

Abstract
Although excessive consumption of deep-fried foods is regarded as 1 of the most important epidemiological factors of lifestyle diseases such as Alzheimer's disease, type 2 diabetes, and obesity, the exact mechanism remains unknown. This review aims to discuss whether heated cooking oil-derived peroxidation products cause cell degeneration/death for the occurrence of lifestyle diseases. Deep-fried foods cooked in ω-6 PUFA-rich vegetable oils such as rapeseed (canola), soybean, sunflower, and corn oils, already contain or intrinsically generate “hydroxynonenal” by peroxidation. As demonstrated previously, hydroxynonenal promotes carbonylation of heat-shock protein 70.1 (Hsp70.1), with the resultant impaired ability of cells to recycle damaged proteins and stabilize the lysosomal membrane. Until now, the implication of lysosomal/autophagy failure due to the daily consumption of ω-6 PUFA-rich vegetable oils in the progression of cell degeneration/death has not been reported. Since the “calpain-cathepsin hypothesis” was formulated as a cause of ischemic neuronal death in 1998, its relevance to Alzheimer's neuronal death has been suggested with particular attention to hydroxynonenal. However, its relevance to cell death of the hypothalamus, liver, and pancreas, especially related to appetite/energy control, is unknown. The hypothalamus senses information from both adipocyte-derived leptin and circulating free fatty acids. Concentrations of circulating fatty acid and its oxidized form, especially hydroxynonenal, are increased in obese and/or aged subjects. As overactivation of the fatty acid receptor G-protein coupled receptor 40 (GPR40) in response to excessive or oxidized fatty acids in these subjects may lead to the disruption of Ca2+ homeostasis, it should be evaluated whether GPR40 overactivation contributes to diverse cell death. Here, we describe the molecular implication of ω-6 PUFA-rich vegetable oil-derived hydroxynonenal in lysosomal destabilization leading to cell death. By oxidizing Hsp70.1, both the dietary PUFA- (exogenous) and the membrane phospholipid- (intrinsic) peroxidation product “hydroxynonenal,” when combined, may play crucial roles in the occurrence of diverse lifestyle diseases including Alzheimer's disease.
Keywords: Alzheimer's disease, calpain-cathepsin hypothesis, cell death, deep-fried food, GPR40, Hsp70.1, hydroxynonenal, lysosomal rupture, polyunsaturated fatty acid, POMC neuron
Introduction
The beneficial outcomes of the dietary consumption of extra-virgin olive oil and fish oil have been shown at the epidemiological level, such as the cardiovascular protective effect of the Mediterranean diet and the Eskimo diet (1–3). Recently, ω-6 PUFA-rich vegetable oils such as rapeseed oil (known also as canola oil), soybean oil, sunflower oil, and corn oil have become appealing choices for cooking deep-fried foods, because they are cheap and are advertised as healthy. Most importantly, however, the majority of the 1000-fold increase in US consumption of vegetable oils such as soybean oil occurred after 1960, coinciding with the increase in obesity, type 2 diabetes, nonalcoholic steatohepatitis (NASH), and other inflammatory disorders (4). Currently, ω-6 PUFA-rich vegetable oils such as soybean and canola oils are the most widely consumed cooking oils in the home and restaurants worldwide, yet very little data are available on the exact risk of daily intake on human health (5).
Surprisingly, a lipid peroxidation product known as hydroxynonenal is generated during deep-frying of vegetable oils made from rapeseed, soybean, and sunflower that contain abundant linoleic acid (Figure 1A). Further, even after incorporation into the body, reactive oxygen species attack ω-6 PUFAs in the membrane phospholipids to generate endogenous hydroxynonenal. As hydroxynonenal forms adducts with 4 different amino acids in proteins, namely cysteine (Cys), histidine (His), lysine (Lys), and arginine (Arg) residues, numerous proteins have the potential to be modified by hydroxynonenal. Accordingly, it can cause dysfunction and degeneration of neurons by modifying membrane-associated glucose and glutamate transporters, ion-motive ATPases, enzymes involved in amyloid metabolism, and cytoskeletal proteins (6). By the genetic deletion of aldehyde dehydrogenase 2 (ALDH2), Aldh2-/- mice cannot detoxify hydroxynonenal and accumulate it in the brain. Previously, these mice were demonstrated to develop memory deficits as well as very similar biochemical and structural pathologies as seen in Alzheimer's patients (7). Although hydroxynonenal-induced accumulation of amyloid β and phosphorylated tau as well as activation of caspases 3 and 6 were demonstrated in the Aldh2-/- mice, its exact role in the neuronal death cascade was not described in detail, except for the implication of the apoptotic pathway. Interestingly, in the neurons after ischemia, a specific oxidative injury, “carbonylation,” was found to occur at the key site Arg469 of heat-shock protein 70.1 (Hsp70.1) in the postischemic monkey hippocampus. Moreover, Hsp70.1, especially after hydroxynonenal-mediated carbonylation, was found to be vulnerable to calpain-mediated cleavage and cause lysosomal membrane destabilization (Figures 2 and 3) (8–10). The consumption of deep-fried foods and high-fat diets (HFDs) is thought to be 1 of the most important epidemiological factors leading to lifestyle (-related) diseases such as type 2 diabetes, obesity (metabolic syndrome), and arteriosclerosis. Accordingly, it is interesting to focus on the causal relation between ω-6 PUFA-rich vegetable oils and cell degeneration/death.
FIGURE 1.

Generation of hydroxynonenal from cooking oils during heating. A: hydroxynonenal generation (ppm) in the 7 cooking oils at 30 and 60 min after 200°C heating. Surprisingly, 2 extra-virgin olive oils (Spain, Australia) generated abundant hydroxynonenal after heating, because they contain ∼10% of linoleic acid to be oxidized. Three canola oil products containing ∼20% of linoleic acid generated a variable amount of hydroxynonenal; the most popular product (Canola oil) generated abundant hydroxynonenal, whereas a trans-fat-free product (Trans-fat-free canola oil) generated very little. Another pure canola oil (Extra-virgin canola oil) and popular rice oil (Rice oil) also generated a moderate amount of hydroxynonenal. Since soybean and sunflower oils have greater amounts of linoleic acid, both oils would produce much more hydroxynonenal than canola oils. In contrast, coconut oil (Coconut oil) generated negligible hydroxynonenal, because it comprises SFAs that are free from oxidative modification. B: diverse oxidative stresses such as heating, radiation, etc., provoke modification of ω-6 PUFAs, leading to the formation of -aldehyde (-CHO: also called -nal). This figure shows generation of hydroxynonenal from linoleic, arachidonic, and γ-linolenic acids by oxidation. Its chemical structure derives from the ω-end 9 ('none' in Greek) carbons of each PUFA. 4-HNE, 4-hydroxy-2-nonenal.
FIGURE 2.

The ‘calpain-cathepsin hypothesis’ explaining the molecular cascade from ω-6 PUFAs to cell death. Hsp70.1 (also called Hsp70 or Hsp72), as a molecular chaperone and lysosomal stabilizer, is a stress-induced protein that confers cell protection against diverse stimuli, but its dysfunction caused by hydroxynonenal induces neurodegeneration via lysosomal rupture and autophagy failure due to the calpain-mediated cleavage of carbonylated Hsp70.1. Lysosomal disintegrity directly causes cell death, whereas autophagy failure contributes to forming vacuoles or microcysts containing garbage proteins or membrane lipid-degradation components. Excessive ω-6 PUFAs lead to ischemic and Alzheimer's neuronal death as well as β cell death in type 2 diabetes, because of the abundant GPR40 expression in the brain and pancreas (11). Despite the negligible GPR40 expression in the liver, hepatocyte and Kupffer cell death may occur in response to ω-6 PUFA-derived hydroxynonenal, because it can induce Ca2+ mobilization via GPR109A (12, 13). (Adapted with permission from reference 14.) GPR40, G-protein coupled receptor 40; Hsp70.1, heat-shock protein 70.1.
FIGURE 3.
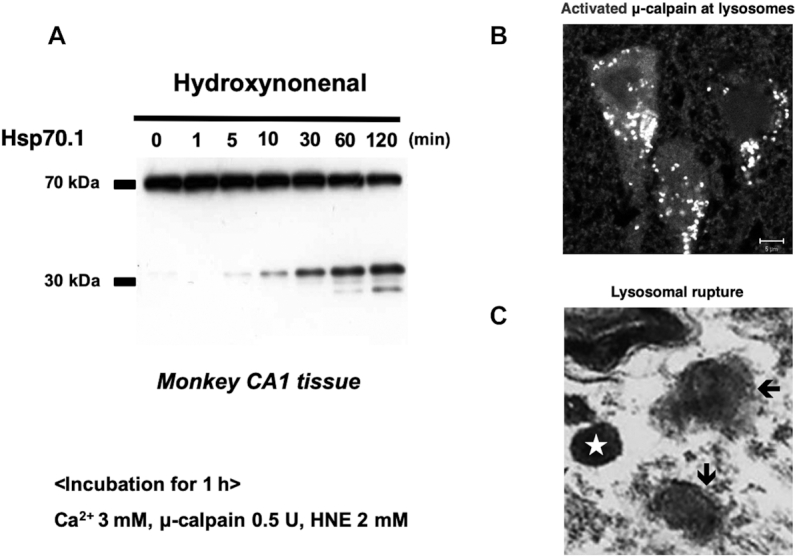
Activated calpain-mediated cleavage of carbonylated Hsp70.1, leading to lysosomal permeabilization/rupture. A: in vitro cleavage of Hsp70.1 by activated μ-calpain in the CA1 tissue of healthy monkeys. Although the calpain-mediated Hsp70.1 proteolysis was negligible before hydroxynonenal treatment (0 min), hydroxynonenal treatment (5–120 min) caused a time-dependent escalation in cleavage. Hsp70.1 is susceptible to cleavage by calpain especially after hydroxynonenal-mediated carbonylation. (Adapted with permission from reference 23). B: activated μ-calpain is immunostained as coarse granules at the lysosomes in the perinuclear cytoplasm of postischemic monkey CA1 neurons. Immunofluorescence staining, using the anti-human activated form of the μ-calpain antibody which recognizes only the activated form but not inactivated calpain. This indicates that μ-calpain is activated at the lysosomal membrane after ischemic insult. Scale bar = 5 μm. (Adapted with permission from reference 21). C: by high-magnification observation with an electron microscope, a cortical neuron in an Alzheimer's patient showed double-contour configuration of the lysosome (arrows), which suggested permeabilization of the limiting membrane. Note the marked contrast in the configuration of intact lysosome (star). Similar lysosomal membrane disintegrity was induced in monkeys by hydroxynonenal injections as shown in Figure 6. (Adapted with permission from reference 21.) HNE, hydroxynonenal; Hsp70.1, heat-shock protein 70.1.
The objective of this review is to describe the implication of hydroxynonenal-mediated lysosomal permeabilization/rupture in cell degeneration/death, and discuss the causality of ω-6 PUFA-rich vegetable oils in the occurrence of diverse lifestyle diseases.
Background
It has recently been accepted that lifestyle diseases are closely related to the occurrence of Alzheimer's disease. Two pathological hallmarks of Alzheimer's disease are formations of 1) extracellular senile plaques composed of insoluble and toxic amyloid-β peptides, and dysfunctional dystrophic neurites, and 2) intracellular neurofibrillary tangles consisting of aggregated, abnormally hyperphosphorylated tau (15–17). Previous studies showed elevated concentrations of hydroxynonenal in association with amyloid-β plaques and/or neurofibrillary tangles (18–20). A possible link between hydroxynonenal and Alzheimer's disease has been suggested by many authors, although the underlying mechanism was not elucidated. Yamashima (21, 22) recently suggested that hydroxynonenal, being derived from ω-6 PUFA-rich vegetable oils, might be the real culprit behind Alzheimer's neuronal death, due to triggering Hsp70.1 dysfunction. Interestingly, the transgenic mouse model of Alzheimer's disease that had consumed canola oil over 6 mo, was found to show an increase of insoluble amyloid-β 42/40, decreased concentrations of postsynaptic density protein-95, and impairments in working memory as well as significant increases in body weight (5). Obesity induced by consuming HFDs is known to be associated with pro-opiomelanocortin (POMC) neuronal injury of the hypothalamic arcuate nucleus which is crucial for body weight control in both rodents and humans. Taken together, it is conceivable that hydroxynonenal can induce degeneration/death of neurons such as in the hippocampus and hypothalamus. However, other than the involvement of Hsp70.1, the detailed molecular mechanism of POMC neuronal injury was unknown until now.
Since the hypothalamic neurons, especially POMC neurons of the arcuate nucleus, are involved in the control of energy homeostasis and body weight control, hypothalamic neuronal loss is believed to play an important role in the development and perpetuation of obesity. When POMC neurons undergo cell death during diet-induced obesity, it provokes loss of the co-ordinated control of caloric intake and energy expenditure (24). The hypothalamus senses information from the adipocyte-derived hormone, leptin, and integrates its signals with those from circulating free fatty acids. In POMC neurons of the hypothalamus, leptin signals are sensed by leptin receptors, whereas fatty acid signals are sensed by G-protein coupled receptor 40 (GPR40). Dietary ω-3 PUFAs have been found to induce neurogenesis of POMC neurons in the hypothalamus, with most of the effect mediated by GPR40 (24). The ω-3 and ω-6 PUFAs not only compete with one another, they also act differently on human tissue, with ω-3 PUFAs lowering the propensity of arachidonic acid cascade overreactions. Accordingly, balancing the proportions of ω-3 and ω-6 PUFAs will undoubtedly contribute to preventing lifestyle diseases, since the balance of ω-3 and ω-6 nutrients in foods eaten by each individual affects both the balance of ω-3 and ω-6 PUFAs in their tissue lipids (25) and generation of hydroxynonenal itself. However, due to the daily intake of deep-fried foods and/or HFDs, signals from excessive ω-6 PUFAs in the blood can overstimulate POMC neurons, and they may gradually develop cell degeneration/death. Therefore, defects in the hypothalamic sensing of adiposity- or nutrient-related signals predispose to a positive energy balance and increased glucose production, which result in pathological body weight gain and insulin resistance. In obesity and metabolic syndrome, concentrations of hydroxynonenal are increased in the blood and organs. In addition, after consumption of deep-fried foods made with ω-6 PUFA-rich vegetable oils, the concentration of hydroxynonenal in the plasma increases rapidly within minutes to hours, and accumulates year by year. Concentrations of hydroxynonenal are elevated in the atherosclerotic plaques of human subjects, because hydroxynonenal covalently modifies LDL and thereby activates macrophages to induce vascular inflammation and atherosclerotic lesions (26).
Currently, hydroxynonenal is increasingly recognized as a particularly important mediator and marker of cellular dysfunction and degeneration in diverse disorders such as arteriosclerosis, cardiovascular disease, stroke, type 2 diabetes, NASH, etc. (18–20, 25, 26). The role of lipid (fatty acid) peroxidation has been widely investigated in the pathogenesis of various neurodegenerative diseases. For example, infusion of hydroxynonenal into the rat brain can damage cholinergic neurons and impair their learning and memory ability (27). Nonetheless, very little is known about how or why hydroxynonenal can play such a detrimental role in cell degeneration.
In 2003, Briscoe et al. (11) found abundant expression of free fatty acid receptor GPR40 (also called free fatty acid receptor 1: FFAR1) in the brain and pancreas. In the brain, GPR40 was ubiquitously expressed, including in the hippocampus, hypothalamus, olfactory bulb, midbrain, medulla oblongata, cerebral cortex, cerebellum, and the spinal cord. Numerous fatty acids, medium- and long-chain or saturated and unsaturated, act as ligands for the GPR40 receptor. For example, DHA, linoleic acid, EPA, arachidonic acid, and linolenic acid show similar pEC50 (the negative logarithm of the EC50) values for GPR40 (11). GPR40 is currently becoming a potential research target in both physiology (insulin secretion and adult neurogenesis) and pathology (Alzheimer's disease, Parkinson's disease, and type 2 diabetes) conditions (28).
