

Abstract
Background
Our primary aim was to evaluate the systematic reanalysis of singleton exome sequencing (ES) data for unsolved cases referred for any indication. A secondary objective was to undertake a literature review of studies examining the reanalysis of genomic data from unsolved cases.
Methods
We examined data from 58 unsolved cases referred between June 2016 and March 2017. First reanalysis at 4–13 months after the initial report considered genes newly associated with disease since the original analysis; second reanalysis at 9–18 months considered all disease‐associated genes. At 25–34 months we reviewed all cases and the strategies which solved them.
Results
Reanalysis of existing ES data alone at two timepoints did not yield new diagnoses. Over the same timeframe, 10 new diagnoses were obtained (17%) from additional strategies, such as microarray detection of copy number variation, repeat sequencing to improve coverage, and trio sequencing. Twenty‐seven peer‐reviewed articles were identified on the literature review, with a median new diagnosis rate via reanalysis of 15% and median reanalysis timeframe of 22 months.
Conclusion
Our findings suggest that an interval of greater than 18 months from the original report may be optimal for reanalysis. We also recommend a multi‐faceted strategy for cases remaining unsolved after singleton ES.
Keywords: exome sequencing, genome sequencing, rare disease, reanalysis
We evaluated the systematic reanalysis of singleton exome sequencing (ES) data for unsolved cases and undertook a literature review of studies examining reanalysis of genomic data from unsolved cases. Our findings suggest that an interval of greater than 18 months from the original report may be optimal for reanalysis. We also recommend a multi‐faceted strategy for cases remaining unsolved after singleton ES.
1. INTRODUCTION
A recent scoping review of the literature on the clinical impact of genomic sequencing in pediatric patients reported a median diagnostic yield of 33.2% (Smith et al., 2019). A “negative” exome in the remaining two‐thirds of cases may be due to a number of factors. There are technical challenges such as imperfect sequencing, variant annotation, and filtering; together with difficulties in variant interpretation owing to incomplete knowledge of gene‐/variant‐disease associations, gene function; and the evolution of patient phenotype with time. One advantage of ES and genome sequencing (GS) is the capacity for reanalysis of existing data in unsolved cases. We applied different reanalysis strategies at two timepoints to evaluate the effectiveness of reanalyzing singleton ES data and compared against other strategies to reach a diagnosis in unsolved cases.
To complement our reanalysis, we undertook a literature review of studies investigating the incremental diagnostic rate achieved via the reanalysis of ES/GS data. There has been no comprehensive literature review of reanalysis studies to date, making this a timely exercise in light of the recent statement by the American College of Medical Genetics and Genomics (ACMG) highlighting the need for reanalysis in undiagnosed cases (Deignan et al., 2019). Understanding the new diagnostic rate afforded by different reanalysis strategies will help to guide laboratories and clinicians considering reanalysis for their patients, as well as inform policy regarding funding schema for systematic reanalysis.
2. MATERIALS AND METHODS
2.1. Ethical compliance
Written consent was provided for each reanalysis case to be included in this research and additional research‐based analysis was undertaken through the Undiagnosed Diseases Program Victoria as approved by The Royal Children's Hospital Ethics Committee (HREC36291A).
2.2. Patient selection
We reanalyzed data from unsolved cases referred for ES to Victorian Clinical Genetics Services (VCGS) Clinical Genomics Laboratory over a 10‐month period between June 2016 and March 2017. Unsolved cases were defined as those with no diagnosis established on ES after analysis of known human Mendelian disorder genes, termed the “Mendeliome,” which comprised 3734 genes in July 2017. Selection criteria included a request for clinical genomic testing irrespective of indication and the provision of informed consent for genomic testing and reanalysis. The following data were collected: demographics, age, alive or deceased, and referral indication.
2.3. Study design
Our first reanalysis (“Tier 1,” Figure 1) involved reviewing only variants in genes newly associated with the disease since the time of original analysis. These genes are added as updates to the Mendeliome every 6 months (“Mendeliome Update”), and are generated from the review of current literature. Reanalysis using this list of updated Mendeliome genes was conducted in July 2017, between 4 and 13 months after the original ES report was generated. There were 570 genes added to the Mendeliome in July 2017, incorporating updates from July 2016, January 2017, and July 2017. Our second reanalysis (“Tier 2”) involved reviewing variants in all disease‐associated genes for each case. Interrogation of the existing exome data for the whole Mendeliome (4304 genes) occurred in November 2017, nine to 18 months after the initial ES report. Finally, we reviewed the status of all cases in April 2019 (25–34 months after the initial report) and collated all strategies that led to a diagnosis.
FIGURE 1.

Study design
2.4. Exome sequencing, variant annotation, and interpretation
Singleton ES was performed at VCGS Pathology, an accredited (National Association of Testing Authorities, Australia) laboratory, using the Nextera Rapid Capture Exome kit (Illumina, San Diego, CA) on HiSeq 4000 sequencers (Illumina, San Diego, CA), with a target mean coverage of 100x and a minimum of 91% of bases sequenced to at least 15x. ES variant calls were reannotated in July 2017 (Tier 1 reanalysis) and November 2017 (Tier 2 reanalysis) using the latest respective version of our custom in‐house bioinformatics pipeline, Cpipe,(Sadedin et al., 2015) in order to generate annotated variant calls within the target region (coding exons ±2 base pairs), via alignment to the reference genome (GRCh37). Variants were annotated against all gene transcripts, with the curation of variants against the HGNC recommended transcript (according to HGVS nomenclature). Variant filtering was performed as previously described (Stark et al., 2016), with the data for each patient assessed and prioritized for classification by two clinical geneticists—the referring geneticist and a study geneticist (N.T.). Variant classification was performed using our in‐house variant classification scheme based on the American College of Medical Genetics and Genomics guidelines for variant interpretation (Richards et al., 2015). Class 3 variants (Variants of Uncertain Significance, VUS) were further sub‐classified as being of potential clinical relevance (class 3A), unknown clinical relevance (class 3B) or low clinical relevance (class 3C) based on the relevance of the variant to the patient phenotype and likelihood of being up‐ or down‐graded with additional evidence (e.g., segregation). Cases were selected for reanalysis if the original report was uninformative—being either a “negative” report (i.e., no clinically reportable variants were identified) or a report with class 3B or 3C variants. The process for patient selection and data analysis is summarized in Figure 2.
FIGURE 2.
