-induced seizure threshold of novel benzodiazepine analogues: 3-substituted 5-(2-phenoxybenzyl)-4H-1,2,4-triazoles and 2-amino-5-(phenoxybenzyl)-1,3,4-oxadiazoles. Bioorg. Med. Chem 2014, 22, 1929–1937. [DOI] [PubMed] [Google Scholar]
- (95).Patel S; Freedman SB The muscarinic receptor agonist L-658,903 modulates the in vivo accumulation of inositol mono-phosphates in mouse brain. Eur. J. Pharmacol., Mol. Pharmacol. Sect 1994, 267, 329–334. [DOI] [PubMed] [Google Scholar]
- (96).Maccioni E; Alcaro S; Cirilli R; Vigo S; Cardia MC; Sanna ML; Meleddu R; Yanez M; Costa G; Casu L; Matyus P; Distinto S 3-Acetyl-2,5-diaryl-2,3-dihydro-1,3,4-oxadiazoles: a new scaffold for the selective inhibition of monoamine oxidase B. J. Med. Chem 2011, 54, 6394–6398. [DOI] [PubMed] [Google Scholar]
- (97).Shook BC; Jackson PF Adenosine A(2A) receptor antagonists and Parkinson’s disease. ACS Chem. Neurosci 2011, 2, 555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Swain CJ; Baker R; Kneen C; Moseley J; Saunders J; Seward EM; Stevenson G; Beer M; Stanton J; Watling K Novel 5-HT3 antagonists. indole oxadiazoles. J. Med. Chem 1991, 34, 140–151. [DOI] [PubMed] [Google Scholar]
- (99).Rajapakse HA; Nantermet PG; Selnick HG; Munshi S; McGaughey GB; Lindsley SR; Young MB; Lai MT; Espeseth AS; Shi XP; Colussi D; Pietrak B; Crouthamel MC; Tugusheva K; Huang Q; Xu M; Simon AJ; Kuo L; Hazuda DJ; Graham S; Vacca JP Discovery of oxadiazoyl tertiary carbinamine inhibitors of β-secretase (BACE-1). J. Med. Chem 2006, 49, 7270–7273. [DOI] [PubMed] [Google Scholar]
- (100).Pitasse-Santos P; Sueth-Santiago V; Lima MEF 1,2,4- and 1,3,4-Oxadiazoles as scaffolds in the development of antiparasitic agents. J. Braz. Chem. Soc 2018, 29, 435–456. [Google Scholar]
- (101).Borg S; Vollinga RC; Labarre M; Payza K; Terenius L; Luthman K Design, synthesis, and evaluation of phe-gly mimetics: heterocyclic building blocks for pseudopeptides. J. Med. Chem 1999, 42, 4331–4342. [DOI] [PubMed] [Google Scholar]
- (102).Wolfe MS γ-Secretase inhibitors and modulators for Alzheimer’s disease. J. Neurochem 2012, 120 (Suppl. 1), 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Wolfe MS Unlocking truths of γ-secretase in Alzheimer’s disease: what is the translational potential? Future Neurol. 2014, 9, 419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).D’Onofrio G; Panza F; Frisardi V; Solfrizzi V; Imbimbo BP; Paroni G; Cascavilla L; Seripa D; Pilotto A Advances in the identification of γ-secretase inhibitors for the treatment of Alzheimer’s disease. Expert Opin. Drug Discovery 2012, 7, 19–37. [DOI] [PubMed] [Google Scholar]
- (105).Gillman KW; Starrett JE Jr.; Parker MF; Xie K; Bronson JJ; Marcin LR; McElhone KE; Bergstrom CP; Mate RA; Williams R; Meredith JE Jr.; Burton CR; Barten DM; Toyn JH; Roberts SB; Lentz KA; Houston JG; Zaczek R; Albright CF; Decicco CP; Macor JE; Olson RE Discovery and evaluation of BMS-708163, a potent, selective and orally bioavailable γ-secretase inhibitor. ACS Med. Chem. Lett 2010, 1, 120–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Koufaki M; Theodorou E; Alexi X; Alexis MN Synthesis of a second generation chroman/catechol hybrids and evaluation of their activity in protecting neuronal cells from oxidative stress-induced cell death. Bioorg. Med. Chem 2010, 18, 3898–3909. [DOI] [PubMed] [Google Scholar]
- (107).Reed CW; Washecheck JP; Quitlag MC; Jenkins MT; Rodriguez AL; Engers DW; Blobaum AL; Jeffrey Conn P; Niswender CM; Lindsley CW Surveying heterocycles as amide bioisosteres within a series of mGlu7 NAMs: discovery of VU6019278. Bioorg. Med. Chem. Lett 2019, 29, 1211–1214. [DOI] [PubMed] [Google Scholar]
- (108).Reed CW; McGowan KM; Spearing PK; Stansley BJ; Roenfanz HF; Engers DW; Rodriguez AL; Engelberg EM; Luscombe VB; Loch MT; Remke DH; Rook JM; Blobaum AL; Conn PJ; Niswender CM; Lindsley CW VU6010608, a novel mGlu7 NAM from a series of N-(2-(1H-1,2,4-triazol-1-yl)-5-(trifluoromethoxy)phenyl)benzamides. ACS Med. Chem. Lett 2017, 8, 1326–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Reed CW; Yohn SE; Washecheck JP; Roenfanz HF; Quitalig MC; Luscombe VB; Jenkins MT; Rodriguez AL; Engers DW; Blobaum AL; Conn PJ; Niswender CM; Lindsley CW Discovery of an orally bioavailable and central nervous system (CNS) penetrant mGlu7 negative allosteric modulator (NAM) in vivo tool compound: N-(2-(1 H-1,2,4-triazol-1-yl)-5-(trifluoromethoxy)phenyl)-4-(cyclopropylmethoxy)-3-me-thoxybenzamide (VU6012962). J. Med. Chem 2019, 62, 1690–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Nordhoff S; Bulat S; Cerezo-Galvez S; Hill O; Hoffmann-Enger B; Lopez-Canet M; Rosenbaum C; Rummey C; Thiemann M; Matassa VG; Edwards PJ; Feurer A The design of potent and selective inhibitors of DPP-4: optimization of ADME properties by amide replacements. Bioorg. Med. Chem. Lett 2009, 19, 6340–6345. [DOI] [PubMed] [Google Scholar]
- (111).Makrilakis K The role of DPP-4 inhibitors in the treatment algorithm of type 2 diabetes mellitus: when to select, what to expect. Int. J. Environ. Res. Public Health 2019, 16, 2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Cao J; Zhou Y; Peng H; Huang X; Stahler S; Suri V; Qadri A; Gareski T; Jones J; Hahm S; Perreault M; McKew J; Shi M; Xu X; Tobin JF; Gimeno RE Targeting Acyl-CoA:diacylglycerol acyltransferase 1 (DGAT1) with small molecule inhibitors for the treatment of metabolic diseases. J. Biol. Chem 2011, 286, 41838–41851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Nakajima K; Chatelain R; Clairmont KB; Commerford R; Coppola GM; Daniels T; Forster CJ; Gilmore TA; Gong Y; Jain M; Kanter A; Kwak Y; Li J; Meyers CD; Neubert AD; Szklennik P; Tedesco V; Thompson J; Truong D; Yang Q; Hubbard BK; Serrano-Wu MH Discovery of an orally bioavailable benzimidazole diacylglycerol acyltransferase 1 (DGAT1) inhibitor that suppresses body weight gain in diet-induced obese dogs and postprandial triglycerides in humans. J. Med. Chem 2017, 60, 4657–4664. [DOI] [PubMed] [Google Scholar]
- (114).Serrano-Wu MH; Coppola GM; Gong Y; Neubert AD; Chatelain R; Clairmont KB; Commerford R; Cosker T; Daniels T; Hou Y; Jain M; Juedes M; Li L; Mullarkey T; Rocheford E; Sung MJ; Tyler A; Yang Q; Yoon T; Hubbard BK Intestinally targeted diacylglycerol acyltransferase 1 (DGAT1) inhibitors robustly suppress postprandial triglycerides. ACS Med. Chem. Lett 2012, 3, 411–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Pasquini S; Ligresti A; Mugnaini C; Semeraro T; Cicione L; De Rosa M; Guida F; Luongo L; De Chiaro M; Cascio MG; Bolognini D; Marini P; Pertwee R; Maione S; Di Marzo V; Corelli F Investigations on the 4-quinolone-3-carboxylic acid motif. 3. synthesis, structure-affinity relationships, and pharmacological characterization of 6-substituted 4-quinolone-3-carboxamides as highly selective cannabinoid-2 receptor ligands. J. Med. Chem 2010, 53, 5915–5928. [DOI] [PubMed] [Google Scholar]
- (116).Pasquini S; Botta L; Semeraro T; Mugnaini C; Ligresti A; Palazzo E; Maione S; Di Marzo V; Corelli F Investigations on the 4-quinolone-3-carboxylic acid motif. 2. synthesis and structure-activity relationship of potent and selective cannabinoid-2 receptor agonists endowed with analgesic activity in vivo. J. Med. Chem 2008, 51, 5075–5084. [DOI] [PubMed] [Google Scholar]
