

Abstract
Triatoma rubrofasciata (T. rubrofasciata), one kind of triatomine insects, is the vector of Trypanosoma cruzi (T. cruzi), which lead to American trypanosomiasis. Although the gut microbiome may play an essential role in the development and susceptibility of triatomine, there is limited research on the gut microbiota of T. rubrofasciata. To elucidate the effect of the vector’s developmental stages and environmental conditions on the gut microbiome, we employed 16S rRNA gene sequencing to profile the gut bacterial community diversity and composition of T. rubrofasciata. Significant shifts were observed in the overall gut microbe diversity and composition across the development of T. rubrofasciata and specific bacteria were detected in different stages. Serratia and Burkholderia-Caballeronia-Paraburkholderia were dominant in the 1st nymphal stage, while the abundance of Staphylococcus was low in the 1st nymphal stage. Oceanicaulis were undetectable in the adult stage and Odoribacter peaked in the 2nd nymphal stage. Moreover, Staphylococcus was correlated negatively with Serratia. Likewise, the total gut microbiota diversity and composition of T. rubrofasciata differentiated significantly by environmental conditions. The ingestion of a bloodmeal increased alpha diversity of gut bacterial communities, and Staphylococcus was more abundant in laboratory-reared bugs whereas Enterococcus enriched in wild-caught bugs. Furthermore, Pantoea was negatively correlated with Staphylococcus, and positively related to Bacillus only. The phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) algorithm showed obvious metagenomic functional differences by environmental conditions, and Chagas disease relevant pathway was enriched in wild-caught T. rubrofasciata.
Keywords: Triatoma rubrofasciata, gut microbiota, developmental stages, environmental conditions, 16S rRNA gene sequencing
Introduction
American trypanosomiasis, also named Chagas disease, is a vector-borne disease for which the causative agent is the protozoan parasite Trypanosoma cruzi (T. cruzi), which is mainly transmitted by triatomines. Chagas disease is also one of the chronic, systemic and neglected tropical diseases (NTDs); with 8 million people infected worldwide, mostly in Latin America, at least 70 million people are at risk of contagion (Gourbière et al., 2012; Orantes et al., 2018; WHO, 2020). Chagas disease had been commonly recorded in Latin American and Caribbean (LAC) region in the past decades, however, it is being increasingly reported as an emerging infectious disease in North America, Europe and the Indo-Pacific region because of the frequent international migration of population and global invasion of the widespread kissing-bug (Hotez et al., 2012; Dujardin et al., 2015). Furthermore, Chagas disease ranks near the top in terms of annual deaths and disability-adjusted life years (DALYs) lost among all NTDs in the Americas because of its highly debilitating chronic course with alteration of the cardiovascular, digestive and nervous systems (Hotez et al., 2012; Sassera et al., 2013). Since a vaccine or effective treatment for Chagas disease is still unavailable, adequate prevention and control of the disease may be achieved by control of the vectors.
Currently, triatomine insects are composed of 151 species that are grouped into 17 genera and organized into 5 tribes, that is, Aberproseniini, Bolboderini, Cavernicolini, Rhodiniini and Triatomini (Galvão and Justi, 2015; Vieira et al., 2018). Most triatomine species occur in the Americas, while six species belonging to the genus Linshcosteus are found in India; moreover, the species of the genus Triatoma are distributed in Africa, the Middle East, South-East Asia and the Western Pacific (Vieira et al., 2018).
Triatoma is one of the most diverse genera, including the species Triatoma rubrofasciata (T. rubrofasciata), which is widespread throughout southern China, such as in Guangdong, Guangxi, Hainan and Taiwan (Ibarra-Cerdeña et al., 2009; Huang et al., 2018). T. rubrofasciata is a domiciliated species with urban characteristics and exists in close association with rodents that act as reservoirs of T. cruzi. It is naturally infected by T. cruzi as well as Trypanosoma conorrhini, which is pathogenic to Rattus rattus but not to humans (Braga et al., 1998; Cortéz and Gonçalves, 1998). Due to the wide distribution of T. rubrofasciata in South China, which suggests the possibility of introduction and transmission of Chagas disease in China, further study of this vector is urgently required.
Triatomines are hemipteran (true) bugs and obligate hematophagous insects with five nymphal stages before the egg reaches adulthood (Gourbière et al., 2012; Oliveira et al., 2018; Vieira et al., 2018). Because both nymphs and adults feed on the blood of vertebrates, so they may become infected and are likely to transmit T. cruzi after they ingest blood from infected mammalian hosts. Once the parasites arrive at the gut of the triatomine, they multiply and are able to infect a new host during a subsequent blood meal; they also come in contact with the local gut microbiota and avoid detrimental interactions with the microbiome to survive and develop inside the bug gut (Azambuja et al., 2005; Gourbière et al., 2012; Oliveira et al., 2018).
Various studies of triatomine gut microbes have been conducted, and to date, more than 57 species of cultivable bacteria have been identified (Lima et al., 2018). Previous studies have shown that the most common bacteria in eight species of wild-caught and laboratory-reared triatomines were Gram-negative rods (Azambuja et al., 2005). Interestingly, bacteria of the genus Rhodococcus in the triatomine gut are believed to play an important role in the metabolism of the vector, such as by participating in the synthesis of group B vitamins or by being digested by the bugs directly to provide missing nutrients (Sassera et al., 2013). Moreover, the most attractive aspect is the host-symbiont relationship between triatomines and Rhodococcus; since Rhodococcus bacteria can be easily cultured and genetically modified to harm the pathogen in vector gut, they are probably suitable tools for the control of trypanosomiasis (Sassera et al., 2013). Another study found that the SM365 and RPH strains of Serratia marcescens, which is a common symbiont of various triatomine species, exhibit trypanolytic activity toward several T. cruzi strains (Azambuja et al., 2004).
However, previous studies of the triatomine gut microbiome have been based mostly on the isolation and identification of cultivable bacteria, did not reflect the relative abundances of these species under natural conditions, and inevitably missed many taxa that could not be cultivated (da Mota et al., 2012; Oliveira et al., 2018). On the other hand, cultivation-independent methods, such as high-throughput DNA sequencing, allow fast and accurate description of bacterial diversity, especially for uncultivable microbes, which are impossible to detect with cultivation-dependent methods (da Mota et al., 2012; Oliveira et al., 2018).
To date, 16S ribosomal RNA (rRNA) gene sequencing has been applied in the characterization of the gut microbiota in some triatomine species (Gumiel et al., 2015; Díaz et al., 2016; Dumonteil et al., 2018; Oliveira et al., 2018; Orantes et al., 2018; Rodríguez-Ruano et al., 2018); to the best of our knowledge, the gut microbiome of T. rubrofasciata has not yet been investigated. Hence, in this study, we applied this technology to determine and compare the relative abundance of both cultivable and uncultivable bacteria in the gut of T. rubrofasciata from 1st instar nymphs to adults for the first time and tried to elucidate the potential interactions between the vector and colonizing bacteria during vector development. In addition, we used 16S rRNA gene sequencing to examine the gut flora of wild-caught and laboratory-reared T. rubrofasciata for the first time to explore and compare the diversity and composition of the gut microbes of triatomines and reveal the correlations between environmental conditions and specific gut microbiota profiles.
Materials and Methods
Bug Collection and DNA Extraction
The four wild adult triatomines used in this study were captured in Huping village (E 117°38′, N 24°39′), Fengshan town, Hua’an county, Zhangzhou city, Fujian province, China, by technicians from the Fujian Center for Disease Control and Prevention, China, in August 2017. A set of 45 randomly selected laboratory-reared triatomines at each developmental stage (1st to 5th instar nymphs plus male and female adults) were reared from eggs of local T. rubrofasciata caught in Huping village between July and August 2017.
DNA extraction from the entire gut of individual bugs was performed by using a QIAamp DNA Mini Kit (cat. no. 51306, QIAGEN, Hilden, Germany) according to the manufacturer’s protocols. The quality and quantity of the DNA were determined by agarose gel electrophoresis and a Nanodrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA), respectively. Then, the extracted DNA was partly diluted to a concentration of 1 ng/μL and stored at -20°C until use.
Detection of T. rubrofasciata Feeding Sources and T. cruzi Infection
To determine the feeding sources of wild-caught and laboratory-reared adult triatomines, we used the primer set BM1/BM2 ( Table 1 ) synthesized by Sangon Biotech (Shanghai, China) to amplify the mitochondrial cytochrome b (cytb) gene of all vertebrates. Polymerase chain reactions (PCRs) were performed in a total volume of 50 μL containing approximately 30 ng of DNA template, 1.1 × Golden Star T6 Super PCR Mix (Tsingke, China) and 0.4 μmol/L each primer. Sterile water was used as a negative control to avoid contamination during PCR. Amplifications were performed in a Bio-Rad PCR C1000 Touch instrument (Bio-Rad, USA), with an initial denaturation at 98°C for 2 min, followed by 35 cycles of denaturation at 98°C for 10 s, annealing at 56°C for 30 s, and extension at 72°C for 15 s and a final extension at 72°C for 3 min.
