

Abstract
Type 1 diabetes mellitus (T1DM) is a chronic autoimmune disease characterized by immune-mediated destruction of pancreatic beta-cells. Multiple microRNAs (miRNAs) have been implicated in T1DM pathogenesis. Although histone deacetylase 3 (HDAC3) has been reported to be involved in T1DM, the underlying mechanisms remain to be further elucidated. This study was designed to investigate the potential regulatory role of Hdac3 on T1DM progression. The expression of miR-296-5p and B-cell leukemia-XL (BCL-XL) was determined using RT-qPCR and Western blot assay in peripheral blood mononuclear cells (PBMCs) of patients with T1DM, tumor necrosis factor-α (TNF-α)- and cycloheximide (CHX)-induced cell model, and streptozotocin (STZ)-induced rat model. The binding affinity between miR-296-5p and Bcl-xl was verified by using dual-luciferase reporter gene assay, and the binding between Hdac3 and the promoter region of miR-296-5p was validated using chromatin immunoprecipitation assay. Western blot analysis and flow cytometry were conducted to assess the apoptotic events of lymphocytes. miR-296-5p expression was downregulated while BCL-XL expression was upregulated in PBMCs of patients with T1DM. An adverse correlation was identified between miR-296-5p and Bcl-xl in mouse TE15 B lymphocytes. Bcl-xl was further validated to be targeted and negatively regulated by miR-296-5p in 293 T cells. Hdac3 inhibited miR-296-5p expression by binding to its promoter region. The effects of overexpressed Hdac3 on lymphocyte apoptosis was counterweighed via downregulation of Bcl-xl or upregulation of miR-296-5p, the mechanism of which was further validated in a rat model of DM. Taken together, the Hdac3-mediated upregulation of Bcl-xl via inhibiting miR-296-5p promoter activity enhanced the anti-apoptotic capacity of lymphocytes to accelerate the occurrence of T1DM.
Keywords: histone deacetylase 3, microRNA-296-5p, B-cell leukemia-XL, type 1 diabetes mellitus, peripheral blood mononuclear cells, apoptosis
Introduction
Type 1 diabetes mellitus (T1DM) is an autoimmune disorder characterized by immune-mediated destruction of pancreatic islet β-cells (Vatanen et al., 2018). The incidence of T1DM has drastically increased since 1950 worldwide (Craig et al., 2019). Patients with T1DM possess a higher risk to suffer from epilepsy compared with healthy individuals (Dafoulas et al., 2017). Additionally, coronary heart disease is the main factor responsible for the mortality of patients with T1DM that is closely related to insulin resistance (Bjornstad et al., 2018). Therefore, it is of great importance to develop novel and effective therapeutic strategies for T1DM treatment.
The occurrence and development of DM have been suggested to be closely correlated with histone deacetylases (HDACs; Zhang et al., 2019b). HDACs have been defined as a group of enzymes that play significant roles in mediating multiple processes at cellular and molecular levels (Merarchi et al., 2019). One of its members, Hdac3, has been reported to manage the gene expression in various tissues related to lipid metabolism (Davalos-Salas et al., 2019). In a mouse model of DM, the inhibition of Hdac3 has been reported to activate NF-E2-related factor 2 to alleviate liver damage caused by DM (Zhang et al., 2018).
More importantly, miR-296 has been revealed as one of the differentially expressed miRNAs in association with Hdac3 according to microarray analysis in a previous study (Wang et al., 2016). Interestingly, miR-296-5p has been elucidated to be functional to diabetic wound healing, suggesting its potential as an effective molecular target in DM (Liu et al., 2019). As revealed from bioinformatics analysis prior to our study, B-cell leukemia-XL (Bcl-xl) was predicted to be targeted by miR-296-5p, suggesting a possible regulatory relation between Bcl-xl and miR-296-5p. Bcl-xl, localized in the mitochondria, belongs to the anti-apoptotic BCL-2 family, which has been reported to be implicated in regulating cell death and cellular functions (Li et al., 2020b). Apoptosis of β-cells has been recognized as a critical pathway underlying T1DM progression where the anti-apoptotic protein BCL-XL was identified as an important player in T1DM progression (Anuradha et al., 2014). The glucose signaling in pancreatic β-cells has been indicated to be restricted by Bcl-xl (Luciani et al., 2013). In addition, upregulation of Bcl-xl has been observed in peripheral blood mononuclear cells (PBMCs) in patients with T1DM (de Oliveira et al., 2012), but the upstream regulators of Bcl-xl remains uncharacterized. Gene expression alternation in PBMCs has also been recognized to provide a novel insight into identifying new biomarkers or treatment modalities for T1DM (Li et al., 2019). Therefore, in this study, we aimed to validate whether Hdac3 could inhibit the apoptotic events of lymphocytes through miR-296-5p or Bcl-xl signaling pathway to increase the occurrence of T1DM.
Materials and Methods
Ethics Statement
All procedures concerning human samples were approved by the Ethics Committee of the Second Hospital of Jilin University. This study was performed based on the Declaration of Helsinki principles. The signed informed consents were obtained from all participants or their family members. All experimental procedures involving animals were approved by the Animal Care and Use Committee of the Second Hospital of Jilin University and in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health.
Study Subjects
PBMCs were obtained from patients with T1DM and age-matched healthy individuals as a control to analyze the expression of miR-296-5p and Bcl-xl. Thirty patients with T1DM (14 males and 16 females; average age 43.07 ± 5.56 years) who met the T1DM classification criteria revised by the American Diabetes Association in 1997 and 30 healthy volunteers (15 males and 15 females; average age 41.77 ± 8.35 years) were recruited in this study. Patients with ketoacidosis and delayed acidosis combined with nephropathy, proliferative retinopathy, diabetic foot syndrome, autonomic neuropathy, cardiovascular disease, and other diabetes complications were excluded from the study. For the controls, alcoholics, smokers, overweight and/or obese, who have a family history of diabetes, infection, hypertension, or long-term medication were also excluded from the study.
Sample Collection and Isolation of PBMCs
An amount of 10 ml peripheral blood sample was collected from participants using EDTA-K2 tubes and processed within 2 h after collection. Ficoll-Hypaque (Sigma, St. Louis, MO, USA) was used to separate PBMCs by density-gradient method (Wang et al., 2018).
Culture and Activation of Lymphocytes in vitro
PBMCs were cultured in Roswell Park Memorial Institute 1,640 medium containing 10% fetal bovine serum (FBS), 2 mmol-L l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells (106 cells/ml) were cultured in a 24-well plate in an incubator containing 5% CO2 at 37°C for 3 days. Cell apoptosis was activated by the treatment with tumor necrosis factor-α (TNF-α) and cycloheximide (CHX; Zhang et al., 2019a; Li et al., 2020a).
Cell Culture
TE15 cells were purchased from American Type Culture Collection (ATCC) and cultured in Dulbecco’s modified eagle medium (DMEM) with 20% FBS and 5% penicillin or streptomycin. The 293 T cells were purchased from ATCC and cultured in DMEM added with 10% FBS and 5% penicillin or streptomycin.
