

Abstract
Background
Congenital muscular dystrophy type 1A (MDC1A), also termed merosin‐deficient congenital muscular dystrophy (CMD), is a severe form of CMD caused by mutations in the laminin α2 gene (LAMA2). Of the more than 300 likely pathogenic variants found in the Leiden Open Variant Database, the majority are truncating mutations leading to complete LAMA2 loss of function, but multiple copy number variants (CNVs) have also been reported with variable frequency.
Methods
We collected a cohort of individuals diagnosed with likely MDC1A and sought to identify both single nucleotide variants and small and larger CNVs via exome sequencing by extending the analysis of sequencing data to detect splicing changes and CNVs.
Results
Standard exome analysis identified multiple novel LAMA2 variants in our cohort, but only four cases carried biallelic variants. Since likely truncating LAMA2 variants are often found in heterozygosity without a second allele, we performed additional splicing and CNV analysis on exome data and identified one splice change outside of the canonical sequences and three CNVs, in the remaining four cases.
Conclusions
Our findings support the expectation that a portion of MDC1A cases may be caused by at least one CNV allele and show how these changes can be effectively identified by additional analysis of existing exome data.
Keywords: CNV analysis, congenital muscular dystrophy, exome sequencing, LAMA2
Exome sequencing analysis can identify both novel single nucleotide variants (SNVs) and copy number variants (CNVs) in LAMA2 in congenital muscular dystrophy cases. When only one SNV allele is found, additional CNV analysis can provide a full genetic diagnosis.
![]()
1. INTRODUCTION
Congenital muscular dystrophies (CMDs) are neuromuscular disorders presenting as muscle weakness from birth or early infancy that are usually associated with severe muscular dystrophy (MD) and variable brain malformations (Kang et al., 2015; O'Grady et al., 2016). Among CMDs, congenital muscular dystrophy type 1A (MDC1A) [MIM: 607855], also termed merosin‐deficient CMD, has a characteristic presentation with congenital or early‐onset MD and often changes in the white matter in the brain (Bönnemann et al., 2014; Geranmayeh et al., 2010). MDC1A is caused by mutations in laminin α2 gene (LAMA2) [MIM:156225] a component of the Laminin‐211 heterotrimer (Helbling‐Leclerc et al., 1995).
Multiple studies on genotype/phenotype correlation of LAMA2 mutations have shown that severe MDC1A is usually associated with truncating mutations and deletions leading to total loss of protein (Geranmayeh et al., 2010; Oliveira et al., 2008, 2018; Pegoraro et al., 1998). While previous studies of large patient cohorts have been able to identify all disease mutations in roughly 80% of cases, recent efforts using whole exome sequencing (WES) and copy number variant (CNV) analysis, have shown that approximately 95% of disease mutations can be identified, leaving about 5% of patients with only one identified heterozygous variant (Oliveira et al., 2018).
According to the Leiden Open Variant Database (LOVD), there are over 300 unique variants in LAMA2 which are disease causing (Oliveira et al., 2018). Of these variants, around half (54%) are protein‐truncating variants (nonsense or frameshift inducing small deletions or insertions) and 25.6% are predicted to affect splicing. Missense SNVs account for a smaller percentage (12.9%) of individuals and tend to fall within specific protein domains in the N‐terminal domain and the C‐terminal G‐like domains. The final group of disease associated variants consists of large duplications and deletions, also known as copy number variants (CNVs). There are 21 unique CNVs reported thus far (6.8%) which are mostly deletions (17/21). The rate of CNVs in LOVD may not be representative of the population, however, as past studies of large patient cohorts has yielded approximately 18% of disease‐causing variants as CNVs (Oliveira et al., 2014; Xiong et al., 2015).
In the present study, we combine standard exome variant analysis with splicing change analysis and CNV prediction identifying likely causative biallelic variants in seven out of eight individuals. We expand the known list of pathogenic genetic variants in LAMA2 that cause CMD by adding three previously unpublished single nucleotide changes. In addition, we report a novel large duplication of exons 6–12. Overall, our study supports the hypothesis that CNVs are underdiagnosed in MDC1A and that combination of WES and CNV analysis can be beneficial in the genetic diagnosis of this disorder.
2. METHODS
2.1. Ethical compliance
This study was performed with approval from the Institutional Review Boards of the George Washington University, Rutgers University, the Children's National Health System, the Hope Generation Genetic Clinic, and the Fondazione IRCCS Istituto Neurologico Carlo Besta.
2.2. Study participants
All study participants presented with severe CMD at birth or within the first 2 years of age. Muscle biopsies for patients (P)1–2 and P5–8 were performed for diagnostic purposes at Children's National Medical Center in Washington DC showing loss of merosin and were banked by the Hoffman laboratory. P3 was enrolled at the Hope Generation Genetic Clinic in Iran with a presentation consistent with MDC1A and P4 was enrolled at the Fondazione IRCCS Istituto Neurologico Carlo Besta in Milan, Italy following a diagnosis of CMD and loss of merosin upon biopsy.