- (117).Pasquini S; De Rosa M; Pedani V; Mugnaini C; Guida F; Luongo L; De Chiaro M; Maione S; Dragoni S; Frosini M; Ligresti A; Di Marzo V; Corelli F Investigations on the 4-quinolone-3-carboxylic acid motif. 4. identification of new potent and selective ligands for the cannabinoid type 2 receptor with diverse substitution patterns and antihyperalgesic effects in mice. J. Med. Chem 2011, 54, 5444–5453. [DOI] [PubMed] [Google Scholar]
- (118).Dundee JW; Halliday NJ; Harper KW; Brogden RN Midazolam. a review of its pharmacological properties and therapeutic use. Drugs 1984, 28, 519–543. [DOI] [PubMed] [Google Scholar]
- (119).Gerecke M Chemical structure and properties of midazolam compared with other benzodiazepines. Br. J. Clin. Pharmacol 1983, 16 (Suppl. 1), 11S–16S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Taghizadeh MJ; Malakpouri G. r.; Javidan A Improved and scalable methods for the synthesis of midazolam drug and its analogues using isocyanide reagents. J. Iran. Chem. Soc 2019, 16, 785–794. [Google Scholar]
- (121).Smith R; Brown J Midazolam for status epilepticus. Aust. Prescr 2017, 40, 23–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Vogel GW; Vogel F Effect of midazolam on sleep of insomniacs. Br. J. Clin. Pharmacol 1983, 16 (Suppl. 1), 103S–108S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Khanderia U; Pandit SK Use of midazolam hydrochloride in anesthesia. Clin. Pharm 1987, 6, 533–547. [PubMed] [Google Scholar]
- (124).Doods H; Arndt K; Rudolf K; Just S CGRP antagonists: unravelling the role of CGRP in migraine. Trends Pharmacol. Sci 2007, 28, 580–587. [DOI] [PubMed] [Google Scholar]
- (125).Bell IM Calcitonin gene-related peptide receptor antagonists: new therapeutic agents for migraine. J. Med. Chem 2014, 57, 7838–7858. [DOI] [PubMed] [Google Scholar]
- (126).Deen M; Correnti E; Kamm K; Kelderman T; Papetti L; Rubio-Beltran E; Vigneri S; Edvinsson L; Maassen Van Den Brink, A. Blocking CGRP in migraine patients - a review of pros and cons. J. Headache Pain 2017, 18, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Paone DV; Shaw AW; Nguyen DN; Burgey CS; Deng JZ; Kane SA; Koblan KS; Salvatore CA; Mosser SD; Johnston VK; Wong BK; Miller-Stein CM; Hershey JC; Graham SL; Vacca JP; Williams TM Potent, orally bioavailable calcitonin gene-related peptide receptor antagonists for the treatment of migraine: discovery of N-[(3R,6S)-6-(2,3-difluorophenyl)-2-oxo-1-(2,2,2-trifluoroethyl)azepan-3-yl]-4- (2-oxo-2,3-dihydro-1H-imidazo-[4,5-b]pyridin- 1-yl)piperidine-1-carboxamide (MK-0974). J. Med. Chem 2007, 50, 5564–5567. [DOI] [PubMed] [Google Scholar]
- (128).Salvatore CA; Hershey JC; Corcoran HA; Fay JF; Johnston VK; Moore EL; Mosser SD; Burgey CS; Paone DV; Shaw AW; Graham SL; Vacca JP; Williams TM; Koblan KS; Kane SA Pharmacological characterization of MK-0974 [N-[(3R,6S)-6-(2,3-difluorophenyl)-2-oxo-1-(2,2,2-trifluoroethyl)azepan-3-yl]-4-(2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-1-yl)piperidine-1-carboxamide], a potent and orally active calcitonin gene-related peptide receptor antagonist for the treatment of migraine. J. Pharmacol. Exp. Ther 2008, 324, 416–421. [DOI] [PubMed] [Google Scholar]
- (129).Paone DV; Nguyen DN; Shaw AW; Burgey CS; Potteiger CM; Deng JZ; Mosser SD; Salvatore CA; Yu S; Roller S; Kane SA; Selnick HG; Vacca JP; Williams TM Orally bioavailable imidazoazepanes as calcitonin gene-related peptide (CGRP) receptor antagonists: discovery of MK-2918. Bioorg. Med. Chem. Lett 2011, 21, 2683–2686. [DOI] [PubMed] [Google Scholar]
- (130).Peese KM; Naidu BN; Patel M; Li C; Langley DR; Terry B; Protack T; Gali V; Lin Z; Samanta HK; Zheng M; Jenkins S; Dicker IB; Krystal MR; Meanwell NA; Walker MA Heterocycle amide isosteres: an approach to overcoming resistance for HIV-1 integrase strand transfer inhibitors. Bioorg. Med. Chem. Lett 2020, 30, 126784. [DOI] [PubMed] [Google Scholar]
- (131).Oliveira M; Ibanescu RI; Anstett K; Mesplede T; Routy JP; Robbins MA; Brenner BG The Montreal Primary HIV (PHI) Cohort Study Group; Selective resistance profiles emerging in patient-derived clinical isolates with cabotegravir, bictegravir, dolutegravir, and elvitegravir. Retrovirology 2018, 15, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Summa V; Petrocchi A; Bonelli F; Crescenzi B; Donghi M; Ferrara M; Fiore F; Gardelli C; Gonzalez Paz O; Hazuda DJ; Jones P; Kinzel O; Laufer R; Monteagudo E; Muraglia E; Nizi E; Orvieto F; Pace P; Pescatore G; Scarpelli R; Stillmock K; Witmer MV; Rowley M Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J. Med. Chem 2008, 51, 5843–5855. [DOI] [PubMed] [Google Scholar]
- (133).Naidu BN; Walker MA; Sorenson ME; Ueda Y; Matiskella JD; Connolly TP; Dicker IB; Lin Z; Bollini S; Terry BJ; Higley H; Zheng M; Parker DD; Wu D; Adams S; Krystal MR; Meanwell NA The discovery and preclinical evaluation of BMS-707035, a potent HIV-1 integrase strand transfer inhibitor. Bioorg. Med. Chem. Lett 2018, 28, 2124–2130. [DOI] [PubMed] [Google Scholar]
- (134).Tracz-Gaszewska Z; Dobrzyn P Stearoyl-CoA desaturase 1 as a therapeutic target for the treatment of cancer. Cancers 2019, 11, 948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Sun S; Zhang Z; Kodumuru V; Pokrovskaia N; Fonarev J; Jia Q; Leung PY; Tran J; Ratkay LG; McLaren DG; Radomski C; Chowdhury S; Fu J; Hubbard B; Winther MD; Dales NA Systematic evaluation of amide bioisosteres leading to the discovery of novel and potent thiazolylimidazolidinone inhibitors of SCD1 for the treatment of metabolic diseases. Bioorg. Med. Chem. Lett 2014, 24, 520–525. [DOI] [PubMed] [Google Scholar]
- (136).Ballatore C; Huryn DM; Smith AB 3rd Carboxylic acid (bio)isosteres in drug design. ChemMedChem 2013, 8, 385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Subramanian V; Knight JS; Parelkar S; Anguish L; Coonrod SA; Kaplan MJ; Thompson PR Design, synthesis, and biological evaluation of tetrazole analogs of Cl-amidine as protein arginine deiminase inhibitors. J. Med. Chem 2015, 58, 1337–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Witalison EE; Thompson PR; Hofseth LJ Protein arginine deiminases and associated citrullination: physiological functions and diseases associated with dysregulation. Curr. Drug Targets 2015, 16, 700–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Leshner M; Wang S; Lewis C; Zheng H; Chen XA; Santy L; Wang Y PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front. Immunol 2012, 3, 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Khandpur R; Carmona-Rivera C; Vivekanandan-Giri A; Gizinski A; Yalavarthi S; Knight JS; Friday S; Li S; Patel RM; Subramanian V; Thompson P; Chen P; Fox DA; Pennathur S; Kaplan MJ NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med 2013, 5, 178ra40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Han D; Handelman G; Marcocci L; Sen CK; Roy S; Kobuchi H; Tritschler HJ; Flohe L; Packer L Lipoic acid increases de novo synthesis of cellular glutathione by improving cystine utilization. BioFactors 1997, 6, 321–338. [DOI] [PubMed] [Google Scholar]
- (142).Podda M; Tritschler HJ; Ulrich H; Packer L α-lipoic acid supplementation prevents symptoms of vitamin E deficiency. Biochem. Biophys. Res. Commun 1994, 204, 98–104. [DOI] [PubMed] [Google Scholar]
