

Summary
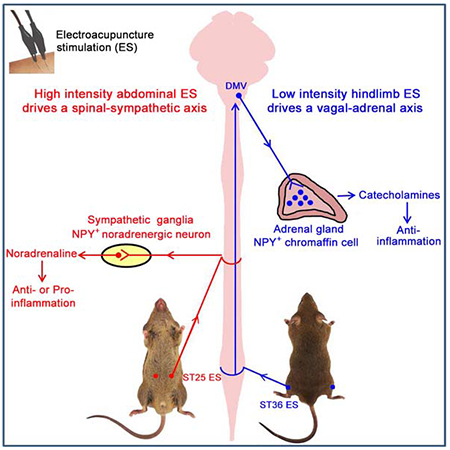
The neuroanatomical basis behind acupuncture practice is still poorly understood. Here we used intersectional genetic strategy to ablate NPY+ noradrenergic neurons and/or adrenal chromaffin cells. Using endotoxin-induced systemic inflammation as a model, we found that electroacupuncture stimulation (ES) drives sympathetic pathways in somatotopy- and intensity-dependent manners. Low intensity ES at hindlimb regions drives the vagal-adrenal axis, producing anti-inflammatory effects that depend on NPY+ adrenal chromaffin cells. High intensity ES at the abdomen activates NPY+ splenic noradrenergic neurons via the spinal-sympathetic axis; these neurons engage incoherent feedforward regulatory loops via activation of distinct adrenergic receptors (ARs), and their ES-evoked activation produces either anti- or pro-inflammatory effects due to disease state-dependent changes in AR profiles. The revelation of somatotopic organization and intensity dependency in driving distinct autonomic pathways could form a road map for optimizing stimulation parameters to improve both efficacy and safety in using acupuncture as a therapeutic modality.
Graphical Abstract
eToc Blurb:
Liu et al. reveals a neuroanatomical basis for acupuncture practice, showing that electroacupuncture stimulation can drive distinct autonomic pathways and modulate systemic inflammation in somatotopy-, stimulation intensity- and disease state-dependent manners.
INTRODUCTION
The goal of this study is to investigate organizational rules on how stimulation at specific body regions drives distinct autonomic nervous pathways, with a particular focus on prevention and treatment of systemic inflammation. Inflammatory responses need to be fine-tuned when animals and humans fight with pathogens. For patients suffering sepsis, the high fatality rate is often caused by excessive release of pro-inflammatory cytokines, referred to as the cytokine storm (Iskander et al., 2013; van der Poll et al., 2017). During past two decades, nerve stimulation has been emerging as a potential therapeutic regimen to treat systemic inflammation (Pavlov and Tracey, 2017; Ulloa et al., 2017). It began with the discovery that electric stimulation of cervical vagal efferents could suppress systemic inflammation (Borovikova et al., 2000). This pathway involves with activation of splenic sympathetic neurons (Pavlov and Tracey, 2017; Ulloa et al., 2017), although the routes leading to this activation are still debated (Komegae et al., 2018; Martelli et al., 2016). As vagal nerve stimulation involves invasive surgical procedure, alternative methods, such as the use of electroacupuncture to drive somatosensory-autonomic pathways, are being explored.
Acupuncture stimulation is an ancient practice to treat human diseases. One core idea is that stimulation at specific somatic tissues (acupoints) can long distantly modulate internal organ physiology. The primitive meridian channel theory is formulated to make such somato-internal organ connections. While modern anatomical studies fail to support the physical presence of such “channels” (Longhurst, 2010), somatosensory autonomic pathways could exactly serve this role. In 1950s, electric stimulation of somatic nerves was shown to be capable of activating sympathetic pathways (Sell et al., 1958). Since 1970s, pinch or electroacupuncture stimulation at abdominal and hindlimb regions was reported to differentially drive sympathetic versus vagal efferent pathways associated with gastric motility control (Li et al., 2007; Sato, 1997). Inspired by the discovery of cholinergic anti-inflammatory pathways, several studies show that limb area acupoint stimulation can suppress systemic inflammation, mainly or partly via activation of vaga efferents (Gu et al., 2011; Huang et al., 2007; Lim et al., 2016; Song et al., 2012; Torres-Rosas et al., 2014; Zhao et al., 2012), including the evoking of the vagal-adrenal anti-inflammatory axis (Torres-Rosas et al., 2014).
Despite this progress, many challenges and knowledge gaps remain in studying somatosensory autonomic pathways. Firstly, sympathetic neurons are molecularly and functionally heterogeneous (Furlan et al., 2016; Janig and Habler, 2000). However, we still lack tools to manipulate sympathetic neuron subtypes. Secondly, somatosensory neurons themselves are heterogeneous. For example, sensory axons are of distinct diameters and myelination degrees, two features inversely correlated with activation thresholds in response to electric stimulation (John et al., 2018). It remains unclear if different electric intensities could drive distinct somato-autonomic pathways. Nor is known if there is somatotopic organization in driving autonomic pathways associated with inflammation control. Thirdly, noradrenaline released from sympathetic nerves can suppress or promote the release of pro-inflammatory cytokines, via activation of distinct adrenergic receptors (Huang et al., 2012; Spengler et al., 1994; Szelenyi et al., 2000; Vida et al., 2011). As such, it remains to be determined if ES-activated noradrenergic sympathetic neurons could produce anti- or pro-inflammatory effects if different disease states cause a change in AR profiles, addressing of which is warranted since it touches both efficacy and safety issues of acupuncture practice.
In this study, we developed an intersectional genetic strategy to manipulate sympathetic cells expressing neuropeptide Y (NPY) in sympathetic ganglia and/or adrenal glands. Using lipopolysaccharide (LPS)-induced endotoxemia as a model, we then demonstrated that ES can activate distinct sympathetic pathways and modulate systemic inflammation in somatotopy-, stimulation intensity-, and disease state-dependent manners.
RESULTS
Genetic marking of NPY-expressing sympathetic neurons and adrenal chromaffin cells
To selectively manipulate peripheral NPY-expressing sympathetic cells, we developed an intersectional genetic strategy (Figure S1A), analogous to the one we used to map spinal circuits (Bourane et al., 2015; Duan et al., 2014). We first labeled these neurons by the expression of the fluorescent tdTomato protein, involving three mouse lines (Figure S1A). One is the Rosa26LSL-FSF-tdTomato (Ai65) intersectional reporter line, in which activation of tdTomato expression requires removal of two stop cassettes by Cre and Flpo recombinases. The other two lines are NPY-Cre (Gerfen et al., 2013) and DBH-p2a-Flpo (Sun and Ray, 2016), in which Cre and Flpo are driven from the loci encoding NPY and DBH (dopamine beta-hydroxylase), respectively, with DBH being a marker for noradrenergic neurons. In resulted triple heterozygous mice, referred to as NPYDBH-tdTomato, tdTomato expression was restricted to NPYDBH cells defined by coexpression of Cre and Flpo (Figure S1A).
Two sets of sympathetic cells have been implicated in systemic inflammation control: splenic noradrenergic neurons located mainly in suprarenal ganglia and chromaffin cells in adrenal glands (Bratton et al., 2012; Torres-Rosas et al., 2014). In suprarenal ganglia, 94.9% of NPY mRNA+ neurons coexpressed tdTomato, and 98.1% of tdTomato+ cells coexpressed NPY (Figure 1A), indicating that NPYDBH faithfully marks NPY-expressing neurons. Double staining showed that tdTomato was detected in 83.3% of DBH mRNA+ noradrenergic neurons in suprarenal ganglia (Figure 1B), and 79.8% of TH+ (tyrosine hydroxylase) chromaffin cells in adrenal glands (Figure 1B). In paravertebral sympathetic chain ganglia, NPY expression is confined to two of five noradrenergic neuron subtypes (Furlan et al., 2016); consistently, tdTomato was expressed in ~50% of DBH mRNA+ neurons and none of cholinergic cells in these ganglia (Figure 1C). Importantly, tdTomato+ cell bodies were not detected in dorsal root ganglia (DRG), nodose ganglia, and the colon, indicating that NPYDBH does not mark somatosensory, visceral sensory or enteric neurons (Figure S1B). Furthermore, cell sorting analyses did not detect tdTomato signal in immune cells (Figure S1C). Within the central nervous system, NPYDBH additionally labels A1/C1 catecholaminergic neurons in the hindbrain (Figures S1D and S1E), and we had a strategy to overcome this limitation (see below, Figure 2).
Figure 1. Intersectional marking of NPYCre-expressing sympathetic cells.
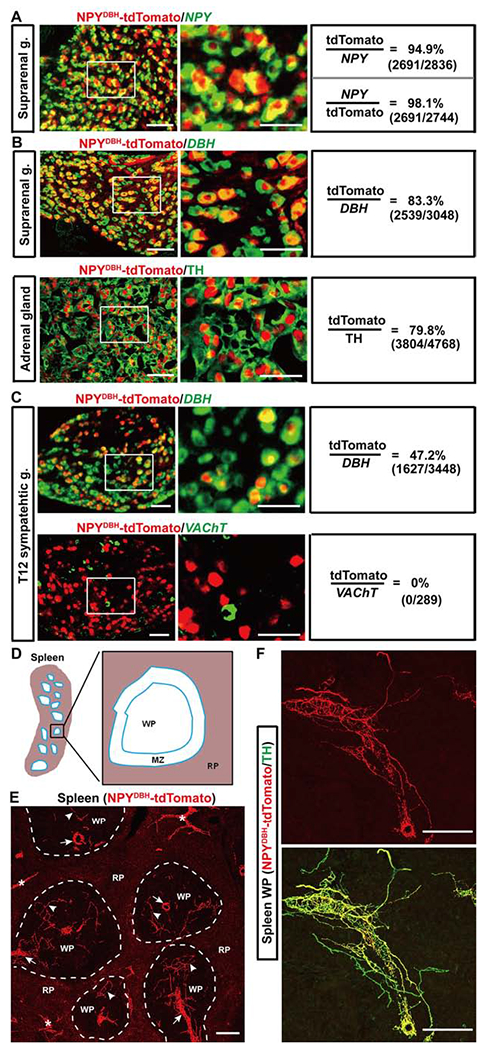
(A-C) Colocalization of tdTomato with NPY, DBH or VAChT mRNA or the TH protein in sections through sympathetic ganglia (“g.”) and adrenal glands of adult NPYDBH-tdTomato mice.
(D) Schematic illustration of splenic subcapsular regions. RP: red pulp, MZ: marginal zone, WP: white pulp.
(E) Splenic innervations by NPYDBH-tdTomato+ fibers. Arrows and arrowheads in WPs: fibers along the arterioles and the periarteriolar lymphatic sheath, respectively. “*”: fibers innervating the trabeculae.
(F) A representative image showing tdTomato overlapping with TH in a WP.
n = 5–6 mice for all groups. Scale bars, 100 μm.
Figure 2. Intersectional ablation of peripheral and adrenal NPYDBH cells.
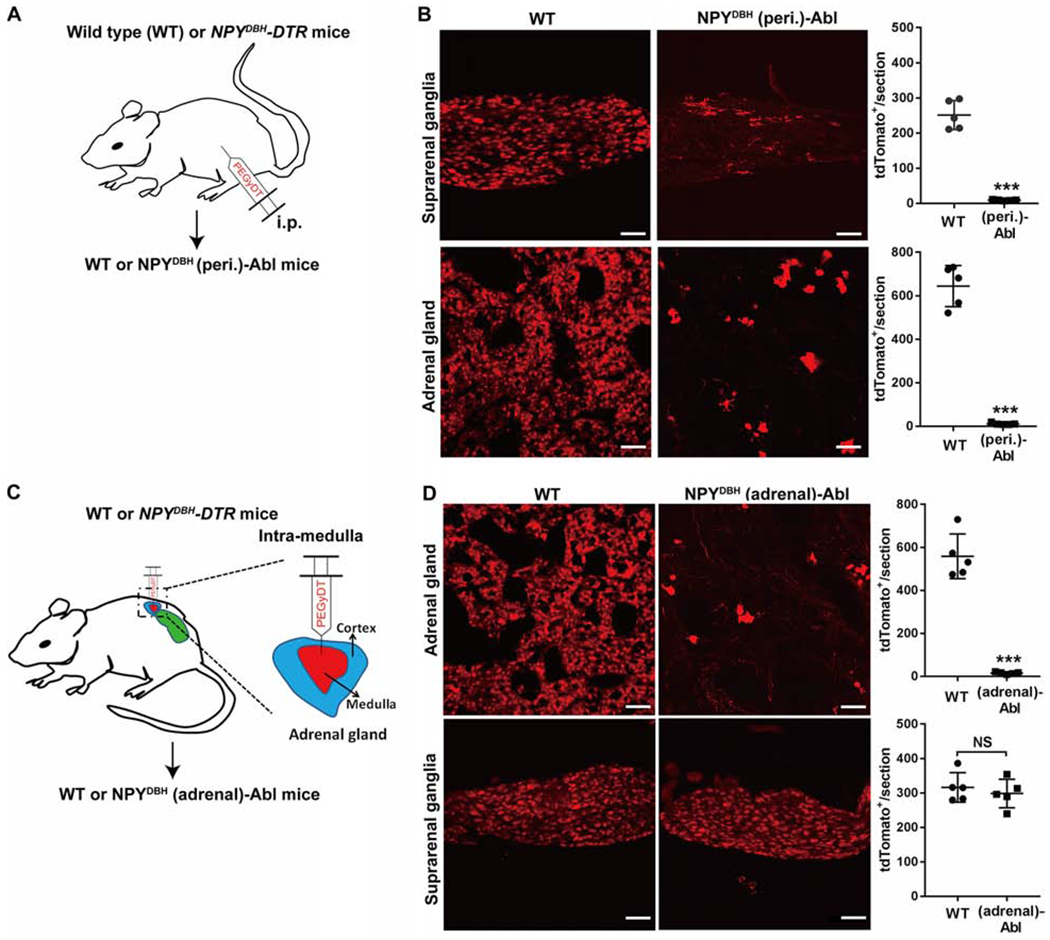
(A) Intraperitoneal injection (i.p.) of PEGyDT to create NPYDBH (peri.)-Abl [“(peri.)-Abl”] mice. “WT”: wild type littermates. All NPYCre cells were labeled by tdTomato from an unshown reporter allele. (B) Ablation of NPYCre-tdTomato+ cells in suprarenal ganglia (t8 = 13.1, ***P < 0.001) and adrenal glands (t8 = 14.96, ***P < 0.001).