2.3. Muscle biopsy histological analysis
Muscle biopsies were flash frozen and cryosectioned for histological examination and immunohistochemistry. Samples were stained with hematoxylin and eosin (H&E) and read by an expert muscle pathologist to identify signs of MD and the level of muscle disease severity. A C‐terminal laminin α2 antibody (MAB1922, Millipore Sigma) was used to test for merosin expression. P4 did not receive a muscle biopsy.
2.4. Whole exome sequencing analysis
DNA was extracted from the subject's muscle (P1–2, 5–8) or blood (P3, 4). Whole exome sequencing was performed at the Broad Institute Genomic Services (Broad Institute, Cambridge, MA) through their Germline Exome pipeline. Sequencing reads were aligned to reference genome hg19 using Burrows Wheeler Aligner (Li & Durbin, 2010). The Genome Analysis Toolkit was used to call variants using the best practices protocol for variant analysis (McKenna et al., 2010; Van der Auwera et al., 2013). Variants were annotated using Annovar (Wang, Li, & Hakonarson, 2010), loaded into an SQL database, and filtered with custom queries to identify rare, homozygous, and compound heterozygous nonsynonymous mutations. Variants were filtered for frequency lower than 1% in the Genome Aggregation Database (gnomAD; Lek et al., 2016) and for coding changes resulting in missense, truncating, splicing changes, or small deletions and duplications. Candidate variants were then examined for frequency in the sequencing reads to remove sequencing errors using Integrative Genomics Viewer (Robinson et al., 2011). Previously reported clinical information for each variant was gathered from Clinvar (Landrum et al., 2018), Leiden Open Variation Database (Fokkema, 2011), and the Human Gene Mutation Database (Stenson et al., 2017). Variants were scored according to the guidelines from the American College of Medical Genetics and Genomics (Richards et al., 2015) using InterVar (Li & Wang, 2017).
2.5. Bioinformatic analysis of missense and splicing LAMA2 variants
All candidate missense and splicing variants in LAMA2 were analyzed for predicted effect on protein function and splicing where applicable. SIFT and Polyphen2 were used for pathogenicity prediction for missense variants as part of the variant annotation with Annovar (Adzhubei et al., 2010; Sim et al., 2012). CADD (v1.4) was used as an additional pathogenicity prediction tool using the CADD web resource (Rentzsch, Witten, Cooper, Shendure, & Kircher, 2019). The online server for Human Splicing Finder (HSF, version 3.1), and NNSplice was used to predict possible effects on splicing (Desmet et al., 2009; Reese, Eeckman, Kulp, & Haussler, 1997). All variants were tested in HSF for possible splicing enhancers and silencers. Only variants that were less than four bases into the exon or less than 21 bases into the intron were run through NNSplice, as this tool only evaluates the original donor and acceptor sites. Scores for NNSplice are from the maximum entropy model (Yeo & Burge, 2004).
2.6. Copy number variant (CNV) analysis and multiplex ligation‐dependent probe amplification (MLPA) validation
All samples were interrogated for CNVs. Normalized coverage profiles were analyzed for copy number variation using ExomeDepth as described (Plagnol et al., 2012) and using recommended parameters. CNV calls were annotated against common CNVs (Conrad et al., 2010). MLPA was used to validate in silico CNV analysis using the SALSA MLPA P391 LAMA2 mix 1 probemix version 2 (MRC‐Holland). FAM‐labeled PCR products were generated using approximately 200 ng of genomic DNA and analyzed as outlined in the MLPA general protocol (MRC‐Holland). PCR products were quantified via capillary electrophoresis using an ABI 3500 Genetic Analyzer (Applied Biosystems) by adding 1 µL PCR product to a mixture of 9.75 µL Hi‐Di™ Formamide and 0.25 µL GeneScan™ 500 LIZ™ dye Size Standard (Thermo Fisher). Peak intensities were analyzed using the Coffalyser software (MRC‐Holland) to detect deviations from the standard DNA copy number of 12 reference samples and one negative control.
3. RESULTS
3.1. Study cohort
All study participants were referred to a neuromuscular clinic before 2 years of age presenting with hypotonia and likely muscle disease, but additional clinical information was fragmentary (Table 1). Muscle biopsy information was available for all patients for H&E staining showing muscle fiber replacement with changes in fiber size, fibrosis, variable presentation of fat infiltration, and inflammation, leading to a congenital muscular dystrophy diagnosis by a pathologist. Brain imaging information was also available for three cases in the form of either computerized tomography (CT) scan (P5) or magnetic resonance imaging (MRI) (P3, 4). P5 showed normal brain imaging. P3 was reported to have cerebral atrophy, which was more severe frontally, white matter changes, and posterior lissencephaly and severe developmental delay. P4 showed diffuse white matter alteration, but no cognitive impairment was present.
