

Abstract
Approximately 10%‐20% of patients with clinically localized clear cell renal cell carcinoma (ccRCC) at time of surgery will subsequently experience metastatic progression. Although considerable progression was seen in the systemic treatment of metastatic ccRCC in last 20 years, once ccRCC spreads beyond the confines of the kidney, 5‐year survival is less than 10%. Therefore, significant clinical advances are urgently needed to improve overall survival and patient care to manage the growing number of patients with localized ccRCC. We comprehensively evaluated expression of 388 candidate genes related with survival of ccRCC by using TCGA RNAseq (n = 515), Total Cancer Care (TCC) expression array data (n = 298), and a well characterized Moffitt RCC cohort (n = 248). We initially evaluated all 388 genes for association with overall survival using TCGA and TCC data. Eighty‐one genes were selected for further analysis and tested on Moffitt RCC cohort using NanoString expression analysis. Expression of nine genes (AURKA, AURKB, BIRC5, CCNE1, MK167, MMP9, PLOD2, SAA1, and TOP2A) was validated as being associated with poor survival. Survival prognostic models showed that expression of the nine genes and clinical factors predicted the survival in ccRCC patients with AUC value: 0.776, 0.821 and 0.873 for TCGA, TCC and Moffitt data set, respectively. Some of these genes have not been previously implicated in ccRCC survival and thus potentially offer insight into novel therapeutic targets. Future studies are warranted to validate these identified genes, determine their biological mechanisms and evaluate their therapeutic potential in preclinical studies.
Keywords: biomarkers, clear cell renal cell carcinoma, gene expression, survival
Our study identified nine genes as prognostic biomarkers of ccRCC survival. Some of these genes have not been previously implicated in ccRCC survival and thus potentially offer insight into novel therapeutic targets.

1. INTRODUCTION
Renal cell carcinoma (RCC) is one of the most common renal malignancies, with an estimated 73,750 new cases and 14,830 deaths in US in 2020. 1 Recent studies showed that incidence and mortality rates of RCC are increasing in the United States. 2 These increased rates may, in part, be due to increasing obesity rates and incidental detection during increased abdominal imaging for nonspecific reasons. 2 , 3 Interestingly, obesity also provides an improved survival. 4 The majority of RCC subtypes are classified as clear cell renal cell carcinoma (ccRCC), which account for almost 70‐75% of all RCCs. 5 Cancer‐specific survival rate at 5 years for ccRCC patients is 68.9% and ccRCC has a poorer prognosis compared with other RCC such as papillary and chromophobe RCC (p < 0.001). 6 , 7
The standard of care for localized ccRCC remains surgical excision, and if detected early, ccRCC patients can be cured by surgery. However, about 10%‐20% of ccRCC patients develop metastasis or recurrence following surgical treatment and ultimately die. 7 , 8 Although considerable progression was seen in the systemic treatment of metastatic ccRCC in last 20 years, once ccRCC spreads beyond the confines of the kidney, 5‐year survival is less than 10%. Therefore, the identification of reliable biomarkers for ccRCC progression is greatly needed. We and others reported candidate biomarkers, such as long non‐coding RNAs, 9 gene expression signatures, 10 , 11 , 12 , 13 , 14 , 15 epigenetics 16 for ccRCC progression and/or survival. 17 , 18 , 19 However, there is currently no clinically accepted molecular biomarker for ccRCC progression.
In this study, we determined the potential expression signature of 388 candidate genes in predicting survival in patients with ccRCC. The association between the expression of candidate genes and overall survival in ccRCC patients was first evaluated in a discovery phase comprising two independent datasets: a cohort of 515 ccRCC patients from The Cancer Genome Atlas (TCGA) and 298 patients from the Total Cancer Care (TCC) data from Moffitt Cancer Center. Eighty‐one genes identified from the discovery dataset were further evaluated as independent predictors of ccRCC survival specifically in a different cohort of 248 ccRCC cases from Moffitt Cancer Center.
2. MATERIAL AND METHODS
2.1. Discovery datasets
2.1.1. The Cancer Genome Atlas (TCGA)
TCGA KIRC RNAseq data were downloaded from https://gdc.cancer.gov/about‐data/publications/pancanatlas and log2 transformed. Overall Survival (OS) for the TCGA KIRC samples was retrieved from the publication by Liu et al. 20 This resulted in 515 ccRCC tumor samples with overall survival (OS) data (Table 1). Methylation data was downloaded as RAW IDAT files and normalized.
Table 1.
Clinical and pathological characteristics of participants for TCC and Moffitt validation.
| TCGA | TCC | Moffitt | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Dead (n = 169) | Alive (n = 346) | p value | Dead (n = 141) | Alive (n = 157) | p value | Short‐term survivors (n = 72) | Long‐term survivors (n = 176) | p value | |
| Gender | 0.65 | 0.24 | 0.045 | ||||||
| Female | 63 (37%) | 122 (35%) | 49 (35%) | 60 (38%) | 16 (22%) | 62 (35%) | |||
| Male | 106 (63%) | 224 (65%) | 92 (65%) | 97 (62%) | 56 (78%) | 114 (65%) | |||
| Race | 0.04 | 0.139 | 0.008 | ||||||
| Black | 11 (7%) | 44 (13%) | 3 (2%) | 6 (4%) | 8 (11%) | 5 (3%) | |||
| White | 155 (92%) | 290 (85%) | 137 (97%) | 145 (92%) | 64(89%) | 171 (97%) | |||
| Other | 1 (1%) | 7 (2) | 1 (1%) | 6 (4%) | |||||
| SEER Stage | <0.0001 | <0.0001 | 0.004 | ||||||
| Localized | 53 (31%) | 254 (73%) | 46 (33%) | 126 (80%) | 38 (53%) | 120 (68%) | |||
| Reginal | 50 (30%) | 76 (22%) | 39 (28%) | 21 (13%) | 21 (29%) | 21 (12%) | |||
| Distant met | 66 (39%) | 16 (5%) | 53 (38%) | 10 (7%) | 13 (18%) | 35 (20%) | |||
| Unknown | 3 (2%) | 0 (0%) | |||||||
| Stage | <0.0001 | <0.0001 | <0.0001 | ||||||
| 1 | 40 (24%) | 212 (61%) | 33 (23%) | 105 (69%) | 5 (7%) | 110 (62%) | |||
| 2 | 12 (7%) | 42 (12%) | 14 (10%) | 14 (9%) | 9 (12%) | 13 (7%) | |||
| 3 | 49 (29%) | 74 (21%) | 32 (23%) | 25 (16%) | 40 (56%) | 31 (18%) | |||
| 4 | 67 (40%) | 16 (5%) | 55 (39%) | 10 (6%) | 18 (25%) | 22 (13%) | |||
| Unknown | 1 (1%) | 2 (1%) | 7 (5%) | 3 (2%) | |||||
| Vital Status | <0.0001 | <0.0001 | <0.0001 | ||||||
| Alive | 0 (0%) | 346 (100%) | 0 (0%) | 157 (100%) | 0 (0%) | 137 (78%) | |||
| Dead | 169 (100%) | 0 (0%) | 141 (100%) | 0 (0%) | 72 (100%) | 39 (22%) | |||
2.1.2. Total Cancer Care (TCC)
Under the TCC protocol, 298 ccRCC tumor tissue samples were collected from patients treated at the Moffitt Cancer Center in 1991‐2009 (Table 1). Tumor RNA was extracted at the centralized Moffitt Tissue Core. Global gene expression assays were performed using a custom Affymetrix HuRSTA (Affymetrix, Santa Clara, CA) GeneChips (HuRSTA‐2a520709, ~60,607 probesets). The expression profile for each sample was extracted from the IRON [32] normalized and de‐batched TCC gene expression database, managed by the Cancer informatics Core (CIC). CIC performs strict quality control and pre‐processing of ccRCC samples, including normalization and removal of RNA‐quality dependent batch effect. Sequence based gene annotation of all probesets on the HuRSTA chip was also obtained from CIC.
