

Abstract
Sudden cardiac death (SCD), or sudden loss of life-sustaining systemic and cerebral perfusion, is most often due to left ventricular (LV) dysfunction secondary to ischemic or structural cardiac disease or channelopathies. Degeneration of sinus rhythm into ventricular tachycardia and ultimately ventricular fibrillation is the final common pathway for most heart failure patients. Right ventricular (RV) dysfunction is recognized as an independent contributor to worsening heart failure. There is emerging evidence that RV dysfunction may also be an independent predictor of SCD. This review examines the role of RV dysfunction on modifying long term risk of SCD, and explores possible mechanisms that may underlie SCD. The RV has unique anatomy and physiology compared to the LV. Subsequently, we begin with a review of cardiac embryology, focusing on the chambers, valves, coronary arteries, and cardiac conduction system to understand the origins of RV dysfunction. Static and dynamic physiology of the RV is contrasted with that of the LV. Particular emphasis is placed on ventriculo-arterial coupling, mechanical cardiac constraint, and ventricular interdependence. The epidemiology of SCD is briefly reviewed to highlight how causes of SCD are age-specific. In turn, the age-specific causes of RV dysfunction are presented, including those which predominate in childhood and adolescence [arrhythmogenic RV dysplasia (ARVD) and hypertrophic cardiomyopathy (HCM)] and older adulthood (cardiac ischemia, chronic congestive heart failure and post-capillary pulmonary hypertension, and pulmonary hypertension). There is a clear need for additional studies on the independent contribution of RV dysfunction to overall functional capacity, SCD-associated mortality, and non-SCD-associated mortality. Discovery would be aided by the development of prospective cohorts with excellent RV phenotyping, coupled with deeper biologic measurements linking mechanisms to clinically relevant outcomes.
Keywords: Right ventricle, sudden cardiac death (SCD), arrhythmogenic right ventricular dysplasia (ARVD), hypertrophic cardiomyopathy (HCM), pulmonary hypertension
Introduction
Arrhythmogenic deterioration of cardiac function may be an acute event or the consequence of end-stage heart failure. In the case of sudden cardiac death (SCD), the conversion from a stable to unstable ventricular rhythm induces a total loss of cardiac and cerebral perfusion. Most often, SCD is ascribed to left ventricular (LV) dysfunction, primary LV dysrhythmias from channelopathies, ischemic or structural cardiac disorders. By contrast, degeneration of sinus rhythm to ventricular tachycardia and, ultimately, ventricular fibrillation is the final common pathway for the vast majority of chronic heart failure patients. In both scenarios, LV dysfunction has garnered the majority of academic and clinical attention. This is fitting, since there is a well-established inverse correlation between LV ejection fraction and SCD, and ischemic heart disease due to atherosclerotic coronary arterial remodeling and myocardial infarction are the most common causes of decreased ejection fraction in industrialized nations.
However, right ventricular (RV) dysfunction also plays a critical role in the pathogenesis of heart failure-associated multi-systemic organ failure and mortality (Figure 1) (1). It is well-established, for example, that a decrease in RV systolic function is an independent predictor of adverse outcome, including heart failure-associated hospitalization and mortality. Changes in RV cavitary dimension, contractility, and diastolic function occur most commonly as a consequence of LV dysfunction, but are also well-documented in isolated right coronary artery myocardial infarction, primary pulmonary circulatory diseases such as pulmonary arterial hypertension, and less so in patients with primary RV cardiomyopathies including arrhythmogenic RV dysplasia (ARVD). Thus, the overall mode of death in patients harboring RV dysfunction is heart failure-associated degeneration of normal cardiac conduction linked to LV dysfunction. More recently, the possibility that RV dysfunction is an emergent independent risk factor for future SCD has been suggested by some observational studies and other reports. This review examines the unique role of RV dysfunction modifying long-term risk of adverse outcomes, and explores the potential mechanisms that may underlie SCD.
Figure 1.

Systemic consequences of pulmonary hypertension and right heart failure in multiple organ systems. Reproduced with permission from (1).
Nomenclature
The right heart system extends beyond the RV itself, and is comprised of structures between the systemic venous network and pulmonary circuit. The systemic circuit encompasses the capacitance (conduit) veins, right atrium, coronary sinus, tricuspid valve, RV free wall, RV outflow tract, and pulmonic valve. The pulmonary circuit includes the main pulmonary artery after the pulmonic valve and secondary and tertiary branches thereof, and completes at the capillary bed. We first present a discussion of RV embryology, anatomy, and physiology to understand the basis, causes, and immediate effects of RV dysfunction. We then discuss long-term risk of adverse outcomes in the context of RV dysfunction, dividing conditions which predominate in childhood and adolescence from those prevalent in adulthood. Finally, we highlight areas of uncertainty, which should prompt investigation to help improve care of patients with RV dysfunction generally, and those at increased risk for mortality in specific circumstances.
Cardiac embryology
Nine steps occur during human cardiac development, in sequence and in parallel, to form a mature heart by post-conception day 50 (2). Developmental errors occurring at different time points can, therefore, have varying effects on distinct anatomic portions of the heart.
Chambers and valves
After the linear heart tube has looped and wedged, the internal structures take shape. The adjacent septum primum and septum secundum divide the right and left atria incompletely due to the ostium primum and ostium secundum. If the ostium primum remains open near the inferior convergence of the septum primum and endocardial cushion, it is termed a primum atrial septal defect (ASD). Right-to-left inter-atrial shunting of blood through the ostium secundum, now termed the patent foramen ovale (PFO), is specifically maintained because the septum primum and septum secundum are not fused into a single membrane until after birth and chamber pressure is higher on the right side of the heart in utero due to high pulmonary vascular resistance (PVR) in the uninflated fetal lungs. As the fetal lungs inflate within minutes after birth, PVR rapidly falls, blood flows preferentially into the RV and pulmonary artery instead of across the PFO, and pressure rises on the left side of the heart, closing the PFO over hours to days (3,4).