TABLE 1.
Muscle biopsy report and clinical findings in the study cohort
| Pat | Muscle biopsy | Immunohistochemistry | Other motor findings | Brain imaging | |||
|---|---|---|---|---|---|---|---|
| Dystrophic | DYS | LAMA2 | SCGA | DYSF | |||
| P1 | Yes, severe | + | − | + | + | n/a | n/a |
| P2 | Yes, severe | + | − | + | + | n/a | n/a |
| P3 | n/a | n/a | n/a | n/a | n/a | Hypotonia | MRI: Cerebral atrophy, leukodystrophy, posterior lissencephaly |
| P4 | Yes, severe | + | − | n/a | n/a | Hypotonia | MRI: Diffuse white matter changes |
| P5 | Yes, severe | + | Partial | Partial | n/a | Congenital weakness, high CK | CT:normal |
| P6 | Yes, severe | n/a | − | n/a | n/a | Hypotonia, decreased deep tendon reflexes | n/a |
| P7 | Yes, severe | n/a | − | n/a | n/a | Hypotonia | n/a |
| P8 | Yes, severe | + | − | + | + | n/a | n/a |
Abbreviations: −, absent; +, present; DYS, dystrophin; DYSF, dysferlin; LAMA2, laminin α2; n/a, not available; Pat, patient; SCGA, α‐sarcoglycan.
3.2. Identification of pathogenic single nucleotide variants in LAMA2
Initial exome sequencing analysis filtered for rare protein‐altering variants (<1% frequency in gnomAD) in known CMD genes. Likely pathogenic variants in LAMA2 were identified in seven individuals, leading to the identification of three novel variants (Table 2, Figure 1). However, only four out of eight patients had biallelic LAMA2 changes. Protein‐truncating mutations were found in three individuals. All variants reported below were mapped on transcript NM_000426.3 and protein NP_000417.2. In P1 we found a novel heterozygous one base pair (bp) deletion c.8038delG, p.(Gly2682Alafs*46), in exon 57, in combination with a heterozygous nonsense variant which was previously reported to be pathogenic c.5476C>T, p.(Arg1826*) (Naom et al., 1998). P2 had a homozygous splicing variant c.396+1G>T altering the canonical splice site and previously reported in several MDC1A patients as both a homozygous and compound heterozygous pathogenic mutation (Oliveira et al., 2018). P4 had a homozygous nonsense variant c.3085C>T, p.(Arg1029*), which also had been previously reported(Allamand & Guicheney, 2002; Oliveira et al., 2008). P3 had a novel homozygous missense variant c.2540G>T (p.Cys847Phe). Pathogenicity prediction tools consistently indicated the variant likely has a profound impact on protein function: SIFT (0.000), PolyPhen (1.000), and CADD (32.0).
TABLE 2.
Genetic variants identified in the study cohort
| Patient | Analysis |
DNA change |
Genomic location hg19 |
Exon |
Protein change |
Interpretation | Publication |
|---|---|---|---|---|---|---|---|
| P1 | 1 | het c.5476C>T | chr6:129722399 | 38 | p.(Arg1826*) | Pathogenic | Naom et al. (1998) |
| 1 | het c.8038delG | chr6:129813191 | 56 | p.(Gly2682fs*46) | Pathogenic | Novel | |
| P2 | 1 | hom c.396+1G> T | chr6:129381042 | Int 3 | Splicing | Pathogenic | Oliveira et al. (2018) |
| P3 | 1 | hom c.2540G>T | chr6:129608994 | 19 | p.(Cys847Phe) | VUS | Novel |
| P4 | 1 | hom c.3085C>T | chr6:129621928 | 22 | p.(Arg1029*) | Pathogenic | Allamand & Guicheney (2002) |
| P5 | 1 | het c.2230C>T | chr6:12588272 | 16 | p.(Arg744*) | Pathogenic | Di Blasi (2001) |
| 1 | het c.4960‐17C>A | chr6:129704250 | Int 34 | Splicing | VUS | Oliveira et al. (2018) | |
| P6 | 1 | het c.4710_4711delGT | chr6:129674495‐129674496 | 32 | p.(Cys1571fs*5) | Pathogenic | Novel |
| 2 | het c.820_1782dup | chr6:129468105‐129513998 | 6–12 | p.(274_594)dup | Likely pathogenic | Novel | |
| P7 | 1 | het c.6955C>T | chr6:129781432 | 49 | p.(Arg2319*) | Pathogenic | Pegoraro et al. (1998) |
| 2 | het c.7893_8069del | chr6:129807620‐129807767 | 56 | p.(Ala2584Hisfs*8) | Pathogenic | Oliveira et al. (2008) | |
| P8 | 2 | het c.397_639del | chr6:129419319‐129419560 | 4 | p.(133–213)del | Likely pathogenic | Xiong et al. (2015) |
| ? | ? | ? | ? | ? |
Abbreviations: het, heterozygous; hom, homozygous; int, intron; VUS, variant of uncertain significance.
