

Abstract
The molecular alterations that initiate the development of multiple myeloma (MM) are not fully understood. Our results revealed that TJP1 was downregulated in MM and positively related to the overall survival of MM patients in The Cancer Genome Atlas (TCGA) database and patient samples. In parallel, cell adhesion capacity representing MM metastasis was decreased in MM patients compared with healthy samples, together with the significantly activated epithelial-to-mesenchymal transition (EMT) transcriptional-like patterns of MM cells. Further analyses demonstrated that TJP1 negatively regulated EMT and consequently positively regulated cell adhesion in MM from TCGA database and MM1s cells. Furthermore, the methylation level of each CpG site on the TJP1 promoter was negatively correlated with TJP1 expression levels. Quantitative real-time PCR and western blot assays demonstrated that methylase DNMT1 regulated the methylation of TJP1. Finally, treatment with a combination of the MM clinical medicine bortezomib, methylation inhibitor, or TJP1 overexpression significantly suppressed the viability and progression of tumor cells of MM orthotopic models. In summary, our results indicate that DNMT1 promotes the methylation of TJP1 promoter, thereby decreasing its expression and regulating the development of EMT-inhibited MM cell adhesion. Therefore, methylation of TJP1 is a potential therapeutic agent to prevent the progression of MM disease.
Keywords: multiple myeloma, epithelial-to-mesenchymal-transition, TJP1, methylation, DNMT1, Cell adhension
Graphical Abstract
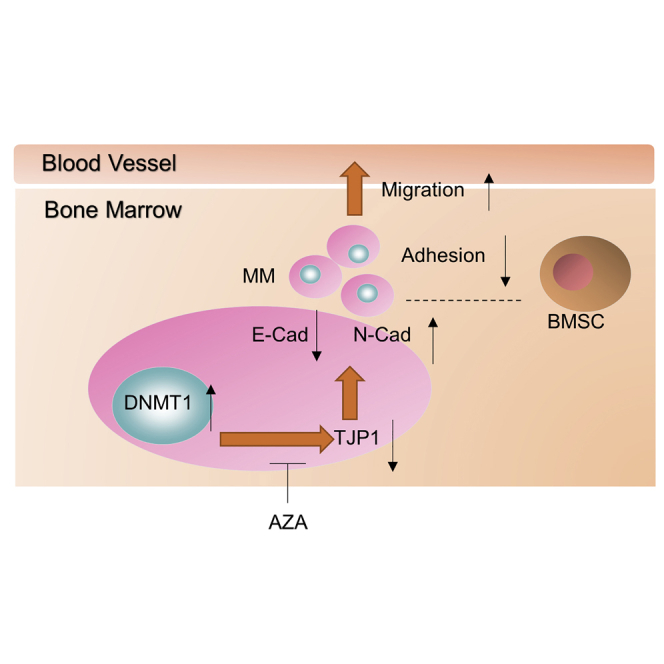
We demonstrate that DNA methyltransferases contribute to the downregulation of TJP1 by DNMT1, thus subsequently promoting the development of EMT-mediated MM progression and the BM microenvironment, leading to MM metastasis. We believe that TJP1 expression and methyltransferase inhibitors would facilitate therapies combined with bortezomib in clinical practice.
Introduction
Multiple myeloma (MM) is an incurable disorder, characterized by clonal multiplication of malignant plasma cells. Despite the recent development of multiple anti-MM agents with better overall survival rates,1 MM still poses a great challenge in its clinical management. With the rapid increase in the incidence of MM, novel and molecular therapies against MM are urgently needed.
Epithelial-to-mesenchymal transition (EMT) features are fundamental for embryonic development and involve changes such as the loss of cell-cell adhesion and the acquisition of migratory and invasive properties.2,3 In cancer, the acquisition of EMT features has been associated with carcinogenesis, invasion, metastasis, stem cell perturbation, and tumor recurrence.4,5 Whereas EMT was a well-recognized phenomenon in solid tumors, recent studies have reported existence of EMT-like features in MM.6,7 For instance, differential expression of EMT-related markers in aberrant conditions induces dissemination of MM cells and thus metastasis to the new bone marrow (BM) regions.8 EMT-mediated C-X-C motif chemokine receptor 4 (CXCR4) mediates migration and homing of MM cells into the BM to enhance metastasis in both solid tumors and MM.6,8, 9, 10 The MM cells with migration activity triggered by EMT thus ultimately translate into the development of disease, drug resistance, metastasis, and relapse of MM.11,12 However, the impact on initiating EMT features to activate the metastasis and invasion in MM and whether it can be therapeutically targeted to improve the survival of MM patients remains poorly explored.
On the other hand, the expression of tight junction protein 1 (TJP1) is suppressed in activated EMT, triggered by transforming growth factor-β (TGF-β).13 TJP1 belongs to the membrane guanylate kinase family, and it encodes a tight junction protein involved in cell proliferation and differentiation.14,15 In addition, TJP1 forms barriers between adjacent cells to control the transport of water, ions, and macromolecules.16 Loss of tight junction barrier function due to TJP1 dysfunction has been associated with progression and metastasis of many cancers, including MM.17, 18, 19, 20, 21 Furthermore, TJP1 promotes actin stress fiber assembly during EMT.22 Our previous study and that of others showed that TJP1 expression is a biomarker for proteasome inhibitor sensitivity in myeloma.23,24 We then hypothesized that downregulation of TJP1 might suppress MM development via inhibition of EMT. Data on whether TJP1 expression is aberrant and the relative mechanisms in MM remain scant.
In addition, aberrant DNA methylation has been shown to be a marker of cancer.25, 26, 27, 28, 29 Under normal conditions, DNA methylation patterns ensure proper regulation of gene expression and stable gene silencing. However, in the development of cancer, several tumor-suppressor genes were shown to be silenced by abnormal DNA methylation.30,31 Furthermore, aberrant hypermethylation of the promoter region of TJP1 was observed in murine leukemia cell lines accompanied by the concomitant suppression of the TJP1 expression.27 How the TJP1 is aberrantly methylated in MM is still not fully understood. DNA methyltransferases (DNMTs) are responsible for the establishment and maintenance of methylation patterns. For example, inhibition of DNMT1 could trigger re-expression of tumor suppressor genes, which were silenced by methylation, and induce cell proliferation in human cancers.32, 33, 34, 35 DNMT1-mediated MM progression via methylation has been reported.36 The relationship between DNA methyltransferases and methylation of TJP1 needs further interrogation.
Here, we speculated that DNA methyltransferases contribute to the downregulation of TJP1, thus regulating the development of EMT-mediated MM progression. We show that TJP1 plays an important role in activating EMT in the BM microenvironment, leading to MM metastasis. Tumor progression was significantly suppressed by the MM clinical medicine bortezomib, methylation inhibitor 5-Aza-2′-deoxycytidine (AZA), or TJP1 cDNA, or their combination. These results highlight important molecular aspects that would facilitate the development of therapies for MM.
