

Graphical abstract
Keywords: SARS-CoV-2, Multi-targeting drugs, Organosulfur compounds, Molecular docking analysis, Molecular dynamics simulation, MM-PBSA, Main protease (Mpro), Papain-like protease (PLpro), Spike protein (Spro), Helicase, RNA-dependent RNA polymerase (RdRp)
Abstract
The emerging paradigm shift from ‘one molecule, one target, for one disease’ towards ‘multi-targeted small molecules’ has paved an ingenious pathway in drug discovery in recent years. We extracted this idea for the investigation of drugs for COVID-19. Perceiving the importance of organosulfur compounds, seventy-six known organosulfur compounds were screened and studied for the interaction with multiple SARS-CoV-2 target proteins by molecular dynamics simulation. Lurasidone and its derivatives displayed substantial binding affinity against five proteins (Mpro, PLpro, Spro, helicase and RdRp). The pharmacokinetics, ADMET properties and target prediction studies performed in this work further potentiates the effectiveness against SARS-CoV-2.
1. Introduction
The deplorable situation of the present world aroused by the dreadful behavior of an RNA virus named the Severe Acute Respiratory Syndrome Corona Virus-2 (SARS-CoV-2) is originated in China in late 2019. The Corona Virus Disease (COVID-19) pandemic caused by SARS-CoV-2 massacred about one million lives leaving more than thirty-six million people in infection across 216 countries. Similar infections were reported in 2012 by the Middle East Respiratory Syndrome Corona Virus (MERS-CoV) and the Severe Acute Respiratory Syndrome Corona Virus (SARS-CoV) in 2003 but they were less contagious [1]. According to the World Health Organization (WHO), there are no approved medicines or vaccines for COVID-19 and the leading approach for the development of curative medication is drug repurposing as it allows for the rapid acceptance, known and optimized synthetic route and often facile to leapfrog the preliminary stages of clinical trial [2].
SARS-CoV-2 is an RNA virus which consists mainly of four structural proteins (spike protein, envelope protein, membrane protein and nucleocapsid protein) and sixteen non-structural proteins which are responsible for the viral multiplication and other specific purposes for infection. As the mutation rate and thus evolution rate is more for RNA viruses [3], multiple target binding increases the efficiency of the drug by reducing the effect of viral resistance against one protein [4]. The five targets which play a pivotal role in the viral action are Chymotrypsin Like Protease (3CLpro) otherwise called Main Protease (Mpro), Papain Like Protease (PLpro) which help in virus replication, Spike protein (Spro), promotes the entry of the virus into the human cell by binding with the type 1 transmembrane metallocarboxypeptidase known as Angiotensin Converting Enzyme-2 (ACE-2), RNA-dependent RNA polymerase (RdRp) which helps in RNA synthesis and also helicase which plays a vital role in replication and the central dogma of the virus.
Organosulfur compounds are an important class of molecules with the sulfur-containing functional groups such as sulfones, sulfonamides, disulfides, sulfoxides, thiophene, thiazole etc. and its impact in the pharmaceutical sector is impeccable right from the example of penicillin [5]. Organosulfur compounds are known to have exceptional properties as free radical scavengers, thus antioxidant along with anti-inflammatory and anti-microbial properties. Now, about 25% of all the pharmaceutical drugs are organosulfur compounds and sulfur is the most appeared heteroatom in the drugs after nitrogen and oxygen. Clinical trial of a range of organosulfur compounds such as ritonavir, arbidol [6], baricitinib [7] etc. is currently underway against SARS-CoV-2. We have selected a library of organosulfur compounds which includes FDA approved drug, drug candidates for various diseases and also the reported drugs for SARS-CoV which has 89.1% genomic similarity to that of SARS-CoV-2. The inhibitory action against the druggable targets of SARS-CoV-2 is studied to investigate the possibility of multiple targets binding. Therefore, molecular docking study of the library is carried out with five different target proteins and the molecular dynamics simulation is performed with the hit compounds. The ADMET properties, target prediction and Lipinski’s rule are also predicted to explore about the pharmacokinetics and druggability.
2. Materials and methods
2.1. Ligand preparation
The structure of the approved organosulfur drug compounds was generated in UCSF Chimera [8] through their PubChem ID. The compounds whose structures are not appeared in PubChem are drawn in ChemDraw and the 3D structure was generated in UCSF Chimera from the SMILE string. All the structures were energy minimized through the same software and converted the PDB structure to PDBQT format by using AutoDock Tools.
2.2. Molecular docking by virtual screening
Molecular docking study was carried out through virtual screening by using AutoDock Vina [9] with Perl script for the integration executables to explore the binding affinity and the involved interactions in between all organosulfur compounds in the library and the five druggable protein targets of SARS-CoV-2 namely Main proteases (Mpro, Chain A), Papain-like proteases (PLpro, Chain A), Spike-protein (Spro, Chain B), Helicase protein (Chain A) and RNA-dependent RNA polymerase (RdRp, Chain A). The crystal structure of Mpro (PDB ID: 6Y84), PLpro (PDB ID: 6W9C), Spro (PDB ID: 6LZG), RdRp (PDB ID: 6M71) and helicase (PDB ID: 6ZSL) were retrieved from the protein databank (http://www.rcsb.org). The hydrogen atoms and gasteiger charges were added to each protein, subsequently, all the proteins were saved in PDBQT format by using the AutoDock v4.2 program [10]. For Mpro protein grid box (30 Å × 30 Å × 30 Å) centered at (X12, Y-8, Z20 Å), for PLpro grid box (30 Å × 30 Å × 30 Å) centered at (X-42, Y29, Z30 Å), Spro grid box (30 Å × 30 Å × 30 Å) centered at (X-36, Y33, Z12 Å), RdRp grid box (30 Å × 30 Å × 30 Å) centered at (X120, Y122, Z127 Å at 0.375 Å spacing) and for helicase protein grid box (46 Å x 46 Å x 46 Å) centered at (X-29, Y27, Z-73 Å at 0.375 Å spacing) were prepared and saved the output grid file in txt format. A docking run was given from the command prompt. Best docked conformation and minimum binding energy were considered for further analysis. UCSF chimera was used for the visualization of the docked conformation and results. PyMol was used to get the pdb format of the docked structures.