RNA Isolation and Quantitation
Total RNA from tissues or cells was extracted using the miRNeasy Mini kit (QIAGEN, GmbH, Hilden, Germany) and subsequently quantified using NanoDrop ND-1000 Spectrophotometer (NanoDrop Products, Wilmington, DE, USA), whereas RNA integrity was evaluated by microfluidic electrophoresis. For reverse transcription (RT) of mRNA, 1 μg of RNA was synthesized into cDNA at 42°C for 50 min by using the TaqMan RT reagent (Roche, Canton of Basel, Switzerland). For the RT of miRNA, specific stem-loop primers were used to synthesize cDNA. The primers used in PCR were all synthesized by Sigma (Santa Clara, CA, USA; Table 1). Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) was used as the internal reference primer for Bcl-xl and Hdac3 genes, whereas U6 was used for miR-296-5p. The relative transcription level of the target gene mRNA was calculated using the relative quantitative method (2−△△CT method; Ayuk et al., 2016).
Table 1.
Primer sequences used in RT-qPCR analysis.
| Primer | Primer sequences (5'-3') | |
|---|---|---|
| Gapdh | F: TGATGGGTGTGAACCACGAG | R: TCAGTGTAGCCCAAGATGCC |
| U6 | F: CTCGCTTCGGCAGCACA | R: AACGTTCACGAATTTGCGT |
| Bcl-xl | F: CTGAATCGGAGATGGAGACC | R: TGGGATGTCAGGTCACTGAA |
| Hdac3 | F: CACCCTATGAAGCCCCATCG | R: GAGACCGTAATGCAGGACCAG |
| miR-296-5p | F: CGACGAGGGCCCCCCCT | R: GTATCCAGTGCAGG GTCCGA |
RT-qPCR, reverse transcription-quantitative polymerase chain reaction; Gapdh, glyceraldehyde-3-phosphate dehydrogenase; Bcl-xl, B-cell leukemia-XL; Hdac3, histone deacetylase 3; miR-296-5p, microRNA-296-5p; F, forward; R, reverse.
Western Blot Assay
PBMCs were added with 1 ml of cell lysis solution for the total protein isolation, and the protein concentration of each sample was measured using a bicinchoninic acid kit (20201ES76, Shanghai Yeasen Bio Technologies Co., Ltd., Shanghai, China). The sample was mixed with loading buffer and boiled at 100°C for 5 min. Following the ice bath and centrifugation, the protein was separated by lauryl sulfate-polyacrylamide gel electrophoresis and electro-transferred onto a nitrocellulose membrane. After it was blocked with 5% skimmed milk powder at 4°C overnight, the membrane was incubated with primary antibody rabbit anti-mouse BCL-XL (ab32370, 1: 2000), fasL (ab15285, 1: 2000), Bad (ab32445, 1: 2000), cleaved-caspase3 (C-caspase3; ab49822, 1: 500), Bcl-2 (ab59348, 1: 1000), and HDAC3 (ab32369, 1: 1000) at 4°C overnight. The membrane was washed thrice with tris-buffered saline tween, each time for 5 min. Horseradish peroxidase-labeled goat anti-rabbit immunoglobulin G (IgG; ab6721, 1: 5000) was then added and incubated with the membrane at room temperature for 1 h. Finally, the membrane was developed using enhanced chemiluminescence reagents. The above antibodies were purchased from Abcam Inc. (Cambridge, UK). The gray value of each band was analyzed by Quantity One software. GAPDH was used as an internal reference for the relative quantitative analysis of the target protein.
Cell Transfection
PBMCs were seeded into a 6-well plate 24 h prior to transfection until cell reached about 70% confluence. Lipofectamine 2000 (11,668,019, Thermo Fisher Scientific, Waltham, MA, USA) was diluted to 20 μl in 500 μl serum-free medium and incubated for 5 min at room temperature. The plasmids and liposomes were gently mixed and incubated at room temperature for 20 min. Cells were washed three times with serum-free medium, followed by the addition of 2 ml of serum-free medium and then incubated with the above-mentioned sequence liposome mixture for 5–24 h. Next, 20% antibiotic-free DMEM was added and incubated for 48 h. The medium was renewed after 6 h of transfection and the cells were collected after 48 h of culture for subsequent experiments. The TE15 lymphocytes were transfected with miR-296-5p inhibitor, or the mimic and negative control (NC) plasmids. After that, the cells were transfected with miR-296-5p inhibitor, small interfering RNA (siRNA) targeting Bcl-xl (siBcl-xl), and the corresponding NCs. Finally, the PBMCs were transfected with vectors pCMV-hdac3 and siBcl-xl, or the corresponding NC plasmids. The PBMCs were transfected with vectors and pCMV-Bcl-xl plasmids, respectively. Afterward, PBMCs were transfected with vector and pCMV-Hdac3 and siHdac3 and siNC plasmids.
Chromatin Immunoprecipitation Assay
The enrichment of Hdac3 in the promoter region of miR-296-5p was determined using the ChIP kit (Millipore, Boston, MA, USA). Cells in the logarithmic growth phase were fixed using 1% formaldehyde for 10 min at room temperature to allow the DNA-protein crosslink formation. Next, cells were ultrasonicated and centrifuged at 13000 rpm at 4°C (some DNA fragments were retained as INPUT), and the supernatant was collected and aliquoted into three tubes, followed by the addition of IgG and target protein-specific antibody HDAC3 (ab32369, 1: 1000) and incubated at 4°C overnight. The above antibodies were purchased from Abcam Inc. (Cambridge, UK). Next, Protein Agarose or Sepharose was used to precipitate the endogenous DNA-protein complex, followed by centrifugation with the supernatant discarded. The non-specific complexes were washed off and the retained complex was allowed to stay overnight at 65°C for the de-crosslinking process. Subsequently, the DNA fragments were collected by phenol/extraction and purification, with INPUT as an internal reference. The specific primers in the promoter region of miR-296-5p (Table 1) were used to examine the binding between Hdac3 and the promoter region of miR-296-5p.
Dual-Luciferase Reporter Gene Assay
Wild type (WT) of miR-296-5p 3'-untranslated region (3'-UTR) was synthesized and the WT target site was mutated. The synthetic target gene fragments WT and mutant (MUT) were subsequently inserted into the pmiR-RB-REPORT™ vector (Guangzhou RiboBio Co., Ltd., Guangzhou, China). At the same time, empty plasmids were transfected as control and the correct luciferin enzyme reporter plasmids WT and MUT were co-transformed with NC mimic, miR-296-5p mimic, inhibitor NC, or miR-296-5p inhibitor into HEK293T cells. Thereafter, the cells were collected and lysed after 48 h of transfection, followed by centrifugation for 3–5 min with the supernatant collected. The relative light unit value was measured using Renilla luciferase detection kit (YDJ2714, Shanghai Yuduo Biotechnology Co., Ltd., Shanghai, China) with firefly luciferase as the internal reference, followed by analysis using dual-luciferase reporting analysis system (Promega Co., Madison, WI, USA).