The atrioventricular septum, atrioventricular valves, and interventricular septum are formed as four endocardial cushions elongate toward the center of the heart, fuse, and undergo cellular differentiation and coordinated cell death. Inappropriate formation of the chambers and/or valves can cause hypoplasia, obstruction, septal defects, and frank cyanosis, which are associated with various structural heart defects (Figure 2). Congenital lesions that impact the RV anatomically or alter pressure or volume loading conditions indirectly via effects on the LV may ultimately underlie RV dysfunction.
Figure 2.

Three-dimensional characterization of an anomalous right upper pulmonary vein (RUPV)-superior vena cava (SVC) communication. A 67-year-old man presented for the first time with unexplained dyspnea. Advanced imaging indicated that this was due to undiagnosed congenital heart disease causing anomalous pulmonary venous return to the superior vena cava. (A) Still frame image acquired during 3-dimensional transesophageal echocardiography demonstrates contiguous blood flow between the right upper pulmonary vein (outlined by arrowheads) and SVC. Asterisks (*) designate the margins of the anomalous channel between these structures. (B) Multislice 3-dimensional reconstructive computed tomographic angiography (acquired at right anterior oblique of 84°, caudal of 31°) reveals an abnormal RUPV-SVC communication (provided at increased magnification in the inset) with normal insertion of the RUPV into the left atrium (LA). (C) Multislice 3-dimensional reconstructed computed tomographic angiography shows a superior course of the anomalous RUPV from the lung parenchyma without meandering before communication with the SVC. Asterisk (*) indicates RUPV. IVC indicates inferior vena cava; and RA, right atrium. Reproduced with permission from Clarke et al., Circ Cardiovasc Imaging 2013;6(2):349-351.
Coronary arteries and the cardiac conducting system
Developmental defects in coronary artery formation per se are an uncommon cause of SCD in childhood and adolescence. Although mechanisms underlying normal, common variant, and rare variant coronary artery development are poorly understood, clinical observation has revealed that single coronary vascular perfusion has implications for limited RV ischemic reserve under pathophysiological conditions, which influences morbidity and mortality in the setting of RV ischemia. Similar to coronary anatomy, the RV develops a narrow electrical supply via the right bundle branch. Chronic scarring of the right bundle branch is associated with ARVD, which primarily involves the RV.
Static and dynamic anatomy of the RV vs. LV
The geometry, compaction, and contractility of the RV and LV are shaped by the fetal circulation and differences in the magnitude and vector of the load experienced by each ventricle during development. Once the distinct pulmonary and systemic circulations are formed, under normal conditions, the RV pushes blood through the low resistance pulmonary vasculature, while the LV pushes blood through the higher resistance systemic vasculature. Subsequently, the interventricular septum normally curves inward toward the RV to accommodate higher LV chamber pressure, and the RV wall is necessarily thinner (Figure 2) (5). It is generally accepted that the LV has three layers of myofibers while the RV has two layers (5). Most driving pressure of the LV is generated by the circumferential middle layer contracting inward. Torsion from apex to base further serves to eject blood toward the outlet in a “wringing” motion. Because the RV lacks a middle layer, contraction is not dependent on torsion. RV work is accomplished through a reduction in RV free wall surface area and a reduction in RV free wall-septal distance, as well as bringing the inlet (tricuspid valve) toward the outlet (pulmonic valve) (6,7). The motion is peristaltic across the RV from inlet to apical trabeculae to outlet. This results in a large change in volume for a relatively small change in free wall area. Although the RV and LV are typically thought of and discussed as separate chambers, they share fibers and have common walls, and functionally interact as a complex unit. For example, more than half of the RV systolic force is generated by LV contraction through the free wall interconnection of fibers (8). In total, RV pressure development occurs from interactions of the RV free wall, the LV free wall, and the interventricular septum (9,10).
Physiology of the RV vs. LV
Because the long-term risk of SCD and heart failure-associated mortality varies by underlying physiology, it is important to understand the physiologic relationships that influence RV cardiac output to understand how RV dysfunction can modulate risk of SCD. Similar to the LV, RV cardiac output is determined by heart rate and stroke volume. Beyond simple equation determinants of cardiac output, though, RV cardiac output is particularly influenced by ventriculo-arterial coupling, ventricular interdependence, and pericardial mechanical constraint.
Ventriculo-arterial coupling
Normally, the pulmonary circulation has lower resistance (i.e., afterload) than the systemic circulation. To maintain constant flow through the RV and LV, lower PVR in the pulmonary circulation allows for a lower pulmonary arterial pressure experienced by the RV compared to the systemic arterial pressure experienced by the LV. Interestingly, when the LV is transposed onto the pulmonary circulation, such as with congenitally corrected transposition of the great arteries, the pressure-volume loop of the LV becomes trapezoidal and identical to the normal RV (11). Alternatively, if PVR gradually increases, such as in pulmonary arterial hypertension, and the RV can adapt to work against pulmonary vascular pressures which mirror systemic pressures, the RV pressure-volume loop changes toward a square-wave LV pattern (11).
Coupling describes the transfer of energy, via the forward motion of blood, between the RV and the pulmonary circulation (12). When afterload increases, pulmonary arterial, and then RV, elastance must increase to preserve energy transfer through the circulation (Figure 3). If afterload increases gradually, such as in pulmonary arterial hypertension, RV elastance increases first through muscle hypertrophy. If afterload continues to increase, continued hypertrophic remodeling becomes maladaptive by eventually stiffening the ventricle and/or decreasing chamber size, ultimately reducing stroke volume and cardiac output. RV cavitary dilation is a harbinger of impending RV failure, a structural indication that RV elastance fails to match increased pulmonary arterial elastance. The RV uncouples from the pulmonary vascular system. RV failure and cor pulmonale will eventually occur. If afterload acutely increases, such as with massive pulmonary embolism, and the RV is not afforded time to hypertrophy as a compensatory mechanism to maintain coupling, it follows that uncoupling can occur rapidly and is signaled by acute RV dilation. The extent to which this process is irreversible is not fully understood, as RV recovery is often observed following lung transplant and pulmonary endarterectomy in patients with end-stage pulmonary arterial hypertension or chronic thromboembolic pulmonary hypertension, respectively.