Calpain (EC 3.4.22.17) is an intracellular, nonlysosomal, Ca2+-dependent, papain-like protease. The most abundant brain calpains are 2 major isoforms; μ-calpain (calpain 1) and m-calpain (calpain 2), which differ in their sensitivity to Ca2+. The half in vitro maximal Ca2+ requirement for the autolytic activation of μ- and m-calpain is 3–50μM and 0.4–0.8mM, respectively (29). Since the Ca2+ requirement is significantly higher than the cytosolic concentration in living cells (<0.05μM), other supporting mechanisms for lowering the Ca2+ requirement are involved in pathological states. Calpains are heterodimers, consisting of 2 different polypeptide subunits. The larger 80-kDa subunit has catalytic activity, whereas the smaller 30-kDa subunit has a regulatory function. When Ca2+ binds to the EF-hands, the N-terminus 4 kDa of calpain undergoes autolytic cleavage, and its molecular weight changes from 80 kDa to 76 kDa. This is accompanied by a conformational change and the catalytic domain becomes activated. Calpain exists in the cytosol as an inactive enzyme and translocates to membranes in response to an increase in cellular Ca2+ concentrations (Figure 3B). Activated calpain cleaves substrate proteins into 2 large fragments with retaining intact domains. Calpain is regarded as a biomodulator, as properties of the substrate proteins are often modulated upon hydrolysis by calpain (30).
Focusing on the roles of these 3 players “hydroxynonenal,” “GPR40,” and “calpain” in the development of cell degeneration/death, the present review discusses 3 major topics: 1) the formation of hydroxynonenal during deep-frying of vegetable oils and occurrence of calpain activation, especially in GPR40-positive neurons in response to excessive fatty acids, 2) activated calpain-mediated cleavage of carbonylated (oxidized) Hsp70.1 with the resultant lysosomal membrane destabilization, and 3) whether the “calpain-mediated cleavage of carbonylated Hsp70.1” can explain the molecular mechanism of diverse cell degeneration/death associated with lifestyle diseases (Figure 2).
Dual Roles of Stress-Induced Protein, Hsp70.1
The control of protein turnover is particularly important in postmitotic cells such as neurons, in which accumulation of misfolded/damaged/aged proteins may be highly detrimental for cell survival (31). Hsp70.1 (also called Hsp70 or Hsp72) is the most structurally and functionally conserved chaperone protein that plays a central role in cellular protein quality control and degradation for cytoprotection under a number of different conditions. Hsp70.1 is crucial not only as a molecular chaperone but also as a stabilizer of the lysosomal-limiting membrane (32–34). Lysosomal defects disturb the balance between the accumulation of damaged proteins and their proteolytic clearance, ultimately resulting in the formation of autophagic vacuoles (35). Proper functioning of lysosomes is crucial for the survival of neurons, whereas lysosomal failure is 1 of the key factors influencing the development of neurodegeneration. The dual roles of Hsp70.1, as a molecular chaperone and lysosomal stabilizer, will first be described in detail.
Molecular chaperone
A balance between protein folding and degradation is fundamental for protein quality control and intracellular hom-eostasis. The basal activity of autophagy is essential in postmitotic cells like neurons which are unable to dilute garbage proteins and noxious components through cell division (36). Since neurons maintain large volumes of membrane and cytoplasm, they continually traffic autophagy-related garbage long distances from the distal ends of dendrites and axons back to the cell body where lysosomes are gathered as the most active organelle for substrate clearance and/or recycling (37). Heat shock proteins represented by Hsp70.1 modulate protein folding and repair, whereas the ubiquitin-proteasome system, autophagy, and other lysosome-dependent systems function in the degradation of dysfunctional proteins (38, 39).
In eukaryotic cells, proteasomes usually degrade 80–90% of cellular proteins including many regulated, short-lived, abnormal, denatured, or, in general, damaged proteins. In contrast, autophagy is primarily responsible for the degradation of long-lived or aggregated proteins and cellular organelles including mitochondria, peroxisomes, ribosomes, and infectious organisms (38, 40). The initial response to misfolded proteins in an otherwise healthy cell is refolding them back into their native shapes by Hsp70.1 (41, 42). To be fully functional, after translation the protein must be folded into the correct 3-dimensional structure, as it requires assistance to assume its native and functionally physiological conformation. Oxidative stress, mitochondrial collapse, hypoxia, irradiation, toxic chemicals, endoplasmic reticulum stress, physical trauma, or thermal stimuli upregulate Hsp70.1 synthesis (43, 44). Hsp70.1, “on demand,” binds to denatured protein aggregates, restricts, and breaks up their aggregation using energy generated from the hydrolysis of ATP.
If severely damaged proteins cannot be folded back into their native shapes, they would be ubiquitinated and targeted to the 26S proteasome by Hsp70.1 etc.; the ubiquitin-proteasome system can then degrade the misfolded proteins. A second alternative to failed protein refolding is the process of lysosomal autophagy, in which aggregated proteins, damaged organelles, carbohydrates, and lipids are engulfed by lysosomes via macroautophagy or microautophagy (45–48). These 2 degradative systems constitute essential components of the cellular control of protein quality, and help maintain amino acid pools and energy balance, either during acute starvation for the ubiquitin-proteasome system or in the course of chronic starvation for lysosomal autophagy. A third alternative is chaperone-mediated autophagy; misfolded proteins with a KFERQ motif are guided by Hsp70.1 into the lysosome via translocation through the lysosomal-associated membrane protein (Lamp)-2A (47, 49–52). The complex Hsp70.1-cytosolic components bind to Lamp-2A (53) and, after unfolding, substrate proteins are translocated into the lysosomal lumen for degradation, assisted by a luminal form of Hsp70.1 (53). Accordingly, Hsp70.1 is crucial for cell survival, and in the case of Hsp70.1 dysfunction, both proteasome degradation and chaperone-mediated autophagy are impaired, which can cause cell degeneration and the pathology of a variety of diseases.
Energy interfaces as a possible balance element between the ubiquitin-proteasome system and lysosomal autophagy. Since the ubiquitin-proteasome system requires energy to assemble the proteasomal complex, activate ubiquitin, and unfold the substrate protein, low concentrations of ATP may globally reduce the efficiency of this pathway. Conversely, low concentrations of ATP, as well as amino acids, glucids, or the anabolic hormones insulin and insulin-like growth factor, result in autophagy activation (38). Under physiological conditions, these 3 systems, i.e., the ubiquitin-proteasome system, lysosomal autophagy, and chaperone-mediated autophagy are sufficient to handle mild fluctuations in the environmental milieu (54). Depending on the constituents of the Hsp70.1/co-chaperone, this complex guides aged/damaged proteins to either the proteasome or lysosome for degradation if they are damaged beyond repair. In this sense, Hsp70.1 is a potent survival protein that confers cell protection against diverse stimuli, so its depletion induces cell degeneration via autophagy failure. Under pathological conditions, the 3 systems are overwhelmed, so affected cells begin to die (55). Cell death is primarily characterized by a disorder of protein misfolding, as nonenzymatic protein modifications by various oxidative stresses negatively affect protein structure and function, thereby eliciting severe proteotoxic stress which is transmissible from cell to cell (55, 56).
Guardian of lysosomal integrity
Maintenance of the lysosomal membrane integrity is crucial for cellular homeostasis and survival. The role of Hsp70.1 in stabilizing lysosomal membranes is largely attributed to its ability to enhance sphingolipid catabolism in the lysosomes through its high-affinity binding to bis(monoacyl)glycerophosphate (BMP) (32–34, 57), which is an essential cofactor for lysosomal sphingolipid catabolism (58). Via its negative charge, BMP tethers several sphingolipid-degrading enzymes to the internal lysosomal membranes where their substrates are located, thereby increasing their activity. Hsp70.1-BMP binding at the luminal side of the lysosomal-limiting membrane protects Hsp70.1 from lysosomal degradation, facilitates the binding of BMP with several sphingolipid-degrading enzymes, and enhances the activity of acid sphingomyelinase which mediates sphingolipid degradation to generate ceramide (58–60). An increase of ceramide protects the lysosomal-limiting membrane from rupturing (32–34), presumably due to a Hsp70.1-induced decrease in sphingomyelin concentrations increasing the fluidity of lysosomal membranes, and because the increased concentration of lysosomal ceramide can facilitate fusion of lysosomes with other intracellular vesicles and membranes to reinforce the lysosomal-limiting membranes (61).
In addition to stabilizing the lysosomal membrane by regulating lipid catabolism, Hsp70.1 can protect the membrane from oxidative stress. Inside the lysosomes, iron can generate reactive oxygen species through Fenton-type chemical reactions, which may lead to oxidization and destabilization of membrane lipids (62, 63). In spite of high oxygen consumption in the brain, however, concentrations of lower molecular weight and enzymatic antioxidants are relatively low. Accordingly, the brain with poor antioxidant defense is particularly susceptible to lipid peroxidation by reactive oxygen species (64). The protective effect of Hsp70.1 against oxidative stress is preserved inside the lysosomes, although it remains unknown whether Hsp70.1 has a direct antioxidant effect or indirect effects by changing the lipid composition of the membranes. In the case of photo-oxidation of acridine orange-loaded lysosomes, for example, real-time high-resolution imaging demonstrated that Hsp70.1 localized in the lysosomal lumen effectively protects lysosomal membranes from local oxidative stress (32). Further, Hsp70 is cytoprotective in other lysosomal oxidative stress models such as age-related macular degeneration and lysosomal iron accumulation (65–67).
Implication of Hydroxynonenal in Ischemic Neuronal Death
Deep-frying of vegetable oils generates hydroxynonenal
Fatty acids are the integral components of cells and parti-cipate in various vital functions such as providing energy, maintaining cell membrane integrity, and serving as signaling molecules. PUFAs represent a class of lipids that contain 2 or more carbon double (unsaturated) bonds (C = C), being classified as ω-3, ω-6, and ω-9. The ω-3 PUFAs such as DHA and ω-6 PUFA such as arachidonic acid, are found in abundance in the brain, although second to adipose tissues. Both are of great importance for neurobiology, and perform significant functions for boosting synaptogenesis and neurogenesis. For example, DHA is linked with vision and memory (68, 69), and is essential for neuronal development in infants and preservation of brain function in adults. In addition, the role of arachidonic acid in the development of infant brains is highly significant (70). Arachidonic acid is found in abundance in the gray matter of the brain as a main component of the synaptic membrane, and serves as an intercellular messenger to activate protein kinase C. It modulates receptors, transporters, and ion channels after its release from the phospholipids of the neuronal membranes (71–73).
Despite the fact that PUFAs are vital for normal biological processes, humans can only achieve very little de novo synthesis of DHA, its precursor EPA, and arachidonic acid. For the human brain the chief source of these fatty acids is dietary, and dietary PUFAs are incorporated into the brain. Accordingly, an adequate lipid environment is vital for the normal functioning of neuronal membrane proteins such as ion channels, enzymes, ion pumps, and receptors. However, since the brain and neurons are regularly exposed to different kinds of acute and chronic environmental stress, they are susceptible to lipid peroxidation due to the high content of PUFAs. Arachidonic acid, although indispensable for brain development, function, and protection, can be enzymatically cleaved by cyclo-oxygenases and lipoxygenases to form 4-hydroxy-2-nonenal (called here simply “hydroxynonenal”) as a final product (74). Hydroxynonenal is an α, β-unsaturated lipophilic aldehyde formed from the lipid peroxidation of ω-6 PUFAs such as arachidonic and linoleic acids. Due to the 3 reactive groups: an aldehyde, a double bond at carbon-2, and a hydroxy group at carbon-4 (Figure 1B), hydroxynonenal is well-known for its cytotoxic and mutagenic effects (75, 76).
Exogenous hydroxynonenal is generated from ω-6 PUFAs, especially linoleic acid, during deep-frying of vegetable oils made from rapeseed (canola), soybean, sunflower, corn, etc. (77). Surprisingly, even extra-virgin olive oil generates abundant hydroxynonenal when heated to ∼200°C (Figure 1A), as it contains a small amount (∼10%) of linoleic acid. Most restaurants and food manufacturers do not typically use expensive extra-virgin olive oil for deep-frying, but may use less-expensive pure-virgin olive oil, canola oil, soybean oil, sunflower oil, corn oil, etc. After consumption of high-fat “fast food” cooked with such ω-6 PUFA-rich vegetable oils, the concentration of hydroxynonenal in the plasma increases significantly and rapidly; within minutes to hours (78). Although consumption of ω-6 PUFA-rich vegetable oils has increased worldwide in recent years due to their lower cost, little basic data are available on the adverse effect of ω-6 PUFA-rich vegetable oils on human health. Deep-frying in ω-6 PUFA-rich vegetable oils such as soybean, canola, sunflower, or corn oil also generates exogenous hydroxynonenal which is incorporated into the body immediately upon consumption. Likewise, common Western foods such as mayonnaise, tub and stick margarine, and Italian salad dressing, contain significant amounts of linoleic acid, even if not fried, and this will be incorporated into the cell membrane and LDL. Accordingly, another common source of hydroxynonenal is endogenous hydroxynonenal, which is produced by peroxidation of ω-6 PUFAs in the membrane phospholipids or LDL in the plasma in response to diverse oxidative stresses. For example, hydroxynonenal generation in the brain has been associated with exposure to drugs, ethanol, or irradiation, and with ischemia or chronic inflammation. Although the differential quantification of the harm from exogenous or intrinsic hydroxynonenal is difficult, ω-6 PUFA-rich vegetable oils are much more harmful by generating more hydroxynonenal, compared with ω-3 PUFAs.
Lipid peroxidation is the oxidative deterioration of PUFAs that contain 2 or more carbon-carbon double bonds (C = C). Initiation of lipid peroxidation can be caused by the addition of reactive oxygen species or by abstraction of a hydrogen atom from a methylene (-CH2-) group; in both cases, a carbon radical results. For example, the hydroxyl radical ●OH can react by addition (-HC = CH- + ●OH → -HC(OH)-●CH-) or by H● abstraction (-CH2- + ●OH → -●CH- + H2O) (79). Hydroxyl radicals readily initiate peroxidation of fatty acids, lipoproteins, and membranes. A single initiation event has the potential to generate multiple peroxide molecules by a chain reaction. Following lipid peroxidation, hydroxynonenal and malondialdehyde are the most abundant aldehydes produced, whereas acrolein is the most reactive (77, 79, 80).
By its multiple impacts on protein regulation, hydroxynonenal has a role in the maintenance of cellular homeostasis at low concentrations of reactive oxygen species. However, severe and long-standing oxidative stress can lead to accumulation of intrinsic hydroxynonenal, thereby compromising cellular functions (81). To understand the effects of hydroxynonenal precisely, it is necessary to highlight its dose-dependent effect. Low concentrations of hydroxynonenal facilitate susceptibility of proteins to proteolysis and removal by the proteasomal system. Under normal conditions, the latter is able to remove the majority of oxidatively damaged and modified proteins. However, under conditions of severe oxidative stress, accumulation of modified proteins occurs due to high concentrations of hydroxynonenal, due to protein crosslinking or malfunction of the proteolytic machinery (80). Further, apoptosis is induced at low doses of hydroxynonenal, whereas necrosis is induced at high doses. For example, in experiments of HepG2 cells exposed to 5–40μM of hydroxynonenal, the cells die by apoptosis, but the same cells exposed to 80–100μM hydroxynonenal die by necrosis (82). Similarly, necrosis was induced by 100μM hydroxynonenal in HeLa cells (83). Although the detailed mechanism still remains unknown except for the calpain-mediated cleavage of carbonylated Hsp70.1 (84), hydroxynonenal has been increasingly recognized as a particularly important mediator and marker of cellular dysfunction and degeneration in diverse disorders including cardiovascular disease (85), stroke (86), Alzheimer's disease (27, 87), arthritis (88), and asthma (89).
