

SUMMARY PARAGRAPH
Transcription factors (TF) recognize specific genomic sequences to regulate complex gene expression programs. Although it is well established that TFs bind specific DNA sequences using a combination of base readout and shape recognition, some fundamental aspects of protein-DNA binding remain poorly understood1,2. Many DNA-binding proteins induce changes in the DNA structure outside the intrinsic B-DNA envelope. However, how the energetic cost associated with distorting DNA contributes to recognition has proven difficult to study because the distorted DNA exists in low-abundance in the unbound ensemble3–9. Here, we use a novel high-throughput assay called SaMBA (Saturation Mismatch-Binding Assay) to investigate the role of DNA conformational penalties in TF-DNA recognition. In SaMBA, mismatches are introduced to pre-induce DNA structural distortions much larger than those induced by changes in Watson-Crick sequence. Strikingly, approximately 10% of mismatches increased TF binding, and at least one mismatch was found that increased the binding affinity for each of 22 examined TFs. Mismatches also converted non-specific sites into high-affinity sites, and high-affinity sites into super-sites stronger than any known canonical binding site. Determination of high-resolution X-ray structures, combined with NMR measurements and structural analyses revealed that many of the mismatches that increase binding induce distortions similar to those induced by protein binding, thus pre-paying some of the energetic cost to deform the DNA. Our work indicates that conformational penalties are a major determinant of protein-DNA recognition, and reveals mechanisms by which mismatches can recruit TFs and thus modulate replication and repair activities in the cell10,11.
TF proteins distort the DNA upon binding
A comprehensive survey of high-resolution structures of TF-bound DNA revealed that more than 40% of the complexes contain base pairs (bp) with geometries that significantly deviate from the B-form envelope of naked DNA duplexes (Extended Data Fig. 1, Methods). The energy cost required to distort DNA must come from favorable intermolecular interactions that form upon complex formation12,13. This energetic cost could vary with sequence and contribute to protein-DNA binding affinity and selectivity1,14,15. Assessing conformational penalties experimentally has proven difficult because it requires accurately measuring the abundance of these distorted DNA conformations in the unbound ensemble3,4, which are difficult to even detect using existing biophysical methods.
Mismatches distort the DNA
Like proteins, mismatches can also induce distortions to the DNA ensemble much greater than those attained in naked Watson-Crick sequences (Fig. 1a–c, Extended Data Fig. 2). For example, purine-purine mismatches such as G-G and G-A widen the bp and can also flip the base into the syn conformation; pyrimidine-pyrimidine mismatches such as C-T and T-T constrict the bp; wobble G-T and T-T mismatches change the shear, while A-A and C-C with only a single hydrogen-bond can adopt a variety of conformations including partially melted states (Extended Data Fig. 2a). Mismatches can also affect the DNA minor/major groove geometry and base-step parameters, albeit to a smaller extent (Extended Data Fig. 2c). In addition, mismatches destabilize the DNA duplex, by amounts (3.5–10 kBT; Extended Data Fig. 2b) that are comparable to the typical energetic cost of distorting the DNA upon protein binding (3–8 kBT)16.
Figure 1. SaMBA measures the effects of mismatches on protein-DNA binding in high throughput.

(a-c) Mismatches change the local DNA geometry (a), affect global features such as the minor groove width (b), and destabilize the DNA (c).
(d) SaMBA is a chip-based assay for testing TF binding to thousands of DNA mismatches and Watson-Crick sequences (Methods). DNA hybridization and protein-DNA binding are quantified using fluorophore-labeled oligos and antibodies, respectively.
(e) Reproducibility of SaMBA data, for technical replicates of Ets1 at 125nM. Axes show the base 2 logarithm of the median fluorescent intensity signal corresponding to the bound Ets1 protein (n=12 replicate spots for Watson-Crick sequence, and 8 for mismatched sequences).
(f) Protein binding levels measured by SaMBA correlate linearly with independent Kd measurements from a variety of experimental methods, allowing calibration of SaMBA data. Similarly to related array-based techniques20, median values over replicate DNA spots are shown for SaMBA (error bars: median absolute deviation). Average values over replicates are shown for the orthogonal methods (error bars: standard deviation, when available). See Methods for the number of replicates (n>=3) for each experiment. Red shaded region: 95% confidence interval for Pearson’s correlation.
SaMBA: Saturation Mismatch-Binding Assay
We developed SaMBA, a high-throughput approach that leverages mismatch-induced distortions to gain insights into the role of DNA conformational penalties in protein-DNA recognition. We reasoned that different types of mismatches could redistribute the unbound DNA ensemble in various ways and lead, in some cases, to increased abundance of distorted DNA states that are recognized by TFs. By pre-paying some of the energetic cost of deforming the DNA, mismatches could in turn increase the TF-DNA binding affinity, provided that the reduction in conformational penalty outweighs any effects due to the potential loss of protein-DNA contacts. A conceptually similar strategy was recently used to assess conformational penalties in RNA-RNA association9.
In SaMBA experiments, mismatches are generated by introducing every possible single-base variation in known DNA binding sites of TF proteins in a high-throughput manner on a high-density DNA chip (Fig. 1d, Extended Data Fig. 3a–d; Methods). Mismatches are introduced by changing the sequence on one strand at a time (e.g. G-C → A-C, T-C, and C-C). Protein binding measurements are then conducted directly on the chip, with excellent reproducibility (Fig. 1e). The SaMBA binding signal intensities can be calibrated to equilibrium dissociation constants (Kd) using binding measurements from a variety of independent experimental methods (Fig. 1f), thus providing a route for determining binding energetics in high throughput (Methods, Extended Data Fig. 3e–h, Supplementary Table 3).
Here, we leverage SaMBA to investigate the role of conformational penalties in TF-DNA recognition. More broadly, SaMBA can be used to examine the impact of mismatches on protein-DNA binding landscapes, and their proposed roles in mutagenesis10,17–19, including in cases in which mismatches enhance binding by creating or reinforcing favorable interactions involving hydrogen bonding, electrostatics, and stacking, as discussed below.
Mismatches enhance TF binding to DNA
For twenty-two TFs representing fifteen distinct protein families, we used SaMBA to obtain saturation mismatch binding profiles showing quantitative changes in protein-binding signal induced by the introduction of every possible mismatch to known TF binding sites and their flanking regions (Fig. 2a, Supplementary Table 1). Whereas two thirds of the mismatches introduced within TF binding sites significantly weakened binding, ~10% increased binding. Strikingly, for each of the twenty-two TFs examined, at least one mismatch was found that increased the binding affinity when introduced within the binding site (Fig. 2a). In some cases, single mismatches created “super sites” stronger than the best canonical Watson-Crick binding sites (e.g. p53, Supplementary Table 1b). In other cases, mismatches introduced in non-specific DNA sites increased TF binding (Supplementary Table 1d) to levels similar to those observed for specific binding sites, thus effectively creating novel binding sites within non-specific DNA. For Ets1, the protein with the largest mismatch-driven effects outside of specific binding sites, we validated that mismatches can indeed increase TF binding beyond the distribution of non-specific binding affinities (defined here as the 99th percentile of random sites) and toward high affinities characteristic of specific binding sites (defined here as sites with Ets1-bound NMR or crystal structures) (Methods, Fig. 2c, Supplementary Table 2).
Figure 2. The effects of DNA mismatches on TF binding.

(a) SaMBA profiles for the 22 tested TFs. Heatmaps show the effects of mismatches on TF binding, normalized so −1 corresponds to the largest decrease (Methods).
(b) SaMBA profile for Ets1, with a representative mismatch-induced binding increase that was independently validated by fluorescence anisotropy (FA). Y-axis: log2 fold-change in median signal intensity, relative to the Watson-Crick site. Colored circles: significant changes (p-value < 0.05, one-sided Mann-Whitney U-test with Benjamini-Hochberg correction). Boxplots show median signals over replicate DNA spots for SaMBA (n=8 or 12 for the mismatch and Watson-Crick site, respectively) and replicate experiments for EMSA (n=3). Boxes extend to the 25th and 75th percentiles. Whiskers extend to the most extreme data points.
(c) Five validated examples of mismatches in non-specific sequences that increase Ets1 binding to levels similar to specific sites (Methods). Each arrow corresponds to one mismatch in a particular non-specific sequence (Supplementary Table 2c). In some cases, Watson-Crick mutations also increase binding affinity, albeit to a smaller extent, indicating that the identity of the newly introduced base is important for enhanced binding affinity (Supplementary Table 2, Extended Data Fig. 5).
(d) Comparison of mismatch versus mutations effects for the Ets1 site in (b), for mismatches on the upper strand. Values represent medians over replicate spots (n=8).
(e) The energetic effects of base pair mutations (diagonal) are different from the sum of the energetic effects of the two corresponding mismatches, demonstrating deviations from an additive model.
We verified representative examples of mismatch-induced enhancements in TF binding sites using fluorescence anisotropy (FA) and electrophoretic mobility shift assays (EMSA), and found binding increases of 0.7–2.3 kBT relative to consensus Watson-Crick binding sites (Fig. 2b, Extended Data Fig. 3e). Overall, the magnitude of mismatch-induced effects on TF binding was comparable to the magnitude of the effects of mutations in Watson-Crick binding sites (Extended Data Fig. 4a), although the directionality of these effects is sometimes opposite for mismatches versus their nearest mutations (e.g. C-G → G-G increases binding, while C-G → G-C decreases binding) (Fig. 2d,e, Extended Data Fig. 4b). This shows that mismatches can provide an additional layer of information about TF-DNA interactions, beyond what can be learned from analyzing the effects of mutations in Watson-Crick DNA using traditional high-throughput methods20–24.
Mismatches versus Watson-Crick mutations
The simplest explanation for the observed mismatch-induced increase in TF binding affinity is that the mutated base forms more favorable interactions with the TF and in a manner independent of the mismatch shape. In this simple additive model, each base in a base pair contributes independently to the TF binding energetics. Such a model predicts that the sum of the energetic gains/losses from the two individual single-base mutations to be equal to the binding energy change due to the double mutant (e.g., ΔΔGCG->CT + ΔΔGCG->AG = ΔΔGCG->AT). On other hand, any mismatch shape-dependent contribution to increased TF binding, including changes in the DNA ensemble that may help offset the energetic cost for DNA deformation, could lead to deviations from the additive model. We tested this simple model for the seven TFs with available calibration data in our study (Methods, Extended Data Fig. 4c, Supplementary Table 4). Indeed, we found that additivity holds, within experimental error, in ~42% of cases where mismatches significantly affect TF binding (e.g. for Ets1 we found that ΔΔGAT->AG + ΔΔGAT->CT ≈ ΔΔGAT->CG for position 7 in the binding site; Extended Data Fig. 4c). For the remaining ~58% of cases, Watson-Crick mutations had a different energetic effect on TF binding compared to the sum of the two corresponding mismatches (Fig. 2e, Extended Data Fig. 4c, Supplementary Table 4), indicating non-additive contributions of the mis-paired bases. While non-additive models have been previously tested for base pairs in Watson-Crick binding sites (e.g. 25,26), our TF-mismatch binding data provides a unique opportunity to investigate dependencies between bases in a base pair.
