

Abstract
Polymer-drug conjugates are promising as strategies for drug delivery, because of their high drug loading capacity and low premature release profile. However, the preparation of these conjugates is often tedious. In this paper, we report an efficient method for polymer-drug conjugates using an ultrafast and reversible click reaction in a post-polymerization functionalization strategy. The reaction is based on the rapid condensation of aryl boronic acid functionalities with salicylhydroxamates. The polymer, bearing the latter functionality, has been designed such that the reaction with boronic acid bearing drugs induces an in situ self-assembly of the conjugates to form well-defined nanostructures. We show that this method is not only applicable for molecules with an intrinsic boronic acid group, but also for the other molecules that can be linked to aryl boronic acids through a self-immolative linker. The linker has been designed to cause traceless release of the attached drug molecules, the efficiency of which has been demonstrated through intracellular delivery.
Keywords: Reaction induced self-assembly, Polymer-drug conjugates, Reversible click reaction, Self-immolation, Nanomedicine
Graphical Abstract
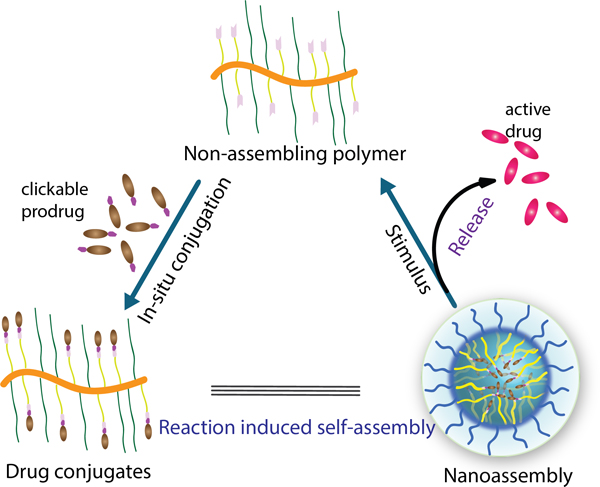
In-situ reversible click reaction induced polymer-drug conjugates and formation of nanoassembly with stimuli-induced pristine drug release process.
Nanosized molecular carriers offer the ability to improve drug pharmacokinetics and enhance the accumulation of therapeutics at the tumor site, a potentially promising approach for tissue targeting in cancer therapy.[1–18] The drug molecules can either be non-covalently encapsulated or covalently conjugated to these nanoparticles. The non-covalent approach is convenient in that a broad range of drugs can be incorporated into the hydrophobic interiors of an amphiphilic assembly.[5–8,18–22] In many of the systems, however, the instability in encapsulation causes the molecules to be prematurely released in biological systems, when undergoing distribution-induced dilution.[23–28] Also, many of these systems also exhibit low drug loading efficiency. The covalent conjugation strategy has the inherent capability of addressing both drug loading efficiency and encapsulation stability issues. Covalent polymer-drug conjugates are obtained either through direct polymerization of drug-containing monomers or with a post-polymerization modification process.[29–32] Among the two, because of the inherently low efficiency of the post-polymerization method, the direct polymerization approach is more commonly used for generating polymer-drug conjugates.[29–32] However, syntheses of functional monomers, that are compatible with the polymerization and self-assembly conditions, make the direct polymerization method quite tedious.[29–32] Moreover, in general, the covalently conjugated systems also require a mechanism by which the drug is released from the nanoparticle in its active form.
In sum, non-covalent encapsulation has the advantage of simply mixing the drug and nanoassembly components to prepare drug-containing nanoparticles, while the covalent approach provides high thermodynamic stability of the conjugates. A useful strategy would combine the advantages of procedural ease of the non-covalent method with the stability and efficiency of encapsulation offered by the covalent conjugation method. Additionally, it is critical that the strategy also allows for the drug to be efficiently released in its pristine active form at the target location.