Hydroxynonenal-mediated Hsp70.1 carbonylation
Hydroxynonenal is an amphiphilic compound with both water-soluble and lipophilic properties. For the biological effects of hydroxynonenal, the lipophilic properties are more important than the hydrophilic properties. Therefore, hydroxynonenal tends to concentrate in biomembranes rather than in the aqueous space of cells. However, hydroxynonenal can be easily transferred from the membrane to both the cytosol and extracellular space, and its shuttling to either the extra- or intra-cellular space occurs very quickly, in the microsecond range. Due to these characteristics of the hydroxynonenal-membrane interaction, hydroxynonenal has reactivity with proteins inside and outside the cell (79). Hydroxynonenal forms adducts with 4 different side chains in proteins, namely Cys, His, Lys, and Arg. Cys residues display by far the highest reactivity, and the order of the molar hydroxynonenal/amino acid ratio is Cys (0.6) >> His (1 × 10–3) > Lys (3 × 10–4) >> Arg (4 × 10–5) (90). Accordingly, numerous proteins are modified by hydroxynonenal, including plasma membrane ion and nutrient transporters, receptors for growth factors and neurotransmitters, mitochondrial electron transport chain proteins, protein chaperones, proteasomal proteins, and cytoskeletal proteins (91, 92).
For example, in a rat model of chronic alcohol intake-induced oxidative stress, Carbone et al. (93) showed by mass spectral (MS) analysis that Hsp72 treated with 10 and 100μM hydroxynonenal caused adduct formation at Cys267 in the ATPase domain of the chaperone. Hydroxynonenal-adducted Hsp70 has also been identified in G93A-SOD1 transgenic mice, a model of familial amyotrophic lateral sclerosis (94). Further, in response to hydroxynonenal being generated by oxidative stress, a specific oxidative injury, “carbonylation,” was found to occur at the key site Arg469 of Hsp70.1 in the postischemic monkey brain. A decrease of its molecular weight from 157.20 to 113.12 indicated a special oxidative injury: carbonylation (8, 9). Two-dimensional carbonyl immunoblots of monkey hippocampal tissues after immunoprecipitation with anti-Hsp70.1 antibody revealed >10-fold upregulation of carbonylated Hsp70.1 in the CA1 tissue after transient ischemia. In addition, DHA (C22:6) and oleic acid (18:1), which are involved in the chemistry of BMP, showed a significant decrease on days 1 and 3 after the ischemic insult, compared with nonischemic controls (95). Such a decrease would cause impaired fusion between limiting and internal membranes in lysosomes, which may lead to lysosomal membrane destabilization. Since Hsp70.1-BMP binding is indispensable for activating acid sphingomyelinase and producing ceramide to stabilize lysosomal membranes, modifications of Hsp70.1 and BMP, if combined, cause storage of sphingomyelin and deficiency of ceramide at the lysosomal membrane, leading to its destabilization (95).
Calpain-mediated cleavage of carbonylated Hsp70.1
Calpain is a family of cysteine proteases that are ubiquitously expressed in most cell types. It is highly expressed in neurons and involved in many brain functions. Intracellular Ca2+ concentrations are stored mainly in the endoplasmic reticulum and mitochondria under normal conditions. Cerebral ischemia or reactive oxygen species can lead to the loss of their membrane integrity, causing a release of Ca2+ into the cytoplasm. As calpain is located in the cytosol as an inactive precursor, it translocates to the intracellular membranes (Figure 3B) in response to increased cytosolic Ca2+, and is activated by autocatalytic hydrolysis. Several forms of evidence have emerged since the early 1990s to support the implication of calpain in postischemic neurodegeneration (96–99). Not only acute, extensive ischemia during stroke but also chronic, mild ischemia due to aging may cause calpain activation (100). Since its discovery (101, 102), however, the exact substrate of μ-calpain in vivo had remained unknown for more than half a century. Despite multiple attempts at substrate sequence analysis (103, 104), no definitive method has existed to identify calpain substrates in the living brain. Intriguingly, however, Sahara and Yamashima (84) suggested from an in vitro experiment using monkey brain tissues that the calpain substrate in neurons should be carbonylated Hsp70.1 (Figures 2 and 3A). Subsequently, also in a mouse photoreceptor cell death model, calpain-mediated cleavage of carbonylated Hsp70 was confirmed by hydroxynonenal generation after methylnitrosourea injection (105).
Prior to hydroxynonenal treatment, in vitro cleavage of Hsp70.1 in response to activated μ-calpain was negligible not only in monkey brain tissues but also in the recombinant Hsp70.1 proteins. However, Hsp70.1 cleavage increased time-dependently following hydroxynonenal-mediated carbonylation not only in the CA1 tissue (Figure 3A) but also in the recombinant Hsp70.1 protein. Calpain-mediated cleavage of carbonylated Hsp70.1 was also demonstrated to occur in various brain tissues including the hippocampal dentate gyrus, cerebral cortex, cerebellar cortex, and substantia nigra (95). Since Hsp70.1 cleavage was dose-dependently blocked by a specific calpain inhibitor 1 (ALLN, AC-LL-norleucinal), N-acetyl-Leu-Leu-Nle-CHO, it is likely that Hsp70.1 can be more efficiently cleaved by activated μ-calpain especially after hydroxynonenal-induced carbonylation (95, 100, 105). The accessibility and reactivity of activated μ-calpain to the Hsp70.1 protein is determined by its tertiary structure. Hydroxynonenal can covalently attach Cys, His, and Lys residues of proteins, which can alter protein structure (106, 107). Hydroxynonenal-induced modification can lead to alteration of the tertiary structure of proteins together with altered subunit association (80). It is conceivable that hydroxynonenal-induced carbonylation of Hsp70.1, presumably due to its conformational changes, exaggerated calpain-mediated cleavage time-dependently by facilitating calpain accessibility.
The lysosomal membrane is a single lipid bilayer protected from intrinsic acidic hydrolases by the expression of heavily glycosylated proteins such as Lamp-1 and Lamp-2, which can resist digestion and form a protective glycocalyx (108). Lamp-2A is an abundant lysosomal membrane protein with an important role in chaperone-mediated autophagy. Lamp-2A aids lysosomal uptake of modified and oxidatively damaged proteins directly into the lumen of lysosomes for degradation and protein turnover (109). The μ-calpain substrates comprise a wide variety of proteins, especially in vitro, but the mechanisms of substrate recognition and cleavage by calpains are not well known, making the task of in vivo identification of calpain substrates very difficult. It is likely that Hsp70.1 is not the sole calpain substrate during oxidative stress. Interestingly, Rodriguez and Torriglia (110) found that Lamp-2A is also a μ-calpain substrate that participates in neuronal cell death. The mechanism by which μ-calpain cleaves Lamp-2A was not elucidated until now. They speculated that μ-calpain cleaves Lamp-2A when it forms the chaperone-mediated autophagy pore (110). Lamp-2A has 3 domains: luminal, transmembrane, and cytoplasmic, and the calpain cleavage site is thought to be at the luminal domain. Therefore, under normal conditions, μ-calpain cannot access the Lamp-2A luminal domain, however, when chaperone-mediated autophagy is activated, Lamp-2A oligomerizes and forms a pore that allows the transport of μ-calpain (110, 111).
Xue et al. (112) demonstrated that carbonylation of myofibrillar proteins by oxygen radicals increased μ-calpain-mediated cleavage of myosin heavy chain and α-actinin. Importantly, the cleavage occurred only in the presence of calpain activation, and the increasing oxidative level caused a stepwise escalation in cleavage, which coincides well with the stepwise carbonyl increase. However, in the absence of calpain activation, even maximum protein oxidation could not cause a spontaneous breakdown of myofibrillar proteins. Taken together, μ-calpain activation at the lysosomal membranes (Figure 3B) may be the principal factor affecting Hsp70.1 susceptibility.
Lysosomal destabilization due to Hsp70.1 dysfunction
Lysosomal destabilization has been recognized as a cause of oxidative stress-induced cell damage, because reactive oxygen species can induce lysosomal leakage (113–116). Lysosomes do not possess catalase or glutathione peroxidase which can degrade H2O2. Accordingly, in response to high levels of oxidative stress, excess H2O2 diffuses into lysosomes and generates hydroxyl radicals. Since lysosomes are enriched in iron by the degradation of iron-containing molecules, Fe2+ can catalyze the homolytic cleavage of H2O2 to form highly reactive hydroxyl radicals via the Fenton reaction (117).
Hydroxyl radicals can destabilize the lysosomal membrane by inducing lipid peroxidation and damaging lysosomal membrane proteins. Under severe oxidative stress conditions, the accumulation of modified proteins occurs, due to protein crosslinking or malfunction of the proteolytic machinery of the cell (80). Endogenous factors capable of causing lysosomal membrane destabilization include sphingosine, oxidized lipids or lipoproteins, reactive oxygen species, calpains, certain caspases, amyloid-β, and apo E (118). The first evidence of lysosomal destabilization was reported over 40 y ago when cultured cells were treated with lysosomotropic detergents (119). However, interest in implicating lysosomes for necrotic cell death faded during the following decades, and concomitantly, lysosomal involvement in cell death was overlooked. This was largely because the ability of caspase inhibitors, such as zVAD-fmk, to inhibit cell death was considered as proof of not “necrotic” but “apoptotic” cell death, and previous researchers had failed to confirm evidence of lysosomal membrane rupture by electron microscopy (120, 121). However, Yamashima et al. (122) ultrastructurally confirmed evidence of lysosomal membrane rupture in postischemic CA1 neurons after transient brain ischemia. Surprisingly, in the cortical neurons of an Alzheimer's patient, double-membrane structures presumably indicating lysosomal membrane permeabilization/rupture (Figure 3C) can be, although extremely difficult to observe, confirmed by electron microscopy under high-magnification observation.
As mentioned above, BMP downregulation, combined with Hsp70.1 cleavage, presumably provoked impairment of acid sphingomyelinase with the accumulation of sphingomyelin and deficiency of ceramide at the lysosomal membrane (10, 35, 95, 100), which may lead to lysosomal permeabilization and/or rupture in both ischemic and Alzheimer's neurons. As Hsp70.1 is involved in maintaining lysosomal membrane integrity, reactive oxygen species-induced Hsp70.1 carbonylation and calpain-mediated cleavage, in combination, cause lysosomal membrane disintegrity. The “calpain-cathepsin hypothesis” (9, 21, 122) can explain how impairments in proteolysis contribute to the development of neuronal dysfunction, degeneration, and death from low-species animals to primates. The pathogenic synergism of calpain activation and Hsp70.1 carbonylation works in concert to destabilize lysosomal membranes via BMP downregulation.
By redox proteomic analysis, elevated levels of Hsp70.1 carbonylation were identified in the brains of patients with mild cognitive impairment (MCI) (123). As a cause of Hsp70.1 carbonylation, elevated concentrations of hydroxynonenal were found in the plasma of Alzheimer's patients (124), whereas increased concentrations of hydroxynonenal were found in the brains of patients with MCI and early Alzheimer's disease (20). These findings, taken together, reinforce involvement of Hsp70.1 alteration in the occurrence of Alzheimer's neuronal death.
Cerebral Ischemia May Cause Alzheimer's Neuronal Death
Alzheimer's disease is a neurodegenerative disorder primarily characterized by progressive cognitive dysfunction and impaired short-term memory, which are followed by personality and behavior changes, and finally by language loss (125, 126). Genetic aberrations account for only a small proportion (<5%) of early-onset/familial Alzheimer's cases. More than 95% of Alzheimer's cases are sporadic with late-onset/age-related occurrence as a result of long-standing oxidative stress and/or decreased cerebral blood flow with aging. The Alzheimer's brain histopathologically exhibits 2 hallmark lesions, i.e., extracellular senile plaques made of amyloid-β42 peptides and intracellular neurofibrillary tangles composed of hyperphosphorylated tau proteins. The primary pathological cause of Alzheimer's disease is neuronal death which particularly affects the medial temporal lobe, especially the hippocampus and its associated areas of the cerebral cortex. Widespread neuronal death results in a decline of cognition and memory, as well as changes of personality. Although extremely important, surprisingly, the underlying mechanisms of Alzheimer's neuronal death were not elucidated for more than a century (127). The amyloid-β hypothesis, first proposed by Hardy and Higgins (128), has been the most dominant theory to explain the etiology and pathogenesis of Alzheimer's disease. Its core concept is that the accumulation of insoluble amyloid-β is an early event leading to neurodegeneration (129). However, over the past decade, the amyloid-β hypothesis became doubtful, because positron emission tomography studies showed no significant correlations between the extent of brain amyloid-β depositions and either degree of dementia or neuronal loss (130). In opposition to the amyloid-β hypothesis, the tangles hypothesis is rather accepted as the central pathogenic feature of Alzheimer's disease (131). Tau hyperphosphorylation decreases the binding of tau to microtubules, which results in increased tau self-aggregation and leads to loss of cytoskeletal function and cell degeneration. As the presence of tangles correlates better with the status of disease than senile plaques (132), the tangles hypothesis, although unable to explain the mechanism of Alzheimer's neuronal death, rather prevails over the amyloid-β hypothesis at present.
Since activated caspase-3 immunoreactivity has been reported in both the human Alzheimer's brain (133) and amyloid transgenic mice (134–136), apoptosis was hypothesized to be involved in amyloid-β42-mediated neuronal death in Alzheimer's disease (137–139). However, morphological evidence of apoptosis such as apoptotic bodies can be only infrequently observed in degenerating neurons of the patient brain. Amyloid-β42 was shown to induce cytochrome c release from mitochondria (140), which activates caspase-3 and causes apoptosis, thus providing a potential mechanism for amyloid-β42-mediated neuronal death. Eimer and Vassar (136) also suggested that intraneuronal amyloid-β42 accumulation in the endosomal-lysosomal pathway may lead to activation of caspase-3 and apoptotic neuron death in 5XFAD transgenic mice. However, the exact roles played by accumulations of amyloid-β plaques and tau protein spindles are not completely understood, nor are how and why neurons die in Alzheimer's disease. Investigating neuronal death pathways in an Alzheimer's brain is considerably complicated by the slow evolution of neurodegeneration in which prodeath and prosurvival factors within a given neuron may battle over months or years, and proteolytic systems may crosstalk extensively, yielding a complex picture (118). Only a tiny percentage of the neurons may be degenerating at any given time of the disease process. Neurons with dystrophic axons containing activated calpain and caspases, and massive accumulations of autophagic vacuoles, may persist for months or years (141–143).
To explain the pathogenesis of Alzheimer's disease, 3 representative causative factors such as oxidative stress, excitotoxicity, and chronic inflammation have been proposed (123, 144–148). Among them, oxidative damage, especially occurring at the protein level, is 1 of the widely-accepted mechanisms of this disease (19, 22, 100, 144, 149–151). The accumulation of oxidatively modified proteins disrupts cellular functions either by a loss of catalytic ability and/or an interruption of regulatory pathways. Oxidative stress may cause reversible or irreversible protein modifications including carbonylation, nitration, and protein-protein crosslinking. It is impossible to assess the extent of oxidative stresses in the living human brain. However, there is strong postmortem and experimental evidence concerning hydroxynonenal-induced oxidative damage occurring in Alzheimer's patients. For instance, elevated concentrations of hydroxynonenal, compared with age-matched controls, have been reported in the affected brain regions that are closely related to memory functions (19), ventricular fluid (87), the amyloid component of senile plaques (152), and in the plasma (124). Hydroxynonenal covalently modifies amyloid-β (1-40) and hastens its aggregation and toxicity, which in turn causes further oxidative stress that leads to the formation of even more lipid peroxidation products, such as hydroxynonenal and more toxic amyloid-β oligomers (153). Intriguingly, analysis of the protein carbonyls by derivatization with 2,4-dinitrophenylhydrazine showed that the cerebral cortex of Alzheimer's patients was affected by more severe membrane protein carbonylation, compared with nondemented aged subjects (154).