Mismatches pre-pay distortion penalty
Deviations from the simple additive model can arise from various mechanisms, including non-native interactions with the newly formed mismatch-dependent DNA shape (including the bases) and from the reinforcement of native interactions due to mismatch-specific changes in the DNA ensemble that offset the conformational penalties associated with distorting the DNA upon TF binding. For the latter case, we would expect the mismatches to be located in regions that are distorted in the protein-bound DNA structure. Indeed, for the subset of twelve TF proteins with available PDB structures, we found that the binding site positions where mismatches enhanced TF binding affinity were significantly more distorted than the rest of binding site positions, either in terms of the magnitude of the distortions (p=0.017) or in terms of the number of distorted features (p=0.015) (Methods; Supplementary Table 5).
If mismatches increase binding affinity in part by pre-paying conformational penalties, we would also expect mismatches to bias local or global aspects of the DNA structural ensemble to better mimic the DNA structure when bound to the TF. Because such ensembles are difficult to obtain, we used high-resolution crystal structure data, available for twelve TF proteins in our study, to compare distortions in the TF-bound DNA against distortions induced by mismatches (Methods). We observed some form of structural mimicry in 66% of cases (Supplementary Table 6). Returning to the example of Ets1, we found that the G-A mismatch at position 6, which increases binding by ~2.3 kBT (Fig. 2b) mimics the stretch, C1′-C1′ distance, and minor groove width of Ets1-bound DNA (Supplementary Table 6d, Extended Data Fig. 5b). At the same time, based on MD simulations of the bound mismatched and Watson-Crick DNA for this and other mismatches that increase TF binding (Extended Data Fig. 5c, Supplementary Table 7), the formation of new protein-DNA contacts might also contribute to the enhanced binding affinity, indicating that a single mismatch can affect the energetics of multiple types of interactions, including base readout, shape readout, and conformational penalties.
To better isolate contributions from the energetic penalty, we focused on mismatches that enhanced binding of TFs p53 and TBP, as these mismatches showed structural mimicry in base parameter features that deviate most strongly from the B-form envelope, and occurred at positions lacking H-bonds with the bases (Supplementary Table 5). In the case of p53, two positions in each p53 half-site have a preference to adopt non-canonical Hoogsteen conformations27,28 (Fig. 3a). Hoogsteen base pairs represent an example of alternative lowly-populated conformations in apo-DNA ensemble, that form with abundance <1%, at an estimated energetic cost of 3–7 kBT4,29,30. The Hoogsteen pairing is achieved by flipping the purine base from an anti to syn conformation, followed by a reduction of ~2 Å in the helical diameter and C1′-C1′ distance. This reduction in the DNA diameter at the p53 binding site allows closer proximity of p53 monomers, stabilizing the p53 tetramer compared to Watson-Crick pairs28. Remarkably, our SaMBA data revealed that replacing the A-T at Hoogsteen sites by T-T or C-T mismatches, which also reduce the C1′-C1′ distance (Fig. 3a), enhanced p53 binding affinity by ~0.4–1.8 kBT (Supplementary Tables 3, 4), comparable to the magnitude of p53 binding affinity changes due to base-pair mutations (Extended Data Fig. 4a).
Figure 3. DNA mismatches that exhibit geometries similar to distorted base pairs in TF-bound DNA lead to increased binding affinity.

(a) p53-DNA crystal structure shows a constricted Hoogsteen conformation at the positions marked in red. C-T and T-T mismatches, which increase p53-DNA binding affinity, mimic Hoogsteen base pairing by constricting the C1′-C1′ distance and minor groove width. Violin plots show the distributions of the C1′-C1′ distance and minor groove width according to MD simulation data (Methods).
(b) NMR results confirm that T-T and C-T mismatches mimic Hoogsteen A-T geometry. Plot shows the chemical shift differences in the sugar C1’/C3’/C4’ carbons for T-T and C-T mismatches versus a locked Hoogsteen conformation (using N1-methyladenosine30), relative to the Watson-Crick base-paired duplex (Methods). Blue shaded region: 95% confidence interval for Pearson’s correlation.
(c) TBP-DNA crystal structure shows destabilization at an ApG base pair step (positions 7–8) critical for TBP binding8,31,32. βh = bending magnitude (Methods).
(d) C-C mismatch destabilizes the DNA and has the lowest stacking propensity33.
(e) High correlation between TBP binding levels (medians over 9 replicate spots) and DNA duplex stability (Methods), computed over all base-pair variants at position 8 in the TBP site suggests that pre-paying the energetic cost for melting this base-pair modulates TBP binding affinity. Blue shaded region: 95% confidence interval for Pearson’s correlation.
(f) Structural overlay of six TBP-DNA complex structures demonstrates nearly identical structures for all complexes. Green: 1QNE, Watson-Crick site 5’-TATAAAAG-3’. Cyan: TBP-CC(2), 5’-TATAAAAG-3’ with CC at position 8. Orange: TBP-AC, 5’-TATAAAAG-3’ with AC at position 7. Yellow: 6NJQ, Watson-Crick site 5’-TATAAACG-3’. Purple: TBP-CC(1a) and pink: TBP-CC(1b), 5’-TATAAACG-3’ with CC at position 7.
(g) Overlay of the TBP-DNA interfaces (for 1QNE and TBP-CC(2)) demonstrates that interactions are highly similar between Watson-Crick and mismatched sites, including Phe interactions at the position of the mismatch (black rectangle).
NMR analysis confirmed that the perturbations induced by A-T Hoogsteen base pairs30 are indeed similar to those induced by T-T and C-T (Fig. 3b, Extended Data Fig. 6c,d). The T-T and C-T mismatches also induced narrowing of the minor groove width, thus resulting in enhanced negative electrostatic potential12, and C-T induced over-twisting of the DNA helix, mimicking the p53-bound Watson-Crick structure (Fig. 3a, Extended Data Fig. 6e). These results indicate that T-T and C-T mismatches effectively mimic structural features of the p53-preferred Hoogsteen pairing in naked DNA, thus pre-paying some of the energetic penalty to form the preferred bound structure. Since T-T and C-T mismatches do not increase the binding energetics to the same extent as the cost of forming Hoogsteen, it is possible that the mismatches do not mimic all aspects of the Hoogsteen conformation, and/or that the Hoogsteen conformation is not fully populated in the protein-bound state of the Watson-Crick DNA.
To evaluate the predictive power of the above trend, we used not only single-base variations, which are typical for SaMBA assays, but also double-base variations to measure the effects of all mismatches at the four HG positions in the p53 binding site (Methods, Supplementary Table 4). As expected, pyrimidine-pyrimidine mismatches (C-T, T-C, T-T, and C-C) enhanced p53 binding affinity, while all other mismatches at these positions either decreased binding or had non-significant effects (Extended Data Fig. 6f), consistent with our hypothesis. These findings are in excellent agreement with a recent study in which modified bases were shown to induce Hoogsteen and increase p53 binding affinity in a similar manner28.
For TBP, prior studies have shown that partial intercalation of Phe residues at the first and last base-steps of the TATAAAAG binding site leads to loss of base stacking and formation of a sharp kink as a key feature of the bound DNA8,31,32 (Fig. 3c). Mismatches also destabilize the DNA duplex, with C-C having the least favorable stacking interactions33 (Fig. 3d). Remarkably, introducing mismatches at position 8 in the TBP binding site, which is one of the highly unstacked positions, resulted in increased TBP binding affinity, with C-C having the largest effect (Extended Data Fig. 7a). This indicates that mismatches increase affinity by prepaying the energetic cost to partially melt the base pairs. If this were true, we would expect an inverse correlation between the increase in binding affinity and the stability of the mismatch. To test this prediction we performed additional TBP binding measurements for all mismatches and base-pair mutations at each position in the TBP binding site using a modified SaMBA protocol (Methods, Supplementary Table 4). We compared these binding measurements to predicted destabilization energies (Methods) and observed a remarkable correlation: R2=0.765 (Fig. 3e). Analysis of the other positions in the TBP site revealed high correlations between destabilization energies and TBP binding (R2>0.4) at three of the four unstacked positions (Extended Data Fig. 7b). No significant correlations were observed at other positions in the binding site, consistent with our hypothesis.
To further examine how mismatches impact protein-DNA binding, we solved four X-ray structures of TBP bound to DNA (resolution 2.0–2.5 Å, Fig. 3f, Extended Data Fig. 7c) containing C-C and A-C mismatches at the unstacked positions 7 and 8, which increase TBP binding affinity by 0.8–1.4 kBT (Supplementary Table 4). These are the first structures of mismatch-containing DNA bound by a TF protein, and can provide a glimpse into how mismatches might increase binding affinity. Remarkably, the heavy atoms of the structures superimpose with root mean square deviations (RMSD) of 0.29–0.49 Å, indicating nearly identical modes of TBP interaction with mismatched versus Watson-Crick sites, including in and around the mismatches (Fig. 3f, Extended Data 7c, Supplementary Discussion). Notably, the four TBP-DNA structures were obtained from distinct crystal forms (Extended Data Table 1), indicating that packing was not a factor in the similar DNA conformations. In all cases, no evidence was found for new contacts with the mismatches that would explain the large increases in TBP binding. This provides additional compelling evidence that mismatch-induced enhancements in protein binding can arise from pre-paying energetic penalties that are invisible to X-ray structure-based detection.
Native and non-native interactions
In addition to pre-paying conformational penalty and thus reinforcing native interactions (i.e. H-bonds, water-mediated interactions, electrostatic and other interactions that would also form in Watson-Crick DNA), our MD simulation data also suggests that mismatches can enhance TF binding by promoting non-native interactions with the mismatched DNA due to changes in both the base identify and the DNA conformation at the mismatch and/or neighboring sites. For example, in the case of the T-G mismatch at position 6 in the Ets1 binding site, for which no structural mimicry was identified in our analyses (Supplementary Table 6), MD simulations of protein-bound mismatched and Watson-Crick DNA revealed that the wobble conformation positions the mismatched T base to form non-native contacts with protein side chains (Extended Data Fig. 5e, Supplementary Table 7). Non-native interactions were also observed in MD simulations of non-specific sites that are rendered high-affinity Ets1 binding sites by specific mismatches (Extended Data Fig. 5i,j, Supplementary Table 7). In addition, a combination of non-native interactions and structural mimicry is observed in the case of A-G at position 6 in the Ets1 binding site (Extended Data Fig. 5b,e,h). Structure determination of these complexes may help reveal the nature of the non-native interactions, which can also include water-mediated H-bonds and electrostatic interactions that may enhance the binding energetics (Extended Data Fig. 8).