Results
TJP1 and TJP1-Regulated EMT Were Linked to MM Cell Adhesion and Overall Survival of MM Patients
We previously demonstrated that TJP1 expression is a biomarker of proteasome inhibitor sensitivity in myeloma.24 However, the regulation mechanism of TJP1 in MM progression has yet to be well understood. We analyzed the relationship between TJP1 expression and overall survival in MM patients from the TCGA database (https://hgserver1.amc.nl/cgi-bin/r2/main.cgi). Notably, we found that upregulation of TJP1 expression reflected a high overall survival rate; thus, TJP1 could be a tumor suppressor gene to MM (p < 0.01) (Figures 1A and S1A). Furthermore, mRNA expression of the TJP1 in healthy donors was higher than in MM patients (p < 0.01) (Figure 1B). Generally, much lower levels of TJP1 were detectable in the MM tissues with higher pathologic stages (Table S1). We also found that the ability of MM cells to adhere to bone marrow stroma cells (BMSCs) was lower than the healthy cells (p < 0.01) (Figure 1C). Furthermore, we checked the correlation between the expression level of TJP1 to the adhesion of MM cells to BMSCs in MM cells derived from 40 patients, and the result showed that TJP1 expression was positively correlated with the adhesion of patients’ MM cells to BMSCs (Figure 1D). Since TJP1 has played an important role in EMT, we further investigated the potential expression differences of EMT-like features in MM patients against the normal cells. To investigate the biological functions of TJP1, we analyzed the gene expression profile in MM patients from the GEO database by Gene Set Enrichment Analysis (GSEA). Interestingly, downregulated EMT-like gene sets were significantly enriched in the cells with high TJP1 expression (p < 0.01) (Figures 1E and S1B). Besides, we found positive correlation between TJP1 and E-cadherin mRNA expression in MM patients (Figure 1F). Furthermore, we randomly picked 1 control sample from healthy donors and 2 samples from MM patients to check the mRNA and protein expression of the EMT-related makers. We found that TJP1 and E-cadherin expression were downregulated while the expression of N-cadherin was increased in MM (p < 0.01) (Figures 1G–1I). Together, these results showed that EMT was activated by the downregulation of TJP1 to suppress cell adhesion in MM patients.
Figure 1.

TJP1 and TJP1-Regulated EMT Were Linked to Cell Adhesion and Overall Survival of MM Patients
(A) Relationship between TJP1 expression (Probe 202011_at) and overall survival in MM patients was analyzed in TCGA database on R2 website. (B) mRNA expression of TJP1 between plasma cells from 5 healthy participants and tumor cells from 40 MM patients as assessed by quantitative real-time PCR. The y axis is plotted on a logarithmic scale. Relative mRNA expression of TJP1 in corresponding normal plasma cells was normalized to 1. (C) The adhesion ability of MM cells to BMSCs was compared to normal healthy cells. The y axis is plotted on a logarithmic scale. The adhesion ability of cells in corresponding normal plasma cells was normalized to 1. (D) Correlation between the expression level of TJP1 to the adhesion of 40 patients’ MM cells to BMSCs. (E) The enrichment of EMT-like gene sets between TJP1High and TJP1Low expression in MM patients from the GEO database (GEO: GSE4581) in differentiation signatures (gene sets: JECHLINGER_EPITHELIAL_TO_MESENCHYMAL_TRANSITION,GOTZMANN_EPITHELIAL_TO_MESENCHYMAL_TRANSITION_DN) was determined with GSEA analysis. (F) Correlation between E-cadherin and TJP1 expression in MM patients as determined by ggstatsplot. T (557) = 3.60, p < 0.0001, RPearson = 0.15, CI95%[0.07, 0.23], Npairs = 559. (G) Protein expression of EMT-related markers were measured in 1 control sample and 2 MM samples by western blot. Relative intensity of EMT-related markers in control sample were normalized to 100%. (H) mRNA expression of EMT-related markers were measured in 1 control sample and 2 MM samples by quantitative real-time PCR. Relative mRNA expression of EMT-related markers in control sample were normalized to 100%. (I) Representative images of immunofluorescence staining for EMT-related markers in 1 normal plasma control sample and 2 MM samples. All data were presented as mean ± SD from 3 independent experiments. Statistical analysis was performed with a paired Student’s t test. ∗∗p < 0.01 comparing with control group.
TJP1 Improves Adhesion Capacity of MM Cells to the BM Stroma via EMT
To further investigate whether TJP1 regulated EMT and cell adhesion to BMSCs in MM, we tested the expression of EMT-related proteins after knockdown or overexpression of TJP1 in the MM1s cells. The results showed that the knockdown of TJP1 resulted into silencing of both the TJP1 and E-cadherin mRNA or protein expression while upregulating the mRNA and protein expression of N-cadherin in MM1s cells (p < 0.01) (Figures 2A, 2B, and 2E). On the other hand, overexpression of TJP1 increased the mRNA and protein expression of both TJP1 and E-cadherin but inhibited the expression of N-cadherin in MM1s cells (p < 0.01) (Figures 2C, 2D, and 2F). The results of the relationship between TJP1 and EMT were consistent with the data in MM patients. In addition, to better understand the role of the TJP1 gene on suppressing adhesion through the EMT axis, we tested the adhesion capacity of MM cells to BMSCs. The data showed that the adhesion of MM1s was inhibited by TJP1 knockdown but was increased by TJP1 overexpression (p < 0.01) (Figures 2G and 2H). Furthermore, we used EMT inhibitor (C19) to treat MM1s cells bearing silence of TJP1 and showed that the adhesion capacity was rescued by EMT inhibitor (C19). We also used EMT activator TGF-β to confirm in TJP1-overexpressed cells, and these results confirmed that TJP1 regulated adhesion via EMT (Figures 2G and 2H).
Figure 2.

TJP1 Improved Adhesion of MM Cells to BM Stroma via EMT In Vitro
(A–D) The mRNA and protein levels of EMT-related genes after knockdown (A and B) and overexpression (C and D) of TJP1 as measured by real-time PCR and western blot (n = 3, ∗∗p < 0.01). (E) The expression of EMT-related protein E-cadherin after knockdown of TJP1 as measured by IF staining (60×). (F) The expression of EMT-related protein E-cadherin after overexpression of TJP1 as measured by IF staining (60×). (G) With treatment of MM1s cells with EMT inhibitor (C19), the adhesion of MM1s cells to BMSCs was detected with TJP1 knockdown. (H) With treatment of MM1s cells with EMT activator TGF-β, the adhesion of MM1s cells to BMSCs was detected with TJP1 overexpression. All data were presented as the mean ± SD from 3 independent experiments. Statistical analysis was performed with a paired Student’s t test. ∗∗p < 0.01 comparing with control group, ##p < 0.01 comparing with treatment group.