2.2. Validation dataset
2.2.1. Moffitt RCC cohort
Patients in the validation dataset were 248 ccRCC patients who were surgically treated at the Moffitt Cancer Center between 1992 and 2009 (Table 1). We selected 72 short‐term survivors (<2 years survival after treatment) and 176 long‐term survivors (minimum 5 years survival after treatment). A pathologist (SD) carried out a blinded comprehensive review of all primary tumors to confirm histological subtype (1997 AJCC/UICC classification), tumor stage, 2012 ISUP tumor grade, tumor size, and coagulative tumor necrosis. Representative formalin‐fixed paraffin embedded (FFPE) block with the highest grade was chosen from each resected tumor and tumor region was demarcated for histologic macro‐dissection, which was performed on 10 μm sections. Total RNA was extracted from FFPE tissue sections using the AllPrep DNA/RNA FFPE kit reagents (Qiagen) following the vendor's standard protocols. RNA integrity was assessed via the 260/280 ratio using nanodrop. Gene expression profiling was performed using NanoString platform as described below. Medical records were abstracted and clinical data including age at diagnosis, stage, tumor grade. and metastatic tissue site were recorded. This study was approved by the Moffitt Institutional Review Board.
2.3. NanoString platform for gene expression:
The NanoString platform was used to quantify gene expression of genes selected for follow‐up analysis. HADHA, MAEA RBM4, and TRIM39 were used as house‐keeping genes. Two hundred nanograms of total RNA from each sample was used for the expression according to the manufacturer's instructions. We determined background hybridization using spiked‐in negative controls. Signals were considered to be below the limits of detection if they were lower than two standard deviations above the mean background. Gene expression was quantified and normalized (positive control normalization and housekeeping gene normalization using geometric mean) using NanoString nSolverTM 4.0 software. Expression values were log2 transformed and exported to MATLAB R2020b software for further analysis.
2.4. Gene selection
Candidate ccRCC‐related genes were selected based on previous published literature., 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 including our review article 19 and pathways described in the Cancer Genome Anatomy project. A total of 388 candidate genes were evaluated in the discovery sets. A subset of 81 genes were selected from the discovery sets for further evaluation in the validation set based on the reproducibility in the two discovery datasets (n = 52 genes), prognostic value, and biological relevance (n = 29 genes, e.g. known function in ccRCC or other cancers) (Table S2).
2.5. Statistical analysis
Participants’ demographic and clinical characteristics were summarized using descriptive statistics, counts, and percentages for categorical variables and means and standard deviations or median and ranges for numeric variables. All genes in the discovery datasets were tested for association with overall survival using Kaplan‐Meier (K‐M). For the K‐M, the median expression was used as a cutpoint to dichotomize expression. In the validation dataset, we compared differential gene expression (of the identified genes in the discovery datasets) between aggressive and indolent cases of ccRCC using t‐test. P values were used to select candidate genes. Genes with p < 0.01 in both sets were considered to be differentially expressed. Univariate and multivariate Cox proportional hazards regression models were sequentially built with genes and clinical prognostic factors (tumor stage and grade) for TCGA and TCC data set and followed by time‐dependent receiver‐operating characteristic curve (ROC) analysis at year 5. Logistic regression was used along with ROC analysis for Moffitt dataset analysis. Area under ROC curve (AUC) were calculated to compare prognostic values of the models.
3. RESULTS
3.1. Results from Discovery phase using TCGA and TCC data
We screened 388 ccRCC‐related candidate genes for their association with overall survival in two independent discovery datasets. A total of 148 genes were associated with OS using RNA‐seq data of 515 ccRCC patients from the TCGA data portal and 99 genes using Affymetrix data for 298 ccRCC patients from TCC (log rank p‐value <0.01). A subset of 81 genes was selected for further evaluation using NanoString in the Moffitt validation set. The selection of these genes was based on overlap between the two discovery datasets (n = 52) and manual curation that integrated statistical and biological information (n = 29), with function in ccRCC or other cancers were favored known).
3.2. Results from validation set, Moffitt cohort using NanoString assay
The demographic and clinical characteristics of participants are in Table 1. The expression of the resulting 81 genes were measured using NanoString on 72 short (<2 years survival after treatment) and 176 long‐term survivors (minimum 5 years survival) (Figure 1). Nine genes (AURKA, AURKB, BIRC5, CCNE1, MK167, MMP9, PLOD2, SAA1, and TOP2A) were confirmed based on expression level between short‐ and long‐term survivor cases (Figure 2). All the nine genes were overexpressed in short survivors compared to long‐term survivors. SAA1 (Log2 fold change (FC)=3.52, p = 1.01E‐10) was the most highly overexpressed, followed by MMP9 (FC=1.82, p = 3.82E‐09), BIRC5 (FC=1.40, p = 2.39E‐14), PLOD2 (FC=1.42, p = 9.79E‐10), TOP2A (FC=1.24, p = 7.32E‐14), MKI67 (FC=1.13, p = 7.41E‐11), AURKB (FC=0.99, p = 2.86E‐11) CCNE1 (FC=0.71, p = 8.01E‐09), and AURKA (FC=0.49, p = 1.90E‐9) (Table 2). Overexpression of these nine genes were associated with poor overall survival in the TCGA and TCC datasets with hazard ratios (HRs) ranging from 1.49‐2.99 (Figure 3). The association between expression of AURKB, BIRC5, CCNE1, MMP9, SAA1, TOP2A, and overall survival was slightly lower in the TCC dataset compared to that of TCGA, while the association was higher in TCC compared to TCGA for AURKA, MKI67, and PLOD2.
Figure 1.

Outline of overall study design. Data from 515 and 298 patients, respectively, were obtained from TCGA and TCC. A COX regression analysis identified genes with expression levels associated with overall survival. Expression levels of 81 genes were further evaluated using NanoString in 248 cases
Figure 2.

Boxplots of nine genes. Nine genes (AURKA, AURKB, BIRC5, CCNE1, MK167, MMP9, PLOD2, SAA1, and TOP2A) were confirmed based on expression level between short‐ and long‐term survivor cases in validation set. All the nine genes were overexpressed in short‐term survivors (aggressive) compared to long‐term survivors (indolent). ****p < 0.0001
Table 2.