FIGURE 1.
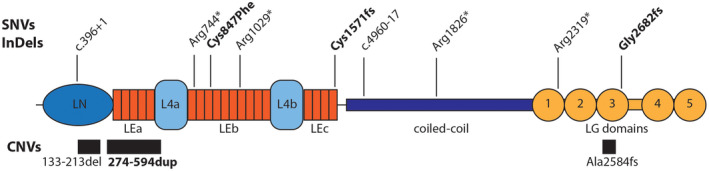
Schematic representation of protein variant, splicing changes, and CNVs identified in LAMA2. Protein changes generated by single nucleotide variants (SNVs) and small deletions and duplications (InDels) are shown above the protein schematic. Location of splicing changes is indicated by labels starting with “c.” showing the corresponding location in the coding sequence. Copy number variants (CNVs) are shown below the protein as black boxes. Domain abbreviations are as follows: LN, Laminin N‐terminal; LE, Laminin‐type‐EGF‐like; LG, Laminin‐type G. Two globular L4 domains are also shown in light blue and coiled‐coil domain in purple. Protein sequence: GenBank NP_000417.2
Three individuals showed only one likely deleterious allele. P5 had a heterozygous nonsense mutation c.2230C>T (p.Arg744*) that was previously reported to create an in frame skipping of exon 15 and a truncated protein product in a case of mild MDC1A (Di Blasi, 2001). P6 had a novel heterozygous 2bp deletion c.4710_4711delGT leading to a frameshift (p.Val1572Phefs*5). P7 had a heterozygous variant c.6955C>T (p.Arg2319*) and had been previously sequenced for LAMA2 by Pegoraro et al. who reported only this variant and were not able to identify a second one (Pegoraro et al., 1998).
Additional analysis of the intronic regions captured by exome sequencing did not identify any candidate variants for P6 and P7, but showed a second heterozygous intronic variant (c.4960‐17C>A) in P5 (Table 2). This variant had been previously reported by Oliveira et al. (2018), but had never been evaluated for its possible effect on splicing. We performed splicing prediction analysis on this variant and included the missense variant in P3 using the canonical splice variant in P2 as a positive control. Since splicing prediction is based on probabilistic approaches, we used a combination of algorithms using different models which analyze the surrounding sequence to determine whether a variant is likely to generate a strong new splice donor or acceptor (Table 3). Whenever applicable, we tested the variants with Human Splicing Finder (HSF) which combines multiple prediction models and prediction of splicing branch point (Desmet et al., 2009), and NNSplice which is based on a machine learning approach where a neural network is trained to detect splicing events (Reese et al., 1997). As expected the canonical splice variant in P2 was predicted to break the splice donor. Both splicing evaluation tools predicted that the c.4960‐17C>A variant in P5 would change the acceptor site in intron 34, with HSF predicting the loss of the canonical site, and both HSF and NNSplice predicting the creation of a new acceptor at the variant location instead. Additional experimental confirmation is still needed to determine how mRNA splicing is disrupted.
TABLE 3.
Bioinformatic analysis of missense and splicing mutations
| Pat | DNA Variant | Protein variant | Nearest junction | Pathogenicity prediction | Splicing prediction | |||
|---|---|---|---|---|---|---|---|---|
| SIFT | Polyphen2 | CADD | HSF |
NNSplice Ref‐>Mut (alt) |
||||
| P2 | c.396+1G>T | Intronic |
+1 bp, Intron 3 donor |
— | — | — | ++ broken donor site | 0.71 ‐>0.00 |
| P5 | c.4960‐17C>A | Intronic |
−17 bp, Intron 34 acceptor |
— | — | — | + new acceptor site | 0 ‐>0.72 |
| P3 | c.2540G>T | p.(Cys847Phe) |
+3 bp, Intron 18 acceptor |
0.000 | 1.000 | 32.0 | + new ESS | 0.93 ‐>0.94 |
DNA and Protein GenBank Accession: NM_000426.3, NP_000417.2.
Abbreviations: +, potentially affects splicing; 1, probably damaging; 1, tolerated; 10, top 1%; 20, top 0.1%; bp, base pairs; CADD: phred scores; ESE, exonic splicing enhancer; ESS, exonic splicing silencer; HSF: ++, Most probable to effect splicing; Nearest Junction: junction, exon/intron boundary; NNSplice: alt, alternative donor or acceptor site. Score 0 > 1; Pat, patient; Polyphen2: 0, benign; SIFT: 0, deleterious.