We were inspired by reaction induced self-assembly (RISA) approaches via tuning the hydrophilic-lipophilic balance (HLB), such as the polymerization induced self-assembly (PISA), for preparing nanoparticles.[33–35] While the fundamentals of the self-assembly process have been quite nicely explored, we became interested in developing systems with clear biomaterials implications. In this paper, we use a simple and efficient reversible click reaction as the post-polymerization modification strategy to prepare polymer-drug conjugates (Figure 1). Specifically, we utilize a fast and efficient boronic acid-salicylhydroxamate reversible click reaction for the drug conjugation, which then also induces the in situ self-assembly of the conjugates. In addition to the highly efficient conjugation reaction, the versatility of this approach is further demonstrated with two more features in the drug release process. First, since the click reaction is reversible, the process naturally lends itself to stimulus-induced release of drug molecules from the conjugate. In addition to utilizing boronic acid itself as the handle for causing the stimulus-induced release, we also show that the strategy lends itself to introducing other cue-induced release of drug molecules, using linker engineering. The versatility of the method to accommodate a variety of functional groups demonstrates its broad applicability to drug molecules (beyond boronate-based ones). Similarly, the ability to tune molecular release in response to different stimuli offers to engineer drug release profiles for a variety of biological microenvironments. We disclose these findings in this manuscript.
Figure 1.

Schematic illustration of polymer-drug reaction for conjugates with in-situ induced nanoassembly of drug-conjugates formation and stimuli induced pristine drug release process.
The reaction between salicylhydroxamate and boronic acid functionalities is considered to be highly efficient with the bimolecular rate constant of ~7x106 M−2s−1.[36] With this rate, this reaction would be considered to be the fastest click reaction reported thus far.[36–40] We envisaged that the fast kinetics can be used for in situ conjugation of small molecule drugs onto polymers with high efficiency. Furthermore, the pH-induced reversibility of this reaction should also offer controlled drug release under mild acidic conditions.
To test the utility of such a process, we used a potent boronic acid-based drug. As boronic acid moiety is considered to be a biostere of carboxylic acid, boronic acid-based drugs have been developed, especially as proteasome inhibitors. For instance, bortezomib (BTZ) is an FDA-approved anticancer drug with a boronic acid functionality. (Figure 2a).[41–43] In the present work, BTZ was first chosen as the candidate drug for the study here. To conjugate this molecule to the polymer, the latter must be functionalized with the salicylhydroxamate (SaH) group. Thus, polymer P1 was prepared by an initial RAFT co-polymerization of trityl-protected salicylhyroxymate monomer and PEG-methacrylate monomer (MW=500 Da), followed by the deprotection of the trityl group. The purpose of the PEG-based co-monomer is to make the polymer sufficiently hydrophilic and potentially biocompatible, while the SaH functionality in the other co-monomer offers the handle for drug conjugation. The ratio of the two monomers in P1 was designed to be 1:1, which was confirmed by NMR (See Supplemental Experimental Procedures, Figure S19). For fluorescence microscopy studies, a corresponding rhodamine-labelled polymer P2 was synthesized through copolymerization of a small percentage (0.5% mole ratio) of a third dye-functionalized monomer. Synthetic protocols and characterizations of the polymers P1 (Mw=10.1 kDa, Ɖ=1.37) and P2 (Mw=10.3 kDa, Ɖ=1.46) are detailed in supporting information.
Figure 2.

Characterization of the in-situ reaction induced polymer-drug conjugates. a) Chemical illustration of the formation of polymer P1-BTZ conjugate and stimulus induced drug release, b) DLS for size of nanoassembly of polymer P1 and P1-BTZ conjugate, c) correlation function of P1 and P1-BTZ conjugate, d) Structure of rhodamine b labelled polymer P2-BTZ conjugate, e) Normalized UV absorbance for rhodamine b labelled polymer P2 and P2-BTZ conjugate.