(C) Intra-adrenal medulla injection of PEGyDT to create NPYDBH (adrenal)-Abl mice [“(adrenal)-Abl”].
(D) Ablation of NPYCre-tdTomato+ cells in adrenal glands, but not in suprarenal ganglia compared with WT mice (adrenal, t8 = 11.68, ***P < 0.001; suprarenal, t8 = 0.659, NS, no significant, P = 0.528).
n = 5 mice for all groups. Two-side student’s unpaired t-test in B and D. Data are shown as mean ± SEM. Scale bars, 100 μm.
As described below, we used the modulation of lipopolysaccharide (LPS)-induced systemic inflammation (Remick et al., 2000) to assess autonomic pathways. LPS activates the toll-like receptor 4 (TLR4) complex in immune cells (Salomao et al., 2008; Ulloa et al., 2017), and induces pro-inflammatory cytokine production partly from the spleen (Huston et al., 2006; Rosas-Ballina et al., 2008). We accordingly examined splenic innervations. Structurally, spleen is divided into white pulps, which are enriched with T cells and B cells, as well as marginal zones plus red pulps, where various macrophage subtypes are enriched (Figure 1D) (Borges da Silva et al., 2015; Bronte and Pittet, 2013). In NPYDBH-tdTomato reporter mice, tdTomato+ sympathetic fibers are enriched in white pulps and trabecula structures, but rarely in red pulps (Figure 1E). They are co-localized with a major subset of TH+ sympathetic fibers (Figure 1F), a finding confirmed by retrograde labeling (see below). Thus, NPYDBH-marked sympathetic cells account for a major subset of adrenal chromaffin cells and splenic noradrenergic neurons.
Ablation of NPYDBH-marked sympathetic neurons and/or adrenal chromaffin cells
To ablate peripheral NPYDBH sympathetic cells, we created NPYDBH-DTR mice, in which the expression of human diphtheria toxin receptor (DTR) was confined to NPYDBH cells defined by coexpression of NPY-Cre and DBH-p2A-Flpo (Figure S2A). Meanwhile, all NPY-Cre-expressing cells were labeled with tdTomato to monitor selective ablation of sympathetic cells (Figures 2A and S2A). To avoid ablation of NPYDBH-marked hindbrain neurons, we injected a modified form of diphtheria toxin (PEGyDT) that does not cross the blood-brain barrier (Pereira et al., 2017). A month after PEGyDT injections, 96% of tdTomato+ cells in suprarenal ganglia and adrenal glands were ablated (Figure 2B). Consistently, most splenic TH+ fibers were eliminated in NPYDBH (peri.)-ablated mice (Figure S2B). Importantly, no ablation was observed in the spinal cord or the brain, including TH+ neurons in hindbrain A1/C1 areas (Figure S2C). We referred these mice with selective ablation of NPYDBH-marked peripheral noradrenergic neurons plus adrenal chromaffin cells to as NPYDBH (peri.)-Abl.
Meanwhile, we created mice with selective ablation of NPYDBH adrenal chromaffin cells via bilateral intra-adrenal injections of low dose PEGyDT (Figure 2C). Four weeks later, 97% of tdTomato+ adrenal chromaffin cells were ablated (Figure 2D), without ablating adjacent suprarenal ganglia (Figure 2D). These mice were referred to as NPYDBH (adrenal)-Abl mice. These two sets of ablation mice, plus control littermates that received the same PEGyDT injections, were subsequently used to study the roles of various NPYDBH cells in controlling systemic inflammation.
NPYDBH noradrenergic neurons are part of an endogenous anti-inflammatory system
We adopted LPS-induced endotoxemia as a model to study systemic inflammation (Remick et al., 2000). For wild type C57BL6/J mice, LPS injection caused a marked increase in serum levels of pro-inflammatory cytokines, including tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6, and this cytokine storm led to a lethality rate at ~80% (Figure S3).
Before conducting electroacupuncture studies, we first examined if ablation of NPYDBH sympathetic cells impacted on baseline inflammation and/or LPS-induced systemic inflammation. Without LPS injection, serum or splenic TNF-α levels were barely detectable in both NPYDBH (peri.)-Abl and control mice (Figures 3A and 3B), and there were no changes of splenic immune cell profiles (Figure S2D). Thus, within the one-month period following PEGyDT injection, no spontaneous systemic inflammation developed. However, following LPS injection, NPYDBH (peri.)-Abl mice did show an increase in TNF-α and IL-6, both in serum and in splenic tissues compared with wild type littermates (Figures 3A, 3B, S4A and S4B). To investigate how NPYDBH cell ablation led to exaggerated systemic inflammation, we performed a series of immunostaining in splenic tissues. Without LPS treatment, TNF-α signals were rarely detected, but greatly elevated 1 hour after LPS injection (Figure 3C), as reported previously (Rosas-Ballina et al., 2008). Double immunostaining with macrophage subtype markers (Borges da Silva et al., 2015; Bronte and Pittet, 2013) showed that in LPS-treated wild type littermates, TNF-α was detected in 45.6 ± 2.6% of F4/80+ macrophages in red pulps, 12.9 ± 0.9% of CD169+ metallophilic macrophages in marginal zones (Figure 3D), and 3.5 ± 0.8% of CD68+;F4/80− macrophages in white pulps (Figure 3D). In LPS-treated NPYDBH (peri.)-Abl mice, the percentages become 50.3 ± 1.6%, 36.2 ± 1.5% and 38.0 ± 6.2%, respectively, with significant increase found in both marginal zones and white pulps compared with control littermates (Figure 3D). TNF-α was also detected in some non-macrophage cells, but their identities remain unknown as no TNF-α expression was detected in T cells, B cells or dendritic cells (Figures S4C and S4D).
Figure 3. Ablation of NPYDBH sympathetic cells caused an increase in LPS-induced TNF-α production.
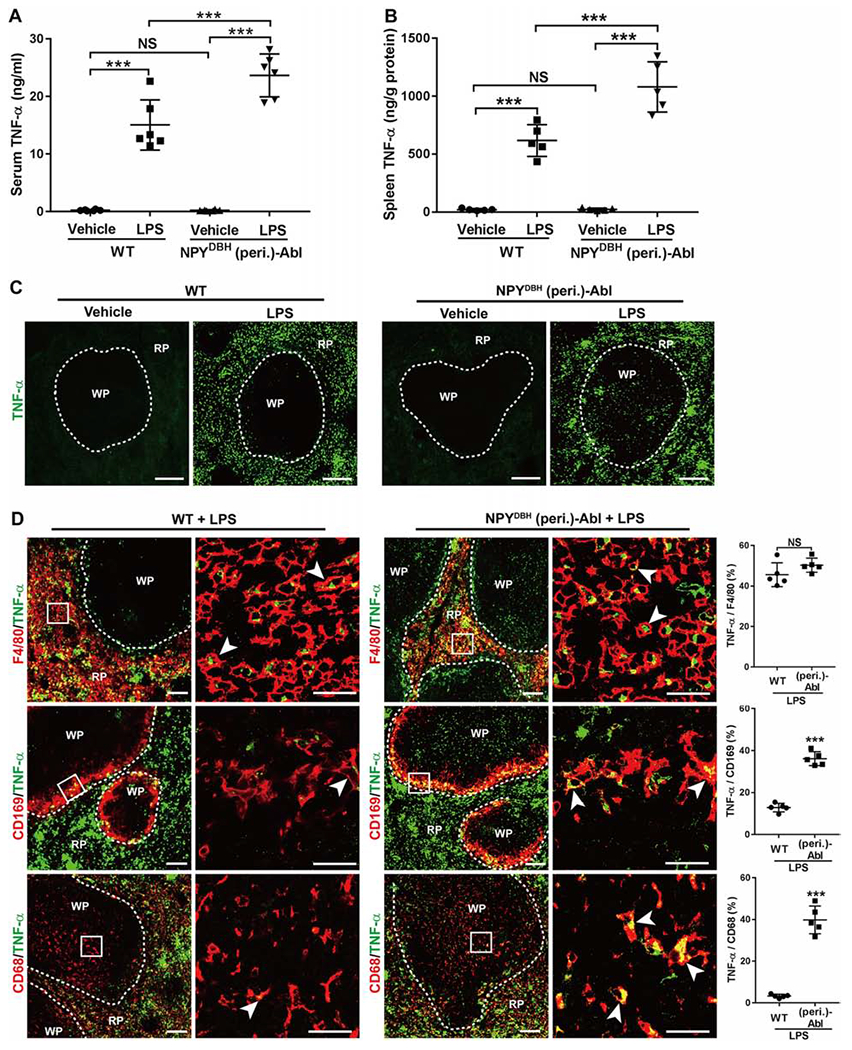
(A-B) LPS induced a larger increase of TNF-α levels in NPYDBH (peri.)-Abl mice compared with WT mice in serum and in spleen. Two-way ANOVA plus post-hoc Tukey test. Serum: F1, 20 = 13.457, P = 0.002; spleen: F1, 16 = 13.174, P < 0.001. ***P < 0.001; NS, not significant, P = 0.913 in A and 0.892 in B.
(C) TNF-α immunostaining images through splenic red pulps (“RP”) and white pulps (“WP”).
(D) TNF-α expression (by immunostaining) in splenic macrophages with indicated markers in LPS-treated wild type (WT) versus NPYDBH (peri.)-Abl mice (two-side student’s unpaired t test; t8 = 1.528 for F4/80, NS, not significant, P = 0.165; t8 = 13.36 for CD169 and 12.22 for CD68, ***P < 0.001). Arrowheads indicate co-localized cells.
N = 5 mice for all groups. Data are shown as mean ± SEM. Scale bars, 100 μm.
In contrast with expanded TNF-α and IL-6 expression in NPYDBH (peri.)-Abl mice, no such expansion was observed in NPYDBH (adrenal)-Abl mice, in which NPYDBH adrenal chromaffin cells were selectively ablated (Figure S4E–G). Thus, in response to LPS exposure, tonic or LPS-induced activation of NPYDBH noradrenergic neurons located in sympathetic ganglia, which were preserved in NPYDBH (adrenal)-Abl mice but eliminated in NPYDBH (peri.)-Abl mice, produced anti-inflammatory effects and sufficiently suppressed TNF-α production in macrophages located in marginal zones and white pulps, under the LPS dose used in this study.
High intensity electroacupuncture stimulation suppressed LPS-induced systemic inflammation via NPYDBH noradrenergic neurons
We next assessed how LPS-induced inflammation was modulated by electroacupuncture stimulation (ES) at the abdominal ST25 acupoint (Figure 4A); this acupoint was chosen based on reported splenic nerve activation evoked by painful pinch stimulation, via both segmental and supraspinal pathways (Sato, 1997). ES was performed for 15 mins, followed by LPS injection. This stimulation mode was referred to as preST25 ES. Electrode insertion at ST25 without current stimulation (0 mA) was used as sham control. We first tested the responses to different ES intensities, and found that serum TNF-α was reduced by 38.5% with 1.0 mA ES and by 71.5% with 3.0 mA ES, whereas 0.5 mA ES did not produce a significant reduction, in comparison with sham ES (Figure 4B). 3.0 mA ES additionally caused a reduction of IL-6 and IL-1β levels (Figure S5A), and was accordingly chosen for subsequent studies.
Figure 4. Requirement of NPYDBH cells for preST25 ES-evoked anti-inflammatory effects.
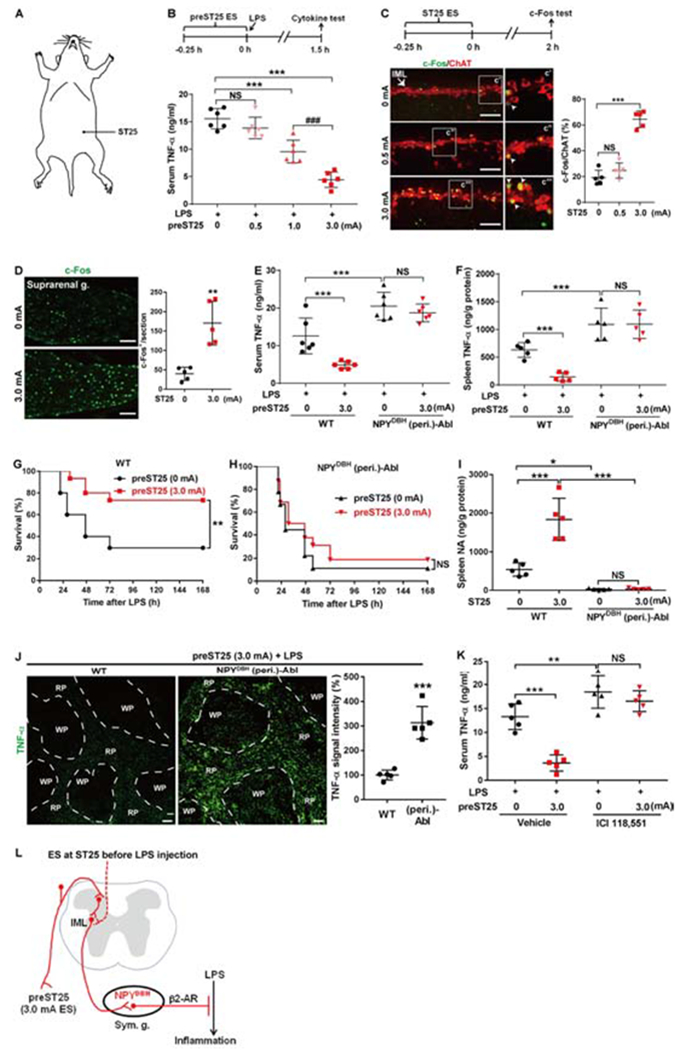
(A) Schematics showing the abdominal ST25 acupoint.
(B) Modulation of LPS-induced TNF-α by ST25 ES (F3, 20 = 43.772, P < 0.001; ***P < 0.001; ###P < 0.001; NS, P = 0.406).
(C) c-Fos induction by ST25 ES in ChAT+ neurons in the spinal intermediolateral nuclei (“IML”). F2, 14 = 86.02, P < 0.001; ***P < 0.001; NS, P = 0.519.