Figure 3.
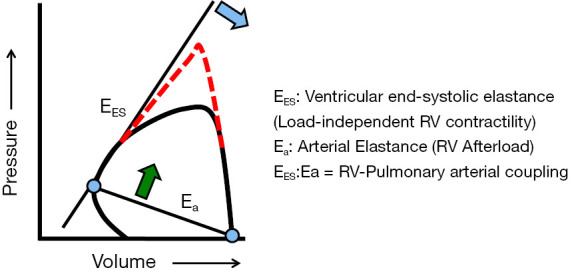
Right ventricular-pulmonary vascular coupling. The pressure-volume diagram is used to calculate right ventricular (RV)-pulmonary vascular coupling. The pressure-volume diagram allows for the determination of RV end-systolic elastance (EES). EES is unaffected by changes to RV afterload (dashed line) and, therefore, is the best possible load-independent measurement of contractility. By contrast, arterial elastance (Ea) is proportional to pulmonary vascular resistance and is a measurement of RV afterload. The ratio of EES to Ea is a measure of the coupling of the respective ventricular and arterial loads. Thus, a decrease in EES (blue arrow) or an increase in Ea (green arrow) due to elevations in pulmonary vascular resistance, for example, disrupts normal coupling. Reproduced with permission from Maron et al., Pulm Circ 2014;4(4):705-716.
Ventricular interdependence and pericardial mechanical constraint
Normally, LV interaction with the RV free wall substantially contributes to RV pressure generation, but the RV does not substantially contribute to LV pressure generation. This was illustrated in a seminal study by Damiano and colleagues in which the ventricles were electrically isolated from each other, and force pressure generation was measured in each ventricle during contralateral electrical stimulation (10). When the RV was stimulated, there was negligible LV intraventricular pressure generation. In contrast, when the LV was stimulated, there was a substantial increase in RV pressure. This demonstrates interdependence convincingly under normal conditions. Hoffman and colleagues extended this work to understand if RV free wall geometry itself, beyond interaction with the LV, was important in determining cardiac output under normal conditions (13). The RV free wall was replaced experimentally with a non-contractile material. Surprisingly, the RV generated similar pressure. But, if the RV free wall was enlarged, mirroring RV dilation under pathophysiologic conditions, LV pressure generation unexpectedly fell. Additional studies have shown that pathophysiologic RV dilation adversely affects LV contractility via ventricular interdependence magnified by pericardial constraint and LV longitudinal strain (1,14,15). In other words, because the inelastic pericardium constrains total cardiac volume, when the RV dilates, particularly acutely, the interventricular septum is forced to bow into the LV, adversely affecting LV preload and contractility. Thus, based on the location (left side vs. right side) and acuity of an initiating lesion, ventricular interdependence mediates the subsequent steps in cardiac adaptation or maladaptation.
Epidemiology of SCD
Factors influencing accuracy and reproducibility of SCD data include differences in endpoint definition [e.g., SCD (excluding survivors) vs. sudden cardiac arrest (SCA, which may include survivors)]; time restrictions (e.g., event occurring within 1, 12, or 24 hours following symptom onset or change in clinical status); event location (e.g., out-of-hospital vs. in-hospital); and specificity of cause (e.g., ischemic or coronary heart disease only vs. general cardiac or cardiovascular disease; non-traumatic vs. traumatic cause of cardiac event). Differentiating between SCD and SCA has particular implications for defining risk factors and pro-survival factors, as well as shifting professional, public, and policy focus toward prevention and survivorship instead of focusing on the death itself. The American Heart Association (AHA) relies on three sources of information to study the epidemiology of SCD: the Resuscitation Outcomes Consortium (ROC), which includes emergency medical services (EMS) assessed and treated out-of-hospital cardiac arrests from multiple regions throughout the United States; the Cardiac Arrest Registry to Enhance Survival (CARES), which estimates EMS-treated, out-of-hospital cardiac arrests of people of any age throughout the United States; and Get with the Guidelines (GWTG)-Resuscitation, an AHA program which applies guideline-based methodology to estimate the number of in-hospital cardiac arrests in the United States from the total number of hospitalized patients.
Based on these data, the AHA estimates that in the US there are approximately 7,000 annual out-of-hospital SCAs in children 18 years old or younger, and approximately 350,000 annual out-of-hospital SCAs in persons older than 18 years of age (16). It is clear that causes of SCA vary by age (Figure 4). Among children, adolescents, and younger adults, SCA is predominantly attributable to congenital structural heart disease and genetically-caused electrical abnormalities. In older adults, ischemic heart disease and chronic congestive heart failure are the primary causes of SCD. It is notable that across all ages, only 5–10% of SCD cases have no underlying CAD or structural heart disease.
Figure 4.

Causes of sudden cardiac arrest by age group. CAD, coronary artery disease; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy. Reproduced with permission from Benjamin EJ, Virani SS, Callaway CW, et al. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation 2018;137(12):e67-e492 (16).
Causes of RV dysfunction in childhood, adolescence, and early adulthood, and long-term risk of SCD
The most common cause of RV dysfunction in childhood, adolescence, and early adulthood is congenital or inherited cardiac defects. Importantly, though, RV dysfunction is common in patients with a primary pathology involving the LV, lungs, or pulmonary vascular system. Thus, RV dysfunction may modulate greater morbidity and mortality by its direct involvement or as a marker of overall worse physiologic dysfunction. For this reason, it is difficult to attribute increased long-term risk of SCD specifically to RV dysfunction in these congenital situations where isolated RV dysfunction is uncommon. The importance of RV dysfunction to long-term risk of SCD in ARVD and HCM is discussed below.