Summary
Our study provides the largest analysis to date of the effects of DNA mismatches on protein binding, and reveals DNA conformational penalties as an important determinant of protein-DNA binding affinity and selectivity. Our new assay can be extended to include distortions in DNA shape induced by multiple mismatches, insertions and deletions, damaged and epigenetically modified nucleotides, and thereby thoroughly investigate these penalties in a high-throughput and unbiased manner. In addition, regardless of the precise mechanisms by which mismatches enhance TF binding, these high-affinity interactions may provide a biophysical mechanism for inhibiting the repair of specific mismatched sites, and consequently contributing to the formation of genetic mutations in the cell11 (Extended Data Fig. 9, Supplementary Discussion).
METHODS
Structural survey of Watson-Crick and mismatched base pairs
We performed a comprehensive survey of DNA base pair (bp) structures deposited in RCSB PDB34. X-ray crystal structures (resolution < 3 Å) and NMR solution structures containing DNA were downloaded from the RCSB webserver and organized into a searchable database35. Base pair parameters (shear, stretch, stagger, buckle, propeller twist, opening, and C1′-C1′ distance) of a given base pair, as well as base step parameters (shift, slide, rise, tilt, roll and twist) were computed using X3DNA-DSSR36 as described previously37. Base pair parameters (except C1′-C1′ distance) and base step parameters of bases with syn conformation (e.g. in Hoogsteen base pairs and G-A and G-G mismatches) were not computed due to incorrect reference frame.
The overall shape of the DNA was characterized by analyzing the following shape parameters: minor groove width, major groove width, local helical bending, bending direction, and local helical twisting. Minor and major groove widths were calculated using the P-P definition38 by X3DNA-DSSR36. A well-established inter-helical Euler angle approach were used to quantify DNA local bending, including the bending magnitude (βh, 0º≤βh≤180º), the bending direction (γh, −180º≤γh≤180º), and the helical twist (ζh, −180º≤ζh≤180º) of two helices across a given base pair junction35,37,39,40. All calculations with poor alignment to the idealized helices (RMSD > 2 Å for sugar and backbone atoms39) were omitted from analysis. Global parameters were analyzed at the mismatch positions as well as ±1 base pair or base step.
A total of 903 A-T and 746 G-C standard Watson-Crick (WC) bps in naked DNA were identified (Supplementary Methods) and used to define the B-DNA envelope (Extended Data Fig. 1a, Supplementary Table 8). A total of 613 TF-DNA structures in PDB34 were used to identify WC bps for which at least one bp parameter deviates from the free B-DNA envelope by 3 standard deviations or is completely outside the envelope. The statistics of these distorted WC bps in TF-bound DNA are summarized in Extended Data Fig. 1 and Supplementary Table 8. To survey the DNA mismatch structure and geometry, all possible single mismatches (excluding modified bases) surrounded by at least two canonical WC bps on both sides were identified and subjected to manual inspection (Supplementary Table 9). Of the 110 identified mismatches, 26 were in free DNA and not mediated by heavy metals (8 G-T, 7 G-A, 5 A-C, 3 T-T, 2 G-G, and 1 C-T) (Supplementary Table 9, Extended Data Fig. 2a).
DNA melting analysis
Thermodynamic parameters for mismatch formation were computed using MELTING 5.2.041 as an average over all possible sequence contexts surrounding each mismatch. Default options for nearest neighbor thermodynamic parameters and ion correction terms were used along with a sodium ion concentration of 150 mM. The energetic terms for helix initiation and symmetry were set to zero in order to mimic the placement of the mismatch within the context of a non-palindromic duplex.
Molecular dynamics (MD) simulations
All MD simulations were performed using the AMBER ff99 force field42 with bsc0 corrections for DNA43, ff14SB corrections for proteins44, and using standard periodic boundary conditions as implemented in the AMBER MD package45. To systematically analyze the ensemble behavior of all mismatches, we performed MD simulations on unbound DNA for all possible WC and mismatched base pairs embedded in constant flanking sequences: 5’-CTCTGCCACGTGGGTCGT-3’ (the variable position is underlined). For G-A and G-G, we simulated two possible geometries: G(anti)-A(anti), G(anti)-A(syn) and G(anti)-G(syn), G(syn)-G(anti), where one of the bases was manually rotated around the glycosidic bond by 180° to generate a syn conformation. Production runs of 500ns were carried out and extended to achieve convergence of the RMSD of the DNA if necessary. Summary descriptions of the ensemble behavior of different mismatches in the unbound DNA simulations are presented in Extended Data Fig. 2c. The dynamics of DNA mismatches in MD simulations are in good agreement with previous work46.
For MD simulations of protein-DNA complexes, starting structures corresponding to the Myc/Max, Ets1, p53, Max/Max, CTCF, Egr1, GR, Elk1, and RelA systems were obtained from PDB entries 1NKP, 2NNY, 3KZ8, 1AN2, 5KKQ, 1P47, 1R4R, 1DUX, and 5U01, respectively (see Supplementary Methods for details). The TFs were chosen due to the availability of TF-DNA structures for DNA sequences similar to the ones tested by SaMBA. Production runs of 200 or 500 ns were carried out and extended to achieve convergence of the RMSD of the protein-DNA complex if necessary. For proteins bound to mismatched DNA sites, we chose not to simulate the mismatches A-A, A-C, and C-C, given the lack of a stable base pairing geometry for A-A47 and the tendency of A-C and C-C to undergo protonation-dependent structural changes in order to form stable base pairing geometries48,49. Protonation-dependent base pairing conformational equilibria are susceptible to being highly influenced by protein binding50, and are also difficult to model computationally51. We simulated one mismatch per protein, focusing on G-T and C-T mismatches, as well as T-T, G-G and G-A in specific cases, given their stable base pairing geometries52–56 and ability to be reliably modeled computationally46. The simulation results were used to analyze protein-DNA contacts and the buried surface area (Supplementary Table 7), as described in Supplementary Methods.
Protein expression and purification
For SaMBA experiments, full-length human proteins Ets1, Elk1, Gabpa, Runx1, E2f1, Six6, Ap2a, Gata1, Myc (c-Myc), Max, Mad (Mad1), human Egr1 residues 335–423, human RelA residues 20–290, human GR residues 418–517, human Stat3 residues 128–715, and full-length S. cerevisiae Cbf1 were expressed and purified as described previously21,57–60. Full-length human p53, TBP, CTCF, Creb1, Crem, and Atf1 were obtained commercially (Supplementary Methods). For X-ray crystallography, A. thaliana TBP DNA-binding domain was produced as in 61. For Ets1 fluorescence anisotropy binding assays, murine Ets1 (residues 280 to 440) was produced as in 62. For EMSA binding affinity measurements, human GR DNA-binding domain (residues 418–506) and human P53 (residues 94–360) were expressed and used as in 63–65.
SaMBA library design and measurements
Saturation Mismatch-Binding Assays (SaMBA) were performed as follows. Five custom DNA libraries (v1-v5), each containing ~15,000 single-stranded 60-base oligonucleotides, were designed computationally based on TF binding site sequences for 22 TF proteins (Supplementary Table 1a,e–j). The binding sites for each TF were selected based on published data showing specific TF binding to these sites. Sites were selected to contain central 8-mers with protein-binding microarray (PBM) enrichment scores (E-score) of 0.35 or higher, which is indicative of specific protein binding20,21. For CTCF, p53 and RelA we selected strong binding sites based on their DNA binding motifs reported in the literature (CTCF66, p5322, RelA67). For GR, we used two identical half-sites of an idealized glucocorticoid response element with the preferred 3-bp spacer, as described and used in 68.
Each DNA library was designed to contain multiple replicates (8–20) of both wild-type binding site sequences and all possible single base variants of the same sites. For SaMBA library v1, we used 14 replicate spots for each wild-type sequence and 8 replicate spots for each mismatch. For SaMBA libraries v2, v3, v4, and v5 we used 20 replicate spots for each wild-type sequence and 10 replicate spots for each mismatch. The DNA libraries were commercially synthesized on DNA microarray chips (Agilent). Next, double stranded DNA binding sites were generated on the chip by hybridization with the wild-type reverse complement oligonucleotides in solution (variant complements were absent from the hybridization solution). For each wild-type sequence, the solution contained ~2.5 μM (large excess) unlabeled HPLC-purified oligonucleotides and ~0.25 μM FAM/Cy3-labelled HPLC-purified oligonucleotides (Integrated DNA Technologies). For the variant sequences on the chip, the absence of perfect complements in solution ensured successful hybridization with the WT complements. The small fraction of fluorescently labeled oligonucleotides allowed us to assess the successful formation of mismatched duplexes on the chip (Extended Data Fig. 3a–d, Supplementary Table 10).
The reaction buffer mixture for the hybridization step was 100 μl 10x reaction buffer (260 mM Tris-HCl, pH 9.5, 65 mM MgCl2) in a total volume of 1000 μl, similarly to 20. The chip was incubated with reaction mixture in a hybridization oven using a pre-warmed stainless-steel chamber and gasket cover slip. After a 5-hour incubation (85 °C for 10 min, 75 °C for 10 min, 65 °C for 60 min, 60 °C for 120 min, and 55 °C for 100 min), the hybridization chamber was disassembled in a glass staining dish in 500 ml phosphate buffered saline (PBS) / 0.01% Triton X-100 at 37°C. The chip was transferred to a second staining dish, washed for 10 min in PBS / 0.01% Triton X-100 at 37°C, washed once more for 3 min in PBS at room temperature, similarly to 20. The fluorescent signal (Cy3/FAM) of hybridized oligonucleotides was measured using a GenePix 4400A microarray scanner to confirm that the hybridization was successful and reproducible, and that no significant cross-hybridization occurred (Extended Data Fig. 3b, Supplementary Table 10).
