

Abstract
There is a paucity in the development of new mechanistic insights and therapeutic approaches for treating psychiatric disease. One of the major challenges is reflected in the growing consensus that risk for these diseases is not determined by a single gene, but rather is polygenic, arising from the action and interaction of multiple genes. Canonically, experimental models in mice have been designed to ascertain the relative contribution of a single gene to a disease by systematic manipulation (e.g. mutation or deletion) of a known candidate gene. Because these studies have been largely carried out using inbred isogenic mouse strains, in which there is no (or very little) genetic diversity among subjects, it is difficult to identify unique allelic variants, gene modifiers, and epigenetic factors that strongly affect the nature and severity of these diseases. Here we review various methods that take advantage of existing genetic diversity or that increase genetic variance in mouse models to (1) strengthen conclusions of single gene function; (2) model diversity among human populations; and (3) dissect complex phenotypes that arise from the actions of multiple genes.
Keywords: strain differences, neuropsychiatric, isogenic, congenic, selective breeding, outbred, quantitative trait locus
1. Introduction
Psychiatric disorders account for approximately 13% of all disease in the world1,2. They are also a leading cause of death and disability3, and incur staggering economic costs, with global estimates for psychiatric illnesses (in 2010) reaching 2.5 trillion U.S. dollars4.
To better understand psychiatric conditions, a standard research method is to genetically alter the function or expression of a candidate gene in model organisms, such as transgenic mice. Testing whether the resulting phenotype in these mice mimics the symptomology of a human condition is used to determine if a gene is ‘causative’ of disease. Results from single-gene studies like these have led to key insights into several fundamental mechanisms underlying basic brain function5 and the identification of genetic insults6 that correlate with the expression of traits consistent with psychiatric disorders.
However, experiments targeting single genes in animal models also have notable limitations. For example, psychiatric diseases are characterized by complex and multifaceted symptoms involving changes in cognition, motivation, and affect, which likely arise from the interaction of multiple different genes (e.g.7). Thus, while a single-gene approach may establish the relative contribution of a single gene to a phenotype in question, it may not fully describe an entire disease state. In addition, many psychiatric disorders (e.g. autism or schizophrenia) are spectrum disorders, in which individuals diagnosed with the same disorder exhibit distinct attributes and impairments. While the different presentations may ultimately be linked to a common underlying mechanism, the final expression is likely modified by a combination of multiple genetic, epigenetic, or environmental factors. Importantly, because of interactions between different allelic variants and/or modifying genes, the genetic background of the mouse model being used, while often overlooked, can profoundly impact the effect of the gene under investigation. There are many examples where similar experiments conducted in transgenic mice harboring the same mutation on different genetic backgrounds have produced discordant results, such as differences in the severity or types of traits expressed (e.g.8–13). Finally, even when the effects of a mutation are solely related to the function of the gene (i.e. not influenced by genetic background), the resulting phenotype(s) produced in a homogenous population of inbred mice may be incongruent with the effects of the same mutation in humans, where genetics and environmental effects vary widely from person to person14–18.
Rodent models have been used very successfully to address these challenges. The approaches we present here have been deployed in both rats and mice, each with their own advantages and disadvantages19. Due to space constraints, we primarily focus this review on mice, but also highlight one example using rats. We will start by presenting a broad overview of inbred isogenic mouse strains, and identify some of the strengths and weaknesses of their use in neuroscience research. We then provide descriptions of alternative strategies and their advantages, including leveraging multiple isogenic strains and generating hybrid or outbred mouse lines. Throughout the review, we highlight exemplar studies which have used these strategies to uncover neurobiological mechanisms underlying the expression of complex phenotypes.
2. Using isogenic strains to establish single-gene function
Isogenic strains are defined as a group of mice resulting from 20 or more generations of full sibling mating (i.e. inbreeding) originating from a single breeding pair. All members of an isogenic strain have less than 2% genetic variance, making each mouse a (near) clone of any other mouse in that strain. The first isogenic mouse strain, known as DBA (diluted brown, or non-agouti) was established by Clarence C. Little in 1909; today over 450 isogenic strains are available20,21. Initially, the use of isogenic mouse strains was largely promulgated by investigators studying tumor immunology and transplantation biology. These strains were key to the discovery that genetic matching between donor and recipient determines the success of tumor and nonneoplastic tissue transplantation17. Since then, the use of isogenic mouse strains has become widespread for the study of single-gene function. Theoretically, because every mouse within an isogenic strain is genetically identical, changes in a phenotype can be attributed directly to an engineered genetic alteration (insertion, deletion, or mutation). While this approach has vast utility and has resulted in a number of important mechanistic insights, the use of isogenic strains to elucidate single-gene function is not always appropriate or sufficient.
One obvious drawback to this approach is that it is generally only useful when the phenotype of interest is determined by the function of a single gene (termed “monogenic”). However, many psychiatric and neurological diseases exhibit complex and multi-faceted symptomology that likely arise from the action and interaction of multiple genes (termed “polygenic”). In support of this assertion, results from genome-wide association studies (GWAS) and genomic structural variation analyses have established that psychiatric disorders are best characterized as a collection of genetic alterations, including both rare and common variants (see22). As a result, the single-gene approach may not fully describe the entire disease state.
Another issue arises from the fact that single isogenic strains cannot recapitulate the modifying effects that different genetic backgrounds have on the gene or genes responsible for a particular disease state. For example, about half the cases of Dravet syndrome (a severe form of early-onset epilepsy) are caused by mutations in a single gene, SCN1A, that encodes a voltage-gated sodium channel, resulting in haploinsufficiency23. However, in mouse models different phenotypes were observed depending on the background strain used; deleting a single copy of the Scn1a gene (Scn1a+/−) on the 129S6/SvEvTac background resulted in no overt phenotype but the same mutation introduced on the C57BL/6J background resulted in spontaneous seizures and premature lethality, similar to the phenotype of human patients with Dravet syndrome24.
While discordant results (as illustrated by the Scn1a example above) may initially seem to confound interpretation of a single gene’s function, these strain-dependent phenotypic variations can be leveraged to reveal important biological mechanisms. For example, these variations may signal the presence of other genes that are important for modifying phenotype expression24,25. While it is possible to make the same mutation in two independent isogenic lines, practical considerations - including the availability of appropriate embryonic stem (ES) cell lines and transgene insertion effects - make this approach impractical. Instead, it is more feasible to generate congenic mice to identify other genes contributing to phenotype expression. We review this approach in the next section.
3. Using congenic strains to reveal gene-modifier effects
A congenic strain is generated through a breeding strategy that introduces genetic material – for example, an engineered transgene – from one isogenic line (donor) into a different isogenic line (recipient). Donor mice (with the transgene) are crossed with wildtype recipient mice to generate F1 hybrid offspring, which will be heterozygous at all loci (having one allele from the donor strain and one allele from the recipient strain); thus each F1 mouse will have one copy of the transgene. Subsequent and successive crossing of generations of offspring (with the transgene) with wildtype mice from the recipient line (called backcrossing) will produce mice that have progressively more genetic material from the recipient line (more than 98% after 5 backcrossed generations), while maintaining the transgene of interest from the donor line (Figure 1 A).
Figure 1: Experimental strategies for leveraging genetic tractability and diversity to understand complex phenotypes.
