

SUMMARY
Canonical Wnt signaling is emerging as a major regulator of endocytosis. Here, we report that Wnt-induced macropinocytosis is regulated through glycogen synthase kinase 3 (GSK3) and the β-catenin destruction complex. We find that mutation of Axin1, a tumor suppressor and component of the destruction complex, results in the activation of macropinocytosis. Surprisingly, inhibition of GSK3 by lithium chloride (LiCl), CHIR99021, or dominant-negative GSK3 triggers macropinocytosis. GSK3 inhibition causes a rapid increase in acidic endolysosomes that is independent of new protein synthesis. GSK3 inhibition or Axin1 mutation increases lysosomal activity, which can be followed with tracers of active cathepsin D, β-glucosidase, and ovalbumin degradation. Microinjection of LiCl into the blastula cavity of Xenopus embryos causes a striking increase in dextran macropinocytosis. The effects of GSK3 inhibition on protein degradation in endolysosomes are blocked by the macropinocytosis inhibitors EIPA or IPA-3, suggesting that increases in membrane trafficking drive lysosomal activity.
Graphical Abstract

In Brief
Albrecht et al. show that macropinocytosis is triggered by GSK3 inhibition or mutation of the tumor suppressor Axin1. Wnt-regulated macropinocytosis leads to acidification and activation of catabolism in lysosomes, increasing the utilization of extracellular macromolecules to fuel cell growth. These findings have implications for novel therapies in cancer.
INTRODUCTION
It is currently emerging that canonical Wnt signaling affects a panoply of cell biological responses. Classically, Wnt growth factors lead to the stimulation of the transcriptional coactivator β-catenin through the inhibition of a destruction complex containing glycogen synthase kinase 3 (GSK3), casein kinase 1 (CK1), and the adaptor proteins axin and adenomatous polyposis coli (APC) (MacDonald et al., 2009; Nusse and Clevers, 2017). Axin and APC are major tumor suppressors and their loss-of-function, which leads to increased nuclear β-catenin, lies at the heart of cancers such as hepatocellular and colorectal carcinomas (Galluzzi et al., 2019; Nusse and Clevers, 2017). GSK3 has many protein substrates in addition to β-catenin, and up to 20% of the human proteome contains three or more consecutive GSK3 consensus motifs (S/T-XXX-S/T) that form phosphodegrons (Taelman et al., 2010). The inhibition of GSK3 by Wnt signaling leads to the stabilization of many proteins in a phenomenon known as Wnt-stabilization of proteins (Wnt-STOP) (Acebron et al., 2014; Koch et al., 2015). Since Wnt signaling is maximal at the G2/M phase of the cell cycle, Wnt-STOP mediates an increase of cell size in preparation for cell division (Acebron et al., 2014). An important yet incompletely understood intersection exists between Wnt signaling and endosomal trafficking. When Wnt ligands bind to their co-receptors low-density lipoprotein (LDL)-receptor-related protein 6 (LRP6) and Frizzled (Fz), the destruction complex binds to the receptor complex and is endocytosed into signalosomes (Bilic et al., 2007). These are then trafficked into multivesicular bodies (MVBs) in late endosomes, causing the sequestration of GSK3 and axin from the cytosol into membrane-bounded organelles via microautophagy mediated by the ESCRT (endosomal sorting complexes required for transport) machinery (Taelman et al., 2010; Vinyoles et al., 2014; Colozza et al., 2020).
To these effects of Wnt on cell physiology, one must now add the regulation of macropinocytosis. Cells can incorporate extracellular liquid-phase macromolecules by pinocytosis (Greek, pinein, to drink). Receptor-mediated endocytosis leads to micropinocytosis of small endocytic vesicles of less than 100 nm (Doherty and McMahon, 2009). However, it was found that Wnt treatment also triggers a large increase of non-receptor-mediated endocytosis (Albrecht et al., 2018), consistent with the view that Wnt signaling may act as regulator of macropinocytosis. Indeed, this notion has been supported by two recent studies (Redelman-Sidi et al., 2018; Tejeda-Muñoz et al., 2019). Wnt signaling triggers massive macropinocytosis, both through the β-catenin transcription network (Redelman-Sidi et al., 2018) and through rapid membrane trafficking stimulation independent of new protein synthesis (Tejeda-Muñoz et al., 2019).
Macropinocytosis is an actin cytoskeleton-based phenomenon driven by the activation of p21-activated kinase-1 (Pak1) that involves large endocytic vesicles (Dharmawardhane et al., 2000; Doherty and McMahon, 2009) and is non-receptor mediated. Through macropinocytosis, serum and a host of extracellular glycoproteins enter the cellular fluid compartment to be either recycled out or directed to lysosomes for degradation to generate key metabolites that fuel cell growth and proliferation (Commisso et al., 2013; Palm and Thompson, 2017; Hodakoski et al., 2019). Some cells such as macrophages display constitutive macropinocytosis, drinking one-third of their volume per hour (Lewis, 1931) and internalizing their entire plasma membrane every 33 min (Steinman et al., 1976). Epidermal growth factor (EGF) treatment can trigger macropinocytosis for about 10 min (Haigler et al., 1979; West et al., 1989). In cancer, Kras-activating mutations can cause sustained macropinocytosis in pancreatic ductal adenocarcinoma (PDAC) (Commisso et al., 2013; Ramirez et al., 2019). The regulatory mechanism by which Wnt elicits sustained macropinocytosis to regulate cell metabolic requirements remains an enigma. Given the widespread role of hyperactivated Wnt signaling in the progression of cancer, investigating the mechanism of Wnt-induced macropinocytosis is important.
How could Wnt signaling have such diverse effects on cell physiology, such as stabilization of nuclear β-catenin, Wnt-STOP, MVB sequestration, and macropinocytosis? Thinking about this question, we realized that one possibility could be the regulation of GSK3, which plays a key role in canonical Wnt signaling. We tested this hypothesis in the present study. We found that inhibition of GSK3 with lithium chloride (LiCl) or with CHIR99021 rapidly triggered macropinocytosis through a mechanism independent of protein synthesis in cultured human cells, as well as in vivo in Xenopus embryos. GSK3 requires the destruction complex to phosphorylate β-catenin. To test the role of the destruction complex, we developed a system using Axin1 mutant hepatocellular carcinoma (HCC) cells (called Alexander cells, also known as PLC/PRF/5 cells), in which a mutation causes skipping of exon 4 containing the GSK3-binding sites (Satoh et al., 2000). We found that these HCC cells displayed constitutive actin-driven macropinocytosis, which could be suppressed by the reconstitution of full-length Axin1 at physiological levels. Together with SW480 colorectal carcinoma cells (CRCs) reconstituted with APC (Faux et al., 2004), Alexander HCC cells ± Axin1 provide a useful model to study the effect of the function of individual Wnt tumor suppressor proteins in cancer cells. From a metabolic standpoint, Wnt-induced macropinocytosis caused a rapid increase in glucose, lactate, and some intracellular amino acid pools. Wnt3a treatment or Axin1 mutation resulted in a striking increase in lysosomal catabolism, marked by increased cathepsin D and β-glucosidase enzyme activity. Similarly, GSK3 inhibition led to strikingly elevated endolysosomal activity that was blocked by macropinocytosis inhibitors, indicating that macropinocytosis drives lysosomal catabolism. The results suggest that GSK3 inhibition stimulates acute changes in macropinocytosis and lysosomal activity.
RESULTS
The Destruction Complex Represses Macropinocytosis
GSK3 activity is promoted by the adaptor protein and tumor suppressor Axin1, which brings GSK3 in contact with substrates such as β-catenin (Ikeda et al., 1998). To investigate the effect of loss of functional Axin1 on macropinocytosis, we used the Alexander HCC cell line (Alexander et. al., 1976; Satoh et. al., 2000) that lacks full-length Axin1 and expresses a defective form of endogenous Axin1, in which exon 4 is skipped, resulting in a shorter protein lacking the GSK3-binding domain. This mutant cell line provides an elegant system to study the relevance of the GSK3-Axin1 axis in destruction complex activity. To generate stable reconstituted cell lines, Axin1 (FLAG-tagged) was restored using vesicular stomatitis virus G (VSV-G) transduction or empty vector, followed by hygromycin B selection. Western blots using anti-Axin1 antibody showed the reconstitution of full-length Axin1 in HCC cells, comparable to or lower than those of endogenous Axin1 in other cell lines, including HeLa and the liver cancer cell line HepG2 (Figure 1A).
Figure 1. Axin1 Mutation Causes Increased Macropinocytosis in Alexander HCC Cells, as Revealed by Axin Reconstitution.
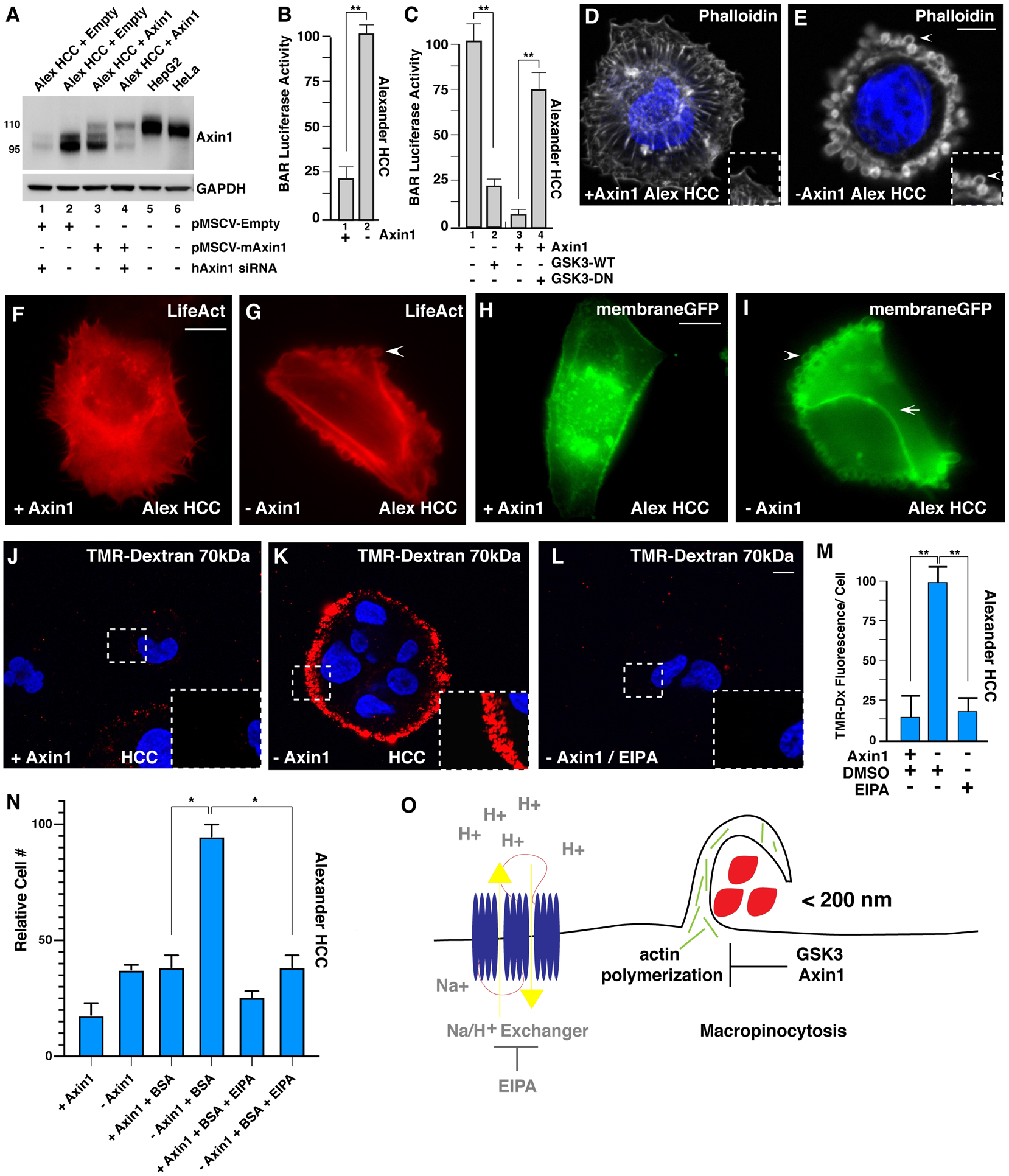
(A) Axin1 protein expression levels assessed after transduction with pMSCV-empty vector (lanes 1 and 2), pMSCV mouse Axin1 in Alexander hepatocellular carcinoma (HCC) cells (lanes 3 and 4), or in HepG2 and HeLa cells (lanes 5 and 6). Alexander cells express an endogenous form of Axin1 that is shorter than the full-length protein (110 kDa) due to the deletion of exon 4 (95 kDa), which is the region that interacts with GSK3 (Satoh et. al., 2000). To show that the lower band was indeed Axin1, cells were transfected with human siRNA that spares mouse Axin1 mRNA (lanes 1 and 4). Note that the expression levels of reconstituted Axin1 were lower than those of the smaller protein skipping exon 4 in Alexander cells or those of endogenous full-length Axin1 in HepG2 or HeLa cells, indicating that Axin1 levels were within the physiological range.