Discovery and Validation of genes associated with survival.
| Gene | Location | TCGA | TCC | Moffitt | |||
|---|---|---|---|---|---|---|---|
| HR | p value | HR | p value | FC* | p value | ||
| AURKA | 20q13 | 2.15 (1.59‐2.91) | 1.30E−06 | 2.42 (1.73‐3.37) | 2.38E−07 | 0.49 | 1.90E−9 |
| AURKB | 17p13.1 | 2.71 (2.00‐3.66) | 8.63E−10 | 1.50 (1.07‐2.08) | 1.68E−02 | 0.99 | 2.86E−11 |
| BIRC5 | 17q25 | 2.51 (1.85‐3.39) | 1.06E−08 | 1.69 (1.22‐2.36) | 1.70E−03 | 1.40 | 2.39E−14 |
| CCNE1 | 19q12 | 2.65 (1.96‐3.59) | 8.01E−10 | 1.89 (1.36‐2.64) | 1.53E−04 | 0.71 | 8.01E−09 |
| MKI67 | 10q26.2 | 1.65 (1.22‐2.23) | 1.29E−03 | 2.05 (1.47‐2.86) | 2.24E−05 | 1.13 | 7.41E−11 |
| MMP9 | 20q11.2‐q13.1 | 1.77 (1.31‐2.39) | 2.44E−04 | 1.49 (1.07‐2.07) | 1.78E−02 | 1.82 | 3.82E−09 |
| PLOD2 | 3q24 | 1.74 (1.29‐2.35) | 4.27E−04 | 2.99 (2.14‐4.17) | 2.31E−10 | 1.42 | 9.79E−10 |
| SAA1 | 11p15.1 | 2.47 (1.82‐3.34) | 1.64E−08 | 2.90 (2.08‐4.04) | 6.24E−10 | 3.52 | 1.01E−10 |
| TOP2A | 17q21‐q22 | 1.87 (1.38‐2.52) | 7.43E−05 | 2.10 (1.51‐2.92) | 1.19E−05 | 1.24 | 7.32E−14 |
Log 2‐fold change.
Figure 3.
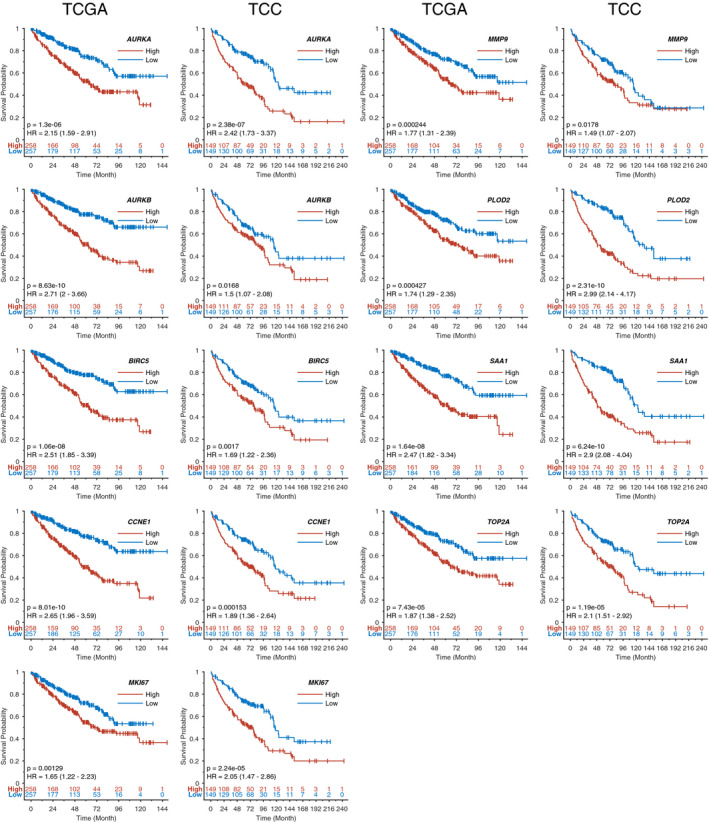
Overexpression of these nine genes was associated with poor overall survival in the TCGA and TCC datasets with hazard ratios (HRs) ranging from 1.49 to 2.99 in discovery set
3.3. Testing the prognostic risk models using ROC analysis
Cox regression models with nine genes, or clinical prognostic factors (tumor stage and grade) showed AUC of 0.731, 0.737 in TCGA (Figure 4A), 0.783, 0.716 in TCC data set (Figure 4B). Incorporating 9 genes into clinical factors regression models yielded non‐significantly increased AUC values of 0.776 in TCGA and 0.821 in TCC. On the other hand, combining nine genes with the clinical factors significantly improved AUC from 0.702 to 0.873 in Moffitt validation dataset (Figure 4C; Table S1).
Figure 4.

Analysis of survival prognostic risk models: three models (nine genes, stage/grade, and combined) for TCGA, TCC, and Moffitt data. 4A. multivariate Cox regression models showed time‐dependent AUC of 0.731, 0.737, and 0.776 in TCGA. 4B. AUC of 0.783, 0.716, and 0.821 in TCC. 4C. logistic regression model for Moffitt data showed AUC of 0.852, 0.702, and 0.873
3.4. Epigenetic regulation of SAA1 and PLOD2
To investigate whether any of these nine genes was epigenetically regulated by methylation, we used the TCGA dataset, combining RNAseq and Illumina 450 K methylation data. Two of the nine genes, SAA1 and PLOD2 showed a negative correlation between methylation level and gene expression level, indicating that these genes are regulated by methylation. Figure 5A shows a box‐plot for each of the 8 CpG‐probes for SAA1 comparing tumor tissue vs normal samples. The data suggested that many tumor samples show hypomethylation compared to normal samples. Three of the probes showed a clear negative correlation between the methylation level and the expression level (Figure 5B) and these probes also showed a high degree of correlation between each other (Figure 5C). The hypo‐methylation of these three probes led to an increased expression (Figure 5D).
Figure 5.
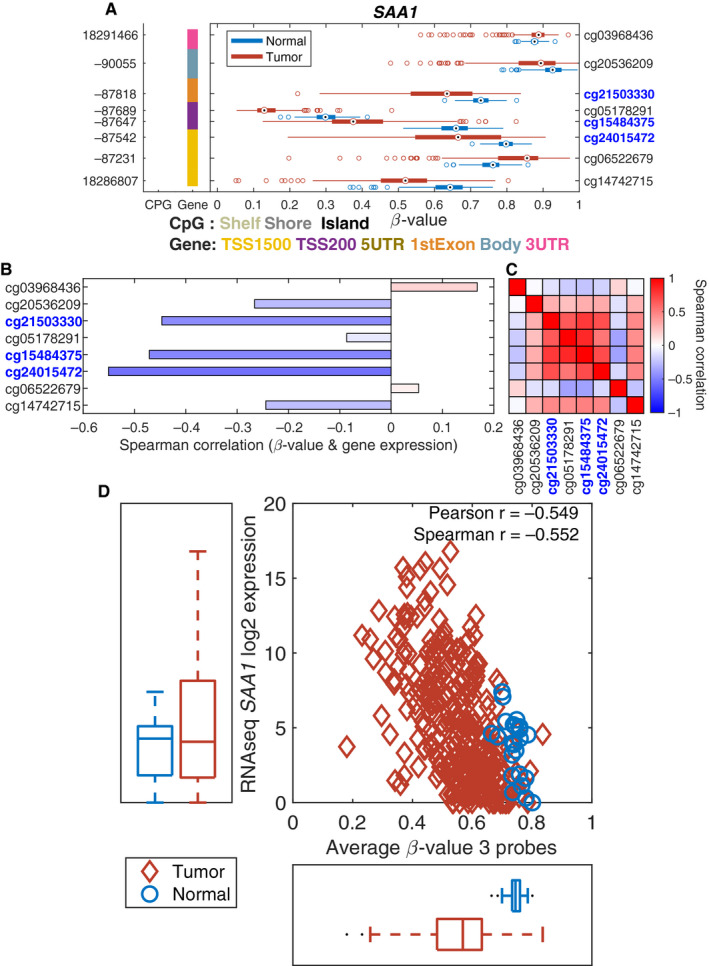
Methylation driven expression of SAA1. 5A. A box‐plot for each of the eight CpG‐probes for SAA1 comparing tumor vs normal samples. 5B. A negative correlation between the methylation and the expression level. 5C. A high degree of correlation between methylation and the expression level. 5D. Hypomethylation of these CpG sites leads to an increased expression
PLOD2 showed a more complicated methylation profile (Figure 6). There are 21 CpG‐probes available for PLOD2 (Figure 6A) with three probes being negatively correlated (r < −0.3) with expression value (Figure 6B). These three CpG‐probes are also correlated (Figure 6C). The average methylation for these CpG‐probes was negatively correlated with the expression level of PLOD2 (Figure 6D).