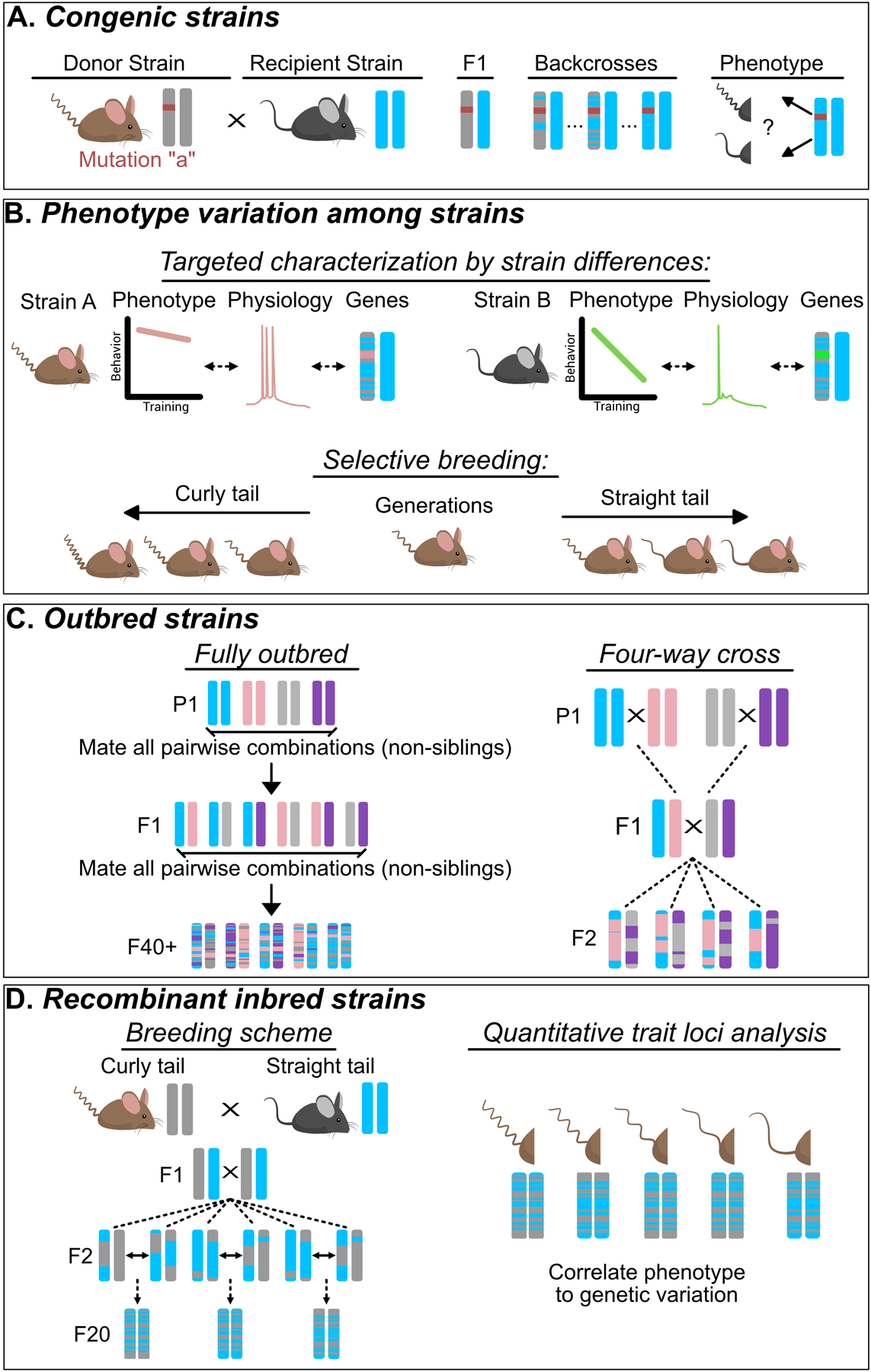
While these strategies can be used to study complex behavior, the physical trait of a curly tail has been used for this illustration. (A) Congenic Strains are used to test for the presence of gene modifiers on a phenotype of interest by determining if the phenotype is maintained in both the donor and recipient strain. (B) Phenotype variation among strains. Targeted characterization by strain differences (top) can be used to link physiological differences among strains to differential phenotypes. When differences between stains are not natively present, selective breeding (bottom) can be employed to generate quantitative divergence for a trait. (C) Outbred strains represent an important technique for testing hypotheses that rely on genetic heterogeneity, such as validating the generalizability of experimental treatments. Distinct outbred breeding strategies yield differences in the amount of genetic diversity and reproducibility. For example, a fully outbred scheme has higher diversity, but lower reproducibility, while a four-way cross has lower diversity, but higher reproducibility. Note that the more unique parental (P1) strains used, the more genetic diversity is generated (D) Recombinant inbred strains have the highest amount of genetic diversity while preserving genetic tractability. Typically, they are used for quantitative trait locus analysis (QTL) which is a technique that matches variation in gene expression to variation in a quantitative trait or phenotype.
Congenic strains are often used to determine whether an observed phenotype is solely dependent upon the gene of interest, or arises due to the interactions between the gene of interest and other genes in an isogenic background. These interactions are collectively referred to as ‘gene modifier effects’, and are manifested in several ways, including changes in the frequency that a phenotype is expressed among individuals, changes in the severity of a phenotype, loss of a phenotype, or even appearance of an entirely new phenotype26. These changes are formally explained as changes in the penetrance, expressivity, dominance, and pleiotropy of a trait and are reviewed in Figure 218.
Figure 2: Effects of modifier genes.
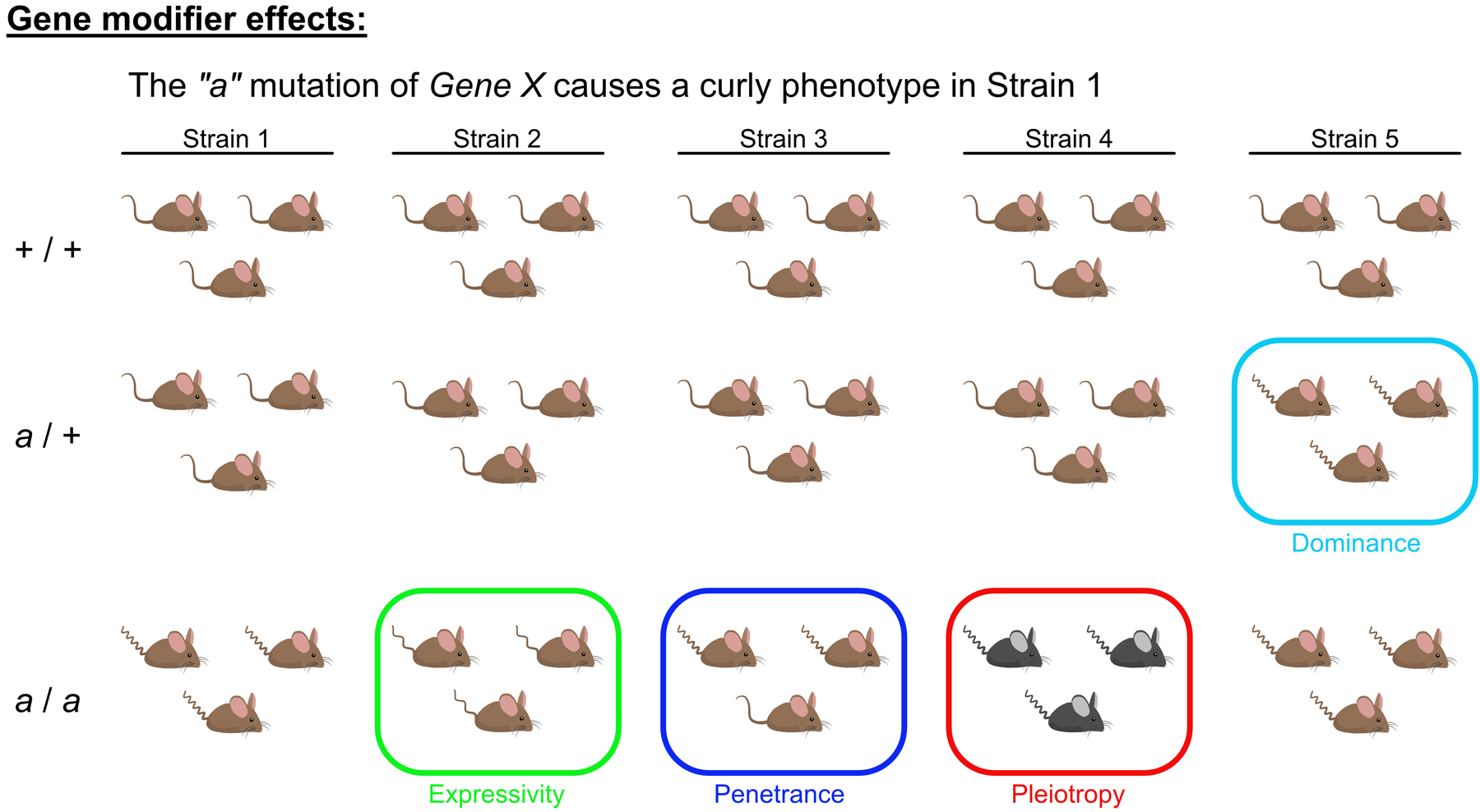
Modifier genes are those that affect the level of expression of other genes. The existence of gene modifier effects is evidenced by changes in the dominance, expressivity, penetrance and pleiotropy of a phenotype when a transgene is expressed in distinct background strains. In this example, homozygous expression of the hypothetical “a” mutation in Gene X causes a curly tail phenotype in mouse Strain 1. The “a” mutation is assessed among 4 additional strains (in columns) for wildtype (mutation null), heterozygous and mutation “a” homozygous populations (in rows). Some gene modifiers will change the dominance (cyan box) of a trait, a measure of the allele dosage needed to cause the curly tail phenotype. For example, in Strains 1–4, a single allele containing the “a” mutation is not sufficient to result in the curly tail phenotype, but in Strain 5 it is. Changes in expressivity (green box), or quantitative differences in the trait, can also be evident: while mice in Strain 2 exhibit a curly tail, there are fewer curls per length. Penetrance (blue box) refers to the proportion of mice that carry the allele (e.g. mutation “a”) that also display the curly tail phenotype; in Strain 3, only 2 out of 3 mutant homozygous mice exhibit a curly tail. Pleiotropy (red box) or the number of phenotypes generated by an allele is also indicative of gene modifiers. In Strain 4, mutant homozygous mice exhibit the curly tail phenotype, but in addition have a change in coat color that is caused by interactions between gene modifier(s) and mutation “a”.
In fact, congenic mice were used to identify gene modifiers of Dravet syndrome (described above); the Scn1a gene deletion was originally carried out in the 129S6/SvEvTac strain and subsequently transferred to C57BL/6J by backcrossing. In addition to finding strain differences in lethality and seizures among Scn1a+/− mice, researchers also found differences in the physiological function of single neurons in the hippocampus27. Altogether, these findings suggested that the C57BL/6J strain carried gene modifiers that altered disease outcomes. Subsequent gene-mapping studies and expression profiling of the 129S6/SvEvTac and C57BL/6J strains identified Gabra2, a gene involved in inhibitory neurotransmission, as a putative regulator of Dravet syndrome penetrance and expressivity24.