The polymer-drug conjugate was prepared by simply mixing BTZ and P1. The polymer itself did not exhibit any self-assembly (or formed ill-defined polymer aggregates), as discerned by the poor correlation function observed in dynamic light scattering (DLS) experiments (Figure 2c). However, in the presence of BTZ, an in-situ formation of a nanoassembly with a size of ~50 nm was observed (Figure 2b and S1). This suggests that the addition of the more hydrophobic drug molecule causes the polymer to attain a hydrophilic-lipophilic balance needed for self-assembly into a nanoparticle. Drug conjugation to the polymer was demonstrated by UV absorbance (Figure 2e). We used the absorption peak from the rhodamine dye in P2 as the internal standard to demonstrate the loading of BTZ onto the polymer assembly, where the P2-BTZ exhibited similar size of 50 nm as P1-BTZ conjugates (Figure 2c and S2). Absorbance spectra of the polymer and the polymer-drug conjugate, normalized at 550 nm, shows a significantly higher absorbance at ~300 nm for the latter. This increase is attributed to the conjugation of BTZ to the polymer. We also confirmed that the drug conjugation does not affect rhodamine fluorescence in P2 (Figure S2c). Further, loading efficiency and capacity of BTZ in P1-BTZ conjugates was quantified by LC-MS/MS, which showed a loading capacity of 25% (246 μg BTZ per mg of P1) and loading efficiency of 50% (Figure S3).
Following the formation of the nanoassembly, we monitored the drug release under different pH values. The P1-BTZ conjugates revealed a pH dependent drug release feature, which exhibited stable encapsulation at physiological pH (7.4) with extended release kinetics under acidic pH (5.0) (Figure S4). Further, the polymer-drug conjugate nanoassembly was evaluated with three different cancer cell lines (HeLa, MDA-MB-231, and MCF-7). Rhodamine-labelled P2 was used to track the location of the conjugate in cells with confocal microscopy, while the cell nucleus was stained with hochest 3332. The conjugate exhibited good cell uptake for all the cell lines as the bright red color was dispersed throughout the cells, around the nucleus after 4 h (Figure 3a). Moreover, the assembled drug conjugate can efficiently kill all of three cancer cell types with clear dose- and time-dependent profiles (Figure 3b), where the polymer itself did not exhibit any toxicity even at a high concentration of 1 mg/mL (Figure S4). We also noted that the conjugates cause the cell death at a slower rate (compare with Figure S5 and Table S1), which is attributed to the controlled release of the drug from the conjugate (Figure S6).
Figure 3.

Cell uptake and toxicity of the polymer-drug conjugates. (a) confocal images for cell uptake of polymer-BTZ conjugate toward different cell lines (BF: bright field). Cells were incubated with rhodamine b labelled polymer-BTZ conjugate for 4 h. (b) Cytotoxicity (MTT assay) of polymer-BTZ conjugate toward different cell lines at different times. The concentration is based on the drug (BTZ).
The results above are promising for the intracellular delivery of drugs that contain an intrinsic boronic acid group. However, most small molecule drugs do not bear a boronic acid functional group. To extend the methodology to other functional groups that are commonly found in drug molecules, we were interested in developing linkers that bridge the boronic acid functional group and the drug molecules. Since chemical modifications might significantly decrease the potency of drugs, we targeted a stimulus-responsive degradable linker that offers to release the drug in its traceless active form at the targeted site. To this end, we first used self-immolative linkers that are responsive to reactive oxygen species (ROS), as highly oxidizing environment is found in certain pathological sites such as in tumor cells, where the concentration is 2 to 3 orders of magnitude higher than in normal cells and can reach up to 100 µM.[44–47] The boronic acid functional group has two functions in that it is the key click reaction handle and is also inherently ROS responsive.