2.3. Molecular dynamics simulation
Herein, top-ranked five compounds complexed with each target protein were subjected to 50 ns molecular dynamics (MD) simulation using the pmemd.cuda module of AMBER18 suite [11]. Top five compounds i.e. lurasidone, lurasidone sulfoxide, lurasidone endo, lurasidone exo and fananserin complexed with Mpro, PLpro, Spro, RdRp and Helicase resulting 25 complexes were simulated for 50 ns to check the stability of the protein-drug complexes. For the validation of our docking results, we also choose 5 different ligands i.e. indinavir for Mpro, darunavir for PLpro, arbidol for Spro, remdesivir for RdRp and ivermectin for Helicase. Configuration files for all set of complexes were generated using leap module of Ambertool19 [12]. Amber ff14SB [13] and updated generalized amber force field (GAFF2) were used in the case of receptor and ligand respectively. AM1-BCC [14] charge scheme was assigned for the ligands using antechamber [15] module of the AMBER 18. Each complex were solvated into octahedron TIP3P water box [16] along with adequate amount Na+ ions to neutralize the systems. Bond lengths involving hydrogen atoms were kept constant using SHAKE algorithm [17]. A non-bonded cut-off of 10 Å was used for calculating the long-range interaction by the particle mesh Ewald method [18]. The temperature of all systems kept constant at 300 K using Langevin thermostat with a collision frequency of 2 ps−1. Similarly, the pressure of the systems was also controlled using Berendsen’s Barostat. All systems were subjected to two-step minimizations followed by a stepwise heating phase and equilibrium simulation before the production run. A detailed description of the simulation protocol was already discussed in our previous work on COVID 19 [19]. Finally, we performed 50 ns production run for all 30 complexes under NPT ensemble. All these trajectories were used to calculate root mean squared deviation (RMSD) to check the binding stability using the cpptraj module of the Ambertool19 [12]. Last 20 ns data were used for the binding free energy calculation which is discussed in the next section.
2.4. Binding free energy (MM-PBSA scheme) calculation
The molecular mechanics/Poisson-Boltzmann surface area (MM/PBSA) scheme is widely used for estimating binding free energies of receptor-inhibitor complexes [20]. The binding free energy () comprises internal energy (desolvation free energy (, and configurational entropy ( which are related by the following equation [21],
| (1) |
| (2) |
| (3) |
is further composed of (bond, dihedral, and angle), (electrostatic) and (van der Waals) terms. The desolvation part of the free energy is composed of polar () and non-polar solvation energy (). A detailed description of the MM-PBSA method is already discussed in our earlier work [22], [23]. We have used 2000 configurations obtained from the final 20 ns trajectory for the MM-PBSA calculation. Due to high computational cost, configurational entropy calculation is avoided in our work. The estimated binding free energy is mostly used for the relative comparison rather than the absolute value.
2.5. Physicochemical properties
The physicochemical properties according to Lipinski’s rule were calculated for all the selected organosulfur compounds to predict the pharmacokinetics property. SwissADME tool was used to calculate the properties from the SMILES structures of each compound. (http://www.swissadme.ch/).
2.6. ADMET studies
Predicting in-silico pharmacokinetic properties of a new drug is very crucial for further studies. ADMET (Absorption, Distribution, Metabolism, Excretion and Toxicity) prediction provides some important information for new compounds. ADMET studies have been carried out by using the computational pkCSM tool (http://biosig.unimelb.edu.au/pkcsm/prediction).
2.7. Molecular target prediction
For the validation of targets, we used molecular target studies by using the Swiss Target Prediction tool (http://www.swisstargetprediction.ch/) which is a web server that predicts the putative targets of the given molecule by utilizing 2D and 3D similarity index with known ligands. The smile formats of the compounds were entered to obtain the targets.
3. Results and discussion
3.1. Molecular docking study
In the current study, a set of organosulfur compounds were selected to carry out the studies to assess their potential against SARS-CoV-2. To examine the possibility of binding with multiple targets, we have selected five SARS-CoV-2 proteins namely Mpro, PLpro, Spro, RdRp and helicase (Fig. 1 ), and screened the compounds by molecular docking method along with their known inhibitors as the reference compounds such as indinavir for Mpro, darunavir for PLpro, arbidol for Spro, remdesivir for RdRp and ivermectin for helicase.
Fig. 1.

Structure of (A) Mpro, (B) PLpro, (C) Spro, (D) RdRp and (E) Helicase with the active sites marked.
The binding energy obtained for the compound library is depicted as the heat map (Fig. 2 ) and we obtained five hit compounds which can potentially inhibit the five targets of the SARS-CoV-2 (Table 1 ).
Fig. 2.

Binding energy of the selected organosulfur compounds against various proteins of SARS-CoV-2 by molecular docking study.
Table 1.
Binding energy in kcal/mol for the top hit compounds along with the reference compounds against each protein target of SARS-CoV-2.
| Sl. No. | Compound name | Binding energy (kcal/mol) against SARS-CoV-2 proteins |
||||
|---|---|---|---|---|---|---|
| Mpro | PLpro | Spro | RdRp | Helicase | ||
| 1 | Lurasidone | −8.4 | −7.3 | −8.1 | −8.2 | −9 |
| 2 | Lurasidone sulfoxide | −9 | −7.4 | −8.1 | −8.3 | −9.3 |
| 3 | Lurasidone endo | −8.2 | −7.4 | −7.7 | −8.4 | −9.3 |
| 4 | Fananserin | −7.9 | −6.6 | −7.7 | −8.1 | −8.3 |
| 5 | Lurasidone exo | −8.2 | −6.8 | −8.1 | −8 | −8.9 |
| Ref | Indinavir | −7.7 | ||||
| Ref | Darunavir | −6.6 | ||||
| Ref | Arbidol | −6.1 | ||||
| Ref | Remdesivir | −7.4 | ||||
| Ref | Ivermectin | −9.5 | ||||
The results obtained from the molecular docking studies are compared with the already reported drug candidates that inhibit the same target protein of SARS-CoV-2 (Table 2 ). There are potential candidates that act against a single protein but the compounds which can inhibit multi targets effectively are minimal. Famotidine was found to be a candidate which inhibits Mpro, PLpro, RdRp and helicase while dutasteride and dihydroergotamine could inhibit both Mpro and RdRp. In this framework, the compounds mentioned in the Table 1 can be a better consideration as they inhibit five target proteins of SARS-CoV-2.
Table 2.
Binding energy (kcal/mol) of the recently published drug candidates against various proteins of SARS-CoV-2 by molecular docking study.
| Sl. No. | Compound name | Binding energy (kcal/mol) against SARS-CoV-2 proteins |
||||
|---|---|---|---|---|---|---|
| Mpro[24] (6Y84) | PLpro[25] (6W9C) | Spro[26] (6LZG) | RdRp[27] (6 M71) | Helicase*[28] (6JYT/6ZSL) | ||
| 1 | Metacycline | −9.1 | ||||
| 2 | Dutasteride | −8.9 | −9.9# | |||
| 3 | Dihydroergotamine | −8.6 | −9.3# | |||
| 4 | Nelfinavir | −8.6 | ||||
| 5 | Tetracycline | −8.1 | ||||
| 6 | Cryptophycin 1 | −7.7 | ||||
| 7 | Cryptophycin 52 | −8.3** | −7.6 | |||
| 8 | Deoxycylindrospermopsin | −8.6** | −7.9 | |||
| 9 | Famotidine | −6.0## | −7.9 | −6.8## | −5.9## | |
| 10 | Tegobuvir | −8.1 | ||||
| 11 | Bromocriptin | −7.7 | ||||
| 12 | Baicalin | −7.6 | ||||
| 13 | Deleobuvir | −7.6 | ||||
| 14 | Dantrolene | −7.6 | ||||
| 15 | VXR | −8.2 | ||||
| 16 | Streptolydigin | −8.1 | ||||
| 17 | Rifabutin | −7.4 | ||||
| 18 | Rifapentine | −6.9 | ||||
| 19 | VRX | −6.5 | ||||
| 20 | Vapreotide | −11.5 | ||||
| 21 | Saquinavir | −7.6 | ||||
| 22 | Colistin | −7.4 | ||||
| 23 | Glecaprevir | −7.4 | ||||
| 24 | Aprepitant | −6.8 | ||||
The previous works have been carried out with the helicase of SARS-CoV (6JYT) and the current results are based on SARS-CoV-2 protein (6ZSL), #6NUR is the protein PDB ID used for the work, **6M03 is the protein PDB ID used, ##PDB ID not mentioned.