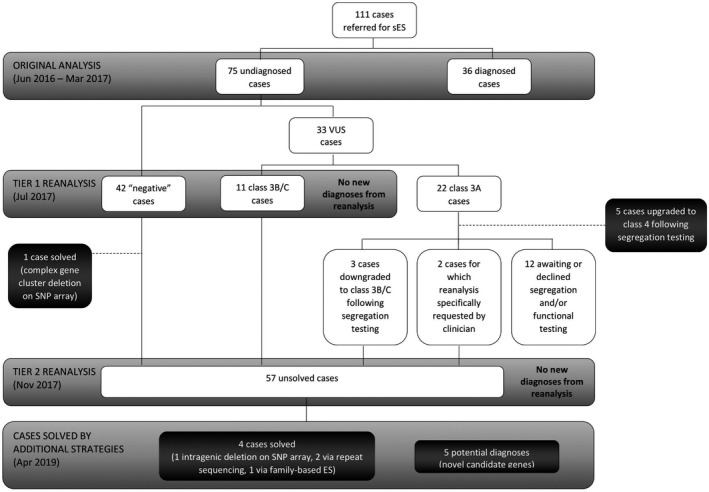
Summary of patient selection
2.5. Literature search
PubMed, Embase, and Medline were searched in December 2019 for articles reporting reanalysis of ES/GS data in unsolved cases. Full search strategies are available as Supplementary Information. Complete reference details for all articles were imported into Endnote (Clarivate Analytics, Boston, MA) and duplicate records removed. Articles published after 1 January 2017 were selected for assessment of eligibility. Two independent reviewers (NT and TT) compared all titles and abstracts against eligibility criteria with any discrepancies resolved by consensus. One author (NT) reviewed each full‐text article and extracted relevant data. Articles that met the following criteria were included in the review: (1) peer‐reviewed original research article; (2) reanalysis was performed of ES or GS data for individuals who received no diagnosis at the time of reporting the original ES or GS result. Studies that reanalyzed patient data derived from targeted gene panels only were excluded. Studies of patients enrolled in a research protocol performing ES or GS for a clinical indication were included. Studies were limited to those published in English. No restrictions were placed on study design or the number of individuals reported. As a result, this literature review encompassed studies utilizing a number of different study methodologies across diverse patient populations. Therefore, results across studies were summarized and narratively described rather than statistically combined in a meta‐analysis.
3. RESULTS
3.1. Characteristics of the study population
One hundred and eleven patients in this cohort were referred for clinical ES during the 10‐month study period; 51 females and 60 males. At the time of the original analysis, pathogenic and likely pathogenic variants were identified for 36 cases, conferring a diagnostic rate of 32% (36/111). The remaining 75 cases received uninformative ES results. The median age at the time of the initial report date was similar for the diagnosed versus uninformative groups (3 vs. 3.5 years). The number of deceased individuals at the time of referral for ES was more than twice as high for the uninformative group than for the diagnosed group (Table 1). Whilst the referral indication varied, the most common phenotype tested was a syndromic neurodevelopmental disorder (56% of all referrals).
TABLE 1.
Characteristics of the study population
| Original ES report outcome | Gender | Median age at initial report date | Deceased at time of referral | Referral indication | ||||
|---|---|---|---|---|---|---|---|---|
| Female | Male | Syndromic Neurodevelopmental Disorder | Non‐Syndromic Neurodevelopmental Disorder | Multiple Congenital Anomalies | Single System Disorder | |||
| Diagnosed (36) | 18 | 18 | 3 years (101 days to 43 years) | 1 (2.8%) | 25 | 2 | 1 | 8 (ophthalmology 3, renal 3, dermatology 1, metabolic 1) |
| Undiagnosed (75) | 33 | 42 | 3.5 years (31 days to 47 years) | 5 (6.7%) | 37 | 3 | 7 | 28 (neurology 11, gastroenterology 4, ophthalmology 4, dermatology 2, oncology 2, respiratory 1, renal 1, immunology 1, endocrinology 1, growth 1) |
3.2. Tier 1 reanalysis
Of the 75 original ES cases that were uninformative, 53 (42 negative cases and 11 class 3B/3C cases) underwent reanalysis applying only the Mendeliome Update genes in July 2017 (Figure 2). Class 3A cases were not examined at this stage as the majority were undergoing or had segregation testing performed, leading to the reclassification of these variants (Figure 2). Eighteen additional variants in 12 negative cases were curated, with no pathogenic or likely pathogenic variants ultimately identified. No additional variants of interest were proposed for curation in the 11 cases initially reported with 3B/3C variants. One case was solved at this stage via SNP array testing that identified an ATAD3 gene cluster deletion (patient S3 in Desai et al., 2017).
3.3. Tier 2 reanalysis
Fifty‐seven unsolved cases were fully reanalyzed after the Mendeliome was updated in November 2017. These cases comprised: 41 of the original 42 negative cases (owing to the solved ATAD3 case); the 11 cases with class 3B/3C variants; three cases that were originally reported with class 3A variants that were later downgraded to class 3B variants following segregation studies; and two cases where the referring clinician requested reanalysis despite class 3A variants being reported on the original ES. Forty‐nine variants in 20 cases were curated, the majority of which were found to be not diagnostic. One patient was identified as a heterozygous carrier of a pathogenic variant in GJB2, which is associated with autosomal recessive deafness (OMIM#220290). This condition was not associated with the patient's phenotype and was, therefore, an incidental carrier status finding. We identified two class 3A missense variants and one splice variant of interest in SYNE1 in a patient with congenital arthrogryposis. Segregation studies are being considered for these variants, as the phenotype is compatible with biallelic SYNE1 pathogenic variants (Attali et al., 2009). In a majority of cases (54%), no variants of interest were identified following this reanalysis.
3.4. Cases solved by additional strategies
By April 2019, 15 of the original 75 uninformative ES cases had been solved, conferring an additional diagnostic rate of 20%. The strategies leading to a resolution of these 15 cases were: (i) completion of segregation studies; (ii) completion of functional studies; (iii) enrolment in a gene discovery research program for trio ES/GS; and (iv) interrogation for genomic lesions either not tractable by ES or not well identified due to technical limitations of sequencing and variant filtration.
Four diagnoses were established following segregation studies. Initial analysis of these cases had identified class 3A variants of potential clinical significance which were all upgraded to class 4 likely pathogenic variants following segregation testing. The segregation studies helped to establish: if a variant was de novo in a proband (NAA10, OMIM*300013); if a variant was inherited from an affected parent (CHD7, OMIM *608892); and if compound heterozygous variants were biparentally inherited (ADSL, OMIM*608222; SLC6A5, OMIM*604159). One case underwent functional studies (sedimentation profiling of mitochondrial protein complexes), together with segregation testing, to upgrade a homozygous variant in SLC25A46 (OMIM*610826) from class 3A to class 4 (Abrams et al., 2018).
In five cases, variants in novel candidate genes were identified via the Undiagnosed Diseases Program Victoria (UDP‐Vic), a gene discovery research project in collaboration with the Broad Institute Center for Mendelian Genomics. Families recruited to this program underwent research‐based trio ES (of the proband and both biological parents). Twenty‐one of the 58 unsolved cases (36%) were enrolled in UDP‐Vic with seven diagnoses achieved (33%). Five of these potential diagnoses are in novel candidate genes that are strongly considered to be causative in light of preliminary functional studies and identification of two or more unrelated individuals with concordant phenotypes via the Matchmaker Exchange (Sobreira et al., 2017) initiative. Two diagnoses were made in established genes following family‐based ES via UDP‐Vic (cases 4 and 5 in Table 2).
TABLE 2.
Results of solved cases—published genes
| Case | Mechanism to achieve diagnosis | Gene (*OMIM) | Genomic variant | Phenotype (#OMIM) | Reason pathogenic variant not detected/selected in original singleton ES |
|---|---|---|---|---|---|
| 1 | SNP array (Illumina Human CytoSNP‐12 v2.1) | ATAD3B/3A (*612316/*612317) | ATAD3B/3A 38,667 bp deletion | Cerebellar dysfunction with altered mitochondrial DNA and cholesterol metabolism (Desai et al., 2017) | Not tractable by ES (gene cluster deletion) |
| 2 | High‐density array (Illumina HumanOmni5 Exome‐4 v1.2) | NIPBL (*608667) | arr[hg19] 5p13.2(36791812_36895270)x1~2 | Cornelia de Lange syndrome (#122470) | Not tractable by ES (mosaic intragenic deletion) |
| 3 | Trio GS | SCN8A (*600702) |
NM_014191.3(SCN8A):c.2549G>A; NP_055006.1(SCN8A):p.(Arg850Gln) |
Early infantile epileptic encephalopathy 13 (#614558) | Low coverage |
| 4 | Trio ES | CHD7 (*608892) |
NM_017780.3(CHD7):c.5405‐7G>A; NP_060250.2(CHD7):p.(=) |
CHARGE syndrome (#214800) | Low coverage |
| 5 | Trio ES | WDR45 (*300526) |
NM_007075.3(WDR45):c.411_416delGTTTGA; NP_009006.2(WDR45):p.(Glu137_Phe138del) |
Neurodegeneration with brain iron accumulation 5 (#300894) | Low variant fraction |
Abbreviations: ES, exome sequencing; GS, genome sequencing; SNP, single nucleotide polymorphism.