- (143).Rochette L; Ghibu S; Richard C; Zeller M; Cottin Y; Vergely C Direct and indirect antioxidant properties of α-lipoic acid and therapeutic potential. Mol. Nutr. Food Res 2013, 57, 114–125. [DOI] [PubMed] [Google Scholar]
- (144).Koufaki M; Kiziridi C; Nikoloudaki F; Alexis MN Design and synthesis of 1,2-dithiolane derivatives and evaluation of their neuroprotective activity. Bioorg. Med. Chem. Lett 2007, 17, 4223–4227. [DOI] [PubMed] [Google Scholar]
- (145).Dani JA Neuronal nicotinic acetylcholine receptor structure and function and response to nicotine. Int. Rev. Neurobiol 2015, 124, 3–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Feduccia AA; Chatterjee S; Bartlett SE Neuronal nicotinic acetylcholine receptors: neuroplastic changes underlying alcohol and nicotine addictions. Front. Mol. Neurosci 2012, 5, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Buckingham SD; Jones AK; Brown LA; Sattelle DB Nicotinic acetylcholine receptor signalling: roles in Alzheimer’s disease and amyloid neuroprotection. Pharmacol. Rev 2009, 61, 39–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Woodruff-Pak DS; Gould TJ Neuronal nicotinic acetylcholine receptors: involvement in Alzheimer’s disease and schizophrenia. Behav. Cogn. Neurosci. Rev 2002, 1, 5–20. [DOI] [PubMed] [Google Scholar]
- (149).Beinat C; Reekie T; Hibbs D; Xie T; Olson TT; Xiao Y; Harvey A; O’Connor S; Coles C; Tsanaktsidis J; Kassiou M Investigations of amide bond variation and biaryl modification in analogues of α7 nAChR agonist SEN12333. Eur. J. Med. Chem 2014, 84, 200–205. [DOI] [PubMed] [Google Scholar]
- (150).Haydar SN; Ghiron C; Bettinetti L; Bothmann H; Comery TA; Dunlop J; La Rosa S; Micco I; Pollastrini M; Quinn J; Roncarati R; Scali C; Valacchi M; Varrone M; Zanaletti R SAR and biological evaluation of SEN12333/WAY-317538: novel alpha 7 nicotinic acetylcholine receptor agonist. Bioorg. Med. Chem 2009, 17, 5247–5258. [DOI] [PubMed] [Google Scholar]
- (151).Beinat C; Banister SD; van Prehn S; Doddareddy MR; Hibbs D; Sako M; Chebib M; Tran T; Al-Muhtasib N; Xiao Y; Kassiou M Consequences of linker length alteration of the α7 nicotinic acetylcholine receptor (nAChR) agonist, SEN12333. Bioorg. Med. Chem. Lett 2012, 22, 2380–2384. [DOI] [PubMed] [Google Scholar]
- (152).Graham TH Prolylcarboxypeptidase (PrCP) inhibitors and the therapeutic uses thereof: a patent review. Expert Opin. Ther. Pat 2017, 27, 1077–1088. [DOI] [PubMed] [Google Scholar]
- (153).Shen HC; Ding FX; Zhou C; Xiong Y; Verras A; Chabin RM; Xu S; Tong X; Xie D; Lassman ME; Bhatt UR; Garcia-Calvo MM; Geissler W; Shen Z; Chen D; Sinharoy R; Hale JJ; Tata JR; Pinto S; Shen DM; Colletti SL Discovery of benzimidazole pyrrolidinyl amides as prolylcarboxypeptidase inhibitors. Bioorg. Med. Chem. Lett 2011, 21, 1299–1305. [DOI] [PubMed] [Google Scholar]
- (154).Wu Z; Yang C; Xiong Y; Feng Z; Lombardo M; Verras A; Chabin RM; Xu S; Tong X; Xie D; Lassman ME; Bhatt UR; Garcia-Calvo MM; Geissler W; Shen Z; Chen Q; Sinharoy R; Hale JJ; Tata JR; Pinto S; Shen DM; Colletti SL Discovery of a new class of potent prolylcarboxypeptidase inhibitors derived from alanine. Bioorg. Med. Chem. Lett 2012, 22, 1774–1778. [DOI] [PubMed] [Google Scholar]
- (155).Debenham JS; Graham TH; Verras A; Zhang Y; Clements MJ; Kuethe JT; Madsen-Duggan C; Liu W; Bhatt UR; Chen D; Chen Q; Garcia-Calvo M; Geissler WM; He H; Li X; Lisnock J; Shen Z; Tong X; Tung EC; Wiltsie J; Xu S; Hale JJ; Pinto S; Shen DM Discovery and optimization of orally active cyclohexane-based prolylcarboxypeptidase (PrCP) inhibitors. Bioorg. Med. Chem. Lett 2013, 23, 6228–6233. [DOI] [PubMed] [Google Scholar]
- (156).Graham TH; Shu M; Verras A; Chen Q; Garcia-Calvo M; Li X; Lisnock J; Tong X; Tung EC; Wiltsie J; Hale JJ; Pinto S; Shen DM Pyrazoles as non-classical bioisosteres in prolylcarboxypeptidase (PrCP) inhibitors. Bioorg. Med. Chem. Lett 2014, 24, 1657–1660. [DOI] [PubMed] [Google Scholar]
- (157).Pacher P; Batkai S; Kunos G The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev 2006, 58, 389–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Pertwee RG Targeting the endocannabinoid system with cannabinoid receptor agonists: pharmacological strategies and therapeutic possibilities. Philos. Trans. R. Soc., B 2012, 367, 3353–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Adam JM; Cairns J; Caulfield W; Cowley P; Cumming I; Easson M; Edwards D; Ferguson M; Goodwin R; Jeremiah F; Kiyoi T; Mistry A; Moir E; Morphy R; Tierney J; York M; Baker J; Cottney JE; Houghton AK; Westwood PJ; Walker G Design, synthesis, and structure-activity relationships of indole-3-carboxamides as novel water soluble cannabinoid CB1 receptor agonists. MedChemComm 2010, 1, 54–60. [Google Scholar]
- (160).Moir EM; Yoshiizumi K; Cairns J; Cowley P; Ferguson M; Jeremiah F; Kiyoi T; Morphy R; Tierney J; Wishart G; York M; Baker J; Cottney JE; Houghton AK; McPhail P; Osprey A; Walker G; Adam JM Design, synthesis, and structure-activity relationship study of bicyclic piperazine analogs of indole-3-carboxamides as novel cannabinoid CB1 receptor agonists. Bioorg. Med. Chem. Lett 2010, 20, 7327–7330. [DOI] [PubMed] [Google Scholar]
- (161).Morrison AJ; Adam JM; Baker JA; Campbell RA; Clark JK; Cottney JE; Deehan M; Easson AM; Fields R; Francis S; Jeremiah F; Keddie N; Kiyoi T; McArthur DR; Meyer K; Ratcliffe PD; Schulz J; Wishart G; Yoshiizumi K Design, synthesis, and structure-activity relationships of indole-3-heterocycles as agonists of the CB1 receptor. Bioorg. Med. Chem. Lett 2011, 21, 506–509. [DOI] [PubMed] [Google Scholar]
- (162).Visse R; Nagase H Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ. Res 2003, 92, 827–839. [DOI] [PubMed] [Google Scholar]
- (163).Loffek S; Schilling O; Franzke CW Series “matrix metalloproteinases in lung health and disease”: biological role of matrix metalloproteinases: a critical balance. Eur. Respir. J 2011, 38, 191–208. [DOI] [PubMed] [Google Scholar]
- (164).Jackson BC; Nebert DW; Vasiliou V Update of human and mouse matrix metalloproteinase families. Hum. Genomics 2010, 4, 194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Sheppard GS; Florjancic AS; Giesler JR; Xu L; Guo Y; Davidsen SK; Marcotte PA; Elmore I; Albert DH; Magoc TJ; Bouska JJ; Goodfellow CL; Morgan DW; Summers JB Aryl ketones as novel replacements for the C-terminal amide bond of succinyl hydroxamate MMP inhibitors. Bioorg. Med. Chem. Lett 1998, 8, 3251–3256. [DOI] [PubMed] [Google Scholar]
- (166).Sheppard GS; Pireh D; Carrera GM Jr.; Bures MG; Heyman HR; Steinman DH; Davidsen SK; Phillips JG; Guinn DE; May PD; Conway RD; Rhein DA; Calhoun WC; Albert DH; Magoc TJ; Carter GW; Summers JB 3-(2-(3-Pyridinyl)thiazolidin-4-oyl)indoles, a novel series of platelet activating factor antagonists. J. Med. Chem 1994, 37, 2011–2032. [DOI] [PubMed] [Google Scholar]
- (167).Sheth S; Brito R; Mukherjea D; Rybak LP; Ramkumar V Adenosine receptors: expression, function and regulation. Int. J. Mol. Sci 2014, 15, 2024–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Carpenter B; Lebon G Human adenosine A2A receptor: molecular mechanism of ligand binding and activation. Front. Pharmacol 2017, 8, 898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Pinna A Adenosine A2A receptor antagonists in Parkinson’s disease: progress in clinical trials from the newly approved istradefylline to drugs in early development and those already discontinued. CNS Drugs 2014, 28, 455–474. [DOI] [PubMed] [Google Scholar]