Table 1.
Primers used to amplify the specific genes in this study.
| Gene target | Primer name | Forward primer (5′→3′) | Reverse primer (5′→3′) | Expected length (bp) |
|---|---|---|---|---|
| Vertebrate mtDNA cytb | BM1/BM2 | CCCCTCAGAATGATATTTGTCCTCA (Boakye et al., 1999) | CCATCCAACATCTCAGCATGATGAAA (Boakye et al., 1999) | 358 |
| Trypanosoma cruzi nDNA satellite repeat | TCZ1/TCZ2 | CGAGCTCTTGCCCACACGGGTGCT (Moser et al., 1989) | CCTCCAAGCAGCGGATAGTTCAGG (Moser et al., 1989) | 188 |
| Trypanosoma cruzi minicircle kDNA | Tc121/Tc122 | AAATAATGTACGGG(T/G)GAGATGCATGA (Wincker et al., 1994) | GGTTCGATTGGGGTTGGTGTAATATA (Wincker et al., 1994) | 330 |
To determine the natural infection status of wild-caught triatomines, the T. cruzi-specific TCZ1/TCZ2 nuclear satellite repeat and Tc121/Tc122 minicircle fragment were amplified with the primers listed in Table 1 . The mixture and conditions used to amplify these two targets were the same as those for cytb except that the annealing temperature of the nuclear satellite repeat was different and was set as 60°C.
All PCR products were observed by electrophoresis on 2% agarose gels stained with ethidium bromide, and then the products of all positive reactions were purified using an Agarose Gel DNA Extraction Kit (Takara, Japan). Sequencing reactions were performed using the primers listed in Table 1 in an Automated DNA Analyzer (ABI 3730XL, Applied Biosystems, Foster City, CA, USA) with the BigDye Terminator v3.1 Cycle Sequencing Kit (cat. no. 4337457, Applied Biosystems, Foster City, CA, USA) by Tsingke Biotechnology Ltd., Co. (Guangzhou, China). Then, the sequences were submitted to the GenBank database for homology searches using BLAST 1 .
16S rRNA Gene Amplification, Library Construction, and Next-Generation Sequencing
The V3-V4 variable regions of the 16S rRNA gene were amplified with the primers 343F (5’-TACGGRAGGCAGCAG-3’) and 798R (5’-AGGGTATCTAATCCT-3’) in combination with the barcoded primers and Takara Ex Taq DNA polymerase (cat. no. RR001Q, Takara, Japan), while the diluted DNA was used as a template. The quality of the PCR amplicon products and the relative intensity of the bands were determined by agarose gel electrophoresis, and then, Agencourt AMPure XP beads (cat. no. A63881, Beckman Coulter, Brea, CA, USA) were applied to purify the amplicon, and another round of PCR was subsequently performed. After purification with Agencourt AMPure XP beads once again, the final amplicon was quantified by a Qubit® dsDNA HS Assay Kit (cat. no. Q32852, Life Technologies, Waltham, MA, USA). Purified amplicons from each sample were pooled together in equal amounts for next-generation sequencing on the Illumina MiSeq platform following the manufacturer’s guidelines.
Quality Control of Sequencing Data and Bacterial Classification
Raw sequencing data were obtained in FASTQ format. Paired-end reads were then preprocessed with Trimmomatic software 2 (Bolger et al., 2014) to detect and cut off ambiguous bases from the N terminus. Low quality sequences with average quality scores lower than 20 were also removed using the sliding window trimming approach. Then, paired-end reads were assembled by Fast Ligation-Based Automatable Solid-Phase High-Throughput (FLASH) software (version 1.2.11) (Reyon et al., 2012). The parameters for assembly were set as 10 base pairs (bp) of minimum overlap, 200 bp of maximum overlap and 20% maximum mismatch rate. Furthermore, reads with ambiguous, homologous sequences and a total length of less than 200 bp were abandoned, while reads with 75% bases above Q20 were retained. Next, reads with chimeras were detected and removed by Quantitative Insights Into Microbial Ecology (QIIME) software (version 1.8.0) (Caporaso et al., 2010).
Clean reads were subjected to primer sequence removal and clustering to generate operational taxonomic units (OTUs) (Blaxter et al., 2005) using Vsearch software 3 (Rognes et al., 2016) with a 97% sequence similarity cutoff (equal to the bacterial species level). The representative read was chosen from each OTU by selecting the most abundant sequence using the QIIME package. All high-quality representative sequences were annotated and blasted against the Silva database (version 123) on the basis of the Ribosomal Database Project (RDP) classifier with the confidence threshold set as 70% (Wang et al., 2007).
Sequences identified as DNA from non-bacterial sources, such as chloroplasts, mitochondria, Archaea, and Eukarya, as well as singletons, were filtered out. Then, the rarefied OTU table was applied to illustrate the composition of the gut bacteria in each bug and for further analysis.
Alpha Diversity and Beta Diversity
To estimate within-sample diversity, alpha diversity estimators, such as the number of observed species, Chao1 richness estimator, Shannon-Wiener index, Good’s coverage estimator and phylogenetic diversity index, were obtained by using Mothur 4 (Schloss et al., 2009) for each individual. The number of observed species indicated the number of OTUs that were actually observed, while the Chao1 richness estimator was used to estimate the number of OTUs that were actually present in the bacterial community. The Shannon-Wiener index reflected both the richness and the evenness of species in the community; the higher the value was, the higher the diversity of the community. Sample coverage was revealed by the Good’s coverage estimator, and the depth of sequencing substantially covered all the species in the sample if the value was close to one. The phylogenetic diversity index based on random sampling of OTUs indicated the evolutionary distance relationship among OTUs; the higher the value was, the greater the evolutionary distance of the species.
Beta diversity, that is, between-sample diversity, was then monitored with principal coordinate analysis (PCoA) and nonmetric multidimensional scaling (NMDS), two kinds of ordination analyses, based on weighted UniFrac distance metrics or Bray-Curtis distance metrics. Moreover, one-way analysis of similarity (ANOSIM) was performed to assess whether the differences among the groups identified by NMDS were significant with 1,000 Monte Carlo permutation tests. In addition, the relative levels of gut bacterial community dispersal from each group were visualized by bar plots at both the phylum level and the genus level.
Statistical Analysis
One-way analysis of variance (ANOVA) or Student’s t test was performed to distinguish differences in gut microbial communities among multiple groups or between two groups, respectively, and differences were considered statistically significant at a p-value lower than 0.05. Linear discriminant analysis (LDA) coupled with effect size (LEfSe) measurement 5 (Segata et al., 2011) was applied to identify gut microbiome that were distinct from different groups. To identify the key signature gut flora of triatomines, a random forest (RF) classification model was built to estimate the importance of the top 30 dominant genera with the mean decrease gini, a measure applied to rank each genus in the model. Finally, Spearman correlation coefficients among the top 30 predominant bacterial genera were calculated to demonstrate the relationships among these genera. Unless otherwise stated, statistical analyses and plotting were carried out using R software (version 3.5.1).
Functional Profile Prediction for Gut Bacterial Communities
The functional profiles of gut bacterial communities were predicted with Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) 6 (Langille et al., 2013) on the basis of 16S sequencing data annotated by the Greengenes reference database 7 (DeSantis et al., 2006). Then, the predicted functions in combination with functional categorization were obtained by mapping the normalized OTU data in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database 8 . The differences between samples and groups were calculated by the Kruskal-Wallis (KW) rank sum test and visualized by a heatmap in level 3 of KEGG pathways.
Results
Identification of Feeding Sources and Natural Infection
Using the primer set BM1/BM2, we successfully amplified the cytb gene of vertebrates from the digestive tract of laboratory-reared adult triatomines, obtaining the characteristic 358-bp product, but this amplification failed in wild-caught triatomines. The feeding source of the laboratory-reared triatomines was identified as Mus musculus, as the triatomines were fed on the blood of ICR mice in our laboratory. Additionally, neither the T. cruzi-specific TCZ1/2 nuclear satellite repeat nor the Tc121/122 minicircle fragment was amplified from the gut of wild-caught triatomines, which suggested that the four bugs did not host T. cruzi.
Summary of High-Throughput Sequencing Data
A total of 1,479,154 high-quality sequences were yielded using 16S rRNA gene amplicon sequencing from 49 extracted DNA samples of the triatomine gut, with an average of 30,186 reads per sample after quality and abundance filtering, and the average sequence length was 438 bp. Among these reads, 1,765 OTUs were identified at a sequence similarity threshold of 97% ( Supplementary Table 1 ). The high value of Good’s coverage index in all the triatomine samples indicated that the sequencing depth was sufficient for profiling of the bacterial communities present ( Supplementary Table 2 ).