3.1.1. Docking studies of the organosulfur compounds with the SARS-CoV-2 Mpro
SARS-CoV-2 Mpro, also known as 3C-like proteins is a 33.8 kDa cysteine protease consists of three domains (domain I-III) [29]. Mpro involved in the cleavage of polyprotein at eleven conserved sites yielding mature and intermediate non-structural proteins [30]. SARS-CoV-2 Mpro exhibits noncanonical dyad of Cys145-His41 in between domain I and II, connected via a loop with domain III [31]. Cys145 and His41 plays crucial role for substrate recognition [32]. Docking grid was made around these amino acid residues for virtual screening of the selected organosulfur compounds with the Mpro of SARS-CoV-2 (6Y84). Indinavir, a well-known drug that has already been reported to inhibit Mpro of the SARS-CoV-2 [23] was studied as a reference compound. Docking score of the organosulfur compounds along with the reference compound is tabulated in Table 1. The docking result reveals that among seventy-six organosulfur compounds, the lurasidone sulfoxide has a highest binding affinity with −9.0 kcal/mol against Mpro. Based on virtual screening results of SARS-CoV-2 Mpro, lurasidone (−8.4 kcal/mol), lurasidone endo (−8.2 kcal/mol), lurasidone exo (−8.2 kcal/mol), ziprasidone (−8.2 kcal/mol), enzalutamide (−8.0 kcal/mol) and fananserin (−7.9 kcal/mol) were predicted to be the best organosulfur compounds.
3.1.2. Docking studies of the organosulfur compounds with the SARS-CoV-2 PLpro
Papain-like protease or PLpro of SARS-CoV-2 shares 83% sequence similarity with SARS-CoV but distant from MERS-CoV PLpro [33]. PLpro is a multifunctional cysteine protease that process viral polyproteins to a functional replicase complex leading to viral spread [34]. PLpro also involved in deubiquitination, de-ISGylation which obstruct the important signaling pathways causing viral invasion of the innate immune response by the expression of type I interferon [35]. All this evidence strongly suggests that inhibition of PLpro activity can block the viral replication which makes it a vital anti-viral drug target. The molecular docking study of organosulfur compounds with the SARS-CoV-2 PLpro showed that the lurasidone sulfoxide and lurasidone endo generates highest binding energy −7.4 kcal/mol. Whereas, lurasidone (−7.3 kcal/mol), lurasidone exo (−6.8 kcal/mol), ziprasidone (−6.6 kcal/mol), and fananserin (−6.6 kcal/mol) showed the higher binding affinity towards SARS-CoV-2 PLpro protein.
3.1.3. Docking studies of the organosulfur compounds with the SARS-CoV-2 spike protein (Spro)
In order to investigate the potential antiviral drugs targeting spike protein of SARS-CoV-2, virtual screening assisted molecular docking was carried out with seventy-six organosulfur compounds on the binding pocket of spike proteins. It is very well reported that viral entry into the cellular system of host with the assistance of ACE-2 receptor [36]. Glycosylated spike 1 (S1) binds to the ACE-2 receptor of the human host cell surface and mediate entry of virions [37]. The docking scores of organosulfur compounds selected for the study of inhibition of Spro of the SARS-CoV-2 are shown in Table 1. Antiviral organosulfur drug for Influenza virus, arbidol, which has been repurposed against the SARS-CoV-2, is taken as the reference compound and is already in the clinical trial [38]. The docking score of arbidol with Spro is −6.1 kcal/mol. However, lurasidone, lurasidone sulfoxide, lurasidone exo, all three compounds exhibited the best docked score (−8.1 kcal/mol) with SARS-CoV-2 spike proteins. While lurasidone endo (−7.7 kcal/mol), fananserin (−7.7 kcal/mol) and ziprasidone (−7.4 kcal/mol) also showed the higher binding affinity. Analyzing these results, it can be observed that organosulfur compounds exhibited considerably low binding energy with Spro of the SARS-CoV-2 warranting further in vitro and in vivo investigation to consider them as potential drugs for COVID-19.
3.1.4. Docking studies of the organosulfur compounds with the SARS-CoV-2 RdRp
RNA-dependent RNA polymerase (RdRp) also known as nsp12 is composed of two additional subunits, nsp7 and nsp8 [39]. Looking towards the structural insight of RdRp is consisting of N-terminal nidovirus RdRp-associated nucleotidyltransferase (NiRAN) domain, an interface domain and a C-terminal end domain [40]. The central function of RdRp is to catalyzing the viral replication, starting from 3′-poly-A end RdRp catalyzes the copy of RNA genome by utilizing (+) RNA strand as template to synthesize a complementary (−) RNA strand [31]. The active site of RdRp composed of α-helices, antiparallel β-strand and catalytic aspartate [40]. Due to its important role in replication cycle of coronaviruses, RdRp considered as important antiviral drug target [41]. We performed molecular docking studies of organosulfur compound library against the RdRp protein of the SARS-CoV-2 revealed that lurasidone endo exhibited lowest binding energy with −8.4 kcal/mol binding energy whereas lurasidone sulfoxide (−8.3 kcal/mol), lurasidone (−8.2 kcal/mol), fananserin (−8.1 kcal/mol), lurasidone exo (−8.0 kcal/mol), enzalutamide (−7.7 kcal/mol) and ziprasidone (−7.6 kcal/mol), also showed promising binding affinity. Based on these observations, the above-mentioned organosulfur compounds can be potential RdRp inhibitors to combat the SARS-CoV-2 infection
3.1.5. Docking studies of the organosulfur compound with the SARS-CoV helicase
Helicase protein of coronavirus carries out NTP dependent unwinding of double-stranded DNA and RNA in a 5′-3′ direction[42]. Moreover, helicase protein shown to be conserved among the species such as SARS-CoV helicase protein which shares almost 99.83% similarity over the complete length of sequences with the helicase protein of SARS-CoV-2 virus [31]. Several binding domain of the helicase protein have been known such as zinc-binding domain, 1A domain 1B domain and 2A domain [31]. However, domain 1A has been known to involve in the unwinding process. Targeting helicase protein offers potential therapeutic strategy to combat COVID-19 [43]. We took recently solved crystal structure of helicase protein of SARS-CoV-2 (6ZSL) for the molecular docking studies. The docking scores of the selected organosulfur compounds and the reference compound are represented in Table 1. Lurasidone sulfoxide and lurasidone endo showed the highest binding affinity against SARS-CoV-2 helicase protein with −9.3 kcal/mol binding energy. While, lurasidone (−9.0 kcal/mol), lurasidone exo (−8.9 kcal/mol), ziprasidone (−8.8 kcal/mol), enzalutamide (−8.4 kcal/mol) and fananserin (−8.3 kcal/mol) also showed good binding affinity.