A microtubule-associated protein, tau, plays an important role in the stabilization of microtubules in axons. However, hyperphosphorylated tau sequesters normal tau and disrupts microtubules. In the Alzheimer's brain, hyperphosphorylated tau dissociates from microtubules and self-associates to form both fibrillar and prefibrillar oligomeric aggregates (155, 156). Aggregation of tau forms neurofibrillary tangles which inhibit kinesin-dependent fast axonal transport. However, Hsp70.1 attenuates tau toxicity by maintaining tau in a soluble, nonaggregated state and by facilitating degradation of aggregated tau (157, 158). Previously, Patterson et al. (159) demonstrated that Hsp70.1 directly inhibits tau aggregation by a preferential association with soluble, monomeric, and prefibrillar oligomeric tau species to facilitate anterograde fast axonal transport. Accordingly, Hsp70.1 disorders such as carbonylation and cleavage may cause tau aggregation and neurofibrillary tangle formation (100, 159).
Fischer et al. (160) reported a statistically significant reduction in net vascular density specifically in the basal forebrain region and the hippocampus of Alzheimer's brains, compared with control brains. Diminished blood flow to the brain results from an impaired microcirculation due to pathological changes and/or reduced numbers of capillaries with aging. Although a significant reduction of the capillary network occurs during the acute ischemia of stroke, it is more or less inhibited by knockout of the GPR40 gene (161). Free fatty acid receptor GPR40 is currently known to have dual functions (162), and appears to be crucial for both physiology (adult neurogenesis in the hippocampus and energy control in the hypothalamus) and pathology (for example, stroke, Alzheimer's disease, Parkinson's disease, and type 2 diabetes) conditions (28). It is probable that in response to excessive ω-6 PUFAs and/or oxidized fatty acids, abnormal Ca2+ mobilization may occur within vascular endothelia to cause cell degeneration/death via calpain activation (161), and contribute to the reduction of the capillary network.
The potential contribution of calpain to the aging process and Alzheimer's disease is obvious as reviewed by Nixon (163). Taniguchi et al. (164) found that calpain is activated >7-fold in Alzheimer's brains compared with age-matched brains. Calpains can be activated by Alzheimer's Aβ42 in primary rat neurons (165), as well as by ischemic injury in primate neurons (166). Not only acute, extensive ischemia during stroke but also long-standing, mild ischemia due to arteriosclerosis with aging may induce calpain activation, which can lead to the cleavage of lysosome membrane proteins such as Hsp70.1 (84), Lamp-2 (167, 168), and subunit b2 of v-ATPase (169). This facilitates lysosomal membrane permeabilization/rupture causing release of lysosome content into the cytosol and the degradation of cell constituents. The “calpain-cathepsin hypothesis” suggests a role for calpain and cysteine proteases in lysosomal destabilization, and further expands the lysosomal theory of neurodegeneration in Alzheimer's disease (21, 22, 100). Gerónimo-Olvera et al. (168) also demonstrated, in cultured cortical neurons with glucose deprivation/reintroduction, that calpain-mediated degradation of Lamp-2 results in lysosomal membrane permeabilization, causing the release of lysosome content, cathepsins, and neuronal death. Lysosomes are the main catabolic organelles of a cell, so their disintegrity should be associated with many lifestyle- and/or age-related diseases.
Can Hydroxynonenal Cause Diverse Cell Death in Lifestyle Disease?
Excessive ω-6 PUFAs can induce abnormal calpain activation via GPR40
When ω-6 PUFAs interact with GPR40 coupled to a Gq heterotrimeric G protein, it results in the binding of an α-subunit of Gq to phospholipase C (PLC) which is located at the plasma membrane. Activated PLC hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) into inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (170). IP3 binds to the Ins3P receptor (Ins3PR) on the ligand-gated Ca2+ channel that is found at the surface of the endoplasmic reticulum. The binding of IP3 to Ins3PR triggers opening of the Ca2+ channel, thus releasing Ca2+ into the cytoplasm (162, 171).
ω-3 PUFAs are widely accepted to play a significant role in the generation of newborn neurons in the adult hippocampus for learning and memory. Until recently, however, there was no consensus regarding the exact molecular mechanism of the effect of PUFAs in adult-born neurons. In 2003, 2 groups independently identified GPR40 as a receptor for diverse fatty acids, and found that among human organs, its expression is most abundant in the brain and next in the pancreas (11, 172). Subsequently, Ma et al. (173) found expression of GPR40 in the central nervous system of young adult monkeys including hypothalamic and hippocampal neurons. They also found expression of GPR40 in the neurogenic niche of the adult monkey hippocampus (174). Intriguingly, in rat neural stem cells transfected with GPR40 genes, they found that DHA can induce neuronal differentiation, and neurite growth and branching through acting on GPR40 (175). In addition, Boneva and Yamashima (176) found that GPR40/phosphorylated cAMP response element-binding protein (pCREB) double-positive cells significantly increased in the hippocampal neurogenic niche of postischemic monkeys, and suggested that GPR40 is capable of activating PUFA-CREB signaling and contributing to adult neurogenesis. Thereafter, using a human neuroblastoma cell line and primary mouse central nervous system neurons that express the GPR40 receptor, Zamarbide et al. (177) demonstrated a direct correlation between GPR40 activation and CREB phosphorylation. Based on these data, Yamashima (162) first suggested implications of the “PUFA-GPR40-CREB-brain-derived neurotrophic factor (BDNF) signaling” pathways for adult neurogenesis as well as learning and memory.
CREB belongs to the family of leucine zipper transcription factors. It attaches to DNA sequences called cAMP response elements (CRE) to regulate the transcription of various genes such as BDNF, c-fos, neuropeptides (such as somatostatin, enkephalin, vascular growth factor, and corticotropin-releasing hormone), and tyrosine hydroxylase. CREB plays a significant role in neuronal plasticity and long-term memory formation in the brain (178). When an appropriate amount of nonoxidized PUFA binds with GPR40 to induce optimal Ca2+ mobilization necessary for CREB phosphorylation and BDNF synthesis, the physiological molecular cascade works initially for synaptic plasticity and eventually, for learning and memory. However, GPR40 has dual roles in response to the amount and status of ligands; an appropriate amount of nonoxidized PUFAs can participate in physiological function, whereas excessive and/or oxidized ω-6 PUFAs may induce abnormal Ca2+ mobilization as a trigger of cell degeneration and death (162). Since foods currently eaten by Western people have abundant amounts of essential ω-6 and relatively less ω-3 PUFAs with the average ω-6/ω-3 ratio being ∼6 (179), balancing ω-3 and ω-6 would be indispensable for reducing overactivation of GPR40 by excessive amounts of ω-6 PUFAs and their metabolites.
One representative case is appetite-regulating neurons in the hypothalamus. The arcuate nucleus is situated adjacent to the floor of the third ventricle in the mediobasal hypothalamus, and contains neurons that exert potent effects on food intake, energy expenditure, and glucose homeostasis (Figure 4A) (180). Anabolic neurons coexpress neuropeptide Y (Npy) and agouti-related peptide (Agrp), which potently stimulate food intake and reduce energy expenditure and thereby promote body weight gain (181–183). Npy/Agrp neurons are inhibited by input from leptin and insulin, whereas reduced input from these hormones increases hypothalamic signaling (184). Melanocortin is 1 of 8 active peptides derived from posttranslational processing of the precursor POMC. POMC neurons in the arcuate nucleus innervate the same hypothalamic areas supplied by fibers from Npy/Agrp neurons. Unlike Npy/Agrp neurons, POMC neurons are stimulated by input from insulin, leptin, and free (nonesterified) fatty acids in the blood (Figure 4A) (185, 186), inhibit food intake, and promote body weight loss (187). In POMC neurons, leptin signals are mediated by leptin receptors, whereas fatty acid signals are mediated by GPR40 (Figure 4B). In the short term of HFD consumption, the hypothalamic POMC neurons are affected at the functional level and develop resistance to the adipostatic signals delivered by leptin and insulin (188, 189). However, over time with sustained stimulation of the GPR40 receptor, POMC neurons may become permanently damaged and undergo cell degeneration/death by calpain activation in both rodents and humans (190–192). POMC neuronal loss contributes to promoting hyperphagia and body weight gain with resultant insulin resistance and glucose intolerance.
FIGURE 4.

GPR40-positive POMC neurons of the arcuate nucleus of hypothalamus inhibiting food intake. A: there are 2 populations of centrally projecting neurons in the arcuate nucleus (light-blue area) that can respond to diverse neuropeptides and free fatty acids: Agrp/Npy and POMC/Cart. Agrp (agouti-related protein) and Npy (neuropeptide Y) stimulate food intake and decrease energy expenditure, whereas POMC (pro-opiomelanocortin) and Cart (cocaine- and amphetamine-regulated transcript) inhibit food intake and increase energy expenditure. Free fatty acids in the blood, insulin/leptin from the pancreas/adipose tissues, and ghrelin from the stomach, altogether regulate Agrp/Npy and POMC/Cart neurons via the corresponding receptors. ⊕ shows activation, whereas ⊖ shows inhibition. (Adapted with permission from reference 193.) B: administration of GPR40-agonist GW9508 activates POMC neurons in the arcuate nucleus with the induction of the proto-oncogene c-fos. It is easily suggested that similar GPR40 activation occurs in POMC neurons in response to fatty acids in the blood. (Adapted with permission from reference 194.) Ghsr, growth hormone secretagogue receptor; GPR40, G-protein coupled receptor 40; Lepr, leptin receptor; Mc3r/Mc4r, melanocortin 3/4 receptor; Y1r, neuropeptide Y1 receptor.
Free fatty acids can induce both physiological and pathological Ca2+ mobilization by binding with the GPR40 receptor. Accordingly, intake of the appropriate amount and the right ω-3/ω-6 balance of nonoxidized PUFAs is indispensable for maintaining health, but intake of excessive ω-6 PUFAs and/or hydroxynonenal in deep-fried foods can cause diverse lifestyle diseases through abnormal GPR40 activation (Figures 2 and 4B). The most important cause of harm is the amount of ω-6 PUFAs, not whether it is a high-, medium-, or low-fat diet.
Hsp70.1 dysfunction and POMC neuron degeneration/death in obesity
As mentioned above, the hypothalamus senses and responds to input from adiposity-related (leptin, insulin) and nutrient-related (free fatty acids) circulating signals that convey information regarding both body energy stores and current energy availability (Figure 4) (180). Thus, the hypothalamus regulates energy intake, energy expenditure, and endogenous glucose production. For energy and body weight control, the hypothalamus is capable of continuously producing new neurons during adult life (195), like the subventricular zone of the lateral ventricles and the subgranular zone of the hippocampal dentate gyrus (196). Nascimento et al. (24) previously demonstrated that the hypothalamus, especially the GPR40-positive POMC neuronal population (Figure 4A), presents constitutive neurogenesis, and ω-3 PUFAs are capable of increasing hypothalamic neurogenesis to levels similar or superior to the effect of BDNF. In contrast, hypothalamic neuron death, if occurring, is believed to play an important role in the development and perpetuation of obesity, which is partially due to hypothalamic resistance to leptin and insulin (197, 198). Birth and death of POMC neurons in the adult hypothalamus are thought to play an important role in controlling food intake and energy expenditure. Like the hippocampus, most of the beneficial effect of dietary fats may be mediated by GPR40 in the hypothalamus (Figure 4), and BDNF may be ≥1 of the final effectors of this process. In the hypothalamus, ω-3 PUFAs can induce neurogenesis of not Npy but POMC neurons by increased expression of GPR40 and BDNF. The inhibition of GPR40 was capable of reducing the neurogenic effect of ω-3 PUFAs, whereas inhibition of BDNF resulted in the global reduction of hypothalamic cells (24). Similarly, in response to oxidized PUFAs, GPR40 shows detrimental effects of POMC neuronal loss leading to obesity. POMC neurons play an essential role in protecting against obesity, so loss of these neurons is sufficient to cause excess weight gain in mice (199). The consumption of deep-fried foods and a HFD is regarded as 1 of the most important epidemiological factors leading to the increased prevalence of obesity in modern society. However, even in mice fed a low-fat diet, dietary linoleic acid (the precursor of arachidonic acid) was demonstrated to elevate the endocannabinoids: 2‐arachidonoylglycerol (2‐AG) and anandamide, and promote weight gain, because the endocannabinoid system is also involved in the regulation of energy balance (200, 201). This is true regardless of high-, medium- or low-fat diets, so fatty acid composition rather than the amount of fat, may actually be the key factor inducing obesity. More specifically, ω-6 PUFAs (dietary linoleic acid or arachidonic acid in membrane phospholipids) increase appetite and adipose tissue inflammation, then produce endogenous 2-AG and anandamide to induce obesity. Given the backbone of both 2‐AG and anandamide is the ω‐6 arachidonic acid, ω-6 PUFAs, not ω-3 PUFAs, are the real culprit of obesity. EPA and DHA, on the other hand, reverse the obesogenic effects of ω-6 PUFAs to normalize body tone thereby elevating the importance of balancing these essential fatty acids (200, 201).
Although hypothalamic neurons in both rodents and humans consuming a HFD undergo cell death during the development of obesity, the implication of the molecular chaperone Hsp70.1 had remained grossly unknown. However, Thaler et al. (192) found in rodents that a HFD (60 kcal% fat, containing 245 g lard and 25 g soybean oil in a total weight of 773.85 g) was associated with upregulation of Hsp70.1 and neuronal injury in POMC neurons of the hypothalamus (Figure 5). In the arcuate nucleus of mice and rats, inflammatory signaling was evident as early as 1 to 3 d after the feeding of a HFD, prior to substantial weight gain. Further, both reactive gliosis and Hsp72 (compatible with Hsp70.1 in humans) upregulation (Figure 5B, D), suggestive of neuron injury, were evident in the arcuate nucleus of the hypothalamus within the first week of HFD feeding. Intriguingly, they also noted evidence of autophagy failure: the accumulation of autophagosomes (Figure 5F, G, black arrows) in the POMC neurons within the first week of HFD feeding. In addition, mitochondrial membrane disruption (Figure 5F white arrows, 5H) and accumulation of autophagosomes engulfing damaged mitochondria were observed at 20 wk after feeding. Consequently, after 8 mo of HFD feeding, an ∼25% reduction in the number of POMC neurons was confirmed in the arcuate nucleus of mice, compared with that of controls which were fed a nonpurified diet over the same time period (Figure 5I Chow, 5J, H, F, D). Interestingly, these responses appear to occur selectively in the arcuate nucleus and specifically in POMC neurons not only in rodents but also in humans (192).
FIGURE 5.