Figure 6.
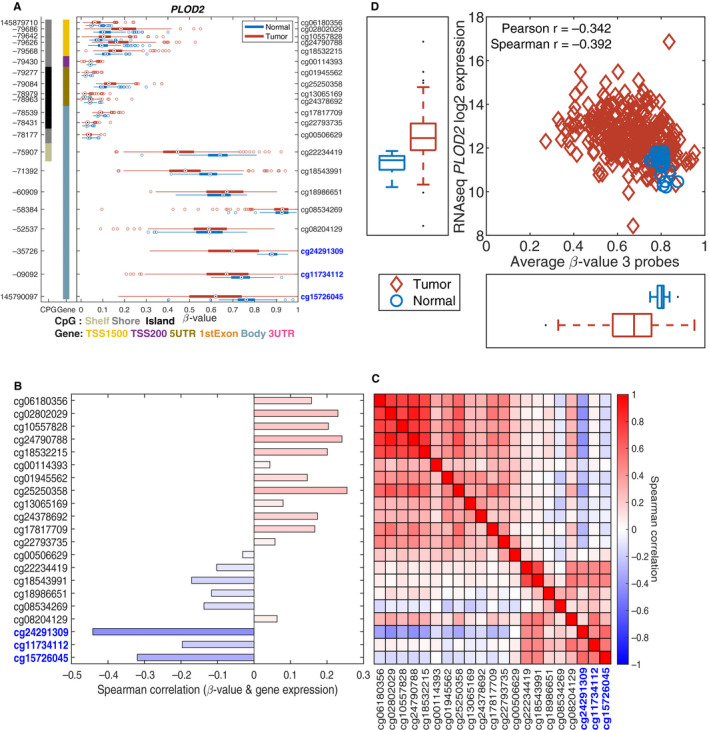
Methylation driven expression of PLOD2. 6A. A box‐plot for each of the 21 CpG‐probes for PLOD2 comparing tumor vs normal samples. 6B. A negative correlation between the methylation and the expression level. 6C. A high degree of correlation between methylation and the expression level. 6D. Hypomethylation of these CpG sites leads to an increased expression
4. DISCUSSION
In this study, we used a multi‐stage design to identify genes associated with ccRCC survival. First, we evaluated, in two independent datasets, the association between overall survival and 388 genes identified through literature search. We then selected 81 genes based on the magnitude of the association with outcome, overlap in the two discovery datasets and biological relevance for validation using the NanoString platform in an independent cohort that included 72 short‐term survivors. Differential expression for nine genes (AURKA, AURKB, BIRC5, CCNE1, MKI67, MMP9, PLOD2, SAA1, and TOP2A) was validated on the NanoString platform. Six of these validated genes (BIRC5, MK167, MMP9, PLOD2, and TOP2A) have previously been implicated ccRCC, 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 while the others have been implicated in other cancers. 35 , 36 , 37 , 38 The prognostic value of models with nine genes and clinical prognostic factors are 0.776, 0.821, and 0.873 in TCGA, TCC and Moffitt data set, respectively.
BIRC5, Baculoviral IAP Repeat Containing 5, or survivin plays a role in apoptotic cell death. Differential expression is associated with survival in ccRCC. 21 Survivin expression is increased in ccRCC compared to adjacent normal renal tissues and the expression is positively correlated with pathological grade and clinical stage. 22 Expression of the survivin protein is associated with ccRCC progression and poor survival. 23 , 24
MKI67, marker of proliferation Ki67, is a nuclear protein associated with cellular proliferation. It has also been implicated in poor outcomes for patient undergoing surgery for localized ccRCC, 25 metastasis of ccRCC, 26 and recurrence of ccRCC after surgery. 39 It is also upregulated in ovarian cancer cell lines treated with estradiol or genistein suggesting a role in drug response. 27
MMP9, Matrix Metallopeptidase 9, plays a role in the breakdown of extracellular matrix during cancer metastasis. It is highly expressed in ccRCC 40 and implicated in the pathogenesis of ccRCC. 28 Upregulation of MMP9 is also associated with migration and invasion 29 and progression of ccRCC. 30 MMP9 was recently included in a 4‐gene prognostic prediction set for predicting the prognosis of ccRCC. 31
PLOD2, Procollagen‐Lysine, 2‐Oxoglutarate 5‐Dioxygenase 2, is a hypoxia‐induced membrane‐bound homodimeric enzyme that is involved in collagen synthesis and extracellular matrix degradation. PLOD2 is overexpressed in ccRCC and downregulation significantly inhibits cell migration and invasion. 32 Overexpression of PLOD2 is also associated with lymph node metastasis and poor recurrence‐free and overall survival in biliary, 41 breast, 42 hepatocellular carcinoma (HCC), 43 cervical, 44 lung. 45 , 46 gastric, 47 glioma 48 and bladder 49 cancers.
TOP2A, DNA Topoisomerase II Alpha, plays an important role in transcription through controlling and altering topologic states of DNA. It is upregulated in ccRCC and associated with progression and prognosis, especially survival among patients undergoing surgery, with a more prominent prognostic value among patients with low‐risk disease. 30 , 33 , 34 It is also important for the progression of prognostic marker for papillary RCC. 50
Aurora‐A (AURKA) and Aurora‐B (AURKB) are kinases that play key roles in the regulation of cell‐cycle progression. 51 , 52 , 53 In addition to cell cycle regulation, Aurora‐A is involved in contributing to epithelial‐mesenchymal transition (EMT) and stem‐like properties of cancer cells. 54 It is implicated in the activation of the mTOR pathway in sarcomatoid RCC. 55 A recent study showed that it is overexpressed in metastasis compared to primary RCC tumors. 56 In addition, AURKA is involved in the pathogenesis or progression of hepatocarcinoma, 35 bladder, 36 breast, 37 liver, 38 gastric, 57 colon, 58 non‐small cell lung, 59 and pancreatic 60 cancers. AURKB modulates drug response in non‐small cell lung, 61 and breast cancers. 62 It is implicated in chronic myelocytic leukemia 63 gastric cancer, 64 and leukemia. 65
CCNE1 and SAA1 have not been previously implicated in RCC, but are implicated in other cancers. CCNE1 is a major G1/S phase cyclin. It is associated with aggressive potential in endometrial cancer. 66 , 67 Upregulation of CCNE1 is associated with worse prognosis of ovarian clear cell carcinoma 68 and treatment resistance and poor outcome in high grade serous ovarian cancer. 69 Upregulation of CCNE1 is also observed in aggressive osteosarcoma 70 and also implicated in cisplatin resistance in bladder cancer. 71 Differential expression of CCNE1 is also observed in other cancers including non‐small cell lung cancer (NSCLC), 72 bladder, 73 breast 74 and hepatocellular carcinoma. 75
Serum Amyloid A1, SAA1, is an apolipoprotein that is highly expressed in response to inflammation and tissue injury. It is overexpressed in glioblastoma (GBM), 76 , 77 cervical carcinoma, 78 NSCLC, 79 AML, 80 and gastric cancer 81 and associated with progression and poor prognosis in these cancers.