Another three cases reached diagnoses in established genes via means other than the reannotation of existing singleton ES data. Two diagnoses were made via high resolution SNP‐based chromosomal microarray (cases 1 and 2 in Table 2). The remaining case was solved through trio GS (case 3 in Table 2) as part of a research study of brain malformations and leukodystrophies. Importantly, of the 37 unsolved cases that were not enrolled in the gene discovery research project, upon review in April 2019 three cases are no longer considered to be likely due to an underlying monogenic condition owing to the resolution of clinical features/symptoms with the passage of time.
3.5. Literature review
The number of articles identified by searches of PubMed, Embase, and Medline was 689, 754, and 1177, respectively. Complete reference details for all 2620 articles were imported into Endnote (Clarivate Analytics, Boston, MA) and duplicate records removed, leaving 1586 articles. Of these, 616 articles published after 1 January 2017 were assessed for eligibility. The full‐text of 27 articles were reviewed and the findings are summarized in Table 3. The strategy of reannotation which, for example, incorporates updated population frequency data and in silico pathogenicity scores, has been employed by a majority of studies identified by our literature review (72%) (Al‐Nabhani et al., 2018; Baker et al., 2019; Epilepsy Genetics, 2019; Bowling et al., 2017; Costain et al., 2018; Ewans et al., 2018; Hiatt et al., 2018; Jalkh et al., 2019; Li et al., 2019; Liu et al., 2019; Machini et al., 2019; Nambot et al., 2018; Ngo et al., 2020; Salmon et al., 2019; Schmitz‐Abe et al., 2019; Shashi et al., 2019; Trinh et al., 2019; Wenger et al., 2017; Wright et al., 2018; Xiao et al., 2018). The median new diagnosis rate via this approach across studies is 13% (0.46‐50; weighted average of 9%). Other reanalysis approaches have included: expansion from singleton to family ES studies (Eldomery et al., 2017; Epilepsy Genetics, 2018); revisiting of ES data only if a pathogenic variant is identified on subsequent GS (Alfares et al., 2018); repeat ES due to loss of original ES data (Shamseldin et al., 2017); GS following or in parallel with ES reanalysis (Shashi et al., 2019); and realignment to a new human genome reference build (Need et al., 2017). The median new diagnosis rate for reanalysis studies adopting any of the above approaches is 15% (0.08–83.33; weighted average of 7%), with a median reanalysis timeframe of 22 months (0–5 years).
TABLE 3.
Comparison of reanalysis studies in the literature
| Publication | Diagnostic platform | Cohort of undiagnosed cases with non‐diagnostic ES/GS | Approach to reanalysis | % New diagnoses (number of diagnoses in published genes OR detection of CNV or SV/number of unsolved cases reanalyzed after initial negative report) | Reasons for new diagnoses | Reanalysis timeframe | Recommendation |
|---|---|---|---|---|---|---|---|
| Exome sequencing (ES) | |||||||
| Wenger et al. (2017) | Singleton ES | 40 probands (mostly paediatric), heterogeneous conditions | Reannotation of ES data | 10 (4/40) | New gene publication, relevant literature located at the time of reanalysis | 20 months (average) | Reanalysis at 2‐ to 3‐year interval could result in a 10% diagnostic yield |
| Shamseldin et al. (2017) | Singleton ES | 33 probands (parental consanguinity in all cases), presumed heterogeneous conditions | Repeat ES as data from original ES not available for reanalysis | 48.48 (16/33) | Improved variant filtration via positional mapping | Not reported | Incorporation of positional mapping in the analysis of ES whenever applicable |
| Need et al. (2017) | Family ES | 6 families, heterogeneous conditions | Realignment to new human genome reference build, increased coverage | 83.33 (5/6) | New gene publication, realignment to new human genome reference build | ~4 years | Multifaceted approach to reanalyzing ES data should be a standard part of clinical diagnostic paradigms |
| Eldomery et al. (2017) | ES (68 family studies, 6 singleton) | 74 families, heterogeneous conditions | Expansion to family ES studies, improved data filtering (SNV prioritization, de novo SNVs), CNV detection from ES data | 36.49 (27/74) | New gene publication, gene discovery publication, putative parental mosaicism, biallelic or hemizygous variants identified via family ES studies, CNV detection from ES data (loss of heterozygosity, uniparental disomy, small CNV), translational research (GeneMatcher) | <3 years | Reanalysis of data coupled with the incorporation of additional family member ES data can improve the molecular diagnostic rate |
| Epilepsy Genetics Initiative (2018) | ES (2 family studies, 1 singleton) | 3 probands, epilepsy conditions | Expansion to family ES studies, reannotation of ES variant data | 0.08 (3/3747) | Newly published alternate exon (updated consensus coding sequence database; CCDS) | ~3 to 4 years for family ES cases, singleton case not reported | Iterative interrogation of ES data, with re‐evaluation of other well‐defined alternative exons in known epilepsy genes |
| Nambot et al. (2018) | Singleton ES | 156 probands, neurodevelopmental disorder with or without congenital anomalies | Reannotation of ES data, CNV detection from ES data | 15.38 (24/156) | New gene publication, gene discovery publication, reclassification of originally reported variant, CNV detection from ES data, translational research (Matchmaker Exchange) | <3 years | Prospective reanalysis of ES data in patients with no diagnosis, with consideration for trio ES (before GS) for cases that remain unsolved despite recurrent reanalyses |
| Xiao et al. (2018) | Singleton ES or targeted sequencing | 19 probands (proportion of singleton ES not reported), neurodevelopmental disorder with or without congenital anomalies | Reannotation of ES data, CNV detection from ES data | 26.32 (5/19) | New gene publication, phenotype expansion publication, CNV detection from ES data | 8–18 months | Re‐evaluation at 1‐ to 2‐year interval |
| Wright et al. (2018) | Family ES | 861 families, neurodevelopmental disorders with or without congenital anomalies, abnormal growth parameters, dysmorphic features, unusual behavioral phenotypes | Reannotation of ES data, CNV detection from ES data | 21.14 (182/861) | New gene publication, improved analysis pipelines (updated annotations and variant filtering thresholds), additional analytical methods (to detect chromosomal aneuploidy, CNV detection from ES data, mosaicism, non‐essential splice variants, uniparental disomy) | ~3 years | Iterative reinterpretation of already reported clinical sequencing data should become routine |
| Ewans et al. (2018) | ES (28 family studies, 9 singleton) | 37 families, heterogeneous conditions | Reannotation of ES data | 15.38 (4/26) | New gene publication, improved analysis pipelines, updated patient phenotype information | 12 months | Reanalysis after 12 months or when instigated by referrers |
| Stark et al. (2019) | Singleton ES | 29 probands, infants with suspected monogenic disorders | Re‐evaluation of existing ES data | 13.79 (4/29) | New gene publication | 6–18 months | Reanalysis at 18 months is a cost‐effective model for the storage and re‐examination of genomic data in clinical service delivery |