Dynamic Alteration in the Gut Microbiota Across T. rubrofasciata Developmental Stages
First, alpha diversity estimators were used to reveal marked differences in the richness and diversity of species in the community during triatomine development. Statistically significant differences were observed in the Chao1 index, Shannon-Wiener index, observed species and phylogenetic diversity index among the bug development stages. All four of the alpha diversity estimates increased in young nymphs (1st-3rd stages) but decreased obviously from 4th stage nymphs, reaching the lowest value in nymphs of the fifth instar; however, the value rebounded to the highest value in adults, especially in the female adults ( Figure 1 , KW test, p < 0.05 for all comparisons), which indicated that the gut bacteria of T. rubrofasciata were affected by aging, and the gut communities in adults were more rich and even than those at earlier stages. Then, PCoA plots based on weighted UniFrac distances demonstrated that the composition of the bug gut flora was related to the development stage, with 65.12% and 18.07% variation explained by principal component (PC) 1 and PC 2, respectively; 1st stage nymphs were distant from the other nymphs and adults, and this result was mainly driven by the largest differences in the abundances of Staphylococcus and Serratia between 1st stage nymphs and the others ( Figure 2A ). Obvious separation of nymphs of the first instar was also found in the NMDS plot on the basis of Bray-Curtis dissimilarities; moreover, the separation of 1st-2nd stage nymphs from older nymph stages and adults became evident, although older nymph stages and adults seemed closely related to each other, samples were still clustered according to developmental stage ( Figure 2B ). The stress value of NMDS in combination with the one-way ANOSIM results (R = 0.1822, p = 0.001) suggested that the grouping of NMDS was reliable. Overall, diversity analyses indicated that the gut microbes of T. rubrofasciata differ with age, in both the richness and evenness of the community in the microbiota structure.
Figure 1.
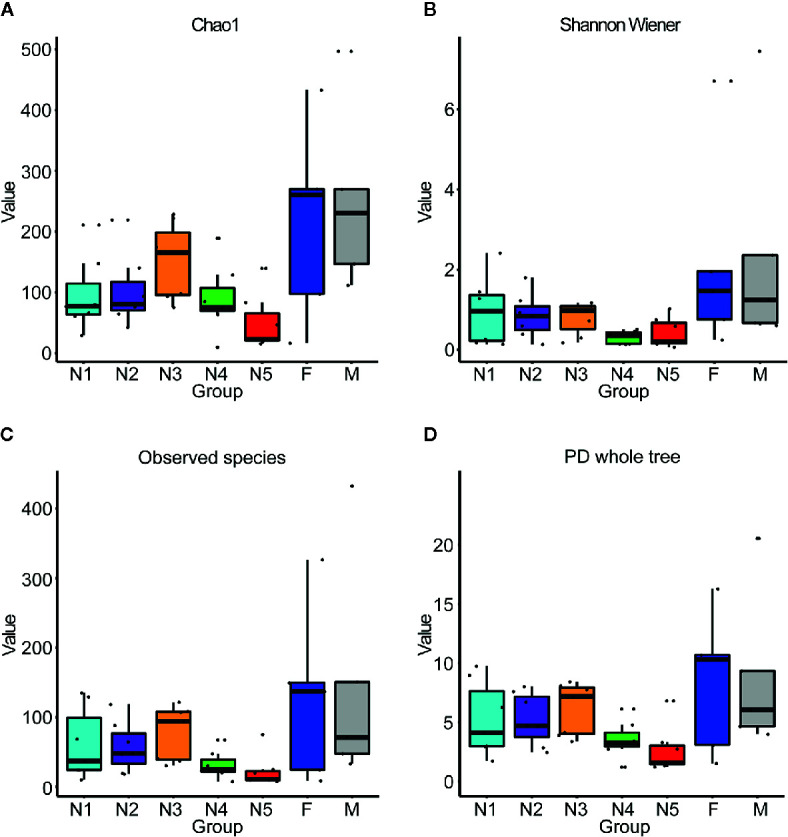
Box plots of 16S rRNA gene sequences obtained from gut samples of T. rubrofasciata across developmental stages based on alpha diversity analyses. Each column represents one group (N1, 1st stage nymphs; N2, 2nd stage nymphs; N3, 3rd stage nymphs; N4, 4th stage nymphs; N5, 5th stage nymphs; F, female adult; M, male adult). The top and bottom whiskers indicate the maximum and minimum values, respectively, and the hyphen represents the median value. (A) Chao1. (B) Shannon Wiener. (C) Observed species. (D) PD whole tree.
Figure 2.

Beta diversity differences in T. rubrofasciata gut samples across developmental stages on the basis of 16S rRNA gene sequencing (N1, 1st stage nymphs; N2, 2nd stage nymphs; N3, 3rd stage nymphs; N4, 4th stage nymphs; N5, 5th stage nymphs; F, female adult; M, male adult). Each point represents an individual. (A) Principal coordinate analysis (PCoA) plot of weighted UniFrac distances. (B) Nonmetric multidimensional scaling (NMDS) plot of Bray-Curtis distance.
Subsequently, bar plots were generated based on the relative abundances of the top 15 dominant gut bacteria at the phylum level ( Figure 3A ) and the genus level ( Figure 3B ) in different groups to observe the alteration in the gut microbiota composition during the development of T. rubrofasciata. Meanwhile, one-way ANOVA was performed to identify significantly altered gut bacterial phyla and genera across the developmental stages. Firmicutes, Proteobacteria, Bacteroidetes and Actinobacteria were the prevalent gut bacterial phyla of the bugs, accounting for more than 95% of the average relative abundances at different ages of T. rubrofasciata. Among these phyla, the average abundances of Firmicutes, Proteobacteria and Actinobacteria were altered significantly (p < 0.05, Supplementary Table 3 ). The average abundance of Proteobacteria was the highest in the 1st nymphal stage, while the average abundance of Firmicutes was the lowest. However, the opposite result was obtained in the 3rd nymphal stage ( Figures 4A, B ). Furthermore, Actinobacteria, which was almost undetectable in nymphs, increased markedly in adults, particularly in male adults ( Figure 4C ). Moreover, Staphylococcus, Serratia, Enterobacter, Burkholderia-Caballeronia-Paraburkholderia and Bacteroides were the 5 most important gut bacterial genera in triatomines ( Figure 3B ); nevertheless, the average relative abundance of Staphylococcus was low in nymphs of the first instar, whereas Serratia was significantly more abundant in 1st stage nymphs ( Figures 5A, B , p < 0.05; see also Supplementary Table 4 ). Interestingly, Burkholderia-Caballeronia-Paraburkholderia, which showed the highest average abundance in the 1st nymphal stage, as well as Oceanicaulis, was undetectable in the adult stage ( Figures 5C, D , p < 0.05; see also Supplementary Table 4 ). Additionally, the average abundance of Odoribacter peaked in the 2nd nymphal stage ( Figure 5E , p < 0.05; see also Supplementary Table 4 ). Taken together, the results showed that not only gut bacterial genera but also gut bacterial phyla changed significantly across T. rubrofasciata developmental stages.
Figure 3.
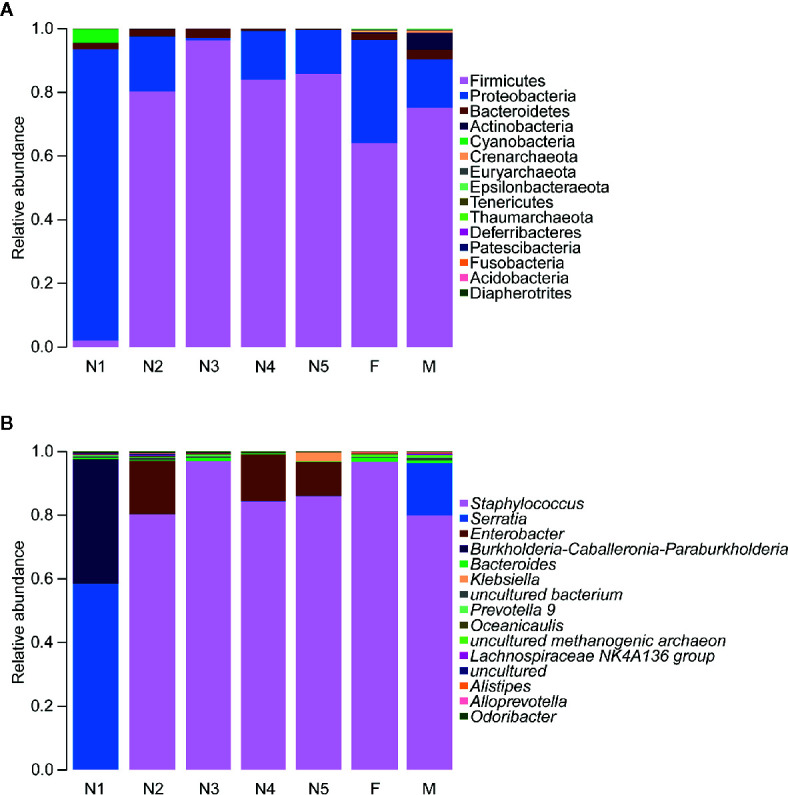
Relative abundances of the 15 most important gut microbiota constituents at the phylum level (A) and genus level (B) across development stages of T. rubrofasciata, as assessed by 16S rRNA sequencing. Each column represents the composition of the microbial taxa in one group (N1, 1st stage nymphs; N2, 2nd stage nymphs; N3, 3rd stage nymphs; N4, 4th stage nymphs; N5, 5th stage nymphs; F, female adult; M, male adult).