Flow Cytometry
After cell transfection for 48 h, cells were detached using 0.25% trypsin and centrifuged. Next, cells were resuspended using annexin-V/propidium iodide (PI) staining solution using annexin-V-fluorescein-5-isothiocyanate kit (556547, Shanghai Solja Technology Co., Ltd., Shanghai, China) and incubated for 15 min. Next, cells were gently mixed and added with 15 ml 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid buffer. The apoptotic events were evaluated using flow cytometer (Bio-Rad ZE5, Bio-Rad Laboratories, Hercules, CA, USA) at a maximum absorption wavelength of 488 nm and excitation wavelength of 525 nm. The maximum absorption and emission wavelengths of the PI-DNA were 535 and 615 nm, respectively. For apoptosis induction, apoptosis inducers (20 ng/μl TNF-α and 10 μg/μl CHX; 1,000: 1) were added to the culture medium 24 h after transfection, and the cells were collected and subjected to flow cytometry for apoptosis detection.
Animal Model Development
Male Sprague Dawley rats (8 weeks, Experimental Animal Center of the Second Hospital of Jilin University) were acclimated for 7 days with free access to food and drinking water in a clean polypropylene cage at (21 ± 2)°C under relative humidity of (50 ± 5)% in a room exposed to artificial 12/12-h light-dark cycles. Eight rats were injected with solvent (62 mg/kg) serving as the sham group. Then, intraperitoneal injection of streptozocin (STZ; 62 mg/kg; Sigma) in citric acid (0.01 M, pH = 4.5) was performed on another 16 rats to induce DM. Thereinto, eight DM rats were injected with lentivirus harboring sh-Hdac3 via tail vein on a daily basis. On the first after inducement, mice were given free access to water containing 5% glucose to avoid hypoglycemia. The glucose and insulin levels of rats were measured after 8 weeks. PBMCs were then isolated from serum of rats for following experiments.
Statistical Analysis
SPSS version 21.0 (IBM Corp., Armonk, NY, USA) was used for statistical analysis. The measurement data were expressed as mean ± standard deviation. Comparison between the two groups complying with the normal distribution and homogeneity of variance was tested using the unpaired t-test. Data comparisons among multiple groups were analyzed using one-way ANOVA, followed by post hoc analysis (Turkey test and Bonferroni correction) to evaluate the statistical significance. Kruskal-Wallis (non-parametric) test was used for data with skewed distribution and then Wilcoxon test with Bonferroni correction was used to compare the mean value in the individual group. A value of p < 0.05 was considered statistically significant.
Results
miR-296-5p Was Downregulated and Bcl-xl Was Upregulated in PBMCs of Patients With T1DM
Initially, a box plot was drawn on the miR-296-5p expression in microarray dataset GSE97123 from GEO database, followed by significant difference analysis using t-test. Results revealed significantly low expression of miR-296-5p in T1DM (Figure 1A). Subsequently, PBMCs from patients with T1DM and healthy individuals were isolated, and the expression of miR-296-5p and Bcl-xl in the samples was determined using reverse transcription quantitative polymerase chain reaction (RT-qPCR). The results showed that miR-296-5p was downregulated and Bcl-xl was upregulated in PBMCs of patients with T1DM compared to healthy individuals (Figures 1B,C). Results from Western blot analysis showed that the protein expression of BCL-XL was significantly increased in PBMCs of patients with T1DM compared to healthy individuals (Figure 1D), suggesting the low expression of miR-296-5p and high expression of Bcl-xl in PBMCs of patients with T1DM.
Figure 1.
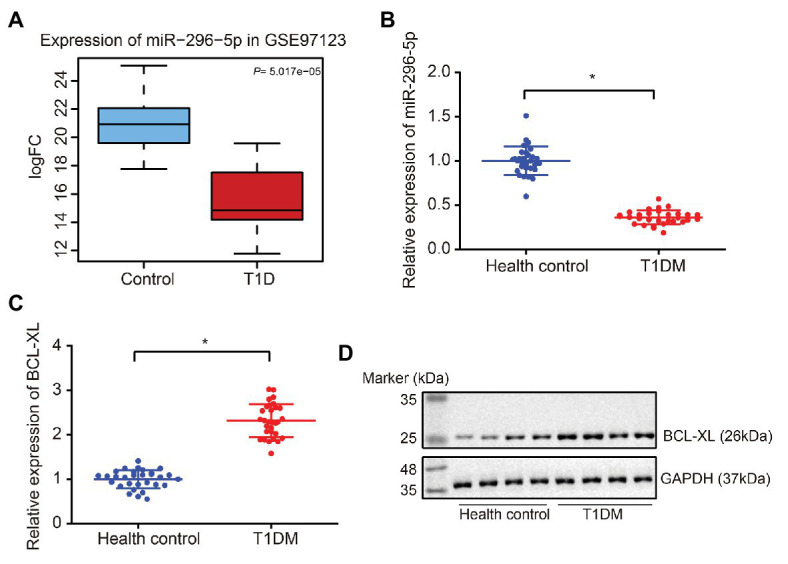
Downregulation of miR-296-5p and upregulation of BCL-XL in peripheral blood mononuclear cells (PBMCs) of patients with Type 1 diabetes mellitus (T1DM) are observed. (A) Box plot of miR-296-5p expression in normal samples (blue box on the left) and in T1DM samples (right box on the right) from microarray dataset GSE97123. (B) The expression of miR-296-5p in PBMCs of patients with T1DM and healthy individuals determined by RT-qPCR. U6 was used as an internal reference. (C) The expression of Bcl-xl in PBMCs of patients with T1DM and healthy individuals determined by RT-qPCR. Gapdh was used as an internal reference. (D) The protein expression of BCL-XL in PBMCs of patients with T1DM and healthy individuals was determined by Western blot assay. GAPDH was used as an internal reference. *p < 0.05 compared with the control. The results were measurement data and expressed as mean ± standard deviation, and data comparison between two groups was performed using unpaired sample t-test, n = 30.
miR-296-5p Targeted and Inhibited the Expression of Bcl-xl
Based on the bioinformatics data obtained from the Starbase website, we predicted that miR-296-5p may target the 3'-UTR of Bcl-xl in both humans and mice (Figure 2A). Therefore, we constructed luciferase reporter vectors and corresponding MUT vectors for the 3'-UTR of Bcl-xl (Figure 2B). The results showed that miR-296-5p directly targeted 3'-UTR of Bcl-xl and diminished the expression of Bcl-xl.
Figure 2.