Previous studies reported differential expressions of various genes in clinical specimens of ccRCC compared to adjacent uninvolved renal tissues and suggested that these genes may serve as promising ccRCC risk stratification biomarkers. 19 , 82 Nuclear HIF expression, elevated expression of Ubiquitin Protein Ligase E3C (UBE3C), reduced expression of phos‐Akt in the nucleus and CAIX and loss of p27 expression are reported as significant independent prognostic factors for poor ccRCC outcomes. 83 , 84 , 85 , 86 , 87 A gene signature with five protein markers (Ki‐67, p53, endothelial VEGFR‐1, epithelial VEGFR‐1, and epithelial VEGF‐D) was proposed to predict survival for ccRCC with AUC of 0.838. 88 CXCL13 was upregulated in ccRCC tumor tissues and CXCL13 expression was associated with advanced stage and poor prognosis in ccRCC. Therefore, CXCL13 expression was proposed as a diagnostic biomarker for ccRCC with AUC of 0.809. 82 Receptor tyrosine kinase (TEK) plays an important role in angiogenesis and remodeling. Downregulation of TEK expression was observed in ccRCC tissues and associated with poor outcome with AUC between 0.637 and 0.839. 89 Recently, seven differentially expressed autophagy‐related genes (PRKCQ, BID, BAG1, BIRC5, ATG16L2, EIF4EBP1, and ATG9B) were included in a recent prognostic survival assessment tool for ccRCC with AUCs of 0.752 and 0.783 for overall and disease free survival, respectively. 90 Similarly, 10 differentially expressed genes (AGR3, CSF2, GAL3ST2, IGLL1, PLG, SAA1, SBSN, SOX2, WFDC13, and ZIC2) were included in a recent prognostic risk assessment tool for ccRCC with AUC of 0.99 without a validation set. 91 The results from previous studies are not consistent with one from the current study. Potential reasons for inconsistency can be small sample sizes, different racial/ethnic background, potential environmental factors and more importantly, lack of validation set.
Dysregulation of gene expression in ccRCC could be caused by various genomic aberrations, such as methylation. We observed that two of the nine genes SAA1 and PLOD2 gene expression is partly regulated by DNA methylation levels in the promoter region. This is similar to what we have seen in our other studies using the TCGA gene expression and methylation data. 92 , 93
Strengths of our study include a multistage design that consisted of two discovery independent discovery datasets and a validation dataset; the use of NanoString platform that allows for accurate and reproducible measurement of RNA from FFPE samples; selection of candidate genes based on a thorough literature search 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 ; and validation of the role of identified genes previously implicated in ccRCC and are potential targets for novel cancer therapies. Indeed, inhibitors of some of these genes are already in early phase I trials. 94 , 95 , 96 We also identified genes that have not been previously identified in ccRCC and represent possible targets for therapy and warrant further evaluation.
A limitation of our study is the selection of the 29 out of the 81 genes for validation based on biological significance. This selection process could potentially introduce selection bias. Thus, candidate gene approach may exclude critical genes for ccRCC survival. In addition, spatial heterogeneity could result in different clonal populations within the same tumor and affect the expression profiling of the evaluated genes in the tumors. Future studies are therefore required to validate our results, use immunohistochemistry to assay the corresponding protein expression, as well demonstrate that therapeutic targeting of the identified genes will result in inhibition of tumor growth. Another limitation is the use of RNA extracted from FFPE tumor tissue. RNA from FFPE may be degraded, but the NanoString platform allows accurate and reproducible measurement of RNA from FFPE samples. The discovery and validation cohorts differed in that the validation cohort was comprised largely of aggressive cases. Furthermore, the gene expression in the discovery datasets was generated on RNA‐seq (for TCGA) and Affymetrix array (for TCC), while the validation study utilized NanoString platform. Despite the differences in the platforms, we identified genes that were consistently differentially expressed in the three different cohorts. In summary, our study identified nine genes as prognostic biomarkers of aggressive ccRCC that are differentially expressed during progression. Some of these genes have not been previously implicated in ccRCC and thus potentially offer insight into novel therapeutic targets. These biomarkers may help to identify aggressive ccRCC patients, who need more intensive treatment. Future studies are warranted to validate the identified genes, determine their biological mechanisms, and evaluate their therapeutic potential in preclinical studies.
CONFLICT OF INTEREST
No author has COI.
AUTHORS CONTRIBUTION
Conception or design of the work was done by AB, EKA, YCK, PES, WJS, BM, RC, CDY, HNL, GDP, AP, and JYP. Data collection was done by AB, PES, WJS, BM, HYP, LW, JC, RC, CDY, HNL, and GDP. Data analysis and interpretation were done by AB, EKA, YCK, PES, WJS, BM, LW, JC, RC, CDY, HNL, GDP, AP, and JYP. Drafting the article was done by AB, EKA, YCK, and JYP. Critical revision of the article was done by AB, EKA, YCK, BM, and JYP. Final approval of the version to be published was done by all authors, AB, EKA, YCK, PES, WJS, BM, HYP, LW, JC, RC, CDY, HNL, GDP, AP, and JYP.
Supporting information
Table S1‐S2
ACKNOWLEDGEMENTS
The authors acknowledge the Moffitt Cancer Center, Tissue Core, and Molecular Genome Core, P30‐CA076292 for their assistance. We thank all patients and their families for their contributions to this study.
Berglund A, Amankwah E K., Kim Y‐C, et al. Influence of gene expression on survival of clear cell renal cell carcinoma. Cancer Med. 2020;9:8662–8675. 10.1002/cam4.3475
Anders Berglund and Ernest K. Amankwah are equally contributed.
Funding information
The development of this manuscript was supported by Florida State James Esther King biomedical program, the National Cancer Institute at the National Institutes of Health (R01CA134466 PI: A. Parker), and Moffitt Cancer Center De Bartolo Family Personalized Medicine Institute (PI: Park).
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author, JYP.
