

Abstract
BACKGROUND
Diffuse intrinsic pontine glioma (DIPG) is a fatal childhood brain tumor and the majority of patients die within 2 yr after initial diagnosis. Factors that contribute to the dismal prognosis of these patients include the infiltrative nature and anatomic location in an eloquent area of the brain, which precludes total surgical resection, and the presence of the blood-brain barrier (BBB), which reduces the distribution of systemically administered agents. Convection-enhanced delivery (CED) is a direct infusion technique to deliver therapeutic agents into a target site in the brain and able to deliver a high concentration drug to the infusion site without systemic toxicities.
OBJECTIVE
To assess the efficacy of enhancer of zeste homolog-2 (EZH2) inhibitor by CED against human DIPG xenograft models.
METHODS
The concentration of EZH2 inhibitor (EPZ-6438) in the brainstem tumor was evaluated by liquid chromatography-mass spectrometry (LC/MS). We treated mice-bearing human DIPG xenografts with EPZ-6438 using systemic (intraperitoneal) or CED administration. Intracranial tumor growth was monitored by bioluminescence image, and the therapeutic response was evaluated by animal survival.
RESULTS
LC/MS analysis showed that the concentration of EPZ-6438 in the brainstem tumor was 3.74% of serum concentration after systemic administration. CED of EPZ-6438 suppressed tumor growth and significantly extended animal survival when compared to systemic administration of EPZ-6438 (P = .0475).
CONCLUSION
Our results indicate that CED of an EZH2 inhibitor is a promising strategy to bypass the BBB and to increase the efficacy of an EZH2 inhibitor for the treatment of DIPG.
Keywords: Convection-enhanced delivery, Diffuse intrinsic pontine glioma, EZH2 inhibitor, Patient-derived xenografts
ABBREVIATIONS
- ABC
ATP-binding cassette
- ANOVA
analysis of variance
- BBB
blood-brain barrier
- BLI
bioluminescence imaging
- CED
convection-enhanced delivery
- CNS
central nervous system
- DIPG
diffuse intrinsic pontine glioma
- DMEM
Dulbecco's modified Eagle's medium
- DMG
diffuse midline glioma
- DMSO
dimethyl sulfoxide
- EZH2
enhancer of zeste homolog-2
- FBS
fetal bovine serum
- GBM
glioblastoma
- HE
hematoxylin and eosin
- IP
intraperitoneal
- LC-MS
liquid chromatography-mass spectrometry
- MRI
magnetic resonance imaging
- PRC2
polycomb repressive complex 2
- PRC2
polycomb repressive complex 2
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TBST
Tris-HCl buffered saline with 0.05% tween 20
- TSM
tumor stem medium
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- WT
wild-type
Diffuse midline glioma (DMG) with H3K27M mutation, resulting in substitution of lysine 27 with methionine (K27M) in histone H3 proteins, is a newly defined entity that comprises all diffusely infiltrating astrocytic neoplasms affecting midline structures of the central nervous system (CNS).1 The pontine category of DMG was previously referred to as diffuse intrinsic pontine glioma (DIPG), which is one of the most devastating childhood cancers; almost all DIPG patients die within 2 yr of diagnosis.2-5 A fundamental limitation in the treatment of brain tumors including DIPG is that many systemically administered therapeutic agents do not cross the blood-brain barrier (BBB). Although some small lipophilic molecules can be delivered systemically to the brain by crossing the BBB, high doses are needed to achieve therapeutic levels, which can lead to substantial toxicity.6-8 The development of new therapeutic approaches to bypass the BBB is a potential path to improve outcomes for DIPG patients.
Convection-enhanced delivery (CED) is a direct drug delivery system to the CNS and can deliver high concentration drug into a target site in the brain without systemic toxicities. CED has shown promising results in both animal models and clinical trials.9-14 We have recently demonstrated that CED of nanoliposomal CPT-11 inhibited tumor growth and increased survival of animals with brainstem tumor xenografts.15
In contrast to adult glioblastoma (GBM), DIPG pathogenesis is uniquely dependent on H3K27M mutation.16,17 The gain-of-function mechanism of K27M mutation has been shown to involve sequestration and inactivation of the polycomb repressive complex 2 (PRC2) K27 methyltransferase leading to a dramatic reduction of K27 methylation in all 16 H3 proteins in the cell.18,19 In addition, allosterically activated PRC2 is particularly sensitive to H3K27M, leading to the failure to spread H3K27 methylation from PRC2 recruitment sites and consequently abrogating PRC2’s ability to establish H3K27 methylation repressive chromatin domains.20 The global reduction of H3K27 methylation is believed to modify cellular gene expression and promote DIPG development. Despite the major loss of H3K27 methylation, however, PRC2 activity is still detected in H3K27M DIPG cells, and the residual PRC2 activity maintains DIPG proliferation by repressing neuronal differentiation and function.21,22 The small molecule inhibitor of enhancer of zeste homolog-2 (EZH2) suppressed DIPG growth by upregulating genes that are normally repressed by PRC2.21,22 Thus, targeted inhibition of EZH2 activity is a potential therapeutic strategy for the treatment of DIPG. EZH2 inhibitor, EPZ-6438, also known as tazemetostat, has been studied in a variety of solid and hematological malignancies,23-28 though EPZ-6438 has not been successful in the treatment of brain tumors. One of the major challenges of EPZ-6438 in treating brain tumors is that the brain penetration of EPZ-6438 upon systemic administration was restricted by ATP-binding cassette (ABC) transporters in Vivo.29
In this study, we tested the anti-tumor activity of EZH2 inhibitor EPZ-6438 using CED and systemic administration in mice-bearing human DIPG xenografts.