Hypermethylation of the Promoter Region Contributes to Aberrant Low Expression of TJP1 in Myeloma Cells
A previous study reported that downregulation of TJP1 was related to methylation.27 Here, we compared the expression of TJP1 in MM patient samples and healthy plasma cells. Our data showed lower mRNA or protein expression of the TJP1 gene in MM patient cells compared to the control group (p < 0.01) (Figures 3A and 3B). In order to identify the CpG islands of TJP1, we obtained the upstream 2,000 bp promoter sequence of TJP1 (selected area: chr15: 29822504–29824503) using the University of California, Santa Cruz (UCSC) Genome Browser (http://genome.ucsc.edu/index.html). Furthermore, we predicted the CpG islands by using the MethPrimer 2.0 (http://www.urogene.org/methprimer2/). Our findings showed that the two CpG islands of TJP1 were located in chr15: 29822791–29823050 (Island 1: 260 bp) and chr15: 29822510–29822715 (Island 2: 206 bp), respectively (Figure S2A). We showed that higher methylation levels existed in 4 MM tissues compared to those in 4 healthy plasma tissues in the upstream TJP1 promoter region in the tested 20 CpG sites (p < 0.01) (Figures 3C, 3D, and S2B). To further confirm whether TJP1 expression was silenced by methylation, we performed a demethylation test in the MM cells by AZA. As shown in Figures 3A and 3B, AZA treatment significantly increased both the mRNA and protein expression of the TJP1 gene. CpG methylation level was inhibited by AZA treatment (p < 0.01) (Figures 3C and 3D). Furthermore, as TJP1 had three transcriptional start sites (TSSs), corresponding to variants 1, 5, and 7, respectively, within the CpG island (chr15: 29821503–29823012, HG38, UCSC Genome Browser), we designed the universal (TSS1-3 primer to amplify sequences of exon 5, which is included in all variants of TJP1) or specific quantitative real-time PCR primers (TSS1 primer for variant 1and TSS2 primer for variant 7) to identify which TSSs were reactivated by AZA (Figure S3A). As shown in Figure S3B, the mRNA level of variant 1 or 7 of TJP1 was significantly upregulated by AZA treatment, as well as the mRNA level of TJP1 measured by universal quantitative real-time PCR primers, suggesting the TSS corresponding to TJP1 transcripts 1 and 7 within the CpG island could become activated after AZA treatment.
Figure 3.

Hypermethylation of the Promoter Region Contributed to the Aberrant Low Expression of TJP1 in Myeloma Cells
(A) The protein expression of the TJP1 gene were tested in healthy plasma cells and MM patient samples by western blot. (B) The mRNA expression of the TJP1 gene were tested in healthy plasma cells and MM patient samples by quantitative real-time PCR. (C) Methylation of multiple CpG sites on CpGs in the upstream promoter region of TJP1 was tested in healthy plasma cells and MM patient samples by AZA by BSP methylation analysis. Black circles signify methylated CpGs, and white circles indicate demethylated CpGs. (D) Methylation rates was analyzed with the methylation of multiple CpG sites on CpGs in the upstream promoter region of TJP1. (E) DNA methylation level of 2 CpG sites (CpG 6 and CpG 12) in the promoter of TJP1 in 40 MM samples and 4 healthy clinical samples was determined by BSP. (F) The correlation between the methylation rates at CpG 6 and the TJP1 mRNA expression was determined in the 40 MM patient samples. (G) The correlation between the methylation rates at CpG 12 and the TJP1 mRNA expression was determined in the 40 MM patient samples. (H) Different deletion mutants were constructed from fragments ranging from 0 to −2,000, as shown in the left panel. These deletion mutants together with pGL3-Basic were individually transfected into HEK293T cells, and their promoter activity was measured. Cells were treated with β-galactosidase for 24 h, after which luciferase activity in cell lysates was determined. All data were presented as mean ± SD from 3 independent experiments. Statistical analysis was performed with a paired Student’s t test. ∗∗p < 0.01 comparing with control group, ##p < 0.01 comparing with treatment group.
In addition, we used bisulfite sequencing PCR (BSP) to measure DNA methylation levels of the 2 CpG sites (CpG 6, CpG 12) in the TJP1 promoter. We observed that MM tissues had greater methylation levels compared with the healthy plasma at CpG 6 and CpG 12 sites (Figure 3E). The methylation levels at each of the CpG sites was found to be negatively correlated with TJP1 expression levels (p < 0.01) (Figures 3F and 3G).
To pinpoint the differential fragment, different deletion mutants were further constructed based on fragments 0 to −2,000. These deletion mutants, together with pGL3-Basic vector, were individually transfected into HEK293T cells, and the promoter activity was measured. We found that the fragments 0 to −2,000, 0 to −566, 0 to −283, and −283 to −2,000 displayed higher promoter activity than other fragments (Figure 3H). Thus, our results demonstrate that the core transcription site of the TJP1 promoter region is located between 0 and −566, which contains 2 CpG islands at the start of the TJP1 promoter. Furthermore, low luciferase activities were observed when the TJP1 promoter was methylated, especially in the fragments in the 2 CpG islands, indicating that methylation suppresses the promoter activity (Figure 3H).
DNMT1-Mediated Hypermethylation of TJP1 Promoter Regulates EMT
To evaluate whether the expression of TJP1 was upregulated by methylase, including DNMT1, DNMT3a, or DNMT3b, we conducted both quantitative real-time PCR and western blot analysis and showed that overexpressed DNMT1 downregulated the mRNA and protein expression of the TJP1 gene as well as upregulated the EMT (p < 0.01) (Figures 4A and 4B). Neither DNMT3a nor DNMT3b affected the expression of TJP1 or EMT (Figure S4A). The CpG methylation level of the TJP1 promoter also increased with the DNMT1 overexpression (p < 0.01) (Figure 4C). To better understand the role of DNMT1 in the regulation of cell adhesion through TJP1, we tested the adhesion capacity of MM cells to BMSCs with DNMT1 overexpression. Our data showed that the adhesion of MM1s cells was inhibited by DNMT1 overexpression but was rescued by TJP1 overexpression (p < 0.01) (Figure 4D). On the other hand, we demonstrated that DNMT1 silencing could upregulate the mRNA and protein expression of TJP1 but downregulate EMT (p < 0.01) (Figures 4E and 4F). The CpG methylation level of TJP1 also decreased with the downregulation of DNMT1 (p < 0.01) (Figure 4G). Furthermore, the adhesion of MM1s was increased by DNMT1 knockdown but was rescued by TJP1 knockdown (p < 0.01) (Figure 4H). Finally, analysis of the relationship between DNMT1 and TJP1 expression in the MM patient database showed significant negative correlation between DNMT1 and TJP1 (p < 0.01) (Figure S4B). Interestingly, we also found that increased DNMT1 expression was associated with low overall survival rate in the MM patients, and upregulated EMT-like gene sets were significantly enriched in the patients with high DNMT1 expression (p < 0.01) (Figures S4C and S4D). These observations suggest that DNMT1 regulates the MM cell adhesion via methylation of the TJP1 gene.