Figure 4.

Relative abundances of the significantly altered 15 most important gut phyla across developmental stages of T. rubrofasciata. Each column represents one group (N1, 1st stage nymphs; N2, 2nd stage nymphs; N3, 3rd stage nymphs; N4, 4th stage nymphs; N5, 5th stage nymphs; F, female adult; M, male adult). The top and bottom whiskers indicate the maximum and minimum values, respectively, and the hyphen represents the median value. (A) Firmicutes. (B) Proteobacteria. (C) Actinobacteria.
Figure 5.

Relative abundances of the significantly altered 15 most important gut genera across developmental stages of T. rubrofasciata. Each column represents one group (N1, 1st stage nymphs; N2, 2nd stage nymphs; N3, 3rd stage nymphs; N4, 4th stage nymphs; N5, 5th stage nymphs; F, female adult; M, male adult). The top and bottom whiskers indicate the maximum and minimum values, respectively, and the hyphen represents the median value. (A) Staphylococcus. (B) Serratia. (C) Burkholderia-Caballeronia-Paraburkholderia. (D) Oceanicaulis. (E) Odoribacter.
Next, LEfSe was applied to identify taxa from the phylum to genus level that were distinct among the developmental stages of triatomines. The cladogram in Figure 6 shows that Burkholderia-Caballeronia-Paraburkholderia, a bacterium from the class Gammaproteobacteria, phylum Proteobacteria, was markedly enriched in the 1st nymphal stage, while Oceanicaulis and Altererythrobacter, bacteria belonging to the class Alphaproteobacteria, phylum Proteobacteria, were significantly associated with the 2nd nymphal stage. Staphylococcus, within the phylum Firmicutes, was identified as the key marker of the 3rd nymphal stage; because it showed higher abundance in nymphs of the third instar than in other stages. Similarly, Enterobacter, a member of the class Gammaproteobacteria, phylum Proteobacteria, was linked to the 4th nymphal stage due to its highest average abundance in that stage. Bacteroides, in addition to some members from the order Clostridiales, phylum Firmicutes, was the significant bacterial genus related to the adult stage. Nevertheless, no discriminative gut microbiome was found in the 5th nymphal stage.
Figure 6.

Cladogram showing discriminated taxa at different ages of T. rubrofasciata (N1, 1st stage nymphs; N2, 2nd stage nymphs; N3, 3rd stage nymphs; N4, 4th stage nymphs; N5, 5th stage nymphs; F, female adult; M, male adult). Regions with different colors represent different groups. Different colored nodes in the branches represent the microbial groups that play an important role in the corresponding groups, whereas yellow nodes indicate bacterial groups that are nonsignificant in all groups.
Finally, the correlation analysis results for the top 30 dominant bacterial genera were plotted ( Figure 7 ). Notably, a majority of the bacteria were significantly positively related to each other, which revealed a symbiotic relationship of the gut microbiota in T. rubrofasciata. Even so, most negative correlations were found between the 5 most abundant gut bacteria and the remaining bacteria; most importantly, Staphylococcus was negatively correlated with Serratia (p < 0.001).
Figure 7.

Correlation plot showing the relationship among the top 30 predominant gut bacteria across developmental stages of T. rubrofasciata. The correlations between the bacteria are indicated by colors; blue indicates positive correlation, while red indicates negative correlation, with a darker color indicating a stronger correlation (*p < 0.05, **p < 0.01, ***p < 0.001).
Comparison of the Gut Microbiota in Wild-Caught and Laboratory-Reared T. rubrofasciata
Based on the boxplot of Chao1, observed species and phylogenetic diversity, we found that the alpha diversity of wild-caught T. rubrofasciata was significantly lower than that of laboratory-reared T. rubrofasciata ( Figures 8A–C , KW test, p < 0.05 for all comparisons); because the wild-caught triatomines had not ingested a blood meal before we captured them. In addition, differences in gut bacterial communities between wild-caught and laboratory-reared insects were visualized by a three-dimensional PCoA plot on the basis of weighted UniFrac distances. A clear separation was observed between the gut microbiome of wild-caught and laboratory-reared insects, with all three variables together explaining 95.92% of the total variance ( Figure 8D ).
Figure 8.

Alpha diversity and beta diversity of laboratory-reared and wild-caught T. rubrofasciata. (A) Chao1. (B) Observed species. (C) Phylogenetic diversity index. (D) Principal coordinate analysis (PCoA) plot of weighted UniFrac distances.
Alterations in the gut flora were determined based on the relative proportions of different taxa, with a sharp reduction in the relative abundance of Proteobacteria (p < 0.05) and an increase in the abundance of Firmicutes, but the difference between laboratory-reared bugs and wild-caught bugs at the phylum level was not significant ( Figure 9A ; see also Supplementary Table 5 ). At the genus level, significant differences in bacterial abundances of the 15 most important genera were observed only in Staphylococcus and Enterococcus. The former was more abundant in laboratory-reared bugs than in wild-caught bugs, while the latter was relatively highly enriched in wild-caught bugs ( Figure 9B , p < 0.05; see also Supplementary Table 6 ).
Figure 9.

Relative abundances of the 15 most important gut microbiota constituents of laboratory-reared and wild-caught T. rubrofasciata at the phylum level (A) and genus level (B), as assessed by 16S rRNA sequencing.
LEfSe identified 20 biomarkers, of which Pantoea, belonging to the phylum Proteobacteria, was markedly associated with wild-caught insects because of its absence in laboratory-reared insects, whereas Staphylococcus, within Firmicutes phylum, was closely linked with laboratory-reared insects ( Figure 10 ). To establish a prediction model to distinguish laboratory-reared triatomines from wild-caught triatomines according to the abundance of genera as measured in the gut, we applied machine learning combined with RF analysis. The results demonstrated that the genera Pantoea and Lactobacillus were the variables with the highest importance in the model, which indicated that these two taxa had the strongest prediction power ( Figure 11 ).
Figure 10.
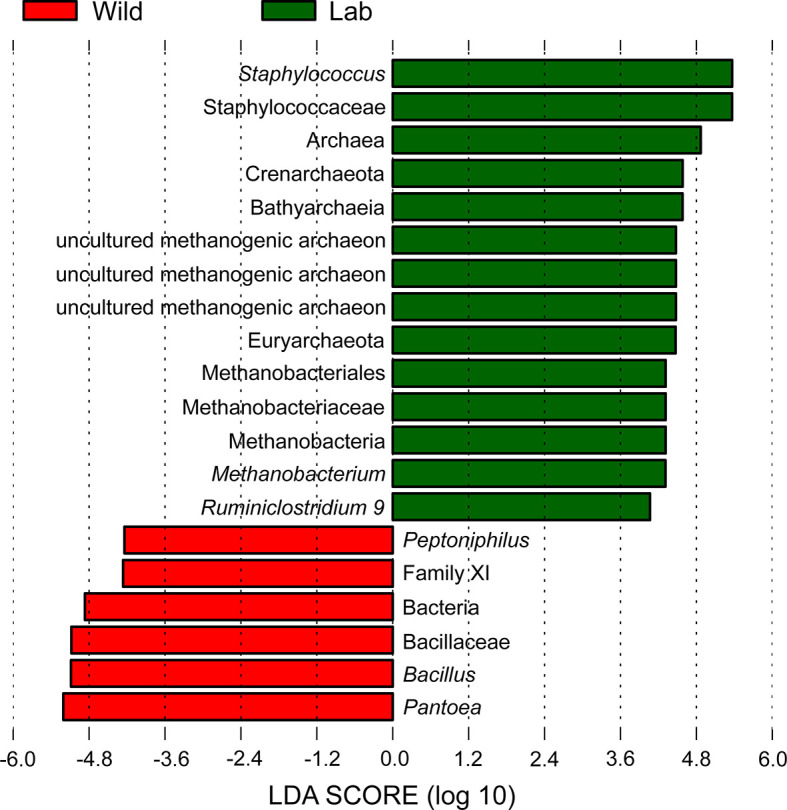
Histogram with LDA scores (threshold > 4) showing differentially abundant gut bacteria between laboratory-reared and wild-caught T. rubrofasciata. Taxa highlighted in green are overrepresented in laboratory-reared bugs, while those in red are overrepresented in wild-caught bugs.
Figure 11.

Random forest model classifying laboratory-reared and wild-caught T. rubrofasciata at the genus level. The importance levels are represented by the data for the mean decrease gini. Various bacterial genera are indicated by corresponding colors.
Using the calculated Spearman correlation coefficients for the top 30 dominant gut microbes at the genus level, we observed both positive and negative correlations among these microbes, with the former being more common ( Figure 12 ). Interestingly, Pantoea was negatively correlated with Staphylococcus and other taxa and positively related to Bacillus only (p > 0.05).
Figure 12.