(B) Axin1 mutant cells showed increased β-catenin activity reporter (BAR- Luciferase, relative to Renilla), in Alexander cells, when compared to Axin1 reconstituted cells.
(C) Wild-type (WT) GSK3-GFP transfection in Axin1-mutated HCC cells decreased BAR-Luciferase activity relative to Renilla in Alexander minus Axin1 cells, while dominant-negative GSK3 (GSK3-DN) expression was sufficient to increase BAR- Luciferase activity in Axin1-reconstituted cells.
(D) Axin1-reconstituted HCC cells displayed actin stress fibers visualized by phalloidin F-actin staining.
(E) Axin1-mutated HCC cells had extensive cortical actin remodeling with large vesicular structures (arrowhead), visualized by phalloidin.
(F) LifeAct F-actin imaging showing actin stress fibers in Axin1-reconstituted HCC cells (see also Movie S1).
(G) Axin1-mutant HCC cells showed macropinosome-like structures by LifeAct F-actin imaging (see also Movie S1).
(H) MembraneGFP transfected Axin1-reconstituted Alexander HCC cells.
(I) Axin1-mutated HCC cells led to dynamic membrane remodeling (arrowhead) and vesicle formation in the apical plasma membrane while not between neighboring cells (arrow) visualized by membraneGFP. H and I represent still images from Movie S2.
(J) Axin1-reconstituted HCC cells do not increase uptake of TMR-dextran (dx) 70 kDa.
(K) Axin1 mutated cells have high levels of TMR-dx marking macropinocytosis. Inset shows higher magnifications to reveal macropinosomes.
(L) TMR-dx uptake is sensitive EIPA treatment in Axin1-mutated cells.
(M) Quantification of increased TMR-dx uptake in Axin1-mutated cells compared to Axin1-reconstituted cells or with treatment with EIPA. See also Figure S1. Error bars denote SEM (n ≥ 3), (*p > 0.05, **p > 0.01) in biological replicates. Scale bars, 10 μm.
(N) Mutation of Axin1 allowed growth of HCC cells in the absence of serum and Glutamine at the expense of 3% BSA in an EIPA-dependent manner (brackets). Alexander HCC cells ±Axin1 were grown in serum-starved medium for three days and the increase in numbers of viable cells counted by trypan blue exclusion assay. The results are consistent with the view that mutation of Axin1 increases cell nutrition through macropinocytosis.
(O) Model showing that macropinocytosis incorporation of extracellular macromolecules is repressed in basal cell conditions by a complex of Axin1 and GSK3, the loss of either one results in actin remodeling and massive macropinocytosis of macromolecules (red) in a process that relies on Na+/H+ exchangers which are the target of the macropinocytosis inhibitor EIPA.
Compared to parental Alexander cells, Axin1-reconstituted cells had decreased β-catenin-activated reporter (BAR) luciferase activity (Figure 1B) and gained robust responsiveness to stimulation by Wnt3a ligand (see below). To eliminate the possibility that these changes were due to the production of endogenous Wnts, cells were preincubated overnight with 2 μM IWP2, a Porcupine inhibitor that blocks palmitoylation and secretion of endogenous Wnt while not interfering with responses to exogenous Wnt3a (Colozza et al., 2020). The Porcupine inhibitor was utilized in all subsequent experiments in this study. Transfection of GSK3 (GFP-tagged) was sufficient to reduce BAR luciferase reporter activity in Axin1-mutated HCC cells (Figure 1C, compare lanes 1 and 2), while dominant-negative (DN) GSK3 expression markedly increased BAR activity in Axin1-reconstituted HCC cells (Figure 1C, lanes 3 and 4).
Staining of F-actin with phalloidin revealed dramatic changes in the cytoskeleton, in which cells lacking Axin1 displayed the loss of actin stress fibers and the formation of large spherical structures reminiscent of macropinosome cups (Condon et al., 2018) (compare Figures 1D and 1E). Transfection of the fluorescent F-actin reporter LifeAct showed the formation of dynamic macropinosome-like vesicles surrounded by actin in Axin1-deficient but not Axin1-reconstituted cells (Figures 1F and 1G; Video S1). These indeed corresponded to plasma membrane vesicles, as they could also be visualized using membrane-targeted (Gap43) GFP (Figures 1H and 1I; Video S2). Interestingly, the active vesicle formation on the cell surface was only observed in the apical region (arrowhead) and not on the basolateral region (arrow) of adjoining cells (Figure 1I). In addition, Axin1 reconstitution had no statistically significant effect on cell proliferation by 5-Ethynyl-2-deoxyuridine (Edu) incorporation or on growth rates compared to parental Alexander HCC cells (Figures S1A and S1B).
Currently, macropinocytosis is defined experimentally by the uptake of a dextran derivative with a hydrated diameter of >200 nm, TMR-dextran 70k (tetramethylrhodamine-dextran), which can be blocked by ethyl-isopropyl amiloride (EIPA). The latter is a derivative of the diuretic amiloride that inhibits the Na+/H+ exchange pump causing cytoplasmic acidification near the plasma membrane, blocking actin polymerization and macropinocytosis (Koivusalo et al., 2010; Commisso et al., 2014). We found that Axin1-deficient, but not Axin1-reconstituted, cells incorporated TMR-dextran 70 kDa in an EIPA-sensitive manner (Figures 1J–1M). Alexander cells grow as small epithelial colonies. Macropinosomes form in the peripheral region, but not in the basolateral region between adjoining cells; this gives the impression of a ring of macropinosomes encircling the cell colony that is composed of many individual macrocytic puncta (Figure 1K, inset). To confirm that TMR-dextran was specifically incorporated into macropinocytic vesicles in Axin1-deficient cells, colocalization analyses were performed with phalloidin at low-power imaging of epithelial HCC cells ± Axin1 colonies (Figures S1D–S1F′). The results illustrated the dependency on EIPA, and endocytosed TMR-dextran was surrounded by F-actin rings in Axin1-depleted HCC cells (Figure S1E′, see inset).
The increased macropinocytosis in Alexander cells lacking Axin1 was reflected in the ability of these cells to proliferate using nutrients derived from 3% BSA, while reconstituted cells did not, in conditions of serum starvation (Figure 1N). Importantly, the growth at the expense of BSA was inhibited by EIPA, as expected, given the differences in macropinocytic uptake (Figure 1N, see brackets). The increase in extracellular BSA utilization by inhibition of Axin1 was confirmed in another cell line, HeLa cells, by transfection of ΔRGS-Axin1-GFP (a DN form of Axin1) (Zeng et. al., 1997). While wild-type (WT) Axin1-GFP did not increase the degradation of BSA-dequenched (DQ), inhibition of Axin1 by overexpression of ΔRGS-Axin1-GFP increased lysosomal degradation of endocytosed BSA (Figures S1G and S1H′).
Collectively, these experiments show that mutation of the destruction complex protein Axin1 in HCC cells triggers actin polymerization on the cell surface, forming macropinosomes that require activity of the Na+/H+ exchanger as summarized in the model in Figure 1O. The increase in macropinocytosis enables Axin1 mutant cells to metabolize extracellular protein in conditions of serum starvation, allowing cell growth.
GSK3 Inhibits Macropinocytosis
We next asked whether the effects of Axin1 loss-of-function were mediated by GSK3 activity. We first used the inhibitor LiCl or NaCl as a control. LiCl stimulated the uptake of TMR-dextran into Alexander cells reconstituted with Axin1 within 20 min of treatment, and this effect was blocked by EIPA (Figures 2A–2D). Since LiCl may have off-target effects, we tested another widely used GSK3 inhibitor, CHIR99021, which also stimulated macropinocytosis within 20 min (Figures 2E–2H). No effects on cell viability were detected under these conditions by trypan blue exclusion assays (Figure S1C). Importantly, GSK3 inhibition was equally effective in the presence of cycloheximide (Figures 2I–2L) at a concentration that blocks protein synthesis (20 μg/mL) (Taelman et al., 2010).
Figure 2. GSK3 Inhibition Increases Macropinocytosis Even in the Absence of Protein Synthesis.

(A) Axin1-reconstituted HCC cells treated with NaCl and stained with TMR-dx 70 kDa.
(B) Axin1-reconstituted HCC cells treated with GSK3 inhibitor, LiCl, increased TMR-dx uptake after 20 min.
(C) Axin1-reconstituted HCC cells treated with EIPA decreased TMR-dx uptake by LiCl.
(D) Quantification of LiCl induced TMR-dx uptake in Axin1-reconstituted HCC cells.
(E) Axin1-reconstituted HCC cells treated with DMSO and stained with TMR-dx.
(F) Axin1-reconstituted HCC cells treated with GSK3 inhibitor, CHIR, increased TMR uptake after 20 min.
(G) Axin1-reconstituted HCC cells treated with EIPA decreased TMR-dx uptake by CHIR.
(H) Quantification of CHIR induced TMR-dx uptake in Axin1-reconstituted HCC cells.
(I) Axin1-reconstituted HCC cells treated NaCl stained with TMR-dx in the absence of new protein synthesis blocked by cycloheximide (20 mg/ml CHX).
(J) Axin1-reconstituted HCC cells treated with LiCl and stained with TMR-dx treated with CHX (4 h).
(K) Axin1-reconstituted HCC cells treated LiCl, CHX, and EIPA stained with TMR-dx.
(L) Quantification of LiCl induced TMR-dx uptake in Axin1-reconstituted HCC cells in the presence of CHX.
