

Abstract
Backgroundd
Sjogren–Larsson syndrome (SLS) is a rare autosomal recessive disorder, characterized by a triad of spastic tetraplegia or diplegia, congenital ichthyosis, and intellectual disability.
Methods
We report a seven‐years‐old female born to consanguineous parents who presented with erythematous dry scaly skin all over the body sparing the face, without collodion membrane which started since birth. There were associated with global developmental delay and seizure disorder. SLS was suspected and hence sequence analysis of the ALDH3A2 gene by next‐generation sequencing was performed for the patient.
Results
A novel nucleotide exchange in homozygous state at position c.1320 in exon 9 of the ALDH3A2 gene (c.1320T>A), leading to a stop of the protein sequence (p.Tyr440) was detected in the patient. Genetic testing of the patient's extended family revealed another four affected family members with the same mutation.
Conclusions
SLS should be suspected in any patient with a triad of ichthyosis, intellectual disability and spastic di/tetraplegia. Molecular genetic testing of the ALDH3A2 gene should be performed to confirm the diagnosis. Extended family screening is highly recommended.
SLS should be suspected in any patient with a triad of ichthyosis, intellectual disability, and spastic di/tetraplegia. Molecular genetic testing of the ALDH3A2 gene should be performed to confirm the diagnosis.

1. INTRODUCTION
Sjogren–Larsson syndrome (SLS); Phenotype MIM number 270200 (SLS; MIM#270200) (Roullet & Rizzo, 2014); is a rare neurocutaneous recessively inherited autosomal disease with spasticity (tetraplegia or diplegia), congenital ichthyosis and intellectual disability (mild to moderate). Less commonly, they are born preterm with retinal perifoveal crystalline inclusions and in some reports retinal pigmentary degeneration in the macular region, short stature, kyphoscoliosis, seizures, and delayed speech (Rizzo, 2016).
The incriminated genetic mutation in SLS is the ALDH3A2 gene mutation; Gene/Locus MIM number 609523. ALDH3A2 gene encodes fatty aldehyde dehydrogenase enzyme (FALDH), an enzyme that catalyzes the oxidation of the fatty aldehyde to fatty acid (Rizzo, 2014).
The ALDH3A2 gene is located on chromosome 17p11.2. More than 70 mutations have been discovered in SLS patients, in form of small deletions or insertions, splicing defects, missense mutations, and complex mutations composed of deletion/insertions and nucleotide substitutions (Engelstad et al., 2011).
Diagnosis of SLS is confirmed by sequence analysis of the ALDH3A2 gene on the locus 17p11.2. and/or measurement of FALDH or fatty alcohol: NAD oxidoreductase in cultured skin fibroblasts (Engelstad et al., 2011). Although some other reports of SLS had been released from Saudi Arabia but mostly without genetic confirmation apart from one report from the Jeddah region, which confirmed a novel large deletion in the ALDH3A2 gene (Gaboon, Jelani, Almramhi, Mohamoud, & Al‐Aama, 2015).
2. METHODS
“Ethical Compliance”: The study was approved by the research and ethics committee of Alhada Armed forces Hospital, Taif, Saudi Arabia.
Here, we report a case series of SLS in a patient and her extended family with a novel genetic mutation. Parents of the patient have provided informed consent for publication of the case.
3. CASE DESCRIPTION
We herein report, a 7‐year‐old Saudi female born to consanguineous parents; first degree cousins. She was born full‐term but small for gestational age by normal vaginal delivery with a birth weight of 1.5 kg. Her parents claimed that pregnancy and delivery have been uneventful. Since birth, she was noted to have erythematous dry scaly skin all over the body sparing the face, without collodion membrane with progressive course.
Parents sought medical advice many times with different dermatologists with good response to abundant hydration with emollient baths, moisturizing creams, and keratolytic agents. Since very early infancy, the patient started to show motor developmental delay and generalized tonic‐clonic seizures developed by the age of 12 months. Epileptogenic discharges were detected by electroencephalography and the seizures were controlled with antiepileptic medications.
It is of note that sitting was at 2 years and crawling was at 3 years of age. Currently, the patient is still not able to stand or walk on her own with delayed speech. She can say 1 sentence of 3 words. Unfortunately, this patient was initially misdiagnosed as spastic cerebral palsy. She received two Botox injections in the lower limbs to relieve muscle spasticity with significant improvement.