The homozygous missense variant in P3 (c.2540G>T) is located in exon 19 3bp downstream from the splice acceptor of intron 18 and HSF and NNSplice indicated limited impact on splicing and considering the high scores in pathogenicity prediction algorithms, these additional findings support the hypothesis that this is a deleterious missense variants with no effect on splicing (Table 3).
3.3. Copy number variant identification in LAMA2
We then analyzed this cohort using ExomeDepth, an in silico CNV analysis approach to identify large duplications and deletions in exome sequencing. We tested all exomes in our cohort to determine how CNVs contribute to the mutation burden in this group and identified three CNVs, two deletions and one duplication (Table 2, Figure 1). We discovered a large heterozygous duplication of exon 6 through 12 as the second allele in P6 c.820_1782dup, (p.274_594dup). While the duplication is in frame, it covers sections of both the N‐terminal domain as well as a Laminin‐type‐EGF‐like domain, which are both critical to the formation of basement membrane structure through self‐polymerization of laminin. Of note, there was a previously reported large duplication of exons 5–12 in a patient with an LGMD/Becker muscular dystrophy‐like phenotype where no second mutation was found (Piluso et al., 2011). We also found a second allele in P7: a heterozygous deletion of exon 56, c.7750_7899del, predicted to cause a truncating frameshift (p.Ala2584Hisfs*8), which is likely the same as a CNV found frequently in the Portuguese population (Oliveira et al., 2008). From break point analysis in previous studies, the frequently reported exon 56 deletion also includes deletion of large portions of the flanking introns (Oliveira et al., 2014).
Finally, a heterozygous large deletion of exon 4, c.397_639del, (p.133_213del) previously reported as a pathogenic CNV and a founder mutation in the Han Chinese population (Ge et al., 2018; Xiong et al., 2015) was identified in P8. Patients with homozygous deletions of exon 4 exhibited the typical signs of MDC1A with loss of muscle laminin α2, CMD, and white matter changes (Xiong et al., 2015). No second variant was discovered in this individual.
Multiplex Ligation‐dependent Probe Amplification (MLPA) Validation was performed to confirm these CNVs (Figure 2) in these cases and in a group of control samples, including normal skeletal muscle and P1, 2, and 5. CNVs were confirmed as a 50% loss of DNA in P7 and P8 and as a 50% increase in P6, indicating that CNV prediction was accurate and can be used to identify large deletions and duplications from exome sequencing data.
FIGURE 2.
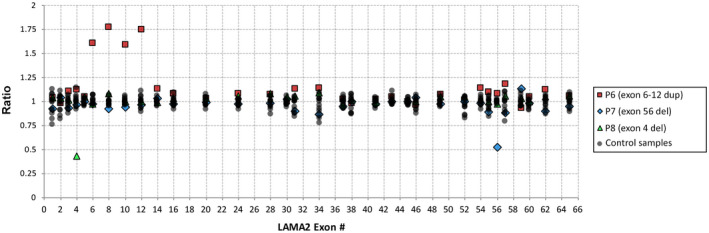
Multiplex Ligation‐dependent Probe Amplification (MLPA) confirms CNVs predicted via exome analysis. CNVs predicted in P6, P7, and P8 were tested using MLPA probes for a subset of LAMA2 exons, in parallel to five control sample, two normal skeletal muscle samples and three cases from this study (P1, P2 and P5). A heterozygous duplication spanning from exon 6 to exon 12 was confirmed in P6 (red squares), as were single exon deletions in P7 (blue diamonds) and P8 (green triangles)
4. DISCUSSION
Here, we present a cohort of MDC1A patients carrying both novel and known single nucleotide variants and CNVs. Due to the high frequency of CNVs among LAMA2 mutations, only one likely pathogenic variant is identified via DNA sequencing in some cases (Geranmayeh et al., 2010; Oliveira et al., 2018; Pegoraro et al., 1998). In fact, only 50% of our cases showed biallelic changes in LAMA2 through exome sequencing and only when additional scrutiny was extended to the intronic regions. By combining variant filtering for rare SNPs and small indels with CNV analysis, we were able to identify two alleles in most individuals and an additional heterozygous deletion in one patient (Table 2, Figure 1). The majority of identified variants (8/12) resulted in a protein truncation via a nonsense (n = 4), splicing (n = 2), or a frameshift change due to a small deletion (n = 2). Of these variants, two were unpublished, while the remaining were previously published as pathogenic either in homozygosity or compound heterozygosity with other variants. In all these cases, complete or partial loss of laminin α2 was identified by immunohistochemistry (Table 1).