- (170).Yang Z; Li X; Ma H; Zheng J; Zhen X; Zhang X Replacement of amide with bioisosteres led to a new series of potent adenosine A2A receptor antagonists. Bioorg. Med. Chem. Lett 2014, 24, 152–155. [DOI] [PubMed] [Google Scholar]
- (171).Stocchi F; Rascol O; Hauser RA; Huyck S; Tzontcheva A; Capece R; Ho TW; Sklar P; Lines C; Michelson D; Hewitt DJ Randomized trial of preladenant, given as monotherapy, in patients with early Parkinson disease. Neurology 2017, 88, 2198–2206. [DOI] [PubMed] [Google Scholar]
- (172).Cho JH; Gregersen PK Genomics and the multifactorial nature of human autoimmune disease. N. Engl. J. Med 2011, 365, 1612–1623. [DOI] [PubMed] [Google Scholar]
- (173).Chang Y; Xu S; Ding K Tyrosine kinase 2 (TYK2) allosteric inhibitors to treat autoimmune diseases. J. Med. Chem 2019, 62, 8951–8952. [DOI] [PubMed] [Google Scholar]
- (174).Liang J; Van Abbema A; Balazs M; Barrett K; Berezhkovsky L; Blair WS; Chang C; Delarosa D; DeVoss J; Driscoll J; Eigenbrot C; Goodacre S; Ghilardi N; MacLeod C; Johnson A; Bir Kohli P; Lai Y; Lin Z; Mantik P; Menghrajani K; Nguyen H; Peng I; Sambrone A; Shia S; Smith J; Sohn S; Tsui V; Ultsch M; Williams K; Wu LC; Yang W; Zhang B; Magnuson S Identification of an imidazopyridine scaffold to generate potent and selective TYK2 inhibitors that demonstrate activity in an in vivo psoriasis model. Bioorg. Med. Chem. Lett 2017, 27, 4370–4376. [DOI] [PubMed] [Google Scholar]
- (175).Baures PW; Ojala WH; Costain WJ; Ott MC; Pradhan A; Gleason WB; Mishra RK; Johnson RL Design, synthesis, and dopamine receptor modulating activity of diketopiperazine peptidomimetics of L-prolyl-L-leucylglycinamide. J. Med. Chem 1997, 40, 3594–3600. [DOI] [PubMed] [Google Scholar]
- (176).Chiu S; Paulose CS; Mishra RK Effect of L-prolyl-L-leucyl-glycinamide (PLG) on neuroleptic-induced catalepsy and dopamine/neuroleptic receptor bindings. Peptides 1981, 2, 105–111. [DOI] [PubMed] [Google Scholar]
- (177).Srivastava LK; Bajwa SB; Johnson RL; Mishra RK Interaction of L-prolyl-L-leucyl glycinamide with dopamine D2 receptor: evidence for modulation of agonist affinity states in bovine striatal membranes. J. Neurochem 1988, 50, 960–968. [DOI] [PubMed] [Google Scholar]
- (178).Baures PW; Ojala WH; Gleason WB; Mishra RK; Johnson RL Design, synthesis, X-ray analysis, and dopamine receptor-modulating activity of mimics of the ″C5″ hydrogen-bonded conformation in the peptidomimetic 2-oxo-3(R)-[(2(S)-pyrrolidinylcarbonyl)amino]-1-pyrrolidineacetamide. J. Med. Chem 1994, 37, 3677–3683. [DOI] [PubMed] [Google Scholar]
- (179).Yu KL; Rajakumar G; Srivastava LK; Mishra RK; Johnson RL Dopamine receptor modulation by conformationally constrained analogues of Pro-Leu-Gly-NH2. J. Med. Chem 1988, 31, 1430–1436. [DOI] [PubMed] [Google Scholar]
- (180).Mishra RK; Srivastava LK; Johnson RL Modulation of high-affinity CNS dopamine D2 receptor by L-pro-L-leu-glycinamide (PLG) analogue 3(R)-(N-L-prolylamino)-2-oxo-1-pyrrolidineacetamide. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 1990, 14, 821–827. [DOI] [PubMed] [Google Scholar]
- (181).Subasinghe NL; Bontems RJ; McIntee E; Mishra RK; Johnson RL Bicyclic thiazolidine lactam peptidomimetics of the dopamine receptor modulating peptide Pro-Leu-Gly-NH2. J. Med. Chem 1993, 36, 2356–2361. [DOI] [PubMed] [Google Scholar]
- (182).Borthwick AD 2,5-Diketopiperazines: synthesis, reactions, medicinal chemistry, and bioactive natural products. Chem. Rev 2012, 112, 3641–3716. [DOI] [PubMed] [Google Scholar]
- (183).Verma K; Zang T; Penning TM; Trippier PC Potent and highly selective aldo-keto reductase 1C3 (AKR1C3) inhibitors act as chemotherapeutic potentiators in acute myeloid leukemia and T-Cell acute lymphoblastic leukemia. J. Med. Chem 2019, 62, 3590–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).de la Fuente Revenga M; Fernandez-Saez N; Herrera-Arozamena C; Morales-Garcia JA; Alonso-Gil S; Perez-Castillo A; Caignard DH; Rivara S; Rodriguez-Franco MI Novel N-acetyl bioisosteres of melatonin: melatonergic receptor pharmacology, physicochemical studies, and phenotypic assessment of their neurogenic potential. J. Med. Chem 2015, 58, 4998–5014. [DOI] [PubMed] [Google Scholar]
- (185).Byrns MC; Penning TM Type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase (AKR1C3): role in breast cancer and inhibition by non-steroidal anti-inflammatory drug analogs. Chem.-Biol. Interact 2009, 178, 221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (186).Ji Q; Chang L; Stanczyk FZ; Ookhtens M; Sherrod A; Stolz A Impaired dihydrotestosterone catabolism in human prostate cancer: critical role of AKR1C2 as a pre-receptor regulator of androgen receptor signaling. Cancer Res. 2007, 67, 1361–1369. [DOI] [PubMed] [Google Scholar]
- (187).Penning TM; Bauman DR; Jin Y; Rizner TL Identification of the molecular switch that regulates access of 5α-DHT to the androgen receptor. Mol. Cell. Endocrinol 2007, 265–266, 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (188).Yin YD; Fu M; Brooke DG; Heinrich DM; Denny WA; Jamieson SM The activity of SN33638, an inhibitor of AKR1C3, on testosterone and 17β-estradiol production and function in castration-resistant prostate cancer and ER-positive breast cancer. Front. Oncol 2014, 4, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (189).Loriot Y; Fizazi K; Jones RJ; Van den Brande J; Molife RL; Omlin A; James ND; Baskin-Bey E; Heeringa M; Baron B; Holtkamp GM; Ouatas T; De Bono JS Safety, tolerability and anti-tumour activity of the androgen biosynthesis inhibitor ASP9521 in patients with metastatic castration-resistant prostate cancer: multi-centre phase I/II study. Invest. New Drugs 2014, 32, 995–1004. [DOI] [PubMed] [Google Scholar]
- (190).Kikuchi A; Furutani T; Azami H; Watanabe K; Niimi T; Kamiyama Y; Kuromitsu S; Baskin-Bey E; Heeringa M; Ouatas T; Enjo K In vitro and in vivo characterisation of ASP9521: a novel, selective, orally bioavailable inhibitor of 17β-hydroxysteroid dehydrogenase type 5 (17βHSD5; AKR1C3). Invest. New Drugs 2014, 32, 860–870. [DOI] [PubMed] [Google Scholar]
- (191).Endo S; Matsunaga T; Kanamori A; Otsuji Y; Nagai H; Sundaram K; El-Kabbani O; Toyooka N; Ohta S; Hara A Selective inhibition of human type-5 17β-hydroxysteroid dehydrogenase (AKR1C3) by baccharin, a component of Brazilian propolis. J. Nat. Prod 2012, 75, 716–721. [DOI] [PubMed] [Google Scholar]
- (192).Zang T; Verma K; Chen M; Jin Y; Trippier PC; Penning TM Screening baccharin analogs as selective inhibitors against type 5 17β-hydroxysteroid dehydrogenase (AKR1C3). Chem.-Biol. Interact 2015, 234, 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Verma K; Zang T; Gupta N; Penning TM; Trippier PC Selective AKR1C3 inhibitors potentiate chemotherapeutic activity in multiple acute myeloid leukemia (AML) cell lines. ACS Med. Chem. Lett 2016, 7, 774–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Pandi-Perumal SR; Srinivasan V; Maestroni GJM; Cardinali DP; Poeggeler B; Hardeland R Melatonin: nature’s most versatile biological signal? FEBS J. 2006, 273, 2813–2838. [DOI] [PubMed] [Google Scholar]