METHODS
Cell Lines and Culture
The human H3K27M DIPG cell lines SF8628 and SF7761, and H3 wild-type (WT) SF9427 GBM cells were obtained from surgical biopsies of tumors from patients admitted to UCSF Medical Center in accordance with an institutionally approved protocol. Establishment of the SF8628 and SF7761 cell cultures and modification for expression of firefly luciferase for in Vivo bioluminescence imaging (BLI) have been described previously.21,30-33 The DIPG007 cell line was kindly provided by Dr Angel Montero Carcaboso (Hospital Sant Joan de Déu, Barcelona, Spain). All DIPG cell lines were confirmed to harboring a H3F3A K27M mutation using DNA sequencing as previously described.18,21,30,34 SF8628 cells were propagated as monolayers in complete medium consisting of Dulbecco's modified Eagle's medium (DMEM, 11965092) supplemented with 10% fetal bovine serum (FBS, A31604-02) and nonessential amino acids (11140-050) from Thermo Fisher. SF7761 cells were grown as neurosphere cells in tumor stem medium (TSM) base medium prepared using neurobasal-A medium (10888-022), DMEM/F-12 medium (11330-032), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid buffer (15630-080), sodium pyruvate (11360-070), minimum essential medium nonessential amino acids (11140-050), GlutaMAX-I supplement (35050-061), antibiotic-antimycotic (15240-096), B-27 supplement minus vitamin A (12587-010) from Thermo Fisher, EGF and FGF (Shenandoah Biotech, 100-26 and 100-146), PDGF-A and PDGF-B (Shenandoah Biotech, 100-16 and 100-18), and 0.2% heparin (STEMCELL Technologies, 07980). DIPG007 and SF9427 cells were grown adherently in TSM base medium with 5% FBS. DNA fingerprints were obtained using Powerplex16HS System (Promega DC2101) to confirm the identities of the cell lines. All cells were cultured in an incubator at 37°C with a humidified atmosphere containing 95% O2 and 5% CO2, and were found to be Mycoplasma-free at the time of testing with Mycoplasma Detection Kit (Invitrogen).
Cell Proliferation and Colony Formation Assay
A total of 30 000 cells were plated in 60-mm dishes and treated with either 0.05% dimethyl sulfoxide (DMSO; Sigma D2650) or with 5 μM of EPZ-6438 (Selleck Chemicals, S7128) as previously described.21,22 Cells were incubated with EPZ-6438 for 16 d, and cell proliferation was assessed by counting viable cells on day 3, 4, 6, 8, 9, 12, and 16. Trypsinized cell suspensions were stained with trypan blue, and viable cells, which were not stained with trypan blue, were counted using a hemocytometer. The colony formation assay was performed by plating 5000 cells in 60-mm dishes, treating the cells with DMSO or 5 μM of EPZ-6438, and incubating them at 37°C for 2 to 3 wk. Then, colonies were stained with 0.05% crystal violet and counted.
Immunoblot Analysis
Cells were cultured as described above and treated with DMSO or 5 μM of EPZ-6438 for 7 d. Asynchronously proliferating cells were lysed in radioimmunoprecipitation assay lysis and extraction buffer (Thermo Fisher 89900) supplemented with 1% protease inhibitor (Thermo Fisher 78430) and 1% phosphatase inhibitor (Thermo Fisher 78420) to obtain total cell lysates. Histone proteins were extracted using Histone Extraction Kit (Abcam, ab13476) and resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The transfer was performed using the Trans-Blot® Turbo™ Transfer System and Midi polyvinylidene difluoride Transfer Packs (Bio-Rad, #1704157). Membranes were blocked in 5% skim milk in Tris-HCl buffered saline with 0.05% tween 20 (TBST) for 1 h at room temperature, probed with primary antibodies (1:1000 dilution) overnight at 4°C, washed 3 times with TBST, incubated with horseradish peroxidase-conjugated secondary antibody (1:2000 dilution), washed with TBST, and visualized using the Pierce ECL western blotting substrate. The primary antibodies histone H3 (#9715), histone H3K27M mutant specific (D3B5T, #74 829), tri-methyl-histone H3 (Lys27, C36B11, #9733), and β-actin (13E5, #4970) and the secondary antibodies (anti-rabbit IgG, #7074P2 and anti-mouse IgG, #7076) were obtained from Cell Signaling Technologies. Anti-CDKN2A/p16INK4a antibody (ab108349) was obtained from Abcam. Purified mouse anti-EZH2 antibody (#612666) was obtained from BD Bioscience.