Figure 4.

DNMT1 Induced Hypermethylation of TJP1 Promoter to Regulate EMT
(A) The western blot assays were performed to determine the expression of protein of DNMT1, TJP1, and E-cadherin after DNMT1 overexpression. (B) The quantitative real-time PCR assays were performed to determine the expression of mRNA of DNMT1, TJP1, and E-cadherin after DNMT1 overexpression. (C) The methylation level of CpG site on TJP1 following DNMT1 overexpression. (D) The adhesion of MM1s cells to BMSCs following DNMT1 overexpression and TJP1 overexpression. (E) The protein expression levels of DNMT1, TJP1, and E-cadherin were detected by western blot analysis after transfection with DNMT1 shRNAs. (F) The mRNA expression levels of DNMT1, TJP1, and E-cadherin were detected by quantitative real-time PCR after transfection with DNMT1 shRNAs. (G) The methylation level of CpG on the TJP1 after DNMT1 knockdown. (H) The adhesive ability of MM1s after DNMT1 inhibition with TJP1 knockdown. All data were presented as mean ± SD from 3 independent experiments. Statistical analysis was performed with a paired Student’s t test. ∗∗p < 0.01 comparing with control group, ##p < 0.01 comparing with treatment group.
Effects Associated with the Use of Methylation Inhibitor and/or TJP1 Overexpression with Bortezomib in MM
To examine whether methylation inhibitor AZA and/or TJP1 overexpression affect drug susceptibility of MM, we used AZA and/or adeno-associated viral (AAV) vector TJP1-cDNA with bortezomib for treatment. MM cells were sorted from patient samples and treated by AZA and/or TJP1 cDNA with bortezomib, which was an anti-cancer agent used in various stages of MM treatment. The results showed that the cell proliferation was inhibited by bortezomib, and AZA or TJP1 cDNA with bortezomib. The combination of AZA, TJP1-cDNA, and bortezomib robustly inhibited cell viability (p < 0.01) (Figure 5A). Furthermore, treatment of tumor-bearing severe combined immunodeficiency (SCID) mice with bortezomib, AZA, or TJP1 cDNA or their combination showed a decrease in the tumor volume in subcutaneous MM models (Figure 5B). We also showed that the use of bortezomib, AZA, or TJP1 cDNA or their combination prolonged the survival time of MM orthotopic models. However, the combination therapy yielded the most significant effect (p < 0.001) (Figure 5C).
Figure 5.

Effects of Combined Treatment of Methylation Inhibitor and/or TJP1 Overexpression with Bortezomib in MM In Vitro and In Vivo
(A) MM cells were obtained from 3 different MM patient samples and plated with 1 × 104 cells/well in 96-well plates for 72 h treated with AZA and/or TJP1-cDNA and bortezomib, and the cell viabilities of MM cells were detected by the WST-1 assay. (B) MM1s cells were injected to SCID mice to establish a subcutaneous MM model. These tumor-bearing mice were treated with bortezomib, AZA, or TJP1-cDNA or their combinations in vivo, and tumor volumes were measured and compared with control group. (C) The survival duration of MM orthotopic mice was recorded after treatment with bortezomib, AZA, or TJP1-cDNA or their combinations and compared with control group. All data in (A) are presented as mean ± SD from 3 independent experiments. A paired Student’s t test was performed for significance testing. ∗∗p < 0.01 comparing with control group, ##p < 0.01 and $$p < 0.01 comparing with treatment group.
Discussion
TJP1 protein is widely used as a maker of tight junctions and is known to promote tumor development in various types of cancer.22,37 Our recent study identified TJP1 as a biomarker for the susceptibility of proteasome inhibitors in MM patients.24 Downregulation of TJP1 expression is associated with disrupted epithelial intercellular junctions alongside E-cadherin during developmental EMT. The adhesion properties are important indicators of malignancy and spread of MM.38 However, data on whether TJP1 or EMT regulates adhesion in the mediation of MM progression are still limited. Here, we show that TJP1 and EMT are regulated by TJP1 and are linked to overall survival of MM patients. Methylation of the promoter region contributes to the aberrant low expression of TJP1, which then increases EMT but decreases the adhesion of MM cells. Furthermore, we investigated the effect of upregulated methylation of the TJP1 promoter by DNMT1 on the low expression of TJP1 in MM cells. Lastly, we treated MM cells and mice models by methylation inhibitor and/or TJP1-cDNA with bortezomib, and the results showed that a combination regimen of methylation inhibitor/TJP1-cDNA with bortezomib was the most effective in MM. It is worth mentioning that TJP1 mRNA and protein expression were much lower in MM compared with healthy samples. Thus, reduced TJP1 expression may be a molecular marker for poor prognosis of MM.
Most of the studies focused on how TJP1 regulates proliferation and cellular permeability to modulate proteasome capacity.39, 40, 41 We previously demonstrated that TJP1 modulates proteasome capacity and proteasome inhibitor sensitivity in MM via EGFR/JAK1/STAT3 signaling.24 However, the mechanism of action of the TJP1 in cancer progression is yet to be investigated. EMT is often observed in primary tumor at the cancer invasion front. Cancer cells lose their adhesive properties and acquire an enhanced mobility by undergoing EMT.42 Recently, junctional adhesion molecules (JAMs), adhesion and transmigration regulatory elements, have been reported to play a critical role in regulating tumor progression.43, 44, 45 MM progression occurs when there is inability to control MM cell adhesion, as well as ability of the MM cells to egress from the BM to the bloodstream and into new BM niches.8,46 TJP1 plays an important role in the EMT machinery, and alternative splicing of TJP1 promotes actin stress fiber assembly during EMT.22 In our study, we demonstrated that TJP1 and cell adhesion were significantly inhibited and EMT was elevated in MM. The EMT activator TGF-β attenuated the induced adhesion of MM by overexpression of TJP1, whereas the EMT inhibitor reversed the inhibition of cell adhesion of MM after TJP1 silencing. Therefore, the TJP1 gene plays an important role in the MM cell adhesion via EMT.