(D) c-Fos induction in suprarenal ganglia (“g.”) by 3.0 mA compared with sham 0 mA ES (two-side student’s unpaired t-test, t8 = 4.97, **P = 0.001).
(E-F) Loss of 3.0 mA preST25 ES-evoked reduction of TNF-α in NPYDBH(peri.)-Abl mice compared with wild type (WT) mice, both in serum (F1, 20 = 19.113, P < 0.001;***P < 0.001; NS, P = 0.715) and in spleen (F1, 16 = 13.147, P = 0.012;***P < 0.001; NS, P = 0.783).
(G, H) 3.0 mA preST25 ES (compared with 0 mA ES) reduced LPS-induced fatality in WT mice but not in (peri.)-Abl mice (n = 24–25 mice per group; log-rank test, **P = 0.007; NS, P = 0.418).
(I) Loss of 3.0 mA preST25 ES-induced splenic noradrenaline release in (peri.)-Abl mice compared with WT mice (F1, 16 = 79.677, P < 0.001; *P < 0.05, ***P < 0.001; NS, P = 0.915).
(J) Splenic TNF-α immunostaining in LPS-treated mice receiving 3.0 mA preST25 ES. Increased signals in NPYDBH (peri.)-Abl mice compared with WT mice (two-side student’s unpaired t-test, t8 = 6.856, ***P < 0.001).
(K) A blockage of 3.0 mA preST25 ES-evoked reduction in serum TNF-α in LPS-treated C57BL/6 mice by ICI 118,551 (a β2-AR antagonist) (F1, 16 = 61.757, P < 0.001; ***P < 0.001; **P = 0.006; NS, P = 0.254).
(L) preST25 ES evokes NPYDBH cell- and β2 AR-dependent anti-inflammatory pathways, via segmental and/or spinal-supra-spinal pathways (“dashed”, to be determined). “Sym. g.”: sympathetic ganglia.
n = 5–6 mice for all mouse groups (except G and H). One-way (B, C) or two-way (E, F, I, K) ANOVA plus post hoc Tukey test. NS, not significant. Data are shown as mean ± SEM. Scale bars, 100 μm.
We next explored how ES modulated systemic information, by focusing on the driving of somatosensory autonomic pathways. 3.0 mA ES at ST25 was able to activate primary sensory neurons in thoracic DRG (Figure S5B) and relay neurons in the spinal cord (Figure S5C), indicating the activation of the somatosensory system. Before assessing sympathetic pathways (see below), we first investigated vagal efferent pathways. Anatomically, there are spinal ascending projections to the nucleus of solitary tract (NTS) in the medulla oblongata (Lima, 2008), and NTS neurons in turn send synaptic outputs to the dorsal motor nuclei of the vagus (DMV), where vagal efferent neurons, marked by the expression of choline acetyltransferase (ChAT), are located (Travagli and Anselmi, 2016). 3.0 mA ES at the abdominal ST25 acupoint, however, failed to induce c-Fos, a marker for neuronal activation (Hunt et al., 1987), in DMV (Figure S5D). As a positive control, ES at the hindlimb acupoint was able to induce c-Fos in these neurons (see below, Figure 6B). Accordingly, neither noradrenaline release (Figure S5E) nor reduction of TNF-α (Figure S5F) or IL-6 (Figure S5G) evoked by 3.0 mA ES at ST25 was affected in mice with subdiaphragmatic vagotomy compared with sham surgery.
Figure 6. 0.5 mA ES at ST36 attenuated ongoing systemic inflammation.
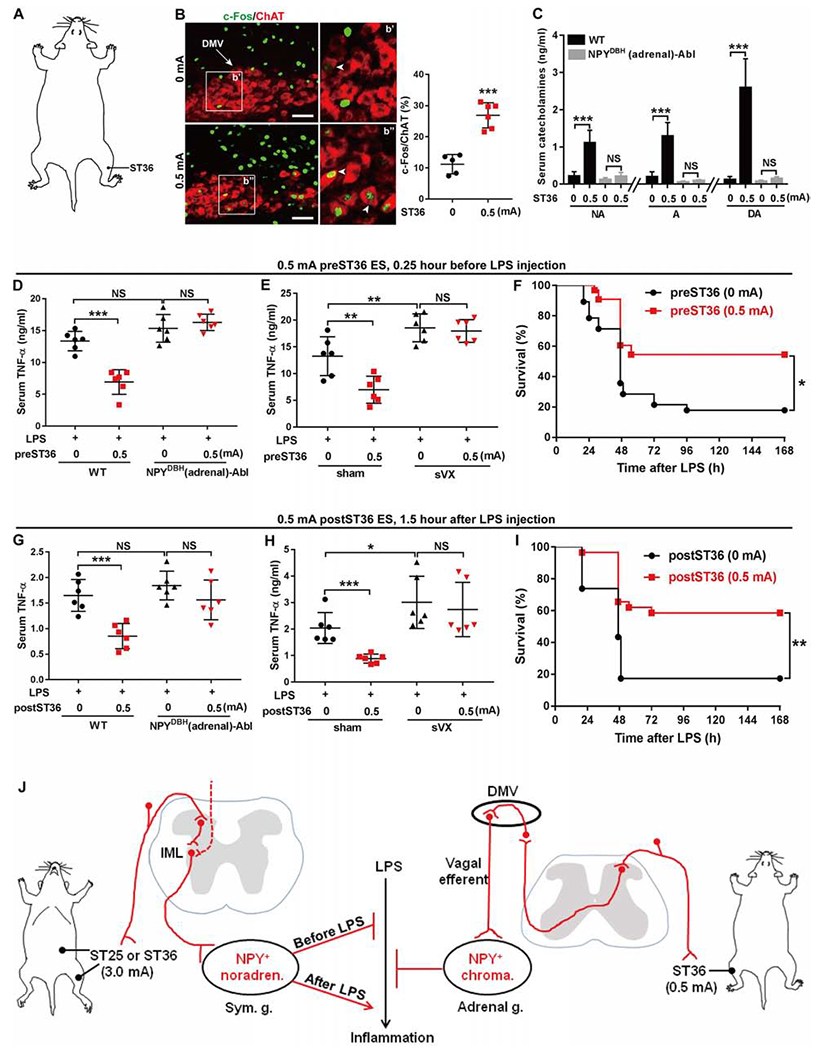
(A) Schematic showing the hindlimb ST36 acupoint.
(B) ES induced c-Fos in ChAT+ neurons in the dorsal motor nuclei of the vagus (“DMV”). Arrowheads: co-expression. Two-side student’s unpaired t-tests, t9 = 7.09, ***P < 0.001.
(C) 0.5 mA ST36 ES increased catecholamine release (“NA” for noradrenaline, “A” for adrenaline, “DA” for dopamine) in WT, but not in NPYDBH (adrenal)-Abl mice [F1, 20 = 40.560 (“NA”), 69.780 (“A”), 60.912 (“DA”), P < 0.001; ***P < 0.001; NS, P = 0.483 (“NA”), 0.734 (“A”), and 0.757 (“DA”)].
(D) 0.5 mA preST36 ES reduced LPS-induced TNF-α in WT, but not in (adrenal)-Abl mice (F1,20 = 20.457, P < 0.001; ***P < 0.001; NS, left, P = 0.064, right, P =0.893).
(E) 0.5 mA preST36 ES reduced LPS-induced TNF-α in mice with sham surgery, but not with subdiaphragmatic vagotomy (“sVX”) (F1, 20 = 72.188, P < 0.001;**P < 0.01; NS, P = 0.628).
(F) 0.5 mA preST36 ES promoted survival compared with 0 mA ES (0 mA, n = 28; 0.5 mA, n = 33, log-rank test, *P = 0.019).
(G) 0.5 mA postST36 ES reduced LPS-induced TNF-α in WT, but not in (adrenal)-Abl mice (F1, 20 = 23.155, P < 0.001; ***P < 0.001; NS, left, P = 0.147, right, P =0.550).
(H) Loss of 0.5 mA postST36 ES-evoked TNF-α reduction by subdiaphragmatic vagotomy (“sVX”) compared with sham surgery (F1, 20 = 81.237, P < 0.001; ***P < 0.001; *P = 0.025; NS, P = 0.315).
(I) 0.5 mA postST36 ES improved survival compared with 0 mA ES (0 mA, n = 23; 0.5 mA, n = 28, log-rank test, **P = 0.001).
(J) ES drives two autonomic pathways and modulates LPS-induced inflammation in somatotopy-, intensity-, and disease state-dependent manners. Sym. g.: sympathetic ganglia. IML: spinal intermediolateral nuclei. Adrenal g.: adrenal gland. Noradren.: Noradrenergic. Chroma.: Chromaffin.
n = 5–6 mice for all groups (except F and I). Two-way ANOVA plus post hoc Tukey’s test (C, D, E, G, and H). NS, not significant. Data are shown as mean ± SEM. Scale bars, 100 μm.
We next assessed spinal sympathetic pathways. Sympathetic preganglionic neurons (PGNs), predominantly located in the intermediolateral (IML) nuclei of the spinal cord, represent the final output neurons linking the central nervous system to peripheral sympathetic ganglia (Deuchars and Lall, 2015). 3.0 mA ES, but not 0.5 mA, drove c-Fos induction in ChAT+ PGNs at thoracic Th6-Th10 levels, compared with sham ES (Figure 4C). Consistently, 3.0 mA ES at ST25 drove marked c-Fos induction in suprarenal ganglia (Figure 4D), 94.3% (3414/3619) of which coexpressed tdTomato in NPYDBH-tdTomato mice (Figure S5H). Retrograde tracing, via intra-spleen Fluoro-gold injections, further showed that among NPYDBH-tdTomato+ cells, c-Fos was detected in 95.1% (2105/2213) of Fluoro-gold-labeled splenic sympathetic neurons (Figure S5E), much higher than 38.2% (1309/3424) detected in Fluoro-gold-negative cells (P < 0.001, Chi-square test). Thus, 3.0 mA ES at the abdominal ST25 acupoint drives somatosensory sympathetic pathways, leading to preferential activation of splenic sympathetic neurons.
We accordingly examined if NPYDBH cells mediated anti-inflammatory effects. 3.0 mA preST25 ES, compared with sham ES, led to marked reduction of TNF-α and IL-6 in wild type littermates, both in serum and in spleen; however, such reduction was abolished in NPYDBH (peri.)-Abl mice (Figures 4E, 4F, S5I and S5J). Consistently, while 3.0 mA preST25 ES led to a 43.3% increase in survival rates in LPS-treated control mice compared with sham ES, no such survival promotion was observed in NPYDBH (peri.)-Abl mice (Figures 4G and 4H). Note that without ES, NPYDBH (peri.)-Abl mice showed a higher fatality rate (87.5%) compared with 68% seen in wild type mice (n = 24-25, log-rank test, *P = 0.034), consistent with exaggerated systemic inflammation upon ablation of NPYDBH neurons (see above, Figure 3).
We also analyzed NPYDBH (adrenal)-Abl mice with selective ablation of adrenal chromaffin cells, and found that anti-inflammatory effects evoked by 3.0 mA preST25 ES were largely unaffected in these ablation mice compared with control littermates (Figures S5K and S5L), despite that this stimulation can also activate NPYDBH adrenal chromaffin cells via spinal pathways, rather than via vagal efferent pathways (data not shown). Thus, NPYDBH noradrenergic neurons located in sympathetic ganglia, spared in NPYDBH (adrenal)-Abl mice, must sufficiently mediate the bulk of anti-inflammatory effects evoked by electric stimulation at the abdomen, whereas NPYDBH adrenal chromaffin cells could play either minimal or redundant, non-essential roles.
We next investigated how NPYDBH neurons worked, by focusing on splenic inflammation. We first confirmed that 3.0 mA preST25 ES led to an increase in splenic noradrenaline release in wild type mice, and both baseline release and ES-evoked increase were eliminated in NPYDBH (peri.)-Abl mice (Figure 4I). Immunostaining on splenic sections showed that while TNF-α was induced in red pulps of LPS-treated wild type mice (see above, Figure 3C), this induction was suppressed by 3.0 mA preST25 ES (Figure 4J, left). Such suppression was, however, not observed in LPS-treated NPYDBH (peri.)-Abl mice, with TNF-α signals still detected in all splenic zones (Figure 4J, middle), leading to elevation of overall splenic TNF-α levels compared with wild type littermates (Figure 4J, right). In other words, while endogenously activated NPYDBH-marked noradrenergic neurons (without ES) are sufficient to suppress TNF-α in marginal zones and white pulps (see above, Figure 3C), the suppression was expanded to red pulps following increased noradrenaline release evoked by 3.0 mA preST25 ES.
Noradrenaline can suppress pro-inflammatory cytokine production in splenic macrophages via activation of β2 adrenergic receptors (ARs) (Agac et al., 2018; Spengler et al., 1994; Vida et al., 2011). We next asked if blockage of β2 ARs by the antagonist ICI 118,551(O’Donnell and Wanstall, 1980) could affect ES-evoked anti-inflammatory effects. We first showed that in cultured splenocytes, pre-exposure to noradrenaline (30 mins before LPS application) can suppress LPS-induced TNF-α production, and this effect can be blocked by ICI 118,551 (Figures S6A and S6B), consistent with reported blockage by other β2 AR antagonists (Vida et al., 2011). We then found that in vivo injection of ICI 118,551 recaptured the phenotypes seen in NPYDBH (peri.)-Abl mice, including i) a loss of effects by 3.0 mA preST25 ES in reducing overall TNF-α and IL-6 production (Figures 4K and S6C), ii) an expansion of TNF-α expression to white pulps in LPS-treated mice without ES (Figure S6D), and iii) failed suppression of TNF-α expression in red pulps in response to 3.0 preST25 ES (Figure S6D). These studies suggest that high intensity ES at the abdominal regions activates NPYDBH sympathetic neurons, whose increased noradrenaline release in turn suppresses LPS-induced systemic inflammation via activation of β2 ARs (Figure 4L). Consistently, acute chemical genetic activation of NPYDBH cells was sufficient to suppress LPS-induced systemic inflammation (Figures S7A and S7B), mimicking the effect evoked by 3.0 mA preST25 ES. Conversely, acute silencing of NPYDBH cells eliminated the anti-inflammatory effects evoked by 3.0 mA preST25 ES (Figures S7A and S7C), mimicking the phenotypes seen in NPYDBH (peri.)-Abl mice (Figures 4E and S4A). A caution is needed in interpreting these chemical genetic data since the whole population of NPYDBH cells, including peripheral and central parts, were affected by this approach (Figure S7).