Isogenic and congenic strains are ideal models for ‘reverse genetics’, or studies where changes in phenotype expression are assessed after the disruption of a gene’s function. However, their use is limited when gene candidates underlying a behavior or disease are unknown. In the following sections we discuss approaches for using distinct pre-existing phenotypes among mouse strains to reveal the underlying neurophysiology and genes that account for trait expression.
4. Using phenotype variation among distinct isogenic strains to reveal underlying neurobiological mechanisms
4a. Targeted neuroanatomical and neurophysiological characterization guided by pre-existing strain-dependent phenotypes
It is clear that distinct isogenic mouse strains exhibit remarkably different behavioral phenotypes. For example, individual strains differ in their spatial learning and memory capabilities, risk-taking, aggression, and stress-responsivity, many of which mimic disease characteristics (see Table 1). Strain-dependent differences in neurophysiology and neuroanatomy have also been identified28–32. Linking these disparate behaviors to neurophysiological mechanisms is now possible with the advent of techniques (e.g. optogenetics33; in vivo calcium imaging34) that enable precise monitoring and/or perturbation of neural circuit function in vivo. Physiological differences between strains can then be used to target gene expression profiling to specific brain regions or cell types (Figure 1 B).
Table 1.
Example references for strain-dependent effects and features
| Strain Dependent Phenotypes: | Citations |
|---|---|
| learning and memory | Colom-Lapetina et al., 2017; Graybeal et al., 2014; Manahan-Vaughan and Schwegler, 2011; Neuner et al., 2016; Turner et al., 2017; Whitehouse et al., 2017 |
| aggression | Kessler et al., 1977; Takahashi et al., 2015 |
| fear and anxiety-like behavior | Gunduz-Cinar et al., 2018; Keum et al., 2016 |
| compulsive behavior | Mitra et al., 2017 |
| locomotor activity | Crawley et al., 1997; Podhorna and Brown, 2002 |
| parental behavior | Carola et al., 2006; Chourbaji et al., 2011 |
| vision | Mattapallil et al., 2012; Mehalow et al., 2003 |
| hearing | Turner et al., 2005; Zheng et al., 1999 |
| responses to pharmaceuticals and substances of abuse | Crabbe et al., 2016; Dockstader and van der Kooy, 2001; Holtz et al., 2015; Mulligan et al., 2008; Surget et al., 2016 |
| non-exhaustive list |
An example of an area which has benefited from leveraging innate differences among strains is the investigation and potential treatment of maladaptive fear and anxiety-related disorders. Distinct isogenic mouse strains display different susceptibility to maladaptive forms of fear, such as persistent fear, which remains elevated even in the absence of the threat, or generalized fear, in which related but non-threatful stimuli still produce a fear response35,36. In humans, the development and expression of fear- or anxiety-related disorders is highly individualized, and is strongly influenced by many interacting factors including environmental variables (i.e. early-life experience or trauma type), and biological variables (i.e. sex, genetic makeup, or epigenetic mechanisms). Not surprisingly, estimates of the heritability of anxiety-related disorders varies widely37,38. Thus, isogenic strains with distinct fear learning phenotypes not only serve as models of differential susceptibility (or resilience) to fear- and anxiety related disorders, but can also be used to elucidate how different factors interact to influence disease expression. To investigate maladaptive fear in the laboratory, researchers commonly use the Pavlovian fear learning paradigm39. During fear conditioning (FC), rodents increasingly exhibit a conditioned fear response to a stimulus (like a tone) when it consistently predicts an aversive event (ie mild electric shock); after this association is learned, the stimulus (now called the conditioned stimulus [CS]) can elicit fear by itself. However, if the CS is repeatedly presented in the absence of the aversive event, rodents learn to gradually diminish their fear responses to CS presentations, a behavior known as fear extinction (FE).
Researchers have leveraged distinct FE learning phenotypes among strains to identify factors that predispose maladaptive fear behavior. For example, some strains like the DBA/2J, are able to learn FE rapidly, but others, such as the 129S1, exhibit profound deficits in FE, such as persistent fear similar to that observed in anxiety-related disorders35,40–42. Work in this area has linked the predisposition to maladaptive fear in 129S1 mice to changes in regulation of the hypothalamic-pituitary-adrenal axis (HPA, a central regulator of the body’s response to stress) along with functional and neuroanatomical changes in cortico-amygdala circuitry35,41,43,44. These results are aligned with work in rodents and humans, which suggests that stress responsivity mediated by release of cortisol under control of the HPA axis and glucocorticoid receptor (GR) signaling may represent a node of dysregulation or vulnerability45–50. In fact, recent work comparing FE recall between strains discovered a novel gene—Ppid (Peptidylprolyl Isomerase D)— that can improve FE learning in a GR-dependent manner28. Interestingly, Ppid belongs to the same family as FKBP5 of tetratricopeptide repeat proteins that influence stress signaling via GRs and are commonly reported as biomarkers for individuals who experience trauma or are diagnosed with post-traumatic stress disorder (PTSD)51–54.
The heterogeneity of symptoms reported by individuals, high level of comorbidities, and variable heritability has made identifying new therapeutic approaches difficult55,56. Further, there is a high non-response or relapse rate to current pharmacological and behavioral treatments57. Strain differences can be exploited to evaluate new pharmacological35,41,58–60, behavioral61, and genetic/epigenetic approaches28,59,60,62 to ameliorate maladaptive fear in mice. For example, we have developed a novel FE protocol, termed Novelty-Facilitated Extinction (NFE), that enhances FE learning. We found that exposure to a fear CS in daily novel environments ameliorates FE deficits in the 129S1 strain61. Another form of NFE training, in which a novel auditory stimulus (instead of a novel context) is paired with a fear CS, is also more effective than standard FE in diminishing conditioned fear in rats63,64. Importantly, NFE has been shown to be effective in human populations. Healthy (control) participants exhibited a diminished conditioned stress response (galvanic skin conductance) to a fear CS paired with a novel stimulus, while exposure therapy carried out in multiple context was more effective therapeutically in phobic patients64–67.
Taken together, these examples provide a powerful illustration of how: (1) phenotype-driven characterization in rodents may engender greater translatability compared to studies focused on a candidate-gene; and (2) how phenotype diversity among strains can be leveraged to elucidate links between behavior, neurophysiology and genetics. These approaches are complementary to “big-data” projects that seek identification of biomarkers and environmental factors that make populations susceptible or resilient to psychiatric disorders (such as GWAS [ie68] and other large consortium studies [ie69]). While “big-data” projects identify important factors, “focal-data” projects using model organisms can reveal how individual differences (i.e. in genes, sex, environment) alter neurophysiological mechanisms which lead to susceptibility or resiliency (for example, by visualizing neural circuit activity during FE learning70). In addition, “focal-data” projects can inform which combination/sequence of treatments may be most effective at altering specific physiology and phenotypes (see59,60,71). However, it is important to note that one limitation of this approach is that it requires at least two mouse strains that natively exhibit significantly distinct phenotypes. In the absence of preexisting phenotypically divergent strains, investigators can often employ selective breeding to generate the desired phenotypic divergence; this is approach is described in the following section.
4b. Use of selective breeding to reveal underlying mechanisms associated with trait expression
Using selective breeding strategies to isolate behavioral phenotypes is another powerful method for studying the neurobiological substrates of complex traits. In this paradigm, offspring in each successive generation that display the most extreme measures of the behavior of interest are selected for inbreeding72. Over many generations, this strategy produces two distinct lines that exhibit vastly different performance for a specific behavior, such as high/low treadmill/wheel running73–76, high/low levels of exploration in novel environments77, high/low alcohol sensitivity78,79, high/low aggression80, and high/low anxiety81, among others (Figure 1 B).
One such example is the selectively-bred high responder (bHR) and low responder (bLR) rats that were differentiated based on their levels of exploration in novel environments. Interestingly, bHR rats also exhibit impulsive, aggressive, and reward-seeking behavior, while bLR rats exhibit anxiety- and depressive-like behaviors, suggesting that these traits may be genetically related to high or low propensity to explore novel environments77,82–84. In addition, it was discovered that bHR rats attend more to a stimulus predictive of a reward rather than the location of the reward delivery (termed “sign-tracking”), but that bLR rats attend more to the location of reward delivery (termed “goal-tracking”)85. In 2011, Flagel and colleagues cleverly utilized the bHR/bLR rats to explore the role of the neurotransmitter dopamine in stimulus reward learning. This study demonstrated that the bHR rats (sign trackers) had increased dopamine release in the nucleus accumbens in response to presentations of the predictive stimulus, whereas bLR did not show preferential dopamine signaling. These results suggest that a stimulus that is predictive of a reward gains greater incentive value for bHR (but not bLR) rats. Interestingly, sign and goal tracking behavior has been characterized in human populations in which sign-tracker individuals are more influenced by stimuli associated with reward and exhibit greater impulsivity86.