- (195).Jockers R; Maurice P; Boutin JA; Delagrange P Melatonin receptors, heterodimerization, signal transduction and binding sites: what’s new? Br. J. Pharmacol 2008, 154, 1182–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (196).Li Y; Li S; Zhou Y; Meng X; Zhang JJ; Xu DP; Li HB Melatonin for the prevention and treatment of cancer. Oncotarget 2017, 8, 39896–39921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (197).Alghamdi BS The neuroprotective role of melatonin in neurological disorders. J. Neurosci. Res 2018, 96, 1136–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (198).Karamitri A; Jockers R Melatonin in type 2 diabetes mellitus and obesity. Nat. Rev. Endocrinol 2019, 15, 105–125. [DOI] [PubMed] [Google Scholar]
- (199).Leclerc V; Fourmaintraux E; Depreux P; Lesieur D; Morgan P; Howell HE; Renard P; Caignard DH; Pfeiffer B; Delagrange P; Guardiola-Lemaitre B; Andrieux J Synthesis and structure-activity relationships of novel naphthalenic and bioisosteric related amidic derivatives as melatonin receptor ligands. Bioorg. Med. Chem 1998, 6, 1875–1887. [DOI] [PubMed] [Google Scholar]
- (200).Morgan PJ; Williams LM; Davidson G; Lawson W; Howell E Melatonin receptors on ovine pars tuberalis: characterization and autoradiographicai localization. J. Neuroendocrinol 1989, 1, 1–4. [DOI] [PubMed] [Google Scholar]
- (201).He P; Ouyang X; Zhou S; Yin W; Tang C; Laudon M; Tian S A novel melatonin agonist Neu-P11 facilitates memory performance and improves cognitive impairment in a rat model of Alzheimer’ disease. Horm. Behav 2013, 64, 1–7. [DOI] [PubMed] [Google Scholar]
- (202).Tian SW; Laudon M; Han L; Gao J; Huang FL; Yang YF; Deng HF Antidepressant- and anxiolytic effects of the novel melatonin agonist Neu-P11 in rodent models. Acta Pharmacol. Sin 2010, 31, 775–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Rami M; Landagaray E; Ettaoussi M; Boukhalfa K; Caignard DH; Delagrange P; Berthelot P; Yous S Novel conformationally constrained analogues of agomelatine as new melatoninergic ligands. Molecules 2013, 18, 154–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).Pala D; Lodola A; Bedini A; Spadoni G; Rivara S Homology models of melatonin receptors: challenges and recent advances. Int. J. Mol. Sci 2013, 14, 8093–8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (205).Jagtap AD; Kondekar NB; Sadani AA; Chern JW Ureas: applications in drug design. Curr. Med. Chem 2017, 24, 622–651. [DOI] [PubMed] [Google Scholar]
- (206).Ghosh AK; Brindisi M Urea derivatives in modern drug discovery and medicinal chemistry. J. Med. Chem 2020, 63 (6), 2751–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Roark WH; Roth BD; Holmes A; Trivedi BK; Kieft KA; Essenburg AD; Krause BR; Stanfield RL Inhibitors of acyl-CoA:cholesterol acyltransferase (ACAT). 2. modification of fatty acid anilide ACAT inhibitors: bioisosteric replacement of the amide bond. J. Med. Chem 1993, 36, 1662–1668. [DOI] [PubMed] [Google Scholar]
- (208).Augelli-Szafran CE; Blankley CJ; Roth BD; Trivedi BK; Bousley RF; Essenburg AD; Hamelehle KL; Krause BR; Stanfield RL Inhibitors of acyl-CoA:cholesterol acyltransferase. 5. identification and structure-activity relationships of novel beta-ketoamides as hypocholesterolemic agents. J. Med. Chem 1993, 36, 2943–2949. [DOI] [PubMed] [Google Scholar]
- (209).Trivedi BK; Holmes A; Stoeber TL; Blankley CJ; Roark WH; Picard JA; Shaw MK; Essenburg AD; Stanfield RL; Krause BR Inhibitors of acyl-Coa:cholesterol acyltransferase. 4. a novel series of urea ACAT inhibitors as potential hypocholesterolemic agents. J. Med. Chem 1993, 36, 3300–3307. [DOI] [PubMed] [Google Scholar]
- (210).Lebreton L; Annat J; Derrepas P; Dutartre P; Renaut P Structure-immunosuppressive activity relationships of new analogues of 15-deoxyspergualin. 1. structural modifications of the hydroxyglycine moiety. J. Med. Chem 1999, 42, 277–290. [DOI] [PubMed] [Google Scholar]
- (211).Chan L; Jin H; Stefanac T; Lavallee JF; Falardeau G; Wang W; Bedard J; May S; Yuen L Discovery of 1,6-naphthyridines as a novel class of potent and selective human cytomegalovirus inhibitors. J. Med. Chem 1999, 42, 3023–3025. [DOI] [PubMed] [Google Scholar]
- (212).Chan L; Jin H; Stefanac T; Wang W; Lavallee JF; Bedard J; May S Isoquinoline-6-carboxamides as potent and selective anti-human cytomegalovirus (HCMV) inhibitors. Bioorg. Med. Chem. Lett 1999, 9, 2583–2586. [DOI] [PubMed] [Google Scholar]
- (213).Chan L; Stefanac T; Lavallee JF; Jin H; Bedard J; May S; Falardeau G Design and synthesis of new potent human cytomegalovirus (HCMV) inhibitors based on internally hydrogen-bonded 1,6-naphthyridines. Bioorg. Med. Chem. Lett 2001, 11, 103–105. [DOI] [PubMed] [Google Scholar]
- (214).Bielawska A; Greenberg MS; Perry D; Jayadev S; Shayman JA; McKay C; Hannun YA (1S,2R)-D-erythro-2-(N-myristoylamino)-1-phenyl-1-propanol as an inhibitor of ceramidase. J. Biol. Chem 1996, 271, 12646–12654. [DOI] [PubMed] [Google Scholar]
- (215).Selzner M; Bielawska A; Morse MA; Rudiger HA; Sindram D; Hannun YA; Clavien PA Induction of apoptotic cell death and prevention of tumor growth by ceramide analogues in metastatic human colon cancer. Cancer Res. 2001, 61, 1233–1240. [PubMed] [Google Scholar]
- (216).Lim S; Ryu JH; Im C; Yim CB Synthesis and cytotoxicity of new 3-alkyl-1-(1-methyl-2-phenylethyl)ureas related to ceramide. Arch. Pharmacal Res 2003, 26, 270–274. [DOI] [PubMed] [Google Scholar]
- (217).Chang YT; Choi J; Ding S; Prieschl EE; Baumruker T; Lee JM; Chung SK; Schultz PG The synthesis and biological characterization of a ceramide library. J. Am. Chem. Soc 2002, 124, 1856–1857. [DOI] [PubMed] [Google Scholar]
- (218).Bieberich E; Hu B; Silva J; MacKinnon S; Yu RK; Fillmore H; Broaddus WC; Ottenbrite RM Synthesis and characterization of novel ceramide analogs for induction of apoptosis in human cancer cells. Cancer Lett. 2002, 181, 55–64. [DOI] [PubMed] [Google Scholar]
- (219).Wang H; Huwaimel B; Verma K; Miller J; Germain TM; Kinarivala N; Pappas D; Brookes PS; Trippier PC Synthesis and antineoplastic evaluation of mitochondrial complex II (succinate dehydrogenase) inhibitors derived from Atpenin A5. ChemMedChem 2017, 12, 1033–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (220).Patrick DA; Wenzler T; Yang S; Weiser PT; Wang MZ; Brun R; Tidwell RR Synthesis of novel amide and urea derivatives of thiazol-2-ethylamines and their activity against Trypanosoma brucei rhodesiense. Bioorg. Med. Chem 2016, 24, 2451–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (221).Patrick DA; Gillespie JR; McQueen J; Hulverson MA; Ranade RM; Creason SA; Herbst ZM; Gelb MH; Buckner FS; Tidwell RR Urea derivatives of 2-aryl-benzothiazol-5-amines: a new class of potential drugs for human African trypanosomiasis. J. Med. Chem 2017, 60, 957–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (222).He JB; Ren YL; Sun QS; You GY; Zhang L; Zou P; Feng LL; Wan J; He HW Design, synthesis and molecular docking of amide and urea derivatives as Escherichia coli PDHc-E1 inhibitors. Bioorg. Med. Chem 2014, 22, 3180–3186. [DOI] [PubMed] [Google Scholar]
- (223).Ghosh AK; Brindisi M Organic carbamates in drug design and medicinal chemistry. J. Med. Chem 2015, 58, 2895–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (224).Eckhardt BJ; Gulick RM Drugs for HIV Infection In Infectious Diseases, 4th ed.; Cohen J, Opal SM, Powderly WG, Ed.; Elsevier Ltd., 2017; pp 1293–1308. [Google Scholar]