Effects of high-fat diet feeding upon hypothalamic neuronal injury in rodents. A, B, C, D: immunohistochemical analysis of Hsp72 (compatible with primate Hsp70.1) in rats fed a nonpurified diet (Chow) (A, C) or HFD (B, D) for 7 d (1W). A remarkable upregulation of Hsp72 is induced by HFD, compared with the nonpurified diet (Chow). Immunofluorescence shows colocalization of Hsp72 (red) with POMC peptide (green) (C, D). Double-staining (merged yellow color) indicates Hsp72 is upregulated at POMC neurons, in response to HFD. E, F: electron micrographs of a POMC neuron from mice fed a nonpurified diet (Chow) (E) or HFD (F) for 20 wk (20W). In HFD mice (F), mitochondrial integrity is disrupted (white arrows), and autophagosomes are being formed (black arrows, inset). G: high-magnification images of a POMC neuron from the HFD mice, show formation of membrane-bound damaged mitochondria; autophagosomes (black arrows). H: high-magnification images of mitochondria in a POMC neuron from mice fed a nonpurified diet (Chow) show parallel-oriented cristae and regular structure, whereas mice fed HFD show nonparallel cristae and irregular structure. Note the remarkable mitochondrial injury induced by HFD, compared with the nonpurified diet (Chow). I, J: representative images of POMC neurons in the arcuate nucleus of mice fed either a nonpurified diet (Chow) (I) or HFD (J) for 8 mo (8M). Compared with the nonpurified diet (Chow) (I), HFD (J) mice showed a significant loss of POMC neurons. Bars = 50 μm (A, B, C, D, I, J); 1 μm (E, F); 500 nm (F; inset); 400 nm (G, H). (Adapted with permission from reference 192.) HFD, high-fat diet; Hsp72, heat-shock protein 72; POMC, pro-opiomelanocortin.
It is of great surprise that a HFD can induce autophagy failure and neuronal injury so fast, however, Thaler et al. (192) did not explain the precise molecular mechanism for this rapid occurrence. Instead, they suggested that the rapid induction of Hsp72 in POMC neurons during HFD feeding might be part of a neuroprotective response, because it was associated with local microglial and astroglial responses. In the hippocampus of the ischemic monkey experimental paradigm, however, Yamashima and colleagues observed >10-fold upregulation of Hsp70.1, but this indicated accumulation of nonfunctional Hsp70.1 because of the calpain-mediated cleavage of carbonylated Hsp70.1 proteins (8, 9, 84). The superficial Hsp70.1 upregulation was far from the neuroprotective response, since the monkey CA1 sector revealed complete neuronal loss after transient brain ischemia. It is conceivable that, in POMC neurons, rapid occurrence of neuronal injury and autophagy failure was caused by high fat-induced abnormal Ca2+ mobilization and calpain activation via GPR40 (Figure 2). Interestingly, in response to oxidized PUFAs such as conjugated linoleic acid (202) or trans isomers of arachidonic acid (203), GPR40 was previously demonstrated in rodents to mediate lipotoxicity in β-cells, neurons, or microvessels via abnormal Ca2+ mobilization, which result in diabetes, retinopathy, stroke, etc., respectively (162). However, it remains undetermined in humans whether and how oxidized, conjugated, or excessive ω-6 PUFAs evoke such remarkable lipotoxicity. We speculate that both deep-fried foods and a HFD contain not only conjugated linoleic acid or trans isomers of arachidonic acid, but also hydroxynonenal which were generated due to reactive oxygen species or inflammation. These are sufficient for excessive GPR40 activation, Ca2+ mobilization, and calpain activation in humans with a high-fat intake. The resultant calpain-mediated cleavage of carbonylated Hsp70.1 and cathepsin leakage by lysosomal membrane permeabilization/rupture, can cause diverse cell death (Figure 2). The extralysosomal release of cathepsin B, D, etc., can depolarize mitochondrial membranes and induce further production of reactive oxygen species (204).
Hydroxynonenal-induced cell death in neurons, hepatocytes, and β-cells
Since overactivation of the GPR40 receptor can, as mentioned above, lead to the disruption of Ca2+ homeostasis and cell death in response to a HFD (192), conjugated linoleic acids (202), or trans isomers of arachidonic acid (203), it should be evaluated whether GPR40 overactivation occurs in response to hydroxynonenal to induce cell death. To the best of our knowledge, there are no previous reports available to answer this question especially in the brain and pancreas where GPR40 are abundant. However, in the liver where GPR40 expression is very little (11), a metabolic sensor GPR109A (also called hydroxycarboxylic acid receptor 2), being coupled with energy, lipid metabolism, and immune system, was recently reported to be expressed in Kupffer cells and hepatocytes (12, 13). In addition, GPR109A was demonstrated to induce Ca2+ mobilization and cell death in retinal pigmented and colon epithelial cells in response to hydroxynonenal (12). It is likely that the same may occur in hepatocytes and Kupffer cells where GPR109A is expressed.
There is evidence that hydroxynonenal contributes to the dysfunction and death of pancreatic β-cells in diabetes (205), and alcohol-induced pancreatic damage (206). Further, the authors recently found that consecutive but sublethal intravenous injections of synthetic hydroxynonenal for 24 wk induced diverse cell death/degeneration in nonpurified diet-fed young adult monkeys (aged 5–6 y) with a body weight of 5–7 kg (207, 14). In particular, the hippocampus, liver, and pancreas microscopically showed remarkable cell death/degeneration due to the permeabilization/rupture of lysosomes (Figure 6G, H, I, stars and red arrows) after the hydroxynonenal injections. The monkey hippocampus after consecutive injections of hydroxynonenal showed neuronal loss (Figure 6D, circles), whereas the liver showed NASH-like pathology (207, 14) which was mainly characterized by necrotic hepatocellular injury and ballooned hepatocytes with the accumulation of lipid debris (Figure 6E, circles) (14). In addition, the Langerhans islets of the pancreas showed numerous microcystic degeneration by light microscopy (Figure 6F, white rectangles), which were revealed to be β-cell degeneration using electron microscopy (Figure 6F, inset). As observed in the cortical neurons of an Alzheimer's patient (Figure 3C), the degenerating hippocampal CA1 neurons (Figure 6G, red arrow) and the hepatocytes (Figure 6H, red arrow) ultrastructurally showed evidence of lysosomal membrane permeabilization/rupture. In addition, lysosomal membrane permeabilization was observed in the β-cells (Figure 6I, red arrow). It is extremely difficult to identify the exact causative substance that has negative effects upon membrane integrity, especially if its adverse effects are weak and many years are needed for progression. However, the monkey experimental paradigm of the authors’ group has clearly shown that the highly reactive α, β-unsaturated aldehyde, “hydroxynonenal” can cause lysosomal membrane disintegrity within a few months, so it must be the real culprit of diverse cell death (14).
FIGURE 6.

Cell degeneration/death and lysosomal rupture in hippocampal CA1 neurons, hepatocytes, and pancreatic β-cells. A, B, C: hematoxylin-eosin staining of the hippocampus (A), liver (B), and pancreas (C) of a control monkey without hydroxynonenal injections. D, E, F: in monkeys after sublethal doses of hydroxynonenal injections, neuronal loss was observed in the CA1 (D, circles), whereas ballooning and cell death (E, circles) of hepatocytes, and microcystic degenerations of the Langerhans cells (F, white rectangles) were seen. The cells with microcystic degenerations were confirmed by electron microscopy to be degenerating β-cells, because of the presence of insulin granules with halo (F, inset). G, H, I: the degenerating CA1 neurons (G) and hepatocytes (H) showed lysosomal permeabilization/rupture with spillage of the lysosomal content (red arrows) from the lysosome (star), whereas β-cells (I) with insulin granules (g) showed permeabilization (red arrow) of lysosome (star) in the process of fusing with an autophagosome. Bar = 400 nm (G, H, I). Note that such lysosomal disintegrity of the hydroxynonenal monkeys was quite similar to that of a human patient with Alzheimer's disease as shown in Figure 3C. (Adapted with permission from reference 14.)
It is likely that an appropriate amount of nonoxidized PUFAs, especially ω-3, increases the serum BDNF concentration through the physiological GPR40 signaling pathway and contributes to synaptogenesis and brain development as well as learning and memory (162, 174–177). On the contrary, an excessive amount of PUFAs, especially ω-6 or hydroxynonenal may cause not only neurodegeneration but also cell degeneration of hepatocytes, and β-cells via GPR40/GPR109A overactivation and the subsequent abnormal Ca2+ mobilization. By focusing on the dual aspects of “brain-lipid sensing,” further detailed studies are required to clarify the precise molecular cascade by which hydroxynonenal causes diverse cell death in lifestyle diseases. Evidence has accumulated in favor of the “membrane sensor hypothesis” which predicts that the concentration of heat shock proteins can be changed as a result of alterations to the plasma membrane (208). According to this hypothesis, it was proposed that besides denatured/misfolded proteins, the actual membrane lipid composition/microheterogeneity are also key factors for regulating the mechanism of cellular stress sensing, signaling and, ultimately, the entire heat shock protein response including Hsp70.1 concentration. In support of the “membrane sensor hypothesis,” when dietary lipids and/or particular membrane intercalating drug candidates are used to alter membrane properties, the simultaneous normalization of dysregulated expression of heat shock proteins would cause beneficial responses to disease states (208).
Summary
GPR40 is a G-protein-coupled receptor that is expressed abundantly in certain regions of the brain, including the hippocampus and hypothalamus as well as in the pancreas and liver. Previous studies (24, 162, 173, 174, 176) have shown the potential neurogenic actions of GPR40 in the hippocampus and hypothalamus in response to ω-3 PUFAs, especially to DHA. GPR40-positive neurons are related to learning and memory in the hippocampus, and related to appetite, energy, and body weight control in the hypothalamus. However, GPR40 may show detrimental roles in response to excessive and/or peroxidized ω-6 PUFAs. Accordingly, here the authors focused on the dual aspects of “brain-lipid sensing” for health and diseases.
According to the “calpain-cathepsin hypothesis” (Figure 2) (9, 21, 122), enzymatically active calpains destabilize the lysosome by cleaving Hsp70.1, which normally works as a molecular chaperone and protects the integrity of the lysosomal membrane. After oxidative stress-induced carbonylation, Hsp70.1 becomes extremely vulnerable to calpain-mediated cleavage (84, 23). Accordingly, the resultant Hsp70.1 disorder can induce lysosomal rupture, which releases cathepsins from lysosomes to cause cell death (9, 21, 100). By covalently modifying Hsp70.1, both dietary ω-6 PUFA- (exogenous) and the membrane lipid- (intrinsic) peroxidation product “hydroxynonenal” conceivably play particularly sinister roles in the occurrence of lifestyle diseases such as Alzheimer's disease, stroke, NASH, type 2 diabetes, obesity, and associated disease processes.
One should always keep in mind that hydroxynonenal alone can induce diverse cell death in the brain, liver, and pancreas in nonpurified diet-fed monkeys (207, 14). The consumption of vegetable oils has significantly increased in non-Mediterranean countries especially after 1960, coinciding with the increase in various lifestyle diseases. Lastly, 3 concepts should be stressed as regards ω-6 PUFAs. First, ω-6 PUFAs are essential fatty acids, however, they have a “very narrow therapeutic window.” Second, too much ω-6 PUFAs shifts normal physiological function to pathophysiological, thereby causing harm in a dose-response manner. Third, “excessive ω-6 PUFA” intake can be described as merely above 0.4% of food energy (0.4 en%) (179). Accordingly, decreasing ω-6 PUFA-rich vegetable oils along with increasing ω-3 PUFA-rich fish oils would be necessary to avoid overactivation of GPR40 and thereby preventing various lifestyle diseases.
In conclusion, it is reasonable to pinpoint ω-6 PUFA-rich vegetable oil-derived “hydroxynonenal” as the real culprit behind cell degeneration/death causing Alzheimer's disease and related lifestyle diseases.
ACKNOWLEDGEMENTS
The authors’ responsibilities were as follows—TeY, TO, and EM: contributed to monkey experiments; HN and YY: analyzed the oils; TaY and MK: analyzed the toxicity of hydroxynonenal; TeY and SK: were responsible for design, writing, and final content; and all authors: read and approved the final manuscript.
Notes
Supported by a grant from Kiban-Kenkyu (B) (19H04029) from the Japanese Ministry of Education, Culture, Sports, Science, and Technology.
Author disclosures: The authors report no conflicts of interest.
Abbreviations used: AgRP, agouti-related peptide; ALDH2, aldehyde dehydrogenase 2; BDNF, brain-derived neurotrophic factor; BMP, bis(monoacyl)glycerophosphate; GPR40, G-protein coupled receptor 40; HFD, high-fat diet; Hsp70.1, heat-shock protein 70.1; Ins3PR, Ins3P receptor; IP3, inositol 1,4,5-triphosphate; Lamp-2, lysosomal-associated membrane protein-2; MCI, mild cognitive impairment; NASH, nonalcoholic steatohepatitis; NPY, neuropeptide Y; pCREB, phosphorylated cAMP response element-binding protein; PLC, phospholipase C; POMC, pro-opiomelanocortin; 2-AG, 2‐arachidonoylglycerol.
Contributor Information
Tetsumori Yamashima, Departments of Psychiatry and Behavioral Science; Cell Metabolism and Nutrition.
Tsuguhito Ota, Cell Metabolism and Nutrition.
Eishiro Mizukoshi, Gastroenterology.
Hiroyuki Nakamura, Environmental and Preventive Medicine.
Yasuhiko Yamamoto, Biochemistry and Molecular Vascular Biology, Kanazawa University Graduate School of Medical Sciences, Kanazawa, Japan.
Mitsuru Kikuchi, Departments of Psychiatry and Behavioral Science.
Tatsuya Yamashita, Cell Metabolism and Nutrition; Gastroenterology.
Shuichi Kaneko, Gastroenterology.