In summary, native trait differences between isogenic mouse strains and/or selective breeding to generate differential phenotypes can be used to model maladaptive or pathological conditions resembling disease features in humans. These methods are powerful because they can facilitate the identification of genes and neurobiological mechanisms underlying complex phenotypes which are likely polygenic and thus impractical to study using single-gene models or reverse genetics. Further, this approach also facilitates the discovery of genetically related traits and the ability to define physiological properties associated with the studied behavior. However, it is important to note that these approaches are still limited in the amount of genetic diversity accounted for in an experiment, because the comparisons are made between few (typically two) isogenic rodent lines. In the next section we review outbred strains, wherein genetic diversity is maximized, and how the resulting genetic variance can be used to assess the effectiveness of therapeutics and robustness of candidate disease mechanisms.
5. Using the genetic diversity of outbred strains to establish disease mechanisms and potential therapeutics
Outbred strains, which avoid sibling-to-sibling mating, maximize phenotypic variation because each individual animal is genetically unique. The advantage of using outbred rodents is apparent when considered in the context of human clinical trials where genetic background heterogeneity is inherent. Obviously, testing therapeutic efficacy in only one subject (or even many clones of the same subject, which is analogous to isogenic replicates) would not effectively represent the range of potential outcomes and/or drug interactions that may arise in different individuals (with unique genetic backgrounds). Thus, in studies developing or testing potential therapeutic interventions, utilizing outbred rodents as test subjects can maximize the content validity of experimental results.
One strategy to generate an outbred strain is to use many (often 4–8, or more) inbred isogenic parental strains, mated in every pairwise combinations, to produce a variety of F1 hybrid mice; then a complex, rotational breeding scheme, which avoids inbreeding, is followed for 40 or more generations to result in genetically unique mosaic mice (Figure 1 C). In theory, outbred strains can be generated by individual laboratories; however, the cost and organizational effort associated with the number of breeders and unique mating crosses required to generate these lines makes this impractical for most investigators. A more feasible option is to obtain outbred rodents from commercial facilities, and then maintain the line by continuous non-sibling mating of a large number (20 or more) of mice. In either case, it is important to remember that all individuals in every outbred line are completely unique and cannot be reproduced at any future time; therefore, it is impossible to fully replicate any study using outbred lines. To maximize both genetic diversity and reproducibility, a number of groups have used specific breeding strategies (such as the four-way cross) to generate mice (or rats) that are genetically heterogeneous at the level of the individual animal while maintaining a fixed gene distribution across the population; these populations can be reproduced by starting with the same parental lines and following the same breeding scheme (Figure 1 C).
An example of the power of this type of approach is provided by the Interventions Testing Program (ITP), developed by the National Institutes on Aging, which aims to identify compounds that can extend lifespan and reduce multiple forms of late-life disease87. Testing a candidate compound in a single isogenic strain could lead to spurious strain-specific conclusions, but testing in multiple isogenic strains would be prohibitive in terms of time, effort, and cost. Instead, the ITP employs a four-way cross breeding strategy. In this strategy, the F1 hybrid offspring from C57BL/6J × BALB/cJ matings are bred with the F1 hybrid offspring from C3H/HeJ × DBA/2J matings. This F1 × F1 breeding scheme produces the experimental strain (called UMHET3) in which all mice have 25% of their genetic material from each original strain, but with unique combination profiles88. Thus, using UMHET3 mice to evaluate the efficacy of a proposed intervention lessens the chance of missing a truly effective agent because it failed to work in one single isogenic strain (false negative) and likewise reduces the possibility of identifying a compound that is only efficacious in a single strain and does not generalize to other strains or organisms (false positive).
One of the first compounds evaluated by the ITP (resveratrol) illustrates how the outbred strain strategy has lessened the emphasis on treatments that may only be beneficial in specific conditions or isogenic mouse strains. Resveratrol modulates levels of sirtuin proteins, which are thought to mediate anti-aging effects via several mechanisms, including maintaining DNA integrity and reducing oxidative stress (for review, see89. Assessing the effect of resveratrol on lifespan in model organisms has produced conflicting conclusions. Some studies have reported beneficial effects on lifespan in nematodes and flies90,91, but these results were not replicated in other studies92. In C57BL/6Nia mice fed a high fat diet, resveratrol was reported to extend median lifespan93, but failed to affect the lifespan of these mice fed normal (control) chow94. The lack of an effect in the control mice was attributed to the late timepoint of treatment initiation (12 months of age)94. Studies using the UMHET3 mice also failed to find an extension of median or maximum lifespan (in either males or females at three different test sites), regardless of whether resveratrol treatment started at 12 months of age, or even earlier at 4 months of age95,96. These results suggest that resveratrol-mediated upregulation of sirtuin function may help to ameliorate deleterious effects in obese mice, but diminishes the enthusiasm for further testing and development of resveratrol as a general anti-aging therapeutic.
On the other hand, treatments resulting in anti-aging effects in isogenic strains that also extend lifespan in UMHET3 mice greatly bolsters their significance because the efficacy of those interventions is more likely to be generalizable to other populations and organisms, including humans97. To date, seven interventions have been successful in ITP studies, including rapamycin98,99 and acarbose100,101 (to see all compounds evaluated in the ITP, see https://www.nia.nih.gov/research/dab/interventions-testing-program-itp). The increased interest in these efficacious interventions is evidenced by ensuing studies (both within the ITP and from other researchers) that are evaluating different treatment regimens24,102, investigating the diverse cellular pathways engaged103,104 and assessing myriad ancillary age-related phenotypes105,106.
In summary, outbred strains maximize genetic diversity among test subjects and are ideally suited for making robust and generalizable conclusions about the effects of experimental perturbations or effectiveness of treatments. The strong experimental support provided by studies utilizing outbred strains is thus more likely to be yield interventions that successfully translate into a therapeutic setting. The advances demonstrated by the ITP in the field of aging research suggest that other areas may also benefit from a similarly-designed program. For example, other government or private foundations could deploy this type of approach to study interventions that modulate diverse physiological phenotypes, such as valence, cognitive, social, and arousal systems107. However, while outbred strains provide a powerful model system that more accurately reflects genetic diversity in human populations, they also have lower genetic tractability, making them less useful for identifying genetic contributions to neurological or psychiatric diseases. A breeding scheme that results in high genetic diversity and high genetic tractability is the recombinant inbred paradigm, which we review in the next section.
6. Using recombinant inbred strains for genome-phenotype association studies
Recombinant inbred (RI) mice are generated by crossing two distinct isogenic strains to produce F1 hybrids; then many individual pairs of F1 hybrids are mated, and each of these pairs become the founders of distinct RI lines. Each line is inbred for 20+ generations to produce distinct isogenic lines that are all each unique genetic mosaics of the parental strains (Figure 1 D). High genetic diversity is achieved because each RI line has a unique genetic mix, and high genetic tractability is achieved because the full sequence of each of the original parental lines is known (meaning each of the RI lines can also be nearly fully sequenced using identified markers through the genome). Therefore, an RI strain can be used to identify genetic sequence(s) that correlate with the expression of a quantitative phenotype (e.g. more or less resiliency to stress). The genetic variants that modulate phenotype expression are termed quantitative trait locus (QTL) (see Figure 1 D). The probability of identifying a QTL depends on the strength of the genetic contribution, which is reflected by the phenotypic variance among the RI lines. The resolution (and statistical power) of the QTL analysis depends on the amount of genetic diversity, which increases with the number of RI lines used108. Further, the inheritance rate of the phenotype, the robustness of the phenotype and the pleiotropy among individual RI lines indicates the extent to which the trait is mono vs polygenic. RI lines have been previously used to identify genes involved in addiction vulnerability109, persistent maladaptive fear28, hyperserotonemia (a biomarker of autism spectrum disorder110), and resiliency to cognitive decline resulting from familial Alzheimer’s mutations111, among others.
A commonly used RI strain is comprised of the BXD lines (from Jackson Laboratories), which are derived from multiple unique crosses of C57BL/6J and DBA/2J isogenic strains112,113. Because the BXDs comprise more than 120 unique lines, they offer greater genetic diversity that better model genetic variance among human populations114,115. They also possess high genetic tractability because they have been profiled for single-nucleotide polymorphisms (SNPs) at over ~470,000 locations (see http://www.genenetwork.org), making high-resolution gene mapping possible without full-genome sequencing113,116.
In an intriguing study, BXD lines were used to identify specific gene sets underlying susceptibility to age-related cognitive deficits. Neuner and colleagues117 measured variance in cognitive aging by testing a cohort of middle-aged (15 months old) mice from 21 BXD lines on a hippocampal-dependent, contextual fear memory task. Utilizing the broad range of cognitive performance among these mouse lines, the investigators were able to demonstrate that genetic alterations in a small region (2.8 Mb) of chromosome 4 are highly correlated with differences in cognitive performance. Importantly, this region did not associate with cognitive performance in young-adult mice118. Out of 10 gene candidates, the researchers first focused on Hp1bp3 (Heterochromatin Protein 1 Binding Protein 3) because they found an age-dependent correlation between hippocampal expression of Hp1bp3 and cognitive performance. The role of Hp1bp3 in cognition was further validated using (1) Hp1bp3 knock-out mice, which exhibited deficits in contextual fear memory117 and (2) by viral-mediated knock-down of Hp1bp3, which resulted in behavioral and transcriptional changes consistent with advanced aging119. Finally, the investigators discovered Hp1bp3 expression was correlated with cognitive performance among elderly adults when they examined human tissue samples collected post-mortem117.