| Al‐Nabhani et al. (2018) | Singleton ES | 50 probands, intellectual disability | Reannotation of ES data | 12 (6/50) | New gene publication, improved analysis pipelines, updated patient phenotype information | 22 months (average) | Reanalysis of negative exomes in this study of intellectual disability cases solved at least 12% of cases |
| Salmon et al. (2019 | Singleton ES | 84 probands, heterogeneous conditions | Reannotation of ES data | 15.48 (13/84) | New gene publication, updated patient phenotype information | Not reported | Reanalysis of exome data can increase the diagnostic yield and reduce the need for additional costly tests such as genome sequencing |
| Baker et al. (2019) | ES (230 family studies, 10 singleton) | 240 probands, heterogeneous conditions | Reannotation of ES data | 15.83 (38/240) | New gene publication, phenotype expansion publication, candidate gene publication, reclassification of originally reported variant | 1.5 years (reported median) | Automated reanalysis methods can facilitate efficient re‐evaluation of non‐diagnostic samples using up‐to‐date literature |
| Jalkh et al. (2019) | Singleton ES | 101 probands, heterogeneous conditions | Reannotation of ES data, variant filtration based on local ES control dataset | 12.87 (13/101) | New gene publication, updated patient phenotype information | Not reported | ES reanalysis should take into consideration updated bioinformatics tools, novel gene discoveries, and new clinical information |
| Li et al. (2019) | Family ES | 76 families, epilepsy with intellectual disability | Reannotation of ES data | 10.53 (8/76) | New gene publication, relevant literature located at the time of reanalysis, updated patient phenotype information, reclassification of synonymous variant affecting splicing | 0–12+ months | Suitable time points for reanalysis might be 6–12 months after the initial report |
| Epilepsy Genetics Initiative (2019) | ES (singleton and family studies) | 137 probands, epilepsy conditions (excludes 2 probands previously reported in Epilepsy Genetics Initiative, 2018) | Reannotation of ES data | 4.38 (6/137) | New gene publication, gene discovery publication, newly published alternate exon (updated consensus coding sequence database; CCDS), new OMIM entry, translational research (GeneMatcher) | 2.3 years (average) | Periodic reinterrogation of unresolved exomes is critical to improving the diagnostic rate |
| Schmitz‐Abe et al. (2019 | ES (singleton and family studies) | 75 probands, heterogeneous conditions | Reannotation of ES data, improved data filtering (SNV calling and prioritization) | 8 (6/75) | New gene publication, improved variant calling using custom‐built pipeline (VExP) | 1.9 ± 1.4 years (average) | Important to reanalyze negative ES data periodically, preferably annually |
| Liu et al. (2019) | Singleton ES | 188 probands (cohort 1) and 1496 probands (cohort 2), presumed heterogeneous conditions | Reannotation of ES data | 31.91 (60/188 cohort 1) and 15.37 (230/1496 cohort 2) | New gene publication, reclassification of originally reported variant, variant‐specific atypical phenotypic presentations, gene‐specific multiple disease inheritance patterns and mechanisms, newly discovered isoforms encompassing previously unknown exons, complex patient phenotypes obscured by multilocus molecular diagnoses | 5 years (sporadic cases performed earlier) | Periodic, cost‐effective reanalysis may benefit patients and their families and physicians |
| Bruel et al. (2019) | Singleton ES | 313 probands, ID/epileptic encephalopathy with or without congenital anomalies | Re‐evaluation of existing ES data, extending variant interpretation to genes not associated with human disease in OMIM | 15.34 (48/313) | Novel gene discovery, relevant literature located at the time of reanalysis, phenotype expansion publication, translational research (GeneMatcher) | Nil (reanalysis performed immediately after original ES result obtained) | Limitations of singleton ES reanalysis could be overcome utilizing trio ES as a second step |
| Trinh et al. (2019) | ES (singleton and family studies) | 3015 probands (heterogeneous conditions) | Reannotation of ES data | 0.46 (14/3015) | Focus on 14 genes recently nominated by the DDD study (new gene publications for 13 of 14 genes) | Not reported | Importance of re‐evaluating ES data in light of new publications |
| Ngo et al. (2020) | ES (singleton and family studies) | 60 probands (ataxic disorders) | Reannotation of ES data, CNV detection from exome data | 8.33 (5/60) | Known pathogenic variant detected, CNV detection from ES data | 5 years | Key focus for undiagnosed cases on repeating bioinformatic analysis at regular intervals, and use of more comprehensive genomic tools and complete methods to identify mutation types currently not observed in ES as they become available |
| Genome sequencing (GS) | |||||||
| Costain et al. (2018) | Singleton GS | 100 patients (paediatric), heterogeneous conditions | Reannotation of GS variant calls | 10.94 (7/64) | New gene publication, phenotype expansion publication, additional case reports | 3 years | Reanalysis every 1–2 years until diagnosis, or sooner if phenotype evolves |
| Machini et al. (2019) | Singleton GS | 100 patients (50 with cardiomyopathy and 50 healthy) | Reannotation of GS variant calls | 22 (22/100) | New gene and variant publications, reclassification of originally reported variant, improved analysis pipelines | 13 months (mean) | Reanalysis on an annual basis, with the frequency and utility of reanalysis to be guided by the presence of new symptoms or availability of new treatments |
| ES and/or GS | |||||||
| Bowling et al. (2017) | ES or GS (singleton and family studies) | 211 families, neurodevelopmental disorders | Reannotation of ES/GS data | 4.74 (10/211) | New gene publication, improved analysis pipelines, updated patient phenotype information, translational research (GeneMatcher) | Not reported | Systematic reanalysis of genomic data should become standard practice |
| Hiatt et al. (2018) | ES or GS (singleton and family studies) | Number of undiagnosed probands not reported, total cohort of 494 individuals with neurodevelopmental disorders | Reannotation of ES/GS data | 1.57 (6/383) | New gene publication, additional analytical methods (to detect uniparental disomy), translational research (GeneMatcher) | Not reported | Datasets first analyzed over two years ago should be prioritised for reanalysis |
| Alfares et al. (2018) | Singleton ES and GS | 108 patients, presumed heterogeneous conditions | Reanalysis of ES data if a pathogenic variant was detected on GS | 30 (3/10) | New gene publication | 5 months (average) | Until the cost of GS approximates that of ES, reanalyzing ES raw data is recommended before performing GS |
| Shashi et al. (2019) | ES and GS (singleton and family studies) | 38 probands (37 ES, 1 GS), heterogeneous conditions | Reannotation of ES data, improved data filtering (in‐house bioinformatics tools), CNV detection from ES data, repeat ES/GS, GS following/in parallel with ES | 50 (19/38) | Deep phenotyping of patients, new gene publication, CNV/SV, translational research (GeneMatcher) | Not reported | GS should be utilized only after ES data have been extensively mined and combined with the phenotypic data to maximize its yield |
Abbreviations: CNV, copy number variation; SNV, single nucleotide variation; SV, structural variant.