References
- 1. Bang HO, Dyerberg J, Nielsen AB. Plasma lipid and lipoprotein pattern in Greenlandic West-coast Eskimos. Lancet. 1971;1:1143–45. [DOI] [PubMed] [Google Scholar]
- 2. Bjerregaard P, Mulvad G, Pedersen HS. Cardiovascular risk factors in Inuit of Greenland. Int J Epidemiol. 1997;26:1182–90. [DOI] [PubMed] [Google Scholar]
- 3. Knoops KT, de Groot LC, Kromhout D, Perrin AE, Moreiras-Varela O, Menotti A, van Staveren WA. Mediterranean diet, lifestyle factors, and 10-year mortality in elderly European men and women: the HALE project. JAMA. 2004;292:1433–9. [DOI] [PubMed] [Google Scholar]
- 4. Blasbalg TL, Hibbeln JR, Ramsden CE, Majchrzak SF, Rawlings RR. Changes in consumption of omega-3 and omega-6 fatty acids in the United States during the 20th century. Am J Clin Nutr. 2011;93:950–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lauretti E, Praticò D. Effect of canola oil consumption on memory, synapse and neuropathology in the triple transgenic mouse model of Alzheimer’s disease. Sci Rep. 2017;7:17134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mattson MP. Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp Gerontol. 2009;44:625–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. D'Souza Y, Elharram A, Soon-Shiong R, Andrew RD, Bennett BM. Characterization of Aldh2 (-/-) mice as an age-related model of cognitive impairment and Alzheimer's disease. Mol Brain. 2015;8:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Oikawa S, Yamada T, Minohata T, Kobayashi H, Furukawa A, Tada-Oikawa S, Hiraku Y, Murata M, Kikuchi M, Yamashima T. Proteomic identification of carbonylated proteins in the monkey hippocampus after ischemia-reperfusion. Free Radic Biol Med. 2009;46:1472–7. [DOI] [PubMed] [Google Scholar]
- 9. Yamashima T, Oikawa S. The role of lysosomal rupture in neuronal death. Prog Neurobiol. 2009;89;343–58. [DOI] [PubMed] [Google Scholar]
- 10. Zhu H, Yoshimoto T, Yamashima T. Heat shock protein 70.1 (Hsp70.1) affects neuronal cell fate by regulating lysosomal acid sphingomyelinase. J Biol Chem. 2014;289:27432–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Briscoe CP, Tadayyon M, Andrews JL, Benson WG, Chambers JK, Eilert MM, Ellis C, Elshourbagy NA, Goetz AS, Minnick DT et al. The orphan G protein-receptor GPR40 is activated by medium and long chain fatty acids. J Biol Chem. 2003;278:11303–11. [DOI] [PubMed] [Google Scholar]
- 12. Gautam J, Banskota S, Shah S, Jee JG, Kwon E, Wang Y, Kim DY, Chang HW, Kim JA. 4-Hydroxynonenal-induced GPR109A (HCA2 receptor) activation elicits bipolar responses, Gαi-mediated anti-inflammatory effects and Gβγ-mediated cell death. Brit J Pharmacol. 2018;175:2581–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jadeja RN, Jones MA, Fromal O, Powell FL, Khurana S, Singh N, Martin PM. Loss of GPR109A/HCAR2 induces aging-associated hepatic steatosis. Aging. 2019;11:386–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yamashima T, Boontem P, Shimizu H, Ota T, Kikuchi M, Yamashita T, Mizukoshi E, Kaneko S. Vegetable oil-derived ‘hydroxynonenal’ causes diverse cell death possibly leading to Alzheimer's and related lifestyle diseases. J Alzheimers Dis Parkinsonism. 2020;10:483. [Google Scholar]
- 15. Dickson TC, King CE, McCormack GH, Vickers JC. Neurochemical diversity of dystrophic neurites in the early and late stages of Alzheimer's disease. Exp Neurol. 1999;156:100–10. [DOI] [PubMed] [Google Scholar]
- 16. Armstrong RA. The molecular biology of senile plaques and neurofibrillary tangles in Alzheimer's disease. Folia Neuropathol. 2009;47:289–99. [PubMed] [Google Scholar]
- 17. Iqbal K, Liu F, Gong CX, Grundke-Iqbal I. Tau in Alzheimer's disease and related tauopathies. Curr Alzheimer's Res. 2010;7:656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG, Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer's disease. J Neurochem. 2002;68;2092–7. [DOI] [PubMed] [Google Scholar]
- 19. Markesbery WR, Lovell MA. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer's disease. Neurobiol Aging. 1998;19:33–6. [DOI] [PubMed] [Google Scholar]
- 20. Williams TI, Lynn BC, Markesbery WR, Lovell MA. Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in Mild Cognitive Impairment and early Alzheimer's disease. Neurobiol Aging. 2006;27:1094–9. [DOI] [PubMed] [Google Scholar]
- 21. Yamashima T. Can ‘calpain-cathepsin hypothesis’ explain Alzheimer's neuronal death?. Ageing Res Rev. 2016;32:169–79. [DOI] [PubMed] [Google Scholar]
- 22. Yamashima T. Vegetable oil: the real culprit behind Alzheimer's disease. J Alzheimers Dis Parkinsonism. 2017;7:410. [Google Scholar]
- 23. Liang H, Kurimoto S, Shima KR, Shimizu D, Ota T, Minabe Y, Yamashima T. Why is hippocampal CA1 especially vulnerable to ischemia?. SOJ Biochem. 2016;; 2(2):7. [Google Scholar]
- 24. Nascimento LF, Souza GF, Morari J, Barbosa GO, Solon C, Moura RF, Victório SC, Ignácio-Souza LM, Razolli DS, Carvalho HF et al. n-3 fatty acids induce neurogenesis of predominantly POMC-expressing cells in the hypothalamus. Diabetes. 2016;65:673–86. [DOI] [PubMed] [Google Scholar]
- 25. Bibus D, Lands B. Balancing proportions of competing omega-3 and omega-6 highly unsaturated fatty acids (HUFA) in tissue lipids. Prostaglandins, Leukotrienes Essent Fatty Acids. 2015;99:19–23. [DOI] [PubMed] [Google Scholar]
- 26. Hoff HF, O'Neil J, Chisolm 3rd GM, Cole TB, Quehenberger O, Esterbauer H, Jürgens G. Modification of low density lipoprotein with 4-hydroxynonenal induces uptake by macrophages. Arteriosclerosis. 1989;9:538–49. [DOI] [PubMed] [Google Scholar]
- 27. Bruce-Keller AJ, Li YJ, Lovell MA, Kraemer PJ, Gary DS, Brown RR, Markesbery WR, Mattson MP. 4-Hydroxynonenal, a product of lipid peroxidation, damages cholinergic neurons and impairs visuospatial memory in rats. J Neuropathol Exp Neurol. 1998;57:257–67. [DOI] [PubMed] [Google Scholar]
- 28. Khan MZ, He L. The role of polyunsaturated fatty acids and GPR40 receptor in brain. Neuropharmacology. 2017;113(Pt B):639–51. [DOI] [PubMed] [Google Scholar]
- 29. Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–80. [DOI] [PubMed] [Google Scholar]
- 30. Suzuki K, Hata S, Kawabata Y, Sorimachi H. Structure, activation, and biology of calpain. Diabetes. 2004;53(Suppl 1):S12–18. [DOI] [PubMed] [Google Scholar]
- 31. Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–30. [DOI] [PubMed] [Google Scholar]
- 32. Kirkegaard T, Roth AG, Petersen NH, Mahalka AK, Olsen OD, Moilanen I, Zylicz A, Knudsen J, Sandhoff K, Arenz C et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature. 2010;463:549–53. [DOI] [PubMed] [Google Scholar]
- 33. Petersen NH, Kirkegaard T. Hsp70 and lysosomal storage disorders: novel therapeutic opportunities. Biochem Soc Trans. 2010;38:1479–83. [DOI] [PubMed] [Google Scholar]
- 34. Petersen NH, Kirkegaard T, Olsen OD, Jäättelä M. Connecting Hsp70, sphingolipid metabolism and lysosomal stability. Cell Cycle. 2010;9:2305–9. [DOI] [PubMed] [Google Scholar]
- 35. Yamashima T. Hsp70.1 and related lysosomal factors for necrotic neuronal death. J Neurochem. 2012;120:477–94. [DOI] [PubMed] [Google Scholar]
- 36. Funderburk SF, Marcellino BK, Yue Z. Cell “self-eating” (autophagy) mechanism in Alzheimer's disease. Mt Sinai J Med. 2010;77:59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee S, Sato Y, Nixon RA. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer's-like axonal dystrophy. J Neurosci. 2011;31:7817–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lilienbaum A. Relationship between the proteasomal system and autophagy. Int J Biochem Mol Biol. 2013;4:1–26. [PMC free article] [PubMed] [Google Scholar]
- 39. Dokladny K, Myers OB, Moseley PL. Heat shock response and autophagy – cooperation and control. Autophagy. 2015;11:200–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rock KL, Gramm C, Rothstein L, Clarck K, Stein R, Dick L, Hwang D, Goldberg AL. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78:761–71. [DOI] [PubMed] [Google Scholar]
- 41. Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008;22:1427–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Morimoto RI. The heat shock response: systems biology of proteotoxic stress in aging and disease. Cold Spring Harb Symp Quant Biol. 2011;76:91–9. [DOI] [PubMed] [Google Scholar]
- 43. Lanneau D, Wettstein G, Bonniaud P, Garrido C. Heat shock proteins: cell protection through protein triage. Sci World J. 2010;10:1543–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Aridon P, Geraci F, Turturici G, D'Amelio M, Savettieri G, Sconzo G. Protective role of heat shock proteins in Parkinson's disease. Neurodegener Dis. 2011;8:155–68. [DOI] [PubMed] [Google Scholar]
- 45. Cuervo AM. Autophagy: in sickness and in health. Trends Cell Biol. 2004;14:70–7. [DOI] [PubMed] [Google Scholar]
- 46. Cuervo AM, Bergamini E, Brunk UT, Droge W, Ffrench M, Terman A. Autophagy and aging: the importance of maintaining “clean” cells. Autophagy. 2005;1:131–40. [DOI] [PubMed] [Google Scholar]
- 47. Kaushik S, Cuervo AM. Autophagy as a cell-repair mechanism: activation of chaperone-mediated autophagy during oxidative stress. Mol Aspects Med. 2006;27:444–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wong E, Cuervo AM. Integration of clearance mechanisms: the proteasome and autophagy. Cold Spring Harb Perspect Biol. 2010;2:a006734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Massey A, Kiffin R, Cuervo AM, Pathophysiology of chaperone mediated autophagy. Int J Biochem Cell Biol. 2004;36:2420–34. [DOI] [PubMed] [Google Scholar]
- 50. Orenstein SJ, Cuervo AM. Chaperone-mediated autophagy: molecular mechanisms and physiological relevance. Semin Cell Dev Biol. 2010;21:719–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xilouri M, Stefanis L. Autophagy in the central nervous system: implications for neurodegenerative disorders. CNS Neurol Disord Drug Targets. 2010;9:701–19. [DOI] [PubMed] [Google Scholar]
- 52. Arias E, Cuervo AM. Chaperone-mediated autophagy in protein quality control. Curr Opin Cell Biol. 2011;23:184–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cuervo AM, Dice J. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273:501–3. [DOI] [PubMed] [Google Scholar]
- 54. Cuervo AM. Chaperone-mediated autophagy: selectivity pays off. Trends Endocrinol Metab. 2010;21:142–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Leak RK. Heat shock proteins in neurodegenerative disorders and aging. J Cell Commun Signal. 2014;8:293–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nowotny K, Jung T, Grune T, Hohn A. Accumulation of modified proteins and aggregate formation in aging. Exp Gerontol. 2014;57:122–31. [DOI] [PubMed] [Google Scholar]
- 57. Petersen NH, Olsen OD, Groth-Pedersen L, Ellegaard AM, Bilgin M, Redmer S, Ostenfeld MS, Ulanet D, Dovmark TH, Lønborg A et al. Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell. 2013;24:379–93. [DOI] [PubMed] [Google Scholar]
- 58. Kolter T, Sandhoff K. Principles of lysosomal membrane digestion: stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annu Rev Cell Dev Biol. 2005;21:81–103. [DOI] [PubMed] [Google Scholar]
- 59. Linke T, Wilkening G, Lansmann S, Moczall H, Bartelsen O, Weisgerber J, Sandhoff K. Stimulation of acid sphingomyelinase activity by lysosomal lipids and sphingolipid activator proteins. Biol Chem. 2001;382:283–90. [DOI] [PubMed] [Google Scholar]
- 60. Linke T, Wilkening G, Sadeghlar F, Mozcall H, Bernardo K, Schuchman E, Sandhoff K. Interfacial regulation of acid ceramidase activity. Stimulation of ceramide degradation by lysosomal lipids and sphingolipid activator proteins. J Biol Chem. 2001;276:5760–68. [DOI] [PubMed] [Google Scholar]
- 61. Heinrich M, Wickel M, Winoto-Morbach S, Schneider-Brachert W, Weber T, Brunner J, Saftig P, Peters C, Krönke M, Schütze S. Ceramide as an activator lipid of cathepsin D. Adv Exp Med Biol. 2000;477:305–15. [DOI] [PubMed] [Google Scholar]
- 62. Kurz T, Eaton JW, Brunk UT. Redox activity within the lysosomal compartment: implications for aging and apoptosis. Antioxid Redox Signal. 2010;13:511–23. [DOI] [PubMed] [Google Scholar]
- 63. Kiselyov K, Colletti GA, Terwilliger A, Ketchum K, Lyons CW, Quinn J, Muallem S. TRPML: transporters of metals in lysosomes essential for cell survival?. Cell Calcium. 2011;50:288–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chong ZZ, Li F, Maiese K. Oxidative stress in the brain. Novel cellular targets that govern survival during neurodegenerative disease. Prog Neurobiol. 2005;75:207–46. [DOI] [PubMed] [Google Scholar]
- 65. Nylandsted J, Wick W, Hirt UA, Brand K, Rohde M, Leist M, Weller M, Jäättelä M. Eradication of glioblastoma, and breast and colon carcinoma xenografts by Hsp70 depletion. Cancer Res. 2002;62:7139–42. [PubMed] [Google Scholar]
- 66. Doulias PT, Kotoglou P, Tenopoulou M, Keramisanou D, Tzavaras T, Brunk U, Galaris D, Angelidis C. Involvement of heat shock protein-70 in the mechanism of hydrogen peroxide-induced DNA damage: the role of lysosomes and iron. Free Radic Biol Med. 2007;42:567–77. [DOI] [PubMed] [Google Scholar]
- 67. Subrizi A, Toropainen E, Ramsay E, Airaksinen AJ, Kaarniranta K, Urtti A. Oxidative stress protection by exogenous delivery of rhHsp70 chaperone to the retinal pigment epithelium (RPE), a possible therapeutic strategy against RPE degeneration. Pharm Res. 2015;32:211–21. [DOI] [PubMed] [Google Scholar]
- 68. Carlson SE, Werkman SH. A randomized trial of visual attention of preterm infants feeds docosahexaenoic acid until two months. Lipids. 1996;31:85–90. [DOI] [PubMed] [Google Scholar]
- 69. Birch EE, Garfield S, Hoffman DR, Uauy R, Birch DG. A randomized controlled trial of early dietary supply of long chain polyunsaturated fatty acids and mental development in term of infants. Dev Med Child Neurol. 2000;42:174–81. [DOI] [PubMed] [Google Scholar]
- 70. Crawaford MA, Golfetto I, Ghebremeskel K, Min Y, Moodley T, Poston L, Phylactos A, Cunnane S, Schmidt W. The potential role for arachidonic and docosahexaenoic acids in protection against some central nervous system injuries in preterm infants. Lipids. 2003;38:303–15. [DOI] [PubMed] [Google Scholar]
- 71. Kawaski A, Han MH, Wei JY, Hirata K, Otori Y, Barnstable CJ. Protective effect of arachidonic acid on glutamate neurotoxicity in rat retinal ganglion cells. Invest Opthalmol Vis Sci. 2002;43:1835–42. [PubMed] [Google Scholar]