Protein binding and antibody steps were performed similarly to PBM assays20 (Supplementary Methods). The fluorescent signal of bound TF for each DNA spot was measured using a GenePix 4400A microarray scanner and the GenePix Pro 7.0 software. Multiple replicates of each sequence were used to quantitatively compare the binding signals between sequences and to statistically assess the significance of binding differences using a one-sided Wilcoxon-Mann-Whitney test, corrected for multiple hypotheses testing using the Benjamini-Hochberg procedure. SaMBA profiles (e.g. Fig. 2b) representing the impact on TF binding for each possible mismatch along each parent sequence were produced by calculating the log2 of the ratio between each mismatch and its corresponding wild-type parent sequence (Supplementary Table 1b). Since the magnitudes of these ratios vary widely between proteins, for each parent site all ratios were also divided by the ratio of the largest decrease at the same site and multiplied by −1, so that the largest decrease for each parent sequence became −1 (Fig. 2a).
Validation and calibration of SaMBA data using TF binding affinity measurements
DNA-binding affinity measurements for p53 were performed using electrophoretic mobility shift assays (EMSA) as described in 64,69 (Supplemental Methods). The macroscopic dissociation binding constants for the dominant p53 tetrameric species were computed for ten different hairpin duplexes: four Watson-Crick and six containing mismatches (Supplementary Table 3). Six replicate measurements were performed for each duplex, and the average binding affinities were used in comparisons with SaMBA data (Fig. 1f, Extended Data Fig. 3e).
Binding affinity measurements for Ets1 (residues 280 to 440, termed Ets1ΔN280) were performed using steady-state fluorescence polarization, as described in 70, using a Cy3-labeled DNA probe encoding the Ets1 binding sequence 5’-CGCACCGGATATCGCA-3’. In brief, 0.5 nM of DNA probe and 10 nM Ets1ΔN280 were co-titrated with one of five unlabeled DNA duplexes: two Watson-Crick and three containing a mismatch (Supplementary Table 3). Triplicate measurements were performed for each duplex. The data confirmed both increased and decreased Ets1 binding due to mismatches, as revealed by SaMBA (Fig. 1f, Extended Data Fig. 3e).
GR binding affinity measurements were performed using EMSA, as described in 63 (Supplemental Methods). One Watson-Crick and three mismatched sites were tested (Supplementary Table 3). To avoid self-hybridization of the probes in EMSA, one of the two GR half-sites and the spacer between them were mutated compared to the SaMBA site. Positions known to be critical for GR binding were kept constant. Measurements were performed in triplicate, and the average binding affinities were used in comparisons with SaMBA data (Fig. 1f, Extended Data Fig. 3e).
The measurements described above were used both to validate TF binding increases and decreases due to mismatches, and to calibrate SaMBA data. To calibrate SaMBA data for additional TFs, we leveraged publicly available binding affinity data for Watson-Crick sequences by using a modified SaMBA protocol to test, for each TF of interest, multiple Watson-Crick sites with available affinity measurements (in addition to the wild-type and mismatched binding sites tested in a typical SaMBA assay). In our modified protocol, 60-mer DNA probes were designed to form hairpin duplexes with and without mismatches, and binding measurements were performed similarly to regular SaMBA assays (Supplementary Table 4). The following TF binding affinity data sets were used: surface plasmon resonance (SPR) data for Cbf171, mechanically induced trapping of molecular interactions (MITOMI) data for Cbf1 and Max72, fluorescence anisotropy (FA) data for p5322, k-MITOMI data for Egr173, and EMSA data for TBP (from sites with consistent measurements in 74 and 75).
Calibration of SaMBA data into free energy terms was performed as shown in Extended Data Fig. 3g,h, based on the correlation between the EMSA/FA/SPR/MITOMI-derived affinities and the logarithm of the binding signal obtained in SaMBA (Supplementary Table 3). DNA libraries used for calibration also included all possible mismatches and mutations over a small number of DNA sites: two binding sites for Ets1, Max, and TBP, one binding site for Cbf1, Egr1, p53 and GR, and two non-specific sites for Ets1 (Supplementary Table 4). The data for these 12 sites was used to directly compare the effects of mutations versus mismatches (Extended Data Fig. 4 and related text). When comparing the effect of base-pair mutations versus the sum of the effects of the corresponding one-base mismatch variants (Extended Data Fig. 4c and related text), the significance of the difference between these quantities was assessed using a two-sided t-test with Benjamini-Hochberg correction for multiple hypotheses testing; significant differences were called at a cutoff of 0.05 for the corrected p-value. The effect of mutations on TF binding was also measured using the standard PBM protocol20 (Supplementary Table 1). Consistent with the results obtained using the SaMBA libraries, the PBM libraries show that mutations have different effects on TF binding compared to mismatches (Fig. 2d; Supplementary Table 1). For all analyses presented here, proteins p53, Ets1, and GR were calibrated using new binding measurements for mismatched and Watson-Crick DNA sites, while Cbf1, Max, TBP, and Egr1 were calibrated using data for Watson-Crick binding sites available in the literature (Supplementary Table 3).
Ets1 non-specific binding analysis
Due to the high density of the DNA chips used in our experiments, each SaMBA DNA library can accommodate binding sites for several TFs (Supplementary Table 1). Thus, each TF was tested not only against its specific binding site(s), but also a small number of non-specific sites, which were specific to other TFs (Supplementary Table 1c,d). For all proteins examined, the introduction of mismatches increased binding even at non-specific DNA sites (Supplementary Table 1d) and, surprisingly, in some cases the new binding levels were similar to those observed for specific binding sites, thus effectively creating novel binding sites within non-specific DNA. To further test the significance and the magnitude of such increases, a new DNA sequence library was designed to measure the effects of mismatches that enhanced Ets1 binding at sites that were not originally designed for Ets1 (i.e. sites that were specific to other TFs). This new library (Supplementary Table 2) contained positive and negative control groups of “specific” and “non-specific” sites, respectively, in order to enable accurate assessment of the relative binding strength of each of the sites of interest. The negative control group was composed of a set of 1000 random DNA sequences. Since specific sites can randomly appear among these sequences, we defined the non-specific binding affinity range by excluding the top 1% of the strongest bound sequences in this group. The positive control sequences were selected from crystal and NMR structures of Ets1-DNA complexes in which the Ets1 was shown to specifically bind the ETS binding core GGA(A/T) (PDB IDs: 2NNY, 2STT, 3MFK, 3RI4). Fig. 2c shows five representative examples where mismatches introduced in a non-specific site (i.e. a site with binding affinity below the 99th percentile of random sites) increases the affinity to reach the specific range (i.e. the range observed for sites with Ets1-bound crystal or NMR structures). The full data set is available in Supplementary Table 2.
Nuclear magnetic resonance (NMR) experiments
We prepared A6-DNA duplexes containing A-T, m1A-T, T-T and C-T base pairs. The m1A-containing single strand was purchased from Yale Keck Oligonucleotide Synthesis Facility with HPLC purification, while all other unmodified single strands were purchased from IDT with standard desalting purification. Concentrations were measured using a Nanodrop 3000, with the extinction coefficients for single and double strands obtained using the ADT bio oligo calculator. After resuspension in water, equimolar amounts of single strands were mixed together to form the duplexes. The duplexes were annealed by heating to 95 °C for 5 min and cooling at room temperature for ~1 hr. They were then exchanged into NMR buffer (15 mM sodium phosphate, 25 mM sodium chloride, 0.1 mM EDTA, pH 6.9) using centrifugal concentrators. Duplex samples containing 10% D2O after buffer exchange were lyophilized into 100% D2O. Assignments of the sugar resonances were performed using a combination of 2D 1H-1H NOESY, 2D 1H-1H TOCSY and 2D 1H-13C HSQC experiments. All measured chemical shift differences are available in Supplementary Table 11.
Structural analyses of mismatches that enhance TF binding
We used existing PDB structures of TF-DNA complexes to examine whether DNA mismatches can indeed mimic distorted conformations in native TF-bound DNA, which could explain the increased TF binding affinity to DNA mismatches. Structures of protein-DNA complexes are available in PDB for 15 of the 22 TFs examined by SaMBA. For three of the 15 proteins (Gata1, Mad, and Stat3), the base pair position(s) where mismatches increase TF binding are different in the crystal structure sequence compared to the sequences tested in SaMBA. We thus focused our structural analyses on the remaining 12 proteins (Supplementary Table 5). When multiple structures were available for the same TF, we chose the one with the DNA sequence most similar to the one tested in SaMBA. For the selected structures, we focused on the regions that are in common between the crystal structure and the SaMBA sequence, and at each position we computed the extent to which each structural feature deviates from the B-DNA envelope (Supplementary Table 5). For each position we also computed the largest deviation observed across all structural parameters, as well as the number of structural features with mean values more than one standard deviation above the mean observed for naked B-DNA (Supplementary Table 8a). We applied Mann-Whitney U tests on these summary statistics to ask whether the positions with mismatch-enhanced binding are more distorted than the other positions in TF binding sites (p=0.017 for the largest deviation; p=0.015 for the fraction of distorted features).
Next, focusing on the regions that were identical between the crystal structure and the SaMBA sequence (underlined in Supplementary Table 6a), we identified 23 positions at which we found increased TF binding, due to a total of 32 mismatches (for some positions we found several mismatches that lead to increased TF binding levels). For these 23 positions, we comprehensively annotated all DNA local and global distortions, defined as deviations in a structural parameter that are greater than one standard deviation above the mean of that parameter in free B-DNA structures (Supplementary Table 6b). Next, we examined the mismatch structures to determine whether the mismatches are inducing structural features that mimic bound geometries. Due to lack of available PDB structures of DNA mismatches embedded in Watson-Crick contexts, we systematically performed MD simulations of free DNA containing each mismatch. Similarly to our analyses of distortions in protein-bound DNA, we identified the local and global distortions caused by mismatches by comparing the distributions of structural parameters for mismatched DNA versus Watson-Crick DNA, according to the MD simulations (Supplementary Table 6c). By intersecting the lists of distortions identified in mismatched DNA versus TF-bound DNA, we identified all candidate features that are potentially mimicked by the mismatches that increased TF binding. We found such candidate features for 21 of the 32 mismatches (66%) (Supplementary Table 6d).
Crystallization and structure determination of TBP-mismatch DNA complexes
TBP-DNA complexes were prepared and used for vapor diffusion crystallization screens (Supplementary Methods), resulting in large well diffracting crystals suitable for data collection after optimization of initial hits. Data for all the crystals were collected at the Advanced Light Source (ALS) on beamlines 8.3.1 and 5.0.1. The data were processed with MOSFLM and scaled with SCALA76,77. The structures were solved by molecular replacement (with MolRep) using a prior TBP structure (PDBID: 1QNE) with the waters removed, as a search model. After refinement in Phenix78, the structures were manually rebuilt in O79. MolProbity was used to guide the process of refitting and refinement80. See Extended Data Table 1 for the final data collection and refinement statistics for each structure.