On presentation to our care at the age of 7 years, her height and weight were 103 cm (<3rd percentile) and 19 kg (25th percentile), respectively. She had spasticity in both lower limbs, power grade 4 with hyperactive deep tendon reflex in both knees and ankles with ankle clonus. Babinski's sign was positive bilaterally. Skin examination showed generalized ichthyosis that was prominent mainly in the nape, trunk, flexure areas, extremities, and sparing the face; Figure 1.
FIGURE 1.

(a–e) Generalized ichthyosis, mainly prominent in: (a) the nape, (b) trunk and abdomen, (c and d) flexure areas, (e) extremities, and sparing the face
Brain magnetic resonance imaging (MRI) was performed; Figure 2. It showed faint rim of high T2 and FLAIR signal intensity in the peritrigonal and periventricular areas at both frontal regions suggesting delayed myelination. WISC‐IV intelligence test revealed a low average (80‐89) intellectual coefficient. Her ophthalmological examination showed no retinal crystalline inclusions.
FIGURE 2.
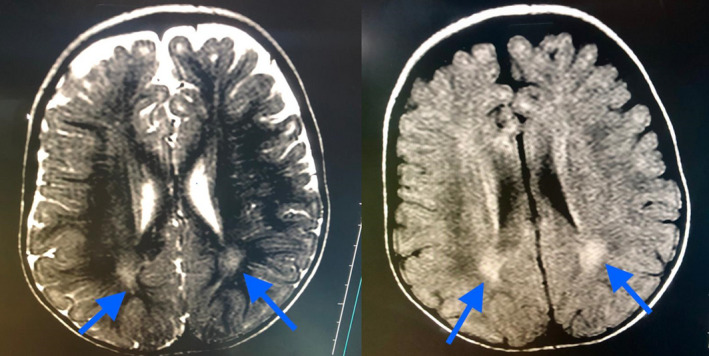
MRI of the brain of the index case
The patient had a youngest sister with the same findings (ichthyosis, developmental delay, and spastic diplegia) and two apparently healthy sisters. Assessment of her extended family revealed the characteristic triad of ichthyosis, spasticity, and developmental delay in one male cousin and two female cousins from maternal aunts.
Constellation of the previous findings suggested the diagnosis of SLS and genetic testing for ALDH3A2 mutation was requested for the index case.
3.1. Genetic analysis of the index case
The coding exons of the ALDH3A2 gene were enriched using Roche/Nimble Gen sequence capture technology and sequenced on an Illumina NextSeq 500 system (next‐generation sequencing, NGS) which revealed homozygous pathogenic variant c.1320T>A p.(Tyr 440*) in the ALDH3A2 gene.
Molecular genetic analysis of the ALDH3A2 gene by NGS showed a nucleotide exchange in homozygous state at position c.1320 in exon 9 of the ALDH3A2 gene (c.1320T>A), leading to a stop of the protein sequence (p.Tyr440), as shown in Figure 3. To the best of our knowledge, this variant has not been described in the literature so far.
FIGURE 3.
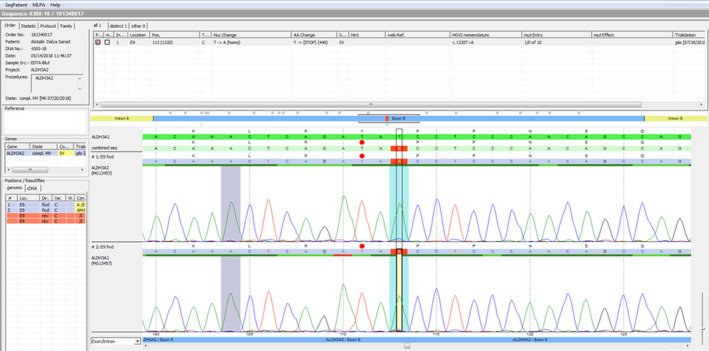
Next‐generation sequencing that shows c.1320T>A mutation in the ALDH3A2 gene
Genetic counseling of the family was provided with a focus on future pregnancies, the expectations of offspring, and the need to perform molecular genetic testing for the parents, and other siblings along with the extended family for the mutation detected in the ALDH3A2 gene.