Whereas previous studies have interrogated TJP1 regulation of downstream pathways like EMT and tumorigenesis,23,24 few studies have evaluated the factors that regulate TJP1 expression. DNA methylation occurs by invertible addition of a methyl group to the cytosine residue of a cytosine-guanine dinucleotide (CpG), which is mainly located in the promoter region of the gene.47 Methylation of cytosines in CpG dinucleotides results in the transcriptional inactivity in mammalian genomes,48 and both DNA and histone methylation may regulate TJP1.27 In this study, we detected DNA hypermethylation in a new regulatory region, chr15: 29822791–29823050 and chr15: 29822510–29822715, within the TJP1 promoter, which was related to lower TJP1 mRNA expression in MM. Further, we proved that the methylation of the promoter region contributed to the aberrant low expression of TJP1 in MM cells and that DNA methylation might affect TJP1 methylation. We also identified a core transcription site of the TJP1 promoter region located between 0 and −566, which contained 2 CpG islands in the start of TJP1 promoter. In vitro methylation with Methyltransferase and S-adenosylmethionine, showed that methylation suppressed promoter activity. In addition, we detected three methyltransferases involved in methylation of the gene. Among them, DNMT1 was the only one shown to modulate TJP1 expression by affecting the CpG methylation of the promoter region in MM. The regulation of TJP1 by DNMT1 affects the EMT pathway, thus regulating MM cell adhesion.
Our data clearly confirmed that epigenetic modifications could affect TJP1 expression. The expression change of TJP1 regulates cell adhesion via EMT in MM. The methylation state of DNA is maintained in a dynamic homeostasis by DNMTs and translocation proteins (TETs). DNMTs catalyze and maintain the methylation state of CpGs in DNA, while the TETs induce demethylation. Although our results found significant upregulation of DNMT expression in MM and direct interactions of DNMT1 with the TJP1 promoter, the role of TETs in regulating TJP1 promoter methylation in MM is still undefined. Therefore, further studies will be necessary to evaluate whether TETs mediate methylation alteration of the TJP1 gene in MM.
In summary, this study robustly demonstrates that TJP1 expression is downregulated in MM clinical samples. TJP1 downregulation modulates EMT, which then suppresses the cell adhesion capacity, thus promoting tumor progression. In addition, the promoter region of the aberrantly expressed TJP1 was shown to be highly methylated by DNMT1 in MM. Methylation rates were negatively correlated with TJP1 mRNA expression levels in both MM cell lines and clinical samples. Methylation inhibitor/activator could rescue and reverse the phenomenon. Moreover, combination of methylation inhibitor and TJP1-cDNA with bortezomib may yield superior outcomes in the treatment of myeloma. Our ongoing studies will further address the inducing factors of DNMT1 in the regulation TJP1 methylation in myeloma.
Materials and Methods
Human Cell Lines and Primary Cells
The human MM cell line MM1s (ATCC: CRL-2974) was cultured in RPMI 1640, supplemented with 10% fetal bovine serum (FBS) (Hyclone, Logan, UT, USA). The study was approved by the Sun Yat-sen University Institutional Review Board, and written informed consent was obtained from all the patients in accordance with the Declaration of Helsinki. The demographic and clinical features of the patients are summarized in Table S1. Primary MM cells were obtained from BM samples using CD138 micro-bead selection (Milteny Biotec, Auburn, CA, USA) with more than 90% purity. The purity was confirmed by flow cytometric analysis with monoclonal antibody against human CD38-PE (BD Biosciences, San Jose, CA, USA). BMSC and plasma cells were obtained from healthy volunteers after Ficoll-Hypaque density sedimentation.
Quantitative Real-Time PCR
To detect mRNA expression in MM and normal cells, we performed quantitative real-time PCR as described previously.24 The qPCR primer TJP1 TSS1-3 primer was used to detected all transcripts of TJP1 located in the exon 5 region (TJP1 TSS1-3 sequence: Forward primer: 5'-ACCAGTAAGTCGTCCTGATCC-3'; Reverse primer: 5'-TCGGCCAAATCTTCTCACTCC-3'). Two other TJP1 TSS1 and TSS2 primers, which were used to detect TJP1 variant 1 (TJP1 TSS1) and variant 7 (TJP1 TSS2) uniquely, are listed in Table S2.
Western Blot
Protein lysates were prepared and analyzed as described previously.49 Primary antibodies anti-GAPDH (Glyceraldehyde-3-Phosphate Dehydrogenase) (1:5,000), anti-DNMT1 (1:1,000), anti-TJP1 (1:500), anti-E-cadherin (1:1,000), and anti-N-cadherin (1:500) from Cell Signaling Technology (Danvers, MA, USA) were used.
GSEA and Pearson Correlation Analysis
To investigate the influence of TJP1 expression on MM progression, we analyzed the public MM GEO dataset (GEO: GSE4581). We then compared 140 samples with high TJP1 expression and another 140 samples with low expression in 414 MM patients. The gene sets were downloaded from the Broad Institute’s MSigDB (https://www.broadinstitute.org/gsea/index.jsp). Pearson correlation analysis was performed by RStudio software with ggstatsplot package to investigate the relationship between the expression of TJP1 and E-cadherin in GEO: GSE4581. A p value <0.05 was considered significant.
DNA Extraction and BSP
Genomic DNA was isolated using a TIANamp Genomic DNA kit (Tiangen Biotech, Beijing, China) and was converted using the EZ DNA Methylation-Gold kit (ZYMO Research, Beijing, China). The methylated CpGs in the TJP1 (number NG_003257) promoter were estimated by MethPrimer (https://www.urogene.org/methprimer/). The primer pair modified by Primer Premier 5 was 5′-GATTTTATTATTAGTTTTAGTTTTGGTAGT-3′ (forward) and 5′-TAAAAAACTTATCCACTTACTCCTC-3′ (reverse). The amplified fragments were purified using PCR Cleanup kit (Axygen, Corning, NY, USA) and then cloned into pEASY-T1 vector (TransGen Biotech, Beijing, China). The clones were sequenced (BGI Tech, Shenzhen, China) and analyzed data for 20 methylation cites using QUMA (http://quma.cdb.riken.jp/).
Lentiviral Techniques
Short hairpin RNA (shRNA)-lentiviral particles against human TJP1, DNMT1, and control lentiviral particles were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). pCDH-MCS-T2A-copGFP-MSCV (System Biosciences, Mountain View, CA, USA) was used as a backbone to make the TJP1 and DNMT1 lentiviral overexpression vector. TJP1 and DNMT1 overexpression recombinant lentiviral particles were produced by transient transfection of HEK293T cells according to standard protocols. Briefly, subconfluent HEK293T cells were transduced with 20 μg of an expression vector, 15 μg of pAX2, and 5 μg of pMD2G-VSVG by calcium phosphate precipitation. After 16 h of transfection, the medium was changed, and recombinant lentivirus was harvested twice at 24 and 48 h later. The raw virus supernatants were concentrated by polyethylene glycol precipitation. For stable expression of knockdown/overexpression of TJP1, DNMT1, and control through lentiviral infection, MM1s were incubated with polybrene (5 μg/mL) for 1 h before the addition of the lentiviral particles. After 24 h, the medium was replaced, and cells were grown for 1 day. Stable shRNA and overexpression cell lines were selected in 2 μg/mL puromycin dihydrochloride.