(M) Dominant-negative GSK3-GFP (DN-GSK3-GFP) induced macropinocytic TMR-dx uptake in Axin1 transfected HCC cells, compared to nontransfected neighboring cells.
(N) DN-GSK3-GFP expression with TMR-dx uptake and DIC phase contrast in Axin1 transfected HCC cells.
(O) Quantification of DN-GSK3-GFP-induced macropinocytic TMR-dx uptake in Axin1 transfected HCC cells, compared to nontransfected neighboring cells.
(P) Wild-type (WT) GSK3-GFP inhibited TMR-dx uptake in transfected minus Axin1 HCC cells, compared to nontransfected neighboring cells.
(Q) WT-GSK3-GFP expression with TMR-dx uptake and DIC phase contrast in Axin1transfected HCC cells.
(R) Quantification of TMR-dx uptake in Axin1 transfected WT-GSK3-GFP HCC cells, compared to nontransfected neighboring cells. Experiments represent biological replicates. Error bars denote SEM (n ≥ 3) (**p > 0.01). Scale bars, 10 μm. See also Figure S2.
HeLa cells have an intact Wnt signaling pathway and, when treated with LiCl, also responded within 20 min, inducing vesicular structures visualized by differential interference contrast (DIC) light microscopy, as well as TMR-dextran endocytosis, in an EIPA-sensitive manner (Figures S2A–S2H). TMR-dextran was also increased by CHIR treatment in Cos7 and HaCaT cells (Figures S2I–S2L), indicating that induction of endocytosis by GSK3 inhibition was effective in other cultured cell types. Thus, the cellular machinery sensitive to GSK3 inhibition is constitutively poised for a rapid macropinocytic response that is independent from the transcriptional program induced by β-catenin stabilization. To rule out pharmacological off-target effects, we transfected a catalytically inactive DN form of GSK3 (DN-GSK3-GFP) into Axin1-reconstituted Alexander cells, which increased TMR-dextran macropinocytosis (Figures 2M–2O). Importantly, overexpression of WT-GSK3 was sufficient to reduce macropinocytic uptake of TMR-dextran in HCC cells lacking Axin1 (Figures 2P–2R). We also tested DN-GSK3-GFP in APC-reconstituted SW480 colorectal carcinoma cells (Tejeda-Muñoz et al., 2019), which also showed increased macropinocytosis (Figures S2M–S2O). As macropinocytosis is also stimulated by a β-catenin transcriptional loop (Redelman-Sidi et. al., 2018), we confirmed these results by transfecting stabilized β-catenin-GFP mutated in its GSK3 sites (Taelman et. al., 2010), which showed increased macropinocytosis in transfected in HCC cells and SW480 cells reconstituted with axin or APC, respectively (Figures S2P–S2U).
Altogether, the results are consistent with a pathway in which the Axin1/GSK3 complex in basal conditions represses the actin machinery that orchestrates macropinocytosis. The suppression of macropinocytosis is a critical function for the GSK3 enzyme.
LiCl Microinjection Induces Macropinocytosis in Xenopus Embryos
To test whether GSK3 inhibition also increases macropinocytosis in an in vivo situation, we used the Xenopus laevis embryo. First, we titrated in detail the optimal amounts of injected TMR-dextran 70 kDa and LiCl. Careful titrations showed that microinjection into the blastula cavity of 8 nL TMR-dextran at 0.5 mg/ml together with 250 mM LiCl or 250 mM NaCl as a control (dissolved in 0.1× Marc’s Modified Ringer’s [MMR] saline solution) was optimal. Albino embryos were injected into the blastocoel at 5 h after fertilization and incubated for a further 1 h before fixation, followed by bisection with a scalpel and visualization under a dissection fluorescent microscope. LiCl, but not NaCl, triggered the accumulation of TMR-dextran in cells surrounding the entire blastula cavity (Figures 3A–3C). Because of the low magnification used, individual puncta are visualized as a haze; the diameter of the blastula embryo is 1.4 mm. To visualize individual macropinocytic puncta, we used the ectodermal explant system. One cell was injected at the animal pole at 4-cell stage with either mGFP alone or with xWnt8 plus mGFP mRNAs, ectodermal explants excised at blastula, and cultured with TMR-dextran 70 kDa in Steinberg’s solution for 1 h. Images taken at higher magnifications showed distinct TMR-dextran puncta not only in the injected cells, but also in the surrounding tissue (in agreement with the short-range effects of the secreted growth factor Wnt8) (Figure 3E). In the controls injected with mGFP mRNA alone, endocytic puncta were not observed (Figure 3D). These in vivo microinjection experiments showed that macropinocytosis induced by GSK3 inhibition occurs not only in cultured cell lines, but also in WT embryos.
Figure 3. In Vivo Inhibition of GSK3 by LiCl Microinjection Causes Increased Macropinocytosis in the Blastula Cavity.

(A) Uninjected Xenopus embryos had no detectable fluorescence at blastula (n = 44).
(A′) Blastocoel in uninjected embryos delineated in the visible light image.
(B) Controls injected with NaCl did not activate macropinocytosis (n = 127; 27% of half-embryos had small fluorescent spots of damaged cells but lacked uniform staining). Embryos were injected at midblastula and cultured for one hour before fixation and bisection.
(B′) Blastocoel in NaCl injected embryos delineated in the visible light image.
(C) LiCl microinjection at midblastula stage increased TMR-dx 70 kDa uptake in blastocoel-lining cells in Xenopus embryos (n = 111 half-embryos, 78% with uniformly increased macropinocytosis in the blastocoel, 2 independent experiments).
(C′) Blastocoel in LiCl injected embryos delineated in the visible light image. Scale bar, 0.35 mm.
(D) Ectodermal explant from a control embryo microinjected into one blastomere with 300 pg mGFP mRNA at the four-cell stage, cultured until late blastula (stage 8.5), and incubated with 1 mg/mL of TMR-dx for 1hr in Steinberg’s solution (n = 13). Note the lack of macropinocytic puncta.
(D′) Contour of the ectodermal explant in control embryo in visible light.
(E) Ectodermal explant co- injected with 10 pg of xWnt8 and mGFP mRNAs and incubated for 1 h with TMR-dx. Note that xWnt8 mRNA stimulates macropinocytosis of TMR-dx in distinct puncta in cells injected with mRNA and in the immediate surrounding cells (n = 20).
(E′) Contour of the ectodermal explant in xWnt8 injected embryo in visible light. Scale bars, 10 μm.
Macropinocytosis Induced by GSK3 Inhibition Increases Lysosomal Activity
Liquid ingested by macropinosomes can be recycled to the outside (Chiasson-MacKenzie et al., 2018) or directed to the lysosomal compartment for the liberation of nutrients (Tejeda-Muñoz et al., 2019). Using a cell-permeable reagent called SirLysosome, containing a peptide that binds only to the active form of cathepsin D (Marciniszyn et al., 1976), we found that HeLa cells treated with LiCl, but not with NaCl, displayed a great increase in active lysosomes, to an extent comparable to that of Wnt3a treatment (Figures 4A–4D). To show that extracellular proteins are digested in endolysosomes, we treated HeLa cells with CHIR99021 or DMSO as a control and found a striking increase in the fluorescence of the reagent ovalbumin-DQ within 20 min of the addition of the GSK3 inhibitor (Figures 4E–4H). Since ovalbumin-DQ fluoresces only when proteolytically degraded, this experiment indicated that GSK3 inhibition leads to increased degradation of extracellular protein to an extent comparable to that caused by the Wnt3a growth factor.
Figure 4. GSK3 Inhibition or Wnt Treatment Cause Increased Lysosomal Activity, Lysosomal Acidification, and Metabolite Changes.

(A) Active lysosomal cathepsin D detected by the cell-permeable SiRLysosome fluorogenic probe in HeLa cells.
(B) SiRLysosome was increased by LiCl treatment after 20 min.
(C) SiRLysosome was increased by Wnt3a protein treatment after 20 min.
(D) Quantification of cathepsin D activity by SiRLysosome fluorescence with LiCl, Wnt3a, or control treatments.
(E) Uptake and lysosomal degradation of extracellular ovalbumin-DQ in DMSO treated HeLa cells.
(F) Ovalbumin-DQ uptake and degradation was increased by CHIR in HeLa cells.
(G) Ovalbumin-DQ uptake and degradation was increased following Wnt3a treatment.
(H) Quantification of ovalbumin-DQ degradation in lysosomes with CHIR or Wnt3a treatment.
(I) Lysosomal pH quantitation using the ratiometric LysoSensor reagent in HeLa cells treated with DMSO. Blue fluorescence indicated pH ~6.0 of pre-existing lysosomes.
(J) LysoSensor pH staining in the yellow channel (pH ~4.5) in DMSO control cells.
(K) LysoSensor staining in control DMSO treated cells with yellow and blue channels merged.
(L) LysoSensor staining in the blue channel was decreased after the addition of CHIR.
(M) LysoSensor staining in the yellow channel (pH ~4.5) was increased by CHIR.
(N) LysoSensor blue and yellow channels in CHIR treated cells indicating that GSK3 inhibition induces lysosomal acidification, which can be visualized as a shift from blue to yellow.
(O) Quantification of LysoSensor pH ratiometric staining in DMSO or CHIR treated cells.
(P) Intracellular glucose following the addition of Wnt3a treatment to HeLa cells for 60 min compared to vehicle control in medium containing U-13C-glucose.
(Q) Intracellular lactate following Wnt3a addition to HeLa cells for 60 min compared to vehicle control in medium containing U-13C-glucose.
(R) Intracellular amino acid pool sizes following Wnt3a addition to HeLa cells for 60 min compared to vehicle control in medium containing U-13C-glucose. Experiments represent biological replicates. Error bars denote SEM (n ≥ 3) (**p > 0.01). Scale bars, 10 μm. See also Figure S3.
To test whether the increase in active lysosomes caused by GSK3 inhibition was related to pre-existing lysosomes, we used the cell-permeable ratiometric probe LysoSensor Yellow/Blue. It fluoresces in blue in lysosomes of higher pH (~6.0) and in yellow in more acidic lysosomes (pH ~4.5). In HeLa cells treated with DMSO, most lysosomes were found to be less acidic and were visualized as blue (Figure 4I), while after 20 min of treatment with the GSK3 inhibitor CHIR99021, they dramatically shifted toward a more acidic state visualized in the yellow channel (Figures 4J–4N; quantitated in Figure 4O). The acidification caused by GSK3 inhibition is best illustrated by comparing Figure 4I to Figure 4M. This indicates that macropinocytosis induced by GSK3 inhibition leads to a rapid and striking acidification of endolysosomes and increased catabolic activity.