3.2. Molecular dynamics simulation
All the MD trajectories of 30 complexes were found to be stable in terms of potential and total energy (data not shown) throughout the 50 ns production simulation. To investigate each protein-ligand complex's stability, we have calculated the root means squared deviation (RMSD) of the backbone atoms of proteins relative to their respective energy minimized conformation. The time evolution of RMSD is displayed in Fig. 3 (A)–(E). It is evident from Fig. 3(A)–(E) that each system has reached equilibrium within the first 10 ns. The average RMSD value was estimated for all cases using the last 30 ns of the trajectory and listed in Table 3 . For all cases, the average RMSD value was found to vary between 1.28 Å and 2.90 Å (see Table 3). The highest deviation was observed for helicase/lurasidone endo, while the lowest RMSD was obtained for Spro/fananserin. Overall, a relatively higher degree of stability was observed for all Spro/ligand complexes. On the other hand, RdRp and helicase showed an overall higher RMSD than the other three receptors because of their large structure, including various flexible loops and domains.
Fig. 3.

Time evolution of root mean squared deviation (RMSD) of backbone atoms for (A) Mpro, (B) PLpro, (C) Spro, (D) RdRp, and (E) helicase complexed with Lurasidone (Lu), Lurasidone Exo (Lu_exo), Lurasidone Endo (Lu_endo), Lurasidone sulfoxide (Lu_s) and Fananserin (Fa) along with the respective reference compounds.
Table 3.
Average backbone RMSD (in Å) of all complexes. The standard errors of the mean (SEM) are listed in the parentheses. The average and SEM values were determined using the block average method.
| System | Reference | Lurasidone | Lurasidone exo | Lurasidone endo | Lurasidone sulfoxide | Fananserin |
|---|---|---|---|---|---|---|
| Mpro | 2.08 (0.03) | 1.54 (0.04) | 1.59 (0.03) | 2.11 (0.07) | 2.28 (0.03) | 2.50 (0.06) |
| PLpro | 1.75 (0.07) | 1.58 (0.03) | 1.79 (0.3) | 1.48 (0.02) | 2.15 (0.04) | 1.62 (0.02) |
| Spro | 1.33 (0.02) | 1.45 (0.02) | 1.60 (0.03) | 1.45 (0.02) | 1.70 (0.02) | 1.28 (0.02) |
| RdRp | 2.37 (0.04) | 2.31 (0.05) | 1.99 (0.02) | 2.55 (0.04) | 2.11 (0.02) | 1.86 (0.02) |
| Helicase | 1.74 (0.03) | 2.78 (0.12) | 2.71 (0.06) | 2.90 (0.07) | 2.07 (0.06) | 2.55 (0.06) |
3.3. Binding free energy analysis
The binding free energy for each complex was calculated using the MM-PBSA scheme and shown in Fig. 4 . The total binding free energy, along with various interactions contributing to the binding free energy, was recorded in Table 4 . It is evident from Table 4 that for all complexes, and contributed favorably to the complexation, whereas was found to disfavor the complex formation. Further, Table 4 revealed that was overcompensated by . In all cases, the binding was mainly driven by the intermolecular van der Waals interactions.
Fig. 4.

Relative binding free energy (kcal/mol) all 30 complexes. Each inhibitor is shown in a different color.
Table 4.
Component of binding free energy for all complexes in kcal/mol. The standard error of the mean (SEM) is listed in the parenthesis.
| Inhibitors | ΔEvdW | ΔEelec | ΔGpol | ΔGnp | ΔEMMa | ΔGsolvb | ΔGTotalc |
|---|---|---|---|---|---|---|---|
| Mpro | |||||||
| Reference | −34.90 (0.25) | −17.02 (0.24) | 31.16 (0.26) | −3.95 (0.02) | −51.92 (0.45) | 27.21 (0.25) | −24.71 (0.22) |
| Lurasidone | −41.38 (0.08) | −16.15 (0.14) | 33.23 (0.10) | −4.07 (0.01) | −57.53 (0.14) | 29.16 (0.10) | −28.37 (0.10) |
| Lurasidone exo | −30.14 (0.18) | −7.05 (0.12) | 22.30 (0.18) | −3.32 (0.02) | −37.19 (0.25) | 18.97 (0.17) | −18.22 (0.12) |
| Lurasidone endo | −28.14 (0.13) | −10.06 (0.17) | 24.13 (0.23) | −3.17 (0.01) | −38.21 (0.24) | 20.96 (0.22) | −17.25 (0.10) |
| Lurasidone sulfoxide | −35.34 (0.08) | −25.15 (0.16) | 54.24 (0.18) | −3.82 (0.01) | −60.50 (0.20) | 50.42 (0.17) | −10.07 (0.11) |
| Fananserin | −20.08 (0.10) | −9.93 (0.11) | 17.12 (0.13) | −2.40 (0.01) | −30.02 (0.17) | 14.72 (0.12) | −15.30 (0.08) |
| PLpro | |||||||
| Reference | –32.73 (0.11) | –32.89 (0.24) | 42.68 (0.18) | −3.68 (0.01) | −65.62 (0.27) | 38.99 (0.18) | −26.62 (0.13) |
| Lurasidone | −24.87 (0.20) | −5.61 (0.16) | 16.67 (0.18) | −2.58 (0.02) | −30.48 (0.30) | 14.09 (0.17) | −16.31 (0.15) |
| Lurasidone exo | −34.41 (0.07) | −11.35 (0.13) | 32,09 (0.13) | −3.26 (0.01) | −45.75 (0.14) | 28.83 (0.13) | −16.93 (0.08) |
| Lurasidone endo | –22.56 (0.13) | −6.36 (0.18) | 16.74 (0.18) | −2.51 (0.01) | −28.92 (0.24) | 14.23 (0.17) | −14.69 (0.10) |
| Lurasidone sulfoxide | −14.97 (0.16) | −10.03 (0.28) | 18.16 (0.30) | −1.66 (0.01) | −25.00 (0.35) | 16.50 (0.29) | −8.50 (0.11) |
| Fananserin | −24.90 (0.09) | −5.78 (0.14) | 20.35 (0.17) | −2.71 (0.01) | −30.68 (0.16) | 17.64 (0.17) | −13.04 (0.09) |
| Spro | |||||||