4. DISCUSSION
Reanalysis of existing genomic data in unsolved cases increases diagnostic yield in the setting of well‐defined disease cohorts (e.g., epilepsy) as well as unselected cohorts with heterogeneous conditions (Table 3). The first publication to address reanalysis systematically (Wenger et al., 2017) did so by reanalyzing negative exome cases for 40 mostly pediatric patients via the reannotation of ES data using updated bioinformatics pipelines. Re‐evaluation of existing ES data conducted without reannotation using updated software and/or reference databases has achieved an incremental diagnosis rate of 14% via reanalysis at six to 18 months after the original analysis (Stark et al., 2019). This approach was also applied by Bruel et al. (2019), with the authors performing reanalysis immediately after obtaining the original ES result. The particular focus of the Bruel study was to extend variant interpretation to genes not currently associated with disease in OMIM for the purpose of research. As such, the need to wait for the benefit of newly published data (e.g., new gene discovery, functional studies) and updated resources (e.g., gnomAD) was not essential, making immediate re‐evaluation of existing ES data feasible in this setting.
Whilst Nambot et al. (2018) reported annual reanalysis over three years, we applied different reanalysis strategies at two timepoints to elucidate the impact of each approach. We elected to reanalyze only genes updated to the Mendeliome in the first instance (Tier 1 reanalysis) in an effort to target newly published genes given that more than three‐quarters of reanalysis studies demonstrate that new diagnoses are achieved via this mechanism (Alfares et al., 2018; Al‐Nabhani et al., 2018; Baker et al., 2019; Epilepsy Genetics, 2019; Bowling et al., 2017; Costain et al., 2018; Eldomery et al., 2017; Ewans et al., 2018; Hiatt et al., 2018; Jalkh et al., 2019; Li et al., 2019; Liu et al., 2019; Machini et al., 2019; Nambot et al., 2018; Need et al., 2017; Salmon et al., 2019; Schmitz‐Abe et al., 2019; Shashi et al., 2019; Stark et al., 2019; Trinh et al., 2019; Wenger et al., 2017; Wright et al., 2018; Xiao et al., 2018). We did not see a benefit from this approach alone, most likely due to the short period of time between initial analysis and our first reanalysis time point (4–13 months). Our second approach looked at all Mendeliome genes (Tier 2 reanalysis) to evaluate if new diagnoses would be delivered by new gene publications, newly reported genotype‐phenotype associations (“expanded phenotype” literature), or in light of new/evolving patient features. It was also hypothesized that this comprehensive reanalysis may reveal diagnoses that were “missed” at original analysis. No new diagnoses were made via this strategy at 9–18 months after the initial analysis. Once again, this may be due to reanalysis being performed too soon after initial analysis given that the median timeframe for reanalysis across all published studies has been 22 months (where reported; Table 3).
New diagnoses in this study were identified using approaches other than the reanalysis of singleton ES data. The successful strategies delineate several important lessons: (i) the need for other testing methodologies to detect genomic lesions not tractable by ES; (ii) consideration for repeated/updated ES for improved coverage; (iii) expansion to family sequencing studies to highlight de novo variants in an affected proband; and (iv) utilizing translational research to pursue novel gene discovery.
In our study, there were two instances of copy number variation (CNV) that were not tractable by ES. One case was published after the initial ES as part of a discovery publication describing complex gene cluster deletions involving the ATAD3 gene locus (Desai et al., 2017). ATAD3 cluster deletions were not a recognized entity at the time of reporting the original microarray and this CNV was not tractable by ES. The second case had a mosaic intragenic deletion of NIPBL that was detected on a high‐density SNP‐based array (Illumina HumanOmni5 Exome‐4 v1.2) that afforded better probe coverage of the gene compared to the array platform utilized for the original microarray (Illumina Human CytoSNP‐12 v2.1). Both genomic lesions lay outside of the ES data generated. Examples of other variants that fall into this category include intronic variants, deep intronic splice variants, nucleotide repeat expansions, and structural variants such as chromosomal rearrangements. Improvements in genomic data analysis to detect CNVs (Pfundt et al., 2017) and repeat expansions (Tankard et al., 2018) may mitigate the need for some forms of adjunctive testing in the future.
There were two cases for which pathogenic missense variants were identified following repeat sequencing with improved coverage of the genes of interest (SCN8A, CHD7). In both cases, the variant was detected on original ES in only five reads. Both cases underwent repeat research‐based ES or GS as part of family sequencing studies in different research projects. The improved coverage helped to bring these variants to clinical attention. The CHD7 variant was also of poor sequencing quality and was filtered out of the variant callset by the standard analysis pipeline used in the original analysis. The SCN8A variant was present in the original exome data and of reasonable sequencing quality and thus should have been detectable as a variant of clinical interest.
One patient was diagnosed owing to a de novo in‐frame deletion in WDR45, which is associated with the condition neurodegeneration with brain iron accumulation 5 (NBIA5). This variant was detected on the original ES with a low variant fraction of 0.21. If the variant fraction is particularly low, as in this case, then it is possible to dismiss these calls as being indicative of somatic mosaicism or artifactual. The WDR45 in‐frame deletion was on this occasion brought to clinical attention when highlighted as de novo in the proband via family‐based ES studies. The variant fraction on repeat ES was 0.41.
One‐third of the diagnoses in this study have been derived from novel gene discovery, illustrating the significance of pursuing translational research when attempting to solve the unsolved. In particular, international data sharing through platforms such as GeneMatcher (Sobreira et al., 2015) has been a successful strategy employed by several reanalysis studies to date, with collaborations between research groups resulting in numerous diagnoses and novel gene publications (Epilepsy Genetics, 2019; Bowling et al., 2017; Bruel et al., 2019; Eldomery et al., 2017; Hiatt et al., 2018; Nambot et al., 2018).
One of the limitations of reanalyzing ES cases is the time commitment required. In this study, reanalysis was particularly labor‐intensive for the referring clinician who was asked to participate in the prioritization of variants for classification as they are best positioned to identify variants of relevance to the phenotype. This familiarity with their patients may introduce clinician bias against the selection of promising variants, with variants dismissed as being not of clinical relevance when the patient may have an atypical presentation of a condition that the clinician had yet to consider. Another limitation of this study is that we did not examine the full impact of family‐based ES studies on the new diagnosis rate as not all cases proceeded to family‐based ES. However, we previously evaluated the diagnostic efficacy of trio versus singleton ES and found that the incremental diagnostic yield of trio ES is low in our local context (Tan et al., 2019).
There are several reasons why a case may remain negative after ES. The variant‐disease association may not yet be established in which case serial reanalysis of existing ES data may disclose a diagnosis in the fullness of time. This approach needs to be balanced against the time and cost associated with reanalysis, a labor‐intensive exercise for laboratory scientists and clinicians. The workload required for iterative reanalysis of unsolved ES cases was recently modeled by Baker and colleagues, with an estimated variant analysis burden of five novel annotations per singleton ES case per month (Baker et al., 2019). Whilst their study provides a benchmark to assist clinical laboratories in determining the frequency with which to perform reanalysis, further studies are required to assess optimal strategies for cost‐effectiveness and scalability of routine reanalysis. A case will also remain unsolved if the gene‐disease association has not yet been well established. Recent statistics suggest that the rate of gene discovery may be slowing (Boycott et al., 2017), and thus the time taken for new gene publications to translate to new diagnoses may mean longer reanalysis timeframes are required before a diagnosis is reached. Another reason may be that the disease is not Mendelian in origin, but rather due to a mitochondrial DNA defect, oligogenic or more complex genetic disorder, or the result of an epigenetic or somatic variant. Finally, as seen in this study, a case will remain negative after ES if the underlying cause is not genetic, as with exposures to teratogenic, environmental or infectious agents.