- (225).Bursavich MG; Rich DH Designing non-peptide peptidomimetics in the 21st century: inhibitors targeting conformational ensembles. J. Med. Chem 2002, 45, 541–558. [DOI] [PubMed] [Google Scholar]
- (226).Ghosh AK; Thompson WJ; McKee SP; Duong TT; Lyle TA; Chen JC; Darke PL; Zugay JA; Emini EA; Schleif WA; Huff JR; Anderson PS 3-Tetrahydrofuran and pyran urethanes as high-affinity P2-ligands for HIV-1 protease inhibitors. J. Med. Chem 1993, 36, 292–294. [DOI] [PubMed] [Google Scholar]
- (227).Thompson WJ; Ghosh AK; Holloway MK; Lee HY; Munson PM; Schwering JE; Wai J; Darke PL; Zugay J; Emini EA; Schleif WA; Huff JR; Anderson PS 3′-Tetrahydrofuranylglycine as a novel, unnatural amino acid surrogate for asparagine in the design of inhibitors of the HIV protease. J. Am. Chem. Soc 1993, 115, 801–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (228).Ghosh AK; Thompson WJ; Holloway MK; McKee SP; Duong TT; Lee HY; Munson PM; Smith AM; Wai JM; Darke PL; Zugay JA; Emini EA; Schleif WA; Huff JR; Anderson PS Potent HIV protease inhibitors: the development of tetrahydrofuranylglycines as novel P2-ligands and pyrazine amides as P3-ligands. J. Med. Chem 1993, 36, 2300–2310. [DOI] [PubMed] [Google Scholar]
- (229).Morsy A; Trippier PC Current and emerging pharmacological targets for the treatment of Alzheimer’s disease. J. Alzheimer’s Dis 2019, 72, S145–S176. [DOI] [PubMed] [Google Scholar]
- (230).Wong GT; Manfra D; Poulet FM; Zhang Q; Josien H; Bara T; Engstrom L; Pinzon-Ortiz M; Fine JS; Lee HJ; Zhang L; Higgins GA; Parker EM Chronic treatment with the γ-secretase inhibitor LY-411,575 inhibits β-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J. Biol. Chem 2004, 279, 12876–12882. [DOI] [PubMed] [Google Scholar]
- (231).Peters JU; Galley G; Jacobsen H; Czech C; David-Pierson P; Kitas EA; Ozmen L Novel orally active, dibenzazepinone-based γ-secretase inhibitors. Bioorg. Med. Chem. Lett 2007, 17, 5918–5923. [DOI] [PubMed] [Google Scholar]
- (232).Fransson R; Nordvall G; Bylund J; Carlsson-Jonsson A; Kratz JM; Svensson R; Artursson P; Hallberg M; Sandstrom A Exploration and pharmacokinetic profiling of phenylalanine based carbamates as novel substance p 1–7 analogues. ACS Med. Chem. Lett 2014, 5, 1272–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (233).Kamenecka TM; Park YJ; Lin LS; de Laszlo S; McCauley ED; Van Riper G; Egger L; Kidambi U; Mumford RA; Tong S; Tang W; Colletti A; Teffera Y; Stearns R; MacCoss M; Schmidt JA; Hagmann WK Amidines as amide bond replacements in VLA-4 antagonists. Bioorg. Med. Chem. Lett 2004, 14, 2323–2326. [DOI] [PubMed] [Google Scholar]
- (234).Hagmann WK; Durette PL; Lanza T; Kevin NJ; de Laszlo SE; Kopka IE; Young D; Magriotis PA; Li B; Lin LS; Yang G; Kamenecka T; Chang LL; Wilson J; MacCoss M; Mills SG; Van Riper G; McCauley E; Egger LA; Kidambi U; Lyons K; Vincent S; Stearns R; Colletti A; Teffera J; Tong S; Fenyk-Melody J; Owens K; Levorse D; Kim P; Schmidt JA; Mumford RA The discovery of sulfonylated dipeptides as potent VLA-4 antagonists. Bioorg. Med. Chem. Lett 2001, 11, 2709–2713. [DOI] [PubMed] [Google Scholar]
- (235).Choudhary A; Raines RT An evaluation of peptide-bond isosteres. ChemBioChem 2011, 12, 1801–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (236).Thorarensen A; Wakefield BD; Romero DL; Marotti KR; Sweeney MT; Zurenko GE; Rohrer DC; Han F; Bryant GL Jr. Preparation of novel anthranilic acids as antibacterial agents. extensive evaluation of alternative amide bioisosteres connecting the A- and the B-rings. Bioorg. Med. Chem. Lett 2007, 17, 2823–2827. [DOI] [PubMed] [Google Scholar]
- (237).Volonterio A; Bravo P; Zanda M Synthesis of partially modified retro and retroinverso ψ[NHCH(CF3)]-peptides. Org. Lett 2000, 2, 1827–1830. [DOI] [PubMed] [Google Scholar]
- (238).Sani M; Volonterio A; Zanda M The trifluoroethylamine function as peptide bond replacement. ChemMedChem 2007, 2, 1693–1700. [DOI] [PubMed] [Google Scholar]
- (239).Zanda M Trifluoromethyl group: an effective xenobiotic function for peptide backbone modification. New J. Chem 2004, 28, 1401–1411. [Google Scholar]
- (240).Gauthier JY; Chauret N; Cromlish W; Desmarais S; Duong LT; Falgueyret JP; Kimmel DB; Lamontagne S; Leger S; LeRiche T; Li CS; Masse F; McKay DJ; Nicoll-Griffith DA; Oballa RM; Palmer JT; Percival MD; Riendeau D; Robichaud J; Rodan GA; Rodan SB; Seto C; Therien M; Truong VL; Venuti MC; Wesolowski G; Young RN; Zamboni R; Black WC The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K. Bioorg. Med. Chem. Lett 2008, 18, 923–928. [DOI] [PubMed] [Google Scholar]
- (241).Grabowskal U; Chambers TJ; Shiroo M Recent developments in cathepsin K inhibitor design. Curr. Opin. Drug Discovery Dev 2005, 8, 619–630. [PubMed] [Google Scholar]
- (242).Falgueyret JP; Desmarais S; Oballa R; Black WC; Cromlish W; Khougaz K; Lamontagne S; Masse F; Riendeau D; Toulmond S; Percival MD Lysosomotropism of basic cathepsin K inhibitors contributes to increased cellular potencies against off-target cathepsins and reduced functional selectivity. J. Med. Chem 2005, 48, 7535–7543. [DOI] [PubMed] [Google Scholar]
- (243).Li CS; Deschenes D; Desmarais S; Falgueyret JP; Gauthier JY; Kimmel DB; Leger S; Masse F; McGrath ME; McKay DJ; Percival MD; Riendeau D; Rodan SB; Therien M; Truong VL; Wesolowski G; Zamboni R; Black WC Identification of a potent and selective non-basic cathepsin K inhibitor. Bioorg. Med. Chem. Lett 2006, 16, 1985–1989. [DOI] [PubMed] [Google Scholar]
- (244).Black WC; Bayly CI; Davis DE; Desmarais S; Falgueyret JP; Leger S; Li CS; Masse F; McKay DJ; Palmer JT; Percival MD; Robichaud J; Tsou N; Zamboni R Trifluoroethylamines as amide isosteres in inhibitors of cathepsin K. Bioorg. Med. Chem. Lett 2005, 15, 4741–4744. [DOI] [PubMed] [Google Scholar]
- (245).Mullard A Merck &Co. drops osteoporosis drug odanacatib. Nat. Rev. Drug Discovery 2016, 15, 669. [DOI] [PubMed] [Google Scholar]
- (246).Butler CR; Ogilvie K; Martinez-Alsina L; Barreiro G; Beck EM; Nolan CE; Atchison K; Benvenuti E; Buzon L; Doran S; Gonzales C; Helal CJ; Hou X; Hsu MH; Johnson EF; Lapham K; Lanyon L; Parris K; O’Neill BT; Riddell D; Robshaw A; Vajdos F; Brodney MA Aminomethyl-derived beta secretase (BACE1) inhibitors: engaging Gly230 without an anilide functionality. J. Med. Chem 2017, 60, 386–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (247).Gaudette F; Raeppel S; Nguyen H; Beaulieu N; Beaulieu C; Dupont I; Macleod AR; Besterman JM; Vaisburg A Identification of potent and selective VEGFR receptor tyrosine kinase inhibitors having new amide isostere headgroups. Bioorg. Med. Chem. Lett 2010, 20, 848–852. [DOI] [PubMed] [Google Scholar]
- (248).Kim K; Kang J; Kim S; Choi S; Lim S; Im C; Yim C Synthesis and cytotoxicity of new aromatic ceramide analogs with alkylsulfonamido chains. Arch. Pharmacal Res 2007, 30, 570–580. [DOI] [PubMed] [Google Scholar]
- (249).Donelson JL; Hodges-Loaiza HB; Henriksen BS; Hrycyna CA; Gibbs RA Solid-phase synthesis of prenylcysteine analogs. J. Org. Chem 2009, 74, 2975–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (250).Majmudar JD; Hahne K; Hrycyna CA; Gibbs RA Probing the isoprenylcysteine carboxyl methyltransferase (Icmt) binding pocket: sulfonamide modified farnesyl cysteine (SMFC) analogs as Icmt inhibitors. Bioorg. Med. Chem. Lett 2011, 21, 2616–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (251).Koehn FE; Carter GT The evolving role of natural products in drug discovery. Nat. Rev. Drug Discovery 2005, 4, 206–220. [DOI] [PubMed] [Google Scholar]