Extended Data
Extended Data Figure 1.

(a) Distributions of base pair parameters in free and TF-bound DNA, from PDB34 survey. Solid lines denote the median value of each parameter. Dashed lines denote the upper and lower bounds of the distribution for free (pink) and bound (green) DNA. 613 TF-bound structures and 409 free B-DNA structures, all with resolution < 3 Å, were used in the analysis (Methods).
(b) Percentage of structures with base pairs outside the B-DNA envelope. Among the 613 TF-bound structures, 41.1% (i.e. 252) contain severe distortions of at least one base pair outside the free B-DNA envelope, with the envelope defined as at most 3 standard deviations above or below the mean. Only 16% (i.e. 65) of the free B-DNA structures satisfy this criterion. (Using a less stringent definition of the B-DNA envelope, by considering 2 standard deviations above or below the mean, we found that 80.8% of the TF-bound structures contain at least one base pair outside the free B-DNA envelope, approximately twice the frequency observed in free DNA, which was 41.8%.) Considering the full range of base pair parameter values as defining the free B-DNA envelope, we found that 11.3% (i.e. 69) of the TF-bound structures contain at least one base pair with an extreme deformation that was never observed in any free DNA structure.
(c) Local deformations of base pairs observed in diverse TF-DNA complex structures. Left: 3D structures with the distorted base pairs highlighted in black boxes. Upper right: enlarged view of the base pair structures with their base pair parameters labeled. Lower right: schematic diagram of the corresponding base pair parameters.
Extended Data Figure 2.

(a) Base pairing geometry of Watson-Crick base pairs and mismatches, obtained from a survey of crystal structures in the PDB34. Mismatches with modified bases and those that were metal-mediated were excluded from analysis (Methods). Predominant base pairing geometries under neutral pH conditions are shown in black. Minor geometries are shown in grey.
(b) Melting energies for DNA mismatches relative to G-C and A-T Watson-Crick base pairs. See Methods for details.
(c) Distributions of structural parameters in Watson-Crick and mismatched DNA, from MD simulations. Solid lines denote the median value of each parameter. Observations from the MD simulation results: (1) G-T retains wobble geometry during the MD simulation, with sheared conformation (|shear| around 2 Å) accompanied by a slight stretch. (2) T-T shows wobble geometry with sheared conformation (|shear| around 2 Å). Different from G-T, the T-T mismatch shows rapid dynamic equilibrium of both wobble geometries with either one of the Ts shifted to the minor groove direction. Despite this rapid dynamic equilibrium, the T-T base pair is still constricted with C1′-C1′ distance 8–9.5 Å. (3) Similar to T-T, the C-T mismatch is also constricted with two H-bonds stably formed for most of the time. However, C-T mismatch can transiently adopt a high-energy conformation with only one H-bond and is not constricted anymore (C1′-C1′ distance ~10 Å), potentially due to the close contact between T-O2 and C-O2. The entire C-T MD trajectory is comprised of approximately 5% of these high-energy species. (4) C-C is partially constricted with C1′-C1′ distance around 9.8 Å due to unstable H-bonding. (5) All pyrimidine-pyrimidine mismatches are stacked in the helix without swing out of the helix in the MD trajectories. (6) G-G does not experience anti-syn equilibrium during the simulation. The C1′-C1′ distance of G-G (G(syn)-G(anti) or G(anti)-G(syn)) is around 11.2–11.5 Å, which is larger than the canonical G-C base pair. (7) G(anti)-A(syn) is not constricted (C1′-C1′ distance around 11Å) and G(anti)-A(anti) reveals large C1′-C1′ distance around 12.8 Å. Base pair and base step parameters of bases with syn conformation (marked with *) were not computed, and are thus greyed out, due to an ill-defined coordinate frame (Methods). The C1’-C1’ distance is shown, since it is not affected by the change of coordinate frame.
(d) Mismatches can mimic distorted base-pair geometries observed in protein-bound DNA. Figure shows overlays of distorted (colored) and idealized WC (grey) base pairs from 3DNA (top); mismatches (colored) and idealized WC (gray) base pairs (middle); and mismatched and distorted WC base pairs (right). The mismatched conformations are of free DNA and were obtained from MD simulations (Methods). The C-T mismatch can mimic an A-T Hoogsteen base pair by constricting the C1′-C1′ distance (taken from PDB: 3KZ8). The G-T mismatch can mimic a sheared A-T base pair by shifting the T to the major groove direction (taken from PDB: 4MZR).
Extended Data Figure 3. Validation and calibration of SaMBA measurements.

(a) Schematic representation of our experimental workflow to detect cross-hybridization. To check whether certain oligonucleotides hybridize with non-target complementary oligonucleotides, we designed an experiment in which only certain oligonucleotides (red) were labeled. If significant cross-hybridization occurred, we would have detected fluorescent signal on the chip even for sequences without fluorescent complements in the hybridization solution (i.e. for the sequences shown in blue).
(b) No significant cross-hybridization was detected. Bottom: list of 12 sequences used in the hybridization solution of one SaMBA experiment (red: fluorescently-labeled oligonucleotides; blue: unlabeled). Top: fluorescent signal from the hybridization of these 12 sequences on the chip. For the sequences on the chip for which their complement is not labeled, the fluorescent signal is practically undetectable (blue), and it is several orders of magnitude lower than the sequences with a labeled complementary strand (red). Boxplots show median signals over replicate DNA spots, with the bottom and top edges of each box indicating the 25th and 75th percentiles, respectively. The whiskers extend to the most extreme data points not considered outliers.
(c) The effect of mismatches on hybridization. To estimate the efficiency of our hybridization protocol, we measured the hybridization signal of one specific sequence (sequence #3 for library v1; see Methods, Supplementary Table 10), to different sequences containing multiple mismatches (0 to ~40), and a completely different sequence (‘60*’). As expected, the hybridization was less efficient for sequences with large numbers of mismatches. However, for small numbers of mismatches the hybridization was highly efficient. Longer incubation time, higher oligonucleotide concentration, and normalization of the signal could enable the use of SaMBA for larger numbers of mismatches. Plot shows medians and standard deviations over all sequences containing the same number of mismatches, with 6 replicate spots per sequence. Mismatches were introduced randomly by generating N random base changes (N=1–5,10,15,25,35,45) to sequence #3, and repeating the procedure ten times for each N. This led to duplexes with 1 to 37 mismatches compared to the original sequence.
(d) Hybridization signal is highly reproducible. The correlation of hybridization signals between two replicate experiments was very high (R2=0.99). Plot shows median values, computed over 6 replicate spots, based on data shown in panel (c).
(e) Validation of mismatch effects by orthogonal methods. For p53, Ets1, and GR proteins, the log-transformed SaMBA binding intensities correlate with independent affinity measurements performed on mismatched and non-mismatched DNA sites (Methods). Similarly to PBM experiments, median values over all replicates were used for SaMBA (n=10 replicate spots); error bars show the median absolute deviation. Average values over replicates were used for the orthogonal methods (n=6 independent measurements for p53, and 3 independent measurements for Ets1 and GR), with error bars showing the standard deviation. Red shaded region: 95% confidence interval for Pearson’s correlation. Binding free energy differences (ΔΔG) are shown between native Watson-Crick binding sites and the highest increase in binding due to a mismatch. Two SaMBA sites were tested for GR (see Methods).
(f) Correlation between binding data obtained by SaMBA versus independent methods. For SaMBA data the plots show the median values over replicate spots (n=10 replicate spots), with error bars showing the median absolute deviation. For independent data (Methods) the plots show the binding affinities as reported in the respective papers. Red shaded region: 95% confidence interval for Pearson’s correlation.
(g) Standard equilibrium thermodynamics equations demonstrate that the logarithm of the dissociation constant (KD) of the TF:DNA complex is linearly proportional to the logarithm of the TF:DNA complex florescence signal, under certain conditions in which the TF concentration and the free DNA concentration are in excess compared to the concentration of the bound complex (and those remain constant during the reaction). (h) Similar to (g), for cases in which the DNA-bound species is a dimer.
Extended Data Figure 4. Comparing the effects of mutations versus mismatches on TF binding.

(a) The magnitude of the energetic effects of mutations (light colors) and mismatches (dark colors) is similar. The effects were computed for all 7 proteins with available calibration data in our study, and for a total of 12 DNA sites (Methods). The effects of mismatches were calculated relative to the two closest Watson-Crick sequences (e.g. for a G-T mismatch the closest Watson-Crick base pairs are G-C and A-T; the mismatch plots include both ΔΔG(G-C -> G-T) and ΔΔG(A-T -> G-T)).
(b) Mismatches and their corresponding mutations have different, even opposite effects on TF binding. Each mutation is compared to the two closest mismatches (e.g. G-C -> A-T is compared to both G-C -> A-C and G-C -> G-T). Upper left quadrant: mutations increase binding, mismatches decrease binding. Upper right quadrant: both mutations and mismatches decrease binding. Lower left quadrant: both mutations and mismatches increase binding. Lower right quadrant: mutations decrease binding, mismatches increase binding. X-axis and Y-axis show calibrated binding measurements computed from the median SaMBA signal intensities (over n=10 replicate spots).
(c) Comparing the effect of mutations versus the cumulative effects of the two closest mismatches. Points close to the diagonal correspond to cases where the effect of the mutation is approximately equal (within experimental noise) to the sum of the effects of the two mismatches. Points above the diagonal correspond to cases where Watson-Crick mutations have either a more beneficial or a less detrimental effect on TF binding compared to the cumulative effect of the two mismatches. Points below the diagonal correspond to cases where Watson-Crick mutations have either a less beneficial or a more detrimental effect on TF binding compared to the cumulative effect of the two mismatches. X-axis and Y-axis show calibrated binding measurements computed from the median SaMBA signal intensities (over n=10 replicate spots). Please see Supplementary Table 4 for the raw binding data used to compute the measurements shown in this figure.
Extended Data Figure 5. The effects of mismatches on Ets1-DNA binding.

(a) SaMBA profile for an Ets1 binding site, highlighting the G-A mismatch at position 6, which shows the largest increase in binding affinity.
(b) Distortions. In the bound Ets1-DNA complex (PDB ID: 1K79), the positions where the recognition helix is inserted into the DNA major groove are significantly distorted, with bending (βh=23°) towards the major groove, local unwinding (ζh=23°), and minor groove widening. Position 6, the middle position of the GGA core binding region, is highlighted to show the expanded C1’-C1’ distance. The G-A mismatch at this position mimics the C1’-C1’ distance of the bound DNA. Violin plots of the MD simulation data show that the G-A mismatch in anti-anti configuration also mimic the minor groove width of the bound G-C.