Given that our study did not achieve any additional diagnoses via singleton ES reanalysis, we are unable to draw conclusions from this data regarding the optimal timing of reanalysis. However, the review of literature suggests considering the reanalysis of singleton ES data for unsolved cases after 18 months from the time of the original report. As all additional diagnoses in this study were derived from strategies other than reanalysis of singleton ES data, this illustrates the need to implement a multi‐faceted strategy for cases remaining unsolved after singleton ES, any of which may be employed ahead of reanalysis.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
AUTHOR CONTRIBUTIONS
All authors have made substantial contributions to conception and design, or acquisition of data, or analysis and interpretation of data; NBT, ZS, MBD, AY, MFH, DJA, NJB SMW, SL, and TYT have been involved in drafting the manuscript or revising it critically for important intellectual content; and all authors have given final approval of the version to be published.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
We thank the Broad Institute Center for Mendelian Genomics for their research collaboration with this project. The authors would like to acknowledge Poh Chua for her assistance with the design of the literature review search strategies. NBT acknowledges the Murdoch Children’s Research Institute and the Australian NHMRC Center for Research Excellence in Neurocognitive Disorders for their support.
Tan NB, Stapleton R, Stark Z, et al. Evaluating systematic reanalysis of clinical genomic data in rare disease from single center experience and literature review. Mol Genet Genomic Med. 2020;8:e1508 10.1002/mgg3.1508
Sebastian Lunke and Tiong Y. Tan should be considered joint senior authors.
REFERENCES
- Abrams, A. J. , Fontanesi, F. , Tan, N. B. L. , Buglo, E. , Campeanu, I. J. , Rebelo, A. P. , Kornberg, A. J. , Phelan, D. G. , Stark, Z. , & Zuchner, S. (2018). Insights into the genotype‐phenotype correlation and molecular function of SLC25A46. Human Mutation, 39(12), 1995–2007. 10.1002/humu.23639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfares, A. , Aloraini, T. , subaie, L. A. , Alissa, A. , Qudsi, A. A. , Alahmad, A. , Mutairi, F. A. , Alswaid, A. , Alothaim, A. , Eyaid, W. , Albalwi, M. , Alturki, S. , & Alfadhel, M. (2018). Whole‐genome sequencing offers additional but limited clinical utility compared with reanalysis of whole‐exome sequencing. Genetics in Medicine, 20(11), 1328–1333. 10.1038/gim.2018.41 [DOI] [PubMed] [Google Scholar]
- Al‐Nabhani, M. , Al‐Rashdi, S. , Al‐Murshedi, F. , Al‐Kindi, A. , Al‐Thihli, K. , Al‐Saegh, A. , Al‐Futaisi, A. , Al‐Mamari, W. , Zadjali, F. , & Al‐Maawali, A. (2018). Reanalysis of exome sequencing data of intellectual disability samples: Yields and benefits. Clinical Genetics, 94(6), 495–501. 10.1111/cge.13438 [DOI] [PubMed] [Google Scholar]
- Attali, R. , Warwar, N. , Israel, A. , Gurt, I. , McNally, E. , Puckelwartz, M. , Glick, B. , Nevo, Y. , Ben‐Neriah, Z. , & Melki, J. (2009). Mutation of SYNE‐1, encoding an essential component of the nuclear lamina, is responsible for autosomal recessive arthrogryposis. Human Molecular Genetics, 18(18), 3462–3469. 10.1093/hmg/ddp290 [DOI] [PubMed] [Google Scholar]
- Baker, S. W. , Murrell, J. R. , Nesbitt, A. I. , Pechter, K. B. , Balciuniene, J. , Zhao, X. , Yu, Z. , Denenberg, E. H. , DeChene, E. T. , Wilkens, A. B. , Bhoj, E. J. , Guan, Q. , Dulik, M. C. , Conlin, L. K. , Abou Tayoun, A. N. , Luo, M. , Wu, C. , Cao, K. , Sarmady, M. , & Santani, A. B. (2019). Automated clinical exome reanalysis reveals novel diagnoses. The Journal of Molecular Diagnostics, 21(1), 38–48. 10.1016/j.jmoldx.2018.07.008 [DOI] [PubMed] [Google Scholar]
- Berkovic, S. F. , Goldstein, D. B. , Heinzen, E. L. , Laughlin, B. L. , Lowenstein, D. H. , Lubbers, L. , Stewart, R. , Whittemore, V. , Angione, K. , Bazil, C. W. , Bier, L. , Bluvstein, J. , Brimble, E. , Campbell, C. , Cavalleri, G. , Chambers, C. , Choi, H. , Cilio, M. R. , Ciliberto, M. , & Stong, N. (2019). The epilepsy genetics initiative: Systematic reanalysis of diagnostic exomes increases yield. Epilepsia, 60(5), 797–806. 10.1111/epi.14698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowling, K. M. , Thompson, M. L. , Amaral, M. D. , Finnila, C. R. , Hiatt, S. M. , Engel, K. L. , Cochran, J. N. , Brothers, K. B. , East, K. M. , Gray, D. E. , Kelley, W. V. , Lamb, N. E. , Lose, E. J. , Rich, C. A. , Simmons, S. , Whittle, J. S. , Weaver, B. T. , Nesmith, A. S. , Myers, R. M. , & Cooper, G. M. (2017). Genomic diagnosis for children with intellectual disability and/or developmental delay. Genome Medicine, 9(1), 43 10.1186/s13073-017-0433-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boycott, K. M. , Rath, A. , Chong, J. X. , Hartley, T. , Alkuraya, F. S. , Baynam, G. , Brookes, A. J. , Brudno, M. , Carracedo, A. , den Dunnen, J. T. , Dyke, S. O. M. , Estivill, X. , Goldblatt, J. , Gonthier, C. , Groft, S. C. , Gut, I. , Hamosh, A. , Hieter, P. , Höhn, S. , & Lochmüller, H. (2017). International cooperation to enable the diagnosis of all rare genetic diseases. American Journal of Human Genetics, 100(5), 695–705. 10.1016/j.ajhg.2017.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruel, A.‐L. , Nambot, S. , Quéré, V. , Vitobello, A. , Thevenon, J. , Assoum, M. , Moutton, S. , Houcinat, N. , Lehalle, D. , Jean‐Marçais, N. , Chevarin, M. , Jouan, T. , Poë, C. , Callier, P. , Tisserand, E. , Philippe, C. , Them, F. T. M. , Duffourd, Y. , Faivre, L. , & Thauvin‐Robinet, C. (2019). Increased diagnostic and new genes identification outcome using research reanalysis of singleton exome sequencing. European Journal of Human Genetics, 27, 1519–1531. 10.1038/s41431-019-0442-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costain, G. , Jobling, R. , Walker, S. , Reuter, M. S. , Snell, M. , Bowdin, S. , Cohn, R. D. , Dupuis, L. , Hewson, S. , Mercimek‐Andrews, S. , Shuman, C. , Sondheimer, N. , Weksberg, R. , Yoon, G. , Meyn, M. S. , Stavropoulos, D. J. , Scherer, S. W. , Mendoza‐Londono, R. , & Marshall, C. R. (2018). Periodic reanalysis of whole‐genome sequencing data enhances the diagnostic advantage over standard clinical genetic testing. European Journal of Human Genetics, 26(5), 740–744. 