| Reference | –32.58 (0.08) | −6.59 (0.12) | 23.62 (0.10) | −3.00 (0.01) | −39.18 (0.15) | 20.62 (0.10) | −18.56 (0.09) |
| Lurasidone | −15.97 (0.13) | −7.39 (0.18) | 14.30 (0.15) | −1.87 (0.01) | –23.37 (0.23) | 12.43 (0.14) | −10.94 (0.11) |
| Lurasidone exo | −42.25 (0.08) | −20.47 (0.19) | 44.49 (0.13) | −4.14 (0.00) | −62.72 (0.18) | 40.35 (0.13) | –22.37 (0.11) |
| Lurasidone endo | −21.83 (0.12) | −11.36 (0.21) | 22.87 (0.25) | −2.33 (0.01) | –33.19 (0.29) | 20.54 (0.24) | −12.66 (0.10) |
| Lurasidone sulfoxide | –23.97 (0.16) | −7.92 (0.25) | 20.06 (0.27) | −2.77 (0.01) | −31.88 (0.30) | 17.59 (0.23) | −14.30 (0.13) |
| Fananserin | –22.49 (0.12) | −10.51 (0.24) | 22.71 (0.29) | −2.47 (0.01) | –33.00 (0.32) | 20.24 (0.27) | −12.76 (0.10) |
| RdRp | |||||||
| Reference | −36.84 (0.09) | −80.50 (0.31) | 100.55 (0.27) | −5.14 (0.01) | −117.33 (0.31) | 95.41 (0.27) | −21.92 (0.16) |
| Lurasidone | −36.84 (0.15) | −17.43 (0.26) | 38.78 (0.23) | −4.04 (0.01) | −54.27 (0.24) | 34.74 (0.23) | −19.53 (0.10) |
| Lurasidone exo | −29.47 (0.09) | −16.42 (0.16) | 34.92 (0.16) | −3.74 (0.01) | −45.89 (0.20) | 31.17 (0.16) | −14.72 (0.11) |
| Lurasidone endo | −28.80 (0.08) | −24.83 (0.17) | 42.76 (0.17) | −3.68 (0.01) | −53.63 (0.19) | 39.08 (0.16) | −14.55 (0.11) |
| Lurasidone sulfoxide | −42.27 (0.12) | −8.94 (0.16) | 35.72 (0.22) | −4.43 (0.01) | −51.21 (0.24) | 31.28 (0.22) | −19.94 (0.09) |
| Fananserin | −18.26 (0.17) | −2.59 (0.16) | 12.21 (0.20) | −2.05 (0.02) | −20.85 (0.26) | 10.17 (0.19) | −10.69 (0.11) |
| Helicase | |||||||
| Reference | −50.31 (0.15) | −14.77 (0.13) | 45.41 (0.19) | −5.59 (0.01) | −65.09 (0.21) | 39.08 (0.21) | −25.26 (0.11) |
| Lurasidone | −53.22 (0.10) | −19.08 (0.14) | 43.24 (0.13) | −5.53 (0.01) | −72.30 (0.19) | 37.71 (0.13) | −34.60 (0.11) |
| Lurasidone exo | −47.74 (0.12) | −20.82 (0.19) | 44.06 (0.12) | −4.97 (0.01) | −68.56 (0.27) | 39.09 (0.23) | −29.47 (0.12) |
| Lurasidone endo | −55.62 (0.09) | –33.73 (0.14) | 63.20 (0.15) | −5.32 (0.00) | −89.34 (0.18) | 57.88 (0.15) | −31.47 (0.12) |
| Lurasidone sulfoxide | −53.61 (0.13) | −29.68 (0.13) | 57.21 (0.15) | −5.29 (0.01) | −83.29 (0.18) | 51.92 (0.15) | −31.37 (0.09) |
| Fananserin | −50.86 (0.07) | −27.82 (0.13) | 55.44 (0.13) | −4.64 (0.00) | −78.69 (0.15) | 50.80 (0.13) | −27.89 (0.10) |
ΔEvdW + ΔEelec.
ΔGnp + ΔGpol.
ΔEvdW + ΔEelec + ΔGnp + ΔGpol.
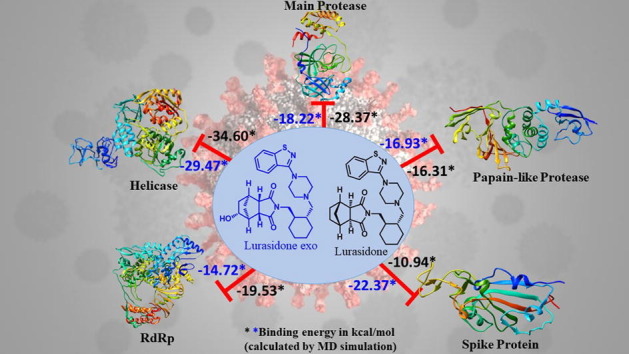
In the case of Mpro, the estimated binding free energy for indinavir, Lurasidone (Lu), Lurasidone exo (Lu_exo), Lurasidone endo (Lu_endo), Lurasidone sulfoxide (Lu_s), and Fananserin (Fa) was −24.71, −28.37, −18.22, −17.25, −10.07, and −15.30 kcal/mol, respectively. Lu displayed a better binding affinity compared to the reference compound (indinavir) as well as other inhibitors. In all cases, the contribution of was found to be nearly 2–3 times more favorable than , indicating a crucial role of the hydrophobic interaction in the binding. Overall, in the case of Mpro, Lu was found to be the most suitable candidate for the binding, followed by Lu_exo.
Similarly, for the target protein PLpro, these two inhibitors (Lu and Lu_exo) also showed the highest binding. Both Lu and Lu_exo bind PLpro with similar free energy, −16.31 kcal/mol and −16.93 kcal/mol, respectively. It was revealed from Table 4 that although both and interactions were more favorable for Lu_exo than Lu; both ligands displayed similar affinity to PLpro. This was because of an increased contribution of unfavorable for Lu_exo compared to Lu. In the case of Spro, Lu_exo displayed binding free energy of –22.37 kcal/mol, which is lower than the binding free energy of the reference inhibitor, arbidol (−18.56 kcal/mol). Both the van der Waals and electrostatic interactions were more favorable for Lu_exo than arbidol, resulting in stronger binding of Lu_exo than arbidol. The other four inhibitors bind with Spro weakly.
Along with Lu, the sulfur derivative of lurasidone (Lu_s) was the top compound in the case of RdRp. The estimated binding free energies were −19.53 and −19.94 kcal/mol for Lu and Lu_s, respectively, while the reference compound (remdesivir) displayed binding free energy of −21.92 kcal/mol. Interestingly, in the helicase case, all five inhibitors showed a higher binding affinity than ivermectin (reference). The predicted binding free energy for Lu, Lu_exo, Lu_endo, and Lu_s was −34.60 kcal/mol, −29.47 kcal/mol, −31.47 kcal/mol, and −31.37, respectively. For the top compound in the case of helicase, i.e. Lu, the net solvation energy disfavored the binding to a lesser extent (−37.71 kcal/mol) than others, making it the best hit. Overall, among all the inhibitors, lurasidone (Lu) was found to have a strong binding affinity against most of the target proteins and come out as the lead molecule in cases of all but Spro.