The molecular design, which allows for the tertiary alcohol of camptothecin (CPT) to be used as the handle for conjugation, is shown in Figure 4a (see SI for synthetic details). When the nanoconjugate is subjected to ROS conditions, the aryl boronic acid moiety would be oxidized to the corresponding phenol, which would induce a 1,6-benzyl self-immolation cascade[48–52] to release the CPT molecule in its original form. CPT is an anticancer drug with a well-established pharmacology profile.[53] The procedure for preparation of the prodrug with a boronic acid terminal functional group is detailed in the Supporting Information. The formation of the nanocarrier P1-CPTROS (Figure 4a) was achieved through an in-situ reaction between the salicylhydroxamate group of the polymer P1 and boronic acid group of prodrug in aqueous media (see SI for details). The resultant polymer-drug conjugate self-assembled to nanosized particles with a diameter of ~70 nm (Figure 4c and 4d). Transmission electron microscopy (TEM) studies suggest that the nanoaggregate had a spherical shape with the size of ~50 nm (Figure 4e). The smaller size in TEM, compared to that found with dynamic light scattering (DLS), is likely due to the shrinkage of the particles upon drying in the former technique. Here too, note that the polymer itself exhibits ill-defined aggregates, as ascertained by the bad correlation function (Figure 2d). Again, this suggests that the in-situ conjugation-induced self-assembly of polymeric nanoparticles can be achieved. The characteristic absorption peak of CPT at ~366 nm (Figure S7) further confirmed the drug conjugation. This distinct peak allowed for the quantification of the drug loading, which was found to be as high as ~70% for this post-polymerization conjugation method (Figure S7 and S8). In comparison, the physical encapsulation of CPT drug without boronic acid functionalization exhibited very poor loading capacity (~2%) and loading efficiency (~5%) (Figure S9), further demonstrating the advantages of the RISA process. Further, we tested the scope of the methodology by tuning the ratio of the SaH functional group to PEG unit in polymers (10%, 25% and 75% SaH group). The reaction between polymers and boronic acid containing prodrug (CPTROS) resulted in good nanosized assemblies for conjugates, except P3-CPTROS with the smallest amount of the hydrophobic functional group (10%). Also, all polymers (P1-P5), by themselves, exhibited ill-defined aggregates (Figure S10–S12). These results were consistent with the RISA process for nanoassembly formation, through alterations in the polymer HLB. It is also to be noted that all these polymers exhibited very high loading drug efficiency (70–90%) (Figure S13).
Figure 4.

Polymer-prodrug conjugates formation. a) structure of ROS sensitive polymer-drug conjugates (P1-CPTROS), b) structure of redox sensitive polymer-drug conjugates (P1-CPTredox), c) DLS of the assemblies from polymer-drug conjugates, d) correlation functions for these assemblies from DLS data, e) TEM image of in-situ formed assembly of ROS sensitive polymer-drug conjugates (P1-CPTROS), f) TEM image of in-situ formed assembly of redox sensitive polymer-drug conjugates (P1-CPTredox).
We hypothesized that the linker would offer to release CPT in its pristine form, upon encountering the ROS stimulus. To investigate this possibility, the nature of the released molecules was analyzed in the presence of H2O2, using LC-MS/MS (see SI for details, Figure S14–S23). The elution profile shows the evolution of the release of pristine drug, suggesting that the self-immolation reaction indeed causes traceless drug release. In control, the linker is hydrolytically stable under different pH values (7.4 and 5.0) (Figure S24). Also, the drug release kinetics was understandably dependent on the ROS concentration (Figure 5c and S25). Due to the easy formation of polymer-drug conjugates with well-defined nanostructure and triggerable release profile, the cytotoxicity of the drug conjugates was assessed toward different cancer cell lines. The polymer-drug conjugate also exhibited good cell killing ability with a concentration (encapsulated CPT drug) and time-dependent profile (Figure 5e), which is comparable to the free CPT (Figure S28 and Table S2).
Figure 5.

Drug release under stimuli. a) schematic illustration of the drug release from ROS sensitive polymer-drug conjugates, b) schematic illustration of the drug release from redox sensitive polymer-drug conjugates, c) LC-MS/MS MRM chromatogram for monitoring CPT release from CPTROS under 1 mM H2O2, d) LC-MS/MS MRM chromatogram for monitoring CPT release from CPTredox under 10 mM DTT, e) cytotoxicity studies for ROS sensitive polymer-drug conjugates toward different cells after different time incubation, f) cytotoxicity studies for redox sensitive polymer-drug conjugates toward different cells after different time incubation. The concentration is based on the equivalent CPT drug.