ES switched to promote inflammation following α2 adrenergic receptor induction by LPS pre-exposure
All above studies reveal that abdominal acupoint stimulation, performed before LPS exposure, can prevent systemic inflammation. Nonetheless, a more urgent clinical challenge is to treat ongoing systemic inflammation. We accordingly assessed the impact of 3.0 mA ST25 ES that started 1.5 hours after LPS injection, when the release of TNF-α and IL-6 had largely reached the peak (Figure S3), and then measured cytokine levels 2.5-4.5 hours later. Note that at these late time points, TNF-α had already been reduced to low levels in wild type mice (Figure S3). We referred ES after LPS exposure to as postST25 ES to distinguish it from preST25 ES (ES before LPS exposure). Surprisingly, following 3.0 mA postST25 ES, the survival rate of LPS-treated wild type mice was reduced to 0%, compared with 21.7% with sham ES (Figure 5A). Consistently, this stimulation caused an increase in serum TNF-α and IL-6 levels (Figure S8A). Thus, 3.0 mA postST25 ES switches to promote inflammation and produce a detrimental effect. This detrimental effect still depended on NPYDBH sympathetic cells, since the increase of TNF-α and IL-6 evoked by 3.0 mA postST25 ES, observed in wild type littermates, did not occur in NPYDBH (peri.)-Abl mice (Figures 5B and S8B).
Figure 5. Activation of NPYDBH sympathetic cells switched to promote inflammation following LPS pre-exposure.
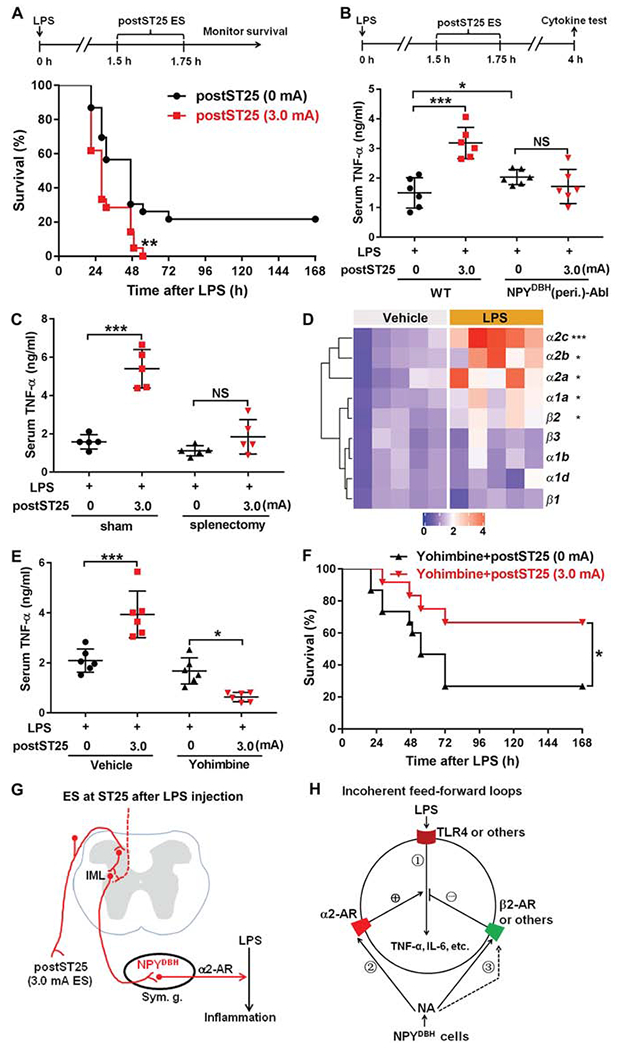
(A) 3.0 mA postST25 ES, performed 1.5 hours after LPS exposure, increased lethality in LPS-treated C57BL/6J mice (log-rank test; 0 mA, n = 23; 3.0 mA, n = 22; **P = 0.006).
(B) Loss of 3.0 mA postST25 ES-evoked increase in serum TNF-α in NPYDBH (peri.)-Abl mice compared with wild type (WT) mice (F1, 20 = 5.602, P < 0.05; ***P < 0.001; NS, P = 0.271).
(C) Splenectomy, compared with sham surgery, blocked postST25 ES-evoked increase in serum TNF-α (F1, 16 = 39.871, P < 0.001; ***P < 0.001; NS, P = 0.125).
(D) Heat map of quantitative RT-PCR results, showing changes in various adrenergic receptor (AR) expression 1 hour after LPS injection (two-side student’s unpaired t-test; β1: t8 = −0.049, P = 0.962; β2: t8 = −3.840, **P = 0.015; β3: t8 = −0.671, P = 0.521; α1a: t8 = −3.764, **P = 0.016; α1b: t8 = −0.876, P = 0.406; a1d: t8 = −0.270, P = 0.794; α2a: t8 = −2.957, *P = 0.018; α2c: t8 = −6.328, ***P < 0.001; α2b: Mann-Whitney Rank Sum test, *P = 0.016).
(E) α2-AR antagonist Yohimbine blocked postST25 ES-evoked increase of TNF-α (F1, 20 = 59.709, P < 0.001;***P < 0.001, *P = 0.016).
(F) Co-treatment with Yohimbine allowed 3.0 mA postST25 ES to promote survival for LPS-treated C57BL/6J mice (log-rank test, *P = 0.045).
(G) postST25 ES drive NPYDBH cell-dependent pro-inflammatory pathways, via activation of α2 ARs. (H) Schematic description of incoherent feed-forward loops. ES-evoked noradrenaline release from NPYDBH cells modulates LPS-induced TNF-α production (via activation of TLR4) in splenic cells, via activation of β2 and a2 ARs that produce the negative and positive modulatory legs, respectively.
n = 22–24 mice (A, F) and 5–6 mice for all other groups. NS: not significant. Data are shown as mean ± SEM.
We next investigated the mechanisms underlying this switch. We first found that postST25 ES-evoked pro-inflammatory effects operated mainly in the spleen, as indicated by a lack of ES-evoked increase in serum TNF-α and IL-6 levels upon surgical removal of spleen compared with sham surgery (Figures 5C and S8C). Previous studies reported that noradrenaline can promote the release of pro-inflammatory cytokines from macrophages via activation of α2 ARs (Huang et al., 2012; Spengler et al., 1994). Real-time polymerase chain reaction analyses showed that 1 hour after LPS injection, expression of all three α2 ARs (α2a, α2b, α2c) was increased markedly, with modest induction for β2 and α1a ARs, and no induction for other tested receptors (Figure 5D). We then found that in mice receiving pretreatment with Yohimbine, a generic antagonist for α2 ARs (Rouot et al., 1982), 3.0 mA postST25 ES no longer promoted, but instead switched back to suppress the production of TNF-α (Figure 5E) and IL-6 (Figure S8D). Consistently, 3.0 mA postST25 ES combined with Yohimbine treatment improved the survival rate to 70% (Figure 5F), compared with 0% for mice receiving 3.0 mA postST25 ES alone (Figure 5A) or 41.6% for mice receiving Yohimbine plus sham ES (Figure 5F). Such switch can be recaptured in cultured splenocytes. Two hours after LPS exposure, noradrenaline application promoted TNF-α production (Figure S8E); following application of Yohimbine with increasing doses, noradrenaline now progressively switched to suppress TNF-α production (Figure S8F). Thus, 3.0 mA postST25 ES produces a net pro-inflammatory effect via α2 AR signaling (Figure 5G), engaging bidirectional modulation of systemic inflammation under different disease states (Figure 5H).
Low intensity ES at the hindlimb ST36 acupoint attenuated ongoing systemic inflammation
While pharmacological blockage of α2 ARs offers a way to allow postST25 ES to manage ongoing systemic inflammation (see above), we additionally explored non-pharmacological approaches to bypass this pro-inflammatory sympathetic pathway, by focusing on the hindlimb ST36 acupoint (Figure 6A). Torres-Rosas et al. reported that ES at the hindlimb ST36 acupoint, using a constant voltage stimulation parameter (4 V), suppresses inflammation in a disease state-independent manner (Torres-Rosas et al., 2014). On the other hand, painful pinch stimulation at both abdominal and hindlimb regions is able to activate the splenic sympathetic nerve (Sato, 1997), which should dynamically modulate systemic inflammation according to above studies. With this apparent discrepancy, we envisioned a scenario that ST36 ES at different intensities might drive different pathways. In agreement with this hypothesis, high intensity stimulation (3.0 mA ES) at ST36 was able to activate spinal sympathetic pathways, as indicated by marked c-Fos induction in sympathetic preganglionic neurons (Figure S9A). As such, 3.0 mA ES at ST36 produced opposite effects at different disease states: anti inflammatory for ES performed before LPS exposure and pro-inflammatory for ES performed after LPS exposure, which in turn caused an increase and a decrease in survival rates, respectively (Figure S9B–G), exactly as 3.0 mA postST25 ES does.
By titrating down stimulation intensity, we then found that 0.5 mA ES at ST36 was sufficient to induce c-Fos in hindbrain ChAT+ vagal efferent neurons located in dorsal motor nuclei of the vagus (Figure 6B), but insufficient to do so in spinal sympathetic preganglionic neurons (Figure S9A). This low intensity stimulation caused an increase in serum catecholamine levels in wild type littermates, particularly dopamine (Figure 6C), and this increase was eliminated in NPYDBH (adrenal)-Abl mice (Figure 6C). Thus, 0.5 mA ST36 ES apparently drove the reported vagal-adrenal axis, leading to activation of NPYDBH chromaffin cells. Importantly, 0.5 mA ST36 ES attenuated TNF-α and IL-6 production by ~50%, irrespective of ES conducted before LPS injection (0.5 mA preST36 ES; Figures 6D and S9H) or 1.5 hours after LPS exposure (0.5 mA postST36 ES; Figures 6G and S9L). This anti-inflammatory effect occurred mainly in non-splenic tissues, since 0.5 mA postST36 ES did not reduce splenic inflammation (Figures S9J and S9K), and was lost in NPYDBH (adrenal)-Abl mice (Figures 6D, 6G, S9H and S9L) and in mice with subdiaphragmatic vagotomy (Figure 6E and 6H, and Figure S9I and S9M). Accordingly, both 0.5 mA preST36 ES and 0.5 mA postST36 ES led to ~40% increase in survival rates in LPS-treated wild type mice, compared with sham ES (Figures 6F and 6I). Thus, low intensity electroacupuncture stimulation at the hindlimb ST36 acupoint drives the vagal-adrenal anti-inflammatory pathway that operates in a disease state-independent manner, thereby offering an alternative way to treat pre-established systemic inflammation.
Discussion
Technical considerations on sympathetic neuron manipulations
To date, sympathetic neuron manipulations rely on chemical or surgical approaches, which face complexities and limitations. The commonly used reserpine depletes transmitter release from sympathetic neurons via blockage of vesicular monoamine transporters; however, this drug can cross the brain-blood barrier and inhibit the release of dopamine, noradrenaline, serotonin and histamine in the brain (Yaffe et al., 2018). 6-hydroxydopamine and guanethidine more selectively block peripheral sympathetic neurons (Nielsen, 1977; Thoenen and Tranzer, 1973), but are unable to target noradrenergic neuron subtypes. Nor do they have major impact on adrenal sympathetic chromaffin cells (Johnson and O’Brien, 1976; Kolibal-Pegher et al., 1994). Here we developed an intersectional genetic strategy to manipulate sympathetic neurons defined by coexpression of NPY-Cre and DBH-Flpo. NPYDBH preferentially marks adrenal chromaffin cells, as well as noradrenergic neurons located in prevertebral ganglia that innervate visceral organs, including most splenic sympathetic neurons. In contrast, NPYDBH marks a smaller subset of noradrenergic neurons and none of cholinergic sympathetic neurons located in paravertebral ganglia. Importantly, NPYDBH does not mark somatosensory, visceral sensory or enteric neurons. Adrenaline, whose synthesis requires DBH, has been reported to be released from immune cells (Flierl et al., 2007; Staedtke et al., 2018); our intersectional NPYDBH, however, does not mark any immune cells (Figure S1C). One limitation is that NPYDBH additionally marks the A1/C1 cluster of hindbrain catecholaminergic neurons, whose activation is capable of modulating inflammation (Abe et al., 2017). This limitation was overcome by using the modified diphtheria toxin that does not cross the brain-blood barrier (Pereira et al., 2017). Moreover, NPYDBH-marked adrenal chromaffin cells can be selectively ablated via intra-adrenal medulla injection of toxin. Our approaches could in principle be extended to study other molecularly defined sympathetic neuron subtypes in controlling body physiology (Furlan et al., 2016; Janig and Habler, 2000).
Somatotopic organization and intensity dependence for electroacupuncture to drive distinct autonomic pathways
We first reveal a somatotopic organization in driving the previously reported vagal-adrenal axis (Torres-Rosas et al., 2014). This axis can be evoked by low intensity stimulation (0.5 mA) at the hindlimb ST36 acupoint, producing anti-inflammatory effects that depend on vagal efferents and NPYDBH-marked adrenal chromaffin cells (Figure 6 and Figure S9). While 0.5 mA ST36 ES can induce c-Fos in vagal efferent neurons in the hindbrain (Figure 6), ES at the abdominal ST25 acupoint, even with high intensity (3.0 mA), fails to do so (Figure S5). Moreover, 0.5 mA ES at both ST25 and ST36 is insufficient to induce c-Fos in spinal sympathetic preganglionic neurons (Figure 4C and Figure S9A). As such, anti-inflammatory effects evoked by low intensity ES can occur at ST36 (Figure 6), but not at ST25 (Figure 4B).