Xenograft Models
Six-week-old female athymic mice (rnu/rnu genotype, BALB/c background) were purchased from Envigo and housed under aseptic conditions. Pontine injection of tumor cells was performed as previously described.21,30-33 Briefly, each mouse was injected with 1 μL of a cell suspension (10 000 cells/μL) into the pontine tegmentum at 1.5 mm to the right of the midline, posterior to the lambdoid suture, and at a depth of 5 mm from the inner base of skull using free hand technique with 26-gauge Hamilton syringe (Hamilton Company, #80300). All procedures were carried out under sterile conditions.
Convection-Enhanced Delivery
CED of therapeutics was performed by infusing a volume of 10 μL as described previously.15,35 Briefly, the infusion cannula with a 1-mm stepped design was placed through the existing hole for cell implantation in the skull. EPZ-6438 (1 mM) was infused at a rate of 1 μL/min for 10 min using a microinfusion pump (BeeHive, Bioanalytical Systems). The cannula was removed 2 min after completion of the infusion to prevent reflux.
Magnetic Resonance Imaging
To confirm the distribution of the infused agent in the brain, magnetic resonance imaging (MRI) was performed immediately after CED of gadolinium (0.1 mM, 15 μL, Gadoteridol, MedchemExpress) into naïve mice. Two mice were imaged using a Bruker Siemens Clinscan 7T MRI under anesthesia by 1.5% to 2.0% isoflurane inhalation (1.0 L O2/min).
Analysis of the Drug Concentration in the Brainstem Tumor
Mice-bearing human DIPG xenografts received 350 mg/kg of EPZ-6438 for 3 d by intraperitoneal (IP) injection, and euthanized 3 h after the third administration. Brainstem tumors and contralateral normal cerebral hemispheres were dissected, and serums were collected by cardiac puncture, snap frozen, and stored at −80°C. EPZ-6438 was extracted from homogenized tissue using Bullet Blender (Next Advance Inc). Homogenates were extracted with organic solvent and transferred to autosampler for liquid chromatography-mass spectrometry (LC/MS) analysis (Shimadzu VP Series 10 System) for determination of EPZ-6438 contents (Integrated Analytical Systems).
In Vivo Therapy Response Study
SF8628 DIPG cells (10 000 cells/μL) were implanted into the pontine tegmentum of athymic mice (day 0). Animals were randomized into 4 treatment groups: (1) CED of DMSO control (1% DMSO, n = 7), 2) IP injection of DMSO control (100% DMSO, n = 6), (3) CED of EPZ-6438 (10 μL at 286.37 μg/kg of EPZ-6438, n = 7), and (4) IP injection of EPZ-6438 (350 mg/kg of EPZ-6438 3 days a week for 2 wk, n = 6). EPZ-6428 treatment by CED or IP administration was initiated on day 23. Biweekly BLI was used to monitor tumor growth and the response to therapy as described previously.21,30-33 Mice were monitored daily and euthanized at the endpoint, which was defined by an irreversible neurological deficit or a body condition score less than 2.
Immunohistochemistry
Brains were collected from mice (n = 2) at 3 h after completion of the treatment. Resected brains were fixed in paraformaldehyde, paraffin-embedded, and sectioned (10 μm) for hematoxylin and eosin (HE), anti-Ki67 antibody (Ventana Inc), anti-H3K27 trimethylation (K27me3) (#9733, Cell Signaling), and anti-CDKN2A/p16INK4a (ab54210, Abcam) staining. To assay the apoptotic response to treatment, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed using the DeadEnd Colorimetric TUNEL system (Promega) according to the manufacturer's protocol.
Statistics
Survival plots were generated and analyzed using the Kaplan-Meier method and GraphPad Prism v7.0 software. Differences between survival plots were estimated using a log-rank test. For other analyses, a 2-tailed unpaired t-test and analysis of variance (ANOVA) with Tukey's multiple comparison were applied using Prism software.
Study Approval
All animal protocols were approved by Institutional Animal Care and Use Committee.
RESULTS
Effects of EPZ-6438 on Cell Growth and H3K27 Methylation in DIPG Cells
We and others have shown that inhibition of EZH2 activity reduces proliferation of DIPG cells.21,22 We first evaluated cytotoxicity of EPZ-6438 in 3 H3K27M DIPG cell lines (SF8628, DIPG007, and SF7761). Cells were treated with 5 μM of EPZ-6438, and the effect on proliferation was determined by counting cells on day 3, 4, 6, 8, 9, 12, and 16. EPZ-6438 inhibited the growth of all H3K27M DIPG cell lines (Figure 1A, left, SF8628: DMSO 2.10 × 106 ± 3.61 × 105 vs EPZ-6438 6.33 × 105 ± 2.08 × 105, P = .0073 on day 12, DIPG007: DMSO 1.97 × 106 ± 1.53 × 105 vs EPZ-6438 3.70 × 105 ± 8.72 × 105, P = .0004 on day 16, SF7761: DMSO 1.47 × 106 ± 2.52 × 105 vs EPZ-6438: 4.10 × 105 ± 1.40 × 105, P = .0069 on day 16), which is consistent with the results described previously.21,22 Treatment with EPZ-6438 also reduced colony formation in H3K27M DIPG cells (Figure 1A, right). Next, we analyzed the effects of EPZ-6438 on H3K27 methylation and p16 expression in DIPG cells using immunoblot analysis. H3K27me3 expression was detected in H3K27M DIPG cell lines with a long exposure time (120 s), whereas the basal level of K27me3 in H3K27M DIPG cells was lower than that in H3 WT SF9427 GBM cells (Figure, Supplemental Digital Content 1). EPZ-6438 treatment reduced expression of H3K27me3 and increased expression of p16, when compared to the DMSO control (Figure 1B).