We next investigated whether the increase in lysosomal activity caused by Wnt3a signaling resulted in intracellular metabolite changes. To test this, HeLa cells were serum starved for 4 h to normalize baseline signaling activity between conditions, then stimulated with purified Wnt3a protein (100 ng/ml) or vehicle control for 60 min in 10% serum-supplemented medium containing 10 mM U-13C6-glucose. Metabolomics were performed in triplicate, and a panel of 150 metabolites was quantified with MZmine2 software. Significant increases in intracellular glucose (Figure 4P) and lactate levels (Figure 4Q) were observed by acute 1-h Wnt3a treatment. Striking increases were also observed in levels of several amino acids that cells derive from external sources including lysine, methionine, tryptophan, valine, and leucine, the most abundant amino acid in albumin (Palm et al., 2015), and in nonessential amino acids such as arginine, glutamine, and tyrosine (Figure 4R). A similar trend was also observed after only 20 min of Wnt stimulation; all metabolomics data are accessible in the public NIH Metabolomics Workbench data repository (NMDR: PR000897). The increases in lactate and glucose were consistent with observations during long-term treatments in transfected or GSK3-inhibited cells (Pate et al., 2014; Momcilovic et al., 2018). To ensure that increased amino acids were not caused by changes in glycolytic-related metabolic flux, the incorporation of labeled glucose was also monitored. After 1 h of Wnt3a treatment, increased amino acids were not labeled by the U-13C6-glucose isotope, suggesting that they were not derived from the increased levels of glucose (Figures S3A–S3C). Treatment with LiCl for 1 h resulted in amino acid changes comparable to those caused by Wnt3a protein in HeLa cells (Figures S3D–S3F). These results support the view that Wnt3a or GSK3 inhibition results in rapid changes in cellular metabolism on a time scale similar to that of Wnt-induced changes in macropinocytosis of serum proteins.
Axin1 Loss Increases Lysosomal Activity
We next analyzed lysosomal activity in HCC cells ± Axin1 cells. Ovalbumin-DQ added to the culture medium was rapidly degraded in axin-deficient cells, even after only 3 min, while cells reconstituted with Axin1 had lower levels of overall proteolytic degradation (Figure 5A). At the 20-min time point, Axin1-deficient cells had copious lysosomal degradation of ovalbumin-DQ, while Axin1-reconstituted cells only had low levels (compare Figures 5B and 5C). Notably, after LiCl treatment, Axin1-reconstituted cells regained proteolytic activity marked by ovalbumin-DQ (Figure 5D, quantified in Figure 5L). Similarly, BSA-DQ was also increased in HCC cells mutant for Axin1 (Figures S4A–S4C). LiCl treatment was sufficient to increase BSA-DQ uptake in Axin1-reconstituted HCC cells (Figures S4D–S4G). Importantly, EIPA inhibited the BSA-DQ lysosomal digestion induced by Axin1 depletion or LiCl treatment (Figures S4B and S4F), supporting a model whereby BSA is internalized via macropinocytosis. Staining with the lysosomotropic dye Lysotracker confirmed that acidic lysosomes were increased by Wnt3a in Axin1-containing HCC cells (Figures 5E, 5F, and 5M). Wnt3a treatment for 20 min also resulted in a marked increase of the active cathepsin D marker SirLysosome (Figures 5G, 5H, and 5N).
Figure 5. Axin Reconstitution in HCC Alexander Cells Represses Macropinocytosis, and Reconstituted Cells Respond to GSK3 Inhibitors or Wnt3a by Increasing Lysosomal Catabolism in a Manner Dependent on Na+/H+ Exchanger Activity.

(A) Axin1 mutated HCC cells rapidly degraded ovalbumin-DQ added to the culture medium, compared to Axin1 reconstituted HCC cells.
(B) Axin1 mutated HCC cells treated with NaCl and stained with ovalbumin-DQ.
(C) Axin1 reconstituted HCC cells treated with NaCl and stained with ovalbumin-DQ.
(D) Axin1 reconstituted HCC cells treated with LiCl (20 min) and stained with ovalbumin-DQ.
(E) Axin1 reconstituted HCC cells stained with Lysotracker in control treatments.
(F) Axin1 reconstituted HCC cells respond to Wnt3a treatment by increasing Lysotracker.
(G) Axin1 reconstituted HCC cells stained with SirLysosome marking active cathepsin D in control cells.
(H) Wnt3a increased active cathepsin D marked by SirLysosome in Axin1 reconstituted HCC cells.
(I) Axin1 reconstituted HCC cells treated with CHX and stained with a substrate of β-glucosidase, which marks enzymatic cleavage activity of lysosomes.
(J) Axin1 reconstituted HCC cells treated with CHX and LiCl and stained with β-glucosidase substrate. LiCl induced lysosomal activity via macropinocytosis independent of protein synthesis.
(K) EIPA treatment in Axin1 reconstituted HCC cells blocked LiCl-induced enzymatic cleavage of the β-glucosidase substrate. (L–O) Quantification of lysosomal fluorescent tracers ovalbumin-DQ (L), lysotracker (M), cathepsin D (N), and β-glucosidase (O).
(P) Lysosomal pH changes measured with the ratiometric cell-permeable LysoSensor reagent in Axin1-mutated HCC cells treated with DMSO in the blue channel (pH ~6.0).
(Q) LysoSensor staining in Axin1-mutated HCC cells treated with DMSO in the yellow channel (pH ~4.5) indicating high lysosomal acidity.
(R) LysoSensor staining in Axin1-mutated HCC cells treated with EIPA in the blue channel (pH ~6.0).
(S) LysoSensor staining in Axin1-mutated HCC cells in the yellow channel (pH ~4.5) showing that EIPA decreased lysosomal acidity.
(T) Quantification of lysosomal pH changes measured with LysoSensor indicated that acidic lysosomes of pH ~4.5 were rapidly reduced following the addition of EIPA, visualized as a shift from yellow to blue (compare Q to R). Experiments represent biological replicates. Error bars denote SEM (n ≥ 3) (**p > 0.01). Scale bars, 10 μm. See also Figure S4 and S5.
The increase in lysosomal activity was not limited to proteases, as a dramatic increase in the enzymatic cleavage of a cell-permeable β-glucosidase fluorogenic substrate (Harlan et al., 2016) was caused by treatment with LiCl in HCC cells reconstituted with Axin1, both in the presence of cycloheximide (Figures 5I and 5J) and in its absence (Figures S4H–S4K). (β-glucosidase is the enzyme mutated in Gaucher disease, the most frequent lysosomal storage disease.) Importantly, the increase in β-glucosidase activity was inhibited by EIPA, indicating that the increase in lysosomal activity is mediated by macropinocytosis (Figures 5K, 5O, and S4J). These results show that the macropinocytosis driven by GSK3 inhibitors is required for the rapid increase in lysosomal activity.
As in the case of HeLa cells, in HCC cells reconstituted with Axin1, GSK3 inhibition with LiCl also induced strong acidification of lysosomes when assayed by Lysosensor ratiometric staining (Figures S4L–S4P). To show that lysosomal acidification required macropinocytosis, we treated Axin1-mutated HCC cells with EIPA and found that acidic lysosomes (in the pH range of ~4.5) were shifted to a higher pH (in the range of about ~6.0) using the ratiometric LysoSensor Yellow/Blue marker (Figures 5P–5T). This remarkable shift is best seen by comparing Figures 5Q and 5R. These results-together with those observed with other lysosome biomarkers such as Lysotracker, SirLysosome, ovalbumin-DQ, BSA-DQ, and β-glucosidase-indicate that GSK3 inhibition or Wnt signaling activation of macropinocytosis drives increased catabolic activity of acidified lysosomes.
Pak1 Is Required for Macropinocytosis Induced by GSK3 Inhibition
Macropinocytosis caused by long-term transcriptional Wnt/β-catenin signaling has been shown to be Pak1 dependent (Redelman-Sidi et al., 2018). To test whether the rapid induction of lysosomal degradation by GSK3 inhibition was also dependent on Pak1 activity, we used the specific inhibitor IPA-3 in Alexander cells reconstituted with Axin1 and treated with CHIR99021. Cells were treated with TMR-dextran 70 kDa (red) together with BSA-DQ (green) added to the culture medium at the same time as the GSK3 inhibitor. The Pak inhibitor IPA-3 blocked the endocytosis of both extracellular tracers (Figures 6A–6I). Interestingly, TMR-dextran was found in macropinocytic vesicles in the cell periphery and became colocalized with BSA-DQ degradation products (yellow) just prior to becoming extinguished within the deeper lysosomal vesicles containing only degraded BSA-DQ (green). Thus, the fluorescence of TMR-dextran 70 kDa is extinguished as macropinosomes acidify. In addition, SW480 cells lacking APC contained large numbers of MVBs/late endolysosomes marked by the intraluminal vesicle marker CD63, which were greatly decreased within 1 h of treatment with IPA-3 (Figure S5; Video S3). We conclude that GSK3 inhibition activates macropinocytosis through the actin-driven machinery downstream of Pak1 (Dharmawardhane et al., 2000).
Figure 6. The Pak1 Inhibitor IPA-3 Blocks Endocytosis of Macropinocytic Vesicles and Traffic into BSA-Degrading Lysosomes.

(A) Macropinocytic uptake of TMR-dx 70 kDa (red) in Axin1 reconstituted HCC cells treated with DMSO.
(B) BSA-DQ (green) in Axin1-reconstituted HCC cells treated with DMSO.
(C) TMR-dx and BSA-DQ merged in Axin1 reconstituted HCC cells treated with DMSO.
(D) CHIR increased macropinocytic uptake of TMR-dx after 20 min in Axin1 reconstituted HCC cells.
(E) CHIR increased BSA-DQ in Axin1 reconstituted HCC cells.
(F) TMR-dx and BSA-DQ merged in Axin1-reconstituted HCC cells treated with CHIR. Note that macropinocytic vesicles are red and that as BSA-DQ is degraded they turn yellow. As lysosomes mature and become acidified, TMR-dx fluorescence becomes extinguished and BSA fluorescence remains. Arrowheads indicate vesicles in which TMR-dx and BSA-DQ are co-localized.
(G) Pak1 inhibitor, IPA-3, blocks TMR-dx uptake induced by CHIR.
(H) CHIR-induced increases in BSA-DQ were blocked by IPA-3.
(I) TMR-dx and BSA-DQ merged in Axin1 reconstituted HCC cells treated with CHIR and IPA-3. Scale bars, 10 μm.
(J) Phenotype of control Xenopus embryos.
(K) Phenotype of Xenopus embryos treated with LiCl (300 mM in 0.1× MMR saline solution for 7 min at 32-cell).
(L) Phenotype of embryos injected with DN-GSK3 mRNA (150 pg into each blastomere at the vegetal pole at 4-cell stage). Note embryo dorsalization with loss of trunk-tail structures and expansion of head structures, indicating hyperactive early Wnt activity. Scale bar, 0.5 mm.
(M) In the same embryos, LiCl treatment or DN-GSK3 mRNA injection stabilized levels of total Pak1 at blastula, which was accompanied by a similar increase in phospho-Pak1 (the active form). Actin was used as loading control.
In Xenopus embryos, GSK3 inhibition by treatment with LiCl solution at 32-cell or microinjection of DN-GSK3 at the 4-cell has very potent phenotypic effects by increasing the early Wnt signal, resulting in an expansion of dorsal-anterior tissues at the tailbud stage, with a loss of tail structures (Figures 6J–6L). Protein extracts of whole embryos at midblastula showed that GSK3 inhibition resulted in stabilization of total Pak1 protein that was paralleled by an increase in its active form phospho-Pak1 (Figure 6M). Interestingly, the Pak family of protein kinases contains three putative GSK3 sites in a row that are highly conserved in evolution (Taelman et. al., 2010), which may serve as phospho-degrons for Wnt-STOP protein stabilization (Acebron et. al., 2014).