One patients had a novel missense variant. The laminin α2 protein is composed of three major protein regions including a globular laminin N‐terminal domain (LN), multiple Laminin‐type‐EGF‐like (LE) domains at the N‐terminus, a central coiled‐coil domain, and five laminin‐type G (LG) globular domains at the C‐terminus (Hohenester & Yurchenco, 2013). Hotspots for missense mutations in LAMA2 are located in the LN and in the three LE domains likely disrupting assembly of the basement membrane and in the LG domains affecting binding to membrane proteins (Figure 1) (Oliveira et al., 2018). The homozygous change p.(Cys847Phe) in P3, which was predicted to be pathogenic by multiple algorithms, would affect one of LE domains (LEb, Figure 1) which binds to other components of the extra‐cellular matrix (ECM) organizing and stabilizing the basement membrane (McKee, Harrison, Capizzi, & Yurchenco, 2007).
Additional analysis for CNVs proved effective in identifying two large deletions and a large duplication and providing genetic information for an additional three cases. The novel exon 6–12 duplication (p.274_594dup), while in frame, affects part of LN and the entire LEa domain. Similarly, the in frame deletion in exon 4 (p.133_213del) would remove part of LN. The heterozygous deletion in exon 56 would instead cause a frameshift at the C‐terminus of the protein starting from the third LG domain (Figure 1).
In addition to identifying novel variants in LAMA2, our study provides further information on the biological importance of previously published variants which were reported without characterization. We also show that analysis of next‐generation sequencing data for CNVs can be a rapid and cost‐effective method for identifying additional variants in cases where only one heterozygous likely pathogenic change is found. Provided sufficient coverage of at least 30 reads per exon (Plagnol et al., 2012), this approach could be effective to inexpensively identify additional alleles in existing exome data and has been validated in multiple large disease cohorts (Ellingford et al., 2017; Marchuk et al., 2018). Overall, our findings show how CNV analysis can further leverage exome data to provide a genetic diagnosis for CMD cases.
CONFLICT OF INTEREST
Dr. E.P. Hoffman is co‐founder and stock holder in AGADA BioSciences, TRiNDS LLC, and ReveraGen BioPharma. No conflict is present with the current study. The other authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
E.S. Cauley and M.C. Manzini conceived the research, analyzed the data and wrote the manuscript. A. Pittman, S. Mummidivarpu, S. Martinez, and D. Podini provided additional data collection and analysis. E.G. Karimiani, I. Moroni, R. Boostani, M. Mora, Y. Jamshidi, and E.P. Hoffman provided samples and clinical information. All authors have read and approved the final version of the manuscript.
ACKNOWLEDGMENTS
First, we would like to thank the families for their participation in this study. We are grateful to Adam Wong at the GWU high‐performance computing cluster Colonial One for technical support, to Anelia Horvath for assistance with bioinformatic analysis, to Jamie Marshall at the Broad Institute, to Drew Bader from the Podini laboratory at GWU for assistance with sample preparation. This research was supported by Research grants from the Muscular Dystrophy Association (#293587), the March of Dimes (#6‐FY14 422), and the National Institutes of Health (R01NS109149) to M.C.M. The EuroBioBank and Telethon Network of Genetic Biobanks are gratefully acknowledged for providing biological samples.
Cauley ES, Pittman A, Mummidivarpu S, et al. Novel mutation identification and copy number variant detection via exome sequencing in congenital muscular dystrophy. Mol Genet Genomic Med. 2020;8:e1387 10.1002/mgg3.1387
Funding information
This study was supported be Muscular Dystrophy Association (#293587), March of Dimes (#6‐FY14 422), National Institutes of Health (R01NS109149) to M.C.M.
DATA AVAILABILITY STATEMENT
The data to support the findings in the study are available from the corresponding author upon reasonable request.