Such somatotopic organization, in terms of producing anti-inflammatory effects from selective acupoints, starts to disappear as stimulation intensity increases, due to recruitment of new autonomic pathways. We found that 3.0 mA ES at the abdominal ST25 acupoint is able to drive a spinal-sympathetic axis (Figure 4), producing anti-inflammatory effects (ES before LPS exposure) that depend on NPYDBH-marked noradrenergic neurons, but is independent of vagal efferents or NPYDBH-marked adrenal chromaffin cells (Figure 4 and Figure S5). 3.0 mA ES at the hindlimb ST36 acupoint, performed before LPS injection, can also drive a spinal-sympathetic axis and produce vagal efferent-independent anti-inflammatory effects (Figure S9B and S9C).
This lack of acupoint selectivity echoes earlier findings, showing that strong pinch stimulation can activate the splenic sympathetic nerve from all body regions tested (Sato, 1997). Other difference exists for the anti-inflammatory pathways evoked by high and low intensity ES at different acupoints. 3.0 mA preST25 ES, but not 0.5 mA ST36 ES, can suppress splenic inflammation (Figures 4 and S9). Also, the anti-inflammatory axis evoked by 0.5 mA ES at ST36 operates via activation of the D1 dopamine receptor but not β2 adrenergic receptors in response to catecholamines released from the adrenal gland (Torres-Rosas et al., 2014; data not shown), whereas the spinal-sympathetic axis evoked by 3.0 mA preST25 ES does the opposite, depending on β2 adrenergic receptors but not the D1 dopamine receptor (Figures 4 and S6, and data not shown). The differential somatotopic organization and intensity dependence in driving these autonomic pathways could explain variable degrees of vagal nerve dependence in modulating systemic inflammation by acupuncture (Gu et al., 2011; Huang et al., 2007; Lim et al., 2016; Song et al., 2012; Torres-Rosas et al., 2014; Villegas-Bastida et al., 2014; Zhao et al., 2012), if different stimulation intensities and/or acupoints were used in different studies.
The presence of somatotopic organization in driving specific autonomic pathways is not unique for inflammatory control. Earlier studies show that vagal and sympathetic pathways associated with gastric motility control can be preferentially evoked from limb and abdominal regions, respectively (Li et al., 2007; Sato, 1997). To some degree, acupoint-selective driving of specific somatosensory autonomic pathways could form a modern neuroanatomical basis for the linking from somatic tissue stimulation to the modulation of internal organ physiology, a role long proposed to be carried out by the traditional, though physically still unfound meridian channels. Future studies will be directed to map somatosensory pathways that drive these distinct autonomic pathways.
Abdominal electroacupuncture stimulation amplifies an endogenous anti-inflammatory system mediated by NPYDBH-marked splenic noradrenergic neurons
The presence of endogenous anti-inflammatory autonomic pathways in response to LPS exposure or bacterial infection was long suggested from increased inflammation following transection of the vagus nerve (Figures S5F and S5G) [(Borovikova et al., 2000; Song et al., 2012), but see also (Martelli et al., 2014)] or the splanchnic nerves (Bratton et al., 2012; Martelli et al., 2016; Martelli et al., 2019). There is a complexity in interpreting these results. For example, about 50% nerve fibers within the splanchnic nerve are sensory afferents derived from DRG (Janig and Habler, 2000). With demonstrated sensory innervations in immune organs (Huang et al., 2019), one alternative possibility is that endogenous anti-inflammation roles might operate via sensory axon reflexes, as suggested from inflammation control in other tissues (Fernandes et al., 2012; Pinho-Ribeiro et al., 2018). Here we reported that NPYDBH marks a subset of sympathetic cells, without marking somatosensory or visceral sensory afferents. We then found that LPS-induced inflammation was exaggerated upon ablation of NPYDBH-marked noradrenergic neurons. In the spleen, these neurons innervate predominantly white pulps, and their tonic activity or LPS-induced activation should produce the highest concentration of noradrenaline in this sub-region. Consistently, with the LPS dose used in our studies, endogenously activated NPYDBH noradrenergic neurons are sufficient to suppress TNF-α in macrophages located in white pulps and marginal zones, but insufficient to do so in red pulps (Figure 3). Following electric stimulation at the abdominal ST25 acupoint, which activates virtually all splenic noradrenergic neurons located in suprarenal ganglia and causes an increase in noradrenaline release, LPS-induced TNF-α can now be suppressed throughout the spleen (Figure 4L). Thus, for preventive interventions, electroacupuncture stimulation at the abdomen acts to amplify an endogenous anti-inflammatory system that involves NPY-expressing splenic noradrenergic neurons.
NPYDBH noradrenergic neurons engage with incoherent regulatory loops and bidirectionally modulate systemic inflammation
Incoherent regulatory loops are recurring regulatory motifs seen from bacteria to mammals (Alon, 2007). A salient feature of such control system is that a given input drives two opposing legs (positive versus negative) to influence a biological process. We found that noradrenaline (NA) released from NPYDBH-marked splenic sympathetic neurons operates via β2 and α2 adrenergic receptors (ARs) to produce the negative and positive legs, respectively, in modulating systemic inflammation (Figure 5H). As such, the net splenic inflammation is determined by the relative strength of three factors (see Figure 5H): i) LPS-activated toll-like receptor signaling that drives TNF-α and IL-6 production (Beutler, 2000; Salomao et al., 2008; Ulloa et al., 2017), ii) the α2-AR-mediated positive modulatory leg, and iii) the β2 AR-mediated negative modulatory leg. For preventive ES, which is delivered before LPS exposure, activation of NPYDBH splenic noradrenergic neurons produces a dominant negative leg, leading to suppression of LPS-induced inflammation. Following LPS exposure, there is a marked induction of α2 ARs in splenic cells. Combined with α2 ARs having a higher affinity to noradrenaline than β2 ARs (Molinoff, 1984), the α2 AR-mediated positive leg progressively gains dominance as the disease progresses. As a result, 3.0 mA ES now becomes detrimental. Following α2 AR blockage, 3.0 mA ES can regain the ability to drive the β2 AR-mediated negative leg, producing anti-inflammatory and beneficial effects. Notably, following LPS exposure, low (0.5 mA) and high (3.0 mA) intensity stimulation at the same ST36 acupoint produces opposite effects: anti- and pro-inflammatory, respectively. Such disease state- and intensity-dependent modulation of systemic inflammation by ES offers an explanation for apparent discrepancy from different studies. For example, Torres-Rosas et al. reported disease state-independent anti-inflammatory effects evoked by ES at ST36 (Torres-Rosas et al., 2014), using the stimulation intensity (4 V, likely equivalent to 0.5 mA used here) that predominantly drives the vagal-adrenal axis. In contrast, Song et al. reported that following LPS exposure, ES at the forelimb regions fails to promote survival (Song et al., 2012), likely reflecting the use of high intensity stimulation (4 mA in rats) that sufficiently drives the spinal-sympathetic axis.
Clinical implications
The findings that acupuncture stimulation modulates systemic inflammation in somatotopy-, intensity-, and disease state-dependent manners (Figure 6J) should help to improve acupuncture practice. For systemic inflammation prevention (analogous to ES before LPS exposure), the vagal-adrenal anti-inflammatory axis evoked by low intensity ES at the hindlimb ST36 acupoint operates in non-splenic tissues, whereas the spinal-sympathetic axis evoked by 3 mA ES at the abdominal ST25 acupoint can suppress splenic inflammation, although it remains to be determined if these two pathways can interact in non-splenic tissues. Accordingly, a combination of 0.5 mA ES at ST36 and 1-3 mA ES at ST25, or high intensity ES at ST36 alone, may concurrently drive both pathways and produce synergistic anti-inflammatory effects. For treating ongoing severe systemic inflammation, after the positive modulatory leg reaches dominance, high intensity stimulation could become contraindicated, making inflammation worsened and fatality rates increased, thereby raising a previously unappreciated safety issue associated with acupuncture practice; under such conditions, we may either choose low intensity ES at the hindlimb region to selectively drive the vagal-adrenal anti-inflammatory axis that operates in a disease state-independent manner, or conduct high intensity ES combined with pharmacological blockage of α2 ARs. The intensity dependence also suggests that electroacupuncture has an advantage in comparison with manual acupuncture that would be more difficult to precisely control the intensity. Thus, the revelation of somatotopic organization and intensity dependence in driving distinct autonomic pathways could help to optimize stimulation parameters and improve both efficacy and safety in using acupuncture to treat systemic inflammation.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Qiufu Ma (qiufu_ma@dfci.harvard.edu).
Materials Availability
The study did not generate new plasmids or unique reagents
The study did not generate new mouse lines; the mouse lines used for intersectional manipulations were all acquired from public resources or from relevant investigators (see below, “Mice” in “EXPERIMENTAL MODEL AND SUBJECT DETAILS”).
Data and Code Availability
This study did not generate any unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All animal experiments were performed with protocols approved by the Institutional Animal Care and Use Committee at Dana-Farber Cancer Institute and followed NIH guidelines. Mice were housed at room temperature with a 12-h/12-h light/dark cycle and had ad libitum access to standard laboratory mouse pellet food and water. The NPYCre transgenic mouse line (RH26) was generated by the Gene Expression Nervous System Atlas (GENSAT) project (Gerfen et al., 2013) and acquired from MMRRC, University of California, Davis (MMRRC:034810-UCD). C57BL/6J male mice (000664), Rosa26-loxp-stop-loxp-tdTomato reporter mice (Ai14, 007908), Rosa26-loxp-stop-loxp-frt-stop-frt-tdTomato mice (Ai65, 021875), Rosa26-loxp-stop-loxp-frt-stop-frt-hM3Dq mice (RC::FL-hM3Dq, 026942) and Rosa26-loxp-stop-loxp-frt-stop-frt-hM4Di mice (RC::FPDi, 029040) were acquired from the Jackson Laboratory (JAX). DBH-p2a-Flpo mice (Sun and Ray, 2016) were acquired from the Russell S. Ray group and these mice are available at MMRRC, University of California, Davis (MMRRC:041575-UCD). Tau-loxp-stop-loxp-frt-stop-frt-DTR mice (Bourane et al., 2015) were acquired from the Martyn Goulding group. 10-12 weeks males and females show differential sensitivity to LPS (data not shown), and we have not yet optimized the dose to achieve the similar lethality rate ranges between males and females; as such, current studies focused on male mice, which produced a LPS-induced lethality rate at the range (~20-30%), allowing us to assess both a suppression and a promotion of LPS-induced systemic inflammation.
METHOD DETAILS
Preparation of PEGyDT
PEGylation of the diphtheria toxin was prepared as previously described (Pereira et al., 2017). Briefly, MS(PEG)4 methyl-PEG-NHS-Ester (22342, Life Technologies), used as a crosslinker, was reconstituted and diluted to 2.5 mM in dimethyl sulfoxide. The diphtheria toxin (DTX) from Corynebacterium diphtheriae (1 mg, D0564, Sigma) was reconstituted with 1 x PBS to the concentration of 1 μg/μl, and PEGylation was performed by mixing the above two with the volume ratio at 1 crosslinker versus 1.97 DTX. The mixture was incubated at room temperature for 4 hours. To remove excess amount of the crosslinker, gel filtration chromatography using the gravity protocol of PD MidiTrapG-25 column (28-9180-07, GE Healthcare) was performed according to manufacturer’s instruction. The concentration of PEGyDT was determined using a NanoDrop ND-2000 UV-Visible spectrophotometer (Fisher Scientific).
Ablation of all peripheral NPYDBH sympathetic cells
To generate NPYDBH (peri.)-Abl mice in which all peripheral NPYDBH sympathetic cells were ablated, 6-8-week old adult male NPYDBH-DTR mice were injected intraperitoneally (i.p.) with PEGyDT (50 μg/kg) twice with a 72-h interval. LPS challenge and histochemical analyses were performed 3-4 weeks after PEGyDT injections. Control littermates that did not carry either the DBHFlpo or the NPYcre allele, thereby lacking DTR expression, received the same PEGyDT injections.
Ablation of adrenal NPYDBH chromaffin cells
To generate NPYDBH (adrenal)-Abl mice, in which NPYDBH chromaffin cells located in the adrenal medulla were selectively ablated, we performed intra-adrenal medulla PEGyDT injections. NPYDBH-DTR mice were anaesthetized with isoflurane (2-5%), and the left and right adrenal glands were exposed through a flank incision. 30 ng/2 μl of PEGyDT was injected into the adrenal medulla using a glass micropipette attached to a 10 μL Hamilton syringe. The abdominal wall and skin were closed via Vicryl sutures. Buprenorphine was administered subcutaneously at the dose of 0.05 mg/kg, one dose every 8-12 hour for 48 hours (total 4 doses post-surgery). Animals were observed during recovery from anesthesia until animals could maintain sternal recumbency. 3-4 weeks after PEGyDT injections, mice were used for experimental studies. Control littermates lacking DTR expression underwent the same surgical intra-adrenal medulla PEGyDT injections.
Acute activation and silencing of NPYDBH cells
For activation experiments, NPYcre/+;DBHFlpo/+;ROSA26oxp-stop-loxp-frt-stop-frt-hM3Dq (NPYDBH-hM3Dq) mice and their wild type (WT) control littermates were used. For the silencing experiments, NPYcre/+;DBHFlpo/+;ROSA26loxp-stop-loxp-frt-stop-frt-hM4Di (NPYDBH-hM4Di) mice and their control WT littermates were used. I.p. injection of 5 mg/kg clozapine-N-oxide (CNO, 10 mg/ml in stock, Sigma-Aldrich, C0832) were used to activate or silence NPYDBH neurons, followed by experimental schemes described in Figure S7.
Compounds
Lipopolysaccharide (LPS) was purchased from Sigma-Aldrich (from E. coli 0111:B4, L2630). The following adrenergic receptor (AR) antagonists were used: for in vivo experiment: ICI 118,551 (a selective β2-AR antagonist; 2 mg/kg; 5 mg/ml in stock; I127; Sigma) was injected (i.p.) 30 min before electroacupuncture stimulation (ES) and Yohimbine (a selective α2-AR antagonist; 0.5 mg/kg; 1 mg/ml in stock; Y3125; Sigma) was administered (i.p.) 15 mins before ES, using the doses as reported previously (Archer and Fredriksson, 2000; Vranjkovic et al., 2012). For in vitro experiment: noradrenaline (A7257, Sigma) was dissolved in sterile 0.9% saline chloride and was added to cultured splenocytes (to the final concentration at 50 μM) 30 mins before or 2 hours after LPS (100 ng/ml) addition. ICI 118,551 or Yohimbine was administrated at the concentrations indicated in figures 30 mins and 15 mins prior to noradrenaline addition, respectively.