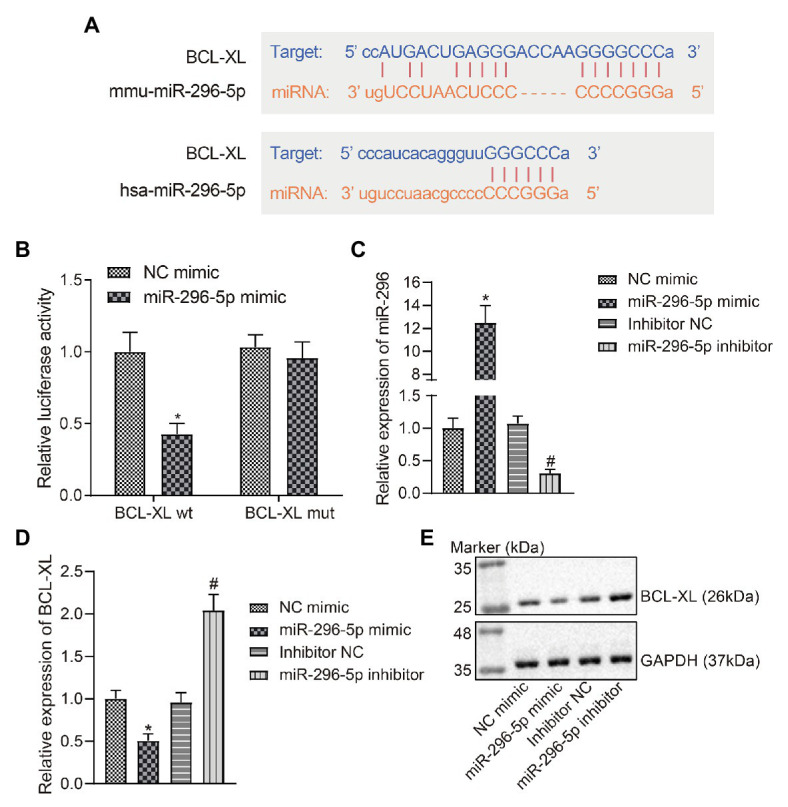
miR-296-5p directly targets Bcl-xl. (A) The binding site of hsa-miR-296-5p and Bcl-xl 3'-UTR predicted by Starbase software. (B) The luciferase activity of Bcl-xl in 293 T cells transfected with miR-296-5p mimic or NC mimic. (C) The expression of miR-296-5p in lymphocytes determined by RT-qPCR. (D) The mRNA expression of Bcl-xl in lymphocyte series determined by RT-qPCR. (E) The protein expression of BCL-XL normalized to GAPDH in lymphocyte series determined by Western blot assay. *p < 0.05 compared with cells transfected with NC mimic, #p < 0.05 compared with cells transfected with inhibitor NC. The results were measurement data and expressed as mean ± standard deviation, and data comparison between two groups was performed using an unpaired sample t-test. The cell experiment was repeated three times independently.
In order to further investigate whether the expression of miR-296-5p and Bcl-xl showed a negative correlation in vitro, TE15 mouse B lymphocytic cell line was selected and the change regarding expression of miR-296-5p and BCL-XL was determined using RT-qPCR and Western blot assay after B lymphocytic series were transfected with NC mimic and miR-296-5p mimic (Figures 2C–E). The results showed that the expression of miR-296-5p and BCL-XL in vitro was significantly correlated in a negative manner, which was consistent with the results from PBMC samples. The expression of miR-296-5p in PBMCs was inhibited using miR-296-5p inhibitor, and the results showed that BCL-XL was upregulated at mRNA and protein levels (Figures 2C–E). To conclude, these results highlighted the inverse relation between miR-296-5p and Bcl-xl.
miR-296-5p Promoted Lymphocyte Apoptosis by Targeting Bcl-xl
The expression of proteins related to apoptosis in PBMCs of patients with T1DM was tested using Western blot analysis (Figure 3A). The results showed that the expression of pro-apoptosis proteins Bad and fasL was significantly decreased, and the expression of anti-apoptosis proteins BCL-XL and BCL-2 was significantly increased in PBMCs of patients with T1DM. The apoptosis of PBMCs in patients with T1DM was evaluated by flow cytometry (Figure 3B), and the results showed that the percentage of apoptotic cells was remarkably decreased in PBMCs of patients with T1DM compared with healthy individuals.
Figure 3.

miR-296-5p overexpression enhances lymphocyte apoptosis by targeting Bcl-xl. (A) The expression of pro-apoptotic and anti-apoptotic proteins normalized to GAPDH in PBMCs of patients with T1DM determined by Western blot assay (n = 30). (B) Quantitative analysis of apoptosis of PBMCs in patients with T1DM compared with healthy individuals analyzed using flow cytometry. (C) The protein expression of BCL-XL and C-caspase3 normalized to GAPDH determined by Western blot assay after lymphocytes transfected with vector and pCMV-Bcl-xl. (D) Quantitative analysis of apoptosis of lymphocytes transfected with vector and pCMV-Bcl-xl analyzed using flow cytometry. (E) The protein expression of BCL-XL and C-caspase3 normalized to GAPDH determined by Western blot assay after lymphocytes transfected with miR-296-5p inhibitor or inhibitor NC. (F) Quantitative analysis of apoptosis of lymphocytes after transfected with miR-296-5p inhibitor or inhibitor NC evaluated by flow cytometry. (G) Rescue experiments on the protein expression of BCL-XL and C-caspase3 normalized to GAPDH determined by Western blot assay. (H) Rescue experiments on the apoptosis of lymphocytes evaluated by flow cytometry. *p < 0.05 compared with cells transfected with control, vector, or inhibitor NC, #p < 0.05 compared with cells transfected with miR-296 inhibitor + si-NC. The results were measurement data and expressed as mean ± standard deviation. Data comparison between two groups was performed using unpaired sample t-test, and data comparisons among multiple groups were performed using one-way analysis of variance. The cell experiment was repeated three times independently.
Next, the effects of Bcl-xl on apoptotic-related proteins and apoptosis levels in lymphocytes were evaluated (Figures 3C,D), and the results showed that the upregulation of Bcl-xl could inhibit apoptosis induced by TNF-α and CHX. Subsequently, the effects of miR-296-5p on apoptotic-related proteins and apoptosis levels in lymphocytes were determined using Western blot and flow cytometry (Figures 3E,F), and effect of downregulated miR-296-5p on lymphocyte apoptosis was consistent with the effect of upregulated Bcl-xl. Therefore, we speculated that miR-296-5p could target and diminish the expression of Bcl-xl to regulate lymphocyte apoptosis. To verify this hypothesis, we performed apoptosis-related rescue experiments (Figures 3G,H), and Bcl-xl was able to rescue the miR-296-5p-induced apoptosis. Taken together, these results suggested that miR-296-5p could promote lymphocyte apoptosis by targeting Bcl-xl.
Hdac3 Downregulated miR-296-5p by Binding to Its Promoter Region
It has been reported that dysfunctional histone and transfer factors acetylation is correlated with pathogenesis of diabetes (Gray and De Meyts, 2005). HDACs, including 18 genes, can mediate acetylation of histone (Blixt et al., 2017). To our knowledge, there are seven studies reporting the correlation between Hdac3 and T1DM, ranking the first among the 18 genes. Meanwhile, Hdac3 contributes to the occurrence of T1DM (Meier and Wagner, 2014). Results from the gene expression dataset have revealed that Hdac3 is negatively correlated with miR-296-5p and can inhibit the occurrence of diabetes (Wang et al., 2016; Liu et al., 2019). Therefore, we inferred that Hdac3 might be the most relevant Hdac with T1DM and there may be an interaction between Hdac3 and miR-296-5p. Then, Hdac3 was overexpressed or knocked down, followed by quantification of resultant expression patterns. Results showed that HDAC3 expression was elevated in response to pCMV- Hdac3 yet diminished in response to siHdac3 (Figures 4A,B). Here, we found that the expression of HDAC3 was significantly increased in PBMCs of patients with T1DM than corresponding controls (Figures 4C,D). For verification purpose, the expression of miR-296-5p was evaluated when Hdac3 was upregulated and downregulated, respectively. The results indicated that there was a significant negative correlation between Hdac3 and miR-296-5p expression (Figure 4E).