Correlation plot showing the relationship among the top 30 predominant gut bacteria in laboratory-reared and wild-caught T. rubrofasciata. The correlations between the bacteria are indicated by colors; blue indicates positive correlation, while red indicates negative correlation, and a darker color indicates a stronger correlation (*p < 0.05, **p < 0.01, ***p < 0.001).
In addition, to predict the functional differences in the gut bacterial community between laboratory-reared and wild-caught T. rubrofasciata, a hierarchical clustering heat map was plotted by utilizing the PICRUSt algorithm to map the KEGG pathways ( Supplementary Figure 1 ). The results demonstrated that pathways associated with metabolism of amino acids (D-arginine and D-ornithine metabolism, ko00472), metabolism of lipids (primary bile acid biosynthesis, ko00120; and secondary bile acid biosynthesis, ko00121), metabolism of terpenoids and polyketides (carotenoid biosynthesis, ko00906) and infectious disease (Staphylococcus aureus infection, ko05150) were upregulated in laboratory-reared triatomines (p < 0.05). In contrast, pathways such as those associated with glycan biosynthesis and metabolism (N-glycan biosynthesis, ko00510; glycosphingolipid biosynthesis - ganglio series, ko00604; and lipopolysaccharide biosynthesis, ko00540), carbohydrate metabolism (starch and sucrose metabolism, ko00500; and ascorbate and aldarate metabolism, ko00053), amino acid metabolism (glutathione metabolism, ko00480), lipid metabolism (alpha-linolenic acid metabolism, ko00592), terpenoid and polyketide metabolism (biosynthesis of siderophore group nonribosomal peptides, ko01053), the immune system (RIG-I-like receptor signaling pathway, ko04622; antigen processing and presentation, ko04612; and NOD-like receptor signaling pathway, ko04621), cell motility (flagellar assembly, ko02040; and bacterial chemotaxis, ko02030), membrane transport (bacterial secretion system, ko03070), and infectious disease (Chagas disease, ko05142; and African trypanosomiasis, ko05143) were enriched in wild-caught triatomines (p < 0.05).
Discussion
Based on the adaptation of triatomines to human dwellings, these insects have traditionally been classified into four categories: sylvatic species, intrusive species, domiciliary species and domestic species (Carbajal-de-la-Fuente et al., 2019). T. rubrofasciata, as one kind of domiciliary species, has recently been frequently captured in human houses or near living areas in southern China (Liu et al., 2017; Huang et al., 2018; Hu et al., 2019; Shi et al., 2020), and residents who were reportedly bitten by this insect showed some clinical symptoms, including an urticaria-like systemic skin response or anaphylactic shock (Huang et al., 2018; Shi et al., 2020). Although T. cruzi has not been detected in wild-caught T. rubrofasciata in China to date, increased attention should be paid to the prevention and control of this vector due to the living habit of T. rubrofasciata. In addition, the feeding sources of wild-caught T. rubrofasciata were not identified in the present study, possibly because the triatomines that we caught were starved at that time, so there was limited blood in their guts, making it difficult to extract DNA and perform PCRs. Therefore, we could not draw the conclusion that these wild-caught triatomines did not ingest blood meals, because they have high resistance to starvation, and the volume of blood ingested by adults was proportionally lower than that ingested by nymphs (Cortéz and Gonçalves, 1998; Orantes et al., 2018; Hieu et al., 2019).
In this study, we demonstrated continuous dramatic changes in the gut microbes of T. rubrofasciata during development with 16S rRNA sequencing. Diversity analyses revealed a relative increase in alpha diversity, whereas a relative reduction in beta diversity was observed during bug development, which indicated that the composition of the gut microbiota in T. rubrofasciata became increasingly similar with age, which is consistent with a previous study of Triatoma sordida (T. sordida) (Oliveira et al., 2018). Notably, the triatomines that we used for this part of the study were laboratory reared. Although the environmental conditions during feeding, such as temperature, humidity and blood meal sources, were all the same, the differences in the gut microbiome across developmental stages were still distinct, which indicated that some significant microbes were tightly linked to the development and growth of T. rubrofasciata, such as maturation of the immune system, selection of specific bacterial taxa, supplementation of nutrient or digestion (Oliveira et al., 2018).
The predominant phylum among the gut microbes of T. rubrofasciata was Firmicutes, which was consistent with the bacterial community in the salivary glands of Rhodnius prolixus (R. prolixus) (Lima et al., 2018). In addition, the abundance of this phylum increased markedly after the 1st nymphal stage, and the same phenomenon was also observed in T. sordida (Oliveira et al., 2018). Conversely, the phylum Proteobacteria, which had the highest relative abundance in the salivary glands of Triatoma brasiliensis, Triatoma infestans (T. infestans), Triatoma rubrovaria and Rhodnius milesi (Lima et al., 2018), as well as in the gut of Triatoma maculata and Rhodnius pallescens (Montoya-Porras et al., 2018; Kieran et al., 2019), was highly depleted upon reaching the 2nd nymphal stage of T. rubrofasciata, this result was opposite to that for T. sordida (Oliveira et al., 2018). Furthermore, the variance in gut bacteria between T. rubrofasciata and T. sordida was also reflected in the abundance of the phylum Actinobacteria, which increased significantly through the development of the former. The gut microbes found in different triatomines form a symbiotic relationship, and the presence of symbiotic organisms can affect the development and survival of both hosts and parasites (Garcia et al., 2010; de Fuentes-Vicente et al., 2018; Salcedo-Porras and Lowenberger, 2019). Among these symbionts, Actinobacteria can provide B complex vitamins, extracellular enzymes, secondary metabolites and antimicrobial bioactive compounds to the host, while Proteobacteria can inhibit the growth of pathogens transmitted by insect vectors (Gumiel et al., 2015; Oliveira et al., 2018). In addition, the results of LEfSe demonstrated that Proteobacteria was the main biomarker in the nymphal stage of T. rubrofasciata, whereas Firmicutes that produce antimicrobial molecules including polyketides and lipopeptides (Aleti et al., 2015) became dominant in the adult stage. Thus, further studies of the complex and dynamic gut microbes may be helpful in finding their potential effects on the host fitness.
Previous studies of the gut bacteria in Triatoma dimidiata showed that Staphylococcus was identified as the main bacterial genus (Dumonteil et al., 2018), and it was common to all the developmental stages of T. sordida but without obvious differences in abundance (Oliveira et al., 2018); however, our study found that Staphylococcus had a high relative abundance from the 1st nymphal stage onwards. Interestingly, its abundance was associated with that of Serratia. Moreover, Serratia and Enterobacter were prevalent in the gut microbiome of T. rubrofasciata, and the result is consistent with that of a prior study of triatomines, including T. infestans, Triatoma pseudomaculata, Panstrongylus megistus, and R. prolixus (Azambuja et al., 2004). Both Serratia and Enterobacter are members of the family Enterobacteriaceae, which appears frequently in insect vectors, particularly in those whose diets are restricted to a few food sources, and Enterobacteriaceae may play an important role in host fitness by resisting pathogenic microbes. Some members of Enterobacteriaceae are able to kill closely related bacteria to reduce competition for essential nutrients (da Mota et al., 2012; da Mota et al., 2018; Montoya-Porras et al., 2018). Nevertheless, various studies have focused on Serratia because it is thought to decrease the population of T. cruzi by attacking the parasite’s membrane to impede the establishment of this pathogen (Flores-Villegas et al., 2015). The trypanocidal activity of Serratia was reported only under in vitro conditions, and its protective effect, preventing colonization by T. cruzi in vivo, was not distinct. It was hypothesized that the expression of bacterial cytotoxic genes varied among specific regions of the digestive tract (Gumiel et al., 2015; da Mota et al., 2018). Moreover, the diversity of Serratia species defines the influence of this genus on the pathogen or the host. While Serratia Y1 has the ability to inhibit the development of Plasmodium berghei in Anopheles by activating the mosquito immune system, Serratia odorifera enhances the susceptibility of Aedes aegypti to DENV-2 infection, and Serratia marcescens is pathogenic to honey bees (Raymann et al., 2018; Bai et al., 2019; Wu et al., 2019). Our results revealed that Serratia was enriched in 1st stage nymphs, and as we could not artificially infect T. rubrofasciata with T. cruzi, the relationship between Serratia and T. cruzi in vivo was not clear. Likewise, Enterobacter was the common bacterium in the gut of Aedes, Anopheles, and Culex, and it could inhibit invasion by and the development of Plasmodium falciparum via the generation of reactive oxygen species (ROS) in Anopheles (Cirimotich et al., 2011; Jayakrishnan et al., 2018), but its impact on T. cruzi has not yet been studied. The abundance of Enterobacter in T. rubrofasciata did not change significantly, which suggested that this bacterium may play a vital role in the biology of the vector or in the transmission of pathogens. Enterobacter species have shown strong hemolytic activity and are capable of performing red blood cell lysis to accelerate blood meal digestion (Muturi et al., 2019). More importantly, the potential of Enterobacter and Serratia in the paratransgenic control of malaria has been reported widely (Eappen et al., 2013; Koosha et al., 2019a; Koosha et al., 2019b).