FIGURE 1.

Effects of EPZ-6438 on cell proliferation and expression of H3K27 methylation and p16. A, Left: Cells were plated then treated with 5 μM of EPZ-6438 or 0.05% DMSO control, then were collected and counted after 3 to 16 d of incubation. Results for each time point are based on triplicate samples. Note that y-axes are log scale, and mean and upper standard deviation (SD) are shown. Statistical analysis was performed using a 2-tailed unpaired t-test: SF8628, **P = .0073 (day 12); DIPG007, ***P = .0004 (day16); SF7761, **P = .0069 (day16). The results showing in (A) are consistent with previously published results.21,22 Right: Colony-formation assay in DIPG cell lines treated with 5 μM of EPZ-6438 or DMSO control. B, Western blot results showing the effects of 5 μM of EPZ-6438 on expression of H3K27me3 and p16 in DIPG cell lines.
Concentration of EPZ-6438 in the Brainstem Tumor Upon Systemic Administration
The brain penetration of EZH2 inhibitors upon systemic administration is restricted by ABC transporters in Vivo.29 We analyzed the concentration of EPZ-6438 in DIPG xenografts after systemic administration. EPZ-6438 was IP injected into tumor-bearing mice, and the mice were euthanized 3 h after the treatment was completed. The normal brain tissues and brainstem tumors were immediately resected, and serums were collected by cardiac puncture. LC/MS analysis of tissue extracts and serums showed a low concentration of EPZ-6438 in normal brain tissues (0.24 ± 0.22 μg/mL, 1.52 ± 1.16%) and brainstem tumors (0.50 ± 0.27 μg/mL, 3.74 ± 1.70%) relative to the concentration in serums (12.78 ± 3.20 μg/mL; Table). These data indicate limited distribution of EPZ-6438 in the brain upon systemic administration. To increase drug distribution in the brainstem tumor, we investigated whether EPZ-6438 can be delivered directly into the brainstem by CED.
TABLE.
EPZ-6438 Concentration in Serum, Normal Brain, and Brainstem Tumor
| EPZ-6438 concentration (μg/mL) | |
|---|---|
| Serum | 12.78 ± 3.20 |
| Normal brain | 0.24 ± 0.22 |
| Brainstem tumor | 0.50 ± 0.27 |
| EPZ-6438 brain penetration ratio (%) | |
| Normal brain/serum | 1.52 ± 1.16 |
| Tumor/serum | 3.74 ± 1.70 |
CED of EPZ-6438 in Human DIPG Xenografts
We previously performed CED of nanoliposomal irinotecan into brainstem tumors using a microinfusion pump.15 To verify that the CED system works, we delivered trypan blue (15 μL) and gadolinium (0.1 mM, 15μL) into the brainstems of naïve mice using CED. Trypan blue staining was observed along the needle tract and in the brainstem 2 h following CED (Figure 2A, upper right). Gadolinium was detected in the right pons of the mouse brain by MRI 1 h following CED (Figure 2A, red arrow). To evaluate in Vivo anti-tumor activity of EPZ-6438, we treated DIPG xenografted mice with EPZ-6438 by CED or IP injection. The treatment schedule is depicted in Figure 2B. CED of EPZ-6438 (1 mM) inhibited tumor growth (Figure 3A) and prolonged survival of the DIPG xenografted mice when compared to DMSO control (P = .0006, Figure 3B). However, IP injection of EPZ-6438 (350 mg/kg) had no effect on tumor growth (Figure 3A) and survival of the DIPG xenografted mice (Figure 3B).
FIGURE 2.

Convection-enhanced delivery (CED) of EPZ-6438 for the treatment of orthotopic human DIPG xenografts. A, Schematic overview of CED into the brainstem (upper left), tissue distribution of trypan blue by CED administration (upper right) and MRI following CED of gadolinium to the brainstem (lower left: axial view, lower right: sagittal view). Red arrows; gadolinium signals at infusion site. B, Experimental design for in Vivo efficacy study of EZH2 inhibitor (EPZ-6438) using CED or intraperitoneal (IP) administration in SF8628 DIPG xenografted models.
FIGURE 3.