(c) Base readout. According to MD simulation results, G-A (anti/anti) and G-T mismatches increase the overall number of H-bonds and the buried surface area at the Ets1-DNA interface, compared to the Watson-Crick G-C pair (Methods).
(d) Ets1-DNA interface in the GGAA core binding region. Contacting residues in the recognition helix are shown in magenta. Direct H-bond contacts with the bases are highlighted; such contacts occur only at the GGA bases, on the “lower” strand of the shown Watson-Crick DNA site.
(e,f) Representative snapshots of different H-bond interactions between Arg391 and the base pair at position 6, from molecular dynamics (MD) simulations. The G-T mismatch shows an additional H-bond compared to G-C and G-A. (g) In a non-specific site where G-A increases the affinity to reach the specific range, MD simulations show that the G-A mismatch forms H-bonds similar to those formed in specific sites (shown in panel f). (h) Non-native H-bond at position 4, due to the G-A mismatch at position 6 in the specific Ets1 binding site. (i,j) Non-native H-bond interactions created in a non-specific site (panel g) at positions neighboring the positions of the mismatch, either with the base (i) or the backbone (j).
(k) SaMBA profiles for additional Ets1 binding sites. We measured the effect of mismatches in four Ets1 binding sites in addition to the one shown in panel a. Although the profiles for different sites are quantitatively different and dependent on the flanks, the trends for increased binding due to mismatches are similar. For all cases, the A-G mismatch at position 6 significantly increases Ets1 binding.
(l) Structural features at the mismatch position. Violin plots show the local twisting and kinking at position 6, and the minor and major groove width at position 5–6 of Ets1-bound DNA, as well as the naked DNA for different base-pairs, according to MD.
Extended Data Figure 6. The effects of mismatches on p53-DNA binding.

(a) Mismatch profile for p53 reveals that increased TF binding occurs only due to C-T and T-T mismatches (red rectangle) at the same positions where the Hoogsteen conformation is observed in p53-DNA complexes (PDB ID: 3KZ8).
(b) MD simulation-based violin plots of C1’- C1’ distance at position 2, as well as the minor grove width (at position 0–1), for p53-bound DNA and naked DNA (wild-type and mismatched) reveals that the minor groove for C-T and T-T mismatches is more similar to the bound form compared to the free A-T base pair. Plot also shows that the G-T mismatch, which reduces p53 binding, does not mimic these distortions seen in the bound DNA. Notably, a narrower minor grove at position 0–1 was previously suggested to be important for the interaction of the DNA with the Arg248 residue in p5327.
(c,d) NMR validation showing that T-T and C-T mimic the reduced C1’-C1’ distance observed in p53-bound DNA27,28. (c) Chemical shift overlays of the 2D HSQC NMR spectra of the C1’-H1’, C4’-H4’ and C3’-H3’ regions for A6-DNA m1A in which the m1AT base pair is in the Hoogsteen conformation30 (left, green), A6-DNA TT (middle, blue) and A6-DNA CT (right, red) with unmodified A6-DNA (black) at pH 6.9, 25 °C. (d) Bar plots of the individual chemical shift differences (relative to unmodified A6-DNA) of the C1’/C3’/C4’ carbons of A6-DNA m1A (top), A6-DNA TT (middle) and A6-DNA CT (bottom). Similarity between the Hoogsteen induced chemical shift differences and mismatch shifts (relative to the Watson-Crick wild-type) are observed for both T-T and C-T.
(e) Additional comparisons of global features (twisting angle, local kinking, and kinking direction at position 2 and major groove width at position 0–1) reveal additional mimicry between C-T mismatch and the Hoogsteen conformation local twisting angle.
(f) Pyrimidine-pyrimidine mismatches (C-T, T-C, T-T and C-C) in all 4 positions in which Hoogsteen conformation is observed (n=16 mismatches total), increased p53 binding. However, all other mismatches at these positions (n=32 mismatches total) decreased p53 binding, or had non-significant effects. ΔΔG represent the differences between the p53-DNA binding energy of each mismatch versus the WT sequence, and were estimated using the calibration with EMSA measurements (Methods). Boxplots show median signals over all mismatches, with the bottom and top edges of each box indicating the 25th and 75th percentiles, respectively. The whiskers extend to the most extreme data points not considered outliers.
(g) Number of p53-DNA H-bonds and buried surface area at p53-DNA interface, obtained from MD simulations, failed to explain the observed increase in p53 binding, consistent with the pre-paying mechanism being an important determinant for binding in this case.
(h) DNA hairpin with 4 mismatches (in the 4 positions for which the Hoogsteen conformation was previously observed), strongly binds p53: 3–6 kBT stronger (depending on the data used for validation, Supplementary Tables 3, 4) compared to the highest-affinity p53 binding sites previously reported22. Notably, we expect the difference in binding affinity to other genomic p53 sites (ΔΔG) to be even larger since most p53 binding sites in the genome are of lower binding affinities22.
Extended Data Figure 7. The effects of mismatches on TBP-DNA binding.

(a) Mismatch profile for TBP.
(b) Correlations between TBP binding levels and DNA duplex stability were computed over all 16 base-pair variants at positions 1 to 8 in the TBP site. Bar-plots (left) represent the squared Pearson correlation coefficient (R2) at each position. For the only three positions with significant correlations (positions 2, 7, and 8) the scatter plot correlation is presented (right), with binding signals representing medians over 9 replicate spots. Blue shaded regions: 95% confidence interval for Pearson’s correlation. The sequences of the Watson-Crick and mismatched base pairs are shown in each scatter plot (e.g. for position 8, GC stands for the wild-type G-C base-pair underlined in the TBP site TATAAAAG, CC stands for C-C at this position, etc.). Remarkably, these high correlations are observed only in the unstacked base step positions.
(c) Left: structural overlays between TBP-DNA complexes with DNA mismatches (TBP-AC, orange; TBP-CC(2), cyan; TBP-CC(1a), purple; TBP-CC(1b), pink) and their corresponding Watson-Crick counterparts with single base substitutions (1QNE, green; 6NJQ, yellow). The base steps at position 7–8 are zoomed in and highlighted in black boxes. The structural overlay of the mismatch and the Watson-Crick base pairs are shown below each box, with their DNA sequences. Right: overlays of protein-DNA interfaces of TBP-DNA complexes, comparing mismatched and Watson-Crick sites. Four Phenylalanines, as well as other amino acids that are discussed in Supplementary Discussion are highlighted with dashed circles.
(d) Comparisons of the effects of Watson-Crick mutations versus the cumulative effects of the two closest mismatches, shown for the mismatches with new crystal structures. In all three cases the mismatches have significantly larger effects than the Watson-Crick mutations (see also Methods and Supplementary Table 4). ΔΔG values for TBP_site_1 in Supplementary Table 4 were used in these comparisons.
(e) Example of a Watson-Crick mutation whose effect is similar (within experimental error, Supplementary Table 4) to the sum of the two closest mismatches. ΔΔG values for TBP_site_1 in Supplementary Tables 4 were used in these comparisons.
Extended Data Figure 8. Potential mechanisms for mismatch-enhanced TF binding.

(a) TF-DNA complex formation involves creation of intermolecular interactions, as well as DNA conformational changes. Thermodynamically, these processes can be separated into two independent events, and thus an increase in binding affinity could stem from additional interactions (decrease of ΔGinteraction), and/or reduction in the penalty to change the DNA conformation (decrease of ΔGpenalty).
(b) A reduction in the energetic penalty to distort the DNA (ΔGpenalty) could originate from DNA conformational changes due to the mismatch, i.e. prior to binding (for example p53 and TBP, as described in the main text).
(c) A reduction in the energetic penalty for DNA distortion (ΔGpenalty) could also originate from changes in the bound DNA. For example, molecular dynamics simulations of the DNA conformations in free form and in the Myc-DNA complex (for the wild-type A-T and the mismatch G-T) suggest that the reduced penalty in this case is primarily due to changes in the mismatched bound form. The extent of overlap of the kinking direction (γh) obtained from the MD simulations was: Ω=0.34 (WT) versus Ω=0.15 for the G-T mismatch, and was analyzed using a revised Jensen-Shannon divergence score (Ω)81. Representative structures of the DNA sites are shown for WT free (pink), WT bound (orange), G-T free (green), G-T bound (blue). The Myc/Max heterodimer is shown as a gray surface.
(d) Mismatches could lead to the formation of non-native interactions such as hydrogen bonds (left), electrostatic potential and shape sensing (center), and water-mediated interactions (right). Red empty arrows point to the locations of the change. These changes could occur directly at the position of the mismatched base (for example the G-T mismatch for Ets1), as well as at the positions of other bases and/or the backbone, due to non-native structures (for example the G-A mismatch for Ets1). Notably, mismatches not only alter the potential interacting chemical groups of the replaced base, but can also alter the relative orientation of the interacting bases (as observed for the T in the Wobble geometry on the left).
Extended Data Figure 9. DNA mismatches in the cell.

(a) Mismatches can result from misincorporation of bases during DNA replication by DNA polymerases. The average rate at which replication errors are generated and escape proofreading is low in healthy cells (~10−9), but high in certain cancers and cells with Pol-ε/Pol-δ mutations. Even in healthy cells, the rates of generation of individual mismatches vary by more than a million-fold17 depending on the sequence context and the type of mismatch.
(b) Mismatches result from genetic recombination. A characteristic feature of homologous recombination is the exchange of DNA strands, which results in the formation of heteroduplex DNA. Mismatches can result from genetic recombination when the parental chromosomes contain non-identical sequences. In addition, mismatches can arise during DNA synthesis associated with recombination repair. The repair of these mismatches might be less efficient since it was previously shown82 that there is a strong temporal coupling between DNA replication and mismatch repair but a lack of temporal coupling for heteroduplex rejection82.
(c) Spontaneous deamination is common and estimated to occur 100–500 times per cell per day in humans83. G-T mismatches generated by deamination of 5-Methylcytosine (5-meC) are not repaired by the MMR pathway and have considerably lower repair efficiency83. The high rate of 5-meC deamination combined with their relatively slow repair in mammalian cells, contribute to making 5-meC a preferential target for point mutations (about 40-fold) compared to other nucleotides in the genome84, and one of the major sources of the frequent C to T mutations observed in human cells18.