Endotoxemia
LPS was administered to mice corresponding to an LD80 dose (8 mg/kg for C57BL/6J male mice and 12 mg/kg for mix genetic background male mice, i.p.), and prepared in sterile PBS (Gibco®, Life Technologies, Grand Island, NY). Serum was collected at the indicated time point. For survival rate monitoring, mice exhibiting any one of the following criteria (a, b and c) were euthanized immediately and counted into the fatality group: a) loss of 15% body weight, b) body temperature below 32° C, and c) the total clinical symptom score reaching 2 (Kadl et al., 2007). At indicated time points, animals were assessed for symptom scores by grading the severity of conjunctivitis, diarrhea, ruffled fur, and lethargy on a three-point scale (0, 1, 2). Means of the three assessments were used for grading. Conjunctivitis: score 0-eyes closed or bleared with serous discharge; score 1-eyes opened with serous discharge; score 2-normal, no conjunctivitis. Stool consistency: score 0-diarrhea; score 1-loose stool; score 2-normal stool. Hair coat: score 0-rough and dull fur, ungroomed; score 1-reduced grooming, rough hair coat; score 2-well groomed, shiny fur. Activity upon moderate stimulation: score 0-lethargic, only lifting of the head after moderate stimulation; score 1-inactive, less alert, <2 steps after moderate stimulation; score 2-normal locomotion and reaction, >2 steps after moderate stimulation. The total maximum score is 8, which indicates a normal health condition.
Electroacupuncture stimulation (ES)
Animals were anesthetized by inhaled isoflurane (0.5-1.5%) via a precision vaporizer and laced on a heating pad to maintain body temperature. ES was performed with a continuous-mode stimulation for 15 min, with the electrical current range of 0-3.0 mA, a pulse width of 50 μs and a frequency of 10 Hz by using a stimulator (Model 3800, A-M Systems) and 4 isolators (Model 3820, A-M Systems). ES was performed at either the hindlimb ST36 (Zusanli) or the abdominal ST25 (Tianshu) acupoint by inserting the 0.16 X 7 mm unipolar stainless steel acupuncture needle electrode about 3 mm deep in each site. The ST36 Zusanli acupoint is located around 4 mm away from the knee joint, 2 mm lateral to the anterior tubercle of the tibia, and in the proximity of the common peroneal and tibial branches of the sciatic nerve (Torres-Rosas et al., 2014). The bilateral ST25 acupoints are located 5 mm lateral to the intersection between the upper 2/3 and the lower 1/3 of the line between the xiphoid process and the pubic symphysis upper border (Gao et al., 2016). For studying ES-evoked pERK induction in DRG, ES was performed for 15 mins and 10 mins later, the mice were perfused and thoracic Th10 DRG were collected. For studying ES-evoked c-Fos induction in spinal sympathetic preganglionic neurons (PGNs), dorsal motor nuclei of the vagus (DMV) and sympathetic ganglia, ES was performed for 15 mins and 2 hours later, the mice were perfused for tissue collection. For studying ES effects on LPS-induced systemic inflammation and mortality, ES was performed either 15 mins before or 1.5 hours after LPS injection. The electrode insertion into the same acupoint without electrical stimulation (0 mA) was used as the sham control.
Subdiaphragmatic vagotomy
Animals were anesthetized by inhaled isofluorane (2-5%) via a precision vaporizer. Depth of anesthesia was assessed by absence of corneal and hindpaw withdrawal reflexes. Animals were laced on a heating pad to maintain body temperature. After an upper midline laparotomy, the stomach was gently retracted inferiorly to expose the esophagus. Both anterior and posterior trunks of the vagal nerves were identified between the diaphragm and the gastric cardia, and then transected. For sham controls, the vagal trunks were exposed, but not ligated or excised. The muscle and skin were closed via Vicryl sutures. Buprenorphine was administered subcutaneously at 0.05 mg/kg every 8-12 hour for 48 hours (total 4 post-surgery doses). 5 days after subdiaphragmatic vagotomy, mice were then used for experimental testing.
Splenectomy
Animals were anesthetized by inhaled isoflurane (2-5%) via a precision vaporizer. Depth of anesthesia was assessed by absence of corneal and hindpaw withdrawal reflexes. Animals were laced on a heating pad to maintain body temperature. After an abdominal incision (nearly 3 cm) above the epigastrium and the mesogastrium, the stomach was retracted slightly to the right side to expose the spleen. The three main branches of the spleen artery were stabilized with the nylon thread, ligated and cut at the middle of the ligations, and the spleen was then removed. For sham surgery control, the spleen was just exposed, without artery ligation and spleen removal. The abdominal wall and the skin were closed via Vicryl sutures. Buprenorphine was administered at 0.05 mg/kg subcutaneous one dose every 8-12 hour for 48 hours. Animals were splenectomized 5 days before experimental testing.
Fluorogold retrograde tracing
Adult NPYDBH-tdTomato mice were anaesthetized with isoflurane (2-5%). The spleen was exposed through a left flank incision and we performed 10 injections of 2 μl of fluorogold (fluorochrome, 2% in sterilized water) into the parenchyma via a glass micropipette driven by a picospritzer III (Parker Hannifin). The abdominal wall and the skin were closed via Vicryl sutures. Buprenorphine was administered at 0.05 mg/kg subcutaneous one dose every 8-12 hour for 48 hours. Mice were allowed to recover for seven days before electroacupuncture stimulation and tissue collection.
Immunohistochemistry and In Situ Hybridization (ISH)
Animals were euthanized by CO2 and then perfused transcardially with 4% paraformaldehyde (PFA). The DRG, spinal cord, brain, nodose ganglia, sympathetic ganglia, adrenal glands and the colon were dissected and post-fixed in the same fixatives overnight at 4 °C. The spleen and the colon were dissected and post-fixed in the Zambone’s solution overnight at 4 °C. These tissues were cryopreserved in 30% sucrose in PBS overnight, and then embedded in Tissue-Tek OCT compound (Sakura Finetek). For immunohistochemistry on sections through DRG, spinal cord, hindbrain, sympathetic ganglia, adrenal glands, spleen and colon, 25 μm (for immunohistochemistry) and 12 μm (for ISH)-thick serial sections were made with a cryostat and mounted on SuperFrost Plus slides (Fisher). Sections were blocked with 0.3% of Triton X-100 plus 5% of normal goat or donkey serum in PBS for 1 hour and then incubated with the following primary antibodies overnight at 4 °C: rabbit anti-c-Fos (1:500, ABE457, Millipore), goat anti-ChAT (1:500, AB144P, Millipore), rabbit anti-TH (1:1000, AB152, Millipore), rabbit anti-dsRed (1:1000, 632496, Clontech), rabbit anti-pERK (1:500, 4370S, Cell signaling technology), goat anti-TNF-α (1:100, AF-410-NA, R&D systems), rat anti-F4/80 (1:100, ab6640, Abcam), rat anti-CD3 Alexa 488 (1:200, clone GK1.5; Biolegend), rat anti-B220 Alexa 488 (1:200, clone RA3-6B2; Biolegend), rat anti-CD68 (1:200, MCA1957, Bio-Rad), rat anti-CD169 (1:200, clone 3D6.112; Biolegend), rat anti-CD11c (1:200, clone N418; Biolegend), rat anti-Ly6G Alexa 488 (1:200, clone 1A8; Biolegend). After washing with 0.3% of Triton X-100 in PBS, the sections were incubated with corresponding secondary antibodies: Alexa Fluor 405, 488, 594 donkey anti-goat, rabbit, sheep, rat IgG (1:500; 711-545-152, 705-585-003, 705-545-003, 705-475-147, 711-585152 and 712-476-150; Jackson ImmunoResearch) for 1 hour at room temperature. Images were acquired using a wild-field fluorescence (Zeiss, AXIO IMAGER Z1) and confocal microscope (Zeiss LSM700).
ISH combined with immunohistochemistry procedures was performed as previously described (Liu et al., 2010; Liu et al., 2008). Both fluorescent and ISH signals were collected using a fluorescent microscope. The tdTomato fluorescent signal was photographed from the slices, followed by ISH. The pseudo fluorescent ISH signals were converted from bright field images, and then merged onto the tdTomato images via the Photoshop software.
To characterize and quantify i) NPYDBH-tdTomato+ neurons in different sympathetic ganglia/adrenal glands and ii) splenic sympathetic neurons retrogradely labeled via Fluoro-gold injection into the spleen, we analyzed 5-10 sections through each sympathetic ganglion or the adrenal medulla, and five adult male NPYDBH-Ai65 mice were used. To test if ST25 and ST36 ES at various intensities activated distinct neural pathways, we quantified pERK+ or c-Fos+ cells in sections through DRG, spinal cord, and sympathetic ganglia, or determined the percentages of ChAT+ preganglionic neurons that were c-Fos+ in parasagittal sections through the intermediate lateral nuclei of the thoracic T6-T10 spinal cord or in coronal hindbrain sections through the dorsal motor nuclei of the vagus (DMV), with five adult male mice per group. To test ablation efficiency, we used 5 pairs of adult control and ablated mice and analyzed 25-50 sections through a ganglion or a particular spinal or brain region per mouse. To test how peripheral or adrenal NPYDBH cell ablation impacted on LPS-induced splenic TNF-α expression, we analyzed 5-8 nonconsecutive sections from each mouse to check the percentages of different macrophages that expressed TNF-α, or measured the TNF-α pixel intensity by Image J software; five paired mice for each group were analyzed. Mice were randomly assigned into treatment groups, with investigators blinded to the genotypes.
Cytokine analyses
Concentrations of TNF-α (BMS607, Thermofisher,), IL-1β (MLB00C, R&D Systems), and IL-6 (M6000B, R&D Systems) were analyzed by ELISA kit following manufacturer’s instructions. Briefly, 100 μl of standard and diluted samples in duplicated wells were incubated at room temperature for 2 hours. After the wells were washed 5 times with 1x wash solution, 100 μl of enzyme conjugate reagent was added into each well, followed by 2-hour incubation at room temperature. 100 μl of substrate solution was then added and after 30-min incubation, 100 μl of stop solution was added to terminate the reaction. The formed color was assayed with a microtiter plate reader (SunriseTM, Tecan, Switzerland).
For in vivo serum cytokine analyses, blood was collected from the penetrated submandibular vein at the indicated time points, allowed to clot for 1.5 hours at room temperature, and then centrifuged at 2500 g for 15 min at 4 °C. The supernatant serum was collected and stored at −80 °C before use. For splenic cytokine analyses, 1 hour after LPS injection, spleen was collected and was then homogenized in 1 ml of PBS containing a cock-tail of protease inhibitors (P2714, Sigma-Aldrich) using a tissue tearing homogenizer. The cytoplasmic fraction was isolated as the supernatant fraction following centrifugation at 10, 000 rpm for 10 min at 4 °C. Protein levels of all samples were quantified with a BCA Protein Assay Kit (Thermo Scientific, Waltham, MA). 250 μg of protein was used to assess cytokines by ELISA.
For in vitro studies on cultured splenocytes, spleens were mechanically disrupted with a 70-μm pore cell strainer (BD Falcon), erythrocytes were lysed in a lyse solution (420302, Biolegend) for 6 mins on the ice, and remaining intact splenic cells were washed twice in PBS. Isolated splenocytes were added into 96-well plates (2.0 × 105/well) in 200 μl of RPMI complete medium containing 10% FBS. Cells were cultured at 37 °C in a humidified incubator with 5% CO2. Splenocytes were incubated with 50 μM noradrenaline at 30 mins before or at 2 hours after giving LPS (100 ng/ml, E. coli 0111:B4; Sigma). TNF-α was analyzed in the conditioned supernatant at 3 hours or 4 hours after the LPS challenge, respectively. Cytokine analyses in the conditioned media were performed using the same reagents as reported above.
Catecholamine measurements
For serum catecholamine measurements, bloods were collected 15 mins after ES at ST25 or T36, allowed to clot for 1.5 hours at room temperature, and centrifuged at 2500 g for 15 mins at 4 °C. The supernatant serum was collected and transferred to regular tubes. The samples were stored at −80 °C until analysis using the catecholamine ELISA kit (BA E-5600, Rocky Mountain Diagnostics), following the manufacturer’s instructions.
For measuring splenic noradrenaline concentrations in response to ST25 ES, animals were euthanized (CO2 inhalation) 15 mins after ES. Spleens were collected in solution containing EDTA (1 mM) and sodium metabisulfite (4 mM) to prevent catecholamine degradation. Samples were then homogenized using Dounce homogenizer, debris was pelleted by centrifugation at 10,000 rpm for 10 mins, and supernatants were stored at −80°C until further use. Noradrenaline was assessed by using the noradrenaline ELISA kit (BA E-5200, Rocky Mountain Diagnostics), following the manufacturer’s instructions.