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. [DOI] [PubMed] [Google Scholar]
- 2. Zheng T, Zhu C, Bassig BA, et al. The long‐term rapid increase in incidence of adenocarcinoma of the kidney in the USA, especially among younger ages. Int J Epidemiol. 2019;48(6):1886–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Capitanio U, Bensalah K, Bex A, et al. Epidemiology of Renal Cell Carcinoma. Eur Urol. 2019;75(1):74–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sanchez A, Furberg H, Kuo F, et al. Transcriptomic signatures related to the obesity paradox in patients with clear cell renal cell carcinoma: a cohort study. Lancet Oncol. 2020;21(2):283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shuch B, Amin A, Armstrong AJ, et al. Understanding pathologic variants of renal cell carcinoma: distilling therapeutic opportunities from biologic complexity. Eur Urol. 2015;67(1):85–97. [DOI] [PubMed] [Google Scholar]
- 6. Cheville JC, Lohse CM, Zincke H, Weaver AL, Blute ML. Comparisons of outcome and prognostic features among histologic subtypes of renal cell carcinoma. Am J Surg Pathol. 2003;27(5):612–624. [DOI] [PubMed] [Google Scholar]
- 7. Matsuda T, Hori M. Five‐year relative survival rate of kidney and renal pelvis cancer in the USA, Europe and Japan. Jpn J Clin Oncol. 2015;45(1):136. [DOI] [PubMed] [Google Scholar]
- 8. Nguyen DP, Vertosick EA, Corradi RB, et al. Histological subtype of renal cell carcinoma significantly affects survival in the era of partial nephrectomy. Urol Oncol. 2016;34(6):259.e1–259.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zeng J‐H, Lu W, Liang L, et al. Prognosis of clear cell renal cell carcinoma (ccRCC) based on a six‐lncRNA‐based risk score: an investigation based on RNA‐sequencing data. J Transl Med. 2019;17(1):281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cao D‐L, Dai W‐X, Huang Y‐Q, et al. Development and validation of a robust multigene signature as an aid to predict early relapse in stage I‐III clear cell and papillary renal cell cancer. J Cancer. 2020;11(5):997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu J, Jin S, Gu W, et al. Construction and Validation of a 9‐Gene Signature for Predicting Prognosis in Stage III Clear Cell Renal Cell Carcinoma. Front Oncol. 2019;9:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hakimi AA, Voss MH, Kuo F, et al. Transcriptomic profiling of the tumor microenvironment reveals distinct subgroups of clear cell renal cell cancer: data from a randomized phase III trial. Cancer Discov. 2019;9(4):510–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Büttner F, Winter S, Rausch S, et al. Clinical utility of the S3‐score for molecular prediction of outcome in non‐metastatic and metastatic clear cell renal cell carcinoma. BMC Med. 2018;16(1):108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Morgan TM, Mehra R, Tiemeny P, et al. A multigene signature based on cell cycle proliferation improves prediction of mortality within 5 yr of radical nephrectomy for renal cell carcinoma. Eur Urol. 2018;73(5):763–769. [DOI] [PubMed] [Google Scholar]
- 15. Luo J, Xie YI, Zheng Y, et al. Comprehensive insights on pivotal prognostic signature involved in clear cell renal cell carcinoma microenvironment using the ESTIMATE algorithm. Cancer Med. 2020;9(12):4310–4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hsieh JJ, Le VH, Oyama T, Ricketts CJ, Ho TH, Cheng EH. Chromosome 3p Loss‐Orchestrated VHL, HIF, and Epigenetic Deregulation in Clear Cell Renal Cell Carcinoma. J Clin Oncol. 2018;36(36):3533–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mytsyk Y, Dosenko V, Skrzypczyk MA, et al. Potential clinical applications of microRNAs as biomarkers for renal cell carcinoma. Cent European J Urol. 2018;71(3):295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang L, Zha Z, Qu W, et al. Tumor necrosis as a prognostic variable for the clinical outcome in patients with renal cell carcinoma: a systematic review and meta‐analysis. BMC Cancer. 2018;18(1):870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Di Pietro G, Luu HN, Spiess PE, et al. Biomarkers and new therapeutic targets in renal cell carcinoma. Eur Rev Med Pharmacol Sci. 2018;22(18):5874–5891. [DOI] [PubMed] [Google Scholar]
- 20. Liu J, Lichtenberg T, Hoadley KA, et al. An Integrated TCGA Pan‐Cancer Clinical Data Resource to Drive High‐Quality Survival Outcome Analytics. Cell. 2018;173(2):400–16 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kosari F, Parker AS, Kube DM, et al. Clear cell renal cell carcinoma: gene expression analyses identify a potential signature for tumor aggressiveness. Clin Cancer Res. 2005;11(14):5128–5139. [DOI] [PubMed] [Google Scholar]
- 22. Shi ZG, Li SQ, Li ZJ, Zhu XJ, Xu P, Liu G. Expression of vimentin and survivin in clear cell renal cell carcinoma and correlation with p53. Clin Transl Oncol. 2015;17(1):65–73. [DOI] [PubMed] [Google Scholar]
- 23. Parker AS, Kosari F, Lohse CM, et al. High expression levels of survivin protein independently predict a poor outcome for patients who undergo surgery for clear cell renal cell carcinoma. Cancer. 2006;107(1):37–45. [DOI] [PubMed] [Google Scholar]
- 24. Weber T, Meinhardt M, Zastrow S, Wienke A, Fuessel S, Wirth MP. Immunohistochemical analysis of prognostic protein markers for primary localized clear cell renal cell carcinoma. Cancer Invest. 2013;31(1):51–59. [DOI] [PubMed] [Google Scholar]
- 25. Gayed BA, Youssef RF, Bagrodia A, et al. Ki67 is an independent predictor of oncological outcomes in patients with localized clear‐cell renal cell carcinoma. BJU Int. 2014;113(4):668–673. [DOI] [PubMed] [Google Scholar]
- 26. Dagher J, Kammerer‐Jacquet SF, Dugay F, et al. Clear cell renal cell carcinoma: a comparative study of histological and chromosomal characteristics between primary tumors and their corresponding metastases. Virchows Arch. 2017;471(1):107–115. [DOI] [PubMed] [Google Scholar]
- 27. Parker LP, Taylor DD, Kesterson S, Gercel‐Taylor C. Gene expression profiling in response to estradiol and genistein in ovarian cancer cells. Cancer Genomics Proteomics. 2009;6(3):189–194. [PubMed] [Google Scholar]
- 28. Qian H, Li X, Zhang W, et al. Caspase‐10, matrix metalloproteinase‐9 and total laminin are correlated with the tumor malignancy of clear cell renal cell carcinoma. Oncology letters. 2018;16(2):2039–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu QH, Wang Y, Yong HM, et al. XRCC1 serves as a potential prognostic indicator for clear cell renal cell carcinoma and inhibits its invasion and metastasis through suppressing MMP‐2 and MMP‐9. Oncotarget. 2017;8(65):109382–109392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yuan L, Zeng G, Chen L, et al. Identification of key genes and pathways in human clear cell renal cell carcinoma (ccRCC) by co‐expression analysis. Int J Biol Sci. 2018;14(3):266–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang Y, Chen L, Wang G, et al. Fifteen hub genes associated with progression and prognosis of clear cell renal cell carcinoma identified by coexpression analysis. J Cell Physiol. 2019;234(7):10225–10237. [DOI] [PubMed] [Google Scholar]