(d) Transcription factors bound to mismatched DNA could interfere with Pol-δ strand displacement activity. Left: DNA synthesized by non-proofreading mismatch-prone Pol-α is normally displaced by the proofreading non-error-prone Pol-δ. Right: Reijns et al.10 recently demonstrated that increased mutation signals arise from regions synthesized by Pol-α that contain TF binding sites. They suggested mismatched DNA synthesized by non-proofreading Pol-α is rapidly bound by TFs that act as barriers to Pol-δ displacement of Pol-α-synthesized DNA, resulting in locally increased mutation rates in subsequent rounds of replication.
Extended Data Table 1:
Data collection and refinement statistics for TBP-DNA mismatch structures.
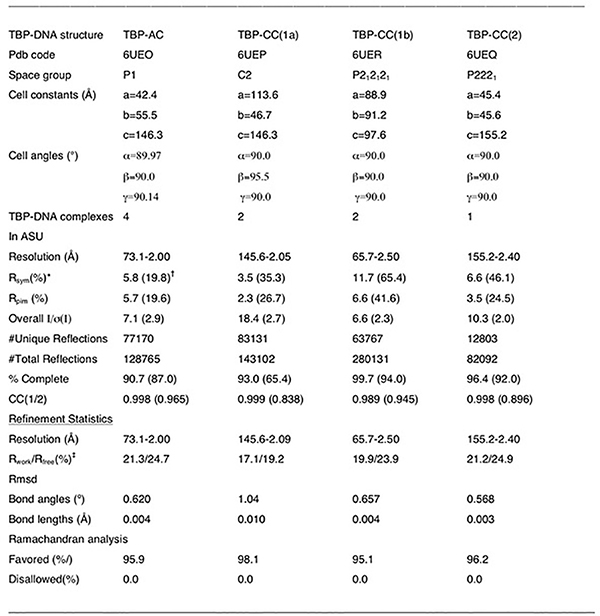 |
Rsym = ΣΣ|Ihkl-Ihkl(j)|/ ΣIhkl, where Ihkl(j) is the observed intensity and Ihkl is the final average value of intensity.
Values in parentheses are for the highest resolution shell.
Rwork = Σ||Fobs| - |Fcalc||/Σ|Fobs| and Rfree = Σ||Fobs| - |Fcalc||/Σ|Fobs|; where all reflections belong to a test set of 5% randomly selected data.
Supplementary Material
Acknowledgements
We dedicate this paper to the memory of Dr. Rosalind E. Franklin, on the occasion of her 100th birthday anniversary. Dr. Franklin’s legacy, including her crucial contribution to the discovery of the molecular structure of DNA, continues to inspire generations of diverse scientists around the world. We thank Dr. Sheera Adar for discussions that initiated this project, Dr. Daniel Herschlag for helpful discussions and comments, Drs. Eyal Arbely, Dmitrij Golovenko, Junji Iwahara, Eric Ortlund, and Richard Young for providing recombinant purified protein, and Dr. Lawrence McIntosh for providing expression plasmids. This work was supported by the National Institutes of Health (NIH) grants R01-GM135658 and R01-GM117106 (to R.G.), R01-GM089846 (to H.M.A.), a Duke University GCB Pilot Grant (to R.G. and H.M.A.), and an Integrated DNA Technologies postdoctoral fellowship award (to A.A.). R.S. and M.A.S. were supported by NIH grant R35-GM130290 (to M.A.S.). A.S. and T.E.H. were supported by the Israel Science Foundation grant 1517/14 (to T.E.H.). S.X. and G.M.K.P. were supported by National Science Foundation (NSF) grant MCB-2028902 (to G.M.K.P.). M.F. and M.A.P. were supported by NSF CAREER award MCB-1552862 (to M.A.P.) High-performance computing was partially supported by the Duke Center for Genomic and Computational Biology. We acknowledge the Advanced Light Source (ALS) at the Lawrence Berkeley National Laboratory for X-ray diffraction data collection on beamlines 8.3.1 and 5.0.1; Beamline 8.3.1 at the Advanced Light Source (ALS) is operated by the University of California Office of the President, Multicampus Research Programs and Initiatives grant MR-15-328599, the National Institutes of Health (R01GM124149 and P30GM124169), Plexxikin Inc. and the Integrated Diffraction Analysis Technologies program of the US Department of Energy Office of Biological and Environmental Research. We also acknowledge beamline 5.0.1. The Pilatus detector on 5.0.1 was funded under NIH grant S10OD021832. The ALS-ENABLE beamlines are supported in part by the National Institutes of Health, National Institute of General Medical Sciences, grant P30 GM124169. The ALS (Berkeley, CA) is a national user facility operated by Lawrence Berkeley National Laboratory on behalf of the US Department of Energy under contract number DE-AC02-05CH11231, Office of Basic Energy Sciences. The Berkeley Center for Structural Biology is supported in part by the Howard Hughes Medical Institute.
Footnotes
Ethics declaration
The authors declare no competing interests.
DATA AND CODE AVAILABILITY STATEMENTS
Data availability
The data that support the findings in this study are available as Supplementary Tables, in Excel format. Coordinates and structure factor amplitudes for the TBP-AC, TBP-CC(1a), TBP-CC(1b) and TBP-CC(2) structures have been deposited in the RCSB Protein Data Bank (PDB) under the accession codes 6UEO, 6UEP, 6UER, and 6UEQ, respectively. The raw SaMBA data has been deposited in the Gene Expression Omnibus (GEO) under accession number GSE156375. The RCSB PDB entries used in this study are available in Extended Data Figures 1, 2, 5, and 7, and Supplementary Tables 5, 6, 7, and 9. High-resolution gel images for the EMSA data are available at https://figshare.com/projects/DNA_mismatches_reveal_conformational_penalties_in_protein-DNA_recognition/83663.
Code availability
The code used for the structural analyses presented in this study is available in GitHub at https://github.com/alhashimilab/TF_MM.
REFERENCES
- 1.Rohs R et al. Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem 79, 233–269, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siggers T & Gordan R Protein–DNA binding: complexities and multi-protein codes. Nucleic Acids Res. 42, 2099–2111, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guéron M, Kochoyan M & Leroy J-L A single mode of DNA base-pair opening drives imino proton exchange. Nature 328, 89, (1987). [DOI] [PubMed] [Google Scholar]
- 4.Nikolova EN et al. Transient Hoogsteen base pairs in canonical duplex DNA. Nature 470, 498, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fischer M, Coleman RG, Fraser JS & Shoichet BK Incorporation of protein flexibility and conformational energy penalties in docking screens to improve ligand discovery. Nat. Chem 6, 575, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fraser JS et al. Hidden alternative structures of proline isomerase essential for catalysis. Nature 462, 669, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lorch Y, Davis B & Kornberg RD Chromatin remodeling by DNA bending, not twisting. Proc. Natl Acad. Sci. USA 102, 1329–1332, (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parvin JD, McCormick RJ, Sharp PA & Fisher DE Pre-bending of a promoter sequence enhances affinity for the TATA-binding factor. Nature 373, 724, (1995). [DOI] [PubMed] [Google Scholar]
- 9.Denny SK et al. High-throughput investigation of diverse junction elements in RNA tertiary folding. Cell 174, 377–390. e320, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reijns MA et al. Lagging-strand replication shapes the mutational landscape of the genome. Nature 518, 502–506, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sabarinathan R, Mularoni L, Deu-Pons J, Gonzalez-Perez A & Lopez-Bigas N Nucleotide excision repair is impaired by binding of transcription factors to DNA. Nature 532, 264–267, (2016). [DOI] [PubMed] [Google Scholar]
- 12.Rohs R et al. The role of DNA shape in protein–DNA recognition. Nature 461, 1248–1253, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeiske T et al. Intrinsic DNA Shape Accounts for Affinity Differences between Hox-Cofactor Binding Sites. Cell Rep. 24, 2221–2230, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Azad RN et al. Experimental maps of DNA structure at nucleotide resolution distinguish intrinsic from protein-induced DNA deformations. Nucleic Acids Res. 46, 2636–2647, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olson WK, Gorin AA, Lu X-J, Hock LM & Zhurkin VB DNA sequence-dependent deformability deduced from protein–DNA crystal complexes. Proc. Natl Acad. Sci. USA 95, 11163–11168, (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Battistini F et al. How B-DNA Dynamics Decipher Sequence-Selective Protein Recognition. J. Mol. Biol 431, 3845–3859, (2019). [DOI] [PubMed] [Google Scholar]
- 17.Kunkel TA & Erie DA Eukaryotic mismatch repair in relation to DNA replication. Annu. Rev. Genet 49, 291–313, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lindahl T Instability and decay of the primary structure of DNA. Nature 362, 709–715, (1993). [DOI] [PubMed] [Google Scholar]
- 19.Pich O et al. Somatic and germline mutation periodicity follow the orientation of the DNA minor groove around nucleosomes. Cell 175, 1074–1087. e1018, (2018). [DOI] [PubMed] [Google Scholar]
- 20.Berger MF et al. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat. Biotechnol 24, 1429–1435, (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen N et al. Divergence in DNA Specificity among Paralogous Transcription Factors Contributes to Their Differential In Vivo Binding. Cell Syst. 6, 470–483. e478, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Veprintsev DB & Fersht AR Algorithm for prediction of tumour suppressor p53 affinity for binding sites in DNA. Nucleic Acids Res. 36, 1589–1598, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jolma A et al. Multiplexed massively parallel SELEX for characterization of human transcription factor binding specificities. Genome Res, gr. 100552.100109, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Warren CL et al. Defining the sequence-recognition profile of DNA-binding molecules. Proc. Natl Acad. Sci. USA 103, 867–872, (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benos PV, Bulyk ML & Stormo GD Additivity in protein–DNA interactions: how good an approximation is it? Nucleic Acids Res. 30, 4442–4451, (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chattopadhyay A, Zandarashvili L, Luu RH & Iwahara J Thermodynamic Additivity for Impacts of Base-Pair Substitutions on Association of the Egr-1 Zinc-Finger Protein with DNA. Biochemistry 55, 6467–6474, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kitayner M et al. Diversity in DNA recognition by p53 revealed by crystal structures with Hoogsteen base pairs. Nat. Struct. Mol. Biol 17, 423, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Golovenko D et al. New Insights into the Role of DNA Shape on Its Recognition by p53 Proteins. Structure, (2018). [DOI] [PubMed] [Google Scholar]
- 29.Alvey HS, Gottardo FL, Nikolova EN & Al-Hashimi HM Widespread transient Hoogsteen base pairs in canonical duplex DNA with variable energetics. Nat. Commun 5, 4786, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi H et al. Atomic structures of excited state A-T Hoogsteen base pairs in duplex DNA by combining NMR relaxation dispersion, mutagenesis, and chemical shift calculations. J. Biomol. NMR 70, 229–244, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim JL, Nikolov DB & Burley SK Co-crystal structure of TBP recognizing the minor groove of a TATA element. Nature 365, 520, (1993). [DOI] [PubMed] [Google Scholar]