In other instances, diversity analyses between wild-caught and laboratory-reared T. rubrofasciata suggested that the gut bacterial populations increased dramatically in the latter, contributing to the ingested blood meal. Previous researches have shown that after blood ingestion, the number of gut bacteria increased rapidly, probably due to the richness of iron and protein in the blood meal (Azambuja et al., 2004; Castro et al., 2012). Hence, the restriction of blood feeding may contribute to the low diversity in the gut microbiota of wild-caught triatomines (Gumiel et al., 2015). Similarly, the PCoA plot suggested that the alterations in bacterial communities were also influenced by the bloodmeal of T. rubrofasciata, which was explained by the most significant phylum Proteobacteria, with the majority of these bacteria belonging to the class Gammaproteobacteria, while the differentiating genus was Staphylococcus, followed by Enterococcus, members of the class Bacilli, and the phylum Firmicutes.
Under laboratory conditions, Staphylococcus demonstrated a stronger colonization capacity in the gut of T. rubrofasciata than other natural bacteria, such as Pantoea, but the opposite result was detected in the gut flora of Aedes albopictus and Culex quinquefasciatus (Gazzoni Araújo Gonçalves et al., 2019). One of the possible reasons is that our wild-caught triatomines had not ingested blood. For the same reason, Serratia was not detected in the gut flora of field-caught T. rubrofasciata. Because Staphylococcus is a common constituent of the natural skin flora of animals, it would proliferate rapidly in the guts of triatomines after a blood meal. Notably, Staphylococcus is also an opportunistic pathogen; once people are bitten by T. rubrofasciata, secondary bacterial infection occurs, since the vector that transports pathogenic microbes could facilitate colonization by and multiplication of opportunistic invaders by providing an ideal environment. An analogous situation was observed in canid species infected with Sarcoptes scabiei mites (DeCandia et al., 2019). Therefore, based on the functional profile of gut bacterial communities predicted by the PICRUSt algorithm, we should pay more attention to the potential of interspecies transmission of the microbiota.
Under natural conditions, Pantoea and Bacillus were the dominant genera in the gut of T. rubrofasciata, which is opposite to the result of a previous study of T. infestans (Waltmann et al., 2019), and interestingly, Pantoea was absent in the gut bacteria of T. rubrofasciata. These phenomena indicated that these genera can be strongly influenced by environmental changes and the consequent immune responses of triatomines. Moreover, Pantoea and Bacillus are common in Anopheles as well as in natural and laboratory-reared sand flies; these bacteria are able to regulate the immune responses of sand fly larvae and could be used as paratransgenic tools against malaria or leishmaniasis (Dinparast Djadid et al., 2011; Akhoundi et al., 2012; Heerman et al., 2015; Karimian et al., 2019). Nevertheless, due to the limited number of natural T. rubrofasciata analyzed in the present study, the variations in the gut microbiome composition of wild populations across a wider geographic area and their correlation with T. cruzi infection will require further investigation.
In summary, we profiled the gut microbiome alterations of T. rubrofasciata across developmental stages, as well as its gut microbiota succession, under natural and laboratory conditions. Importantly, we observed significant differences in both the diversity and composition of the gut microbes of T. rubrofasciata at different ages and environmental statuses. The specific gut bacteria may be modulated by feeding type and may have an effect on T. cruzi infection; however, further exploration is essential for determining whether the microbiota changes identified are causal and identifying the important metabolic pathways of gut microbes using metagenomics. Moreover, understanding the interactions among vectors-T. cruzi-symbionts (including Serratia, Enterobacter, Pantoea and Bacillus), along with understanding the biological functions and potential antiparasitic activity in vivo of gut symbionts might lead to the application of these bacteria in paratransgenic control of American trypanosomiasis.
Data Availability Statement
The sequencing data for the 16S rRNA gene have been deposited in the NCBI Sequence Read Archive under project number PRJNA645287.
Author Contributions
ZL, JY, and HX conceived and designed the experiments. ZL, YH, and HX drafted the manuscript. HX collected the samples. YH and MG performed the experiments. YH, MG, and PH collected and analyzed the data. HZ, YM, MZ, and JL participated in study design, technological guidance, and coordination. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China (grant no. 81572023 and 81371836), the National Parasitic Resources Center of China (grant no. NPRC-2019-194-30), Science and Technology Planning Project of Guangdong Province (grant no. 2019B030316025), Natural Science Foundation of Guangdong Province (grant no. 2019A1515011541), the National Key Research and Development Program of China (grant no. 2016YFC1202000 and 2016YFC1200500), the Open Foundation of Key Laboratory of Tropical Translational Medicine of Ministry of Education, Hainan Medical University (grant no. 2020TTM007), the 111 Project (grant no. B12003), Teaching Reform Project of Guangdong Province (grant no. 2017001) and Construction of Fujian Provincial Scientific and Technological Innovation Platform (2019Y2001).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2020.587708/full#supplementary-material
Hierarchical clustering heatmap of PICRUSt analysis with annotated 16S sequencing data demonstrates significant level 3 KEGG pathways between laboratory-reared and wild-caught T. rubrofasciata. Samples are shown in columns, and KEGG pathways are shown in rows.
OTU tables and taxonomic classifications of the 16S rRNA gene.
Good’s coverage estimators of T. rubrofasciata gut samples (N1, 1st stage nymphs; N2, 2nd stage nymphs; N3, 3rd stage nymphs; N4, 4th stage nymphs; N5, 5th stage nymphs; F, female adult; M, male adult).
One-way ANOVA of relative abundances of gut microbiome constituents across developmental stages of T. rubrofasciata at the phylum level (N1, 1st stage nymphs; N2, 2nd stage nymphs; N3, 3rd stage nymphs; N4, 4th stage nymphs; N5, 5th stage nymphs; F, female adult; M, male adult).
One-way ANOVA of relative abundances of gut microbiome constituents across developmental stages of T. rubrofasciata at the genus level (N1, 1st stage nymphs; N2, 2nd stage nymphs; N3, 3rd stage nymphs; N4, 4th stage nymphs; N5, 5th stage nymphs; F, female adult; M, male adult).
One-way ANOVA of relative abundances of gut microbiome constituents in laboratory-reared and wild-caught T. rubrofasciata at the phylum level.
One-way ANOVA of relative abundances of gut microbiome constituents in laboratory-reared and wild-caught T. rubrofasciata at the genus level.
References
- Akhoundi M., Bakhtiari R., Guillard T., Baghaei A., Tolouei R., Sereno D., et al. (2012). Diversity of the bacterial and fungal microflora from the midgut and cuticle of phlebotomine sand flies collected in North-Western Iran. PloS One 7 (11), e50259. 10.1371/journal.pone.0050259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleti G., Sessitsch A., Brader G. (2015). Genome mining: Prediction of lipopeptides and polyketides from Bacillus and related Firmicutes. Comput. Struct. Biotechnol. J. 13, 192–203. 10.1016/j.csbj.2015.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azambuja P., Feder D., Garcia E. S. (2004). Isolation of Serratia marcescens in the midgut of Rhodnius prolixus: impact on the establishment of the parasite Trypanosoma cruzi in the vector. Exp. Parasitol. 107 (1-2), 89–96. 10.1016/j.exppara.2004.04.007 [DOI] [PubMed] [Google Scholar]
- Azambuja P., Garcia E. S., Ratcliffe N. A. (2005). Gut microbiota and parasite transmission by insect vectors. Trends Parasitol. 21 (12), 568–572. 10.1016/j.pt.2005.09.011 [DOI] [PubMed] [Google Scholar]
- Bai L., Wang L., Vega-Rodríguez J., Wang G., Wang S. (2019). A Gut Symbiotic Bacterium Serratia marcescens Renders Mosquito Resistance to Plasmodium Infection Through Activation of Mosquito Immune Responses. Front. Microbiol. 10, 1580. 10.3389/fmicb.2019.01580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaxter M., Mann J., Chapman T., Thomas F., Whitton C., Floyd R., et al. (2005). Defining operational taxonomic units using DNA barcode data. Philos. Trans. R Soc. Lond. B Biol. Sci. 360 (1462), 1935–1943. 10.1098/rstb.2005.1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boakye D. A., Tang J., Truc P., Merriweather A., Unnasch T. R. (1999). Identification of bloodmeals in haematophagous Diptera by cytochrome B heteroduplex analysis. Med. Vet. Entomol. 13 (3), 282–287. 10.1046/j.1365-2915.1999.00193.x [DOI] [PubMed] [Google Scholar]
- Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30 (15), 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga M. V., Pinto Z. T., Lima M. M. (1998). Life cycle and reproductive patterns of Triatoma rubrofasciata (De Gee ) (Hemiptera: Reduviidae), under laboratory conditions. Mem. Inst. Oswaldo Cruz 93 (4), 539–542. 10.1590/s0074-02761998000400022 [DOI] [PubMed] [Google Scholar]
- Caporaso J. G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F. D., Costello E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7 (5), 335–336. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbajal-de-la-Fuente A. L., Fernández M. D. P., Piccinali R. V., Rodríguez-Planes L. I., Duarte R., Gürtler R. E. (2019). Occurrence of domestic and intrusive triatomines (Hemiptera: Reduviidae) in sylvatic habitats of the temperate Monte Desert ecoregion of Argentina. Acta Trop. 196, 37–41. 10.1016/j.actatropica.2019.04.028 [DOI] [PubMed] [Google Scholar]
- Castro D. P., Moraes C. S., Gonzalez M. S., Ratcliffe N. A., Azambuja P., Garcia E. S. (2012). Trypanosoma cruzi immune response modulation decreases microbiota in Rhodnius prolixus gut and is crucial for parasite survival and development. PloS One 7 (5), e36591. 10.1371/journal.pone.0036591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirimotich C. M., Dong Y., Clayton A. M., Sandiford S. L., Souza-Neto J. A., Mulenga M., et al. (2011). Natural microbe-mediated refractoriness to Plasmodium infection in Anopheles gambiae. Science 332 (6031), 855–858. 10.1126/science.1201618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortéz M. G., Gonçalves T. C. (1998). Resistance to starvation of Triatoma rubrofasciata (De Geer 1773) under laboratory conditions (Hemiptera: Reduviidae: Triatominae). Mem. Inst. Oswaldo Cruz 93 (4), 549–554. 10.1590/s0074-02761998000400024 [DOI] [PubMed] [Google Scholar]
- da Mota F. F., Marinho L. P., Moreira C. J., Lima M. M., Mello C. B., Garcia E. S., et al. (2012). Cultivation-independent methods reveal differences among bacterial gut microbiota in triatomine vectors of Chagas disease. PloS Negl. Trop. Dis. 6 (5), e1631. 10.1371/journal.pntd.0001631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Mota F. F., Castro D. P., Vieira C. S., Gumiel M., de Albuquerque J. P., Carels N., et al. (2018). In vitro Trypanocidal Activity, Genomic Analysis of Isolates, and in vivo Transcription of Type VI Secretion System of Serratia marcescens Belonging to the Microbiota of Rhodnius prolixus Digestive Tract. Front. Microbiol. 9:3205:3205. 10.3389/fmicb.2018.03205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Fuentes-Vicente J. A., Gutiérrez-Cabrera A. E., Flores-Villegas A. L., Lowenberger C., Benelli G., Salazar-Schettino P. M., et al. (2018). What makes an effective Chagas disease vector? Factors underlying Trypanosoma cruzi-triatomine interactions. Acta Trop. 183, 23–31. 10.1016/j.actatropica.2018.04.008 [DOI] [PubMed] [Google Scholar]
- DeCandia A. L., Leverett K. N., vonHoldt B. M. (2019). Of microbes and mange: consistent changes in the skin microbiome of three canid species infected with Sarcoptes scabiei mites. Parasit. Vectors 12 (1), 488. 10.1186/s13071-019-3724-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis T. Z., Hugenholtz P., Larsen N., Rojas M., Brodie E. L., Keller K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72 (7), 5069–5072. 10.1128/aem.03006-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz S., Villavicencio B., Correia N., Costa J., Haag K. L. (2016). Triatomine bugs, their microbiota and Trypanosoma cruzi: asymmetric responses of bacteria to an infected blood meal. Parasit. Vectors 9 (1), 636. 10.1186/s13071-016-1926-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinparast Djadid N., Jazayeri H., Raz A., Favia G., Ricci I., Zakeri S. (2011). Identification of the midgut microbiota of An. stephensi and An. maculipennis for their application as a paratransgenic tool against malaria. PloS One 6 (12), e28484. 10.1371/journal.pone.0028484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujardin J. P., Pham Thi K., Truong Xuan L., Panzera F., Pita S., Schofield C. J. (2015). Epidemiological status of kissing-bugs in South East Asia: A preliminary assessment. Acta Trop. 151, 142–149. 10.1016/j.actatropica.2015.06.022 [DOI] [PubMed] [Google Scholar]
- Dumonteil E., Ramirez-Sierra M. J., Pérez-Carrillo S., Teh-Poot C., Herrera C., Gourbière S., et al. (2018). Detailed ecological associations of triatomines revealed by metabarcoding and next-generation sequencing: implications for triatomine behavior and Trypanosoma cruzi transmission cycles. Sci. Rep. 8 (1), 4140. 10.1038/s41598-018-22455-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eappen A. G., Smith R. C., Jacobs-Lorena M. (2013). Enterobacter-activated mosquito immune responses to Plasmodium involve activation of SRPN6 in Anopheles stephensi. PloS One 8 (5), e62937. 10.1371/journal.pone.0062937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Villegas A. L., Salazar-Schettino P. M., Córdoba-Aguilar A., Gutiérrez-Cabrera A. E., Rojas-Wastavino G. E., Bucio-Torres M. I., et al. (2015). Immune defence mechanisms of triatomines against bacteria, viruses, fungi and parasites. Bull. Entomol. Res. 105 (5), 523–532. 10.1017/s0007485315000504 [DOI] [PubMed] [Google Scholar]
- Galvão C., Justi S. A. (2015). An overview on the ecology of Triatominae (Hemiptera:Reduviidae). Acta Trop. 151, 116–125. 10.1016/j.actatropica.2015.06.006 [DOI] [PubMed] [Google Scholar]
- Garcia E. S., Genta F. A., de Azambuja P., Schaub G. A. (2010). Interactions between intestinal compounds of triatomines and Trypanosoma cruzi. Trends Parasitol. 26 (10), 499–505. 10.1016/j.pt.2010.07.003 [DOI] [PubMed] [Google Scholar]
- Gazzoni Araújo Gonçalves G., Feitosa A. P. S., Portela-Júnior N. C., de Oliveira C. M. F., de Lima Filho J. L., Brayner F. A., et al. (2019). Use of MALDI-TOF MS to identify the culturable midgut microbiota of laboratory and wild mosquitoes. Acta Trop. 200, 105174. 10.1016/j.actatropica.2019.105174 [DOI] [PubMed] [Google Scholar]
- Gourbière S., Dorn P., Tripet F., Dumonteil E. (2012). Genetics and evolution of triatomines: from phylogeny to vector control. Heredity (Edinb). 108 (3), 190–202. 10.1038/hdy.2011.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumiel M., da Mota F. F., Rizzo Vde S., Sarquis O., de Castro D. P., Lima M. M., et al. (2015). Characterization of the microbiota in the guts of Triatoma brasiliensis and Triatoma pseudomaculata infected by Trypanosoma cruzi in natural conditions using culture independent methods. Parasit. Vectors 8, 245. 10.1186/s13071-015-0836-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heerman M., Weng J. L., Hurwitz I., Durvasula R., Ramalho-Ortigao M. (2015). Bacterial Infection and Immune Responses in Lutzomyia longipalpis Sand Fly Larvae Midgut. PloS Negl. Trop. Dis. 9 (7), e0003923. 10.1371/journal.pntd.0003923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hieu H. V., Do L. T., Pita S., Ha H., Khoa P. T., Tuan P. A., et al. (2019). Biological attributes of the kissing bug Triatoma rubrofasciata from Vietnam. Parasit. Vectors 12 (1), 585. 10.1186/s13071-019-3844-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotez P. J., Dumonteil E., Woc-Colburn L., Serpa J. A., Bezek S., Edwards M. S., et al. (2012). Chagas disease: “the new HIV/AIDS of the Americas”. PloS Negl. Trop. Dis. 6 (5), e1498. 10.1371/journal.pntd.0001498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y., Gao M. Z., Huang P., Zhou H. L., Ma Y. B., Zhou M. Y., et al. (2019). Taxonomic integrative and phylogenetic identification of the first recorded Triatoma rubrofasciata in Zhangzhou, Fujian Province and Maoming, Guangdong Province, China. Infect. Dis. Poverty 8 (1), 70. 10.1186/s40249-019-0579-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y. L., Huang D. N., Wu W. H., Yang F., Zhang X. M., Wang M., et al. (2018). Identification and characterization of the causative triatomine bugs of anaphylactic shock in Zhanjiang, China. Infect. Dis. Poverty 7 (1), 127. 10.1186/s40249-018-0509-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra-Cerdeña C. N., Sánchez-Cordero V., Townsend Peterson A., Ramsey J. M. (2009). Ecology of North American Triatominae. Acta Trop. 110 (2-3), 178–186. 10.1016/j.actatropica.2008.11.012 [DOI] [PubMed] [Google Scholar]
- Jayakrishnan L., Sudhikumar A. V., Aneesh E. M. (2018). Role of gut inhabitants on vectorial capacity of mosquitoes. J. Vector Borne Dis. 55 (2), 69–78. 10.4103/0972-9062.242567 [DOI] [PubMed] [Google Scholar]