DISCUSSION
The investigations reported here uncover an unexpected role for GSK3 in the regulation of macropinocytosis. In the absence of Wnt, GSK3, in collaboration with the destruction complex proteins axin and APC, represses macropinocytosis. When canonical Wnt ligands or pharmacological inhibitors downregulate GSK3, macropinocytic uptake of extracellular nutrients is triggered. These macromolecules are trafficked to lysosomes where they are catabolized. Pre-existing lysosomes become much more acidic, and lysosomal enzymes such as cathepsin D and β-glucosidase are activated in a macropinocytosis-dependent way. The linchpin in these cell physiological changes lies in GSK3. The model in Figure 7 seeks to integrate Wnt receptor-mediated endocytosis, macropinocytosis, and the inhibition of GSK3. Overall, the findings support the logic that canonical Wnt/GSK3 signaling orchestrates a growth program through β-catenin signaling, Wnt-STOP, and macropinocytosis.
Figure 7. Model of the Intersection among Receptor-Mediated Endocytosis of Wnt, Macropinocytosis, and Lysosomes.

During Wnt signaling, micropinocytosis of the LRP6/Fz receptor results in the sequestration of GSK3 (blue) inside the intraluminal vesicles of MVBs. As cytosolic levels of GSK3 decrease, Pak1 induces the submembraneous actin machinery to form macropinocytic cups that close and engulf macromolecules (red) from the surrounding medium. The axin/GSK3 complex represses macropinocytosis. When Axin1 is mutated, GSK3 is unable to repress the actin machinery, resulting in a prodigious amount of nutrient uptake by macropinocytosis. When GSK3 is inhibited pharmacologically or with DN-GSK3, macropinocytosis is stimulated to a similar degree than that caused by Wnt3a ligand treatment. The results presented in this study point to a hitherto-unappreciated role for GSK3 and axin in the suppression of macropinocytosis in basal conditions.
GSK3 Inhibition Induces Macropinocytosis and Lysosomal Activity
In this study, we demonstrated that GSK3 inhibition with LiCl or CHIR99021 or DN-GSK3 triggered massive macropinosomes that were rapidly trafficked into lysosomes. Given the multiple roles of GSK3 on metabolism (e.g., through its regulation by Akt via receptor tyrosine kinases; RTKs) (Cohen and Frame, 2001; Jope and Johnson, 2004), the observation that the extent of the effects of GSK3 inhibition on macropinocytosis and lysosomes were comparable to those caused by treatment with the Wnt3a growth factor was surprising. This observation is consistent with the view that Wnt growth factor signaling sequesters active GSK3 from the cytosol (Albrecht et al., 2018; Taelman et al., 2010; Vinyoles et al., 2014). Wnt3a protein caused rapid metabolic changes, increasing intracellular glucose, lactate, and several amino acids within 1 h of treatment. Macropinocytosis resulted in an increase in the enzyme activities of cathepsin D and β-glucosidase in lysosomes and digestion of macropinocytosed ovalbumin. The remarkable increase in lysosomal activity by GSK3 inhibition took place in the absence of protein synthesis and correlated with the acidification of pre-existing lysosomes as measured by the LysoSensor ratiometric probe. The effects of canonical Wnt on β-catenin, Wnt-STOP, nutrient acquisition, metabolism, and membrane trafficking all appear to converge at the level of GSK3.
Axin1 Is Required for the Activity of GSK3 in Membrane Trafficking
A destruction complex regulates the degradation of β-catenin after phosphorylation by GSK3. The central scaffold protein is Axin1, a tumor suppressor that operates as a node for GSK3, CK1, and APC interactions. Here, we introduce a system to compare cells differing only by the presence or absence of Axin1. HCC cells (Alexander cells) were stably transfected with Axin1-FLAG, and dramatic differences in cell behavior were revealed. Reconstitution of Axin1 to Alexander cells, which had been in culture over 40 years (Alexander et al., 1976), was sufficient to suppress the very active macropinocytosis of these cancer cells. The massive increase of macropinocytosis caused by axin loss-of-function suggested that a normal function of the destruction complex is to repress macropinocytosis.
The biochemical activities of Axin1 and GSK3 are interdependent in the destruction complex. GSK3 phosphorylates Axin1 scaffold protein, keeping it in an active “open” form able to bind and phosphorylate β-catenin (Kim et. al., 2013). Conversely, Axin1 binds protein phosphatase 2A (PP2A) in close proximity to the Axin1-GSK3-binding domain (Hsu et. al., 1999; Li et. al., 2001). PP2A, in turn, dephosphorylates GSK3 at serine 9, activating the kinase activity (Mitra et. al., 2012). By using the Alexander HCC cell line, which carries a mutation that causes skipping of the GSK3-binding regions within Axin1 exon 4, our data reveal an exquisite requirement of GSK3 for Axin1 function. Scaffolding by Axin1 may be required for multiple GSK3 functions, such as the Wnt-STOP pathway and the macropinocytosis machinery.
In the presence of axin, Alexander cells responded to Wnt3a, LiCl, or CHIR99021 with robust macropinocytosis within 20 min, even in the absence of new protein synthesis, which was blocked by the macropinocytosis inhibitor EIPA. This indicates that the complex of GSK3 and Axin1 normally represses macropinocytosis in basal conditions. GSK3 inhibition caused a striking increase in active lysosomes. The increased degradation of extracellular protein and activity of lysosomal β-glucosidase was also blocked by treatment with EIPA. This indicates that macropinocytosis drives endolysosomal acidification by Wnt/GSK3 signaling. GSK3b loss-of-function mutations have been reported in some cancers (The UniProt Consortium, 2018); such mutations might contribute to tumor growth.
Macropinocytosis, Lysosomes, and Growth Factor Signaling
Pak1 is the key regulator of macropinocytosis, which, when activated, triggers the formation of macropinocytic cups by the actin machinery (Condon et al., 2018; Swanson and Yoshida, 2019). We found that the Pak1 inhibitor IPA-3 blocked TMR-dextran uptake and BSA-DQ degradation induced by short-term treatment with GSK3 inhibitors. While the mechanism by which GSK3 inhibition or Axin1 mutation activates Pak1 remains unknown, there is a wealth of knowledge on the activation of macropinocytosis through the RTK pathway via EGFR, Ras, PTEN, PI3K, Rac, and Pak (Lee et al., 2019; Palm and Thompson, 2017; Swanson and Yoshida, 2019). In pioneering work, Stanley Cohen showed that EGF triggers not only receptor-mediated endocytosis via coated vesicle micropinocytosis, but also a transient activation of liquid-phase macropinocytosis (Haigler et al., 1979; West et al., 1989). In the case of PDAC, activating mutations in Kras drive sustained micropinocytosis, allowing cells to reprogram their metabolism in order to grow through the acquisition of extracellular macromolecules (Commisso et al., 2013; Davidson et al., 2017; Palm and Thompson, 2017). In multicellular animals, cells prefer to feed from free amino acids and sugars transported from the extracellular milieu through their respective nutrient transporters. However, the vast majority of biomass in plasma is contained in nutrient packages provided by glycoproteins (Palm, 2019). Macropinocytosis provides a way of ingesting concentrated packages of amino acids (proteins) and sugars (glycosylated proteins) at much higher concentrations than when soluble in serum. Plasma and lymph contain 50 mg/ml of serum albumin, which becomes available for degradation when cells activate macropinocytosis (Leak et al., 2004; Tejeda-Muñoz et al., 2019).
High levels of macropinocytosis have been observed in bladder, prostate, and non-small-cell lung cancer (NSCLC) in addition to PDAC (Redelman-Sidi et al., 2018; Kim et al., 2018; Hodakoski et al., 2019; Zhang and Commisso, 2019; Ramirez et al., 2019). Similar to the RTK-Ras-PI3K pathway, the Wnt pathway shares the requirement for Pak1 to activate the downstream actin-macropinocytic machinery. How Wnt regulates Pak1 activity is unknown, but several possibilities can be envisaged. Among the Wnt-STOP target proteins with three putative GSK3 sites stabilized by Wnt are Pak and DOCK proteins (guanine nucleotide exchange factors that activate Cdc42), and these phosphorylation sites were highly conserved in evolution (Taelman et al., 2010). An in vitro study with HeLa cell extracts found that the polyubiquitination of Pak4 and 6 was decreased by GSK3 inhibitors, suggesting they contain phosphodegrons stabilized via Wnt-STOP (Acebron et al., 2014). Ras itself is known to contain two threonine residues phosphorylated by GSK3 that generate a phosphodegron leading to degradation via the β-TrCP-E3 polyubiquitin ligase, in a process that is enhanced by Axin1 (Jeong et al., 2012). Wnt-STOP could play an important role in the effects on macropinocytosis described here. That Ras is a direct substrate of GSK3 is interesting. In a recent study using proximity labeling of LRP6-APEX2, Kras interacted with LRP6 after only 5 or 20 min of Wnt3a treatment (Colozza et al., 2020). Since Kras is a key upstream regulator of Pak1, its molecular interaction with LRP6 could help explain the activation of macropinocytosis. Importantly, a recent study by Grainger et al. (2019) using Frizzled9-APEX2 revealed that phosphorylation of Fz9b by EGFR is required for β-catenin signaling by Wnt9a. The issue of whether a crosstalk between Ras/EGFR and GSK3/Wnt signaling participates in Wnt-induced macropinocytosis warrants further investigation.
Our results suggest that GSK3 activity within the destruction complex is an essential component of the actin machinery-regulated macropinocytosis pathway. Macropinocytosis was found to drive a striking increase in endolysosomal activity. Given this finding, the lysosome may represent a point of vulnerability for targeting the progression of Wnt-driven cancers. Lysosomes can be targeted with hydroxychloroquine and newer derivatives (Rebecca et al., 2017), LLOME (L-leucyl-L-leucine O-methyl ester) (Skowyra et al., 2018), and albumin-bound conjugates (Miele et al., 2009). Further, targeting newly identified molecular players in the Wnt pathway like protein arginine methyltransferase 1 (PRMT1), an enzyme required for Wnt-induced endocytosis (Albrecht et al., 2018, 2019), may represent additional points for targeting Wnt-driven diseases. The relationship between lysosomes and cancer is an active and promising area for cancer therapeutic interventions.
Wnt is an ancient signaling system that plays a fundamental role in development, homeostasis, and regeneration in all meta-zoans (Loh et al., 2016; Steinhart and Angers, 2018). In Xenopus embryos, we found that GSK3 inhibition with LiCl strongly increased macropinocytosis in the blastocoel cavity. Using TMR-dextran uptake, we did not detect regions of differential macropinocytosis in the control embryos, but we only examined blastula stages. Future studies will examine whether areas of differential macropinocytosis and lysosomal activity exist at other stages of normal development and whether they result from Wnt signaling.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Edward M. De Robertis (ederobertis@mednet.ucla.edu).
Materials Availability
No custom code, software, or algorithm central to supporting the main claims of the paper were generated in this manuscript.