3.4. Physicochemical properties study based on the Lipinski’s rule
The physicochemical properties of the compounds were studied to predict the pharmacokinetics of the drug by the Lipinski’s rule. The guidelines for an orally active drug according to the Lipinski’s rule are (i) molecular weight (MW) < 500 Daltons, (ii) octanol-water partition coefficient (clogP) < 5, (iii) polar surface area (PSA) < 150 Å2, (iv) number of hydrogen bond donors (HBD) < 5, (v) number of hydrogen bond acceptors (HBA) < 5 and (vi) Number of rotatable bonds (RB) < 10 [44]. The calculated values for the same for the organosulfur compounds that are shown to possess high activity after simulation studies are tabulated in Table 5 and the result showed that both the compounds follow Lipinski’s rule with only one violation in lurasidone exo where lurasidone showed zero violation. This indicates that the compounds have the potential for drug-like activities.
Table 5.
Physicochemical properties of the lurasidone and lurasidone exo.
| Property | Lurasidone | Lurasidone exo |
|---|---|---|
| MW (Daltons) | 492.68 | 508.68 |
| clogP | 4.13 | 3.22 |
| PSA (Å2) | 84.99 | 105.22 |
| No. of HBD | 0 | 1 |
| No. of HBA | 4 | 5 |
| No. of RB | 5 | 5 |
| Violations | Zero | 1 (MW > 500) |
3.5. Prediction of the absorption, distribution, metabolism, excretion, and toxicity (ADMET) profile
We carried out ADMET property profiling to explore the drug likeliness of the organosulfur compounds. Lurasidone and lurasidone exo exhibited efficient binding energy among the all selected organosulfur compounds against RdRp, PLpro, Mpro, Spro and helicase proteins of SARS-CoV-2 in the molecular docking and MD-simulation study. In-silico pharmacological prediction of lurasidone and lurasidone exo were performed using the pkCSM server to assess the overall ADMET properties (see Table 6 ). A favorable ADMET profile is necessary for the molecules in drug discovery. Lurasidone and lurasidone exo showed water solubility and high Caco-2 permeability, which indicate that these drugs can be absorbed orally. Both the compounds showed good human intestinal absorption and skin permeability. However, lurasidone and lurasidone exo were predicted to be substrate of P-glycoprotein as well as P-glycoprotein I and II inhibitor
Table 6.
Predicted ADMET properties of the lurasidone and lurasidone exo.
| Properties | Model name | Predicted values |
Unit | |
|---|---|---|---|---|
| Lurasidone | Lurasidone exo | |||
| Absorption | Water solubility | −3.924 | −3.449 | log mol/L |
| Caco2 permeability | 1.327 | 0.857 | Log Papp in 10-6 cm/s | |
| Human intestinal absorption | 89.166 | 90.502 | % Absorbed | |
| Skin permeability | −2.833 | −3.039 | log Kp | |
| P-glycoprotein substrate | Yes | Yes | Yes/No | |
| P-glycoprotein I inhibitor | Yes | Yes | Yes/No | |
| P-glycoprotein II inhibitor | Yes | Yes | Yes/No | |
| Distribution | VDss | 0.808 | 0.781 | log L/kg |
| Fraction unbound (human) | 0.072 | 0.14 | Fu | |
| BBB permeability | 0.134 | −0.621 | log BB | |
| CNS permeability | −2.014 | −2.732 | log PS | |
| Metabolism | CYP2D6 substrate | Yes | Yes | Yes/No |
| CYP3A4 substrate | Yes | Yes | Yes/No | |
| CYP1A2 inhibitor | No | No | Yes/No | |
| CYP2C19 inhibitor | No | No | Yes/No | |
| CYP2C9 inhibitor | No | No | Yes/No | |
| CYP2D6 inhibitor | Yes | No | Yes/No | |
| CYP3A4 inhibitor | Yes | Yes | Yes/No | |
| Excretion | Total clearance | 0.44 | 0.4 | log ml/min/kg |
| Renal OCT2 substrate | Yes | Yes | Yes/No | |
| Toxicity | AMES toxicity | No | No | Yes/No |
| Maximum tolerated dose (Human) | −0.325 | −0.526 | log mg/kg/day | |
| hERG I inhibitor | No | No | Yes/No | |
| hERG II inhibitor | Yes | Yes | Yes/No | |
| Oral rat acute toxicity (LD50) | 3.562 | 3.529 | mol/kg | |
| Oral rat chronic toxicity (LOAEL) | 1.548 | 1.911 | log mg/kg_bw/day | |
| Hepatotoxicity | Yes | Yes | Yes/No | |
| Skin sensitivity | No | No | Yes/No | |
| T. pyriformis toxicity | 0.298 | 0.295 | µg/L | |
| Minnow toxicity | 1.471 | 1.851 | log mM | |
The Volume of distribution at steady state (VDss) prediction indicates low theoretical dose of lurasidone and lurasidone exo will be required to get it uniformly distributed in blood plasma. Blood brain barrier (BBB) permeability prediction showed that lurasidone and lurasidone exo readily cross the BBB and drug can penetrate the central nervous system.
It is well known that cytochrome P450s can regulate the metabolism of various drugs. In that respect, it is worth to note that inhibitors of CYP2D6/CYP3A4 can hamper the pharmacological properties of drugs. Lurasidone inhibits CYP2D6 and CYP3A4, whereas it is predicted to be a substrate of CYP2D6 and CYP3A4. Whereas, lurasidone exo predicted to be substrate and it inhibits CYP3A4. Further, it was observed that lurasidone and lurasidone exo are substrate of ROCT-2 which means that these drugs excreted through urine.
We have also assessed the toxicity index of the organosulfur compounds. The toxicity prediction from the Ames test (Salmonella typhimurium reverse mutation assay) revealed that lurasidone and lurasidone exo cannot be considered as a mutagenic agent. Low toxicity was predicted for both the compounds in Tetrahymena pyriformis. Lurasidone and lurasidone exo were shown to inhibit the human ether-a-go-go-related gene II (hERG II). However, both the drugs were found to be associated with hepatotoxicity. The maximum recommended tolerated dose (MRTD) in human prediction shows that both the drugs were not violating MRTD. Lurasidone and lurasidone exo are predicted to be low acute toxic compound as it does not fall under minnow toxicity. Additionally, none of the compounds are associated with skin sensitivity.
3.6. Identification of target class for lurasidone and lurasidone exo via target prediction studies
Most of the drug performs their mechanism of action by interacting with the proteins, enzymes and other biomacromolecules. However, many drugs have more than one target. In-silico predictions of drug targets based on resemblance with known drugs are very useful to find out the number of targets. Here we observed that lurasidone has 8% enzyme and protease as a target, whereas lurasidone exo has 8% protease and 4% enzyme as target. As shown in Fig. 5 , lurasidone and lurasidone exo interacts with broad range of proteins and enzymes. The detailed information on the target, common name, UniProt ID, ChEMBL ID, target class, probability and known actives in 2D/3D are shown in the Tables S1 and S2 in the Supplementary Information .