ARVD
ARVD, in which RV dysfunction often precedes SCD, is of particular note because the origin of the arrhythmia can be more often attributed to the RV. In ARVD, fibrofatty tissue replaces normal myocardium and has a strong predilection toward affecting the RV while sparing the LV. There are four recognized pathophysiological phases: (I) the concealed phase; (II) an overt electrical disorder; (III) RV failure; (IV) an advanced phase often characterized by severe biventricular failure. The majority of cases of SCD occur during the concealed phase when RV dysfunction, but not failure, is present though undiagnosed (17,18).
Hulot et al. collected 130 ARVD patients from a tertiary care center in France from 1977–2000 (19). Mean [± standard deviation (SD)] age at symptom onset was 32 (±14) years, with a range of 10–73 years. After a mean (± SD) follow up of 8.1 (±7.8) years there were 24 deaths, with a mean (± SD) age at death of 54 (±19) years and an annual mortality rate of 2.3%. Of the 24 deaths, 21 were due to cardiovascular disease (14 progressive heart failure, 7 SCD). All patients who died had a history of VT. In a multivariate model predicting death, significant independent predictors included clinical signs of RV failure and LV dysfunction. Ten patients received an implantable cardioverter defibrillator (ICD), and none of them died. Similar outcomes were observed in a more contemporary cohort from Johns Hopkins (20). Although ARVD is typically thought of as a disease in younger patients, these studies illustrate that ARVD patients can be diagnosed in the 8th decade of life. A report from the University Medical Center Utrecht ARVD/C registry (n=110 patients) focused on ARVD patients ≥50 years old [n=29/110, mean (± SD) age 59 (±6) years old] confirms that ARVD can remain concealed even into older adulthood (21).
The 2015 International Task Force Consensus Statement on risk stratification recommends ICD implantation for all high-risk cases (Class Ia recommendation), which includes secondary prevention for aborted SCD and sustained ventricular arrhythmia and primary prevention for cases with severe dysfunction of either or both ventricles (22). Low-risk groups are those with no risk factors and “healthy carriers” (with a negative genetic test for known pathogenic variants). ICD implantation carries a Class IIa recommendation for intermediate risk patients, stratified based on ≥1 major risk factors for syncope and electrical instability with nonsustained VT (NSVT) or moderate dysfunction of LV, RV, or both ventricles. The presence of ≥1 minor risk factor (compound heterozygotes, digenic carriers, electroanatomic scar on RV endocardial voltage mapping per 5% increment, fragmented electrograms, T-wave inversion in inferior leads, T-wave inversion in 2 out of 3 inferior leads, T-wave inversion in ≥3 precordial leads, QRS fragmentation and precordial QRS amplitude ratio <0.48) carries a Class IIb recommendation for consideration of ICD implantation.
HCM
HCM is defined by increased left ventricular wall thickness in the absence of abnormal loading conditions or another cause by which to account for the observed abnormality (23). In some cases it is inherited in an autosomal dominant pattern in relation to mutations in cardiac sarcomere protein genes. Histologically, it is defined by myocardial disarray, fibrosis, and small vessel disease (24). The incidence of SCD in HCM is thought to be <1% per year, but varies by underlying risk factors (25,26), and has likely been influenced by historical selection and diagnostic biases. Maron and colleagues showed that death in HCM is manifested in three modes that trend in prevalence by age: sudden death (51% of cases, mean age 45), progressive heart failure (36% of cases, mean age 56), and HCM-associated stroke due to atrial fibrillation (13% of cases, mean age 73) (25).
HCM is almost always a disease of abnormal LV wall thickness. The first study of RV involvement in HCM using cardiac magnetic resonance imaging (CMR) was additionally revealing (27). Maron and colleagues compared 46 HCM cases without pulmonary hypertension to 22 healthy controls. Fifteen (33%) HCM patients had maximal RV wall thickness ≥8 mm (2 SDs higher than the mean for controls) and 4 (9%) had severe RV wall hypertrophy ≥10 mm. RV hypertrophy was predominantly diffuse involving all or most of the RV wall. RV wall thickness was positively correlated with LV wall thickness and mass. Interestingly, only 1 (2%) patient had RV wall fibrosis. The role of the RV in ventricular arrhythmias in HCM is not known, but is likely to be low if any.
Causes of RV dysfunction in later adulthood and long-term risk of SCD
The most significant causes of chronic RV dysfunction in older adulthood are cardiac ischemia, congestive heart failure, and pulmonary hypertension. Valvular lesions, such as aortic stenosis, mitral regurgitation, or mitral stenosis, typically cause RV dysfunction via development of congestive heart failure and ventricular interdependence, or via World Health Organization Group 2 pulmonary hypertension directly impacting the RV. Isolated structural or functional tricuspid valve disease is increasingly recognized as a cause of RV dysfunction by leading to chronically elevated preload, but the incidence of SCD is thought to be very rare in isolated tricuspid disease. Congenital abnormalities, such as those discussed earlier, can lead to RV dysfunction in older adulthood, but are less prevalent than cardiac ischemia, CHF, or pulmonary hypertension; moreover, isolating the risk of SCD due to RV dysfunction in these cases is quite difficult. Outside the scope of this review is RV dysfunction due to sepsis, perioperative injury, transplant, or cardiac or peripheral shunts, as these are rarer causes of SCD and, with the exception of shunts, typically transiently increase the risk of SCD. Subsequently, we will consider the risk of SCD due to the most prevalent causes of RV dysfunction.