Interestingly, molecular genetic testing revealed that the proband's younger sister and three cousins are homozygous to the same mutation found in the proband establishing the diagnosis of SLS in the five family members. The parents of the affected five children were heterozygous for the same mutation. Four of the other apparently healthy children from the examined extended family were also heterozygous for the same mutation while three of them did not carry any ALDH3A2 gene mutations (Figure 4).
FIGURE 4.

Family Pedigree
Patients are currently on regular follow‐up with multidisciplinary care plan with dermatologist, pediatrician, neurologist, ophthalmologist, genetic specialist, and physiotherapist.
4. DISCUSSION AND CONCLUSIONS
In 1957, Sjogren and Larson described a rare syndrome consisting of congenital ichthyosis associated with spastic pyramidal symptoms (di/or tetraplegia) and intellectual disability which was named under their names [(Mittal, Maini, Gupta, & Gupta, 2011), (Sjogren & Larsson, 1957)].
SLS is an inborn error of lipid metabolism caused by a deficiency of the microsomal enzyme FALDH that is a component of fatty‐alcohol: NAD‐oxidoreductase‐enzyme‐complex. FALDH deficiency may lead to an accumulation of long‐chain‐fatty‐alcohols within structural consequences for cell membrane integrity which disrupts the function of the skin barrier and the white matter of the brain (Mittal et al., 2011).
SLS patients present cutaneous manifestations due to FALDH deficiency that affects epidermal functioning and leaky water barrier causes ichthyosis (Rizzo, 2014). This kind of ichthyosis in SLS is usually present at birth and is the first symptom that brings the patient to medical attention (Rizzo, 2007) as described in our series.
Most patients with SLS are born without a collodion membrane covering the skin. The ichthyosis is generalized in distribution and is prominent in the nape, trunk, flexure areas, extremities, and sparing the face. The skin is mildly erythematous early in life (Rizzo, 2007), as shown in our series who showed congenital ichthyosis all over the body sparing the face since birth.
Neurological manifestations are variable including non‐progressive intellectual disability and delayed or impaired speech which is usually obvious at 1 to 2 years of age and can range from mild to severe [(Carman, Yimenicioglu, Ekici, & Yakut, 2012), (Fuijkschot, Maassen, Gorter, Gerven, & Willemsen, 2009)].
Spasticity may be apparent before the age of 3 years and is more prominent in the lower limbs than in other parts of the body. The majority of SLS patients require a wheelchair for mobility over time and they also develop contractures (Roullet & Rizzo, 2014). That agreed with our case in whom, the developmental delay was noticed at very early infancy evidenced by the sitting at 2 years of age, crawling at 3 years, and not being able to stand and walk. Delayed speech was also observed; however, intelligence was low average with an intellectual co‐efficient of 89. Papathemeli et al reported normal intelligence of 95 in their patient (Papathemeli et al., 2017).
Cerebral palsy (CP) is the most common cause of motor disability in childhood. CP and SLS share the same features of spasticity and a non‐progressive nature in the clinical course (Carman et al., 2012). The reported index case was first misdiagnosed as CP.
MRI of the brain usually shows hypomyelination involving the periventricular white matter may be extended from the frontal to the occipital area and mild ventricular enlargement may be an additional feature. These findings are due to lipid metabolism products accumulation in the central nervous system (Nakayama, Távora, Alvim, Araujo, & Gama, 2006). Similarly, in the reported proband, brain MRI showed a faint rim of high T2 and FLAIR signal intensity in the peritrigonal area and periventricular at both frontal regions most suggestive of delayed myelination.
Ophthalmological manifestations may be caused by the accumulation of long‐chain fatty alcohols or fatty aldehydes (Cho, Shim, & Kim, 2018). The main features are crystalline juvenile macular dystrophy, cystoid foveal atrophy, and lack of macular pigment [(Cho et al., 2018), (van der Veen et al., 2010)]. High refractive errors (Papathemeli et al., 2017) and photophobia may be present (Cho et al., 2018). Those were not detected in our patient who had a normal ophthalmological assessment.
The reported patient seems not to present as a complete form of SLS, being Ocular signs absent. Many articles point attention to incomplete presentation and atypical findings in SLS [(Papathemeli et al., 2017), (Didona et al., 2007)]. Didona et al, reported two Italian patients with absent ocular manifestations (Didona et al., 2007). Similarly, Papathemeli et al reported absence of the pathognomonic ophthalmological findings of SLS in their patient (Papathemeli et al., 2017).