Real-Time PCR
Spleens were dissected, frozen in dry ice, and stored at −80 °C. Total RNA was extracted using Trizol reagent (Invitrogen) according to manufacturer’s instructions. The amount of RNA was measured using a NanoDrop ND-2000 UV-Visible spectrophotometer (Fisher Scientific). Two μg of total RNA was reverse transcribed into cDNA using the M-MLV reverse transcriptase (Promega) and oligo (dT) primers. The PCR reactions were performed using the following pairs of oligonucleotide primers: β1-AR (NM_007419.3): upstream primer, 5-CGGCCTTTCGTGTGTTTAAT-3; and downstream primer, 5-CACACCAAACCTGAGCTGAA-3. β2-AR (NM_007420.3): upstream primer, 5-GAGCACAAAGCCCTCAAGAC-3; and downstream primer, 5-GTTGACGTAGCCCAACCAGT-3. P3-AR (NM_013462.3): upstream primer, 5-GACAGCCTCAAATGCATCCT-3; and downstream primer, 5-CCCAGTCCACACACCTTTCT-3. a1a-AR (NM_001271759.1): upstream primer, 5-ACCATTGTCACCCAGAGGAG-3; and downstream primer, 5-ATGATGGTCAGTGGCACGTA-3. a1b-AR (NM_001284380.1): upstream primer, 5-CGCCCACCAACTACTTCATT-3; and downstream primer, 5-AATGGAGATGGCACATAGGC-3. α1d-AR (NM_013460.5): upstream primer, 5-GCCTCTGAGGTGGTTCTGAG-3; and downstream primer, 5-GGACGAAGAAAAAGGGGAAC-3. α2a-AR (NM_007417.4): upstream primer, 5-TCTGGCTGAGAGGGACA-3; and downstream primer, 5-GGGTGTGGAGGAGATAAT-3. α2b-AR (NM_009633.3): upstream primer, 5-TGGAAGTGGGTTGAGGTG-3; and downstream primer, 5-TGCCTGCTCAATGACAAAG-3. α2c-AR (NM_007418.3): upstream primer, 5-GTACTTCGGGCAAGTGTGGT-3; and downstream primer, 5-CGGTAGAACGAGACGAGAGG-3. The reference gene GAPDH (NM_001289726.1; upstream primer, 5-AAATGGTGAAGGTGAAGGTCGGTGTG-3; and downstream primer, 5-AGGTCAATGAAGGGGTCGTT-3). Relative expression of the AR gene in the spleen obtained from mice treated with LPS was calculated in comparison to vehicle (PBS)-treated control samples using the delta Ct method. 5 mice per groups were analyzed. All samples were run in duplicate.
Flow cytometric analysis
Spleens were harvested in Flow Cytometry Staining Buffer (eBioscience) and cell suspensions were made by pushing the spleen tissue through a 70-μm pore cell strainer using the plunger of a 5 ml syringe. Erythrocytes were lysed in a lysis solution (420302, Biolegend) for 6 min on the ice, and intact cells were washed twice in staining buffer. Splenocytes were counted on a hemocytometer and 1 × 106 cells were incubated with Fc-block (anti-mouse-CD16/CD32 antibody; Biolegend, clone 93). Samples were stained with the following antibodies: anti-CD45-APC/Cy7 (clone 30-F11, Biolegend), anti-CD11b-BV421 (clone M1/70, BD Biosciences), anti-CD45R (B220)-PE-Cy7 (clone RA3-6B2; BD Biosciences) and anti-CD3-AF488 (clone GK1.5; Biolegend). All antibodies were diluted to 0.5 μg for 5 × 105 cells. Splenocytes (10,000 events per sample) were analyzed using a FACSCanto flow cytometer with FACSDiva software (BD Biosciences). Data analyses were conducted with FlowJo software (FlowJo). Dot plots of forward scatter (FSC) versus side scatter (SSC) were used to exclude debris, and then doublets were excluded using FSC-H versus FSC-A plots. For detecting NPYDBH-tdTomato-expressed splenocytes, we use the NPYCre-tdTomato splenocytes as a control to gate tdTomato+ signal. For detecting splenocytes changes after NPYDBH (peri.)-Abl, the number of each subset was calculated as the percentage of labeled cells multiplied by the total number of splenocytes.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
Statistical analyses were done by using the GraphPad Prism7 and SigmaStat 3.5 software. Results are expressed as mean ± SEM. All datasets were tested for normality for t-test, and if the normality failed, the Mann-Whitney rank-sum test was used. Survival rates were expressed by a Kaplan-Meier curve and comparisons of survival curves were performed with a Mantel Cox log-rank test. To compare intensity-dependent inhibition of systemic TNF-α and IL-6 expression and c-Fos induction in the thoracic T6-T10 preganglionic neurons by preST25 and preST36, data were analyzed with a one-way ANOVA followed by a post hoc Tukey test. To compare the noradrenaline, IC1118,551 and Yohimbine treatment on LPS-induced TNF-α expression in cultured splenocytes, data were analyzed with a one-way ANOVA followed by a post hoc Dunnett test. To compare the impact of vagotomy, splenectomy, ablation and pharmacological treatments on ST25 or ST36 ES-evoked modulation on LPS-induced cytokine production and/or on catecholamine release, the data were analyzed with a two-way ANOVA followed by a post hoc Tukey test. No statistical methods were used to predetermine sample sizes. Sample sizes for all histochemical, cytokine and transmitter measurements were chosen according to the common practice reported during past two decades on inflammation modulation by electric stimulation. For survival rate analyses, they normally require large sample sizes, with the power set at 0.8 and the confidence interval set at 95%; due to complex genetic crossing required to produce ablation mice, we realistically aimed to detect major changes, such as a change of survival from 20% to 60-80% or vice versa, which required n = 20-25 per group. Differences were considered significant when P < 0.05.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | INDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-c-Fos | Millipore | Cat# ABE457 RRID:AB_2631318 |
| goat anti-ChAT | Millipore | Cat# AB144P RRID:AB_2079751 |
| rabbit anti-TH | Millipore | Cat# AB152 RRID:AB_390204 |
| rabbit anti-dsRed | Clontech | Cat# 632496 RRID:AB_2333092 |
| rabbit anti-pERK | Cell signaling technology | Cat# 4370S RRID:AB_2315112 |
| goat anti-TNF-α | R&D Systems | Cat# AF-410-NA RRID:AB_354479 |
| rat anti-F4/80 | Abcam | Cat# ab6640 RRID:AB_1140040 |
| rat anti-CD3 Alexa 488 | Biolegend | Cat# 100210 RRID:AB_389301 |
| rat anti-B220 Alexa 488 | Biolegend | Cat# 103225 RRID:AB_389308 |
| rat anti-CD68 | Bio-Rad | Cat# MCA1957 RRID:AB_322219 |
| rat anti-CD169 | Biolegend | Cat# 142401 RRID:AB_10915134 |
| rat anti-CD11c | Biolegend | Cat# 117303 RRID:AB_313772 |
| rat anti-Ly6G Alexa 488 | Biolegend | Cat# 127626 RRID:AB_2561340 |
| Donkey anti-rabbit Alexa 488 | Jackson ImmunoResearch | Cat# 711-545-152 RRID:AB_2313584 |
| Donkey anti-goat Alexa 594 | Jackson ImmunoResearch | Cat# 705-585-003 RRID:AB_2340432 |
| Donkey anti-goat Alexa 488 | Jackson ImmunoResearch | Cat# 705-545-003 RRID:AB_2340428 |
| Donkey anti-goat Alexa 405 | Jackson ImmunoResearch | Cat# 705-475-147 RRID:AB_2340427 |
| Donkey anti-rabbit Alexa 594 | Jackson ImmunoResearch | Cat# 711-585-152 RRID:AB_2340621 |
| Donkey anti-rat Alexa 488 | Jackson ImmunoResearch | Cat# 712-476-150 RRID:AB_2632567 |
| rat anti-CD45 APC/Cy7 | Biolegend | Cat# 103115 RRID:AB_312980 |
| rat anti-mouse-CD16/CD32 | Biolegend | Cat# 101320 RRID:AB_1574975 |
| rat anti-CD11b BV421 | BD Bioscience | Cat# 562605 RRID:AB_11152949 |
| rat anti-CD31 | BD Bioscience | Cat# 562939 RRID:AB_2665476 |
| rat anti-B220 PE-Cy7 | BD Bioscience | Cat# 552772 RRID:AB_394458 |
| anti-Digoxigenin-AP | Sigma | Cat# 11093274910 RRID:AB_514497 |
| Chemicals, Peptides, and
Recombinant Proteins | ||
| Diphtheria toxin | Sigma | Cat# D0564 |
| (PEG)4 methyl-PEG-NHS-Ester | Life Technologies | Cat# 22342 |
| Clozapine-N-oxide | Sigma | Cat# C0832 |
| LPS | Sigma | Cat# L2630 |
| ICI 118,551 | Sigma | Cat# I127 |
| Yohimbine | Sigma | Cat# Y3125 |
| PBS | Life Technologies | Cat# 10010-023 |
| Noradrenaline | Sigma | Cat# A7257 |
| Fluoro-gold | Fluorochrome | N/A |
| Critical Commercial
Assays | ||
| TNF-α ELISA kit | Thermofisher | Cat# BMS607 |
| IL-1β ELISA kit | R&D Systems | Cat# MLB00C |
| IL-6 ELISA kit | R&D Systems | Cat# M6000B |
| Catecholamine ELISA kit | Rocky Mountain Diagnostics | Cat# BA E-5600 |
| Noradrenaline ELISA kit | Rocky Mountain Diagnostics | Cat# BA E-5200 |
| BCA Protein Assay Kit | Sigma | Cat# 71285 |
| SYBR Green Mater Mix | Life Technologies | Cat# 4368706 |
| Experimental Models:
Organisms/Strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | JAX:000664 |
| Mouse: NPY-Cre | MMRRC, University of California, Davis | MMRRC:034810-UCD |
| Mouse: DBH-p2a-Flpo | MMRRC, University of California, Davis | MMRRC:041575-UCD |
| Mouse: Rosa26-loxp-stop-loxp-tdTomato | The Jackson Laboratory | JAX: 007908 |
| Mouse: Rosa26-CAG-loxp-stop-loxp-frt-stop-frt-hM3Dq | The Jackson Laboratory | JAX: 026942 |
| Mouse: Rosa26-CAG-loxp-stop-loxp-frt-stop-frt-hM4Di | The Jackson Laboratory | JAX: 029040 |
| Mouse: Rosa26-loxp-stop-loxp-frt-stop-frt-tdTomato | The Jackson Laboratory | JAX: 021875 |
| Mouse: Tau-loxp-stop-loxp-frt-stop-frt-DTR | Generated by Goulding lab (Bourane et al., 2015) | N/A |
| Software and Algorithms | ||
| GraphPad Prism 7.0 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| G*Power | University of Dusseldorf | http://www.gpower.hhu.de/ |
| FlowJo v10.4 | Tree Star | https://www.flowjo.com/solutions/flowjo |
| ImageJ 1.51 s | National Institute of Health | https://imagej-nih-gov.ezp-prod1.hul.harvard.edu/ij/download.html |
| SigmaStat 3.5 | Systat Software | https://systatsoftware.com/products/sigmastat/ |
| Other | ||
| LSR-II flow cytometer | BD Biosciences | N/A |
| NanoDrop | Fisher Scientific | ND-2000 |
| Electric stimulator | A-M Systems | Model 3800 |
| Isolator | A-M Systems | Model #3820 |
| PD MidiTrapG-25 column | GE Healthcare | 28-9180-07 |
| Microtiter plate reader | Tecan | N/A |
| Picospritzer III | Parker Hannifin | N/A |
| AXIO IMAGER Z1 microscope | Zeiss | N/A |
| LSM700 laser-scanning confocal microscope | Zeiss | N/A |
HIGHLIGHTS:
Intersectional genetic manipulation of NPY+ sympathetic cells
Electroacupuncture stimulation (ES) drives distinct sympathetic pathways
ES operates in somatotopy- and intensity-dependent manners
NPY+ noradrenergic neurons bidirectionally modulate systemic inflammation
Acknowledgements
We thank Drs. Bing Zhu, Isaac M. Chiu and Charles D. Stiles for helpful comments, and thank Martyn Goulding, Susan M. Dymecki, and the Allen Brain Institute/the Jackson Laboratory/MMRRC at University of Davis for genetically modified mice. S.L.’s salary was partly supported by the China Postdoctoral Science Foundation (KLF101846) and by Development Project of Shanghai Peak Disciplines-Integrated Medicine (20150407). Y.S.’s salary was supported by China Scholarship Council (CSC NO. 201609110039). Z.W.’s salary was partly supported from Fujian University of Traditional Chinese Medicine. Histochemical imaging was supported by Boston Children’s Hospital IDDRC (1U54HD090255). The work was supported by NIH grants (R01AT010629), the Harvard/MIT Joint Research Program in Basic Neuroscience, and the Wellcome Trust grant (200183/Z/15/Z) to Q.M..
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
The authors declare no competing interest.