In summary, RI strains coupled with QTL analysis can be used to systematically exploit genetic complexity to identify underlying molecular mechanisms that determine complex trait expression. Importantly, studies using RI rodents can fill gaps where human research is impractical or even impossible. For example, the ability to identify disease “resiliency genes” is limited because asymptomatic individuals rarely enter the clinic for treatment and more typically serve as the control subjects for GWAS111.
7. Combining approaches
The strategies presented above leverage both the genetic tractability of isogenic strains and the genetic (and trait) diversity between strains or in hybrid/outbred mice. Importantly, while we present these techniques as separate approaches, their use for understanding complex phenotypes is by no means mutually exclusive. Below we provide an example where insights into the relationship between the neurotransmitter serotonin (5-HT) and autism spectrum disorder (ASD) were revealed through a series of studies that incorporated phenotype comparisons between multiple isogenic strains, the use of RI lines, QTL analysis, and generation of congenic strains.
ASD is a male-predominant psychiatric condition characterized by repetitive behavior and deficits in social interaction and communication. An alteration in serotonin signaling is viewed as one of the primary candidates underlying disease expression; in fact, hyperserotonemia —or elevated 5-HT levels in the blood— is used as an effective biomarker for predicting ASD incidence120–122. Human linkage studies also implicate the 17q chromosomal region, which contains the SLC6A4 gene encoding the serotonin transporter (SERT), in ASD123–125.
To define the links between SERT gene variation and neurophysiological differences, multiple mouse strains harboring polymorphisms in the Slc6a4 were compared126. These studies found that mouse strains segregated into two distinct haplotypes. The GK haplotype (Gly39/Lys152) exhibited reduced effectiveness for 5-HT uptake as compared to the ER haplotype (Glu39/Arg152)126,127. To further distinguish the specific effects(s) of the haplotypes on complex traits from the effects of genetic background in these strains, researchers turned to a RI strategy. By correlating anatomical, biochemical and behavioral measures across many BXD RI lines (generated by crossing C57BL/6J mice that have the GK haplotype with DBA/2J mice that have the ER haplotype), specific phenotypes were identified that were correlated with either the GK or ER variation in SERT126. The GK haplotype was found to be associated with altered expression of the dopamine transporter (Slc6a3) in the caudate putamen as well as altered expression of the D2 (Drd2) dopamine receptor in the ventral midbrain. The GK mice also exhibited reduced immobility in a tail-suspension test and lower ethanol consumption126. Further, a QTL analysis was performed to test if hyperserotonemia (an ASD biomarker) was associated with SERT function in the BXD line. Interestingly, the same locus identified in humans, SLC6A4 (encoding SERT), was identified as a locus for whole-blood 5-HT levels in mice. Taken together, these results indicate that mice represent a faithful model in which to test the links between SERT polymorphisms and complex behavior128.
One well established SERT polymorphism is the Ala56 variant, which in humans is most commonly associated with rigid-compulsive behavior and sensory aversion. Although SERT Ala56 was found to have strong transmission bias (2:1 affected to unaffected children), many individuals harboring the variant were devoid of any ASD symptoms, suggesting that epistatic factors can modify symptom expression123. Thus, to distinguish the specific contribution of the Ala56 variant from gene modifier effects, researchers evaluated this mutation in congenic mouse lines. Knock-in mice for SERT Ala56 were originally generated in the 129S4/S6 background strain, and exhibited hyperserotonemia, altered social behavior, decreased ultrasonic vocalizations, and repetitive climbing/hanging behavior in the home cage129. Subsequently, the Ala56 129S4/S6 knock-in mice were backcrossed to generate a C57BL/6 congenic strain. Across both lines, the tendency for Ala56 mice to withdraw from social encounters remained consistent; however, other traits such as ultrasonic vocalization, hyperserotonemia, 5-HT receptor hypersensitivity, and repetitive behavior were differentially altered130. Taken together, these data suggest that alteration of SERT function associated with Ala56 primarily affects social behavior, while other traits are likely a result of more complex polygenic interactions130.
8. Summary and Conclusions
There is an increasing consensus that factors altering risk, resiliency, and the expression of many psychiatric and neurological disorders is polygenic. For psychiatric disease, this consensus is supported by numerous GWAS (see131) and is unsurprising considering that many psychiatric disorders, like schizophrenia, are characterized by complex alterations in both cognitive and affective behaviors. However, polygenicity is also an important factor to consider even for many neurological, ‘single-gene’ or ‘Mendelian disorders’ (such as Huntington’s Disease), where the severity of disease and pleiotropy can be altered by gene modifiers (see132).
While animal models cannot fully recapitulate human disease, using the techniques outlined in this review makes them well suited to identify the relationship between specific gene variants and discrete traits (i.e. stress resilience, maladaptive avoidance, cognition). Since 2005, over 1,800 GWAS have been carried out to identify associations between genes and disease or trait expression133. However, while GWAS can confidently link SNPs to a disease, they do not identify which genes relate to which specific traits within the disease. For example, thousands of SNPs have been linked to schizophrenia134; each of these identified gene variants may be altering multiple molecular mechanisms, modulating multiple neural circuits and contributing to multiple behavioral traits.
The present challenge is to establish a context for how collections of SNPs coalesce into mechanisms and may explain discrete alteration in neural function and behavior. The approaches presented in this review, which focus on systematically leveraging genetic and trait heterogeneity in rodents, may be well suited to decipher the complex relationships between identified gene variants and phenotypes. These approaches are synergistic with genetic studies in humans and help unravel the complex relationships between genes, environments and behavior.
Footnotes
Conflict of interest statement: The authors declare no conflict of interests or competing financial interests in relation to the work described.
References
- 1.Collins PY, Patel V, Joestl SS, March D, Insel TR, Daar AS et al. Grand challenges in global mental health. Nature 2011; 475: 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crimmins EM. Trends in the health of the elderly. Annu Rev Public Health 2004; 25: 79–98. [DOI] [PubMed] [Google Scholar]
- 3.Murray CJL, Lopez AD. Measuring the global burden of disease. N Engl J Med 2013; 369: 448–457. [DOI] [PubMed] [Google Scholar]
- 4.Trautmann S, Rehm J, Wittchen HU. The economic costs of mental disorders: Do our societies react appropriately to the burden of mental disorders? EMBO Rep 2016; 17: 1245–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Josselyn SA, Frankland PW. Memory Allocation: Mechanisms and Function. Annu Rev Neurosci 2018; 41: 389–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glineburg MR, Todd PK, Charlet-Berguerand N, Sellier C. Repeat-associated non-AUG (RAN) translation and other molecular mechanisms in Fragile X Tremor Ataxia Syndrome. Brain Research 2018; 1693: 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schizophrenia Working Group of the Psychiatric Genomics Consortium, Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016; 530: 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bailey SJ, Toth M. Variability in the benzodiazepine response of serotonin 5-HT1A receptor null mice displaying anxiety-like phenotype: evidence for genetic modifiers in the 5-HT-mediated regulation of GABA(A) receptors. J Neurosci 2004; 24: 6343–6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dominguez-Salazar E, Bateman HL, Rissman EF. Background matters: the effects of estrogen receptor alpha gene disruption on male sexual behavior are modified by background strain. Horm Behav 2004; 46: 482–490. [DOI] [PubMed] [Google Scholar]
- 10.Errijgers V, Kooy RF. Genetic modifiers in mice: the example of the fragile X mouse model. Cytogenet Genome Res 2004; 105: 448–454. [DOI] [PubMed] [Google Scholar]
- 11.Bruening S, Oh E, Hetzenauer A, Escobar-Alvarez S, Westphalen RI, Hemmings HC et al. The anxiety-like phenotype of 5-HT receptor null mice is associated with genetic background-specific perturbations in the prefrontal cortex GABA-glutamate system. J Neurochem 2006; 99: 892–899. [DOI] [PubMed] [Google Scholar]