Data and Code Availability
Original data have been deposited to NIH Metabolomics Workbench Data Repository with the project identifier NMDR: PR000897.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
Alexander cells (RRID:CVCL_0485), Cos7 (RRID:CVCL_0224), HaCaT (RRID:CVCL_3300), HeLa (RRID: CVCL_0030), SW480 (RRID: CVCL_0546), and HepG2 (RRID: CVCL_1098), were obtained from ATCC and cultured in DMEM (GIBCO) supplemented with 10% fetal bovine serum (FBS), glutamine and antibiotics. For reconstitution of Axin1 in Alexander cells, retroviral particles were prepared and infections were carried out as previously described (Azzolin et al., 2012), and Axin1-Flag or empty vector were cultured in hygromyocin until antibiotic-independent cell lines were established. SW480 cells were maintained in RPMI 1640 (GIBCO), 10% FBS, glutamine and antibiotics, supplemented with 0.025 U/ml insulin (Sigma), 1 μg/ml hydrocortisone (Sigma), 0.01 μg/mL 1-thioglycerol (Sigma), and 1.5 mg/ml G418 (Sigma). Each cell line maintained distinct morphology and nuclear staining. All cell lines tested negative for mycoplasma contamination. ATCC cell line authentication services were performed to confirm identity of cultured cell types.
Xenopus embryo microinjection and animal cap assays
All animal experiments were approved by the University of California Animal Research Committee. Xenopus laevis were purchased from Nasco and embryos fertilized in vitro and cultured according to standard methods (Moriyama and De Robertis, 2018). Staging was performed as previously described (Nieuwkoop and Faber, 1967). NaCl or LiCl (200 mM) together with the high molecular weight marker tetramethylrhodamine (TMR) dextran 70 kDa (0.5 mg/mL) were introduced into the blastula cavity by microinjection using an Oxford micromanipulator and an IM 300 microinjection pump (Narishige International USA, Inc); 8 nL one time at stage 8 was found to be optimal. Fertilized embryos were cultured for 5 hours in 0.1× MMR saline, transferred to 1× MMR for injection, and cultured further for one hour at 0.1× MMR. The embryos were fixed overnight, and bisected with a scalpel blade. The 60× digital magnification zoom of Axio Zoom V.16 Stereo Zoom Zeiss was used for collecting images. For ectodermal explant experiments, Xenopus embryos were injected into the animal pole at 4-cell stage with xWnt8 (10 pg) and mGFP (300 pg) mRNAs. Animal caps were dissected at stage 8.5 and incubated with TMR-dextran (1 mg/mL) diluted in 1x Steinberg’s solution for 1 hr. The caps were washed 3 times and mounted on 35 mm Petri dishes glass with glass bottoms (#0 cover glass, Cell E&G: GBD00003–200), positioned with the inner layer (less pigmented layer) facing downward with Anti-Fade Fluorescence Mounting Medium-Aqueous, Fluoroshield (ab104135). Image acquisition was performed using a Carl Zeiss Axio Observer Z1 Inverted Microscope with Apotome.
METHOD DETAILS
Drug treatments
The optimal concentrations for inhibition of macropinocytosis in cultured cells have been determined previously to be 40 μM for EIPA and 2.5 μM for IPA-3 (Commisso et al., 2013), and were the concentrations used here. GSK3 inhibition was performed with 40 mM LiCl or 40 mM NaCl as a control and with 8 nM CHIR99021 using DMSO added at matched volumes as a control. Wnt3a and control buffer were also used for 20 min incubation periods or less. Wnt3a protein was purchased from PeproTech (Cat# 315–20) and used at 100 ng/mL. For cycloheximide experiments, 20 μg/mL cycloheximide was added to cells 4 hours prior to GSK3 inhibition or Wnt3a addition. In order to inhibit any possible endogenous Wnt signals, the Porcupine inhibitor IWP-2 (2 μM) was added to the culture medium in these studies for 12 hours prior to each experiment as described (Colozza et al., 2020).
Macropinocytosis and lysosome activity
Macropinocytosis and regulated-lysosomal function were examined using tracers of extracellular albumin incorporation and proteolytic activity in living cells essentially according to manufacturer protocols with minor modifications. Macropinocytosis was traced with TMR-Dextran 70 kDa (ThermoFisher Cat# D1818; 1 mg/ml) as performed previously (Commisso et. al., 2014). Extracellular protein uptake and degradation in lysosomes was tracked with Bovine Serum Albumin DeQuenched (BSA-DQ; ThermoFisher Cat# L12051; 5 μg/mL) and Ovalbumin-DQ (ThermoFisher Cat# D12053; 5 μg/mL) (Albrecht et. al., 2018). Lysosomal enzyme activity of Cathepsin D was traced with SiR-lysosome (Cytoskeleton Inc Cat# CY-SC012; 1μM) a cell permeable fluorescent pepstatin A peptide that binds Cathepsin D in live cells and marks lysosomal activity with high specificity. Changes in lysosomal pH were traced using the ratiometric probe Lysosensor Yellow/Blue DND-160 (ThermoFisher Cat# L7545; 1:1000 dilution) that produces blue fluorescence in less acidic vesicles and changes to yellow in more acidic environments. Lysotracker was also used in experiments as it specifically marks acidic lysosomal vesicles (ThermoFisher Cat# L7528; 1:1000 dilution). To assess lysosomal enzymatic activity, we used the β-glucosidase substrate (Marker Gene Tech Cat# M27745; 5 μM) that fluoresces following cleavage by lysosomal enzymes.
For these experiments, cells were first plated on 12-well plates containing circular glass coverslips and grown to 60% confluency in a 5% CO2 incubator at 37°C (Albrecht et al., 2019). GSK3 inhibition was performed for 20–30 min and comparisons were made between LiCl (40 mM) and NaCl (40 mM), or CHIR99021 (8 nM) compared to DMSO added at matched volumes. Wnt3a and control buffer were also used for 20 min incubation periods or less. Transfections of GSK3-WT (0.8 μg), GSK3-DN (0.8 μg), or Stbl-β-catenin (0.8 μg) were performed on cells plated on coverslips for 24 hr. Macropinocytosis and lysosomal tracers were incubated with living cells in 10% FBS for time points ranging from 20 to 60 min. β-glucosidase was added to cells for 25 minutes with or without EIPA or cycloheximide treatments. Following incubation with tracers, cells were washed with ice-cold DPBS three times and fixed in 4% paraformaldehyde (Sigma #P6148).
Fluorescence microscopy
HeLa, Cos7, HaCaT, Alexander ± Axin1, or SW480 ± APC cells were grown on circular coverslips, fixed with 4% paraformaldehyde (Sigma #P6148) for 15 min, permeabilized with 0.2% Triton X-100 in Dulbecco’s phosphate buffered saline (DPBS; GIBCO) for 10 min, and blocked with 5% goat serum and 0.5% BSA in DPBS for 1 hour. Phalloidin (1:1000) staining to visualize F-actin was performed on fixed cells. After three additional washes with DPBS, chambers were removed and coverslips mounted with Fluoroshield Mounting Medium with DAPI (ab104139). Immunofluorescence was analyzed and photographed using a Carl Zeiss Axio Observer Z.1 Inverted Microscope with Apotome and Zen 2.3 imaging software from Zeiss and ImageJ software from NIH. Statistical analysis of the data was performed using the Student t test. A p value of < 0.05 is considered to be statistically significant for n > 25 cells. All experiments were done in biological triplicates.
Immunoblotting
Protein lysates were obtained from cells grown for 2 days at high density after sonication in lysis buffer (20 mM HEPES pH 7.8, 100 mM NaCl, 5% glycerol, 5 mM EDTA, 0.5% NP40, and protease and phosphatase inhibitors) (Azzolin et al., 2012). For Xenopus western blots (n = 4), 6 embryos per condition were collected at blastula, mechanically dissociated in RIPA buffer (0.1% NP40, 20 mM Tris/HCl pH 7.5), 10% Glycerol, protease (Roche #04693132001) and phosphatase inhibitors (Calbiochem #524629), and incubated for 15 mins on ice. The western blot procedure was carried out as described previously (Azzolin et al., 2012). Immunoblotting was performed with antibodies for total Pak1 (Abcam, ab131522), phospho-Pak (T423) (Abcam, ab2477), and Actin (Abcam, ab8227). Secondary antibodies from Li-Cor (IRDye 680 (926–68072) and IRDye 800 (926–32213), 1:3000) were used with the Li-Cor Odyssey system. ImageQuant LAS 4000 1.2 was used to acquire western blot images.
Intracellular metabolite extraction and mass spectrometry-based metabolomics
HeLa cell cultures were initially grown with triplicates of each condition in 6-well plates until the wells were ~70% confluent. Extraction began by placing the 6-well dishes on ice and washing each well with 2 mL of a 150 mM ammonium acetate solution (pH 7.4, 4°C). Post wash, metabolites were extracted from each well on dry ice with 500 μL of a (1:4) dH20, MeOH solution at −80°C. The samples were then incubated for 15 min at −80°C. Afterward, the metabolite solutions were transferred to eppitubes and spun down at 4°C for 10 min at 17,000 g. The supernatants were transferred to new vials and dried with an EZ2-Elite lyophilizer (Genevac). Dried metabolites were re-suspended in 100 μL (1:1) dH20:Acetonitrile solution. After re-suspension, metabolite solutions were spun at 17,000 g for 10 min. The top 70 μL of the supernatant was then transferred to HPLC autosampler vials. 10 μL of these suspensions were injected per analysis. Samples were run on a Vanquish (Thermo Scientific) UHPLC system with mobile phase A (5mM NH4AcO, pH 9.9) and mobile phase B (ACN) at a flow rate of 200 μL/min on a Luna 3 mm NH2 100A (150 × 2.0mm) at 40°C with a gradient going from 15% to 95% A in 18 min followed by an 11 minute isocratic step. The UHPLC was coupled to a Q-Exactive (Thermo Scientific) mass analyzer running in polarity switching mode at 3.5kV with an MS1 resolution of 70,000. Metabolites were identified via exact mass (MS1), retention time, and in some cases by their fragmentation patterns (MS2) at normalized collision energy (NCE). Quantification was performed via area under the curve (AUC) integration of MS1 ion chromatograms with the MZmine 2 software package. Area values were then normalized to cell count averages from triplicate wells treated in parallel for each condition.
Gene knockdown, knockout and overexpression
DNA constructs or siRNA were transfected with Lipofectamine 3000 as described by the manufacturer and cultured for 24 hr. A mixture of two overlapping siRNAs was used for human Axin1 depletion: GGGCATATCTGGATACCTG and GGATACCTGCCGACCTTAA. Transfections of GSK3-WT (0.8 μg), GSK3-DN (0.8 μg), or Stbl-β-catenin (0.8 mg) were performed on cells plated on coverslips for 24 hr. Reconstitution of Axin1-Flag or empty vector into Alexander cells was performed using retroviral particle preparation and infection protocols as previously described (Azzolin et al., 2012). Embryo injections used xWnt8 (10 pg) and mGFP (300 pg) mRNAs.
Luciferase reporter analysis
Alexander cells were plated on 24-well plates at 70% confluency and transfected with 1 μg of β-catenin Activated Reporter (BAR) DNA and 0.05 μg of Renilla reporter DNA (generous gifts of R. Moon). In the case of GSK3 transfections, Alexander ± Axin1 cells were co-transfected with β-catenin Activated Reporter (BAR 0.8 μg), and Renilla reporter (0.05 μg) and either GSK3-WT (0.8 μg) or GSK3-DN (0.8 μg) and incubated for 24 hours. Luciferase activity was measured with the Dual-Luciferase Reporter Assay System (Promega) according to manufacturer’s instructions, using a Glomax Microplate Luminometer (Promega). Luciferase values of each sample were normalized for Renilla activity. Experiments were performed in biological triplicates.