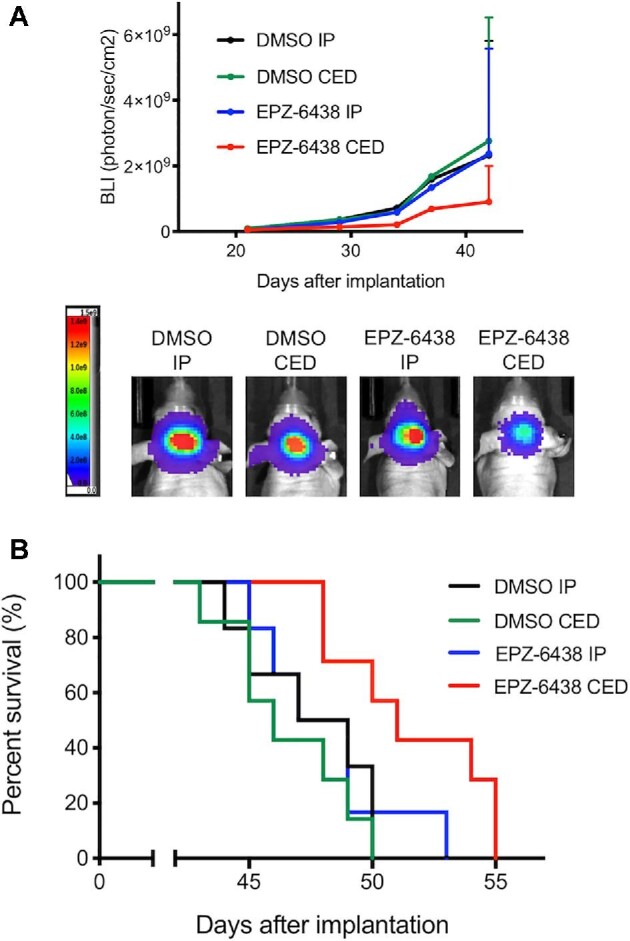
In Vivo anti-tumor activity of EPZ-6438 by CED administration compared to systemic administration in patient-derived DIPG xenografted models. A, Mice with SF8628 intracranial tumor were randomized to 4 treatment groups: 1. DMSO intraperitoneal (IP) injection (N = 6), 2. DMSO CED (N = 7), 3. EPZ-6438 IP (N = 6), 4. EPZ-6438 CED (N = 7). Tumor growth curve for bioluminescence values in each treatment group (upper). Tumor bioluminescence values show mean and upper standard deviation (SD). Representative bioluminescence imaging (BLI) signals in each treatment group (bottom). B, Corresponding survival analysis for each experiment. Median survival: DMSO IP, 48 d; DMSO CED, 46 d; EPZ-6438 IP, 48 d; EPZ-6438 CED, 51 d. Statistical analysis was performed using a log-rank test: DMSO CED vs EPZ-6438 CED, ***P = .0006, EPZ-6438 IP vs EPZ-6438 CED, *P = .0475.
At the end of the treatment, the mice were euthanized to obtain brainstem tumor samples to analyze tumor cell proliferation (Ki-67 staining), apoptosis (TUNEL assay), H3K27me3 and p16 expression. Immunohistochemical analysis of brainstem tumors revealed that CED of EPZ-6438 reduced the number of Ki-67-positive cells compared to DMSO control (P < .0001, Figure 4A). There was no significant difference in the number of Ki-67-positive cells in tumors from mice IP injected with DMSO and EPZ-6438. TUNEL staining showed that the highest proportion of positive cells in tumors from mice received EPZ-6438 by CED compared to DMSO control (P < .0001, Figure 4A). There was no significant difference in the number of TUNEL-positive cells between the tumors from mice treated with DMSO and EPZ-6438 by IP injection. No TUNEL positivity was observed in normal brain tissues surrounding tumors in mice that received either of the EPZ-6438 treatments. At the EPZ-6438 CED infusion sites in the brainstem tumors, we observed inflammatory cell infiltration (Figure 4B, left: HE), as well as an increase in the number of TUNEL-positive cells (Figure 4B, right). In addition, tumor cell H3K27me3 positivity was decreased and p16 positivity was increased, as a result of EPZ-6438 CED (Figure, Supplemental Digital Content 2).
FIGURE 4.

Immunohistochemical analysis of DIPG xenografts treated with EPZ-6438. A, Images of representative Ki-67 and TUNEL staining of intracranial tumor from mice euthanized at the end of treatment. Mean and standard deviation (SD) values represent the average number of positive cells in 3 high-powered fields for each tumor. Statistical analysis was performed ANOVA with Tukey's multiple comparison. Ki-67: CED DMSO vs CED EPZ-6438, ****P < .0001, TUNEL: DMSO CED vs EPZ-6438 CED, ****P < .0001. B, Representative histological changes around the intratumor injection site upon EPZ-6438 administration by CED (left: hematoxylin and eosin (HE) staining, right: TUNEL staining). Black arrow, inflammatory cell infiltration.