- 32.Mondal M, Mukherjee S & Bhattacharyya D Contribution of phenylalanine side chain intercalation to the TATA-box binding protein–DNA interaction: molecular dynamics and dispersion-corrected density functional theory studies. J. Mol. Model 20, 2499, (2014). [DOI] [PubMed] [Google Scholar]
- 33.Peyret N, Seneviratne PA, Allawi HT & SantaLucia J Nearest-neighbor Thermodynamics and NMR of DNA Sequences With Internal A·A, C·C, G·G, and T·T Mismatches. Biochemistry 38, 3468–3477, (1999). [DOI] [PubMed] [Google Scholar]
METHODS REFERENCES
- 34.Berman HM et al. The protein data bank. Nucleic Acids Res. 28, 235–242, (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou H et al. New insights into Hoogsteen base pairs in DNA duplexes from a structure-based survey. Nucleic Acids Res. 43, 3420–3433, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu X-J, Bussemaker HJ & Olson WK DSSR: an integrated software tool for dissecting the spatial structure of RNA. Nucleic Acids Res. 43, e142–e142, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sathyamoorthy B et al. Insights into Watson–Crick/Hoogsteen breathing dynamics and damage repair from the solution structure and dynamic ensemble of DNA duplexes containing m1A. Nucleic Acids Res. 45, 5586–5601, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.El Hassan M & Calladine C Two distinct modes of protein-induced bending in DNA. J. Mol. Biol 282, 331–343, (1998). [DOI] [PubMed] [Google Scholar]
- 39.Bailor MH, Mustoe AM, Brooks CL 3rd & Al-Hashimi HM 3D maps of RNA interhelical junctions. Nat. Protoc. 6, 1536–1545, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bailor MH, Sun X & Al-Hashimi HM Topology links RNA secondary structure with global conformation, dynamics, and adaptation. Science 327, 202–206, (2010). [DOI] [PubMed] [Google Scholar]
- 41.Le Novère N MELTING, computing the melting temperature of nucleic acid duplex. Bioinformatics 17, 1226–1227, (2001). [DOI] [PubMed] [Google Scholar]
- 42.Cheatham TE, Cieplak P & Kollman PA A modified version of the Cornell et al. force field with improved sugar pucker phases and helical repeat. J. Biomol. Struct. Dyn 16, 845–862, (1999). [DOI] [PubMed] [Google Scholar]
- 43.Perez A, Luque FJ & Orozco M Dynamics of B-DNA on the microsecond time scale. J. Am. Chem. Soc 129, 14739–14745, (2007). [DOI] [PubMed] [Google Scholar]
- 44.Maier JA et al. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput 11, 3696–3713, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salomon-Ferrer R, Gotz AW, Poole D, Le Grand S & Walker RC Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput 9, 3878–3888, (2013). [DOI] [PubMed] [Google Scholar]
- 46.Rossetti G et al. The structural impact of DNA mismatches. Nucleic Acids Res. 43, 4309–4321, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arnold FH, Wolk S, Cruz P & Tinoco I Jr. Structure, dynamics, and thermodynamics of mismatched DNA oligonucleotide duplexes d(CCCAGGG)2 and d(CCCTGGG)2. Biochemistry 26, 4068–4075, (1987). [DOI] [PubMed] [Google Scholar]
- 48.Kouchakdjian M, Li BF, Swann PF & Patel DJ Pyrimidine.pyrimidine base-pair mismatches in DNA. A nuclear magnetic resonance study of T.T pairing at neutral pH and C.C pairing at acidic pH in dodecanucleotide duplexes. J. Mol. Biol 202, 139–155, (1988). [DOI] [PubMed] [Google Scholar]
- 49.Boulard Y et al. The pH dependent configurations of the C.A mispair in DNA. Nucleic Acids Res. 20, 1933–1941, (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peng Y & Alexov E Computational investigation of proton transfer, pKa shifts and pH-optimum of protein-DNA and protein-RNA complexes. Proteins 85, 282–295, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen W, Morrow BH, Shi C & Shen JK Recent development and application of constant pH molecular dynamics. Mol. Simul 40, 830–838, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rangadurai A et al. Why are Hoogsteen base pairs energetically disfavored in A-RNA compared to B-DNA? Nucleic Acids Res. 46, 11099–11114, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patel DJ, Kozlowski SA, Ikuta S & Itakura K Deoxyguanosine-deoxyadenosine pairing in the d(C-G-A-G-A-A-T-T-C-G-C-G) duplex: conformation and dynamics at and adjacent to the dG X dA mismatch site. Biochemistry 23, 3207–3217, (1984). [DOI] [PubMed] [Google Scholar]
- 54.Webster GD et al. Crystal structure and sequence-dependent conformation of the A.G mispaired oligonucleotide d(CGCAAGCTGGCG). Proc. Natl Acad. Sci. USA 87, 6693–6697, (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Allawi HT & SantaLucia J Jr. NMR solution structure of a DNA dodecamer containing single G.T mismatches. Nucleic Acids Res. 26, 4925–4934, (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boulard Y, Cognet JA & Fazakerley GV Solution structure as a function of pH of two central mismatches, C. T and C. C, in the 29 to 39 K-ras gene sequence, by nuclear magnetic resonance and molecular dynamics. J. Mol. Biol 268, 331–347, (1997). [DOI] [PubMed] [Google Scholar]
- 57.Gordân R et al. Genomic regions flanking E-box binding sites influence DNA binding specificity of bHLH transcription factors through DNA shape. Cell Rep. 3, 1093–1104, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Frank F, Okafor CD & Ortlund EA The first crystal structure of a DNA-free nuclear receptor DNA binding domain sheds light on DNA-driven allostery in the glucocorticoid receptor. Sci. Rep 8, 13497, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Takayama Y, Sahu D & Iwahara J NMR studies of translocation of the Zif268 protein between its target DNA Sites. Biochemistry 49, 7998–8005, (2010). [DOI] [PubMed] [Google Scholar]
- 60.Belo Y et al. Unexpected implications of STAT3 acetylation revealed by genetic encoding of acetyl-lysine. Biochim. Biophys. Acta, Gen. Subj 1863, 1343–1350, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stelling AL et al. Infrared Spectroscopic Observation of a G-C(+) Hoogsteen Base Pair in the DNA:TATA-Box Binding Protein Complex Under Solution Conditions. Angew. Chem., Int. Ed. Engl 58, 12010–12013, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stephens DC & Poon GM Differential sensitivity to methylated DNA by ETS-family transcription factors is intrinsically encoded in their DNA-binding domains. Nucleic Acids Res. 44, 8671–8681, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang L et al. SelexGLM differentiates androgen and glucocorticoid receptor DNA-binding preference over an extended binding site. Genome Res. 28, 111–121, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vyas P et al. Diverse p53/DNA binding modes expand the repertoire of p53 response elements. Proc. Natl Acad. Sci. USA 114, 10624–10629, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weinberg RL, Veprintsev DB & Fersht AR Cooperative binding of tetrameric p53 to DNA. J. Mol. Biol 341, 1145–1159, (2004). [DOI] [PubMed] [Google Scholar]
- 66.Sandelin A, Alkema W, Engström P, Wasserman WW & Lenhard B JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 32, D91–D94, (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Siggers T et al. Principles of dimer-specific gene regulation revealed by a comprehensive characterization of NF-κB family DNA binding. Nat. Immunol 13, 95–102, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Luisi BF et al. Crystallographic analysis of the interaction of the glucocorticoid receptor with DNA. Nature 352, 497–505, (1991). [DOI] [PubMed] [Google Scholar]
- 69.Beno I, Rosenthal K, Levitine M, Shaulov L & Haran TE Sequence-dependent cooperative binding of p53 to DNA targets and its relationship to the structural properties of the DNA targets. Nucleic Acids Res. 39, 1919–1932, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stephens DC et al. Pharmacologic efficacy of PU.1 inhibition by heterocyclic dications: a mechanistic analysis. Nucleic Acids Res. 44, 4005–4013, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Siggers T, Duyzend MH, Reddy J, Khan S & Bulyk ML Non-DNA-binding cofactors enhance DNA-binding specificity of a transcriptional regulatory complex. Mol. Syst. Biol 7, 555, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maerkl SJ & Quake SR A systems approach to measuring the binding energy landscapes of transcription factors. Science 315, 233–237, (2007). [DOI] [PubMed] [Google Scholar]
- 73.Geertz M, Shore D & Maerkl SJ Massively parallel measurements of molecular interaction kinetics on a microfluidic platform. Proc. Natl Acad. Sci. USA 109, 16540–16545, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Drachkova I et al. Effect of TATA Box polymorphisms in human β-globin gene promoter associated with β-thalassemia on interaction with TATA-binding protein. Russ. J. Genet. Appl. Res 1, 183–188, (2011). [Google Scholar]
- 75.Drachkova I et al. The mechanism by which TATA-box polymorphisms associated with human hereditary diseases influence interactions with the TATA-binding protein. Hum. Mutat 35, 601–608, (2014). [DOI] [PubMed] [Google Scholar]
- 76.Leslie AG The integration of macromolecular diffraction data. Acta Crystallogr., Sect. D: Biol. Crystallogr 62, 48–57, (2006). [DOI] [PubMed] [Google Scholar]
- 77.Potterton E, Briggs P, Turkenburg M & Dodson E A graphical user interface to the CCP4 program suite. Acta Crystallogr., Sect. D: Biol. Crystallogr 59, 1131–1137, (2003). [DOI] [PubMed] [Google Scholar]
- 78.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr 66, 213–221, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jones TA, Zou JY, Cowan SW & Kjeldgaard M Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr., Sect. A: Found. Crystallogr 47 (Pt 2), 110–119, (1991). [DOI] [PubMed] [Google Scholar]
- 80.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr 66, 12–21, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yang S, Salmon L & Al-Hashimi HM Measuring similarity between dynamic ensembles of biomolecules. Nat. Methods 11, 552–554, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hombauer H, Srivatsan A, Putnam CD & Kolodner RD Mismatch repair, but not heteroduplex rejection, is temporally coupled to DNA replication. Science 334, 1713–1716, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Krokan HE, Drabløs F & Slupphaug G Uracil in DNA-occurrence, consequences and repair. Oncogene 21, 8935, (2002). [DOI] [PubMed] [Google Scholar]
- 84.Shen JC, Rideout WM 3rd & Jones PA The rate of hydrolytic deamination of 5-methylcytosine in double-stranded DNA. Nucleic Acids Res. 22, 972–976, (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.