10.1038/s41431-018-0114-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deignan, J. L. , Chung, W. K. , Kearney, H. M. , Monaghan, K. G. , Rehder, C. W. , & Chao, E. C. (2019). Points to consider in the reevaluation and reanalysis of genomic test results: A statement of the American College of Medical Genetics and Genomics (ACMG). Genetics in Medicine, 21(6), 1267–1270. 10.1038/s41436-019-0478-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai, R. , Frazier, A. E. , Durigon, R. , Patel, H. , Jones, A. W. , Dalla Rosa, I. , Lake, N. J. , Compton, A. G. , Mountford, H. S. , Tucker, E. J. , Mitchell, A. L. R. , Jackson, D. , Sesay, A. , Di Re, M. , van den Heuvel, L. P. , Burke, D. , Francis, D. , Lunke, S. , McGillivray, G. , & Spinazzola, A. (2017). ATAD3 gene cluster deletions cause cerebellar dysfunction associated with altered mitochondrial DNA and cholesterol metabolism. Brain, 140(6), 1595–1610. 10.1093/brain/awx094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldomery, M. K. , Coban‐Akdemir, Z. , Harel, T. , Rosenfeld, J. A. , Gambin, T. , Stray‐Pedersen, A. , Küry, S. , Mercier, S. , Lessel, D. , Denecke, J. , Wiszniewski, W. , Penney, S. , Liu, P. , Bi, W. , Lalani, S. R. , Schaaf, C. P. , Wangler, M. F. , Bacino, C. A. , Lewis, R. A. , & Lupski, J. R. (2017). Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Medicine, 9(1), 26 10.1186/s13073-017-0412-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epilepsy Genetics, I . (2018). De novo variants in the alternative exon 5 of SCN8A cause epileptic encephalopathy. Genetics in Medicine, 20(2), 275–281. 10.1038/gim.2017.100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewans, L. J. , Schofield, D. , Shrestha, R. , Zhu, Y. , Gayevskiy, V. , Ying, K. , Walsh, C. , Lee, E. , Kirk, E. P. , Colley, A. , Ellaway, C. , Turner, A. , Mowat, D. , Worgan, L. , Freckmann, M.‐L. , Lipke, M. , Sachdev, R. , Miller, D. , Field, M. , & Roscioli, T. (2018). Whole‐exome sequencing reanalysis at 12 months boosts diagnosis and is cost‐effective when applied early in Mendelian disorders. Genetics in Medicine, 20(12), 1564–1574. 10.1038/gim.2018.39 [DOI] [PubMed] [Google Scholar]
- Hiatt, S. M. , Amaral, M. D. , Bowling, K. M. , Finnila, C. R. , Thompson, M. L. , Gray, D. E. , Lawlor, J. , Cochran, J. N. , Bebin, E. M. , Brothers, K. B. , East, K. M. , Kelley, W. V. , Lamb, N. E. , Levy, S. E. , Lose, E. J. , Neu, M. B. , Rich, C. A. , Simmons, S. , Myers, R. M. , & Cooper, G. M. (2018). Systematic reanalysis of genomic data improves quality of variant interpretation. Clinical Genetics, 94(1), 174–178. 10.1111/cge.13259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalkh, N. , Corbani, S. , Haidar, Z. , Hamdan, N. , Farah, E. , Abou Ghoch, J. , Ghosn, R. , Salem, N. , Fawaz, A. , Djambas Khayat, C. , Rajab, M. , Mourani, C. , Moukarzel, A. , Rassi, S. , Gerbaka, B. , Mansour, H. , Baassiri, M. , Dagher, R. , Breich, D. , & Chouery, E. (2019). The added value of WES reanalysis in the field of genetic diagnosis: lessons learned from 200 exomes in the Lebanese population. BMC Medical Genomics, 12(1), 11 10.1186/s12920-019-0474-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Gao, K. , Yan, H. , Xiangwei, W. , Liu, N. , Wang, T. , Xu, H. , Lin, Z. , Xie, H. , Wang, J. , Wu, Y. E. , & Jiang, Y. (2019). Reanalysis of whole exome sequencing data in patients with epilepsy and intellectual disability/mental retardation. Gene, 700, 168–175. 10.1016/j.gene.2019.03.037 [DOI] [PubMed] [Google Scholar]
- Liu, P. , Meng, L. , Normand, E. A. , Xia, F. , Song, X. , Ghazi, A. , Rosenfeld, J. , Magoulas, P. L. , Braxton, A. , Ward, P. , Dai, H. , Yuan, B. O. , Bi, W. , Xiao, R. , Wang, X. , Chiang, T. , Vetrini, F. , He, W. , Cheng, H. , & Yang, Y. (2019). Reanalysis of clinical exome sequencing data. New England Journal of Medicine, 380(25), 2478–2480. 10.1056/NEJMc1812033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machini, K. , Ceyhan‐Birsoy, O. , Azzariti, D. R. , Sharma, H. , Rossetti, P. , Mahanta, L. , Hutchinson, L. , McLaughlin, H. , Green, R. C. , Lebo, M. , & Rehm, H. L. (2019). Analyzing and reanalyzing the genome: Findings from the MedSeq Project. American Journal of Human Genetics, 105, 177–188. 10.1016/j.ajhg.2019.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambot, S. , Thevenon, J. , Kuentz, P. , Duffourd, Y. , Tisserant, E. , Bruel, A.‐L. , Mosca‐Boidron, A.‐L. , Masurel‐Paulet, A. , Lehalle, D. , Jean‐Marçais, N. , Lefebvre, M. , Vabres, P. , El Chehadeh‐Djebbar, S. , Philippe, C. , Tran Mau‐Them, F. , St‐Onge, J. , Jouan, T. , Chevarin, M. , Poé, C. , & Thauvin‐Robinet, C. (2018). Clinical whole‐exome sequencing for the diagnosis of rare disorders with congenital anomalies and/or intellectual disability: substantial interest of prospective annual reanalysis. Genetics in Medicine, 20(6), 645–654. 10.1038/gim.2017.162 [DOI] [PubMed] [Google Scholar]
- Need, A. C. , Shashi, V. , Schoch, K. , Petrovski, S. , & Goldstein, D. B. (2017). The importance of dynamic re‐analysis in diagnostic whole exome sequencing. Journal of Medical Genetics, 54(3), 155–156. 10.1136/jmedgenet-2016-104306 [DOI] [PubMed] [Google Scholar]
- Ngo, K. J. , Rexach, J. E. , Lee, H. , Petty, L. E. , Perlman, S. , Valera, J. M. , Deignan, J. L. , Mao, Y. , Aker, M. , Posey, J. E. , Jhangiani, S. N. , Coban‐Akdemir, Z. H. , Boerwinkle, E. , Muzny, D. , Nelson, A. B. , Hassin‐Baer, S. , Poke, G. , Neas, K. , Geschwind, M. D. , & Fogel, B. L. (2020). A diagnostic ceiling for exome sequencing in cerebellar ataxia and related neurological disorders. Human Mutation, 41(2), 487–501. 10.1002/humu.23946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfundt, R. , del Rosario, M. , Vissers, L. E. L. M. , Kwint, M. P. , Janssen, I. M. , de Leeuw, N. , Yntema, H. G. , Nelen, M. R. , Lugtenberg, D. , Kamsteeg, E.‐J. , Wieskamp, N. , Stegmann, A. P. A. , Stevens, S. J. C. , Rodenburg, R. J. T. , Simons, A. , Mensenkamp, A. R. , Rinne, T. , Gilissen, C. , Scheffer, H. , & Hehir‐Kwa, J. Y. (2017). Detection of clinically relevant copy‐number variants by exome sequencing in a large cohort of genetic disorders. Genetics in Medicine, 19(6), 667–675. 10.1038/gim.2016.163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , & Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadedin, S. P. , Dashnow, H. , James, P. A. , Bahlo, M. , Bauer, D. C. , Lonie, A. , Lunke, S. , Macciocca, I. , Ross, J. P. , Siemering, K. R. , Stark, Z. , White, S. M. , Taylor, G. , Gaff, C. , Oshlack, A. , & Thorne, N. P. (2015). Cpipe: A shared variant detection pipeline designed for diagnostic settings. Genome Medicine, 7(1), 68 10.1186/s13073-015-0191-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon, L. B. , Orenstein, N. , Markus‐Bustani, K. , Ruhrman‐Shahar, N. , Kilim, Y. , Magal, N. , & Bazak, L. (2019). Improved diagnostics by exome sequencing following raw data reevaluation by clinical geneticists involved in the medical care of the individuals tested. Genetics in Medicine, 21(6), 1443–1451. 10.1038/s41436-018-0343-7 [DOI] [PubMed] [Google Scholar]