REFERENCES
- Adzhubei, I. A. , Schmidt, S. , Peshkin, L. , Ramensky, V. E. , Gerasimova, A. , Bork, P. , … Sunyaev, S. R. . (2010). A method and server for predicting damaging missense mutations. Nature Methods, 7(4), 248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allamand, V. , & Guicheney, P. (2002). Merosin‐deficient congenital muscular dystrophy, autosomal recessive (MDC1A, MIM#156225, LAMA2 gene coding for alpha2 chain of laminin). European Journal of Human Genetics, 10(2), 91–94. [DOI] [PubMed] [Google Scholar]
- Bönnemann, C. G. , Wang, C. H. , Quijano‐Roy, S. , Deconinck, N. , Bertini, E. , Ferreiro, A. , … North, K. N. (2014). Diagnostic approach to the congenital muscular dystrophies. Neuromuscular Disorders, 24(4), 289–311. 10.1016/j.nmd.2013.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad, D. F. , Pinto, D. , Redon, R. , Feuk, L. , Gokcumen, O. , Zhang, Y. , Fitzgerald, T. (2010). Origins and functional impact of copy number variation in the human genome. Nature, 464(7289), 704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmet, F.‐O. , Hamroun, D. , Lalande, M. , Collod‐Béroud, G. , Claustres, M. , & Béroud, C. (2009). Human splicing finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Research, 37(9), e67. 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Blasi, C. (2001). Mild muscular dystrophy due to a nonsense mutation in the LAMA2 gene resulting in exon skipping. Brain, 124(Pt 4), 698–704. 10.1093/brain/124.4.698 [DOI] [PubMed] [Google Scholar]
- Ellingford, J. M. , Campbell, C. , Barton, S. , Bhaskar, S. , Gupta, S. , Taylor, R. L. , … Black, G. C. M. (2017). Validation of copy number variation analysis for next‐generation sequencing diagnostics. European Journal of Human Genetics, 25(6), 719–724. 10.1038/ejhg.2017.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fokkema, I. F. A. C. et al (2011). LOVD vol 2.0: The next generation in gene variant databases Lindblom A., & Robinson P. N. (Eds.), Human mutation (Vol. 32, pp. 557–563). [DOI] [PubMed] [Google Scholar]
- Ge, L. , Liu, A. , Gao, K. , Du, R. , Ding, J. , Mao, B. , … Xiong, H. (2018). Deletion of exon 4 in LAMA2 is the most frequent mutation in Chinese patients with laminin α2‐related muscular dystrophy. Scientific Reports, 8(1), 10.1038/s41598-018-33098-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geranmayeh, F. , Clement, E. , Feng, L. H. , Sewry, C. , Pagan, J. , Mein, R. , … Muntoni, F. (2010). Genotype–phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscular Disorders, 20(4), 241–250. 10.1016/j.nmd.2010.02.001 [DOI] [PubMed] [Google Scholar]
- Helbling‐Leclerc, A. , Zhang, X. , Topaloglu, H. , Cruaud, C. , Tesson, F. , & Weissenbach, J. , … Guicheney, P. (1995). Mutations in the laminin alpha 2‐chain gene (LAMA2) cause merosin‐deficient congenital muscular dystrophy. Nature Genetics, 11(2), 216–218. [DOI] [PubMed] [Google Scholar]
- Hohenester, E. , & Yurchenco, P. D. (2013). Laminins in basement membrane assembly. Cell Adhesion & Migration, 7(1), 56–63. 10.4161/cam.21831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, P. B. , Morrison, L. , Iannaccone, S. T. , Graham, R. J. , Bönnemann, C. G. , & Rutkowski, A. , … Getchius, T. S. (2015). Evidence‐based guideline summary: Evaluation, diagnosis, and management of congenital muscular dystrophy: Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology, 84(13), 1369–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum, M. J. , Lee, J. M. , Benson, M. , Brown, G. R. , Chao, C. , Chitipiralla, S. , … Maglott, D. R. (2018). ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Research, 46(D1), D1062–D1067. 10.1093/nar/gkx1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Minikel, E. V. , Samocha, K. E. , Banks, E. , Fennell, T. … Tukiainen, T. (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2010). Fast and accurate long‐read alignment with Burrows‐Wheeler transform. Bioinformatics, 26(5), 589–595. 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Q. , & Wang, K. (2017). InterVar: Clinical interpretation of genetic variants by the 2015 ACMG‐AMP guidelines. American Journal of Human Genetics, 100(2), 267–280. 10.1016/j.ajhg.2017.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchuk, D. S. , Crooks, K. , Strande, N. , Kaiser‐Rogers, K. , Milko, L. V. , Brandt, A. , … Berg, J. S. (2018). Increasing the diagnostic yield of exome sequencing by copy number variant analysis. PLoS One, 13(12), e0209185 10.1371/journal.pone.0209185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee, K. K. , Harrison, D. , Capizzi, S. , & Yurchenco, P. D. (2007). Role of laminin terminal globular domains in basement membrane assembly. Journal of Biological Chemistry, 282(29), 21437–21447. 10.1074/jbc.M702963200 [DOI] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , … DePristo, M. A. (2010). The genome analysis toolkit: A MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Research, 20(9), 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naom, I. , Dalessandro, M. , Sewry, C. A. , Philpot, J. , Manzur, A. Y. , Dubowitz, V. & Muntoni, F. (1998). Laminin alpha 2‐chain gene mutations in two siblings presenting with limb‐girdle muscular dystrophy. Neuromuscular Disorders, 8(7), 495–501. [DOI] [PubMed] [Google Scholar]