- 12.van den Buuse M, Martin S, Holgate J, Matthaei K, Hendry I. Mice deficient in the alpha subunit of G(z) show changes in pre-pulse inhibition, anxiety and responses to 5-HT(1A) receptor stimulation, which are strongly dependent on the genetic background. Psychopharmacology 2007; 195: 273–283. [DOI] [PubMed] [Google Scholar]
- 13.Petkau TL, Hill A, Leavitt BR. Core neuropathological abnormalities in progranulin-deficient mice are penetrant on multiple genetic backgrounds. Neuroscience 2016; 315: 175–195. [DOI] [PubMed] [Google Scholar]
- 14.Crawley JN, Belknap JK, Collins A, Crabbe JC, Frankel W, Henderson N et al. Behavioral phenotypes of inbred mouse strains: implications and recommendations for molecular studies. Psychopharmacology 1997; 132: 107–124. [DOI] [PubMed] [Google Scholar]
- 15.Silva AJ, Simpson EM, Takahashi JS, Lipp H-P, Nakanishi S, Wehner JM et al. Mutant Mice and Neuroscience: Recommendations Concerning Genetic Background. Neuron 1997; 19: 755–759. [DOI] [PubMed] [Google Scholar]
- 16.Frankel WN. Mouse strain backgrounds: more than black and white. Neuron 1998; 20: 183. [DOI] [PubMed] [Google Scholar]
- 17.Miller RA, Austad S, Burke D, Chrisp C, Dysko R, Galecki A et al. Exotic mice as models for aging research: polemic and prospectus. Neurobiology of Aging 1999; 20: 217–231. [DOI] [PubMed] [Google Scholar]
- 18.Riordan JD, Nadeau JH. From Peas to Disease: Modifier Genes, Network Resilience, and the Genetics of Health. The American Journal of Human Genetics 2017; 101: 177–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellenbroek B, Youn J. Rodent models in neuroscience research: is it a rat race? Dis Model Mech 2016; 9: 1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beck JA, Lloyd S, Hafezparast M, Lennon-Pierce M, Eppig JT, Festing MF et al. Genealogies of mouse inbred strains. Nat Genet 2000; 24: 23–25. [DOI] [PubMed] [Google Scholar]
- 21.Castle WE, Little CC. THE PECULIAR INHERITANCE OF PINK EYES AMONG COLORED MICE. Science 1909; 30: 313–314. [DOI] [PubMed] [Google Scholar]
- 22.Sullivan PF, Daly MJ, O’Donovan M. Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat Rev Genet 2012; 13: 537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meisler MH, Kearney JA. Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest 2005; 115: 2010–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller AR, Hawkins NA, McCollom CE, Kearney JA. Mapping genetic modifiers of survival in a mouse model of Dravet syndrome. Genes, Brain and Behavior 2014; 13: 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci 2006; 9: 1142–1149. [DOI] [PubMed] [Google Scholar]
- 26.Nadeau JH. Modifier genes in mice and humans. Nat Rev Genet 2001; 2: 165–174. [DOI] [PubMed] [Google Scholar]
- 27.Stein RE, Kaplan JS, Li J, Catterall WA. Hippocampal deletion of NaV1.1 channels in mice causes thermal seizures and cognitive deficit characteristic of Dravet Syndrome. Proc Natl Acad Sci USA 2019; 116: 16571–16576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gunduz-Cinar O, Brockway E, Lederle L, Wilcox T, Halladay LR, Ding Y et al. Identification of a novel gene regulating amygdala-mediated fear extinction. Mol Psychiatry 2018; 36: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lenselink AM, Rotaru DC, Li KW, van Nierop P, Rao-Ruiz P, Loos M et al. Strain Differences in Presynaptic Function PROTEOMICS, ULTRASTRUCTURE, AND PHYSIOLOGY OF HIPPOCAMPAL SYNAPSES IN DBA/2J AND C57Bl/6J MICE. J Biol Chem 2015; 290: 15635–15645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moore SJ, Throesch BT, Murphy GG. Of mice and intrinsic excitability: genetic background affects the size of the postburst afterhyperpolarization in CA1 pyramidal neurons. Journal of Neurophysiology 2011; 106: 1570–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sandberg R, Yasuda R, Pankratz DG, Carter TA, Del Rio JA, Wodicka L et al. Regional and strain-specific gene expression mapping in the adult mouse brain. Proc Natl Acad Sci USA 2000; 97: 11038–11043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woo-Hyun Cho J-SH. Differences in the Flexibility of Switching Learning Strategies and CREB Phosphorylation Levels in Prefrontal Cortex, Dorsal Striatum and Hippocampus in Two Inbred Strains of Mice. Front Behav Neurosci 2016; 10: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim CK, Adhikari A, Deisseroth K. Integration of optogenetics with complementary methodologies in systems neuroscience. Nature Reviews Neuroscience 2017; 18: 222–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin MZ, Schnitzer MJ. Genetically encoded indicators of neuronal activity. Nat Neurosci 2016; 19: 1142–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hefner K, Whittle N, Juhasz J, Norcross M, Karlsson R-M, Saksida LM et al. Impaired fear extinction learning and cortico-amygdala circuit abnormalities in a common genetic mouse strain. J Neurosci 2008; 28: 8074–8085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muigg P, Scheiber S, Salchner P, Bunck M, Landgraf R, Singewald N. Differential stress-induced neuronal activation patterns in mouse lines selectively bred for high, normal or low anxiety. PLOS ONE 2009; 4 e5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fenster RJ, Lebois LAM, Ressler KJ, Suh J. Brain circuit dysfunction in post-traumatic stress disorder: from mouse to man. Nature Reviews Neuroscience 2018; 69: 516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smoller JW. The Genetics of Stress-Related Disorders: PTSD, Depression, and Anxiety Disorders. Neuropsychopharmacology 2016; 41: 297–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rescorla RA. Pavlovian conditioning. It’s not what you think it is. Am Psychol 1988; 43: 151–160. [DOI] [PubMed] [Google Scholar]
- 40.Camp M, Norcross M, Whittle N, Feyder M, D’Hanis W, Yilmazer-Hanke D et al. Impaired Pavlovian fear extinction is a common phenotype across genetic lineages of the 129 inbred mouse strain. Genes, Brain and Behavior 2009; 8: 744–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Camp MC, MacPherson KP, Lederle L, Graybeal C, Gaburro S, DeBrouse LM et al. Genetic Strain Differences in Learned Fear Inhibition Associated with Variation in Neuroendocrine, Autonomic, and Amygdala Dendritic Phenotypes. Neuropsychopharmacology 2012; 37: 1534–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Temme SJ, Bell RZ, Pahumi R, Murphy GG. Comparison of inbred mouse substrains reveals segregation of maladaptive fear phenotypes. Front Behav Neurosci 2014; 8: 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fitzgerald PJ, Whittle N, Flynn SM, Graybeal C, Pinard CR, Gunduz-Cinar O et al. Prefrontal single-unit firing associated with deficient extinction in mice. Neurobiology of Learning and Memory 2014; 113: 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flores Á, Valls-Comamala V, Costa G, Saravia R, Maldonado R, Berrendero F. The hypocretin/orexin system mediates the extinction of fear memories. Neuropsychopharmacology 2014; 39: 2732–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Daviu N, Füzesi T, Rosenegger DG, Rasiah NP, Sterley T-L, Peringod G et al. Paraventricular nucleus CRH neurons encode stress controllability and regulate defensive behavior selection. Nat Neurosci 2020; 23: 398–410. [DOI] [PubMed] [Google Scholar]
- 46.Futch HS, McFarland KN, Moore BD, Kuhn MZ, Giasson BI, Ladd TB et al. An anti-CRF antibody suppresses the HPA axis and reverses stress-induced phenotypes. J Exp Med 2019; 216: 2479–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zorn JV, Schür RR, Boks MP, Kahn RS, Joëls M, Vinkers CH. Cortisol stress reactivity across psychiatric disorders: A systematic review and meta-analysis. Psychoneuroendocrinology 2017; 77: 25–36. [DOI] [PubMed] [Google Scholar]
- 48.Wichmann S, Kirschbaum C, Böhme C, Petrowski K. Cortisol stress response in post-traumatic stress disorder, panic disorder, and major depressive disorder patients. Psychoneuroendocrinology 2017; 83: 135–141. [DOI] [PubMed] [Google Scholar]
- 49.Yehuda R, Halligan SL, Bierer LM. Cortisol levels in adult offspring of Holocaust survivors: relation to PTSD symptom severity in the parent and child. Psychoneuroendocrinology 2002; 27: 171–180. [DOI] [PubMed] [Google Scholar]
- 50.Sapolsky RM. Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders. Arch Gen Psychiatry 2000; 57: 925–935. [DOI] [PubMed] [Google Scholar]
- 51.Le-Niculescu H, Roseberry K, Levey DF, Rogers J, Kosary K, Prabha S et al. Towards precision medicine for stress disorders: diagnostic biomarkers and targeted drugs. Mol Psychiatry 2019; 376: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Holmes SE, Girgenti MJ, Davis MT, Pietrzak RH, DellaGioia N, Nabulsi N et al. Altered metabotropic glutamate receptor 5 markers in PTSD: In vivo and postmortem evidence. Proc Natl Acad Sci USA 2017; 114: 8390–8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yehuda R, Cai G, Golier JA, Sarapas C, Galea S, Ising M et al. Gene expression patterns associated with posttraumatic stress disorder following exposure to the World Trade Center attacks. Biological Psychiatry 2009; 66: 708–711. [DOI] [PubMed] [Google Scholar]
- 54.Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB et al. Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA 2008; 299: 1291–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kessler RC, Sonnega A, Bromet E, Hughes M, Nelson CB. Posttraumatic stress disorder in the National Comorbidity Survey. Arch Gen Psychiatry 1995; 52: 1048–1060. [DOI] [PubMed] [Google Scholar]