- 72. Wang J, Wu X, Simonavicius N, Tian H, Ling L. Medium chain fatty acids as ligands for orphan G protein-coupled receptor GPR84. J Biol Chem. 2006;281:34457–64. [DOI] [PubMed] [Google Scholar]
- 73. Wang ZJ, Liang CL, Li GM, Yu CY, Yin M. Neuroprotective effects of arachidonic acid against oxidative stress on rat hippocampal slices. Chem Biol Interact. 2006;163:207–17. [DOI] [PubMed] [Google Scholar]
- 74. Yao Y, Clark CM, Trojanowski JQ, Lee VM, Pratico D. Elevation of 12/15 lipoxygenase products in AD and mild cognitive impairment. Ann Neurol. 2005;58:623–6. [DOI] [PubMed] [Google Scholar]
- 75. Witz G. Biological interactions of α,β-unsaturated aldehydes. Free Radic Biol Med. 1989;7:333–49. [DOI] [PubMed] [Google Scholar]
- 76. Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. [DOI] [PubMed] [Google Scholar]
- 77. Dalleau S, Baradat M, Guéraud F, Huc L. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 2013;20:1615–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Devaraj S, Wang-Polagruto J, Polagruto J, Keen CL, Jialal I. High-fat, energy-dense, fast-food-style breakfast results in an increase in oxidative stress in metabolic syndrome. Metabolism. 2008;57:867–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Schaur RJ, Siems W, Bresgen N, Eckl PM. 4-Hydroxy-nonenal – a bioactive lipid peroxidation product. Biomolecules. 2015;5:2247–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Perluigi M, Coccia R, Butterfield DA. 4-Hydroxy-2-nonenal, a reactive product of lipid peroxidation, and neurodegenerative diseases: a toxic combination illuminated by redox proteomics studies. Antioxid Redox Signal. 2012;17:1590–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Grune T, Davies KJ. The proteasomal system and HNE-modified proteins. Mol Aspects Med. 2003;24:195–204. [DOI] [PubMed] [Google Scholar]
- 82. Chaudhary P, Sharma R, Sharma A, Vatsyayan R, Yadav S, Singhal SS, Rauniyar N, Prokal L, Awasthi S, Awasthi YC. Mechanisms of 4-hydroxy-2-nonenal induced pro- and anti-apoptotic signaling. Biochemistry. 2010;49:6263–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sovic A, Borovic S, Loncaric I, Kreuzer T, Zarkovic K, Vukovic T, Wäg G, Hrascan R, Wintersteiger R, Klinger R et al. The carcinostatic and proapoptotic potential of 4-hydroxynonenal in HeLa cells is associated with its conjugation to cellular proteins. Anticancer Res. 2001;21:1997–2004. [PubMed] [Google Scholar]
- 84. Sahara S, Yamashima T. Calpain-mediated Hsp70.1 cleavage in hippocampal CA1 neuronal death. Biochem Biophys Res Commun. 2010;393:806–11. [DOI] [PubMed] [Google Scholar]
- 85. Leonarduzzi G, Chiarpotto E, Biasi F, Poli G. 4-Hydroxynonenal and cholesterol oxidation products in atherosclerosis. Mol Nutr Food Res. 2005;49:1044–9. [DOI] [PubMed] [Google Scholar]
- 86. Tang SC, Arumugam TV, Cutler RG, Jo DG, Magnus T, Chan SL, Mughal MR, Telljohann RS, Nassar M, Ouyang X et al. Neuroprotective actions of a histidine analogue in models of ischemic stroke. J Neurochem. 2007;101:729–36. [DOI] [PubMed] [Google Scholar]
- 87. Lovell MA, Ehmann WD, Mattson MP, Markesbery WR. Elevated 4-hydroxynonenal in ventricular fluid in Alzheimer's disease. Neurobiol Aging. 1997;18:457–61. [DOI] [PubMed] [Google Scholar]
- 88. Morquette B, Shi Q, Lavigne P, Ranger P, Fernandes JC, Benderdour M. Production of lipid peroxidation products in osteoarthritic tissues: new evidence linking 4-hydroxynonenal to cartilage degradation. Arthritis Rheum. 2006;54:271–81. [DOI] [PubMed] [Google Scholar]
- 89. Johnson JB, Summer W, Cutler RG, Martin B, Hyun DH, Dixit VD, Pearson M, Nassar M, Telljohann R, Maudsley S et al. Alternate day calorie restriction improves clinical findings and reduces markers of oxidative stress and inflammation in overweight adults with moderate asthma. Free Radic Biol Med. 2007;42:665–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Legards JF, des Rosiers C. Assay of 4-hydroxynonenal (HNE) adducts with various polyaminoacids (PAA) using gas chromatography-mass spectrometry (GCMS). In Proceedings of the 3rd International Meeting of the HNE-Club. Genova, Italy: 2006, p. 16–18. [Google Scholar]
- 91. Petersen DR, Doorn JA. Reactions of 4-hydroxynonenal with proteins and cellular targets. Free Radic Biol Med. 2004;37:937–45. [DOI] [PubMed] [Google Scholar]
- 92. Poli G, Biasi F, Leonarduzzi G. 4-Hydroxynonenal-protein adducts: a reliable biomarker of lipid oxidation in liver diseases. Mol Aspects Med. 2008;29:67–71. [DOI] [PubMed] [Google Scholar]
- 93. Carbone DL, Doorn JA, Kiebler Z, Sampey BP, Petersen DR. Inhibition of Hsp72-mediated protein refolding by 4-hydroxy-2-nonenal. Chem Res Toxicol. 2004;17:1459–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Perluigi M, Poon HF, Hensley K, Pierce WM, Klein JB, Calabrese V, de Marco C, Butterfield DA. Proteomic analysis of 4-hydroxy-2-nonenal-modified proteins in G93A-SOD1 transgenic mice – a model of familial amyotrophic lateral sclerosis. Free Radic Biol Med. 2005;38:960–8. [DOI] [PubMed] [Google Scholar]
- 95. Yamashima T, Mathivanan A, Dazortsava MY, Sakai S, Kurimoto S, Zhu H, Funaki N, Liang H, Hullin-Matsuda F, Kobayashi T et al. Calpain-mediated Hsp70.1 cleavage in monkey CA1 after ischemia induces similar ‘lysosomal vesiculosis’ to Alzheimer's neurons. J Alzheimers Dis Parkinsonism. 2014;04:2 10.4172/2161-0460.1000139. [DOI] [Google Scholar]
- 96. Saido TC, Yokota M, Nagao S, Yamaura I, Tani E, Tsuchiya T, Suzuki K, Kawashima S. Spatial resolution of fodrin proteolysis in postischemic brain. J Biol Chem. 1993;268:25239–43. [PubMed] [Google Scholar]
- 97. Roberts-Lewis JM, Savage MJ, Marcy VR, Pinsker LR, Siman R. Immunolocalization of calpain I-mediated spectrin degradation to vulnerable neurons in the ischemic gerbil brain. J Neurosci. 1994;14:3934–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Neumar RW, Hagle SM, DeGracia DJ, Krause GS, White BC. Brain μ-calpain autolysis during global cerebral ischemia. J Neurochem. 2002;66:421–4. [DOI] [PubMed] [Google Scholar]
- 99. Bartus RT, Dean RL, Mennerick S, Eveleth D, Lynch G. Temporal ordering of pathogenic events following transient global ischemia. Brain Res. 1998;790:1–13. [DOI] [PubMed] [Google Scholar]
- 100. Yamashima T. Reconsider Alzheimer's disease by the ‘calpain-cathepsin hypothesis' – a perspective review. Prog Neurobiol. 2013;105:1–23. [DOI] [PubMed] [Google Scholar]
- 101. Guroff G. A neutral, calcium-activated proteinase from the soluble fraction of rat brain. J Biol Chem. 1964;239:149–55. [PubMed] [Google Scholar]
- 102. Murachi T, Tanaka K, Hatanaka M, Murakami T. Intracellular Ca-dependent protease (calpain) and its high-molecular-weight endogenous inhibitor (calpastatin). Adv Enzyme Regul. 1981;19:407–24. [DOI] [PubMed] [Google Scholar]
- 103. Tompa P, Buzder-Lantos P, Tantos A, Farkas A, Szilágyi A, Bánóczi Z, Hudecz F, Friedrich P. On the sequential determinants of calpain cleavage. J Biol Chem. 2004;279:20775–85. [DOI] [PubMed] [Google Scholar]
- 104. Cuerrier D, Moldoveanu T, Davies PL. Determination of peptide substrate specificity for μ-calpain by a peptide library-based approach: the importance of primed side interactions. J Biol Chem. 2005;280:40632–41. [DOI] [PubMed] [Google Scholar]
- 105. Koriyama Y, Sugitani K, Ogai K, Kato S. Heat shock protein 70 induction by valproic acid delays photoreceptor cell death by N-methyl-N-nitrosourea in mice. J Neurochem. 2014;130:707–19. [DOI] [PubMed] [Google Scholar]
- 106. Uchida K, Stadtman ER. Covalent attachment of 4-hydroxynonenal to glyceraldehyde-3-phosphate dehydrogenase. A possible involvement of intra- and intermolecular cross-linking reaction. J Biol Chem. 1993;268:6388–93. [PubMed] [Google Scholar]
- 107. Subramaniam R, Roediger F, Jordan B, Mattson MP, Keller JN, Wang G, Butterfield DA. The lipid peroxidation product, 4-hydroxy-2-trans-nonenal, alters the conformation of cortical synaptosomal membrane proteins. J Neurochem. 2002;69:1161–9. [DOI] [PubMed] [Google Scholar]
- 108. Gómez-Sintes R, Ledesma MD, Boya P. Lysosomal cell death mechanisms in aging. Ageing Res Rev. 2016;32:150–68. [DOI] [PubMed] [Google Scholar]
- 109. Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol. 2008;28:5747–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Rodriguez GEV, Torriglia A. Calpain 1 induce lysosomal permeabilization by cleavage of lysosomal associated membrane protein 2. Biochim Biophys Acta. 2013;1833:2244–53. [DOI] [PubMed] [Google Scholar]
- 111. Sridhar S, Botbol Y, Macian F, Cuervo AM. Autophagy and disease: always two sides to a problem. J Pathol. 2012;226:255–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Xue M, Huang F, Huang M, Zhou G. Influence of oxidation on myofibrillar proteins degradation from bovine via μ-calpain. Food Chem. 2012;134:106–12. [Google Scholar]
- 113. Zdolsek J, Zhang H, Roberg K, Brunk UT. H2O2-mediated damage to lysosomal membranes of J-774 cells. Free Radic Res Commun. 1993;18:71–85. [DOI] [PubMed] [Google Scholar]
- 114. Antunes F, Cadenas E, Brunk UT. Apoptosis induced by exposure to a low steady-state concentration of H2O2 is a consequence of lysosomal rupture. Biochem J. 2001;356:549–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Dare E, Li W, Zhivotovsky B, Yuan X, Ceccatelli S. Methylmercury and H(2)O(2) provoke lysosomal damage in human astrocytoma D384 cells followed by apoptosis. Free Radic Biol Med. 2001;30:1347–56. [DOI] [PubMed] [Google Scholar]
- 116. Persson HL, Yu Z, Tirosh O, Eaton JW, Brunk UT. Prevention of oxidant-induced cell death by lysosomotropic iron chelators. Free Radic Biol Med. 2003;34:1295–305. [DOI] [PubMed] [Google Scholar]
- 117. Terman A, Kurz T, Gustafsson B, Brunk UT. Lysosomal labilization. IUBMB Life. 2006;58:531–9. [DOI] [PubMed] [Google Scholar]
- 118. Nixon RA, Yang DS. Autophagy and neuronal cell death in neurological disorders. Cold Spring Harb Perspect Biol. 2012;4:a008839 pii: a008839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Firestone RA, Pisano JM, Bonney RJ. Lysosomotropic agents. 1. Synthesis and cytotoxic action of lysosomotropic detergents. J Med Chem. 1979;22:1130–3. [DOI] [PubMed] [Google Scholar]
- 120. Aits S, Jäättelä M. Lysosomal cell death at a glance. J Cell Sci. 2013;126:1905–12. [DOI] [PubMed] [Google Scholar]
- 121. Brunk UT, Ericsson JL. Cytochemical evidence for the leakage of acid phosphatase through ultrastructurally intact lysosomal membranes. Histochem J. 1972;4:479–91. [DOI] [PubMed] [Google Scholar]
- 122. Yamashima T, Kohda Y, Tsuchiya K, Ueno T, Yamashita J, Yoshioka T, Kominami E. Inhibition of ischaemic hippocampal neuronal death in primates with cathepsin B inhibitor CA-074: a novel strategy for neuroprotection based on ‘calpain–cathepsin hypothesis’. Eur J Neurosci. 1998;10:1723–33. [DOI] [PubMed] [Google Scholar]
- 123. Sultana R, Perluigi M, Newman SF, Pierce WM, Cini C, Coccia R, Butterfield DA. Redox proteomic analysis of carbonylated brain proteins in mild cognitive impairment and early Alzheimer's disease. Antioxid Redox Signal. 2010;12:327–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. McGrath LT, McGleenon BM, Brennan S, McColl D, McIlroy S, Passmore AP. Increased oxidative stress in Alzheimer's disease as assessed with 4-hydroxynonenal but not malondialdehyde. QJM. 2001;94:485–90. [DOI] [PubMed] [Google Scholar]
- 125. Yankner BA. Mechanisms of neuronal degeneration in Alzheimer's disease. Neuron. 1996;16:921–32. [DOI] [PubMed] [Google Scholar]
- 126. Iulita MF, Cuello AC. Nerve growth factor metabolic dysfunction in Alzheimer's disease and Down syndrome. Trends Pharmacol Sci. 2014;35:338–48. [DOI] [PubMed] [Google Scholar]
- 127. Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010;362:329–44. [DOI] [PubMed] [Google Scholar]
- 128. Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–5. [DOI] [PubMed] [Google Scholar]
- 129. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. [DOI] [PubMed] [Google Scholar]
- 130. Robakis NK. Are Aβ and its derivatives causative agents or innocent bystanders in AD?. Neurodegenerative Dis. 2010;7:32–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Strittmatter WJ, Weisgraber KH, Goedert M, Saunders AM, Huang D, Corder EH, Dong LM, Jakes R, Alberts MJ, Gilbert JR et al. Hypothesis: microtubule instability and paired helical filament formation in the Alzheimer's disease brain are related to apolipoprotein E genotype. Exp Neurol. 1994;; 125:163–71. [DOI] [PubMed] [Google Scholar]
- 132. Nagy Z, Esiri MM, Jobst KA, Morris JH, King EM, McDonald B, Litchfield S, Smith A, Barnetson L, Smith AD. Relative roles of plaques and tangles in the dementia of Alzheimer's disease: correlations using three sets of neuropathological criteria. Dementia. 1995;6:21–31. [DOI] [PubMed] [Google Scholar]
- 133. Gastard MC, Troncoso JC, Koliatsos VE. Caspase activation in the limbic cortex of subjects with early Alzheimer's disease. Ann Neurol. 2003;54:393–8. [DOI] [PubMed] [Google Scholar]
- 134. Uetsuki T, Takemoto K, Nishimura L, Okamoto M, Niinobe TM, Miura M, Yoshikawa K. Activation of neuronal caspase-3 by intracellular accumulation of wild-type Alzheimer's amyloid precursor protein. J Neurosci. 1999;19:6955–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Hwang DY, Chae KR, Kang TS, Hwang JH, Lim CH, Kang HK, Goo JS, Lee MR, Lim HJ, Min SH et al. Alterations in behavior, amyloid β-42, caspase-3, and Cox-2 in mutant PS2 transgenic mouse model of Alzheimer's disease. FASEB J. 2002;16:805–13. [DOI] [PubMed] [Google Scholar]
- 136. Eimer WA, Vassar R. Neuron loss in the 5XFAD mouse model of Alzheimer's disease correlates with intraneuronal Aβ42 accumulation and caspase-3 activation. Mol Neurodegeneration. 2013;8:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Behl C. Apoptosis and Alzheimer's disease. J Neural Transm. 2000;107:1325–44. [DOI] [PubMed] [Google Scholar]
- 138. LeBlanc AC. The role of apoptotic pathways in Alzheimer's disease neurodegeneration and cell death. Curr Alzheimer's Res. 2005;2:389–402. [DOI] [PubMed] [Google Scholar]
- 139. Rohn TT, Head E. Caspase activation in Alzheimer's disease: early to rise and late to bed. Rev Neurosci. 2008;19:383–93. [DOI] [PubMed] [Google Scholar]
- 140. Kim HS, Lee JH, Lee JP, Kim EM, Chang KA, Park CH, Jeong SJ, Wittendorp MC, Seo JH, Choi SH et al. Amyloid beta peptide induces cytochrome C release from isolated mitochondria. Neuroreport. 2002;13:1989–93. [DOI] [PubMed] [Google Scholar]