- Karimian F., Vatandoost H., Rassi Y., Maleki-Ravasan N., Mohebali M., Shirazi M. H., et al. (2019). Aerobic midgut microbiota of sand fly vectors of zoonotic visceral leishmaniasis from northern Iran, a step toward finding potential paratransgenic candidates. Parasit. Vectors 12 (1):10. 10.1186/s13071-018-3273-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieran T. J., Arnold K. M. H., Thomas J. C. T., Varian C. P., Saldaña A., Calzada J. E., et al. (2019). Regional biogeography of microbiota composition in the Chagas disease vector Rhodnius pallescens. Parasit. Vectors 12 (1), 504. 10.1186/s13071-019-3761-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koosha M., Vatandoost H., Karimian F., Choubdar N., Abai M. R., Oshaghi M. A. (2019. a). Effect of Serratia AS1 (Enterobacteriaceae: Enterobacteriales) on the Fitness of Culex pipiens (Diptera: Culicidae) for Paratransgenic and RNAi Approaches. J. Med. Entomol. 56 (2), 553–559. 10.1093/jme/tjy183 [DOI] [PubMed] [Google Scholar]
- Koosha M., Vatandoost H., Karimian F., Choubdar N., Oshaghi M. A. (2019. b). Delivery of a Genetically Marked Serratia AS1 to Medically Important Arthropods for Use in RNAi and Paratransgenic Control Strategies. Microb. Ecol. 78 (1), 185–194. 10.1007/s00248-018-1289-7 [DOI] [PubMed] [Google Scholar]
- Langille M. G., Zaneveld J., Caporaso J. G., McDonald D., Knights D., Reyes J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31 (9), 814–821. 10.1038/nbt.2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima M. S., Laport M. S., Lorosa E. S., Jurberg J., Dos Santos K. R. N., da Silva Neto M. A. C., et al. (2018). Bacterial community composition in the salivary glands of triatomines (Hemiptera: Reduviidae). PloS Negl. Trop. Dis. 12 (9), e0006739. 10.1371/journal.pntd.0006739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., Guo Y. H., Zhang Y., Zhou Z. B., Zhang L. L., Zhu D., et al. (2017). First records of Triatoma rubrofasciata (De Gee ) (Hemiptera, Reduviidae) in Foshan, Guangdong Province, Southern China. Infect. Dis. Poverty 6 (1), 129. 10.1186/s40249-017-0342-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoya-Porras L. M., Omar T. C., Alzate J. F., Moreno-Herrera C. X., Cadavid-Restrepo G. E. (2018). 16S rRNA gene amplicon sequencing reveals dominance of Actinobacteria in Rhodnius pallescens compared to Triatoma maculata midgut microbiota in natural populations of vector insects from Colombia. Acta Trop. 178, 327–332. 10.1016/j.actatropica.2017.11.004 [DOI] [PubMed] [Google Scholar]
- Moser D. R., Kirchhoff L. V., Donelson J. E. (1989). Detection of Trypanosoma cruzi by DNA amplification using the polymerase chain reaction. J. Clin. Microbiol. 27 (7), 1477–1482. 10.1128/JCM.27.7.1477-1482.1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muturi E. J., Dunlap C., Ramirez J. L., Rooney A. P., Kim C. H. (2019). Host blood-meal source has a strong impact on gut microbiota of Aedes aegypti. FEMS Microbiol. Ecol. 95 (1), fiy213. 10.1093/femsec/fiy213 [DOI] [PubMed] [Google Scholar]
- Oliveira J. L., Cury J. C., Gurgel-Gonçalves R., Bahia A. C., Monteiro F. A. (2018). Field-collected Triatoma sordida from central Brazil display high microbiota diversity that varies with regard to developmental stage and intestinal segmentation. PloS Negl. Trop. Dis. 12 (8), e0006709. 10.1371/journal.pntd.0006709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orantes L. C., Monroy C., Dorn P. L., Stevens L., Rizzo D. M., Morrissey L., et al. (2018). Uncovering vector, parasite, blood meal and microbiome patterns from mixed-DNA specimens of the Chagas disease vector Triatoma dimidiata. PloS Negl. Trop. Dis. 12 (10), e0006730. 10.1371/journal.pntd.0006730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymann K., Coon K. L., Shaffer Z., Salisbury S., Moran N. A. (2018). Pathogenicity of Serratia marcescens Strains in Honey Bees. mBio 9 (5), e01649–18. 10.1128/mBio.01649-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyon D., Tsai S. Q., Khayter C., Foden J. A., Sander J. D., Joung J. K. (2012). FLASH assembly of TALENs for high-throughput genome editing. Nat. Biotechnol. 30 (5), 460–465. 10.1038/nbt.2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Ruano S. M., Škochová V., Rego R. O. M., Schmidt J. O., Roachell W., Hypša V., et al. (2018). Microbiomes of North American Triatominae: The Grounds for Chagas Disease Epidemiology. Front. Microbiol. 9, 1167. 10.3389/fmicb.2018.01167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rognes T., Flouri T., Nichols B., Quince C., Mahé F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e2584. 10.7717/peerj.2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salcedo-Porras N., Lowenberger C. (2019). The innate immune system of kissing bugs, vectors of chagas disease. Dev. Comp. Immunol. 98, 119–128. 10.1016/j.dci.2019.04.007 [DOI] [PubMed] [Google Scholar]
- Sassera D., Epis S., Pajoro M., Bandi C. (2013). Microbial symbiosis and the control of vector-borne pathogens in tsetse flies, human lice, and triatomine bugs. Pathog. Glob. Health 107 (6), 285–292. 10.1179/2047773213y.0000000109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss P. D., Westcott S. L., Ryabin T., Hall J. R., Hartmann M., Hollister E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75 (23), 7537–7541. 10.1128/aem.01541-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N., Izard J., Waldron L., Gevers D., Miropolsky L., Garrett W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12 (6), R60. 10.1186/gb-2011-12-6-r60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Wei Y., Feng X., Liu J., Jiang Z., Ou F., et al. (2020). Distribution, genetic characteristics and public health implications of Triatoma rubrofasciata, the vector of Chagas disease in Guangxi, China. Parasit. Vectors 13 (1), 33. 10.1186/s13071-020-3903-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira C. B., Praça Y. R., Bentes K., Santiago P. B., Silva S. M. M., Silva G. D. S., et al. (2018). Triatomines: Trypanosomatids, Bacteria, and Viruses Potential Vectors? Front. Cell Infect. Microbiol. 8, 405. 10.3389/fcimb.2018.00405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltmann A., Willcox A. C., Balasubramanian S., Borrini Mayori K., Mendoza Guerrero S., Salazar Sanchez R. S., et al. (2019). Hindgut microbiota in laboratory-reared and wild Triatoma infestans. PloS Negl. Trop. Dis. 13 (5), e0007383. 10.1371/journal.pntd.0007383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Garrity G. M., Tiedje J. M., Cole J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73 (16), 5261–5267. 10.1128/aem.00062-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2020).Chagas disease (American trypanosomiasis). In: Epidemiology. Available at: https://www.who.int/chagas/en/ (Accessed 16 March 2020).
- Wincker P., Britto C., Pereira J. B., Cardoso M. A., Oelemann W., Morel C. M. (1994). Use of a simplified polymerase chain reaction procedure to detect Trypanosoma cruzi in blood samples from chronic chagasic patients in a rural endemic area. Am. J. Trop. Med. Hyg. 51 (6), 771–777. 10.4269/ajtmh.1994.51.771 [DOI] [PubMed] [Google Scholar]
- Wu P., Sun P., Nie K., Zhu Y., Shi M., Xiao C., et al. (2019). A Gut Commensal Bacterium Promotes Mosquito Permissiveness to Arboviruses. Cell Host Microbe 25 (1), 101–112.e105. 10.1016/j.chom.2018.11.004 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Hierarchical clustering heatmap of PICRUSt analysis with annotated 16S sequencing data demonstrates significant level 3 KEGG pathways between laboratory-reared and wild-caught T. rubrofasciata. Samples are shown in columns, and KEGG pathways are shown in rows.
OTU tables and taxonomic classifications of the 16S rRNA gene.
Good’s coverage estimators of T. rubrofasciata gut samples (N1, 1st stage nymphs; N2, 2nd stage nymphs; N3, 3rd stage nymphs; N4, 4th stage nymphs; N5, 5th stage nymphs; F, female adult; M, male adult).
One-way ANOVA of relative abundances of gut microbiome constituents across developmental stages of T. rubrofasciata at the phylum level (N1, 1st stage nymphs; N2, 2nd stage nymphs; N3, 3rd stage nymphs; N4, 4th stage nymphs; N5, 5th stage nymphs; F, female adult; M, male adult).
One-way ANOVA of relative abundances of gut microbiome constituents across developmental stages of T. rubrofasciata at the genus level (N1, 1st stage nymphs; N2, 2nd stage nymphs; N3, 3rd stage nymphs; N4, 4th stage nymphs; N5, 5th stage nymphs; F, female adult; M, male adult).
One-way ANOVA of relative abundances of gut microbiome constituents in laboratory-reared and wild-caught T. rubrofasciata at the phylum level.
One-way ANOVA of relative abundances of gut microbiome constituents in laboratory-reared and wild-caught T. rubrofasciata at the genus level.
Data Availability Statement
The sequencing data for the 16S rRNA gene have been deposited in the NCBI Sequence Read Archive under project number PRJNA645287.