Figure 4.

HDAC3 restricts the expression of miR-296-5p by binding to with its promoter region. (A) The expression of Hdac3 determined by RT-qPCR. (B) The expression of HDAC3 determined by Western blot assay. (C) The expression of Hdac3 in PBMCs of T1DM patients by RT-qPCR. (D) The expression of HDAC3 in PBMCs of T1DM patients by Western blot assay. (E) The expression of miR-296-5p determined by RT-qPCR when HDAC3 was upregulated and downregulated, respectively. (F) Inhibition of miR-296-5p promoter activity by Hdac3 evaluated using ChIP-qPCR assay. (G) The expression of Bcl-xl determined by RT-qPCR when Hdac3 was upregulated or downregulated, respectively. (H) The expression of BCL-XL determined by Western blot assay when HDAC3 was upregulated or downregulated, respectively. (I) Rescue experiments showed that HDAC3 upregulated the expression of BCL-XL by targeting miR-296-5p. *p < 0.05 compared with cells transfected with vector, #p < 0.05 compared with cells transfected with si-NC. The results were measurement data and expressed as mean ± standard deviation. Data comparison between two groups was performed using unpaired sample t-test, and data comparisons among multiple groups were performed using one-way analysis of variance. The cell experiment was repeated three times independently.
In order to further investigate the effect of HDAC3 on the promoter activity of miR-296-5p, the ChIP-qPCR experiment was performed and the results showed that the promoter activity of miR-296-5p was significantly deteriorated when Hdac3 was upregulated (Figure 4F). Based on the aforementioned study of miR-296-5p and Bcl-xl, the relationship between Hdac3 and Bcl-xl was further explored, and the results showed that the expression of Hdac3 was positively correlated with the expression of Bcl-xl (Figures 4G,H). The results also showed that Hdac3 could regulate the expression of Bcl-xl by targeting miR-296-5p (Figure 4I). These results indicated that Hdac3 could inhibit the expression of miR-296-5p by binding to its promoter region to reinforce the expression of Bcl-xl.
HDAC3 Regulated BCL-XL by Mediating miR-296-5p to Suppress Apoptosis of Lymphocytes
The expression of apoptosis-related proteins was determined when HDAC3 was upregulated (Figure 5A), and the effect on apoptosis was evaluated by flow cytometry. The results showed that the effect of upregulated HDAC3 on apoptosis was consistent with the effect of downregulated miR-296-5p on apoptosis (Figure 5B). Results also showed that BCL-XL expression was restored when HDAC3 was upregulated and BCL-XL downregulated at the same time (Figure 5C), and the results from flow cytometry showed that the protein levels of apoptosis in the presence of both upregulated HDAC3 and downregulated BCL-XL were restored (Figure 5D). At the same time, the expression of BCL-XL was restored when both HDAC3 and miR-296-5p were upregulated (Figure 5E), and protein levels of apoptosis in response to upregulated HDAC3 and downregulated BCL-XL were restored (Figure 5F). These results consistently showed that HDAC3 could regulate BCL-XL by mediating the expression of miR-296-5p to inhibit lymphocyte apoptosis.
Figure 5.
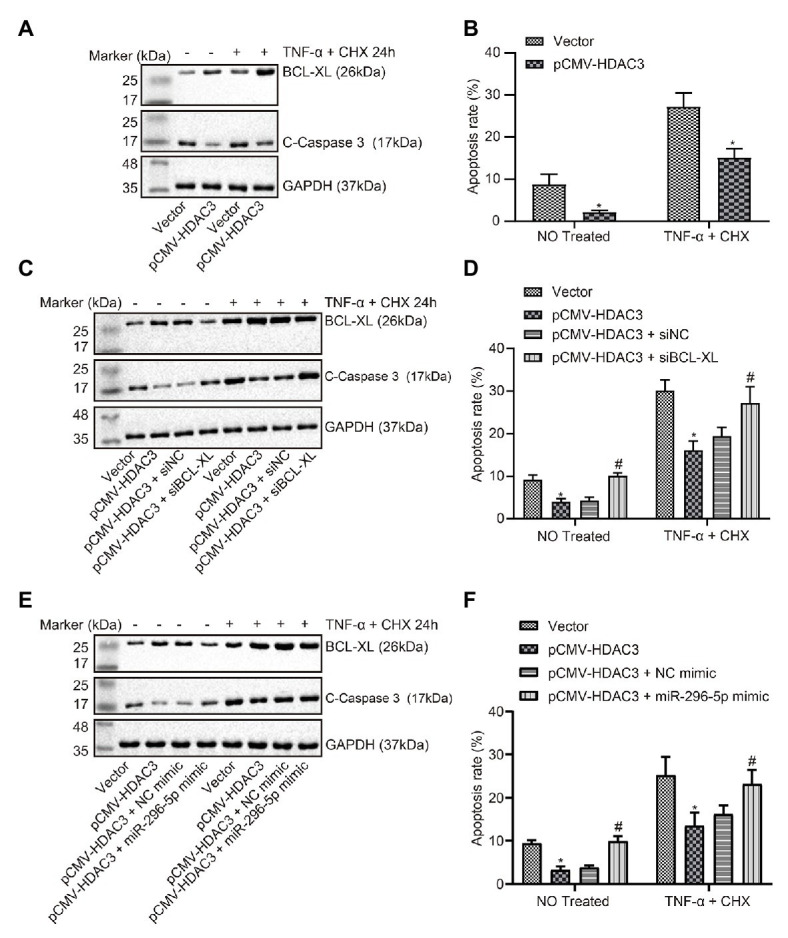
HDAC3-mediated miR-296-5p attenuates the apoptosis of lymphocytes via the regulation of BCL-XL. (A) The expression of BCL-XL and C-caspase3 normalized to GAPDH determined by Western blot assay when HDAC3 was upregulated in lymphocytes. (B) The effect on apoptosis evaluated by flow cytometry when Hdac3 was upregulated in lymphocytes. (C) The effect of BCL-XL on the expression of C-caspase3 normalized to GAPDH after HDAC3 was downregulated in lymphocytes determined by Western blot assay. (D) The effect of Bcl-xl on apoptosis after Hdac3 was upregulated in lymphocytes evaluated by flow cytometry. (E) The effect of miR-296-5p on the expression of C-caspase3 normalized to GAPDH after HDAC3 was upregulated in lymphocytes tested by Western blot assay. (F) The effect of miR-296-5p on apoptosis after Hdac3 was upregulated in lymphocytes tested by flow cytometry. *p < 0.05 compared with cells transfected with vector, #p < 0.05 compared with cells transfected with pCMV-Hdac3 + si-NC or pCMV-Hdac3 + NC mimic. The results were measurement data and expressed as mean ± standard deviation. Data comparison between two groups was performed using unpaired sample t-test, and data comparisons among multiple groups were performed using one-way analysis of variance. The cell experiment was repeated three times independently.