- 32. Kurozumi A, Kato M, Goto Y, et al. Regulation of the collagen cross‐linking enzymes LOXL2 and PLOD2 by tumor‐suppressive microRNA‐26a/b in renal cell carcinoma. Int J Oncol. 2016;48(5):1837–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yuan L, Chen L, Qian K, et al. Co‐expression network analysis identified six hub genes in association with progression and prognosis in human clear cell renal cell carcinoma (ccRCC). Genom Data. 2017;14:132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Parker AS, Eckel‐Passow JE, Serie D, et al. Higher expression of topoisomerase II alpha is an independent marker of increased risk of cancer‐specific death in patients with clear cell renal cell carcinoma. Eur Urol. 2014;66(5):929–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen C, Song G, Xiang J, Zhang H, Zhao S, Zhan Y. AURKA promotes cancer metastasis by regulating epithelial‐mesenchymal transition and cancer stem cell properties in hepatocellular carcinoma. Biochem Biophys Res Commun. 2017;486(2):514–520. [DOI] [PubMed] [Google Scholar]
- 36. Mobley A, Zhang S, Bondaruk J, et al. Aurora Kinase A is a Biomarker for Bladder Cancer Detection and Contributes to its Aggressive Behavior. Sci Rep. 2017;7:40714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zheng F, Yue C, Li G, et al. Nuclear AURKA acquires kinase‐independent transactivating function to enhance breast cancer stem cell phenotype. Nat Commun. 2016;7:10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dauch D, Rudalska R, Cossa G, et al. A MYC‐aurora kinase A protein complex represents an actionable drug target in p53‐altered liver cancer. Nat Med. 2016;22(7):744–753. [DOI] [PubMed] [Google Scholar]
- 39. Haddad AQ, Luo J‐H, Krabbe L‐M, et al. Prognostic value of tissue‐based biomarker signature in clear cell renal cell carcinoma. BJU Int. 2017;119(5):741–747. [DOI] [PubMed] [Google Scholar]
- 40. Hagemann T, Gunawan B, Schulz M, Fuzesi L, Binder C. mRNA expression of matrix metalloproteases and their inhibitors differs in subtypes of renal cell carcinomas. Eur J Cancer. 2001;37(15):1839–1846. [DOI] [PubMed] [Google Scholar]
- 41. Okumura Y, Noda T, Eguchi H, et al. Hypoxia‐Induced PLOD2 is a Key Regulator in Epithelial‐Mesenchymal Transition and Chemoresistance in Biliary Tract Cancer. Ann Surg Oncol. 2018;25(12):3728–3737. [DOI] [PubMed] [Google Scholar]
- 42. Gjaltema RA, de Rond S, Rots MG, Bank RA. Procollagen Lysyl Hydroxylase 2 Expression Is Regulated by an Alternative Downstream Transforming Growth Factor beta‐1 Activation Mechanism. J Biol Chem. 2015;290(47):28465–28476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Noda T, Yamamoto H, Takemasa I, et al. PLOD2 induced under hypoxia is a novel prognostic factor for hepatocellular carcinoma after curative resection. Liver Int. 2012;32(1):110–118. [DOI] [PubMed] [Google Scholar]
- 44. Xu F, Zhang J, Hu G, Liu L, Liang W. Hypoxia and TGF‐beta1 induced PLOD2 expression improve the migration and invasion of cervical cancer cells by promoting epithelial‐to‐mesenchymal transition (EMT) and focal adhesion formation. Cancer Cell Int. 2017;17:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen Y, Terajima M, Yang Y, et al. Lysyl hydroxylase 2 induces a collagen cross‐link switch in tumor stroma. J Clin Invest. 2015;125(3):1147–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Du H, Chen Y, Hou X, et al. PLOD2 regulated by transcription factor FOXA1 promotes metastasis in NSCLC. Cell Death Dis. 2017;8(10):e3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kiyozumi Y, Iwatsuki M, Kurashige J, et al. PLOD2 as a potential regulator of peritoneal dissemination in gastric cancer. Int J Cancer. 2018;143(5):1202–1211. [DOI] [PubMed] [Google Scholar]
- 48. Xu Y, Zhang L, Wei Y, et al. Procollagen‐lysine 2‐oxoglutarate 5‐dioxygenase 2 promotes hypoxia‐induced glioma migration and invasion. Oncotarget. 2017;8(14):23401–23413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Miyamoto K, Seki N, Matsushita R, et al. Tumour‐suppressive miRNA‐26a‐5p and miR‐26b‐5p inhibit cell aggressiveness by regulating PLOD2 in bladder cancer. Br J Cancer. 2016;115(3):354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ye M, He Z, Dai W, Li Z, Chen X, Liu J. A TOP2A‐derived cancer panel drives cancer progression in papillary renal cell carcinoma. Oncology letters. 2018;16(4):4169–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Willems E, Dedobbeleer M, Digregorio M, Lombard A, Lumapat PN, Rogister B. The functional diversity of Aurora kinases: a comprehensive review. Cell Div. 2018;13:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ritchey L, Chakrabarti R. Aurora A kinase modulates actin cytoskeleton through phosphorylation of Cofilin: Implication in the mitotic process. Biochim Biophys Acta. 2014;1843(11):2719–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ritchey L, Ottman R, Roumanos M, Chakrabarti R. A functional cooperativity between Aurora A kinase and LIM kinase1: implication in the mitotic process. Cell Cycle. 2012;11(2):296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yan M, Wang C, He B, et al. Aurora‐A Kinase: A Potent Oncogene and Target for Cancer Therapy. Med Res Rev. 2016;36(6):1036–1079. [DOI] [PubMed] [Google Scholar]
- 55. Pal SK, He M, Tong T, et al. RNA‐seq reveals aurora kinase‐driven mTOR pathway activation in patients with sarcomatoid metastatic renal cell carcinoma. Mol Cancer Res. 2015;13(1):130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ghatalia P, Yang ES, Lasseigne BN, et al. Kinase Gene Expression Profiling of Metastatic Clear Cell Renal Cell Carcinoma Tissue Identifies Potential New Therapeutic Targets. PLoS One. 2016;11(8):e0160924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sehdev V, Katsha A, Arras J, et al. HDM2 regulation by AURKA promotes cell survival in gastric cancer. Clin Cancer Res. 2014;20(1):76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lee DH, Kim CG, Lim Y, Shin SY. Aurora kinase A inhibitor TCS7010 demonstrates pro‐apoptotic effect through the unfolded protein response pathway in HCT116 colon cancer cells. Oncology letters. 2017;14(6):6571–6577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Al‐Khafaji ASK, Marcus MW, Davies MPA, et al. AURKA mRNA expression is an independent predictor of poor prognosis in patients with non‐small cell lung cancer. Oncology letters. 2017;13(6):4463–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Xie Y, Zhu S, Zhong M, et al. Inhibition of Aurora Kinase A Induces Necroptosis in Pancreatic Carcinoma. Gastroenterology. 2017;153(5):1429–43 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Al‐Khafaji ASK, Davies MPA, Risk JM, et al. Aurora B expression modulates paclitaxel response in non‐small cell lung cancer. Br J Cancer. 2017;116(5):592–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ou O, Huppi K, Chakka S, et al. Loss‐of‐function RNAi screens in breast cancer cells identify AURKB, PLK1, PIK3R1, MAPK12, PRKD2, and PTK6 as sensitizing targets of rapamycin activity. Cancer Lett. 2014;354(2):336–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yang J, Ikezoe T, Nishioka C, Udaka K, Yokoyama A. Bcr‐Abl activates AURKA and AURKB in chronic myeloid leukemia cells via AKT signaling. Int J Cancer. 2014;134(5):1183–1194. [DOI] [PubMed] [Google Scholar]
- 64. Enjoji M, Iida S, Sugita H, et al. BubR1 and AURKB overexpression are associated with a favorable prognosis in gastric cancer. Mol Med Rep. 2009;2(4):589–596. [DOI] [PubMed] [Google Scholar]