REFERENCES
- Abe C, Inoue T, Inglis MA, Viar KE, Huang L, Ye H, Rosin DL, Stornetta RL, Okusa MD, and Guyenet PG (2017). C1 neurons mediate a stress-induced anti-inflammatory reflex in mice. Nat Neurosci 20, 700–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agac D, Estrada LD, Maples R, Hooper LV, and Farrar JD (2018). The beta2-adrenergic receptor controls inflammation by driving rapid IL-10 secretion. Brain Behav Immun 74, 176–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon U (2007). Network motifs: theory and experimental approaches. Nat Rev Genet 8, 450–461. [DOI] [PubMed] [Google Scholar]
- Archer T, and Fredriksson A (2000). Effects of clonidine and alpha-adrenoceptor antagonists on motor activity in DSP4-treated mice I: dose-, time- and parameter-dependency. Neurotoxicity research 1, 235–247. [DOI] [PubMed] [Google Scholar]
- Armbruster BN, Li X, Pausch MH, Herlitze S, and Roth BL (2007). Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A 104, 5163–5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutler B (2000). Endotoxin, toll-like receptor 4, and the afferent limb of innate immunity. Curr Opin Microbiol 3, 23–28. [DOI] [PubMed] [Google Scholar]
- Borges da Silva H, Fonseca R, Pereira RM, Cassado Ados A, Alvarez JM, and D’Imperio Lima MR (2015). Splenic Macrophage Subsets and Their Function during Blood-Borne Infections. Front Immunol 6, 480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borovikova L, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, and Tracey KJ (2000). Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 405, 458–462. [DOI] [PubMed] [Google Scholar]
- Bourane S, Grossmann KS, Britz O, Dalet A, Del Barrio MG, Stam FJ, Garcia-Campmany L, Koch S, and Goulding M (2015). Identification of a spinal circuit for light touch and fine motor control. Cell 160, 503–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratton BO, Martelli D, McKinley MJ, Trevaks D, Anderson CR, and McAllen RM (2012). Neural regulation of inflammation: no neural connection from the vagus to splenic sympathetic neurons. Exp Physiol 97, 1180–1185. [DOI] [PubMed] [Google Scholar]
- Bronte V, and Pittet MJ (2013). The spleen in local and systemic regulation of immunity. Immunity 39, 806–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuchars SA, and Lall VK (2015). Sympathetic preganglionic neurons: properties and inputs. Compr Physiol 5, 829–869. [DOI] [PubMed] [Google Scholar]
- Duan B, Cheng L, Bourane S, Britz O, Padilla C, Garcia-Campmany L, Krashes M, Knowlton W, Velasquez T, Ren X, et al. (2014). Identification of spinal circuits transmitting and gating mechanical pain. Cell 159, 1417–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes ES, Liang L, Smillie SJ, Kaiser F, Purcell R, Rivett DW, Alam S, Howat S, Collins H, Thompson SJ, et al. (2012). TRPV1 deletion enhances local inflammation and accelerates the onset of systemic inflammatory response syndrome. J Immunol 188, 5741–5751. [DOI] [PubMed] [Google Scholar]
- Flierl MA, Rittirsch D, Nadeau BA, Chen AJ, Sarma JV, Zetoune FS, McGuire SR, List RP, Day DE, Hoesel LM, et al. (2007). Phagocyte-derived catecholamines enhance acute inflammatory injury. Nature 449, 721–725. [DOI] [PubMed] [Google Scholar]
- Furlan A, La Manno G, Lubke M, Haring M, Abdo H, Hochgerner H, Kupari J, Usoskin D, Airaksinen MS, Oliver G, et al. (2016). Visceral motor neuron diversity delineates a cellular basis for nipple- and pilo-erection muscle control. Nat Neurosci 19, 1331–1340. [DOI] [PubMed] [Google Scholar]
- Gao X, Zhao Y, Su Y, Liu K, Yu X, Cui C, Yang Z, Shi H, Jing X, and Zhu B (2016). beta1/2 or M2/3 Receptors Are Required for Different Gastrointestinal Motility Responses Induced by Acupuncture at Heterotopic or Homotopic Acupoints. PLoS One 11, e0168200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Paletzki R, and Heintz N (2013). GENSAT BAC cre-recombinase driver lines to study the functional organization of cerebral cortical and basal ganglia circuits. Neuron 80, 1368–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu G, Zhang Z, Wang G, Han F, Han L, Wang K, Liu J, and Li W (2011). Effects of electroacupuncture pretreatment on inflammatory response and acute kidney injury in endotoxaemic rats. J Int Med Res 39, 1783–1797. [DOI] [PubMed] [Google Scholar]
- Huang CL, Tsai PS, Wang TY, Yan LP, Xu HZ, and Huang CJ (2007). Acupuncture stimulation of ST36 (Zusanli) attenuates acute renal but not hepatic injury in lipopolysaccharide-stimulated rats. Anesth Analg 104, 646–654. [DOI] [PubMed] [Google Scholar]
- Huang JL, Zhang YL, Wang CC, Zhou JR, Ma Q, Wang X, Shen XH, and Jiang CL (2012). Enhanced phosphorylation of MAPKs by NE promotes TNF-alpha production by macrophage through alpha adrenergic receptor. Inflammation 35, 527–534. [DOI] [PubMed] [Google Scholar]
- Huang S, Ziegler CGK, Austin J, Mannoun N, Vukovic M, Ordovas-Montanes J, Shalek AK, and von Andrian UH (2019). Sensory Neurons Innervate Peripheral Lymph Nodes and Locally Regulate Gene Expression in Postsynaptic Endothelium, Stromal Cells, and Innate Leukocytes. bioRxiv. doi: 10.1101/833509 [DOI] [Google Scholar]
- Hunt SP, Pini A, and Evan G (1987). Induction of c-fos-like protein in spinal cord neurons following sensory stimulation. Nature 328, 632–634. [DOI] [PubMed] [Google Scholar]
- Huston JM, Ochani M, Rosas-Ballina M, Liao H, Ochani K, Pavlov VA, Gallowitsch-Puerta M, Ashok M, Czura CJ, Foxwell B, et al. (2006). Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med 203, 1623–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iskander KN, Osuchowski MF, Stearns-Kurosawa DJ, Kurosawa S, Stepien D, Valentine C, and Remick DG (2013). Sepsis: multiple abnormalities, heterogeneous responses, and evolving understanding. Physiological reviews 93, 1247–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janig W, and Habler HJ (2000). Specificity in the organization of the autonomic nervous system: a basis for precise neural regulation of homeostatic and protective body functions. Prog Brain Res 122, 351–367. [DOI] [PubMed] [Google Scholar]
- Ji RR, Baba H, Brenner GJ, and Woolf CJ (1999). Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci 2, 1114–1119. [DOI] [PubMed] [Google Scholar]
- John L, Nastaran H & Dean M Electrically evoked compound action potential recording in peripheral nerves. Bioelectronics in medicine 2018. 1(1), 71–83. [Google Scholar]
- Johnson EM Jr., and O’Brien F (1976). Evaluation of the permanent sympathectomy produced by the administration of guanethidine to adult rats. J Pharmacol Exp Ther 196, 53–61. [PubMed] [Google Scholar]
- Kadl A, Pontiller J, Exner M, and Leitinger N (2007). Single bolus injection of bilirubin improves the clinical outcome in a mouse model of endotoxemia. Shock 28, 582–588. [DOI] [PubMed] [Google Scholar]
- Kolibal-Pegher S, Edwards DJ, Meyers-Schoy SA, and Vollmer RR (1994). Adrenal medullary adaptations and cardiovascular regulation after 6-hydroxydopamine treatment in rats. J Auton Nerv Syst 48, 113–120. [DOI] [PubMed] [Google Scholar]
- Komegae EN, Farmer DGS, Brooks VL, McKinley MJ, McAllen RM, and Martelli D (2018). Vagal afferent activation suppresses systemic inflammation via the splanchnic antiinflammatory pathway. Brain Behav Immun 73, 441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YQ, Zhu B, Rong PJ, Ben H, and Li YH (2007). Neural mechanism of acupuncture-modulated gastric motility. World J Gastroenterol 13, 709–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HD, Kim MH, Lee CY, and Namgung U (2016). Anti-Inflammatory Effects of Acupuncture Stimulation via the Vagus Nerve. PLoS One 11, e0151882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima D (2008). Ascending Pathways: Anatomy and Physiology. The Senses: A Comprehensive Reference 5, 477–526. [Google Scholar]
- Liu Y, Abdel Samad O, Duan B, Zhang L, Tong Q, Ji RR, Lowell B, and Ma Q (2010). VGLUT2-dependent glutamate release from peripheral nociceptors is required to sense pain and suppress itch. Neuron 68, 543–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Yang FC, Okuda T, Dong X, Zylka MJ, Chen CL, Anderson DJ, Kuner R, and Ma Q (2008). Mechanisms of compartmentalized expression of Mrg class G-protein-coupled sensory receptors. J Neurosci 28, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhurst JC (2010). Defining meridians: a modern basis of understanding. J Acupunct Meridian Stud 3, 67–74. [DOI] [PubMed] [Google Scholar]
- Martelli D, Farmer DG, and Yao ST (2016). The splanchnic anti-inflammatory pathway: could it be the efferent arm of the inflammatory reflex? Exp Physiol 101, 1245–1252. [DOI] [PubMed] [Google Scholar]
- Martelli D, Farmer DGS, McKinley MJ, Yao ST, and McAllen RM (2019). Antiinflammatory reflex action of splanchnic sympathetic nerves is distributed across abdominal organs. Am J Physiol Regul Integr Comp Physiol 316, R235–R242. [DOI] [PubMed] [Google Scholar]
- Martelli D, Yao ST, McKinley MJ, and McAllen RM (2014). Reflex control of inflammation by sympathetic nerves, not the vagus. J Physiol 592, 1677–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinoff PB (1984). Alpha- and beta-adrenergic receptor subtypes properties, distribution and regulation. Drugs 28 Suppl 2, 1–15. [DOI] [PubMed] [Google Scholar]
- Nielsen GD (1977). Guanethidine induced sympathectomy in the adult rat. II. Functional effects following chronic administration. Acta Pharmacol Toxicol (Copenh) 41, 209–217. [DOI] [PubMed] [Google Scholar]
- O’Donnell SR, and Wanstall JC (1980). Evidence that ICI 118, 551 is a potent, highly Beta 2-selective adrenoceptor antagonist and can be used to characterize Beta-adrenoceptor populations in tissues. Life Sci 27, 671–677. [DOI] [PubMed] [Google Scholar]
- Pavlov VA, and Tracey KJ (2017). Neural regulation of immunity: molecular mechanisms and clinical translation. Nat Neurosci 20, 156–166. [DOI] [PubMed] [Google Scholar]
- Pereira MM, Mahu I, Seixas E, Martinez-Sanchez N, Kubasova N, Pirzgalska RM, Cohen P, Dietrich MO, Lopez M, Bernardes GJ, et al. (2017). A brain-sparing diphtheria toxin for chemical genetic ablation of peripheral cell lineages. Nat Commun 8, 14967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho-Ribeiro FA, Baddal B, Haarsma R, O’Seaghdha M, Yang NJ, Blake KJ, Portley M, Verri WA, Dale JB, Wessels MR, et al. (2018). Blocking Neuronal Signaling to Immune Cells Treats Streptococcal Invasive Infection. Cell 173, 1083–1097 e1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remick DG, Newcomb DE, Bolgos GL, and Call DR (2000). Comparison of the mortality and inflammatory response of two models of sepsis: lipopolysaccharide vs. cecal ligation and puncture. Shock 13, 110–116. [DOI] [PubMed] [Google Scholar]
- Rosas-Ballina M, Ochani M, Parrish WR, Ochani K, Harris YT, Huston JM, Chavan S, and Tracey KJ (2008). Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc Natl Acad Sci U S A 105, 11008–11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouot B, Quennedey MC, and Schwartz J (1982). Characteristics of the [3H]-yohimbine binding on rat brain alpha2-adrenoceptors. Naunyn Schmiedebergs Arch Pharmacol 321, 253259. [DOI] [PubMed] [Google Scholar]
- Salomao R, Martins PS., Brunialti MK, Fernandes Mda, L., Martos LS, Mendes ME., Gomes NE, and Rigato O. (2008). TLR signaling pathway in patients with sepsis. Shock 30 Suppl 1, 73–77. [DOI] [PubMed] [Google Scholar]
- Sato A (1997). Neural mechanisms of autonomic responses elicited by somatic sensory stimulation. Neurosci Behav Physiol 27, 610–621. [DOI] [PubMed] [Google Scholar]
- Sell R, Erdelyi A, and Schaefer H (1958). [Effect of peripheral nerve stimulation on sympathetic activity]. Pflugers Arch Gesamte Physiol Menschen Tiere 267, 566–581. [DOI] [PubMed] [Google Scholar]
- Song JG, Li HH, Cao YF, Lv X, Zhang P, Li YS, Zheng YJ, Li Q, Yin PH, Song SL, et al. (2012). Electroacupuncture improves survival in rats with lethal endotoxemia via the autonomic nervous system. Anesthesiology 116, 406–414. [DOI] [PubMed] [Google Scholar]
- Spengler RN, Chensue SW, Giacherio DA, Blenk N, and Kunkel SL (1994). Endogenous norepinephrine regulates tumor necrosis factor-alpha production from macrophages in vitro. J Immunol 152, 3024–3031. [PubMed] [Google Scholar]
- Staedtke V, Bai RY, Kim K, Darvas M, Davila ML, Riggins GJ, Rothman PB., Papadopoulos N, Kinzler KW, Vogelstein B, et al. (2018). Disruption of a self-amplifying catecholamine loop reduces cytokine release syndrome. Nature 564, 273–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JJ, and Ray R (2016). Generation of Two Noradrenergic-Specific Dopamine-Beta-Hydroxylase-FLPo Knock-In Mice Using CRISPR/Cas9-Mediated Targeting in Embryonic Stem Cells. PLoS One 11, e0159474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szelenyi J, Kiss JP, and Vizi ES (2000). Differential involvement of sympathetic nervous system and immune system in the modulation of TNF-alpha production by alpha2- and beta-adrenoceptors in mice. J Neuroimmunol 103, 34–40. [DOI] [PubMed] [Google Scholar]
- Thoenen H, and Tranzer JP (1973). The pharmacology of 6-hydroxydopamine. Annu Rev Pharmacol 13, 169–180. [DOI] [PubMed] [Google Scholar]
- Torres-Rosas R, Yehia G, Pena G, Mishra P, del Rocio Thompson-Bonilla M, Moreno-Eutimio MA, Arriaga-Pizano LA, Isibasi A, and Ulloa L (2014). Dopamine mediates vagal modulation of the immune system by electroacupuncture. Nat Med 20, 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travagli RA, and Anselmi L (2016). Vagal neurocircuitry and its influence on gastric motility. Nat Rev Gastroenterol Hepatol 13, 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulloa L, Quiroz-Gonzalez S, and Torres-Rosas R (2017). Nerve Stimulation: Immunomodulation and Control of Inflammation. Trends Mol Med 23, 1103–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Poll T, van de Veerdonk FL, Scicluna BP, and Netea MG (2017). The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol 17, 407–420. [DOI] [PubMed] [Google Scholar]
- Vida G, Pena G, Kanashiro A, Thompson-Bonilla Mdel R, Palange D, Deitch EA, and Ulloa L (2011). beta2-Adrenoreceptors of regulatory lymphocytes are essential for vagal neuromodulation of the innate immune system. FASEB J 25, 4476–4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villegas-Bastida A, Torres-Rosas R, Arriaga-Pizano LA, Flores-Estrada J, Gustavo-Acosta A, and Moreno-Eutimio MA (2014). Electrical Stimulation at the ST36 Acupoint Protects against Sepsis Lethality and Reduces Serum TNF Levels through Vagus Nerve- and Catecholamine-Dependent Mechanisms. Evid Based Complement Alternat Med 2014, 451674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranjkovic O, Hang S, Baker DA, and Mantsch JR (2012). beta-adrenergic receptor mediation of stress-induced reinstatement of extinguished cocaine-induced conditioned place preference in mice: roles for betal and beta2 adrenergic receptors. J Pharmacol Exp Ther 342, 541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe D, Forrest LR, and Schuldiner S (2018). The ins and outs of vesicular monoamine transporters. J Gen Physiol 150, 671–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao YX, He W, Jing XH, Liu JL, Rong PJ, Ben H, Liu K, and Zhu B (2012). Transcutaneous auricular vagus nerve stimulation protects endotoxemic rat from lipopolysaccharide-induced inflammation. 2012 Evid Based Complement Altemat Med. 2012;2012:627023, 627023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.