- Schmitz‐Abe, K. , Li, Q. , Rosen, S. M. , Nori, N. , Madden, J. A. , Genetti, C. A. , Wojcik, M. H. , Ponnaluri, S. , Gubbels, C. S. , Picker, J. D. , O’Donnell‐Luria, A. H. , Yu, T. W. , Bodamer, O. , Brownstein, C. A. , Beggs, A. H. , & Agrawal, P. B. (2019). Unique bioinformatic approach and comprehensive reanalysis improve diagnostic yield of clinical exomes. European Journal of Human Genetics, 27(9), 1398–1405. 10.1038/s41431-019-0401-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamseldin, H. E. , Maddirevula, S. , Faqeih, E. , Ibrahim, N. , Hashem, M. , Shaheen, R. , & Alkuraya, F. S. (2017). Increasing the sensitivity of clinical exome sequencing through improved filtration strategy. Genetics in Medicine, 19(5), 593–598. 10.1038/gim.2016.155 [DOI] [PubMed] [Google Scholar]
- Shashi, V. , Schoch, K. , Spillmann, R. , Cope, H. , Tan, Q.‐G. , Walley, N. , Pena, L. , McConkie‐Rosell, A. , Jiang, Y.‐H. , Stong, N. , Need, A. C. , & Goldstein, D. B. (2019). A comprehensive iterative approach is highly effective in diagnosing individuals who are exome negative. Genetics in Medicine, 21(1), 161–172. 10.1038/s41436-018-0044-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, H. S. , Swint, J. M. , Lalani, S. R. , Yamal, J.‐M. , de Oliveira Otto, M. C. , Castellanos, S. , Taylor, A. , Lee, B. H. , & Russell, H. V. (2019). Clinical application of genome and exome sequencing as a diagnostic tool for pediatric patients: A scoping review of the literature. Genetics in Medicine, 21(1), 3–16. 10.1038/s41436-018-0024-6 [DOI] [PubMed] [Google Scholar]
- Sobreira, N. L. M. , Arachchi, H. , Buske, O. J. , Chong, J. X. , Hutton, B. , Foreman, J. , Schiettecatte, F. , Groza, T. , Jacobsen, J. O. B. , Haendel, M. A. , Boycott, K. M. , Hamosh, A. , & Rehm, H. L. ; Matchmaker Exchange, C . (2017). Matchmaker exchange. Current Protocols in Human Genetics, 95, 9.31.1–9.31.15. 10.1002/cphg.50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobreira, N. , Schiettecatte, F. , Valle, D. , & Hamosh, A. (2015). GeneMatcher: A matching tool for connecting investigators with an interest in the same gene. Human Mutation, 36(10), 928–930. 10.1002/humu.22844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark, Z. , Schofield, D. , Martyn, M. , Rynehart, L. , Shrestha, R. , Alam, K. , & White, S. M. (2019). Does genomic sequencing early in the diagnostic trajectory make a difference? A follow‐up study of clinical outcomes and cost‐effectiveness. Genetics in Medicine, 21(1), 173–180. 10.1038/s41436-018-0006-8 [DOI] [PubMed] [Google Scholar]
- Stark, Z. , Tan, T. Y. , Chong, B. , Brett, G. R. , Yap, P. , Walsh, M. , Yeung, A. , Peters, H. , Mordaunt, D. , Cowie, S. , Amor, D. J. , Savarirayan, R. , McGillivray, G. , Downie, L. , Ekert, P. G. , Theda, C. , James, P. A. , Yaplito‐Lee, J. , Ryan, M. M. , & White, S. M. (2016). A prospective evaluation of whole‐exome sequencing as a first‐tier molecular test in infants with suspected monogenic disorders. Genetics in Medicine, 18(11), 1090–1096. 10.1038/gim.2016.1 [DOI] [PubMed] [Google Scholar]
- Tan, T. Y. , Lunke, S. , Chong, B. , Phelan, D. , Fanjul‐Fernandez, M. , Marum, J. E. , Kumar, V. S. , Stark, Z. , Yeung, A. , Brown, N. J. , Stutterd, C. , Delatycki, M. B. , Sadedin, S. , Martyn, M. , Goranitis, I. , Thorne, N. , Gaff, C. L. , & White, S. M. (2019). A head‐to‐head evaluation of the diagnostic efficacy and costs of trio versus singleton exome sequencing analysis. European Journal of Human Genetics, 27, 1791–1799. 10.1038/s41431-019-0471-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tankard, R. M. , Bennett, M. F. , Degorski, P. , Delatycki, M. B. , Lockhart, P. J. , & Bahlo, M. (2018). Detecting expansions of tandem repeats in cohorts sequenced with short‐read sequencing data. American Journal of Human Genetics, 103(6), 858–873. 10.1016/j.ajhg.2018.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh, J. , Kandaswamy, K. K. , Werber, M. , Weiss, M. E. R. , Oprea, G. , Kishore, S. , Lohmann, K. , & Rolfs, A. (2019). Novel pathogenic variants and multiple molecular diagnoses in neurodevelopmental disorders. Journal of Neurodevelopmental Disorders, 11(1), 11 10.1186/s11689-019-9270-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger, A. M. , Guturu, H. , Bernstein, J. A. , & Bejerano, G. (2017). Systematic reanalysis of clinical exome data yields additional diagnoses: Implications for providers. Genetics in Medicine, 19(2), 209–214. 10.1038/gim.2016.88 [DOI] [PubMed] [Google Scholar]
- Wright, C. F. , McRae, J. F. , Clayton, S. , Gallone, G. , Aitken, S. , FitzGerald, T. W. , Jones, P. , Prigmore, E. , Rajan, D. , Lord, J. , Sifrim, A. , Kelsell, R. , Parker, M. J. , Barrett, J. C. , Hurles, M. E. , FitzPatrick, D. R. , & Firth, H. V. (2018). Making new genetic diagnoses with old data: Iterative reanalysis and reporting from genome‐wide data in 1,133 families with developmental disorders. Genetics in Medicine, 20(10), 1216–1223. 10.1038/gim.2017.246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, B. , Qiu, W. , Ji, X. , Liu, X. , Huang, Z. , Liu, H. , Fan, Y. , Xu, Y. , Liu, Y. U. , Yie, H. , Wei, W. , Yan, H. , Gong, Z. , Shen, L. , & Sun, Y. U. (2018). Marked yield of re‐evaluating phenotype and exome/target sequencing data in 33 individuals with intellectual disabilities. American Journal of Medical Genetics. Part A, 176(1), 107–115. 10.1002/ajmg.a.38542 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material