Cardiac ischemia
A large proportion of cases of out-of-hospital cardiac arrest have prevalent coronary artery disease (28), and most cases of SCA are directly attributable to cardiac ischemia (29). RV dysfunction can contribute to long-term risk of SCD in the setting of cardiac ischemia in at least three ways: (I) via direct ischemic injury to the RV or RV conduction system, leading to impaired RV contractility and propensity for arrhythmia originating in the RV; (II) via chronic RV dysfunction impairing reserve when another region of the heart becomes dysfunctional; (III) via triggering LV ischemia in patients with severe PH and RV dysfunction, when a dilated pulmonary artery compresses the left coronary artery main stem (left main compression syndrome) (30). Isolated RV ischemia or infarction is rare, and chronic right heart failure due to isolated RV ischemia is also rare. As a lower pressure system, the RV is perfused during systole and diastole, whereas the LV is perfused almost exclusively during diastole. There is greater left to right collateral coronary perfusion, so even though a large portion of the RV has single coronary perfusion, collaterals from the LV help support the RV when ischemia occurs. In a left dominant system, which exists in 15% of the population, more than 50% of the RV free wall can be supplied by the left coronary system. Oxygen demand is also lower in the RV due to lower RV mass and afterload. Subsequently, RV myocardial infarction (RVMI) occurs more often when there is an acute coronary syndrome to the inferior wall of the LV, which involves RV myocardium in 30–50% of cases (31-36).
Isolated RVMI is a high-risk event and associated with increased in-hospital morbidity and mortality compared to myocardial infarction of a similar magnitude that does not involve the RV. Moreover, LVMI which involves the RV has poorer outcomes than an LVMI which does not involve the RV (31-39). A meta-analysis of six studies before the era of widespread percutaneous coronary intervention (PCI), which included 1,198 patients, showed the presence of RV involvement vs. no RV involvement was associated with a higher incidence of short-term death (odds ratio 3.2, 95% confidence interval, 2.4–4.1), cardiogenic shock (OR 3.2, 2.4–3.5), sustained VT (OR 2.7, 2.1–3.5), and advanced AV block (OR 3.4, 2.7–4.2) (40). Increased mortality seemed related to presence of RV involvement and not to infarct size, and was mediated via increased prevalence of hypotension (cardiogenic shock) and arrhythmia. A chronic right coronary occlusion rarely results in significant physiologic impairment at rest or with exercise because of RV remodeling which preserves RV free wall motion and ejection fraction augmentation (37,38,41). Survival after an RVMI is primarily determined by the extent of LV involvement.
Chronic congestive heart failure and post-capillary pulmonary hypertension
SCD is well known in the setting of chronic congestive heart failure, particularly in patients with heart failure with reduced ejection fraction, and is most often due to ventricular arrhythmia. RV dysfunction in the setting of chronic CHF occurs most frequently due to group 2 pulmonary hypertension and chronically increased RV afterload, as well as altered RV geometry and function due to LV remodeling or dysfunction (ventricular interdependence) (42). There is a class I recommendation for ICD implantation for patients with an LV ejection fraction of 35% or less and New York Heart Association Functional Class II or III symptoms, regardless of heart failure cause and phenotype.
Shen and colleagues examined trends in SCD over time in the era of sequential medication development and recommendation (43). They included patients from 12 clinical trials (n=40,195) from 1995 to 2014 who did not have an ICD implanted at the time of trial enrollment. SCD occurred in 3,583 patients, who were more often older, male, with an ischemic cause of heart failure and with worse cardiac function. The rate of SCD within 90 days of trial enrollment declined from 2.4% to 1.0% during the time frame examined. RV dysfunction specifically was not considered in the analysis, though it follows that chronic heart failure due to its common causes (ischemia, systemic hypertension, valvular disease, dilated cardiomyopathy) is likely associated with increased morbidity and mortality when RV dysfunction is present. Using data from a cohort of 5,463 patients seen consecutively at the Mayo Clinic cardiac care unit, Naksuk and colleagues showed that RV dysfunction was associated with increased risk of SCD independent of LV function or use of an ICD (44). Recent work from the Mayo Clinic highlighted progressive changes (dilation and dysfunction) in RV (but not LV) over time in patients with heart failure with preserved ejection fraction (HFpEF), and both prevalent and incident RV dysfunction were associated with increased mortality (45). The association between RV dysfunction and mortality was also shown in other studies (46,47), but the specific impact of SCD in this context has not been thoroughly evaluated. Of note is a recent study by Pandat et al. in which RV dysfunction was associated with SCD independent of reduced LV ejection fraction, and the combination of reduced RV and LV ejection fraction was even more strongly associated with SCD (48).
Pulmonary hypertension
The incidence of SCD in the setting pulmonary hypertension due to chronic lung disease or chronic thromboembolic pulmonary hypertension is low (likely very low), as the majority of mortality in these groups is due to heart failure. Registry data focusing on patients with pulmonary arterial hypertension, as well, suggests that the majority of patients die from heart failure. In the National Institutes of Health registry, 106 deaths were reported in 194 PAH patients (49). The majority (47%) were due to right heart failure, 26% were classified as sudden death, and 27% were due to other causes. In the UZ, Leuven, Belgium registry, there were 99 deaths in a cohort of 316 PAH patients (50). Most deaths were due to right heart failure (N=32, 32%), although sudden death was reported in 18% of patients (N=18). Syncope is a high-risk feature in PAH but is most often from a problem of cerebrovascular perfusion in the setting of heart failure, not from ventricular arrhythmia. When it does occur, SCD is proposed to manifest in these patients due to arrhythmia in general, circulatory collapse, compression of the left main coronary artery, or pulmonary artery rupture or dissection (51,52). It is notable that ventricular arrhythmias are uncommon in PAH and CTEPH. If arrhythmia occurs, it is more often supraventricular, and these events often precede clinical deterioration. Subsequently, there is no strong data to support the use of ICD implantation in PAH or CTEPH. Medical management of supraventricular arrhythmias specifically in the setting of PAH and CTEPH have been proposed (53), but further study is needed. Because clinical trials of PAH specific therapies most frequently use functional class, exercise capacity, and quality of life as endpoints, it is also unknown if incidence of SCD specifically is decreased by PAH specific therapy, such as prostacycline agonists, endothelin receptor antagonists, or nitric oxide-cyclic guanosine monophosphate inhibitors or soluble guanylate cyclase stimulators.