Cell Viability Assays
The tetrazolium reagent WST-1 was used to determine cell viability according to the manufacturer’s protocol and as previously described.50
Adhesion Assay
A confluent monolayer of the BMSCs (passages 2–5) was generated by plating 1 × 104 cells/well in 96-well plates for 24 h. MM1s cells were cultured for 24 h, then labeled with fluorescent calcein-AM (1 μg/mL for 1 h), washed, and a suspension of 0.5 × 106 cells/mL was prepared. MM1s cells or stroma were separately treated with E-cadherin-blocking Ab for 1 h and then washed before being used in the adhesion assay. 100 μL of the MM cell suspension was added to the BMSCs and then incubated for 1 h. Non-adherent cells were washed, and adhesion was detected by measuring the fluorescence intensity in the wells using a plate-reader fluorometer Infiniti M200 (excitement/emission, 485/520 nm; Tecan, Baldwin Park, CA, USA).
Immunofluorescence (IF) Staining
IF staining was performed according to the protocol.51 We incubated the slides with human-specific anti-E-cadherin monoclonal antibody (1:200) (Abcam, Cambridge, MA, USA) for 90 min at room temperature. Thereafter, the appropriate secondary antibodies (1:5,000) were applied and incubated for 30 min. The IF staining was assessed by combining measurements of the intensity and extent of immunopositivity (60×).
Animal Models for MM In Vivo
All animal experiments were approved by the Animal Ethics Committee of Sun Yat-sen University and performed in accordance with the guidelines of the Animal Care and Use Committee of Sun Yat-sen University. In total, 30 male, non-obese diabetic (NOD)-SCID mice (7–9 weeks old, median weight 28 g) were obtained from the Sun Yat-sen University animal center. An in vivo xenograft model was developed in immunodeficient mice by injecting MM1s cells. Myeloma xenograft tumors were generated by injecting 5 × 106 cells resuspended in Matrigel (BD Biosciences) subcutaneously in the right flank of mice. Mice were blindly randomized into five groups (n = 3) of the control, bortezomib, bortezomib/AZA, bortezomib/TJP1-cDNA, or bortezomib/AZA/TJP1-cDNA. For subcutaneous injection of tumor cells to detect the tumor volumes, the tumor volumes were measured every 3 days and calculated as follows (length × width2)/2. Ethical endpoints were defined as a tumor volume of 400 mm3 or 20% loss of original body weight. For studies of the survival time of MM orthotopic models, a total of 3 × 106 MM1s cells were injected into mice via tail veins, and mice were inspected daily for any signs of distress. Mice were blindly randomized into five groups (n = 3) of control, bortezomib, bortezomib/AZA, bortezomib/TJP1-cDNA, or bortezomib/AZA/TJP1-cDNA. The survival duration of MM orthotopic mice was recorded. All mice were euthanized by asphyxiation with CO2 followed by cervical dislocation, away from other animals.
AZA, EMT Inhibitor (C19), and TGF-β Treatment in MM Cells
After growing to about 40%–50% confluency, the cells were treated with inhibitors of DNA methylation AZA (Sigma, Shanghai, China), EMT inhibitor (C19, MedChemExpress, Monmouth Junction, NJ, USA), or EMT activator TGF-β (Sigma), dissolved in PBS, from a serial dilution of 3.0 μM, 10 μM, or 10 ng/mL, respectively. The media exchange and reagent treatments were performed on cells for 2 days. We used untreated cells as the study control.
Statistical Analysis
Data were analyzed and represented using GraphPad Prism 7 software. Error bars represent the mean ± standard deviation (SD) for triplicate measurements from at least 3 independent experiments. Differences between the 2 groups were analyzed by the Student’s t test. Survival curves were created using the Kaplan-Meier method, with the log-rank test used to assess significance. p values <0.05 were considered statistically significant.
Author Contributions
L.Q., Y.Z., and X.D.Z. designed the experiments. M.L., L.Q., S.C., and J.B.X. performed the experiments. X.R.S., Z.X.D., and X.D.Z. analyzed data. Y.Z. provided essential research reagents. J.L., T.L.F., and Z.Y.M. provided and reviewed pathological data. S.N.C. and L.Y.Z. edited the manuscript. L.Q. and X.D.Z. directed the research and wrote the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported in part by the National Natural Science Foundation of China, China (grant nos. 81970191 and 81970193); Guangdong Basic and Applied Basic Research Foundation, China (grant no. 2019A1515011327); Fundamental Research Funds for the Central Universities, Sun Yat-sen University, China (grant nos. 19ykpy149 and 19ykpy39); and Medical Scientific Research Foundation of Guangdong Province, China (grant no. A2018031).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.10.004.
Contributor Information
Lin Qi, Email: qilin23@mail.sysu.edu.cn.
Yongjiang Zheng, Email: zhengyj5@mail.sysu.edu.cn.
Xing-Ding Zhang, Email: zhangxd39@mail.sysu.edu.cn.