The differential diagnosis of SLS includes congenital ichthyosiform erythroderma with neurological signs such as Conradi–Hunermann–Happle syndrome, and Tay syndrome and neurocutaneous disorders such as multiple sulfatase deficiency and neutral lipid storage disease. However, MRI, proton magnetic resonance spectroscopy findings help to differentiate SLS from these disorders (Bindu, 2020). Also, the clinical presentation including ichthyosis and developmental delay with spastic diplegia made SLS the leading differential. Proton magnetic resonance spectroscopy was not available in our center.
The diagnosis is confirmed by the sequence analysis of the ALDH3A2 gene (Engelstad et al., 2011). In our case study, molecular genetic analysis of the ALDH3A2 gene by NGS showed a nucleotide exchange in homozygous state at position c.1320 in exon 9 of the ALDH3A2 gene (c.1320T>A), leading to a stop of the protein sequence (p.Tyr440). Same mutation was detected in the other four affected family members. To the best of our knowledge, this variant has not been described in the literature so far.
We followed the agreed world‐wide symptomatic management plan of SLS (Jagell & Lidén, 2008) in all the five patients. We implemented a multidisciplinary care plan with the involvement of a dermatologist, a pediatrician, a neurologist, an ophthalmologist, a genitologist, and a physiotherapist.
The physiotherapy and frequent soft tissue surgery, for example, hamstring release surgery and braces that will help in controlling the spasticity of limbs and facilitate the patient to move around (Jagell & Lidén, 2008).The symptomatic treatment for ichthyosis is including the topical application of soft paraffin, lanolin, 5% citric acid ointment or mineral oil (Kondremul, Zymenol), also, emollients (Vitamin D‐3 analogs), the skin retinoids, and keratolytic (urea) agents are also recommended for SLS patients as they reduce the increased epidermal turnover and soften the epidermis [(Jagell & Lidén, 2008), (Bindu, 2020)].
The sauna baths and frequent showers help in reducing the pruritic condition of SLS patients. Leukotriene synthesis inhibitors were predicted as curative treatment of the future for the ichthyosis and pruritus of the SLS patients (Bindu, 2020).
There are some prospective potential therapeutic options for SLS including; antioxidants, retinoids and vitamin D analogs, dietary lipid modification, fatty aldehyde scavengers, ALDH activators, modulation of sphingosine signaling pathway, replacement of cutaneous lipids, reduction of fatty alcohol production, inhibition of Leukotriene B4 synthesis, and gene therapy (Bindu, 2020).
In conclusion, SLS should be suspected in any patient presenting by the triad mentioned earlier. Molecular genetic test proves the diagnosis and extended family testing of the detected offending mutation help to identify undiagnosed cases and in the prevention of further cases in the same family by proper genetic counseling to avoid the marriage of relatives.
CONFLICT OF INTEREST
All authors declare no competing interests related to the study.
AUTHORS’ CONTRIBUTIONS
KA: set the idea of the study and designed the study. KA, NK, AB, MA, HA, KB: reviewed literature, drafted the manuscript, critically analyzed the data. All authors reviewed and approved the manuscript for final publication.
ETHICAL APPROVAL AND CONSENT TO PARTICIPATE
The study was approved by the research and ethical committee of the participating hospitals. All parents of enrolled children signed written informed consent for the participation of their children in the current study.
CONSENT FOR PUBLICATION
All parents of enrolled children signed written informed consent for the publication of the current study.
ACKNOWLEDGEMENTS
Not applicable.