Fig. 5.

Molecular target predictions for (A) lurasidone, (B) lurasidone exo obtained from Swiss target prediction report. The frequency of the target classes (top 25) is depicted in the pie chart.
From all the above studies, we projected that the organosulfur compounds are potential class of molecules to inhibit various proteins of SARS-CoV-2 and thus can be potential drugs against the menace. The other types of organic molecules which are capable to serve as drugs against the COVID-19 are plant derived polyphenols [45], corticosteroids [46], flavonoids [47], peptidyl analogues [48] etc. along with other biologicals such as antibodies and therapeutic RNA [49].
4. Conclusion
As viral drug targets are susceptible to mutations at higher rates, our aim was to investigate compounds which could bind multiple-targets of SARS-CoV-2. From the selected list of organosulfur compounds, we identified compounds that interacted with multiple-targets and interestingly lurasidone and lurasidone exo were found to be very effective on inhibiting all five SARS-CoV-2 targets (Mpro, PLpro, Spro, RdRp and helicase) with significant binding affinities. Hence, this compound can be a potential candidate against SARS-CoV-2 for long-term as it is capable of binding with multiple-targets and inhibiting their activity, thus reducing the effect of drug-resistance. We also found that all the compounds are druggable with negligible violations from the Lipinski’s rule. The ADMET profile and target prediction studies suggest that lurasidone and lurasidone exo has promising pharmacokinetic properties as well. Taken together, we believe that these two organosulfur compounds can potentially inhibit SARS-CoV-2 via binding multiple drug-targets and warrants further in vitro and in vivo investigations.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors acknowledge financial supports from the Indian Institute of Technology Palakkad and Indian Institute of Technology Indore. PK is supported by the Department of Biotechnology, Govt. of India (grant number BT/RLF/Re-entry/40/2014, DBT-Ramalingaswami Re-entry Fellowship). This work was supported by the Department of Science and Technology-Science & Engineering Research Board, Govt. of India (ECR/2017/002082 to S. Sadhukhan, and the Ramanujan Fellowship to M. Porel).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.cplett.2020.138193.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.de Wit E., van Doremalen N., Falzarano D., Munster V.J. SARS and MERS: recentinsights into emerging coronaviruses. Nat. Rev. Microbiol. 2016;14:523–534. doi: 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Senanayake S.L. Drug repurposing strategies for COVID-19. Future Sci. 2020 doi: 10.4155/fdd-2020-0010. [DOI] [Google Scholar]
- 3.Sun J., He W.-T., Wang L., Lai A., Ji X., Zhai X., Li G., Suchard M.A., Tian J., Zhou J. COVID-19: epidemiology, evolution, and cross-disciplinary perspectives. Trends Mol. Med. 2020;26:483–495. doi: 10.1038/s41467-019-13940-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Joshi R.S., Jagdale S.S., Bansode S.B., Shankar S.S., Tellis M.B., Pandya V.K., Chugh A., Giri A.P., Kulkarni M.J. Discovery of potential multi-target-directed ligands by targeting host-specific SARS-CoV-2 structurally conserved main protease. J. Biomol. Struct. Dyn. 2020:1–16. doi: 10.1080/07391102.2020.1760137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.K.A. Scott, J.T. Njardarson, Analysis of US FDA-approved drugs containing sulfur atoms, in: Sulfur Chem., Springer, vol. 376, 2019, pp. 1–34. https://doi.org/10.1038/s41598-020-61432-1. [DOI] [PubMed]
- 6.Sheahan T.P., Sims A.C., Leist S.R., Schäfer A., Won J., Brown A.J., Montgomery S.A., Hogg A., Babusis D., Clarke M.O. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat. Commun. 2020;11:1–14. doi: 10.1038/s41467-019-13940-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richardson P., Griffin I., Tucker C., Smith D., Oechsle O., Phelan A., Stebbing J. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet (London, England) 2020;395:e30. doi: 10.1016/S0140-6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 9.Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Case D.A., Ben-Shalom I.Y., Brozell S.R., Cerutti D.S., Cheatham T.E., III, Cruzeiro V.W.D., Darden T.A., Duke R.E., Ghoreishi D., Gilson M.K., Gohlke H., Goetz A.W., Greene D., Harris R., Homeyer N., Izadi S., Kovalenko A., Kurtzman T., Lee T.S., LeGrand S., Li P., Lin C., Liu J., Luchko T., Luo R., Mermelstein D.J., Merz K.M., Miao Y., Monard G., Nguyen C., Nguyen H., Omelyan I., Onufriev A., Pan F., Qi R., Roe D.R., Roitberg A., Sagui C., Schott-Verdugo S., Shen J., Simmerling C.L., Smith J., Salomon-Ferrer R., Swails J., Walker R.C., Wang J., Wei H., Wolf R.M., Wu X., Xiao L., York D.M., Kollman P.A. University of California; San Francisco: 2018. AMBER 2018. [Google Scholar]
- 12.Roe D.R., Cheatham T.E., III PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 13.Maier J.A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K.E., Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jakalian A., Jack D.B., Bayly C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 15.Wang J., Wang W., Kollman P.A., Case D.A. Antechamber: an accessory software package for molecular mechanical calculations. J. Am. Chem. Soc. 2001;222:U403. [Google Scholar]
- 16.Price D.J., Brooks C.L., III A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004;121:10096–10103. doi: 10.1063/1.1808117. [DOI] [PubMed] [Google Scholar]
- 17.Kräutler V., Van Gunsteren W.F., Hünenberger P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001;22:501–508. doi: 10.1002/1096-987X(20010415)22:5<501::AID-JCC1021>3.0.CO;2-V. [DOI] [Google Scholar]
- 18.Darden T., York D., Pedersen L. Particle mesh Ewald: an N⋅ log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- 19.Sk M.F., Roy R., Jonniya N.A., Poddar S., Kar P. Elucidating biophysical basis of binding of inhibitors to SARS-CoV-2 main protease by using molecular dynamics simulations and free energy calculations. J. Biomol. Struct. Dyn. 2020;1:1–21. doi: 10.1080/07391102.2020.1768149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roy R., Ghosh B., Kar P. Investigating conformational dynamics of Lewis Y Oligosaccharides and elucidating blood group dependency of cholera using molecular dynamics. ACS Omega. 2020;5:3932–3942. doi: 10.1021/acsomega.9b03398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kollman P.A., Massova I., Reyes C., Kuhn B., Huo S., Chong L., Lee M., Lee T., Duan Y., Wang W. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 22.Kar P., Lipowsky R., Knecht V. Importance of polar solvation for cross-reactivity of antibody and its variants with steroids. J. Phys. Chem. B. 2011;115:7661–7669. doi: 10.1021/jp201538t. [DOI] [PubMed] [Google Scholar]