Cell proliferation & viability
Alexander cells were plated on fibronectin-coated chamber slides. After 24h, EdU (10 μM) was added to the culture medium and cells were incubated for 90 min at 37°C, 5% CO2. Cells were then fixed with 4% paraformaldehyde (PFA) for 10 minutes at room temperature. The EdU assay was performed according to the manufacturer’s instructions (Click-iT EdU Imaging Kits, Invitrogen). Images were obtained with a Leica TCS SP5 equipped with a CCD camera and analyzed using velocity software (Perkin Elmer). Data are presented as % of EdU-positive cells per nuclei. Cell viability assays were performed using cells plated at 0.3 × 106/well in 12-well plates. 24 hours after plating, Alexander ± Axin1 and HeLa cells were incubated for 30 min with CHIR99021 (8 nM), LiCl (40 mM), or EIPA (up to 80 μM). For serum starved assays, cells were first starved for three days and incubated with 3% bovine serum albumin (BSA) solubilized in serum free media (24 hr) with or without EIPA (40 μM). Cells were trypsinized, and resuspended to a final volume of 1 ml. Cell viability was measured by trypan blue exclusion using a Vi-Cell Beckman-Coulter Viability Analyzer. Treatments had no effect on cell viability after 24 hours.
QUANTIFICATION AND STATISTICAL ANALYSIS
Image quantification
Vesicle quantification by light microscopy of differential interference contrast (DIC) microscopy was performed as previously described (Albrecht et al., 2018). The number of vesicles above a diameter of 200 nm were quantified between experimental conditions using a combination of Zeiss (Zen) imaging software for size measurement and ImageJ for vesicle number counts using the particle counting computer-assisted program. Thresholds were set for the DIC channels using MaxEntropy in the FIJI ImageJ software package and individual vesicles were distinguished using the binary watershed function. Particle function was applied to count number of vesicles 0.2–7 μm, circularity −1. In all cases, the total number of cells counted was more than 25 cells per condition, in triplicate experiments. For quantitatively comparing lysosome and macropinosome fluorescence in control versus treated cells, ImageJ software was used, and analyzed in n > 25 cells per condition. Fluorescence intensity was normalized in images compared, in each triplicate condition.
Statistical analyses
Results from three or more independent experiments are shown as the mean ± SEM. Statistical analyses of computer-assisted particle analyses were performed using two-tailed t tests for two-sided comparisons where statistical significance was defined as *p < 0.05 and **p < 0.01. Images chosen for display in figures were chosen as representative images for the whole experiment that included an n < 25 cells from triplicate experiments, as described previously (Tejeda-Muñoz et al., 2019).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| GAPDH | EMD Millipore | Cat# AB2302; RRID:AB_10615768 |
| Actin | Abcam | Cat# ab8227, RRID:AB_2305186 |
| Axin1 | Cell Signaling | Cat# 2087; RRID:AB_2274550 |
| β-catenin | Santa Cruz | Cat# sc-7963; RRID:AB_626807 |
| IRDye 680 | Li-Cor | Cat# 926-68072; RRID:AB_10953628 |
| IRDye 800 | Li-Cor | Cat# 926-32213; RRID:AB_621848 |
| Goat anti-rabbit IgG, Alexa Fluor 488 conjugate | ThermoFisher | Cat# A-11034; RRID:AB_2536177 |
| Donkey anti-rabbit IgG, Alexa Fluor 568 conjugate | Jackson | Cat# 711-166-152; RRID:AB_2313568 |
| Donkey anti-mouse IgG, Alexa Fluor 488 conjugate | Jackson | Cat# 715-546-150; RRID:AB_2340849 |
| Pak1 | Abcam | Cat# ab131522, RRID:AB_11156726 |
| Phospho-Pak1 | Abcam | Cat# ab2477, RRID:AB_303091 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dextran Tetramethylrhodamine (TMR-Dx) 70,000 | ThermoFisher | Cat# D1818 |
| Bovine Serum Albumin DeQuenched (BSA-DQ) | ThermoFisher | Cat# D12051 |
| SirLysosome | Cytoskeleton Inc | Cat# CYSC012 |
| β-glucosidase substrate | Marker Gene Technologies | Cat# M27745 |
| Lysotracker | ThermoFisher | Cat# L7528 |
| Alexa Fluor 488 Phalloidin | ThermoFisher | Cat# A12379 |
| Alexa Fluor 568 Phalloidin | ThermoFisher | Cat# A12380 |
| Ovalbumin DeQuenched (DQ) | ThermoFisher | Cat# D12053 |
| Cycloheximide | Sigma | Cat# C-7698 |
| Lithium chloride (LiCl) | Sigma | Cat# L4408 |
| Methylbenzothiazolyl-tetrahydro-oxophenylthienopyrimidinthio-acetamide (IWP-2) | Sigma | Cat# IO536 |
| CHIR99021 | Sigma | Cat# SML1046 |
| DMSO | Sigma | Cat# W387520 |
| 5-(N-Ethyl-N-isopropyl) amiloride (EIPA) | Sigma | Cat# A3085 |
| IPA-3 | Sigma | Cat# I2285 |
| Prolong Antifade + DAPI | ThermoFisher | Cat# P36931 |
| Sodium chloride (NaCl) | Sigma | Cat# S9888 |
| Wnt3a | Peprotech | Cat# 315-20 |
| U-13C6-glucose | Cambridge Isotope Laboratories | Cat# CLM-1396-1 |
| Lipofectamine 3000 | ThermoFisher | Cat# L3000001 |
| Lysosensor | ThermoFisher | Cat# L7545 |
| Critical Commercial Assays | ||
| Dual-Luciferase Reporter Assay System | Promega | Cat# E1500 |
| Deposited Data | ||
| Metabolomics workbench data repository; PR000897 | NIH | https://www.metabolomicsworkbench.org |
| Experimental Models: Cell Lines | ||
| Alexander (hepatocellular carcinoma) | ATCC | RRID:CVCL_0485 |
| Cos7 | ATCC | RRID:CVCL_0224 |
| HaCaT | ATCC | RRID:CVCL_3300 |
| HeLa (human cervical adenocarcinoma) | ATCC | RRID: CVCL_0030 |
| HepG2 | ATCC | RRID:CVCL_1098 |
| SW480 | ATCC | RRID:CVCL_0546 |
| Experimental Models: Organisms/Strains | ||
| Xenopus laevis | Nasco | N/A |
| Oligonucleotides | ||
| siRNA targeting sequence: human Axin1 #1: GGGCATATCTGGATACCTG | This paper | N/A |
| siRNA targeting sequence: human Axin1 #2: GGATACCTGCCGACCTTAA | This paper | N/A |
| Recombinant DNA | ||
| LifeAct | IMSR | RRID:IMSR_EM:12427 |
| pCS2-mGFP | Addgene | RRID:Addgene_14757 |
| CD63-RFP | Addgene | RRID:Addgene_62964 |
| Dominant-negative (DN)-GSK3-GFP | Addgene | RRID:Addgene_29681 |
| Stabilized β-catenin mutated in its GSK3 sites | Addgene | RRID:Addgene_29684 |
| Wnt8 | Addgene | RRID:Addgene_16863 |
| Wildtype (WT)-Axin1-GFP | Addgene | RRID:Addgene_21287 |
| ΔRGS-Axin1-GFP | Addgene | RRID:Addgene_16303 |
| Wildtype (WT)-GSK3-GFP | Addgene | RRID:Addgene_29680 |
| β-catenin Activated Reporter (BAR) | Addgene | RRID:Addgene_12456 |
| Renilla reporter | Addgene | RRID:Addgene_62186 |
| MSCVhyg-F-mAxin1 | Addgene | RRID:Addgene_24928 |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Prism 7 | GraphPad | https://www.graphpad.com:443/scientific-software/prism/ |
| MZmine2 software | http://mzmine.github.io | |
| Zen 2.3 imaging software | Zeiss | http://www.zeiss.com |
| Other | ||
| Vi-CELL XR Cell Viability Analyzer | Beckman Coulter | N/A |
| IM 300 microinjection pump | Narishige International USA, Inc | N/A |
| Axio Observer Z1 Inverted Microscope with Apotome | Zeiss | N/A |
Highlights.