How to cite this article: Abidi KT, Kamal NM, Bakkar A. AA, et al. Sjogren–Larsson Syndrome: A case series of five members from an extended family with a novel mutation. Molecular Genetics & Genomic Medicine. 2020;8:e1487. 10.1002/mgg3.1487
References
- Bindu, P. S. (2020). Sjogren‐Larsson Syndrome: Mechanisms and management. The Application of Clinical Genetics, 13, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman, K. B. , Yimenicioglu, S. , Ekici, A. , & Yakut, A. (2012). Sjogren‐larsson syndrome: A rare differential diagnosis of cerebral palsy. International Journal of Clinical Pediatrics, 1(4–5), 133–135. [Google Scholar]
- Cho, K. H. , Shim, S. H. , & Kim, M. (2018). Clinical, biochemical, and genetic aspects of Sjögren‐Larsson syndrome. Clinical Genetics, 93(4), 721–730. [DOI] [PubMed] [Google Scholar]
- Didona, B. , Codispoti, A. , Bertini, E. , Rizzo, W. B. , Carney, G. , Zambruno, G. , … Terrinoni, A. (2007). Novel and recurrent ALDH3A2 mutations in Italian patients with Sjogren‐Larsson syndrome. Journal of Human Genetics, 52, 865–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelstad, H. , Carney, G. , S'Aulis, D. , Rise, J. , Sanger, W. G. , Rudd, M. K. , … Rizzo, W. B. (2011). Large contiguous gene deletions in Sjögren‐Larsson syndrome. Molecular Genetics and Metabolism, 104(3), 356–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuijkschot, J. , Maassen, B. , Gorter, J. W. , Gerven, M. V. , & Willemsen, M. (2009). Speech‐language performance in Sjögren‐Larsson syndrome. Developmental Neurorehabilitation, 12(2), 106–112. [DOI] [PubMed] [Google Scholar]
- Gaboon, N. , Jelani, M. , Almramhi, M. , Mohamoud, H. , & Al‐Aama, J. (2015). Case of Sjögren‐Larsson Syndrome with a large deletion in the ALDH3A2 gene confirmed by single nucleotide polymorphism array analysis. Journal of Dermatology, 42(7), 706–709. [DOI] [PubMed] [Google Scholar]
- Jagell, S. , & Lidén, S. (2008). Ichthyosis in the Sjogren‐Larsson syndrome. Clinical Genetics, 21(4), 243–252. [DOI] [PubMed] [Google Scholar]
- Mittal, A. , Maini, B. , Gupta, S. , & Gupta, S. (2011). Sjogren‐Larsson syndrome: A case report of a rare disease. Indian Dermatology Online Journal, 2(1), 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama, M. , Távora, D. G. F. , Alvim, T. C. L. , Araujo, A. C. B. , & Gama, R. L. (2006). MRI and 1H‐MRS findings of three patients with Sjogren‐Larsson syndrome. Arquivos De Neuro‐Psiquiatria, 64(2b), 398–401. [DOI] [PubMed] [Google Scholar]
- Papathemeli, D. , Mataftsi, A. , Patsatsi, A. , Sotiriadis, D. , Samouilidou, M. , Chondromatidou, S. , & Evangeliou, A. (2017). Atypical presentation of Sjogren‐Larsson syndrome. Case Reports in Pediatrics, 2017, 7981750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo, W. B. (2007). Sjögren‐Larsson syndrome: Molecular genetics and biochemical pathogenesis of fatty aldehyde dehydrogenase deficiency. Molecular Genetics and Metabolism, 90(1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo, W. B. (2014). Fatty aldehyde and fatty alcohol metabolism: Review and importance for epidermal structure and function. Biochimica Et Biophysica Acta (BBA) ‐ Molecular and Cell Biology of Lipids, 1841(3), 377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo, W. B. (2016). Genetics and prospective therapeutic targets for Sjögren‐Larsson Syndrome. Expert Opinion on Orphan Drugs, 4(4), 395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roullet, J.‐B. , & Rizzo, W. B. (2014). Sjögren‐Larsson Syndrome In Daroff R. & Aminoff M. (Eds.), Encyclopedia of the neurological sciences (2nd ed., pp. 179–184). USA: Academic Press. [Google Scholar]
- Sjogren, T. , & Larsson, T. (1957). Oligophrenia in combination with congenital ichthyosis and spastic disorders; a clinical and genetic study. Acta Psychiatrica Neurologica Scandinavica Supplementum., 113, 1–112. [PubMed] [Google Scholar]
- van der Veen, R. L. P. , Fuijkschot, J. , Willemsen, M. A. A. P. , Cruysberg, J. R. M. , Berendschot, T. T. J. M. , & Theelen, T. (2010). Patients with Sjogren‐Larsson syndrome lack macular pigment. Ophthalmology, 117(5), 966–971. [DOI] [PubMed] [Google Scholar]