To further expand the scope of this approach, we were interested in developing a strategy where the conjugating boronic acid moiety itself is not involved in the stimulus-induced release of the drug. To this end, we targeted a glutathione (GSH) responsive linker, as this tripeptide concentration is ~103 times higher inside cells, compared to the extracellular environment.[54–56] The linker is based on a disulfide bond, which is connected to the β-position of the carbonate functionality that connects to the drug, CPT. The relative positioning of these two functionalities would ensure a self-immolative cyclization,[48,57] when the disulfide bond is reductively cleaved to produce the corresponding thiol (Figure 4b and 5b). The other terminus of the linker contains the boronic acid moiety that would connect with the polymer P1 to cause the in-situ reaction induced self-assembly. Similar to the ROS sensitive CPT-based nanoassembly above, a ~70 nm spherical assembly was observed, as discerned from DLS and TEM studies (Figure 4d–4f). The drug loading efficiency was found to be ~74% (Figure S8). The traceless release of CPT drug from the redox sensitive drug conjugates was also demonstrated by LC-MS/MS (details in Supporting Information, Figure S14–S26) in the presence of 10 mM DTT (mimics intracellular reduction conditions) and the release kinetics was monitored (Figure 5d). The drug can be efficiently released, as all of the prodrug can be transformed into CPT within 1 h. In comparison, there is negligible release amount, when 10 µM DTT (mimics extracellular conditions) was used (Figure S25). Further, this linker is also hydrolytically stable under different pH values (7.4 and 5.0) (Figure S27). These results suggest that the CPT drug can be selectively released in response to an intracellular biological trigger. Further, cell toxicity for the nanoassembly of this polymer-drug conjugate was also studied toward three different cell lines (HeLa, MDA-MB-231, MCF-7). The polymer-drug conjugates can efficiently kill the cancer cells with dosage and time dependence (Figure 5f, Figure S28 and Table S2).
In summary, a novel in-situ salicylhydroxamate-boronic acid based reversible click chemistry was used to achieve reaction induced assembly. With the reactive component as drug molecules that inherently contain boronic acid moieties, this approach is effective in producing polymer-drug conjugates that self-assemble into nanoscopic structures. The utility of this approach is further extended to drug molecules that do not contain boronic acid moieties with two different strategies. In one approach, we use the boronic acid moiety’s inherent reactivity with oxidants to generate ROS-responsive release of drug molecules. In a second approach, the linker separates the conjugation chemistry from the responsive release chemistry. In both cases, we have shown that the drug molecules can be released in its traceless form, i.e. without any remnants of the conjugated components that might affect their efficacy. These polymer-drug nanoconjugates also exhibit efficient cellular activity. The simplicity and versatility of the approach highlight the potential utility of this reaction induced self-assembly of polymer-drug conjugates in nanomedicine.
Supplementary Material
Acknowledgements
We thank the support from the National Institutes of Health (GM-128181 and GM-136395).
Footnotes
Supporting information for this article is given via a link at the end of the document.
Contributor Information
Bin Liu, Department of Chemistry, University of Massachusetts, Amherst, Massachusetts 01003, USA.
Ruiling Wu, Department of Chemistry, University of Massachusetts, Amherst, Massachusetts 01003, USA.
Shuai Gong, Department of Chemistry, University of Massachusetts, Amherst, Massachusetts 01003, USA.
Hang Xiao, Department of Food Science, University of Massachusetts, Amherst, Massachusetts 01003, USA; Center for Bioactive Delivery, Institute for Applied Life Sciences, University of Massachusetts, Amherst, Massachusetts 01003, USA; Molecular and Cellular Biology Program, University of Massachusetts, Amherst, Massachusetts 01003, USA.