- (252).Ferreira AK; Tavares MT; Pasqualoto KF; de Azevedo RA; Teixeira SF; Ferreira-Junior WA; Bertin AM; de-Sa-Junior PL; Barbuto JA; Figueiredo CR; Cury Y; Damiao MC; Parise-Filho R RPF151, a novel capsaicin-like analogue: in vitro studies and in vivo preclinical antitumor evaluation in a breast cancer model. Tumor Biol. 2015, 36, 7251–7267. [DOI] [PubMed] [Google Scholar]
- (253).de-Sa-Junior PL; Pasqualoto KF; Ferreira AK; Tavares MT; Damiao MC; de Azevedo RA; Camara DA; Pereira A; de Souza DM; Parise Filho R RPF101, a new capsaicin-like analogue, disrupts the microtubule network accompanied by arrest in the G2/M phase, inducing apoptosis and mitotic catastrophe in the MCF-7 breast cancer cells. Toxicol. Appl. Pharmacol 2013, 266, 385–398. [DOI] [PubMed] [Google Scholar]
- (254).Choy N; Choi H-I; Jung WH; Kim CR; Yoon H; Kim SC; Lee TG; Koh JS Synthesis of irreversible HIV-1 protease inhibitors containing sulfonamide and sulfone as amide bond isosteres. Bioorg. Med. Chem. Lett 1997, 7, 2635–2638. [Google Scholar]
- (255).Shu B; Gong P Structural basis of viral RNA-dependent RNA polymerase catalysis and translocation. Proc. Natl. Acad. Sci. U. S. A 2016, 113, E4005–E4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (256).Venkataraman S; Prasad B; Selvarajan R RNA dependent RNA Polymerases: insights from structure, function and evolution. Viruses 2018, 10, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (257).Abdurakhmanov E; Oie Solbak S; Danielson UH Biophysical mode-of-action and selectivity analysis of allosteric inhibitors of hepatitis C virus (HCV) polymerase. Viruses 2017, 9, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (258).Kati W; Koev G; Irvin M; Beyer J; Liu Y; Krishnan P; Reisch T; Mondal R; Wagner R; Molla A; Maring C; Collins C In vitro activity and resistance profile of dasabuvir, a nonnucleoside hepatitis C virus polymerase inhibitor. Antimicrob. Agents Chemother 2015, 59, 1505–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (259).Pierra Rouviere C; Amador A; Badaroux E; Convard T; Da Costa D; Dukhan D; Griffe L; Griffon JF; LaColla M; Leroy F; Liuzzi M; Loi AG; McCarville J; Mascia V; Milhau J; Onidi L; Paparin JL; Rahali R; Sais E; Seifer M; Surleraux D; Standring D; Dousson C Synthesis of potent and broad genotypically active NS5B HCV non-nucleoside inhibitors binding to the thumb domain allosteric site 2 of the viral polymerase. Bioorg. Med. Chem. Lett 2016, 26, 4536–4541. [DOI] [PubMed] [Google Scholar]
- (260).Rochon K; Proteau-Gagne A; Bourassa P; Nadon JF; Cote J; Bournival V; Gobeil F Jr.; Guerin B; Dory YL; Gendron L Preparation and evaluation at the delta opioid receptor of a series of linear leu-enkephalin analogues obtained by systematic replacement of the amides. ACS Chem. Neurosci 2013, 4, 1204–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (261).Hann MM; Sammes PG; Kennewell PD; Taylor JB On double bond isosters of the peptide bond; an enkephalin analogue. J. Chem. Soc., Chem. Commun 1980, 234–235. [Google Scholar]
- (262).Hann MM; Sammes PG; Kennewell PD; Taylor JB On the double bond isostere of the peptide bond: preparation of an enkephalin analogue. J. Chem. Soc., Perkin Trans 1 1982, 307–314. [Google Scholar]
- (263).Wipf P; Henninger TC; Geib SJ Methyl- and (trifluoromethyl)alkene peptide isosteres: synthesis and evaluation of their potential as β-turn promoters and peptide mimetics. J. Org. Chem 1998, 63, 6088–6089. [DOI] [PubMed] [Google Scholar]
- (264).Donner P; Randolph JT; Huang P; Wagner R; Maring C; Lim BH; Colletti L; Liu Y; Mondal R; Beyer J; Koev G; Marsh K; Beno D; Longenecker K; Pilot-Matias T; Kati W; Molla A; Kempf D High potency improvements to weak aryl uracil HCV polymerase inhibitor leads. Bioorg. Med. Chem. Lett 2013, 23, 4367–4369. [DOI] [PubMed] [Google Scholar]
- (265).Randolph JT; Krueger AC; Donner PL; Pratt JK; Liu D; Motter CE; Rockway TW; Tufano MD; Wagner R; Lim HB; Beyer JM; Mondal R; Panchal NS; Colletti L; Liu Y; Koev G; Kati WM; Hernandez LE; Beno DWA; Longenecker KL; Stewart KD; Dumas EO; Molla A; Maring CJ Synthesis and biological characterization of aryl uracil inhibitors of hepatitis C virus NS5B polymerase: discovery of ABT-072, a trans-stilbene analog with good oral bioavailability. J. Med. Chem 2018, 61, 1153–1163. [DOI] [PubMed] [Google Scholar]
- (266).Durham PL Calcitonin gene-related peptide (CGRP) and migraine. Headache 2006, 46 (Suppl. 1), S3–S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (267).Russell FA; King R; Smillie SJ; Kodji X; Brain SD Calcitonin gene-related peptide: physiology and pathophysiology. Physiol. Rev 2014, 94, 1099–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (268).Stump CA; Bell IM; Bednar RA; Bruno JG; Fay JF; Gallicchio SN; Johnston VK; Moore EL; Mosser SD; Quigley AG; Salvatore CA; Theberge CR; Blair Zartman C; Zhang XF; Kane SA; Graham SL; Vacca JP; Williams TM The discovery of highly potent CGRP receptor antagonists. Bioorg. Med. Chem. Lett 2009, 19, 214–217. [DOI] [PubMed] [Google Scholar]
- (269).Kim JJ; Wood MR; Stachel SJ; de Leon P; Nomland A; Stump CA; McWherter MA; Schirripa KM; Moore EL; Salvatore CA; Selnick HG (E)-Alkenes as replacements of amide bonds: development of novel and potent acyclic CGRP receptor antagonists. Bioorg. Med. Chem. Lett 2014, 24, 258–261. [DOI] [PubMed] [Google Scholar]
- (270).Larsen SD; Hester MR; Craig Ruble J; Kamilar GM; Romero DL; Wakefield B; Melchior EP; Sweeney MT; Marotti KR Discovery and initial development of a novel class of antibacterials: inhibitors of Staphylococcus aureus transcription/translation. Bioorg. Med. Chem. Lett 2006, 16, 6173–6177. [DOI] [PubMed] [Google Scholar]
- (271).Brouwer AJ; Elgersma RC; Jagodzinska M; Rijkers DT; Liskamp RM Delayed fibril formation of amylin(20–29) by incorporation of alkene dipeptidosulfonamide isosteres obtained by solid phase olefin cross metathesis. Bioorg. Med. Chem. Lett 2008, 18, 78–84. [DOI] [PubMed] [Google Scholar]
- (272).Yamamoto Y; Kimachi T; Kanaoka Y; Kato S; Bessho K; Matsumoto T; Kusakabe T; Sugiura Y Synthesis and DNA binding properties of amide bond-modified analogues related to distamycin. Tetrahedron Lett. 1996, 37, 7801–7804. [Google Scholar]
- (273).Arcamone F; Penco S; Orezzi P; Nicolella V; Pirelli A Structure and synthesis of distamycin A. Nature 1964, 203, 1064–1065. [DOI] [PubMed] [Google Scholar]
- (274).Wada M; Doi R; Hosotani R; Higashide S; Ibuka T; Habashita H; Nakai K; Fujii N; Imamura M Effect of a new bombesin receptor antagonist, (E)-alkene bombesin isostere, on amylase release from rat pancreatic acini. Pancreas 1995, 10, 301–305. [DOI] [PubMed] [Google Scholar]
- (275).Mishra CB; Gusain S; Shalini S; Kumari S; Prakash A; Kumari N; Yadav AK; Kumari J; Kumar K; Tiwari M Development of novel carbazole derivatives with effective multifunctional action against Alzheimer’s diseases: design, synthesis, in silico, in vitro and in vivo investigation. Bioorg. Chem 2020, 95, 103524. [DOI] [PubMed] [Google Scholar]