- 65. Goldenson B, Crispino JD. The aurora kinases in cell cycle and leukemia. Oncogene. 2015;34(5):537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Noske A, Brandt S, Valtcheva N, et al. Detection of CCNE1/URI (19q12) amplification by in situ hybridisation is common in high grade and type II endometrial cancer. Oncotarget. 2017;8(9):14794–14805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nakayama K, Rahman MT, Rahman M, et al. CCNE1 amplification is associated with aggressive potential in endometrioid endometrial carcinomas. Int J Oncol. 2016;48(2):506–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ayhan A, Kuhn E, Wu R‐C, et al. CCNE1 copy‐number gain and overexpression identify ovarian clear cell carcinoma with a poor prognosis. Mod Pathol. 2017;30(2):297–303. [DOI] [PubMed] [Google Scholar]
- 69. Au‐Yeung G, Lang F, Azar WJ, et al. Selective Targeting of Cyclin E1‐Amplified High‐Grade Serous Ovarian Cancer by Cyclin‐Dependent Kinase 2 and AKT Inhibition. Clin Cancer Res. 2017;23(7):1862–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ghosh T, Varshney A, Kumar P, et al. MicroRNA‐874‐mediated inhibition of the major G1/S phase cyclin, CCNE1, is lost in osteosarcomas. J Biol Chem. 2017;292(52):21264–21281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kim SH, Ho J‐N, Jin H, et al. Upregulated expression of BCL2, MCM7, and CCNE1 indicate cisplatin‐resistance in the set of two human bladder cancer cell lines: T24 cisplatin sensitive and T24R2 cisplatin resistant bladder cancer cell lines. Investig Clin Urol. 2016;57(1):63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wang X, Sun Q, Chen C, et al. ZYG11A serves as an oncogene in non‐small cell lung cancer and influences CCNE1 expression. Oncotarget. 2016;7(7):8029–8042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chen M, Rothman N, Ye Y, et al. Pathway analysis of bladder cancer genome‐wide association study identifies novel pathways involved in bladder cancer development. Genes Cancer. 2016;7(7–8):229–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Luhtala S, Staff S, Tanner M, Isola J. Cyclin E amplification, over‐expression, and relapse‐free survival in HER‐2‐positive primary breast cancer. Tumour Biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2016;37(7):9813–9823. [DOI] [PubMed] [Google Scholar]
- 75. Zhang X, Hu S, Zhang X, et al. MicroRNA‐7 arrests cell cycle in G1 phase by directly targeting CCNE1 in human hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2014;443(3):1078–1084. [DOI] [PubMed] [Google Scholar]
- 76. Lin CY, Yang ST, Shen SC, et al. Serum amyloid A1 in combination with integrin alphaVbeta3 increases glioblastoma cells mobility and progression. Mol Oncol. 2018;12(5):756–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Knebel FH, Uno M, Galatro TF, et al. Serum amyloid A1 is upregulated in human glioblastoma. J Neurooncol. 2017;132(3):383–391. [DOI] [PubMed] [Google Scholar]
- 78. Ren Y, Wang HE, Lu D, et al. Expression of serum amyloid A in uterine cervical cancer. Diagn Pathol. 2014;9:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Milan E, Lazzari C, Anand S, et al. SAA1 is over‐expressed in plasma of non small cell lung cancer patients with poor outcome after treatment with epidermal growth factor receptor tyrosine‐kinase inhibitors. J Proteomics. 2012;76:91–101. [DOI] [PubMed] [Google Scholar]
- 80. Raza SK, Saleem M, Shamsi T, Choudhary MI, Atta Ur R, Musharraf SG. 5D proteomic approach for the biomarker search in plasma: Acute myeloid leukaemia as a case study. Sci Rep. 2017;7(1):16440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Liu C, Pan C, Shen J, Wang H, Yong L. Identification of serum amyloid A in the serum of gastric cancer patients by protein expression profiling. Oncology Letters. 2012;3(6):1259–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zheng Z, Cai Y, Chen H, et al. CXCL13/CXCR5 Axis Predicts Poor Prognosis and Promotes Progression Through PI3K/AKT/mTOR Pathway in Clear Cell Renal Cell Carcinoma. Front Oncol. 2018;8:682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kroeze SGC, Vermaat JS, van Brussel A, et al. Expression of nuclear FIH independently predicts overall survival of clear cell renal cell carcinoma patients. Eur J Cancer. 2010;46(18):3375–3382. [DOI] [PubMed] [Google Scholar]
- 84. Wen JL, Wen XF, Li RB, et al. UBE3C promotes growth and metastasis of renal cell carcinoma via activating Wnt/beta‐catenin pathway. PLoS One. 2015;10(2):e0115622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Pantuck AJ, Seligson DB, Klatte T, et al. Prognostic relevance of the mTOR pathway in renal cell carcinoma: implications for molecular patient selection for targeted therapy. Cancer. 2007;109(11):2257–2267. [DOI] [PubMed] [Google Scholar]
- 86. Bui MH, Seligson D, Han KR, et al. Carbonic anhydrase IX is an independent predictor of survival in advanced renal clear cell carcinoma: implications for prognosis and therapy. Clin Cancer Res. 2003;9(2):802–811. [PubMed] [Google Scholar]
- 87. Migita T, Oda Y, Naito S, Tsuneyoshi M. Low expression of p27(Kip1) is associated with tumor size and poor prognosis in patients with renal cell carcinoma. Cancer. 2002;94(4):973–979. [PubMed] [Google Scholar]
- 88. Klatte T, Seligson DB, LaRochelle J, et al. Molecular signatures of localized clear cell renal cell carcinoma to predict disease‐free survival after nephrectomy. Cancer Epidemiol Biomarkers Prev. 2009;18(3):894–900. [DOI] [PubMed] [Google Scholar]
- 89. Ha M, Son YR, Kim J, et al. TEK is a novel prognostic marker for clear cell renal cell carcinoma. Eur Rev Med Pharmacol Sci. 2019;23(4):1451–1458. [DOI] [PubMed] [Google Scholar]
- 90. Wan B, Liu B, Yu G, Huang Y, Lv C. Differentially expressed autophagy‐related genes are potential prognostic and diagnostic biomarkers in clear‐cell renal cell carcinoma. Aging (Albany NY). 2019;11(20):9025–9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Xu D, Dang W, Wang S, Hu B, Yin L, Guan B. An optimal prognostic model based on gene expression for clear cell renal cell carcinoma. Oncology letters. 2020;20(3):2420–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Berglund A, Mills M, Putney RM, Hamaidi I, Mule J, Kim S. Methylation of immune synapse genes modulates tumor immunogenicity. J Clin Invest. 2020;130(2):974–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Konno H, Yamauchi S, Berglund A, Putney RM, Mule JJ, Barber GN. Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene. 2018;37(15):2037–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Haddad TC, D’Assoro A, Suman V, et al. Phase I trial to evaluate the addition of alisertib to fulvestrant in women with endocrine‐resistant, ER+ metastatic breast cancer. Breast Cancer Res Treat. 2018;168(3):639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kantarjian HM, Schuster MW, Jain N, et al. A phase 1 study of AMG 900, an orally administered pan‐aurora kinase inhibitor, in adult patients with acute myeloid leukemia. Am J Hematol. 2017;92(7):660–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Gossage DL, Cieslarová B, Ap S, et al. Phase 1b Study of the Safety, Pharmacokinetics, and Disease‐related Outcomes of the Matrix Metalloproteinase‐9 Inhibitor Andecaliximab in Patients With Rheumatoid Arthritis. Clin Ther. 2018;40(1):156–165. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1‐S2
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author, JYP.