DISCUSSION
Effects of EPZ-6438 on Cell Proliferation and H3K27 Methylation in DIPG
We and others have recently shown that the K27M mutation redirects residual EZH2 activity to loci that suppress neural differentiation in H3K27M DIPG cells. Targeted inhibition of EZH2 activity is a potential therapeutic strategy for the treatment of DIPG.21,22 Indeed, EZH2 inhibitor EPZ-6438 significantly suppressed cell growth and colony formation of H3K27M DIPG cells (Figure 1A) and reduced H3K27me3 expression (Figure 1B), even though the basal level of K27me3 in H3K27M DIPG cells was lower than that in H3 WT SF9427 GBM cells (Figure, Supplemental Digital Content 1), which is consistent with previous reports.21,22
Brain Distribution of EPZ-6438 Upon Systemic Administration in DIPG Mice Models
Because of their promising anti-tumor activity, EZH2 inhibitors are being tested in several clinical trials with cancer patients, including childhood CNS tumors (NCT03213665, NCT02875548, NCT02601950, NCT03155620). However, EZH2 inhibitors have not been successful in the treatment of brain tumor. Because EZH2 inhibitors are substrates of efflux ABC transporters (ABCB1 and ABCB2), their brain penetration was severely restricted in Vivo.29 Pharmacokinetic study in ABC transporter knockout mice revealed that brain concentration of EPZ-6438 administered by intravenous injection was significantly higher in ABC transporter knockout mice (Abcb1a/1b−/−) than in WT mice. In addition, the ABC transporter inhibitor elacridar increased the brain concentration of EPZ‐6438 in WT mice indicating that brain penetration of systemically administered EPZ-6438 is restricted by ABC transporters.
Our LC/MS analysis showed that the concentration of EPZ-6438 in brainstem tumors was 3.74 ± 1.70% of the serum concentration in mice IP injected with EPZ-6438 (Table), suggesting limited brain distribution of EPZ-6438 by systemic administration. The therapeutically effective brain concentration of temozolomide, a first-line chemotherapy for GBM, was shown to be ∼30% of the plasma concentration.36 Thus, the limited brain concentration of EPZ-6438 upon systemic administration could be insufficient to achieve therapeutic levels against to brain tumor. In fact, IP injection of EPZ-6438 showed no survival benefit in our DIPG xenografted mice (Figure 3B). To increase the in Vivo efficacy of systemic administration, high dose of EPZ-6438 would be required and it would lead to substantial systemic toxicity. Indeed, serious adverse effects such as hepatic dysfunction, immunosuppression, and secondary cancer development have been observed upon systemic administration of EPZ-6438 in clinical trials (NCT03010982, NCT03217253).37
CED of EPZ-6438 in Patient-Derived DIPG Xenograft Models
Given this fact, the development of a system that bypasses the BBB and directly delivers EPZ-6438 to the brain is highly important for the treatment of DIPG. We evaluated the anti-tumor activity of EPZ-6438 in DIPG xenografted mice upon CED and systemic administration. We previously performed CED to deliver fluorescently labeled nanoliposomes and nanoliposomal irinotecan to brains and brain tumors in mice.15,35,38 The effective CED of gadolinium into the brainstem was confirmed by MRIs (Figure 2A). Gadolinium has been used to mix with therapeutic agents in human CED study as using accurate image tracer and showed similar distribution with the agents confirmed by MRI.39
We finally found that administration of EPZ-6438 by CED inhibited tumor growth (Figure 3A) and significantly prolonged survival of DIPG xenografted mice when compared to mice that received EPZ-6438 by systemic administration (Figure 3B). Administration of EPZ-6438 by CED significantly increased the number of TUNEL-positive cells and decreased the number of Ki-67-positive cells in brainstem tumors when compared to the DMSO control, indicating that treatment with EPZ-6438 by CED promotes apoptosis and cell death (Figure 4A). Furthermore, CED of EPZ-6438 reduced the expression of H3K27me3 and increased expression of p16, which is one of polycomb target gene products, indicating that CED of EPZ-6438 inhibits EZH2 activity in DIPG xenografts (Figure, Supplemental Digital Content 2). To our knowledge, this is the first report to demonstrate the preclinical activity of EPZ-6438 by CED in patient-derived DIPG xenografted models.
Current Limitations and Future Development of CED of EPZ-6438
In this study, we performed single CED treatment, considering that the multiple CED to the mouse brainstem is highly invasive. The survival benefit of EZP-6438 was significant by single CED in compared to systemic administration; however, we are expecting that survival benefit will further increase using longer acting method with implantable device such as ALZET osmotic pumps. In order to increase the therapeutic effect of CED, we would also use the liposomal encapsulation of EPZ-6468, as previously described.15
CED has been tested in clinical trials using nanoliposomal irinotecan (NCT03086616), panobinostat nanoparticle formulation (NCT03566199), and radioactive iodine-labeled monoclonal antibody 8H9 (NCT01502917) to treat children with DIPG. Our results demonstrate the promising anti-tumor activity of EPZ-6438 by CED and support further development of CED of EZH2 inhibitor as a potential therapeutic approach for DIPG patients.
CONCLUSION
CED of EPZ-6438 significantly extended animal survival when compared to systemic administration of EPZ-6438. Our results indicate that CED of EZH2 inhibitor would provide a promising strategy for bypassing the BBB and increasing efficacy of EZH2 inhibitor for the treatment of DIPG.