Supplemental Information
References
- 1.Mimura N., Hideshima T., Anderson K.C. Novel therapeutic strategies for multiple myeloma. Exp. Hematol. 2015;43:732–741. doi: 10.1016/j.exphem.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tai Y.T., Dillon M., Song W., Leiba M., Li X.F., Burger P., Lee A.I., Podar K., Hideshima T., Rice A.G. Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. Blood. 2008;112:1329–1337. doi: 10.1182/blood-2007-08-107292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Acloque H., Thiery J.P., Nieto M.A. The physiology and pathology of the EMT. Meeting on the epithelial-mesenchymal transition. EMBO Rep. 2008;9:322–326. doi: 10.1038/embor.2008.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shenoy A.K., Lu J. Cancer cells remodel themselves and vasculature to overcome the endothelial barrier. Cancer Lett. 2016;380:534–544. doi: 10.1016/j.canlet.2014.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jolly M.K., Ware K.E., Gilja S., Somarelli J.A., Levine H. EMT and MET: necessary or permissive for metastasis? Mol. Oncol. 2017;11:755–769. doi: 10.1002/1878-0261.12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roccaro A.M., Mishima Y., Sacco A., Moschetta M., Tai Y.T., Shi J., Zhang Y., Reagan M.R., Huynh D., Kawano Y. CXCR4 Regulates Extra-Medullary Myeloma through Epithelial-Mesenchymal-Transition-like Transcriptional Activation. Cell Rep. 2015;12:622–635. doi: 10.1016/j.celrep.2015.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ryu J., Koh Y., Park H., Kim D.Y., Kim D.C., Byun J.M., Lee H.J., Yoon S.S. Highly Expressed Integrin-α8 Induces Epithelial to Mesenchymal Transition-Like Features in Multiple Myeloma with Early Relapse. Mol. Cells. 2016;39:898–908. doi: 10.14348/molcells.2016.0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Azab A.K., Hu J., Quang P., Azab F., Pitsillides C., Awwad R., Thompson B., Maiso P., Sun J.D., Hart C.P. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood. 2012;119:5782–5794. doi: 10.1182/blood-2011-09-380410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ullah T.R. The role of CXCR4 in multiple myeloma: Cells’ journey from bone marrow to beyond. J. Bone Oncol. 2019;17:100253. doi: 10.1016/j.jbo.2019.100253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chirillo R., Aversa I., Di Vito A., Salatino A., Battaglia A.M., Sacco A., Di Sanzo M.A., Faniello M.C., Quaresima B., Palmieri C. FtH-Mediated ROS Dysregulation Promotes CXCL12/CXCR4 Axis Activation and EMT-Like Trans-Differentiation in Erythroleukemia K562 Cells. Front. Oncol. 2020;10:698. doi: 10.3389/fonc.2020.00698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bianchi G., Richardson P.G., Anderson K.C. Promising therapies in multiple myeloma. Blood. 2015;126:300–310. doi: 10.1182/blood-2015-03-575365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guang M.H.Z., McCann A., Bianchi G., Zhang L., Dowling P., Bazou D., O’Gorman P., Anderson K.C. Overcoming multiple myeloma drug resistance in the era of cancer ‘omics’. Leuk. Lymphoma. 2018;59:542–561. doi: 10.1080/10428194.2017.1337115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang J., Tian X.J., Xing J. Signal Transduction Pathways of EMT Induced by TGF-β, SHH, and WNT and Their Crosstalks. J. Clin. Med. 2016;5:41. doi: 10.3390/jcm5040041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laing J.G., Koval M., Steinberg T.H. Association with ZO-1 correlates with plasma membrane partitioning in truncated connexin45 mutants. J. Membr. Biol. 2005;207:45–53. doi: 10.1007/s00232-005-0803-2. [DOI] [PubMed] [Google Scholar]
- 15.Hoover K.B., Liao S.Y., Bryant P.J. Loss of the tight junction MAGUK ZO-1 in breast cancer: relationship to glandular differentiation and loss of heterozygosity. Am. J. Pathol. 1998;153:1767–1773. doi: 10.1016/S0002-9440(10)65691-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.González-Mariscal L., Tapia R., Chamorro D. Crosstalk of tight junction components with signaling pathways. Biochim. Biophys. Acta. 2008;1778:729–756. doi: 10.1016/j.bbamem.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 17.Martin T.A., Jiang W.G. Loss of tight junction barrier function and its role in cancer metastasis. Biochim. Biophys. Acta. 2009;1788:872–891. doi: 10.1016/j.bbamem.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 18.Kaihara T., Kawamata H., Imura J., Fujii S., Kitajima K., Omotehara F., Maeda N., Nakamura T., Fujimori T. Redifferentiation and ZO-1 reexpression in liver-metastasized colorectal cancer: possible association with epidermal growth factor receptor-induced tyrosine phosphorylation of ZO-1. Cancer Sci. 2003;94:166–172. doi: 10.1111/j.1349-7006.2003.tb01414.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kleeff J., Shi X., Bode H.P., Hoover K., Shrikhande S., Bryant P.J., Korc M., Büchler M.W., Friess H. Altered expression and localization of the tight junction protein ZO-1 in primary and metastatic pancreatic cancer. Pancreas. 2001;23:259–265. doi: 10.1097/00006676-200110000-00006. [DOI] [PubMed] [Google Scholar]
- 20.Ni S., Xu L., Huang J., Feng J., Zhu H., Wang G., Wang X. Increased ZO-1 expression predicts valuable prognosis in non-small cell lung cancer. Int. J. Clin. Exp. Pathol. 2013;6:2887–2895. [PMC free article] [PubMed] [Google Scholar]
- 21.Smalley K.S., Brafford P., Haass N.K., Brandner J.M., Brown E., Herlyn M. Up-regulated expression of zonula occludens protein-1 in human melanoma associates with N-cadherin and contributes to invasion and adhesion. Am. J. Pathol. 2005;166:1541–1554. doi: 10.1016/S0002-9440(10)62370-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim Y.E., Won M., Lee S.G., Park C., Song C.H., Kim K.K. RBM47-regulated alternative splicing of TJP1 promotes actin stress fiber assembly during epithelial-to-mesenchymal transition. Oncogene. 2019;38:6521–6536. doi: 10.1038/s41388-019-0892-5. [DOI] [PubMed] [Google Scholar]
- 23.Riz I., Hawley R.G. Increased expression of the tight junction protein TJP1/ZO-1 is associated with upregulation of TAZ-TEAD activity and an adult tissue stem cell signature in carfilzomib-resistant multiple myeloma cells and high-risk multiple myeloma patients. Oncoscience. 2017;4:79–94. doi: 10.18632/oncoscience.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X.D., Baladandayuthapani V., Lin H., Mulligan G., Li B., Esseltine D.W., Qi L., Xu J., Hunziker W., Barlogie B. Tight Junction Protein 1 Modulates Proteasome Capacity and Proteasome Inhibitor Sensitivity in Multiple Myeloma via EGFR/JAK1/STAT3 Signaling. Cancer Cell. 2016;29:639–652. doi: 10.1016/j.ccell.2016.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ziller M.J., Gu H., Müller F., Donaghey J., Tsai L.T., Kohlbacher O., De Jager P.L., Rosen E.D., Bennett D.A., Bernstein B.E. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013;500:477–481. doi: 10.1038/nature12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weber M., Davies J.J., Wittig D., Oakeley E.J., Haase M., Lam W.L., Schübeler D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat. Genet. 2005;37:853–862. doi: 10.1038/ng1598. [DOI] [PubMed] [Google Scholar]