- O'Grady, G. L. , Lek, M. , Lamande, S. R. , Waddell, L. , Oates, E. C. , Punetha, J. , … North, K. (2016). Diagnosis and etiology of congenital muscular dystrophy: We are halfway there. Annals of Neurology, 80(1), 101–111. 10.1002/ana.24687 [DOI] [PubMed] [Google Scholar]
- Oliveira, J. , Gonçalves, A. , Oliveira, M. E. , Fineza, I. , Pavanello, R. C. M. , Vainzof, M. , … Sousa, M. (2014). Reviewing large LAMA2 deletions and duplications in congenital muscular dystrophy patients. Journal of Neuromuscular Diseases, 1(2), 169–179. 10.3233/JND-140031. [DOI] [PubMed] [Google Scholar]
- Oliveira, J. , Gruber, A. , Cardoso, M. , Taipa, R. , Fineza, I. , & Gonçalves, A. , … Oliveira, M. E. (2018). LAMA2 gene mutation update: Toward a more comprehensive picture of the laminin‐α2 variome and its related phenotypes. Human Mutation, 39(10), 1314–1337. [DOI] [PubMed] [Google Scholar]
- Oliveira, J. , Santos, R. , Soares‐Silva, I. , Jorge, P. , Vieira, E. , Oliveira, M. E. , … Bronze‐da‐Rocha, E. (2008). LAMA2 gene analysis in a cohort of 26 congenital muscular dystrophy patients. Clinical Genetics, 74(6), 502–512. 10.1111/j.1399-0004.2008.01068.x [DOI] [PubMed] [Google Scholar]
- Pegoraro, E. , Marks, H. , Garcia, C. A. , Crawford, T. , Mancias, P. , & Connolly, A. M. … Stella, A. (1998). Laminin alpha2 muscular dystrophy: Genotype/phenotype studies of 22 patients. Neurology, 51(1), 101–110. [DOI] [PubMed] [Google Scholar]
- Piluso, G. , Dionisi, M. , Del Vecchio Blanco, F. , Torella, A. , Aurino, S. , Savarese, M. , … Nigro, V. (2011). Motor chip: A comparative genomic hybridization microarray for copy‐number mutations in 245 neuromuscular disorders. Clinical Chemistry, 57(11), 1584–1596. 10.1373/clinchem.2011.168898 [DOI] [PubMed] [Google Scholar]
- Plagnol, V. , Curtis, J. , Epstein, M. , Mok, K. Y. , Stebbings, E. , Grigoriadou, S. , … Nejentsev, S. (2012). A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics, 28(21), 2747–2754. 10.1093/bioinformatics/bts526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese, M. G. , Eeckman, F. H. , Kulp, D. , & Haussler, D. (1997). Improved splice site detection in genie. Journal of Computational Biology, 4(3), 311–323. 10.1089/cmb.1997.4.311 [DOI] [PubMed] [Google Scholar]
- Rentzsch, P. , Witten, D. , Cooper, G. M. , Shendure, J. , & Kircher, M. (2019). CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Research, 47(D1), D886–D894. 10.1093/nar/gky1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–423. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, J. T. , Thorvaldsdóttir, H. , Winckler, W. , Guttman, M. , Lander, E. S. , Getz, G. , & Mesirov, J. P. (2011). Integrative genomics viewer. Nature Biotechnology, 29(1), 24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim, N.‐L. , Kumar, P. , Hu, J. , Henikoff, S. , Schneider, G. , & Ng, P. C. (2012). SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Research, 40(W1), W452–W457. 10.1093/nar/gks539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson, P. D. , Mort, M. , Ball, E. V. , Evans, K. , Hayden, M. , Heywood, S. , … Cooper, D. N. (2017). The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next‐generation sequencing studies. Human Genetics, 136(6), 665–677. 10.1007/s00439-017-1779-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera, G. A. , Carneiro, M. O. , Hartl, C. , Poplin, R. , Del Angel, G. , & Levy‐Moonshine, A. , … Banks, E. (2013). From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Current Protocols in Bioinformatics, 43(1), 11–10. 10.1007/s00439-017-1779-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, K. , Li, M. , & Hakonarson, H. (2010). ANNOVAR: Functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Research, 38(16), e164. 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong, H. , Tan, D. , Wang, S. , Song, S. , Yang, H. , Gao, K. , … Wu, X. (2015). Genotype/phenotype analysis in Chinese laminin‐α2 deficient congenital muscular dystrophy patients. Clinical Genetics, 87(3), 233–243. 10.1111/cge.12366 [DOI] [PubMed] [Google Scholar]
- Yeo, G. , & Burge, C. B. (2004). Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. Journal of Computational Biology, 11(2–3), 377–394. 10.1089/1066527041410418 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data to support the findings in the study are available from the corresponding author upon reasonable request.