- 56.Ressler KJ. Translating Across Circuits and Genetics Toward Progress in Fear- and Anxiety-Related Disorders. Am J Psychiatry 2020; 177: 214–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roy-Byrne P Treatment-refractory anxiety; definition, risk factors, and treatment challenges. Dialogues in Clinical Neuroscience 2015; 17: 191–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Whittle N, Hauschild M, Lubec G, Holmes A, Singewald N. Rescue of impaired fear extinction and normalization of cortico-amygdala circuit dysfunction in a genetic mouse model by dietary zinc restriction. J Neurosci 2010; 30: 13586–13596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Whittle N, Schmuckermair C, Gunduz-Cinar O, Hauschild M, Ferraguti F, Holmes A et al. Deep brain stimulation, histone deacetylase inhibitors and glutamatergic drugs rescue resistance to fear extinction in a genetic mouse model. Neuropharmacology 2013; 64: 414–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Whittle N, Maurer V, Murphy C, Rainer J, Bindreither D, Hauschild M et al. Enhancing dopaminergic signaling and histone acetylation promotes long-term rescue of deficient fear extinction. Transl Psychiatry 2016; 6: e974–e974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cazares VA, Rodriguez G, Parent R, Ouillette L, Glanowska KM, Moore SJ et al. Environmental variables that ameliorate extinction learning deficits in the 129S1/SvlmJ mouse strain. Genes, Brain and Behavior 2019; 19: e12575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Murphy CP, Li X, Maurer V, Oberhauser M, Gstir R, Wearick-Silva LE et al. MicroRNA-Mediated Rescue of Fear Extinction Memory by miR-144–3p in Extinction-Impaired Mice. Biological Psychiatry 2017; 81: 979–989. [DOI] [PubMed] [Google Scholar]
- 63.Dunsmoor JE, Campese VD, Ceceli AO, LeDoux JE, Phelps EA. Novelty-facilitated extinction: providing a novel outcome in place of an expected threat diminishes recovery of defensive responses. Biological Psychiatry 2015; 78: 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dunsmoor JE, Kroes MCW, Li J, Daw ND, Simpson HB, Phelps EA. Role of Human Ventromedial Prefrontal Cortex in Learning and Recall of Enhanced Extinction. Journal of Neuroscience 2019; 39: 3264–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hermann A, Stark R, Müller EA, Kruse O, Wolf OT, Merz CJ. Multiple extinction contexts modulate the neural correlates of context-dependent extinction learning and retrieval. Neurobiology of Learning and Memory 2020; 168: 107150. [DOI] [PubMed] [Google Scholar]
- 66.de Jong R, Lommen M, de Jong PJ, Nauta MH. Using multiple contexts and retrieval cues in exposure-based therapy to prevent relapse in anxiety disorders. Cogn Behav Pract 2019; 26: 156–165 [Google Scholar]
- 67.Dunsmoor JE, Campese VD, Ceceli AO, LeDoux JE, Phelps EA. Novelty-Facilitated Extinction: Providing a Novel Outcome in Place of an Expected Threat Diminishes Recovery of Defensive Responses. Biological Psychiatry 2015; 78: 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Duncan LE, Ratanatharathorn A, Aiello AE, Almli LM, Amstadter AB, Ashley-Koch AE et al. Largest GWAS of PTSD (N =20 070) yields genetic overlap with schizophrenia and sex differences in heritability. Mol Psychiatry 2018; 23: 666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McLean SA, Ressler K, Koenen KC, Neylan T, Germine L, Jovanovic T et al. The AURORA Study: a longitudinal, multimodal library of brain biology and function after traumatic stress exposure. Mol Psychiatry 2020; 25: 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grewe BF, Gründemann J, Kitch LJ, Lecoq JA, Parker JG, Marshall JD et al. Neural ensemble dynamics underlying a long-term associative memory. Nature 2017; 543: 670–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sartori SB, Maurer V, Murphy C, Schmuckermair C, Muigg P, Neumann ID et al. Combined Neuropeptide S and D-Cycloserine Augmentation Prevents the Return of Fear in Extinction-Impaired Rodents: Advantage of Dual versus Single Drug Approaches. Int J Neuropsychopharmacol 2016; 19: pyv128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Swallow JG, Garland T. Selection Experiments as a Tool in Evolutionary and Comparative Physiology: Insights into Complex Traits--an Introduction to the Symposium. Integr Comp Biol 2005; 45: 387–390. [DOI] [PubMed] [Google Scholar]
- 73.Zhang P, Rhodes JS, Garland T, Perez SD, Southey BR, Rodriguez-Zas SL. Brain region-dependent gene networks associated with selective breeding for increased voluntary wheel-running behavior. PLOS ONE 2018; 13: e0201773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kolb EM, Rezende EL, Holness L, Radtke A, Lee SK, Obenaus A et al. Mice selectively bred for high voluntary wheel running have larger midbrains: support for the mosaic model of brain evolution. Journal of Experimental Biology 2013; 216: 515–523. [DOI] [PubMed] [Google Scholar]
- 75.Koch LG, Britton SL. Artificial selection for intrinsic aerobic endurance running capacity in rats. Physiol Genomics 2001; 5: 45–52. [DOI] [PubMed] [Google Scholar]
- 76.Thompson Z, Argueta D, Garland T Jr., DiPatrizio N. Circulating levels of endocannabinoids respond acutely to voluntary exercise, are altered in mice selectively bred for high voluntary wheel running, and differ between the sexes. Physiol Behav 2017; 170: 141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stead JDH, Clinton S, Neal C, Schneider J, Jama A, Miller S et al. Selective breeding for divergence in novelty-seeking traits: heritability and enrichment in spontaneous anxiety-related behaviors. Behav Genet 2006; 36: 697–712. [DOI] [PubMed] [Google Scholar]
- 78.de Fiebre NC, Dawson R, de Fiebre CM. The Selectively Bred High Alcohol Sensitivity (HAS) and Low Alcohol Sensitivity (LAS) Rats Differ in Sensitivity to Nicotine. Alcoholism: Clinical and Experimental Research 2002; 26: 765–772. [PubMed] [Google Scholar]
- 79.Thiele TE, Miura GI, Marsh DJ, Bernstein IL, Palmiter RD. Neurobiological responses to ethanol in mutant mice lacking neuropeptide Y or the Y5 receptor. Pharmacology Biochemistry and Behavior 2000; 67: 683–691. [DOI] [PubMed] [Google Scholar]
- 80.Gariépy J-L, Bauer DJ, Cairns RB. Selective breeding for differential aggression in mice provides evidence for heterochrony in social behaviours. Animal Behaviour 2001; 61: 933–947. [Google Scholar]
- 81.Slattery DA, Naik RR, Grund T, Yen YC, Sartori SB, Fuchsl A et al. Selective Breeding for High Anxiety Introduces a Synonymous SNP That Increases Neuropeptide S Receptor Activity. Journal of Neuroscience 2015; 35: 4599–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Clinton SM, Vazquez DM, Kabbaj M, Kabbaj M-H, Watson SJ, Akil H. Individual differences in novelty-seeking and emotional reactivity correlate with variation in maternal behavior. Horm Behav 2007; 51: 655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Perez JA, Clinton SM, Turner CA, Watson SJ, Akil H. A new role for FGF2 as an endogenous inhibitor of anxiety. J Neurosci 2009; 29: 6379–6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Turner CA, Clinton SM, Thompson RC, Watson SJ, Akil H. Fibroblast growth factor-2 (FGF2) augmentation early in life alters hippocampal development and rescues the anxiety phenotype in vulnerable animals. Proc Natl Acad Sci USA 2011; 108: 8021–8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Flagel SB, Robinson TE, Clark JJ, Clinton SM, Watson SJ, Seeman P et al. An Animal Model of Genetic Vulnerability to Behavioral Disinhibition and Responsiveness to Reward-Related Cues: Implications for Addiction. Neuropsychopharmacology 2010; 35: 388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Garofalo S, di Pellegrino G. Individual differences in the influence of task-irrelevant Pavlovian cues on human behavior. Front Behav Neurosci 2015; 9: 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Warner HR, Ingram D, Miller RA, Nadon NL, Richardson AG. Program for testing biological interventions to promote healthy aging. Mech Ageing Dev 2000; 155: 199–207. [DOI] [PubMed] [Google Scholar]
- 88.Miller RA, Burke D, Nadon N. Announcement: four-way cross mouse stocks: a new, genetically heterogeneous resource for aging research. J Gerontol A Biol Sci Med Sci 1999; 54: B358–60. [DOI] [PubMed] [Google Scholar]
- 89.Grabowska W, Sikora E, Bielak-Zmijewska A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology 2017; 18: 447–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M et al. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 2004; 430: 686–689. [DOI] [PubMed] [Google Scholar]
- 91.Viswanathan M, Kim SK, Berdichevsky A, Guarente L. A role for SIR-2.1 regulation of ER stress response genes in determining C. elegans life span. Dev Cell 2005; 9: 605–615. [DOI] [PubMed] [Google Scholar]
- 92.Bass TM, Weinkove D, Houthoofd K, Gems D, Partridge L. Effects of resveratrol on lifespan in Drosophila melanogaster and Caenorhabditis elegans. Mechanisms of Ageing and Development 2007; 128: 546–552. [DOI] [PubMed] [Google Scholar]