- 141. Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6:889–98. [DOI] [PubMed] [Google Scholar]
- 142. Rao MV, Mohan PS, Peterhoff CM, Yang DS, Schmidt SD, Stavrides PH, Campbell J, Chen Y, Jiang Y, Paskevich PA et al. Marked calpastatin (CAST) depletion in Alzheimer's disease accelerates cytoskeleton disruption and neurodegeneration: neuroprotection by CAST overexpression. J Neurosci. 2008;28:12241–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Adalbert R, Nogradi A, Babetto E, Janeckova L, Walker SA, Kerschensteiner M, Misgeld T, Coleman MP. Severely dystrophic axons at amyloid plaques remain continuous and connected to viable cell bodies. Brain. 2009;132:402–16. [DOI] [PubMed] [Google Scholar]
- 144. Markesbery WR. Oxidative stress hypothesis in Alzheimer's disease. Free Radic Biol Med. 1997;23:134–47. [DOI] [PubMed] [Google Scholar]
- 145. Butterfield DA. Amyloid β-peptide (1-42)-induced oxidative stress and neurotoxicity: implications for neurode-generation in Alzheimer's disease brain. Free Radic Res. 2002;36:1307–13. [DOI] [PubMed] [Google Scholar]
- 146. Hynd MR, Scott HL, Dodd PR. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer's disease. Neurochem Int. 2004;45:583–95. [DOI] [PubMed] [Google Scholar]
- 147. Ho GJ, Drego R, Hakimian E, Masliah E. Mechanisms of cell signaling and inflammation in Alzheimer's disease. Curr Drug Targets Inflamm Allergy. 2005;4:247–56. [DOI] [PubMed] [Google Scholar]
- 148. Tuppo EE, Arias HR. The role of inflammation in Alzheimer's disease. Int J Biochem Cell Biol. 2005;37:289–305. [DOI] [PubMed] [Google Scholar]
- 149. Butterfield DA. β-amyloid-associated free radical oxidative stress and neurotoxicity: implications for Alzheimer's disease. Chem Res Toxicol. 1997;10:495–506. [DOI] [PubMed] [Google Scholar]
- 150. Praticò D. Oxidative stress hypothesis in Alzheimer's disease: a reappraisal. Trends Pharmacol Sci. 2008;29:609–15. [DOI] [PubMed] [Google Scholar]
- 151. Reed TT, Pierce WM, Markesbery WR, Butterfield DA. Proteomic identification of HNE-bound proteins in early Alzheimer's disease: insights into the role of lipid peroxidation in the progression of AD. Brain Res. 2009;1274:66–76. [DOI] [PubMed] [Google Scholar]
- 152. Ando Y, Brännström T, Uchida K, Nyhlin N, Näsman B, Suhr O, Yamashita T, Olsson T, El Salhy TM, Uchino M et al. Histochemical detection of 4-hydroxynonenal protein in Alzheimer's amyloid. J Neurol Sci. 1998;156:172–6. [DOI] [PubMed] [Google Scholar]
- 153. Siegel SJ, Bieschke J, Powers ET, Kelly JW. The oxidative stress metabolite 4-hydroxynonenal promotes Alzheimer's protofibril formation. Biochemistry. 2007;46:1503–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Granold M, Moosmann N, Staib-Lasarzik I, Arendt T, del Rey A, Engelhard K, Behl C, Hajieva P. High membrane protein oxidation in the human cerebral cortex. Redox Biol. 2015;4:200–07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Iqbal L, Liu F, Gong CX, Alonso AC, Grundke-Iqbal I. Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 2009;118:53–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Patterson KR, Remmers C, Fu Y, Brooker S, Kanaan NM, Vana L, Ward S, Reyes JF, Philibert K, Glucksman MJ et al. Characterization of prefibrillar tau oligomers in vitro and in Alzheimer's disease. J Biol Chem. 2011;286:23063–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A, Gouras GK, Greengard P, Xu H. Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci USA. 2003;100:721–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A, De Lucia M, McGowan E, Lewis J, Prihar G et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet. 2004;13:703–14. [DOI] [PubMed] [Google Scholar]
- 159. Patterson KR, Ward SM, Combs B, Voss K, Kanaan NM, Morfini G, Brady ST, Gamblin TC, Binder LI. Heat shock protein 70 prevents both tau aggregation and the inhibitory effects of preexisting tau aggregates on fast axonal transport. Biochemistry. 2011;50:10300–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Fischer VW, Siddiqi A, Yusufaly Y. Altered angioarchitecture in selected areas of brains with Alzheimer's disease. Acta Neuropathol. 1990;79:672–9. [DOI] [PubMed] [Google Scholar]
- 161. Honoré JC, Kooli A, Hamel D, Alquier T, Rivera JC, Quiniou C, Hou X, Kermorvant-Duchemin E, Hardy P, Poitout V et al. Fatty acid receptor GPR40 mediates neuromicrovascular degeneration induced by transarachidonic acids in rodents. Arterioscler Thromb Vasc Biol. 2013;33:954–61. [DOI] [PubMed] [Google Scholar]
- 162. Yamashima T. Dual effects of the non-esterified fatty acid receptor ‘GPR40’ for human health. Prog Lipid Res. 2015;58:40–50. [DOI] [PubMed] [Google Scholar]
- 163. Nixon RA. A "protease activation cascade" in the pathogenesis of Alzheimer's disease. Ann N Y Acad Sci. 2000;924:117–31. [PubMed] [Google Scholar]
- 164. Taniguchi S, Fujita Y, Hayashi S, Kakita A, Takahashi H, Murayama S, Saido TC, Hisanaga S, Iwatsubo T, Hasegawa M. Calpain-mediated degradation of p35 to p25 in postmortem human and rat brains. FEBS Lett. 2001;489:46–50. [DOI] [PubMed] [Google Scholar]
- 165. Reifert J, Hartung-Cranston D, Feinstein SC. Amyloid β-mediated cell death of cultured hippocampal neurons reveals extensive tau fragmentation without increased full-length tau phosphorylation. J Biol Chem. 2011;286:20797–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Yamashima T, Saido TC, Takita M, Miyazawa A, Yamano J, Miyakawa A, Nishijyo H, Yamashita J, Kawashima S, Ono T et al. Transient brain ischaemia provokes Ca2+, PIP2 and calpain responses prior to delayed neuronal death in monkeys. Eur J Neurosci. 1996;8:1932–44. [DOI] [PubMed] [Google Scholar]
- 167. Villalpando Rodríguez GE, Torriglia A. Calpain 1 induce lysosomal permeabilization by cleavage of lysosomal associated membrane protein 2. Biochim Biophys Acta. 2013;1833:2244–53. [DOI] [PubMed] [Google Scholar]
- 168. Gerónimo-Olvera C, Montiel T, Rincon-Heredia R, Castro-Obregón S, Massieu L. Autophagy fails to prevent glucose deprivation/glucose reintroduction-induced neuronal death due to calpain-mediated lysosomal dysfunction in cortical neurons. Cell Death Dis. 2017;8:e2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Arnandis T, Ferrer-Vicens I, García-Trevijano ER, Miralle VJ, García C, Torres L, Viña JR, Zaragozá R. Calpains mediate epithelial-cell death during mammary gland involution: mitochondria and lysosomal destabilization. Cell Death Differ. 2012;19:1536–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Biaggioni I, Robertson D. Chapter 9. Adrenoceptor agonists & sympathomimetic drugs. In: Katzung BG, Masters SB, Trevor AJ (eds), Basic & Clinical Pharmacology. New York: McGraw-Hill Education; 2011. p. 11e [Google Scholar]
- 171. Barrett KE, Barman SM, Boitano S, Brooks H. Chapter 2. Overview of cellular physiology in medical physiology. In: Barrett KE, Barman SM, Boitano S, Brooks H (eds). Ganon's Review of Medical Physiology, 23rd Edition New York: McGraw-Hill Medical; 2010. [Google Scholar]
- 172. Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, Ogi K, Hosoya M, Tanaka Y, Uejima H et al. Free fatty acids regulate insulin secretion from pancreatic β cells through GPR40. Nature. 2003;422:173–6. [DOI] [PubMed] [Google Scholar]
- 173. Ma D, Tao B, Warashina S, Kotani S, Lu L, Kaplamadzhiev DB, Mori Y, Tonchev AB, Yamashima T. Expression of free fatty acid receptor GPR40 in the central nervous system of adult monkeys. Neurosci Res. 2007;58:394–401. [DOI] [PubMed] [Google Scholar]
- 174. Ma D, Lu L, Boneva NB, Warashina S, Kaplamadzhiev DB, Mori Y, Nakaya MA, Kikuchi M, Tonchev AB, Okano H et al. Expression of free fatty acid receptor GPR40 in the neurogenic niche of adult monkey hippocampus. Hippocampus. 2008;18:326–33. [DOI] [PubMed] [Google Scholar]
- 175. Ma D, Zhang M, Larsen CP, Xu F, Hua W, Yamashima T, Mao Y, Zhou L. DHA promotes the neuronal differentiation of rat neural stem cells transfected with GPR40 gene. Brain Res. 2010;1330:1–8. [DOI] [PubMed] [Google Scholar]
- 176. Boneva NB, Yamashima T. New insights into ‘GPR40-CREB interaction in adult neurogenesis’ specific for primates. Hippocampus. 2012;22:896–905. [DOI] [PubMed] [Google Scholar]
- 177. Zamarbide M, Etayo-Labiano I, Ricobaraza A, Martínez-Pinilla E, Aymerich MS, Luis Lanciego J, Pérez-Mediavilla A, Franco R. GPR40 activation leads to CREB and ERK phosphorylation in primary cultures of neurons from the mouse CNS and in human neuroblastoma cells. Hippocampus. 2014;24:733–9. [DOI] [PubMed] [Google Scholar]
- 178. Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–23. [DOI] [PubMed] [Google Scholar]
- 179. Clark C, Lands B. Creating benefits from omega-3 functional foods and nutraceuticals. Food and Nutrition Sciences. 2015;6:1613–23. [Google Scholar]
- 180. Schwartz MW, Porte D Jr. Diabetes, obesity, and the brain. Science. 2005;307:375–9. [DOI] [PubMed] [Google Scholar]
- 181. Clark JT, Kalra PS, Kalra SP. Neuropeptide Y stimulates feeding but inhibits sexual behavior in rats. Endocrinology. 1985;117:2435–42. [DOI] [PubMed] [Google Scholar]
- 182. Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen Y, Gantz I, Barsh GS. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science. 1997;278:135–8. [DOI] [PubMed] [Google Scholar]
- 183. Kotz CM, Briggs JE, Pomonis JD, Grace MK, Levine AS, Billington CJ. Neural site of leptin influence on neuropeptide Y signaling pathways altering feeding and uncoupling protein. Am J Physiol. 1998;275(2 Pt 2):R478–484. [DOI] [PubMed] [Google Scholar]
- 184. Schwartz MW, Woods SC, Porte D Jr., Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–71. [DOI] [PubMed] [Google Scholar]
- 185. Schwartz MW, Seeley RJ, Woods SC, Weigle DS, Campfield LA, Burn P, Baskin DG. Leptin increases hypothalamic pro-opiomelanocortin mRNA expression in the rostral arcuate nucleus. Diabetes. 1997;46:2119–23. [DOI] [PubMed] [Google Scholar]
- 186. Cowley MA, Smart JL, Rubinstein M, Cerdán MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–4. [DOI] [PubMed] [Google Scholar]
- 187. Cone RD. The central melanocortin system and energy homeostasis. Trends Endocrinol Metab. 1999;10:211–16. [DOI] [PubMed] [Google Scholar]
- 188. El-Haschimi K, Pierroz DD, Hileman SM, Bjørbaek C, Flier JS. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J Clin Invest. 2000;105:1827–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, Saad MJ, Velloso LA. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology. 2005;146:4192–9. [DOI] [PubMed] [Google Scholar]
- 190. Moraes JC, Coope A, Morari J, Cintra DE, Roman EA, Pauli JR, Romanatto T, Carvalheira JB, Oliveira AL, Saad MJ et al. High-fat diet induces apoptosis of hypothalamic neurons. PLoS One. 2009;4:e5045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. van de Sande-Lee S, Pereira FR, Cintra DE, Fernandes PT, Cardoso AR, Garlipp CR, Chaim EA, Pareja JC, Geloneze B, Li LM et al. Partial reversibility of hypothalamic dysfunction and changes in brain activity after body mass reduction in obese subjects. Diabetes. 2011;60:1699–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, Zhao X, Sarruf DA, Izgur V, Maravilla KR et al. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest. 2012;122:153–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Barsh GS, Schwartz MW. Genetic approaches to studying energy balance: perception and integration. Nat Rev Genet. 2002;3:589–600. [DOI] [PubMed] [Google Scholar]
- 194. Nakamoto K, Nishinaka T, Sato N, Mankura M, Koyama Y, Kasuya F, Tokuyama S. Hypothalamic GPR40 signaling activated by free long chain fatty acids suppresses CFA-induced inflammatory chronic pain. PLoS One. 2013;8:e81563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Kokoeva MV, Yin H, Flier JS. Evidence for constitutive neural cell proliferation in the adult murine hypothalamus. J Comp Neurol. 2007;505:209–20. [DOI] [PubMed] [Google Scholar]
- 196. Ming GL, Song H. Adult neurogenesis in the mammalian central nervous system. Annu Rev Neurosci. 2005;28:223–50. [DOI] [PubMed] [Google Scholar]
- 197. Münzberg H, Myers MG Jr. Molecular and anatomical determinants of central leptin resistance. Nat Neurosci. 2005;8:566–70. [DOI] [PubMed] [Google Scholar]
- 198. Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443:289–95. [DOI] [PubMed] [Google Scholar]
- 199. Gropp E, Shanabrough M, Borok E, Xu AW, Janoschek R, Buch T, Plum L, Balthasar N, Hampel B, Waisman A et al. Agouti-related peptide-expressing neurons are mandatory for feeding. Nat Neurosci. 2005;8:1289–91. [DOI] [PubMed] [Google Scholar]
- 200. Alvheim AR, Malde MK, Osei-Hyiaman D, Lin YH, Pawlosky RL, Madsen L, Kristiansen K, Frøyland L, Hibbeln JR. Dietary linoleic acid elevates endocannabinoids 2-AG and anandamide and induces obesity. Obesity. 2012;20:1984–94. 10.1038/oby.2012.38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Alvheim AR, Torstensen BE, Lin YH, Lillefosse HH, Lock EJ, Madsen L, Frøyland L, Hibbeln JR, Malde MK. Dietary linoleic acid elevates the endocannabinoids 2-AG and anandamide and promotes weight gain in mice fed a low fat diet. Lipids. 2014;49:59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Mancini AD, Poitout V. The fatty acid receptor FFA1/GPR40 a decade later: how much do we know?. Trends Endocrinol Metab. 2013;24:398–407..b9 [DOI] [PubMed] [Google Scholar]
- 203. Balazy M, Chemtob S. Trans-arachidonic acids: new mediators of nitro-oxidative stress. Pharmacol Ther. 2008;119:275–90. [DOI] [PubMed] [Google Scholar]
- 204. Yashin DV, Romanova EA, Ivanova OK, Sashchenko LP. The Tag7-Hsp70 cytotoxic complex induces tumor cell necroptosis via permeabilisation of lysosomes and mitochondria. Biochimie. 2016;123:32–6. [DOI] [PubMed] [Google Scholar]
- 205. Lenzen S. Oxidative stress: the vulnerable beta-cell. Biochem Soc Trans. 2008;36:343–7. [DOI] [PubMed] [Google Scholar]
- 206. Aleynik SI, Leo MA, Aleynik MK, Lieber CS. Alcohol-induced pancreatic oxidative stress: protection by phospholipid repletion. Free Radic Biol Med. 1999;26:609–19. [DOI] [PubMed] [Google Scholar]
- 207. Yamashima T. The scourge of vegetable oil – destroyer of nations. J Alzheimers Dis Parkinsonism. 2018;08(4):1–4. [Google Scholar]
- 208. Török Z, Crul T, Maresca B, Schütz GJ, Viana F, Dindia L, Piotto S, Brameshuber M, Balogh G, Péter M et al. Plasma membranes as heat stress sensors: from lipid-controlled molecular switches to therapeutic applications. Biochim Biophys Acta. 2014;1838:1594–618. [DOI] [PubMed] [Google Scholar]