Disclosures
This work was supported by the National Institutes of Health (R01NS093079 to R.H., and R01CA197313 to O.J.B), Alex's Lemonade Stand Foundation for Childhood Cancer (R.H.), St. Baldrick's Foundation (RH), Rally Foundation (R.H.), Bear Necessities Pediatric Cancer Foundation (R.H.), Segal Family Foundation (R.H.), and John McNicholas Pediatric Brain Tumor Foundation (R.H., S.G.). The authors have no personal, financial, or institutional interest in any of the drugs, materials, or devices described in this article.
Supplementary Material
Acknowledgments
We thank Dr Angel Montero Carcaboso (Hospital Sant Joan de Déu, Barcelona, Spain) for use of the HSJD-DIPG-007 cell line. Histology and MRI services were provided by the Mouse Histology and Phenotyping Laboratory and the Center for Translational Imaging at Northwestern University, respectively.
Notes
We presented a part of the animal study of CED of EZH2 inhibitor at 24th Annual Scientific Meeting and Education Day of Society of Neuro-Oncology in Phoenix, Arizona on November 22, 2019.
Supplemental Digital Content 1. Figure. Immunoblotting analysis of H3K27 trimethylation in H3K27M DIPG cell lines. Immunoblotting showing expression of H3K27 trimethylation (H3K27me3) in H3 wild-type (H3WT) pediatric GBM cell line (SF9427) and H3K27M DIPG cell lines with different exposure times.
Supplemental Digital Content 2. Figure. Immunohistochemical analysis of H3K27 trimethylation and p16 in DIPG xenografts treated with EPZ-6438 using convection-enhanced delivery (CED). Images of representative H3K27 trimethylation (H3K27me3) and p16 staining of intracranial tumor from mice euthanized at the end of treatment. Mean and standard deviation (SD) values represent the average number of positive cells in 3 high-powered fields for each tumor. Statistical analysis was performed with a 2-tailed unpaired t-test. H3K27me3: DMSO CED vs EPZ-6438 CED, **P = .002, p16: DMSO CED vs EPZ-6438 CED, ****P < .0001.
COMMENT
The authors present an investigation into the efficacy of convection-enhanced delivery (CED) of an EZH2 inhibitor (EPZ-6438) in a patient derived xenograft mouse model of DIPG. They performed in vitro treatment studies on patient derived DIPG cell lines with EPZ-6438 and showed that intraperitoneal administration of EPZ-6438 yields only limited tumor penetration in the xenograft mouse model. They confirmed the feasibility of their in vivo CED-protocol and showed a survival benefit for animals treated with CED-based delivery of EPZ-6438 when compared to intraperitoneal administration.
As detailed by the authors, the unique dependency of DIPG on the H3K27M mutation should make it an ideal entity to be targeted with an EZH2 inhibitor. So far, however, the deficient Blood-Brain-Barrier penetration of EPZ-6438 has limited its impact on intrinsic brain tumors. With their work, the authors provide promising data that, in the context of DIPG, CED with EPZ-6438 could effectively circumvent the BBB.
Given the devastating clinical course of DIPG new therapeutic approaches are urgently needed and this study is likely to contribute to the clinical translation of EPZ-6438.
Niklas Antonio von Spreckelsen
Köln, Germany
REFERENCES
- 1. Louis DN, Perry A, Reifenberger G et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803-820. [DOI] [PubMed] [Google Scholar]
- 2. Grimm SA, Chamberlain MC. Brainstem glioma: a review. Curr Neurol Neurosci Rep. 2013;13(5):346. [DOI] [PubMed] [Google Scholar]
- 3. Warren KE. Diffuse intrinsic pontine glioma: poised for progress. Front Oncol. 2012;2:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Clymer J, Kieran MW. The integration of biology into the treatment of diffuse intrinsic pontine glioma: a review of the North American clinical trial perspective. Front Oncol. 2018;8:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jansen MH, van Vuurden DG, Vandertop WP, Kaspers GJ. Diffuse intrinsic pontine gliomas: a systematic update on clinical trials and biology. Cancer Treat Rev. 2012;38(1):27-35. [DOI] [PubMed] [Google Scholar]
- 6. Huynh GH, Deen DF, Szoka FC Jr. Barriers to carrier mediated drug and gene delivery to brain tumors. J Control Release. 2006;110(2):236-259. [DOI] [PubMed] [Google Scholar]
- 7. Banks WA. Delivery of peptides to the brain: emphasis on therapeutic development. Biopolymers. 2008;90(5):589-594. [DOI] [PubMed] [Google Scholar]
- 8. van Tellingen O, Yetkin-Arik B, de Gooijer MC, Wesseling P, Wurdinger T, de Vries HE. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist Updat. 2015;19:1-12. [DOI] [PubMed] [Google Scholar]
- 9. Himes BT, Zhang L, Daniels DJ. Treatment strategies in diffuse midline gliomas with the H3K27M mutation: the role of convection-enhanced delivery in overcoming anatomic challenges. Front Oncol. 2019;9:1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen PY, Ozawa T, Drummond DC et al. Comparing routes of delivery for nanoliposomal irinotecan shows superior anti-tumor activity of local administration in treating intracranial glioblastoma xenografts. Neuro Oncol. 2013;15(2):189-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhou Z, Singh R, Souweidane M. Convection-enhanced delivery for diffuse intrinsic pontine glioma treatment. Curr Neuropharmacol. 2016;15(1):116-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sewing AC, Caretti V, Lagerweij T et al. Convection enhanced delivery of carmustine to the murine brainstem: a feasibility study. J Neurosci Methods. 2014;238:88-94. [DOI] [PubMed] [Google Scholar]