Although unexpected or sudden death is reported in patients with pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension (54), the nature of such deaths is not clear. Primary ventricular arrhythmias, secondary ventricular arrythmias due to the systemic consequences of chronic RV dysfunction leading to renal dysfunction and electrolyte (potassium) disturbances; left main coronary artery compression syndrome; PA dissection; or cardiac tamponade are potential contributors (1). This may become more important in the modern treatment era, as patients live longer and new limiting factors may evolve, including systemic effects of pulmonary arterial hypertension and RV dysfunction (1,55). Furthermore, pulmonary hypertension and right-sided heart failure often involves the combination of systemic congestion and systemic low output, which, in turn, triggers an inflammatory systemic reaction insulting multiple organ systems including kidney (salt and water balance), liver (cardiac cirrhosis), brain (decreased cognitive function), and skeletal muscle (volitional muscle atrophy) (55,56). End-organ injury may directly or indirectly decrease the threshold for hemodynamic instability and sudden cardiac events.
Future directions
RV dysfunction is an increasingly recognized physiologic complication of many other conditions. Additional research should try and fill gaps about the epidemiology and pathophysiology of RV dysfunction and SCD. This would be assisted by the development of longitudinal cohorts of patients with excellent RV phenotyping tied to clinically relevant outcomes. Methods for more accurate RV functional assessment include CMR and echocardiographic strain imaging, also referred to as speckle imaging (57). Further mechanistic research uncovering unique, targetable aspects of the RV system will hopefully discover targets for novel RV-specific therapies which can reduce the risk of SCD associated with RV dysfunction.
Acknowledgments
Funding: NIH grants: R56HL131787, R01HL139613-01, R21HL145420, R01HL153502; National Scleroderma Foundation, Cardiovascular Medical Research Education Foundation, and McKenzie Family Charitable Trust.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Open Access Statement: This is an Open Access article distributed in accordance with the Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License (CC BY-NC-ND 4.0), which permits the non-commercial replication and distribution of the article with the strict proviso that no changes or edits are made and the original work is properly cited (including links to both the formal publication through the relevant DOI and the license). See: https://creativecommons.org/licenses/by-nc-nd/4.0/.
Footnotes
Provenance and Peer Review: This article was commissioned by the Guest Editors (Martin Koestenberger, Harm-Jan Bogaard and Georg Hansmann) for the series “Right Ventricular Dysfunction” published in Cardiovascular Diagnosis and Therapy. The article was sent for external peer review organized by the Editor-in-Chief and the editorial office.
Conflicts of Interest: The authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/cdt-20-450). The series “Right Ventricular Dysfunction” was commissioned by the editorial office without any funding or sponsorship. Dr. MK served as the unpaid Guest Editors of the series. Dr. SR reports personal fees and other from Abbott, grants and personal fees from Actelion, personal fees from Acceleron, grants and personal fees from AstraZeneca, grants and personal fees from Bayer, personal fees from BMS, personal fees from Janssen, personal fees from Merck, grants and personal fees from Novartis, grants and personal fees from Pfizer, grants and personal fees from United Therapeutics, personal fees from Vifor, outside the submitted work. Dr. BAM reports other from Acetelion, during the conduct of the study. The author has no other conflicts of interest to declare.
References
- 1.Rosenkranz S, Howard LS, Gomberg-Maitland M, et al. Systemic Consequences of Pulmonary Hypertension and Right-Sided Heart Failure. Circulation 2020;141:678-93. 10.1161/CIRCULATIONAHA.116.022362 [DOI] [PubMed] [Google Scholar]
- 2.Kloesel B, DiNardo JA, Body SC. Cardiac Embryology and Molecular Mechanisms of Congenital Heart Disease: A Primer for Anesthesiologists. Anesth Analg 2016;123:551-69. 10.1213/ANE.0000000000001451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Emmanouilides GC, Moss AJ, Duffie ER, Jr, et al. Pulmonary Arterial Pressure Changes in Human Newborn Infants from Birth to 3 Days of Age. J Pediatr 1964;65:327-33. 10.1016/S0022-3476(64)80395-4 [DOI] [PubMed] [Google Scholar]
- 4.Dell'Italia LJ. The right ventricle: anatomy, physiology, and clinical importance. Curr Probl Cardiol 1991;16:653-720. 10.1016/0146-2806(91)90009-Y [DOI] [PubMed] [Google Scholar]
- 5.Sheehan F, Redington A. The right ventricle: anatomy, physiology and clinical imaging. Heart 2008;94:1510-5. 10.1136/hrt.2007.132779 [DOI] [PubMed] [Google Scholar]
- 6.Santamore WP, Meier GD, Bove AA. Effects of hemodynamic alterations on wall motion in the canine right ventricle. Am J Physiol 1979;236:H254-62. [DOI] [PubMed] [Google Scholar]
- 7.Sakuma M, Ishigaki H, Komaki K, et al. Right ventricular ejection function assessed by cineangiography--Importance of bellows action. Circ J 2002;66:605-9. 10.1253/circj.66.605 [DOI] [PubMed] [Google Scholar]
- 8.Woodard JC, Chow E, Farrar DJ. Isolated ventricular systolic interaction during transient reductions in left ventricular pressure. Circ Res 1992;70:944-51. 10.1161/01.RES.70.5.944 [DOI] [PubMed] [Google Scholar]