Hdac3 Knockdown Curbed the Apoptosis of PBMCs in Rats With DM
Glucose and insulin levels of rats in each group were measured (Figures 6A,B) and miR-296-5p expression in PBMCs of rats was determined by RT-qPCR (Figure 6C). Results revealed significantly higher glucose level, lower insulin level, and diminished miR-296-5p expression in rats with DM while further addition of sh-Hdac3 reversed the results. PBMC apoptosis was then evaluated by flow cytometric analysis (Figure 6D) and Western blot analysis (Figure 6E). It was found that PBMC apoptosis was potentiated in rats with DM accompanied by upregulation of HDAC3, BCL-XL, and C-caspase-3, all of which were reversed by delivery of sh-Hdac3. Taken together, shRNA-mediated silencing of HDAC3 significantly alleviated DM-induced PBMC apoptosis in rats.
Figure 6.
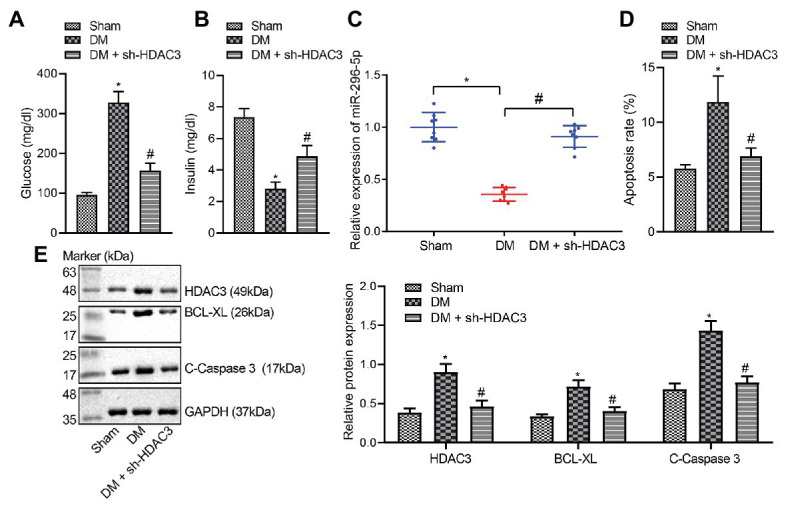
shRNA-mediated silencing of HDAC3 alleviates DM-induced PBMC apoptosis in vivo. (A) Glucose level of rats. (B) Insulin levels of rats. (C) miR-296-5p expression in PBMCs of rats determined by RT-qPCR. (D) Quantitative analysis of PBMC apoptosis identified by flow cytometric analysis. (E) The expression of HDAC3, BCL-XL, and C-caspase-3 in PBMCs normalized to GAPDH determined by Western blot analysis. *p < 0.05 compared with sham-operated rats, #p < 0.05 compared with rats with DM. Data comparisons among multiple groups were performed using one-way analysis of variance. n = 8.
Discussion
DM has been revealed to be closely related to HDACs. Hdac3 inhibitors have been proposed as a potential therapeutic candidate in managing DM (Meier and Wagner, 2014; Khan and Jena, 2015). The present study was designed to explore the effect of Hdac3 on the expression of miR-296-5p and Bcl-xl, as well as to identify its potential role in T1DM. This study provided evidence demonstrating that Hdac3 could inhibit the apoptosis of lymphocytes by restricting miR-296-5p to upregulate Bcl-xl, thereby promoting the occurrence of T1DM.
Initially, our results demonstrated that miR-296-5p was downregulated and BCL-XL was upregulated in PBMCs of patients with T1DM. The regulation of gene expression in PBMCs has been widely indicated to offer new and better treatment modalities for patients with T1DM (Li et al., 2019). Differentially expressed miRNAs in PBMCs have been deciphered to function as promising biomarkers of T1DM, providing new insights into the understanding and the underlying molecular mechanism (Takahashi et al., 2014). Consistently, miR-296-5p expression has been reported to show a significant reduction in DM tissues in comparison to corresponding normal tissues, while healing of diabetic wounds has been shown to be facilitated by the overexpression of miR-296-5p by targeting sodium-glucose transporter 2 (Liu et al., 2019). A similar expression profile of multiple miRNAs has also been identified in patients suffering from T1DM, including miR-150, miR-146a, and miR-424, with great clinical application potential (Wang et al., 2018). In addition, a previous report has documented that Bcl-xl is highly expressed in PBMCs of patients with T1DM, which leads to the development of T1DM (de Oliveira et al., 2012). Moreover, according to experimental data of the present study, it was verified that miR-296-5p could target Bcl-xl and negatively regulate the expression of Bcl-xl, which resulted in enhanced apoptosis of lymphocytes. B lymphocytes have been suggested to participate in β-cell destruction in patients with T1DM (Deng et al., 2016). Notably, targeting dysfunctional β-cell signaling has been proposed as a novel target for the management of T1DM (Fenske and Kimple, 2018). Additionally, lymphocytes have also been demonstrated to be increased in patients with T1DM in association with poor glycemic control related to the occurrence of cardiovascular disease and coronary syndromes (Giubilato et al., 2011), supporting the validation of our findings.
Subsequently, diminished levels of pro-apoptotic proteins, Bad and fasL, were detected in PBMCs collected from patients with T1DM accompanied by elevated expression of anti-apoptotic proteins, BCL-XL and BCL-2. In agreement with our results, previous work has reported that the upregulation of Bad and fasL along with downregulation of BCL-XL has been detected in PBMCs of patients with T1DM, serving as potential biomarkers of clinical remission (de Oliveira et al., 2012). Furthermore, vector and pCMV-Bcl-xl were delivered into lymphocytes to unravel the involvement of Hdac3 in T1DM. It was found that Hdac3 downregulated the expression of miR-296-5p by binding to the promoter region of miR-296-5p. Many studies showed that as HDACs function as part of multi-protein complexes that deacetylate histone tails and modify chromatin structure and gene repression, HDAC inhibitors decreased miRNA promoter methylation (Mazzu et al., 2019), and we thus speculated that Hdac3 also affects the acetylation level of the promoter region of miR-296-5p and to reduce its expression. Additionally, HDAC3 exerted an inhibitory effect on lymphocyte apoptosis by upregulating BCL-XL expression via miR-296-5p. Meanwhile, downregulation of BCL-XL or overexpression of miR-296-5p was shown to override the effects of overexpressed HDAC on protein expression of apoptosis-related factors and flow cytometric analysis of lymphocyte apoptosis. HDAC enzyme plays a regulatory role in chromatin structure and metabolic enzyme acetylation in mitochondria and cytosol, shedding light on the intrinsic role of HDAC inhibitors as therapeutic strategies for DM (Christensen et al., 2011; Makkar et al., 2019). Class I HDAC inhibition has been reported to increase insulin secretion and prevent pancreatic beta cell from apoptosis (Peng et al., 2019). Concordantly, the downregulation of Hdac3 has been considered as a strategy for developing new ways for the treatment of diabetes (Meier and Wagner, 2014). Previous data have shown that inhibition of Hdac3 prevents cytokine-induced beta-cell apoptosis, which is important to the etiology of T1DM (Chou et al., 2012). In addition, Hdac3 can integrate microbiota-derived signals to control intestinal homeostasis in intestinal epithelial cells (IECs), where disruption of Hdac3 causes weight loss and improves metabolic profile, suppressing the diet-induced obesity (Whitt et al., 2018). Likewise, inhibition of HDACs has been highlighted to improve myocardial function in a model of diabetic mice by increasing GLUT1 acetylation and p38 phosphorylation (Chen et al., 2015). Moreover, suppression of Hdac3 has been elucidated to prevent diabetic cardiomyopathy in mouse models through epigenetic regulation of the DUSP5-ERK1/2 pathway (Xu et al., 2017). Therefore, it could be concluded from our collective experimental data both in vitro and in vivo, as well as the previous reports, that HDAC3 could harbor the potential to impede miR-296-5p expression and lead to the aggravation of T1DM.