- 13. Freeman AC, Platt SR, Holmes S et al. Convection-enhanced delivery of cetuximab conjugated iron-oxide nanoparticles for treatment of spontaneous canine intracranial gliomas. J Neurooncol. 2018;137(3):653-663. [DOI] [PubMed] [Google Scholar]
- 14. Bruce JN, Fine RL, Canoll P et al. Regression of recurrent malignant gliomas with convection-enhanced delivery of topotecan. Neurosurgery. 2011;69(6):1272-1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Louis N, Liu S, He X et al. New therapeutic approaches for brainstem tumors: a comparison of delivery routes using nanoliposomal irinotecan in an animal model. J Neurooncol. 2018;136(3):475-484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schwartzentruber J, Korshunov A, Liu XY et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482(7384):226-231. [DOI] [PubMed] [Google Scholar]
- 17. Wu G, Broniscer A, McEachron TA et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44(3):251-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chan KM, Fang D, Gan H et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013;27(9):985-990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lewis PW, Muller MM, Koletsky MS et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340(6134):857-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stafford JM, Lee C, Voigt P et al. Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci Adv. 2018;4(10):eaau5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Piunti A, Hashizume R, Morgan MA et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat Med. 2017;23(4):493-500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mohammad F, Weissmann S, Leblanc B et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med. 2017;23(4):483-492. [DOI] [PubMed] [Google Scholar]
- 23. Friedmann-Morvinski D, Verma IM. Dedifferentiation and reprogramming: origins of cancer stem cells. EMBO Rep. 2014;15(3):244-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fu H, Cheng L, Sa R, Jin Y, Chen L. Combined tazemetostat and MAPKi enhances differentiation of papillary thyroid cancer cells harboring BRAF(V600E) by synergistically decreasing global trimethylation of H3K27. J Cell Mol Med. 2020;24(6):3336-3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Italiano A, Soria J-C, Toulmonde M et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol. 2018;19(5):649-659. [DOI] [PubMed] [Google Scholar]
- 26. Knutson SK, Kawano S, Minoshima Y et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther. 2014;13(4):842-854. [DOI] [PubMed] [Google Scholar]
- 27. Knutson SK, Warholic NM, Wigle TJ et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci USA. 2013;110(19):7922-7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Makita S, Tobinai K. Targeting EZH2 with tazemetostat. Lancet Oncol. 2018;19(5):586-587. [DOI] [PubMed] [Google Scholar]
- 29. Zhang P, de Gooijer MC, Buil LCM et al. ABCB1 and ABCG2 restrict the brain penetration of a panel of novel EZH2-Inhibitors. Int J Cancer. 2015;137(8):2007-2018. [DOI] [PubMed] [Google Scholar]
- 30. Hashizume R, Andor N, Ihara Y et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat Med. 2014;20(12):1394-1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hashizume R, Gupta N. Patient-derived tumor models for diffuse intrinsic pontine gliomas. Curr Neuropharmacol. 2016;15(1):98-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hashizume R, Smirnov I, Liu S et al. Characterization of a diffuse intrinsic pontine glioma cell line: implications for future investigations and treatment. J Neurooncol. 2012;110(3):305-313. [DOI] [PubMed] [Google Scholar]
- 33. Aoki Y, Hashizume R, Ozawa T et al. An experimental xenograft mouse model of diffuse pontine glioma designed for therapeutic testing. J Neurooncol. 2012;108(1):29-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Taylor KR, Mackay A, Truffaux N et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet. 2014;46(5):457-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Serwer L, Hashizume R, Ozawa T, James CD. Systemic and local drug delivery for treating diseases of the central nervous system in rodent models. J Vis Exp. 2010;42:1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Agarwala SS, Kirkwood JM. Temozolomide, a novel alkylating agent with activity in the central nervous system, may improve the treatment of advanced metastatic melanoma. Oncologist. 2000;5(2):144-151. [DOI] [PubMed] [Google Scholar]
- 37. Healio. FDA places partial clinical hold on tazemetostat trials. https://www.healio.com/hematologyoncology/lymphoma/news/online/%7Be01f728c-1b20-4901-8710-2cad7205bbe9%7D/fda-places-partial-clinical-hold-on-tazemetostat-trials. Accessed April 25, 2018. [Google Scholar]
- 38. Weng KC, Hashizume R, Noble CO et al. Convection-enhanced delivery of targeted quantum dot-immunoliposome hybrid nanoparticles to intracranial brain tumor models. Nanomedicine. 2013;8(12):1913-1925. [DOI] [PubMed] [Google Scholar]
- 39. Tosi U, Souweidane MM. Longitudinal monitoring of Gd-DTPA following convection enhanced delivery in the brainstem. World Neurosurg. 2020;137:38-42. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.