- 23.Jonniya N.A., Sk M.F., Kar P. Investigating phosphorylation-induced conformational changes in WNK1 kinase by molecular dynamics simulations. ACS Omega. 2019;4:17404–17416. doi: 10.1021/acsomega.9b02187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gul S., Ozcan O., Asar S., Okyar A., Barıs I., Kavakli I.H. In silico identification of widely used and well-tolerated drugs as potential SARS-CoV-2 3C-like protease and viral RNA-dependent RNA polymerase inhibitors for direct use in clinical trials. J. Biomol. Struct. Dyn. 2020:1–20. doi: 10.1080/07391102.2020.1802346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quimque M.T.J., Notarte K.I.R., Fernandez R.A.T., Mendoza M.A.O., Liman R.A.D., Lim J.A.K., Pilapil L.A.E., Ong J.K.H., Pastrana A.M., Khan A. Virtual screening-driven drug discovery of SARS-CoV2 enzyme inhibitors targeting viral attachment, replication, post-translational modification and host immunity evasion infection mechanisms. J. Biomol. Struct. Dyn. 2020:1–23. doi: 10.1080/07391102.2020.1776639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chandel V., Sharma P.P., Raj S., Choudhari R., Rathi B., Kumar D. Structure-based drug repurposing for targeting Nsp9 replicase and spike proteins of severe acute respiratory syndrome coronavirus 2. J. Biomol. Struct. Dyn. 2020:1–14. doi: 10.1080/07391102.2020.1811773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elkarhat Z., Charoute H., Elkhattabi L., Barakat A., Rouba H. Potential inhibitors of SARS-cov-2 RNA dependent RNA polymerase protein: molecular docking, molecular dynamics simulations and MM-PBSA analyses. J. Biomol. Struct. Dyn. 2020:1–14. doi: 10.1080/07391102.2020.1813628. [DOI] [PubMed] [Google Scholar]
- 28.Borgio J.F., Alsuwat H.S., Al Otaibi W.M., Ibrahim A.M., Almandil N.B., Al Asoom L.I., Salahuddin M., Kamaraj B., AbdulAzeez S. State-of-the-art tools unveil potent drug targets amongst clinically approved drugs to inhibit helicase in SARS-CoV-2. Arch. Med. Sci. AMS. 2020;16:508–518. doi: 10.5114/aoms.2020.94567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin Z., Zhao Y., Sun Y., Zhang B., Wang H., Wu Y., Zhu Y., Zhu C., Hu T., Du X. Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat. Struct. Mol. Biol. 2020;27:529–532. doi: 10.1038/s41594-020-0440-6. [DOI] [PubMed] [Google Scholar]
- 30.Anand K., Palm G.J., Mesters J.R., Siddell S.G., Ziebuhr J., Hilgenfeld R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α-helical domain. The EMBO J. 2002;21:3213–3224. doi: 10.1093/emboj/cdf32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mirza M.U., Froeyen M. Structural elucidation of SARS-CoV-2 vital proteins: computational methods reveal potential drug candidates against main protease, Nsp12 polymerase and Nsp13 helicase. J. Pharm. Anal. 2020 doi: 10.1016/j.jpha.2020.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020:1–5. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 33.Shin D., Mukherjee R., Grewe D., Bojkova D., Baek K., Bhattacharya A., Schulz L., Widera M., Mehdipour A.R., Tascher G. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. 2020:1–10. doi: 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao X., Qin B., Chen P., Zhu K., Hou P., Wojdyla J.A., Wang M., Cui S. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm. Sin. B. 2020 doi: 10.1016/j.apsb.2020.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Báez-Santos M., John S.E.S., Mesecar A.D. The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015;115:21–38. doi: 10.1016/j.antiviral.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 37.Liu Z., Xiao X., Wei X., Li J., Yang J., Tan H., Zhu J., Zhang Q., Wu J., Liu L. Composition and divergence of coronavirus spike proteins and host ACE2 receptors predict potential intermediate hosts of SARS-CoV-2. J. Med. Virol. 2020;92:595–601. doi: 10.1002/jmv.25726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dong L., Hu S., Gao J. Discovering drugs to treat coronavirus disease 2019 (COVID-19) Drug Discov. Ther. 2020;14:58–60. doi: 10.5582/ddt.2020.01012. [DOI] [PubMed] [Google Scholar]
- 39.Shannon A., Le N.T.T., Selisko B., Eydoux C., Alvarez K., Guillemot J.-C., Decroly E., Peersen O., Ferron F., Canard B. Remdesivir and SARS-CoV-2: Structural requirements at both nsp12 RdRp and nsp14 Exonuclease active-sites. Antiviral Res. 2020:104793. doi: 10.1016/j.antiviral.2020.104793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yin W., Mao C., Luan X., Shen D.-D., Shen Q., Su H., Wang X., Zhou F., Zhao W., Gao M. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science. 2020 doi: 10.1126/science.abc1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y., Anirudhan V., Du R., Cui Q., Rong L. RNA-dependent RNA polymerase of SARS-CoV-2 as a therapeutic target. J. Med. Virol. 2020 doi: 10.1002/jmv.26264. [DOI] [PubMed] [Google Scholar]
- 42.Ivanov K.A., Ziebuhr J. Human coronavirus 229E nonstructural protein 13: characterization of duplex-unwinding, nucleoside triphosphatase, and RNA 5′-triphosphatase activities. J. Virol. 2004;78:7833–7838. doi: 10.1128/JVI.78.14.7833-7838.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Habtemariam S., Nabavi S.F., Banach M., Berindan-Neagoe I., Sarkar K., Sil P.C., Nabavi S.M. Should we try SARS-CoV-2 helicase inhibitors for COVID-19 therapy. Arch. Med. Res. 2020 doi: 10.1016/j.arcmed.2020.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997;23:3–25. doi: 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- 45.Singh S., Sk M.F., Sonawane A., Kar P., Sadhukhan S. Plant-derived natural polyphenols as potential antiviral drugs against SARS-CoV-2 via RNA-dependent RNA polymerase (RdRp) inhibition: an in-silico analysis. J. Biomol. Struct. Dyn. 2020:1–16. doi: 10.1080/07391102.2020.1796810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zha L., Li S., Pan L., Tefsen B., Li Y., French N., Chen L., Yang G., Villanueva E.V. Corticosteroid treatment of patients with coronavirus disease 2019 (COVID-19) Med. J. Aust. 2020;212:416–420. doi: 10.5694/mja2.50577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jo S., Kim S., Shin D.H., Kim M.-S. Inhibition of SARS-CoV 3CL protease by flavonoids. J. Enzyme Inhib. Med. Chem. 2020;35:145–151. doi: 10.1080/14756366.2019.1690480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bhattacharya M., Sharma A.R., Patra P., Ghosh P., Sharma G., Patra B.C., Lee S.S., Chakraborty C. Development of epitope-based peptide vaccine against novel coronavirus 2019 (SARS-COV-2): immunoinformatics approach. J. Med. Virol. 2020;92:618–631. doi: 10.1002/jmv.25736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang S., Hillyer C., Du L. Neutralizing antibodies against SARS-CoV-2 and other human coronaviruses. Trends Immunol. 2020 doi: 10.1016/j.it.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.