- (276).Mishra CB; Kumari S; Manral A; Prakash A; Saini V; Lynn AM; Tiwari M Design, synthesis, in-silico and biological evaluation of novel donepezil derivatives as multi-target-directed ligands for the treatment of Alzheimer’s disease. Eur. J. Med. Chem 2017, 125, 736–750. [DOI] [PubMed] [Google Scholar]
- (277).Mishra CB; Manral A; Kumari S; Saini V; Tiwari M Design, synthesis and evaluation of novel indandione derivatives as multifunctional agents with cholinesterase inhibition, anti-β-amyloid aggregation, antioxidant and neuroprotection properties against Alzheimer’s disease. Bioorg. Med. Chem 2016, 24, 3829–3841. [DOI] [PubMed] [Google Scholar]
- (278).Fu Y; Bieschke J; Kelly JW E-olefin dipeptide isostere incorporation into a polypeptide backbone enables hydrogen bond perturbation: probing the requirements for Alzheimer’s amyloidogenesis. J. Am. Chem. Soc 2005, 127, 15366–15367. [DOI] [PubMed] [Google Scholar]
- (279).Shue YK; Tufano MD; Carrera GM Jr.; Kopecka H; Kuyper SL; Holladay MW; Lin CW; Witte DG; Miller TR; Stashko M; Nadzan AM Double bond isosteres of the peptide bond: synthesis and biological activity of cholecystokinin (CCK) C-terminal hexapeptide analogs. Bioorg. Med. Chem 1993, 1, 161–171. [DOI] [PubMed] [Google Scholar]
- (280).Meanwell NA Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem 2018, 61, 5822–5880. [DOI] [PubMed] [Google Scholar]
- (281).Pasternak GW; Pan YX Mu opioids and their receptors: evolution of a concept. Pharmacol. Rev 2013, 65, 1257–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (282).Weinberger SB; Martinez JL Jr. Characterization of hydrolysis of [leu]enkephalin and D-ala2-[L-leu]enkephalin in rat plasma. J. Pharmacol. Exp. Ther 1988, 247, 129–135. [PubMed] [Google Scholar]
- (283).Thompson SE; Audus KL Leucine-enkephalin metabolism in brain microvessel endothelial cells. Peptides 1994, 15, 109–116. [DOI] [PubMed] [Google Scholar]
- (284).Karad SN; Pal M; Crowley RS; Prisinzano TE; Altman RA Synthesis and opioid activity of Tyr1-ψ[(Z)CF=CH]-Gly2 and Tyr1-ψ[(S)/(R)-CF3CH-NH]-Gly2 leu-enkephalin fluorinated peptidomimetics. ChemMedChem 2017, 12, 571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (285).Altman RA; Sharma KK; Rajewski LG; Toren PC; Baltezor MJ; Pal M; Karad SN Tyr1- ψ[(Z)CF=CH]-Gly2 fluorinated peptidomimetic improves distribution and metabolism properties of leu-enkephalin. ACS Chem. Neurosci 2018, 9, 1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (286).Nadon JF; Rochon K; Grastilleur S; Langlois G; Dao TT; Blais V; Guerin B; Gendron L; Dory YL Synthesis of Gly-ψ[(Z)CF=CH]-Phe, a fluoroalkene dipeptide isostere, and its incorporation into a leu-enkephalin peptidomimetic. ACS Chem. Neurosci 2017, 8, 40–49. [DOI] [PubMed] [Google Scholar]
- (287).Lajoie G; Lepine F; Lemaire S; Jolicoeur F; Aube C; Turcotte A; Belleau B Synthesis and biological activity of monothionated analogs of leucine-enkephalin. Int. J. Pept. Protein Res 1984, 24, 316–327. [DOI] [PubMed] [Google Scholar]
- (288).Proteau-Gagne A; Bournival V; Rochon K; Dory YL; Gendron L Exploring the backbone of enkephalins to adjust their pharmacological profile for the δ-opioid receptor. ACS Chem. Neurosci 2010, 1, 757–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (289).Lee C; Ma H; Hang JQ; Leveque V; Sklan EH; Elazar M; Klumpp K; Glenn JS The hepatitis C virus NS5A inhibitor (BMS-790052) alters the subcellular localization of the NS5A nonstructural viral protein. Virology 2011, 414, 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (290).Chang W; Mosley RT; Bansal S; Keilman M; Lam AM; Furman PA; Otto MJ; Sofia MJ Inhibition of hepatitis C virus NS5A by fluoro-olefin based γ-turn mimetics. Bioorg. Med. Chem. Lett 2012, 22, 2938–2942. [DOI] [PubMed] [Google Scholar]
- (291).Evelyn CR; Bell JL; Ryu JG; Wade SM; Kocab A; Harzdorf NL; Showalter HDH; Neubig RR; Larsen SD Design, synthesis and prostate cancer cell-based studies of analogs of the Rho/MKL1 transcriptional pathway inhibitor, CCG-1423. Bioorg. Med. Chem. Lett 2010, 20, 665–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (292).Canales E; Carlson JS; Appleby T; Fenaux M; Lee J; Tian Y; Tirunagari N; Wong M; Watkins WJ Tri-substituted acylhydrazines as tertiary amide bioisosteres: HCV NS5B polymerase inhibitors. Bioorg. Med. Chem. Lett 2012, 22, 4288–4292. [DOI] [PubMed] [Google Scholar]
- (293).Trippier PC Selecting good ‘drug-like’ properties to optimize small molecule blood-brain barrier penetration. Curr. Med. Chem 2016, 23, 1392–1407. [DOI] [PubMed] [Google Scholar]
- (294).Wuitschik G; Rogers-Evans M; Müller K; Fischer H; Wagner B; Schuler F; Polonchuk L; Carreira EM Oxetanes as promising modules in drug discovery. Angew. Chem., Int. Ed 2006, 45, 7736–7739. [DOI] [PubMed] [Google Scholar]
- (295).Bull JA; Croft RA; Davis OA; Doran R; Morgan KF Oxetanes: recent advances in synthesis, reactivity, and medicinal chemistry. Chem. Rev 2016, 116, 12150–12233. [DOI] [PubMed] [Google Scholar]
- (296).McLaughlin M; Yazaki R; Fessard TC; Carreira EM Oxetanyl peptides: novel peptidomimetic modules for medicinal chemistry. Org. Lett 2014, 16, 4070–4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (297).Powell NH; Clarkson GJ; Notman R; Raubo P; Martin NG; Shipman M Synthesis and structure of oxetane containing tripeptide motifs. Chem. Commun 2014, 50, 8797–8800. [DOI] [PubMed] [Google Scholar]
- (298).Wuitschik G; Carreira EM; Wagner B; Fischer H; Parrilla I; Schuler F; Rogers-Evans M; Muller K Oxetanes in drug discovery: structural and synthetic insights. J. Med. Chem 2010, 53, 3227–3246. [DOI] [PubMed] [Google Scholar]
- (299).Beadle JD; Powell NH; Raubo P; Clarkson GJ; Shipman M Synthesis of oxetane- and azetidine-containing spiro-cycles related to the 2,5-diketopiperazine framework. Synlett 2015, 27, 169–172. [Google Scholar]
- (300).Lovering F; Bikker J; Humblet C Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
- (301).Trippier PC; McGuigan C Boronic acids in medicinal chemistry: anticancer, antibacterial and antiviral applications. MedChemComm 2010, 1, 183–198. [Google Scholar]
- (302).Fernandes GFS; Denny WA; Dos Santos JL Boron in drug design: recent advances in the development of new therapeutic agents. Eur. J. Med. Chem 2019, 179, 791–804. [DOI] [PubMed] [Google Scholar]
- (303).Malde AK; Khedkar SA; Coutinho EC The B(OH)–NH analog is a surrogate for the amide bond (CO–NH) in peptides: an ab initio Study. J. Chem. Theory Comput 2007, 3, 619–627. [DOI] [PubMed] [Google Scholar]
- (304).Ramesh R; Reddy DS Quest for novel chemical entities through incorporation of silicon in drug scaffolds. J. Med. Chem 2018, 61, 3779–3798. [DOI] [PubMed] [Google Scholar]
- (305).Chen CA; Sieburth SM; Glekas A; Hewitt GW; Trainor GL; Erickson-Viitanen S; Garber SS; Cordova B; Jeffry S; Klabe RM Drug design with a new transition state analog of the hydrated carbonyl: silicon-based inhibitors of the HIV protease. Chem. Biol 2001, 8, 1161–1166. [DOI] [PubMed] [Google Scholar]
- (306).Madsen AS; Kristensen HME; Lanz G; Olsen CA The effect of various zinc binding groups on inhibition of histone deacetylases 1–11. ChemMedChem 2014, 9, 614–626. [DOI] [PubMed] [Google Scholar]
- (307).Slusarczyk M; Serpi M; Pertusati F Phosphoramidates and phosphonamidates (ProTides) with antiviral activity. Antivir. Chem. Chemother 2018, 26, 1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]