GSK3 and Axin1 normally repress macropinocytosis in basal cellular conditions
GSK3 inhibition or Axin1 mutation triggers macropinocytosis and metabolite changes
Macropinocytosis induces acidification and catabolic activity of lysosomes
Lysosomal activation by Wnt signaling is independent of new protein synthesis
ACKNOWLEDGMENTS
We are grateful to Dr. M. Faux and the Ludwig Institute for SW480APC cells; Dr. R. Moon for plasmid constructs; and Emilie Marcus, Alyssa Dsousa, and Pooja Sheladiya for comments on the manuscript. L.V.A. was supported by postdoctoral fellowships NIH F32 GM123622 and by A.P. Giannini; N.T.-M. was supported by a joint University of California Institute for Mexico and the United States (UC MEXUS) and Consejo Nacional de Ciencia y Tecnologia (CONACYT) postdoctoral fellowship (FE-17-65); and M.H.B. was supported by a UCLA Clinical and Translational Institute undergraduate fellowship (CTSI UL1TR000124). This work was supported by NIH R01 CA215185, Fondazione AIRC under 5 per Mille 2019 -ID. 22759 program to S.P., the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (DENOVOSTEM grant agreement 670126), and the Norman Sprague Endowment for Molecular Oncology and the Howard Hughes Medical Institute.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107973.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Acebron SP, Karaulanov E, Berger BS, Huang Y-L, and Niehrs C (2014). Mitotic wnt signaling promotes protein stabilization and regulates cell size. Mol. Cell 54, 663–674. [DOI] [PubMed] [Google Scholar]
- Albrecht LV, Ploper D, Tejeda-Muñoz N, and De Robertis EM (2018). Arginine methylation is required for canonical Wnt signaling and endolysosomal trafficking. Proc. Natl. Acad. Sci. USA 115, E5317–E5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht LV, Bui MH, and De Robertis EM (2019). Canonical Wnt is inhibited by targeting one-carbon metabolism through methotrexate or methionine deprivation. Proc. Natl. Acad. Sci. USA 116, 2987–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander J, Bey E, Whitcutt JM, and Gear JH (1976). Adaptation of cells derived from human malignant tumours to growth in vitro. S. Afr. J. Med. Sci 41, 89–98. [PubMed] [Google Scholar]
- Azzolin L, Zanconato F, Bresolin S, Forcato M, Basso G, Bicciato S, Cordenonsi M, and Piccolo S (2012). Role of TAZ as mediator of Wnt signaling. Cell 151, 1443–1456. [DOI] [PubMed] [Google Scholar]
- Bilic J, Huang Y-L, Davidson G, Zimmermann T, Cruciat C-M, Bienz M, and Niehrs C (2007). Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science 316, 1619–1622. [DOI] [PubMed] [Google Scholar]
- Chiasson-MacKenzie C, Morris ZS, Liu CH, Bradford WB, Koorman T, and McClatchey AI (2018). Merlin/ERM proteins regulate growth factor-induced macropinocytosis and receptor recycling by organizing the plasma membrane:cytoskeleton interface. Genes Dev. 32, 1201–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, and Frame S (2001). The renaissance of GSK3. Nat. Rev. Mol. Cell Biol 2, 769–776. [DOI] [PubMed] [Google Scholar]
- Colozza G, Jami-Alahmadi Y, Dsouza A, Tejeda-Muñoz N, Albrecht LV, Sosa E, Wohlschlegel JA, and De Robertis EM (2020). Wnt-inducible Lrp6-APEX2 interacting proteins identify ESCRT machinery and Trk-fused gene as components of the Wnt signaling pathway. bioRxiv. 10.1101/2020.05.03.072579. [DOI] [PMC free article] [PubMed]
- Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, et al. (2013). Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commisso C, Flinn RJ, and Bar-Sagi D (2014). Determining the macropinocytic index of cells through a quantitative image-based assay. Nat. Protoc 9, 182–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condon ND, Heddleston JM, Chew T-L, Luo L, McPherson PS, Ioannou MS, Hodgson L, Stow JL, and Wall AA (2018). Macropinosome formation by tent pole ruffling in macrophages. J. Cell Biol 217, 3873–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson SM, Jonas O, Keibler MA, Hou HW, Luengo A, Mayers JR, Wyckoff J, Del Rosario AM, Whitman M, Chin CR, et al. (2017). Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nat. Med 23, 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharmawardhane S, Schürmann A, Sells MA, Chernoff J, Schmid SL, and Bokoch GM (2000). Regulation of macropinocytosis by p21-activated kinase-1. Mol. Biol. Cell 11, 3341–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty GJ, and McMahon HT (2009). Mechanisms of endocytosis. Annu. Rev. Biochem 78, 857–902. [DOI] [PubMed] [Google Scholar]
- Faux MC, Ross JL, Meeker C, Johns T, Ji H, Simpson RJ, Layton MJ, and Burgess AW (2004). Restoration of full-length adenomatous polyposis coli (APC) protein in a colon cancer cell line enhances cell adhesion. J. Cell Sci 117, 427–439. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Spranger S, Fuchs E, and López-Soto A (2019). WNT Signaling in Cancer Immunosurveillance. Trends Cell Biol. 29, 44–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainger S, Nguyen N, Richter J, Setayesh J, Lonquich B, Oon CH, Wozniak JM, Barahona R, Kamei CN, Houston J, et al. (2019). EGFR is required for Wnt9a-Fzd9b signalling specificity in haematopoietic stem cells. Nat. Cell Biol 21, 721–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigler HT, McKanna JA, and Cohen S (1979). Rapid stimulation of pinocytosis in human carcinoma cells A-431 by epidermal growth factor. J. Cell Biol 83, 82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlan FK, Lusk JS, Mohr BM, Guzikowski AP, Batchelor RH, Jiang Y, and Naleway JJ (2016). Fluorogenic substrates for visualizing acidic organelle enzyme activities. PLoS ONE 11, e0156312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodakoski C, Hopkins BD, Zhang G, Su T, Cheng Z, Morris R, Rhee KY, Goncalves MD, and Cantley LC (2019). Rac-mediated macropinocytosis of extracellular protein promotes glucose independence in non-small cell lung cancer. Cancers (Basel) 11, cancers11010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu W, Zeng L, and Costantini F (1999). Identification of a domain of Axin that binds to the serine/threonine protein phosphatase 2A and a self-binding domain. J. Biol. Chem 274, 3439–3445. [DOI] [PubMed] [Google Scholar]
- Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, and Kikuchi A (1998). Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J. 17, 1371–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong WJ, Yoon J, Park JC, Lee SH, Lee SH, Kaduwal S, Kim H, Yoon JB, and Choi KY (2012). Ras stabilization through aberrant activation of Wnt/β-catenin signaling promotes intestinal tumorigenesis. Sci. Signal 5, ra30. [DOI] [PubMed] [Google Scholar]
- Jope RS, and Johnson GVW (2004). The glamour and gloom of glycogen synthase kinase-3. Trends Biochem. Sci 29, 95–102. [DOI] [PubMed] [Google Scholar]
- Kim SE, Huang H, Zhao M, Zhang X, Zhang A, Semonov MV, MacDonald BT, Zhang X, Garcia Abreu J, Peng L, and He X (2013). Wnt stabilization of β-catenin reveals principles for morphogen receptor-scaffold assemblies. Science 340, 867–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SM, Nguyen TT, Ravi A, Kubiniok P, Finicle BT, Jayashankar V, Malacrida L, Hou J, Robertson J, Gao D, et al. (2018). PTEN deficiency and AMPK activation promote nutrient scavenging and anabolism in prostate cancer cells. Cancer Discov. 8, 866–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch S, Acebron SP, Herbst J, Hatiboglu G, and Niehrs C (2015). Post-transcriptional Wnt Signaling Governs Epididymal Sperm Maturation. Cell 163, 1225–1236. [DOI] [PubMed] [Google Scholar]
- Koivusalo M, Welch C, Hayashi H, Scott CC, Kim M, Alexander T, Touret N, Hahn KM, and Grinstein S (2010). Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J. Cell Biol 188, 547–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leak LV, Liotta LA, Krutzsch H, Jones M, Fusaro VA, Ross SJ, Zhao Y, and Petricoin EF III. (2004). Proteomic analysis of lymph. Proteomics 4, 753–765. [DOI] [PubMed] [Google Scholar]
- Lee S-W, Zhang Y, Jung M, Cruz N, Alas B, and Commisso C (2019). EGFR-Pak signaling selectively regulates glutamine deprivation-induced macropinocytosis. Dev. Cell 50, 381–392.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis WH (1931). Pinocytosis. Bull Johns Hopkins Hosp; 49, 17–27. [Google Scholar]
- Li X, Yost HJ, Virshup DM, and Seeling JM (2001). Protein phosphatase 2A and its B56 regulatory subunit inhibit Wnt signaling in Xenopus. EMBO J. 20, 4122–4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh KM, van Amerongen R, and Nusse R (2016). Generating cellular diversity and spatial form: Wnt signaling and the evolution of multicellular animals. Dev. Cell 38, 643–655. [DOI] [PubMed] [Google Scholar]
- MacDonald BT, Tamai K, and He X (2009). Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniszyn J Jr., Hartsuck JA, and Tang J (1976). Mode of inhibition of acid proteases by pepstatin. J. Biol. Chem 251, 7088–7094. [PubMed] [Google Scholar]
- Miele E, Spinelli GP, Miele E, Tomao F, and Tomao S (2009). Albumin-bound formulation of paclitaxel (Abraxane ABI-007) in the treatment of breast cancer. Int. J. Nanomedicine 4, 99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra A, Menezes ME, Pannell LK, Mulekar MS, Honkanen RE, Shevde LA, and Samant RS (2012). DNAJB6 chaperones PP2A mediated dephosphorylation of GSK3β to downregulate β-catenin transcription target, osteopontin. Oncogene 31, 4472–4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momcilovic M, Bailey ST, Lee JT, Fishbein MC, Braas D, Go J, Graeber TG, Parlati F, Demo S, Li R, et al. (2018). The GSK3 signaling axis regulates adaptive glutamine metabolism in lung squamous cell carcinoma. Cancer Cell 33, 905–921.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama Y, and De Robertis EM (2018). Embryonic regeneration by relocalization of the Spemann organizer during twinning in Xenopus. Proc. Natl. Acad. Sci. USA 115, E4815–E4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwkoop PD, and Faber J (1967). Normal Table of Xenopus laevis (Daudin): A Systematic and Chronological Survey of the Development from the Fertilized Egg till the End of Metamorphosis. (Garland Publishing, Inc.). [Google Scholar]
- Nusse R, and Clevers H (2017). Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 169, 985–999. [DOI] [PubMed] [Google Scholar]
- Palm W (2019). Metabolic functions of macropinocytosis. Philos. Trans. R. Soc. Lond. B Biol. Sci 374, 20180285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm W, Park Y, Wright K, Pavlova N, Tuveson D, and Thompson C (2015). The Utilization of Extracellular Proteins as Nutrients Is Suppressed by mTORC1. Cell 162, 259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm W, and Thompson CB (2017). Nutrient acquisition strategies of mammalian cells. Nature 546, 234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pate KT, Stringari C, Sprowl-Tanio S, Wang K, TeSlaa T, Hoverter NP, McQuade MM, Garner C, Digman MA, Teitell MA, et al. (2014). Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. 33, 1454–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez C, Hauser AD, Vucic EA, and Bar-Sagi D (2019). Plasma membrane V-ATPase controls oncogenic RAS-induced macropinocytosis. Nature 576, 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebecca VW, Nicastri MC, McLaughlin N, Fennelly C, McAfee Q, Ronghe A, Nofal M, Lim C-Y, Witze E, Chude CI, et al. (2017). A unified approach to targeting the lysosome’s degradative and growth signaling roles. Cancer Discov. 7, 1266–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redelman-Sidi G, Binyamin A, Gaeta I, Palm W, Thompson CB, Romesser PB, Lowe SW, Bagul M, Doench JG, Root DE, and Glickman MS (2018). The canonical Wnt pathway drives macropinocytosis in cancer. Cancer Res. 78, 4658–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T, Kawasoe T, Ishiguro H, Fujita M, Tokino T, et al. (2000). AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat. Genet 24, 245–250. [DOI] [PubMed] [Google Scholar]
- Skowyra ML, Schlesinger PH, Naismith TV, and Hanson PI (2018). Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 360, eaar5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhart Z, and Angers S (2018). Wnt signaling in development and tissue homeostasis. Development 145, dev.146589. [DOI] [PubMed] [Google Scholar]
- Steinman RM, Brodie SE, and Cohn ZA (1976). Membrane flow during pinocytosis. A stereologic analysis. J. Cell Biol 68, 665–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson JA, and Yoshida S (2019). Macropinosomes as units of signal transduction. Philos. Trans. R. Soc. Lond. B Biol. Sci 374, 20180157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taelman VF, Dobrowolski R, Plouhinec J-L, Fuentealba LC, Vorwald PP, Gumper I, Sabatini DD, and De Robertis EM (2010). Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell 143, 1136–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejeda-Muñoz N, Albrecht LV, Bui MH, and De Robertis EM (2019). Wnt canonical pathway activates macropinocytosis and lysosomal degradation of extracellular proteins. Proc. Natl. Acad. Sci. USA 116, 10402–10411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium. (2018). UniProt: the universal protein knowledge-base. Nucleic Acids Res. 46, 2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinyoles M, Del Valle-Pérez B, Curto J, Viñas-Castells R, Alba-Castellón L, García de Herreros A, and Duñach M (2014). Multivesicular GSK3 sequestration upon Wnt signaling is controlled by p120-catenin/cadherin interaction with LRP5/6. Mol. Cell 53, 444–457. [DOI] [PubMed] [Google Scholar]
- West MA, Bretscher MS, and Watts C (1989). Distinct endocytotic pathways in epidermal growth factor-stimulated human carcinoma A431 cells. J. Cell Biol 109, 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Fagotto F, Zhang T, Hsu W, Vasicek TJ, Perry WL 3rd, Lee JJ, Tilghman SM, Gumbiner BM, and Costantini F (1997). The mouse Fused locus encodes Axin, an inhibitor of the Wnt signaling pathway that regulates embryonic axis formation. Cell 90, 181–192. [DOI] [PubMed] [Google Scholar]
- Zhang Y, and Commisso C (2019). Macropinocytosis in Cancer: A Complex Signaling Network. Trends Cancer 5, 332–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original data have been deposited to NIH Metabolomics Workbench Data Repository with the project identifier NMDR: PR000897.