Conclusion
In conclusion, the present study demonstrates that Hdac3 could upregulate Bcl-xl by repressing miR-296-5p expression, leading to inhibited apoptosis of lymphocytes and subsequently promoted occurrence of T1DM (Figure 7). This study provides new insights into mechanisms underlying T1DM and novel potential therapeutic targets for patients with T1DM. Despite these promising results, the molecular mechanisms underlying apoptosis of PBMCs in the context of T1DM are still not well-characterized. Therefore, more in-depth investigations are required to reveal the molecular mechanisms underlying T1DM development, as a pre-requisite for better application in a clinical setting in the future.
Figure 7.
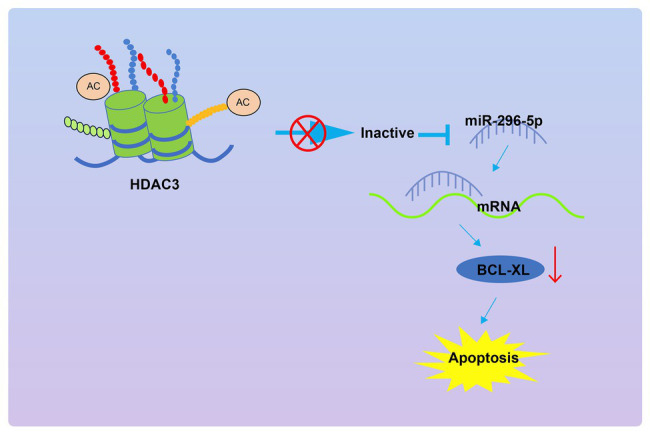
The graphical summary of the function and mechanism of Hdac3 on the apoptosis of PBMCs in T1DM via the miR-296-5p/Bcl-xl axis.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
All procedures concerning human samples were approved by the ethics committee of the Second Hospital of Jilin University. The written informed consents were obtained from all participants or their family members.
Author Contributions
QH and GC designed the study. YY and HX collated the data, carried out data analyses, and produced the initial draft of the manuscript. QH and JT contributed to drafting the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to give our sincere appreciation to the colleagues for their helpful comments on this article.
References
- Anuradha R., Saraswati M., Kumar K. G., Rani S. H. (2014). Apoptosis of beta cells in diabetes mellitus. DNA Cell Biol. 33, 743–748. doi: 10.1089/dna.2014.2352, PMID: [DOI] [PubMed] [Google Scholar]
- Ayuk S. M., Abrahamse H., Houreld N. N. (2016). The role of photobiomodulation on gene expression of cell adhesion molecules in diabetic wounded fibroblasts in vitro. J. Photochem. Photobiol. B 161, 368–374. doi: 10.1016/j.jphotobiol.2016.05.027, PMID: [DOI] [PubMed] [Google Scholar]
- Bjornstad P., Schafer M., Truong U., Cree-Green M., Pyle L., Baumgartner A., et al. (2018). Metformin improves insulin sensitivity and vascular health in youth with type 1 diabetes mellitus. Circulation 138, 2895–2907. doi: 10.1161/CIRCULATIONAHA.118.035525, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blixt N. C., Faulkner B. K., Astleford K., Lelich R., Schering J., Spencer E., et al. (2017). Class II and IV HDACs function as inhibitors of osteoclast differentiation. PLoS One 12:e0185441. doi: 10.1371/journal.pone.0185441, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Du J., Zhao Y. T., Zhang L., Lv G., Zhuang S., et al. (2015). Histone deacetylase (HDAC) inhibition improves myocardial function and prevents cardiac remodeling in diabetic mice. Cardiovasc. Diabetol. 14:99. doi: 10.1186/s12933-015-0262-8, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou D. H., Holson E. B., Wagner F. F., Tang A. J., Maglathlin R. L., Lewis T. A., et al. (2012). Inhibition of histone deacetylase 3 protects beta cells from cytokine-induced apoptosis. Chem. Biol. 19, 669–673. doi: 10.1016/j.chembiol.2012.05.010, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen D. P., Dahllof M., Lundh M., Rasmussen D. N., Nielsen M. D., Billestrup N., et al. (2011). Histone deacetylase (HDAC) inhibition as a novel treatment for diabetes mellitus. Mol. Med. 17, 378–390. doi: 10.2119/molmed.2011.00021, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig M. E., Kim K. W., Isaacs S. R., Penno M. A., Hamilton-Williams E. E., Couper J. J., et al. (2019). Early-life factors contributing to type 1 diabetes. Diabetologia 62, 1823–1834. doi: 10.1007/s00125-019-4942-x, PMID: [DOI] [PubMed] [Google Scholar]
- Dafoulas G. E., Toulis K. A., McCorry D., Kumarendran B., Thomas G. N., Willis B. H., et al. (2017). Type 1 diabetes mellitus and risk of incident epilepsy: a population-based, open-cohort study. Diabetologia 60, 258–261. doi: 10.1007/s00125-016-4142-x, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos-Salas M., Montgomery M. K., Reehorst C. M., Nightingale R., Ng I., Anderton H., et al. (2019). Deletion of intestinal Hdac3 remodels the lipidome of enterocytes and protects mice from diet-induced obesity. Nat. Commun. 10:5291. doi: 10.1038/s41467-019-13180-8, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng C., Xiang Y., Tan T., Ren Z., Cao C., Huang G., et al. (2016). Altered peripheral B-lymphocyte subsets in type 1 diabetes and latent autoimmune diabetes in adults. Diabetes Care 39, 434–440. doi: 10.2337/dc15-1765, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira G. L., Malmegrim K. C., Ferreira A. F., Tognon R., Kashima S., Couri C. E., et al. (2012). Up-regulation of fas and fasL pro-apoptotic genes expression in type 1 diabetes patients after autologous haematopoietic stem cell transplantation. Clin. Exp. Immunol. 168, 291–302. doi: 10.1111/j.1365-2249.2012.04583.x, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenske R. J., Kimple M. E. (2018). Targeting dysfunctional beta-cell signaling for the potential treatment of type 1 diabetes mellitus. Exp. Biol. Med. 243, 586–591. doi: 10.1177/1535370218761662, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giubilato S., Liuzzo G., Brugaletta S., Pitocco D., Graziani F., Smaldone C., et al. (2011). Expansion of CD4+CD28null T-lymphocytes in diabetic patients: exploring new pathogenetic mechanisms of increased cardiovascular risk in diabetes mellitus. Eur. Heart J. 32, 1214–1226. doi: 10.1093/eurheartj/ehq499, PMID: [DOI] [PubMed] [Google Scholar]