- 9.Chow E, Farrar DJ. Effects of left ventricular pressure reductions on right ventricular systolic performance. Am J Physiol 1989;257:H1878-85. [DOI] [PubMed] [Google Scholar]
- 10.Damiano RJ, Jr, La Follette P, Jr, Cox JL, et al. Significant left ventricular contribution to right ventricular systolic function. Am J Physiol 1991;261:H1514-24. [DOI] [PubMed] [Google Scholar]
- 11.Redington AN, Rigby ML, Shinebourne EA, et al. Changes in the pressure-volume relation of the right ventricle when its loading conditions are modified. Br Heart J 1990;63:45-9. 10.1136/hrt.63.1.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vonk Noordegraaf A, Westerhof BE, Westerhof N. The Relationship Between the Right Ventricle and its Load in Pulmonary Hypertension. J Am Coll Cardiol 2017;69:236-43. 10.1016/j.jacc.2016.10.047 [DOI] [PubMed] [Google Scholar]
- 13.Hoffman D, Sisto D, Frater RW, et al. Left-to-right ventricular interaction with a noncontracting right ventricle. J Thorac Cardiovasc Surg 1994;107:1496-502. 10.1016/S0022-5223(12)70150-2 [DOI] [PubMed] [Google Scholar]
- 14.Brookes C, Ravn H, White P, et al. Acute right ventricular dilatation in response to ischemia significantly impairs left ventricular systolic performance. Circulation 1999;100:761-7. 10.1161/01.CIR.100.7.761 [DOI] [PubMed] [Google Scholar]
- 15.Kishiki K, Singh A, Narang A, et al. Impact of Severe Pulmonary Arterial Hypertension on the Left Heart and Prognostic Implications. J Am Soc Echocardiogr 2019;32:1128-37. 10.1016/j.echo.2019.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benjamin EJ, Virani SS, Callaway CW, et al. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation 2018;137:e67-e492. 10.1161/CIR.0000000000000558 [DOI] [PubMed] [Google Scholar]
- 17.Bennett RG, Haqqani HM, Berruezo A, et al. Arrhythmogenic Cardiomyopathy in 2018-2019: ARVC/ALVC or Both? Heart Lung Circ 2019;28:164-77. 10.1016/j.hlc.2018.10.013 [DOI] [PubMed] [Google Scholar]
- 18.Gandjbakhch E, Redheuil A, Pousset F, et al. Clinical Diagnosis, Imaging, and Genetics of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: JACC State-of-the-Art Review. J Am Coll Cardiol 2018;72:784-804. 10.1016/j.jacc.2018.05.065 [DOI] [PubMed] [Google Scholar]
- 19.Hulot JS, Jouven X, Empana JP, et al. Natural history and risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation 2004;110:1879-84. 10.1161/01.CIR.0000143375.93288.82 [DOI] [PubMed] [Google Scholar]
- 20.Dalal D, Nasir K, Bomma C, et al. Arrhythmogenic right ventricular dysplasia: a United States experience. Circulation 2005;112:3823-32. 10.1161/CIRCULATIONAHA.105.542266 [DOI] [PubMed] [Google Scholar]
- 21.van der Pols MJ, Mast TP, Loh P, et al. Clinical characterisation and risk stratification of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy >/=50 years of age. Neth Heart J 2016;24:740-7. 10.1007/s12471-016-0886-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corrado D, Wichter T, Link MS, et al. Treatment of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: An International Task Force Consensus Statement. Circulation 2015;132:441-53. 10.1161/CIRCULATIONAHA.115.017944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2008;29:270-6. 10.1093/eurheartj/ehm342 [DOI] [PubMed] [Google Scholar]
- 24.Varnava AM, Elliott PM, Sharma S, et al. Hypertrophic cardiomyopathy: the interrelation of disarray, fibrosis, and small vessel disease. Heart 2000;84:476-82. 10.1136/heart.84.5.476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maron BJ, Olivotto I, Spirito P, et al. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation 2000;102:858-64. 10.1161/01.CIR.102.8.858 [DOI] [PubMed] [Google Scholar]
- 26.Elliott PM, Poloniecki J, Dickie S, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol 2000;36:2212-8. 10.1016/S0735-1097(00)01003-2 [DOI] [PubMed] [Google Scholar]
- 27.Maron MS, Hauser TH, Dubrow E, et al. Right ventricular involvement in hypertrophic cardiomyopathy. Am J Cardiol 2007;100:1293-8. 10.1016/j.amjcard.2007.05.061 [DOI] [PubMed] [Google Scholar]
- 28.Chelly J, Mongardon N, Dumas F, et al. Benefit of an early and systematic imaging procedure after cardiac arrest: insights from the PROCAT (Parisian Region Out of Hospital Cardiac Arrest) registry. Resuscitation 2012;83:1444-50. 10.1016/j.resuscitation.2012.08.321 [DOI] [PubMed] [Google Scholar]
- 29.Deo R, Albert CM. Epidemiology and genetics of sudden cardiac death. Circulation 2012;125:620-37. 10.1161/CIRCULATIONAHA.111.023838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McLaughlin VV, Hoeper MM, Channick RN, et al. Pulmonary Arterial Hypertension-Related Morbidity Is Prognostic for Mortality. J Am Coll Cardiol 2018;71:752-63. 10.1016/j.jacc.2017.12.010 [DOI] [PubMed] [Google Scholar]
- 31.Andersen HR, Falk E, Nielsen D. Right ventricular infarction: frequency, size and topography in coronary heart disease: a prospective study comprising 107 consecutive autopsies from a coronary care unit. J Am Coll Cardiol 1987;10:1223-32. 10.1016/S0735-1097(87)80122-5 [DOI] [PubMed] [Google Scholar]
- 32.Goldstein JA. Pathophysiology and management of right heart ischemia. J Am Coll Cardiol 2002;40:841-53. 10.1016/S0735-1097(02)02048-X [DOI] [PubMed] [Google Scholar]
- 33.Isner JM, Roberts WC. Right ventricular infarction complicating left ventricular infarction secondary to coronary heart disease. Frequency, location, associated findings and significance from analysis of 236 necropsy patients with acute or healed myocardial infarction. Am J Cardiol 1978;42:885-94. 10.1016/0002-9149(78)90672-0 [DOI] [PubMed] [Google Scholar]
- 34.Isner JM. Right ventricular myocardial infarction. JAMA 1988;259:712-8. 10.1001/jama.259.5.712 [DOI] [PubMed] [Google Scholar]