S. Thayumanavan, Department of Chemistry, University of Massachusetts, Amherst, Massachusetts 01003, USA; Center for Bioactive Delivery, Institute for Applied Life Sciences, University of Massachusetts, Amherst, Massachusetts 01003, USA; Molecular and Cellular Biology Program, University of Massachusetts, Amherst, Massachusetts 01003, USA
References
- [1].Blanco E, Shen H, Ferrari M, Nat. Biotechnol 2015, 33, 941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Roemeling C, Jiang W, Chan CK, Weissman IL, Kim BYS, Trends Biotechnol 2017, 35, 159–171. [DOI] [PubMed] [Google Scholar]
- [3].Chen H, Zhang W, Zhu G, Xie J, Chen X, Nat. Rev. Mater 2017, 2, 17024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Tibbitt MW, Dahlman JE, Langer R, J. Am. Chem. Soc 2016, 138, 704–717. [DOI] [PubMed] [Google Scholar]
- [5].Li Y, Maciel D, Rodrigues J, Shi X, Tomás H, Chem. Rev 2015, 115, 8564–8608. [DOI] [PubMed] [Google Scholar]
- [6].Mura S, Nicolas J, Couvreur P, Nat. Mater 2013, 12, 991–1003. [DOI] [PubMed] [Google Scholar]
- [7].Elsabahy M, Heo G, Lim S, Sun G, Wooley KL, Chem. Rev 2015, 115, 10967–11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cheetham AG, Chakroun RW, Ma W, Cui H, Chem. Soc. Rev 2017, 46, 6638–6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Li Y, Wang Y, Huang G, Gao J, Chem. Rev 2018, 118, 5359–5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Srinivasarao M, Low PS, Chem. Rev 2017, 117, 12133–12164. [DOI] [PubMed] [Google Scholar]
- [11].Pelaz B, Alexiou C, Alvarez-Puebla RA et al. , ACS Nano 2017, 11, 2313–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Webber MJ, Langer R, Chem. Soc. Rev 2017, 46, 6600–6620. [DOI] [PubMed] [Google Scholar]
- [13].Sun T, Zhang Y, Pang B, Hyun D, Yang M, Xia Y, Angew. Chem. Int. Ed 2014, 53, 12320–12364. [DOI] [PubMed] [Google Scholar]
- [14].Torchilin V, Adv. Drug Delivery Rev 2011, 63, 131–135. [DOI] [PubMed] [Google Scholar]
- [15].Wong PT, Choi S, Chem. Rev 2015, 115, 3388–3432. [DOI] [PubMed] [Google Scholar]
- [16].Kim KT, Cornelissen JJLM, Nolte RJM, van Hest JCM, Adv. Mater 2009, 21, 2787–2791. [Google Scholar]
- [17].Chali SP, Ravoo BJ, Angew. Chem. Int. Ed 2020, 59, 2962–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Uhrich KE, Cannizzaro SM, Langer RS, Shakesheff KM, Chem. Rev 1999, 99, 3181–3198. [DOI] [PubMed] [Google Scholar]
- [19].Cabral H, Miyata K, Osada K, Kazunori K, Chem. Rev 2018, 118, 6844–6892. [DOI] [PubMed] [Google Scholar]
- [20].Pattni BS, Chupin VV, Torchilin VP, Chem. Rev 2015, 115, 10938–10966. [DOI] [PubMed] [Google Scholar]
- [21].Lv S, Wu Y, Cai K, He H; Li Y, Lan M, Chen X, Cheng J, Yin L, J. Am. Chem. Soc 2018, 140, 1235–1238. [DOI] [PubMed] [Google Scholar]
- [22].Cheetham AG, Zhang P, Lin Y, Lock LL, Cui H J. Am. Chem. Soc 2013, 135, 2907–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Miller T, Breyer S, van Colen G, Mier W, Haberkorn U, Geissler S, Voss S, Weigandt M, Goepferich A, Int. J. Pharm 2013, 445, 117–124. [DOI] [PubMed] [Google Scholar]
- [24].Liu B, Thayumanavan S, J. Am. Chem. Soc 2017, 139, 2306–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Owen SC, Chan DPY, Shoichet MS, Nano Today 2012, 7, 53–65. [Google Scholar]
- [26].Kim S, Shi Y, Kim JY, Park K, Cheng JX, Expert Opin. Drug Delivery 2010, 7, 49–62. [DOI] [PubMed] [Google Scholar]
- [27].Liu B, Thayumanavan S, Chem 2019, 5, 3166–3183. [Google Scholar]