- Gray S. G., De Meyts P. (2005). Role of histone and transcription factor acetylation in diabetes pathogenesis. Diabetes Metab. Res. Rev. 21, 416–433. doi: 10.1002/dmrr.559, PMID: [DOI] [PubMed] [Google Scholar]
- Khan S., Jena G. (2015). The role of butyrate, a histone deacetylase inhibitor in diabetes mellitus: experimental evidence for therapeutic intervention. Epigenomics 7, 669–680. doi: 10.2217/epi.15.20, PMID: [DOI] [PubMed] [Google Scholar]
- Li C., Chen L., Song M., Fang Z., Zhang L., Coffie J. W., et al. (2020a). Ferulic acid protects cardiomyocytes from TNF-alpha/cycloheximide-induced apoptosis by regulating autophagy. Arch. Pharm. Res. 43, 863–874. doi: 10.1007/s12272-020-01252-z, PMID: [DOI] [PubMed] [Google Scholar]
- Li L., Pan Z., Yang X. (2019). Identification of dynamic molecular networks in peripheral blood mononuclear cells in type 1 diabetes mellitus. Diabetes Metab. Syndr. Obes. 12, 969–982. doi: 10.2147/DMSO.S207021, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., Wang D., He J., Chen L., Li H. (2020b). Bcl-XL: a multifunctional anti-apoptotic protein. Pharmacol. Res. 151:104547. doi: 10.1016/j.phrs.2019.104547 [DOI] [PubMed] [Google Scholar]
- Liu X., Wang Y., Zhang X., Zhang X., Guo J., Zhou J., et al. (2019). MicroRNA-296-5p promotes healing of diabetic wound by targeting sodium-glucose transporter 2 (SGLT2). Diabetes Metab. Res. Rev. 35:e3104. doi: 10.1002/dmrr.3104, PMID: [DOI] [PubMed] [Google Scholar]
- Luciani D. S., White S. A., Widenmaier S. B., Saran V. V., Taghizadeh F., Hu X., et al. (2013). Bcl-2 and Bcl-xL suppress glucose signaling in pancreatic beta-cells. Diabetes 62, 170–182. doi: 10.2337/db11-1464, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makkar R., Behl T., Arora S. (2019). Role of HDAC inhibitors in diabetes mellitus. Curr. Res. Transl. Med. 68, 45–50. doi: 10.1016/j.retram.2019.08.001, PMID: [DOI] [PubMed] [Google Scholar]
- Mazzu Y. Z., Yoshikawa Y., Nandakumar S., Chakraborty G., Armenia J., Jehane L. E., et al. (2019). Methylation-associated miR-193b silencing activates master drivers of aggressive prostate cancer. Mol. Oncol. 13, 1944–1958. doi: 10.1002/1878-0261.12536, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier B. C., Wagner B. K. (2014). Inhibition of HDAC3 as a strategy for developing novel diabetes therapeutics. Epigenomics 6, 209–214. doi: 10.2217/epi.14.11, PMID: [DOI] [PubMed] [Google Scholar]
- Merarchi M., Sethi G., Shanmugam M. K., Fan L., Arfuso F., Ahn K. S. (2019). Role of natural products in modulating histone deacetylases in cancer. Molecules 24:1047. doi: 10.3390/molecules24061047, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X., Liao G., Sun P., Yu Z., Chen J. (2019). An overview of HDAC inhibitors and their synthetic routes. Curr. Top. Med. Chem. 19, 1005–1040. doi: 10.2174/1568026619666190227221507, PMID: [DOI] [PubMed] [Google Scholar]
- Takahashi P., Xavier D. J., Evangelista A. F., Manoel-Caetano F. S., Macedo C., Collares C. V., et al. (2014). MicroRNA expression profiling and functional annotation analysis of their targets in patients with type 1 diabetes mellitus. Gene 539, 213–223. doi: 10.1016/j.gene.2014.01.075, PMID: [DOI] [PubMed] [Google Scholar]
- Vatanen T., Franzosa E. A., Schwager R., Tripathi S., Arthur T. D., Vehik K., et al. (2018). The human gut microbiome in early-onset type 1 diabetes from the TEDDY study. Nature 562, 589–594. doi: 10.1038/s41586-018-0620-2, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Frank D. B., Morley M. P., Zhou S., Wang X., Lu M. M., et al. (2016). HDAC3-dependent epigenetic pathway controls lung alveolar epithelial cell remodeling and spreading via miR-17-92 and TGF-beta signaling regulation. Dev. Cell 36, 303–315. doi: 10.1016/j.devcel.2015.12.031, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G., Gu Y., Xu N., Zhang M., Yang T. (2018). Decreased expression of miR-150, miR146a and miR424 in type 1 diabetic patients: association with ongoing islet autoimmunity. Biochem. Biophys. Res. Commun. 498, 382–387. doi: 10.1016/j.bbrc.2017.06.196, PMID: [DOI] [PubMed] [Google Scholar]
- Whitt J., Woo V., Lee P., Moncivaiz J., Haberman Y., Denson L., et al. (2018). Disruption of epithelial HDAC3 in intestine prevents diet-induced obesity in mice. Gastroenterology 155, 501–513. doi: 10.1053/j.gastro.2018.04.017, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z., Tong Q., Zhang Z., Wang S., Zheng Y., Liu Q., et al. (2017). Inhibition of HDAC3 prevents diabetic cardiomyopathy in OVE26 mice via epigenetic regulation of DUSP5-ERK1/2 pathway. Clin. Sci. 131, 1841–1857. doi: 10.1042/CS20170064, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Chen Y., Jiang Q., Song W., Zhang L. (2019b). Therapeutic potential of selective histone deacetylase 3 inhibition. Eur. J. Med. Chem. 162, 534–542. doi: 10.1016/j.ejmech.2018.10.072, PMID: [DOI] [PubMed] [Google Scholar]
- Zhang J., Niu H., Zhao Z. J., Fu X., Wang Y., Zhang X., et al. (2019a). CRISPR/Cas9 knockout of Bak mediates Bax translocation to mitochondria in response to TNFalpha/CHX-induced apoptosis. Biomed. Res. Int. 2019:9071297. doi: 10.1155/2019/9071297, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Xu Z., Gu J., Jiang S., Liu Q., Zheng Y., et al. (2018). HDAC3 inhibition in diabetic mice may activate Nrf2 preventing diabetes-induced liver damage and FGF21 synthesis and secretion leading to aortic protection. Am. J. Physiol. Endocrinol. Metab. 315, E150–E162. doi: 10.1152/ajpendo.00465.2017, PMID: [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.