- 35.Cabin HS, Clubb KS, Wackers FJ, et al. Right ventricular myocardial infarction with anterior wall left ventricular infarction: an autopsy study. Am Heart J 1987;113:16-23. 10.1016/0002-8703(87)90004-4 [DOI] [PubMed] [Google Scholar]
- 36.Kinch JW, Ryan TJ. Right ventricular infarction. N Engl J Med 1994;330:1211-7. 10.1056/NEJM199404283301707 [DOI] [PubMed] [Google Scholar]
- 37.Berger PB, Ruocco NA, Jr, Ryan TJ, et al. Frequency and significance of right ventricular dysfunction during inferior wall left ventricular myocardial infarction treated with thrombolytic therapy (results from the thrombolysis in myocardial infarction [TIMI] II trial). The TIMI Research Group. Am J Cardiol 1993;71:1148-52. 10.1016/0002-9149(93)90637-R [DOI] [PubMed] [Google Scholar]
- 38.Sakata K, Yoshino H, Kurihara H, et al. Prognostic significance of persistent right ventricular dysfunction as assessed by radionuclide angiocardiography in patients with inferior wall acute myocardial infarction. Am J Cardiol 2000;85:939-44. 10.1016/S0002-9149(99)00905-4 [DOI] [PubMed] [Google Scholar]
- 39.Zehender M, Kasper W, Kauder E, et al. Right ventricular infarction as an independent predictor of prognosis after acute inferior myocardial infarction. N Engl J Med 1993;328:981-8. 10.1056/NEJM199304083281401 [DOI] [PubMed] [Google Scholar]
- 40.Mehta SR, Eikelboom JW, Natarajan MK, et al. Impact of right ventricular involvement on mortality and morbidity in patients with inferior myocardial infarction. J Am Coll Cardiol 2001;37:37-43. 10.1016/S0735-1097(00)01089-5 [DOI] [PubMed] [Google Scholar]
- 41.Lim ST, Marcovitz P, Pica M, et al. Right ventricular performance at rest and during stress with chronic proximal occlusion of the right coronary artery. Am J Cardiol 2003;92:1203-6. 10.1016/j.amjcard.2003.07.032 [DOI] [PubMed] [Google Scholar]
- 42.Rosenkranz S, Gibbs JS, Wachter R, et al. Left ventricular heart failure and pulmonary hypertension. Eur Heart J 2016;37:942-54. 10.1093/eurheartj/ehv512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shen L, Jhund PS, Petrie MC, et al. Declining Risk of Sudden Death in Heart Failure. N Engl J Med 2017;377:41-51. 10.1056/NEJMoa1609758 [DOI] [PubMed] [Google Scholar]
- 44.Naksuk N, Tan N, Padmanabhan D, et al. Right Ventricular Dysfunction and Long-Term Risk of Sudden Cardiac Death in Patients With and Without Severe Left Ventricular Dysfunction. Circ Arrhythm Electrophysiol 2018;11:e006091. 10.1161/CIRCEP.117.006091 [DOI] [PubMed] [Google Scholar]
- 45.Obokata M, Reddy YNV, Melenovsky V, et al. Deterioration in right ventricular structure and function over time in patients with heart failure and preserved ejection fraction. Eur Heart J 2019;40:689-97. 10.1093/eurheartj/ehy809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ghio S, Guazzi M, Scardovi AB, et al. Different correlates but similar prognostic implications for right ventricular dysfunction in heart failure patients with reduced or preserved ejection fraction. Eur J Heart Fail 2017;19:873-9. 10.1002/ejhf.664 [DOI] [PubMed] [Google Scholar]
- 47.Gorter TM, Hoendermis ES, van Veldhuisen DJ, et al. Right ventricular dysfunction in heart failure with preserved ejection fraction: a systematic review and meta-analysis. Eur J Heart Fail 2016;18:1472-87. 10.1002/ejhf.630 [DOI] [PubMed] [Google Scholar]
- 48.Pandat S, Nagaura T, Nair SG, et al. An association between right ventricular dysfunction and sudden cardiac death. Heart Rhythm 2020;17:169-74. 10.1016/j.hrthm.2019.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.D'Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med 1991;115:343-9. 10.7326/0003-4819-115-5-343 [DOI] [PubMed] [Google Scholar]
- 50.Delcroix M, Naeije R. Optimising the management of pulmonary arterial hypertension patients: emergency treatments. Eur Respir Rev 2010;19:204-11. 10.1183/09059180.00004910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.European Heart Rhythm Association ; Heart Rhythm Society, Zipes DP, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death). J Am Coll Cardiol 2006;48:e247-346. 10.1016/j.jacc.2006.07.010 [DOI] [PubMed] [Google Scholar]
- 52.Demerouti EA, Manginas AN, Athanassopoulos GD, et al. Complications leading to sudden cardiac death in pulmonary arterial hypertension. Respir Care 2013;58:1246-54. 10.4187/respcare.02252 [DOI] [PubMed] [Google Scholar]
- 53.Cirulis MM, Ryan JJ, Archer SL. Pathophysiology, incidence, management, and consequences of cardiac arrhythmia in pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension. Pulm Circ 2019;9:2045894019834890. 10.1177/2045894019834890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tonelli AR, Arelli V, Minai OA, et al. Causes and circumstances of death in pulmonary arterial hypertension. Am J Respir Crit Care Med 2013;188:365-9. 10.1164/rccm.201209-1640OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Banerjee D, Grammatopoulos TN, Palmisciano A, et al. Alternative Splicing of the Cardiac Sodium Channel in Pulmonary Arterial Hypertension. Chest 2020. doi: . 10.1016/j.chest.2019.12.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maron BA, Opotowsky AR, Landzberg MJ, et al. Plasma aldosterone levels are elevated in patients with pulmonary arterial hypertension in the absence of left ventricular heart failure: a pilot study. Eur J Heart Fail 2013;15:277-83. 10.1093/eurjhf/hfs173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tadic M, Pieske-Kraigher E, Cuspidi C, et al. Right ventricular strain in heart failure: Clinical perspective. Arch Cardiovasc Dis 2017;110:562-71. 10.1016/j.acvd.2017.05.002 [DOI] [PubMed] [Google Scholar]