- 93.Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006; 444: 337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pearson KJ, Baur JA, Lewis KN, Peshkin L, Price NL, Labinskyy N et al. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab 2008; 8: 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci 2011; 66: 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Strong R, Miller RA, Astle CM, Baur JA, de Cabo R, Fernandez E et al. Evaluation of resveratrol, green tea extract, curcumin, oxaloacetic acid, and medium-chain triglyceride oil on life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci 2013; 68: 6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nadon NL, Strong R, Miller RA, Harrison DE. NIA Interventions Testing Program: Investigating Putative Aging Intervention Agents in a Genetically Heterogeneous Mouse Model. EBioMedicine 2017; 21: 3–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009; 460: 392–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wilkinson JE, Burmeister L, Brooks SV, Chan C-C, Friedline S, Harrison DE et al. Rapamycin slows aging in mice. Aging Cell 2012; 11: 675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Harrison DE, Strong R, Allison DB, Ames BN, Astle CM, Atamna H et al. Acarbose, 17-α-estradiol, and nordihydroguaiaretic acid extend mouse lifespan preferentially in males. Aging Cell 2014; 13: 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Harrison DE, Strong R, Alavez S, Astle CM, DiGiovanni J, Fernandez E et al. Acarbose improves health and lifespan in aging HET3 mice. Aging Cell 2019; 18: e12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Reid JJ, Linden MA, Peelor FF, Miller RA, Hamilton KL, Miller BF. Brain Protein Synthesis Rates in the UM-HET3 Mouse Following Treatment With Rapamycin or Rapamycin With Metformin. J Gerontol A Biol Sci Med Sci 2020; 75: 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Drake JC, Peelor FF, Biela LM, Watkins MK, Miller RA, Hamilton KL et al. Assessment of mitochondrial biogenesis and mTORC1 signaling during chronic rapamycin feeding in male and female mice. J Gerontol A Biol Sci Med Sci 2013; 68: 1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sadagurski M, Cady G, Miller RA. Anti-aging drugs reduce hypothalamic inflammation in a sex-specific manner. Aging Cell 2017; 16: 652–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zaseck LW, Miller RA, Brooks SV. Rapamycin Attenuates Age-associated Changes in Tibialis Anterior Tendon Viscoelastic Properties. J Gerontol A Biol Sci Med Sci 2016; 71: 858–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Altschuler RA, Kanicki A, Martin C, Kohrman DC, Miller RA. Rapamycin but not acarbose decreases age-related loss of outer hair cells in the mouse Cochlea. Hearing research 2018; 370: 11–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stoyanov D, Telles-Correia D, Cuthbert BN. The Research Domain Criteria (RDoC) and the historical roots of psychopathology: A viewpoint. Eur Psychiatry 2019; 57: 58–60. [DOI] [PubMed] [Google Scholar]
- 108.Mackay TF. The genetic architecture of quantitative traits. Annu Rev Genet 2001; 35: 303–339. [DOI] [PubMed] [Google Scholar]
- 109.Zhou Z, Blandino P, Yuan Q, Shen P-H, Hodgkinson CA, Virkkunen M et al. Exploratory locomotion, a predictor of addiction vulnerability, is oligogenic in rats selected for this phenotype. Proc Natl Acad Sci USA 2019; 116: 13107–13115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Muller CL, Anacker AMJ, Veenstra-VanderWeele J. The serotonin system in autism spectrum disorder: From biomarker to animal models. Neuroscience 2016; 321: 24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Neuner SM, Heuer SE, Huentelman MJ, O’Connell KMS, Kaczorowski CC. Harnessing Genetic Complexity to Enhance Translatability of Alzheimer’s Disease Mouse Models: A Path toward Precision Medicine. Neuron 2019; 101: 399–411.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Taylor BA. Recombinant inbred strains. Use in gene mapping, p. 423–438. In Morse h. C. iii ,[ed.] Origins of inbred mice. Ny: 1978. [Google Scholar]
- 113.Peirce JL, Lu L, Gu J, Silver LM, Williams RW. A new set of BXD recombinant inbred lines from advanced intercross populations in mice. BMC Genet 2004; 5: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ashbrook DG, Arends D, Prins P, Mulligan MK, Roy S, Williams EG et al. The expanded BXD family of mice: A cohort for experimental systems genetics and precision medicine. biorxiv.org 2019. doi: 10.1101/672097. [DOI] [Google Scholar]
- 115.International HapMap Consortium, Daly MJ. The International HapMap Project. Nature 2003; 426: 789–796. [DOI] [PubMed] [Google Scholar]
- 116.Chintalapudi SR, Maria D, Di Wang X, Bailey JNC, NEIGHBORHOOD consortium, International Glaucoma Genetics consortium et al. Systems genetics identifies a role for Cacna2d1 regulation in elevated intraocular pressure and glaucoma susceptibility. Nat Commun 2017; 8: 1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Neuner SM, Garfinkel BP, Wilmott LA, Ignatowska-Jankowska BM, Citri A, Orly J et al. Systems genetics identifies Hp1bp3 as a novel modulator of cognitive aging. Neurobiology of Aging 2016; 46: 58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Philip VM, Duvvuru S, Gomero B, Ansah TA, Blaha CD, Cook MN et al. High-throughput behavioral phenotyping in the expanded panel of BXD recombinant inbred strains. Genes, Brain and Behavior 2010; 9: 129–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Neuner SM, Ding S, Kaczorowski CC. Knockdown of heterochromatin protein 1 binding protein 3 recapitulates phenotypic, cellular, and molecular features of aging. Aging Cell 2019; 18: e12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Schain RJ, Freedman DX. Studies on 5-hydroxyindole metabolism in autistic and other mentally retarded children. The Journal of Pediatrics 1961; 58: 315–320. [DOI] [PubMed] [Google Scholar]
- 121.Piven J, Tsai GC, Nehme E, Coyle JT, Chase GA, Folstein SE. Platelet serotonin, a possible marker for familial autism. J Autism Dev Disord 1991; 21: 51–59. [DOI] [PubMed] [Google Scholar]
- 122.Mulder EJ, Anderson GM, Kema IP, de Bildt A, van Lang ND, Boer den JA et al. Platelet Serotonin Levels in Pervasive Developmental Disorders and Mental Retardation: Diagnostic Group Differences, Within-Group Distribution, and Behavioral Correlates. Journal of the American Academy of Child & Adolescent Psychiatry 2004; 43: 491–499. [DOI] [PubMed] [Google Scholar]
- 123.Sutcliffe JS, Delahanty RJ, Prasad HC, McCauley JL, Han Q, Jiang L et al. Allelic Heterogeneity at the Serotonin Transporter Locus (SLC6A4) Confers Susceptibility to Autism and Rigid-Compulsive Behaviors. The American Journal of Human Genetics 2005; 77: 265–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.International Molecular Genetic Study of Autism Consortium (IMGSAC). A genomewide screen for autism: strong evidence for linkage to chromosomes 2q, 7q, and 16p. The American Journal of Human Genetics 2001; 69: 570–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cantor RM, Kono N, Duvall JA, Alvarez-Retuerto A, Stone JL, Alarcón M et al. Replication of Autism Linkage: Fine-Mapping Peak at 17q21. The American Journal of Human Genetics 2005; 76: 1050–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Carneiro AMD, Airey DC, Thompson B, Zhu C-B, Lu L, Chesler EJ et al. Functional coding variation in recombinant inbred mouse lines reveals multiple serotonin transporter-associated phenotypes. Proc Natl Acad Sci USA 2009; 106: 2047–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ellegood J, Yee Y, Kerr TM, Muller CL, Blakely RD, Henkelman RM et al. Analysis of neuroanatomical differences in mice with genetically modified serotonin transporters assessed by structural magnetic resonance imaging. Molecular Autism 2013 4:1 2018; 9: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ye R, Carneiro AMD, Airey D, Bush ES, Williams RW, Lu L et al. Evaluation of heritable determinants of blood and brain serotonin homeostasis using recombinant inbred mice. Genes, Brain and Behavior 2014; 13: 247–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Veenstra-VanderWeele J, Muller CL, Iwamoto H, Sauer JE, Owens WA, Shah CR et al. Autism gene variant causes hyperserotonemia, serotonin receptor hypersensitivity, social impairment and repetitive behavior. Proc Natl Acad Sci USA 2012; 109: 5469–5474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kerr TM, Muller CL, Miah M, Jetter CS, Pfeiffer R, Shah C et al. Genetic background modulates phenotypes of serotonin transporter Ala56 knock-in mice. Molecular Autism 2013 4:1 2013; 4: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Geschwind DH, Flint J. Genetics and genomics of psychiatric disease. Science 2015; 349: 1489–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Holmans PA, Massey TH, Jones L. Genetic modifiers of Mendelian disease: Huntington’s disease and the trinucleotide repeat disorders. Hum Mol Genet 2017; 26: R83–R90. [DOI] [PubMed] [Google Scholar]
- 133.Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res 2014; 42: D1001–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Duncan LE, Ostacher M, Ballon J. How genome-wide association studies (GWAS) made traditional candidate gene studies obsolete. Neuropsychopharmacology 2019; 44: 1518–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]