- [28].Liu B, Thayumanavan S, Biomacromolecules 2017, 18, 4163–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Liu J, Liu W, Weitzhandler I, Bhattacharyya J, Li X, Wang J, Qi Y, Bhattacharjee S, Chilkoti A, Angew. Chem. Int. Ed 2015, 54, 1002–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhang Y, Yin Q, Yin L, Ma L, Tang L, Cheng J, Angew. Chem. Int. Ed 2013, 52, 6435–6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Liao L, Liu J, Dreaden EC, Morton SW, Shopsowitz KE, Hammond PT, Johnson JA, J. Am. Chem. Soc 2014, 136, 5896–5899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hu X, Hu J, Tian J, Ge Z, Zhang G, Luo K, Liu S, J. Am. Chem. Soc 2013, 135, 17617–17629. [DOI] [PubMed] [Google Scholar]
- [33].Wong S, Zhao J, Cao C, Wong CK, Kuchel RP, Luca SD, Hook JM, Garvey CJ, Smith S, Ho J, Stenzel MH, Nat. Commun 2019, 10, 582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Li S, Wang J, Shen J, Wu B, He Y, ACS Macro Lett 2018, 7, 437–441. [DOI] [PubMed] [Google Scholar]
- [35].Warren NJ, Armes SP, J. Am. Chem. Soc 2014, 136, 10174–10185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Shin SBY, Almeida RD, Gerona-Navarro G, Bracken C, Jaffrey SR, Chem. Biol 2010, 17, 1171–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Arzt M, Seidler C, Ng DYW, Weil T, Chem. Asian J 2014, 9, 1994–2003. [DOI] [PubMed] [Google Scholar]
- [38].Ng DYW, Arzt M, Wu Y, Kuan SL, Lamla M, Weil T Angew. Chem. Int. Ed 2014, 53, 324–328. [DOI] [PubMed] [Google Scholar]
- [39].Knall A, Slugovc C, Chem. Soc. Rev 2013, 42, 5131–5142. [DOI] [PubMed] [Google Scholar]
- [40].Liu B, Ianosi-Irimie M, Thayumanavan S, ACS Nano 2019, 13, 9408–9420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Su J, Chen F, Cryns VL, Messersmith PB, J. Am. Chem. Soc 2011, 133, 11850–11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wu S, Qi R, Kuang H, Wei Y, Jing X, Meng F, Huang Y, ChemPlusChem 2013, 78, 175–184. [Google Scholar]
- [43].Stubelius A, Lee S, Almutairi A, Acc. Chem. Res 2019, 52, 3108–3119. [DOI] [PubMed] [Google Scholar]
- [44].Kwon S, Ko H, You DG, Kataoka K, Park JH, Acc. Chem. Res 2019, 527, 1771–1782. [DOI] [PubMed] [Google Scholar]
- [45].Ye H, Zhou Y, Liu X, Chen Y, Duan S, Zhu R, Liu Y, Yin L, Biomacromolecules 2019, 20, 2441–2463. [DOI] [PubMed] [Google Scholar]
- [46].Chance B, Sies H, Physiol. Rev 1979, 59, 527–605. [DOI] [PubMed] [Google Scholar]
- [47].Szatrowski TP, Nathan CF, Cancer Res 1991, 51, 794–798. [PubMed] [Google Scholar]
- [48].Blencowe CA, Russell AT, Greco F, Hayes W, Thornthwaite DW, Polym. Chem 2011, 2, 773–790. [Google Scholar]
- [49].Jourden JLM, Cohen SM, Angew. Chem. Int. Ed 2010, 49, 6795–6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Broaders KE, Grandhe S, Fréchet JMJ, J. Am. Chem. Soc 2011, 133, 756–758. [DOI] [PubMed] [Google Scholar]
- [51].Jourden JLM, Daniel KB, Cohen SM, Chem. Commun 2011, 47, 7968–7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Mahoney KM, Goswami PP, Syed A, Kolker P, Shannan B, Smith EA, Winter AH, J. Org. Chem 2014, 79, 11740–11743. [DOI] [PubMed] [Google Scholar]
- [53].Pommier Y, Nat. Rev. Cancer 2006, 6, 789. [DOI] [PubMed] [Google Scholar]
- [54].Bernkop-Schnürch A, Adv. Drug Delivery Rev 2005, 57, 1569–1582. [DOI] [PubMed] [Google Scholar]
- [55].Sivakumar S, Bansal V, Cortez C, Chong S-F, Zelikin AN, Adv. Mater 2009, 21, 1820–1824. [Google Scholar]
- [56].Ryu J, Chacko RT, Jiwpanich S, Bickerton S, Babu RP, Thayumanavan S, J. Am. Chem. Soc 2010, 132, 17227–17235. [DOI] [PubMed] [Google Scholar]
- [57].Riber CF, Smith AAA, Zelikin AN, Adv. Healthcare Mater 2015, 4, 1887–1890. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.