- 27.Cen J., Shen J., Wang X., Kang H., Wang L., Sun L., Li Y., Yu L. Association between lymphoma prognosis and aberrant methylation of ID4 and ZO-1 in bone marrow and paraffin-embedded lymphoma tissues of treatment-naive patients. Oncol. Rep. 2013;30:455–461. doi: 10.3892/or.2013.2450. [DOI] [PubMed] [Google Scholar]
- 28.Easwaran H., Tsai H.C., Baylin S.B. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell. 2014;54:716–727. doi: 10.1016/j.molcel.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahuja N., Easwaran H., Baylin S.B. Harnessing the potential of epigenetic therapy to target solid tumors. J. Clin. Invest. 2014;124:56–63. doi: 10.1172/JCI69736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Böck J., Appenzeller S., Haertle L., Schneider T., Gehrig A., Schröder J., Rost S., Wolf B., Bartram C.R., Sutter C., Haaf T. Single CpG hypermethylation, allele methylation errors, and decreased expression of multiple tumor suppressor genes in normal body cells of mutation-negative early-onset and high-risk breast cancer patients. Int. J. Cancer. 2018;143:1416–1425. doi: 10.1002/ijc.31526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith E.M., Boyd K., Davies F.E. The potential role of epigenetic therapy in multiple myeloma. Br. J. Haematol. 2010;148:702–713. doi: 10.1111/j.1365-2141.2009.07976.x. [DOI] [PubMed] [Google Scholar]
- 32.Wu X.Y., Chen H.C., Li W.W., Yan J.D., Lv R.Y. DNMT1 promotes cell proliferation via methylating hMLH1 and hMSH2 promoters in EGFR-mutated non-small cell lung cancer. J. Biochem. 2020;168:151–157. doi: 10.1093/jb/mvaa034. [DOI] [PubMed] [Google Scholar]
- 33.Chen T., Hevi S., Gay F., Tsujimoto N., He T., Zhang B., Ueda Y., Li E. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat. Genet. 2007;39:391–396. doi: 10.1038/ng1982. [DOI] [PubMed] [Google Scholar]
- 34.Yoo J., Kim J.H., Robertson K.D., Medina-Franco J.L. Molecular modeling of inhibitors of human DNA methyltransferase with a crystal structure: discovery of a novel DNMT1 inhibitor. Adv. Protein Chem. Struct. Biol. 2012;87:219–247. doi: 10.1016/B978-0-12-398312-1.00008-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wong K.K., Lawrie C.H., Green T.M. Oncogenic Roles and Inhibitors of DNMT1, DNMT3A, and DNMT3B in Acute Myeloid Leukaemia. Biomark. Insights. 2019;14 doi: 10.1177/1177271919846454. 1177271919846454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harada T., Ohguchi H., Grondin Y., Kikuchi S., Sagawa M., Tai Y.T., Mazitschek R., Hideshima T., Anderson K.C. HDAC3 regulates DNMT1 expression in multiple myeloma: therapeutic implications. Leukemia. 2017;31:2670–2677. doi: 10.1038/leu.2017.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsai K.W., Kuo W.T., Jeng S.Y. Tight Junction Protein 1 Dysfunction Contributes to Cell Motility in Bladder Cancer. Anticancer Res. 2018;38:4607–4615. doi: 10.21873/anticanres.12765. [DOI] [PubMed] [Google Scholar]
- 38.Zeisberg M., Neilson E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Invest. 2009;119:1429–1437. doi: 10.1172/JCI36183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wan L., Yu W., Shen E., Sun W., Liu Y., Kong J., Wu Y., Han F., Zhang L., Yu T. SRSF6-regulated alternative splicing that promotes tumour progression offers a therapy target for colorectal cancer. Gut. 2019;68:118–129. doi: 10.1136/gutjnl-2017-314983. [DOI] [PubMed] [Google Scholar]
- 40.Zhang X.D., Wang Y., Wang Y., Zhang X., Han R., Wu J.C., Liang Z.Q., Gu Z.L., Han F., Fukunaga K., Qin Z.H. p53 mediates mitochondria dysfunction-triggered autophagy activation and cell death in rat striatum. Autophagy. 2009;5:339–350. doi: 10.4161/auto.5.3.8174. [DOI] [PubMed] [Google Scholar]
- 41.Balda M.S., Garrett M.D., Matter K. The ZO-1-associated Y-box factor ZONAB regulates epithelial cell proliferation and cell density. J. Cell Biol. 2003;160:423–432. doi: 10.1083/jcb.200210020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Wever O., Pauwels P., De Craene B., Sabbah M., Emami S., Redeuilh G., Gespach C., Bracke M., Berx G. Molecular and pathological signatures of epithelial-mesenchymal transitions at the cancer invasion front. Histochem. Cell Biol. 2008;130:481–494. doi: 10.1007/s00418-008-0464-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bradfield P.F., Scheiermann C., Nourshargh S., Ody C., Luscinskas F.W., Rainger G.E., Nash G.B., Miljkovic-Licina M., Aurrand-Lions M., Imhof B.A. JAM-C regulates unidirectional monocyte transendothelial migration in inflammation. Blood. 2007;110:2545–2555. doi: 10.1182/blood-2007-03-078733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kolaczkowska E., Kubes P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013;13:159–175. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 45.Zhou D., Tang W., Zhang Y., An H.X. JAM3 functions as a novel tumor suppressor and is inactivated by DNA methylation in colorectal cancer. Cancer Manag. Res. 2019;11:2457–2470. doi: 10.2147/CMAR.S189937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ghobrial I.M. Myeloma as a model for the process of metastasis: implications for therapy. Blood. 2012;120:20–30. doi: 10.1182/blood-2012-01-379024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stelzer Y., Shivalila C.S., Soldner F., Markoulaki S., Jaenisch R. Tracing dynamic changes of DNA methylation at single-cell resolution. Cell. 2015;163:218–229. doi: 10.1016/j.cell.2015.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bird A.P. The relationship of DNA methylation to cancer. Cancer Surv. 1996;28:87–101. [PubMed] [Google Scholar]
- 49.Qi L., Sun X., Li F.E., Zhu B.S., Braun F.K., Liu Z.Q., Tang J.L., Wu C., Xu F., Wang H.H. HMGB1 Promotes Mitochondrial Dysfunction-Triggered Striatal Neurodegeneration via Autophagy and Apoptosis Activation. PLoS ONE. 2015;10:e0142901. doi: 10.1371/journal.pone.0142901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qi L., Wang Z.Y., Shao X.R., Li M., Chen S.N., Liu X.Q., Yan S., Zhang B., Zhang X.D., Li X. ISL2 modulates angiogenesis through transcriptional regulation of ANGPT2 to promote cell proliferation and malignant transformation in oligodendroglioma. Oncogene. 2020;39:5964–5978. doi: 10.1038/s41388-020-01411-y. [DOI] [PubMed] [Google Scholar]
- 51.Steinberg Z., Myers C., Heim V.M., Lathrop C.A., Rebustini I.T., Stewart J.S., Larsen M., Hoffman M.P. FGFR2b signaling regulates ex vivo submandibular gland epithelial cell proliferation and branching morphogenesis. Development. 2005;132:1223–1234. doi: 10.1242/dev.01690. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.